
A simple and direct atomic absorption spectrometry method for the direct determination of Hg in dried blood spots and dried urine spots prepared using various microsampling devices†
Flávio V.
Nakadi
a,
Raúl
Garde
a,
Márcia A. M. S.
da Veiga
b,
Julio
Cruces
c and
Martín
Resano
 *a
*a
aDepartment of Analytical Chemistry, University of Zaragoza, 12 Pedro Cerbuna St., Zaragoza, 50009, Spain. E-mail: mresano@unizar.es
bDepartamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, Universidade de São Paulo, 3900 Bandeirantes Av., Ribeirão Preto, 14040-901, Brazil
cComercial Rafer SL, Uncastillo 19, bajos, 50008 Zaragoza, Spain
First published on 27th November 2019
Abstract
The use of dried matrix spots, such as dried blood spots (DBSs) and dried urine spots (DUSs), is becoming more popular in clinical analyses, beyond their traditional use for screening newborns. Currently, there are new types of microsampling devices that have been designed to collect a low and known volume of biological fluids, regardless of the hematocrit level, thus making it feasible to develop fully quantitative methods for their analysis. In this study, three of the most promising microsampling devices (Capitainer B, Hemaxis DB 10 and Mitra) were evaluated aiming at the direct determination of Hg using a dedicated “analyzer” based on atomic absorption spectrometry. Whole blood and urine reference materials were used to evaluate the methods developed. This Hg analyzer possesses a gold trap to enhance the sensitivity and minimize matrix effects, enabling direct analysis of the solid DBS and DUS samples using calibration with Hg aqueous solutions, with an instrumental limit of detection (LOD) and quantification (LOQ) of 3.4 and 11 pg, respectively. LODs when using the three microsampling devices were similar, ranging between 2.5 and 3.2 μg L−1. It is also demonstrated that the simultaneous analysis of four Mitra derived DBSs in a quartz boat results in a decrease of the LOD to 0.32 μg L−1.
1. Introduction
The toxicity of Hg and its species is well established.1–3 Human exposure to Hg should be monitored, and blood and urine are the typical target samples.4 While urinary Hg is often used to measure occupational exposure, blood Hg is used to study the exposure of the general population.5 It is accepted that a person who does not eat fish usually shows values around 2 μg L−1 total Hg in blood,6 but this value may increase due to frequent fish ingestion. This was corroborated by a recent study in Spain, which reported that people who consumed fish less than once a week show a significantly lower Hg level in the blood (geometric mean = 3.71 μg L−1 for total Hg) than the ones who ate fish more than 5 times a week (geometric mean = 8.38 μg L−1 for total Hg).7 But it is important to stress that fish consumption is beneficial in many other aspects, so it should not be discouraged. That is why the best solution is to measure Hg exposure to learn from it and provide useful recommendations to high risk populations (women of reproductive age and children4).Many papers in the literature have aimed at Hg determination in clinical samples using voltammetry,8 inductively coupled plasma mass spectrometry (ICP-MS)9–12 or atomic absorption spectrometry (AAS).5,13 However, most of these methods required some degree of sample preparation. Moreover, they are based on the use of liquid blood obtained after venipuncture.
The development of alternative, much less invasive approaches for sampling biological fluids is a current trend in clinical analysis. For instance, the use of dried blood spots (DBSs) was already proposed by Guthrie and Susi back in 1963.14 This approach, based on the deposition of a few droplets of blood onto clinical filter paper and a subsequent drying step, is now employed in numerous newborn screening programs throughout the world.15
The advantages of DBSs are numerous.16,17 They are minimally invasive, which is essential for newborns for whom venipuncture is traumatic. The use of DBSs is also a very intriguing alternative when the goal is to carry out large-scale monitoring, as people may be less reluctant to participate when only a few droplets of blood are required.18,19 In fact, the subjects of the study or their relatives can actually prepare DBSs at home and send them to the lab after minimal training. Finally, DBSs are much more stable and easy to store than whole blood samples, which is relevant for hospitals and biobanks. These advantages justify the interest in the development of new analytical methods based on the use of DBSs15 and also the expansion of the approach to other biological fluids, most often urine, giving rise to dried matrix spots (DMSs) in general, and dried urine spots (DUSs) in particular.
Obviously, the use of DMSs also results in some disadvantages and challenges. One of the main ones is related to the difficulty in producing a DMS of an accurately known volume, while preserving the ease of use which enables patients to prepare it at home. For instance, the spreading of a blood sample onto a clinical filter paper depends on its particular physical properties (e.g., viscosity, which depends on the hematocrit level20), which vary for different people. This may not be a big issue when the aim is just to detect some metabolites associated with some disorder or when sampling can be done in the hospital using a pipette to deposit an exact volume onto the filter (providing the whole spot is analyzed, and not just a portion of it). But it is a problem when quantitative analysis is aimed at and ease of operation is to be preserved.
This situation has changed in the last lustrum, as several new types of devices have been introduced.21 The philosophy behind them is similar. In comparison with traditional approaches, where a relatively large amount of blood is deposited (50–100 μL), they all require only between 10 and 30 μL of blood, and they are designed to produce a DBS of a known and reproducible volume regardless of the hematocrit level. And they have been designed in a way that anybody can puncture himself/herself with a mini-lancet and produce the DBS.
For this study, three of these devices have been selected. The first one performs what has been termed volumetric absorptive microsampling (VAMS), and it is the most studied device of the trio. After its introduction in 2014,22 this device has been tested for several applications, as described in two recent reviews.23,24 However, to the best of the authors' knowledge, the use of VAMS in the context of elemental analysis has been reported in four articles, but always after extracting the analytes and never targeting Hg.25–28 A device based on this VAMS concept is commercially available with the name Mitra®29 and will be tested in this work.
The second device was introduced more recently, in 2016,30,31 and it is a microfluidic-based volumetric sampling system commercially termed Hemaxis DB 10.32 There is still little information on its performance in different applications and none on elemental analysis that we could find. Finally, the third device, Capitainer B, is another type of microfluidic device33–35 that has been produced very recently based on the concepts described by Lenk et al.36 It also remains untested for elemental analysis, as far as these authors know. Details on the principle of operation of these devices will be provided in Section 2.3. What is relevant to emphasize at this point is that the literature on elemental analysis of DMSs is still scarce and is even more scarce when focusing on methods for the direct analysis of such DMSs.17 In fact, no paper has reported the direct determination of Hg in DMSs of any kind.
This fact is surprising, since DMSs are liquids that are transformed into complex solid samples, and their direct analysis should provide higher sample throughput and fewer contamination problems, besides avoiding poor extraction issues.37,38
This work aims at the development of a simple and fast method that maximizes the advantages of DMSs for Hg determination, that simplifies calibration and minimizes matrix effects and that is robust enough to be used by technicians in clinical labs. For this purpose, a dedicated instrument for Hg monitoring based on atomic absorption spectrometry will be tested.
2. Experimental
2.1. Instrumentation
All the measurements were carried out using a Hg “analyzer” Hydra IIC (Teledyne Leeman Labs, Hudson, USA). A solid or liquid sample is introduced in a nickel or quartz boat into a pyrolysis (combustion) furnace. Two heating steps are usually programmed, drying and pyrolysis, which eliminate the solvent and the organic matter, respectively, with an O2 (99.9999%) flux. All the gases generated from the pyrolysis pass through a heated catalyst furnace to remove halogens, nitrogen oxides and sulfur oxides. The remaining species are carried through a Nafion® dryer for water vapor removal, and, finally, to a gold trap that amalgamates the analyte (as Hg0). This trap is heated to release the Hg0 in its gaseous form and the O2 flow transports it through an external dryer tube filled with Mg(ClO4)2, for further water vapor removal, and ultimately to the spectrometer based on AAS. Hg is measured at its main atomic line, 253.7 nm. The spectrometer possesses two optical cells, a high-sensitivity cell (total optical length = 25.4 cm) and a low-sensitivity cell (optical length = 2.54 cm). The instrumental parameters finally selected are shown in Table 1.| Wavelength/nm | 253.65 |
| O2 flow rate/mL min−1 | 350 |
| Detection time/s | 100 |
| Temperature program | ||
|---|---|---|
| Step | Temperature/°C | Time/s |
| Drying | 150 | 120 |
| Pyrolysis | 600 | 120 |
| Catalyst | 600 | 60 |
| Amalgamator | 600 | 30 |
2.2. Reagents, standards and samples
All the reagents were of analytical grade or higher. An Hg aqueous standard 1000 mg L−1 (Merck, Darmstadt, Germany) was used for preparing the calibration curve. Hydrochloric acid 37% m/m (Merck) was diluted to 1% v/v and used to dilute the Hg standard. Whole blood L-1 (lot 1406263), L-2 (lot 1406264) and L-3 (lot 1509408), and urine L-2 (lot 1403081) reference materials (RM, Sero, Billingstad, Norway) were used as samples for producing DBSs and DUSs, respectively, prior to the subsequent analysis. The volume of the Mitra device for producing DUSs was evaluated using the urine level II control lyophilized ClinChek® (lot 923) reference material.The Hydra IIC instrument was also evaluated using different solid certified RMs (CRMs): trace elements in calcareous loam soil (BCR 141R), San Joaquin soil (NIST 2709), trace elements in spinach leaves (NIST 1570a), tomato leaves (NIST 1573a), human hair (NCS ZC 81002b), trace elements in bituminous coal (NIST 1632d), and elements in polyethylene (ERM-EC680).
2.3. Sample preparation
DBSs and DUSs were produced using three microsampling devices: Capitainer B (Capitainer AB, Stockholm, Sweden), Hemaxis™ DB 10 (DBS System SA, Gland, Switzerland) and Mitra® (Neoteryx, Torrance, USA). Blood and urine RMs were reconstituted according to the instructions provided by the manufacturer.There are three Mitra devices available for purchase. They are all similar but their sizes and consequently the volume of biological fluid absorbed vary: 10, 20 and 30 μL. Mitra devices of approximately 20 μL were evaluated in this study. An aliquot of each sample, ca. 500 μL, was transferred to a 1.5 mL Eppendorf tube and the tip of the device was placed in contact with the fluid surface for 5 s. Fig. 1 shows Mitra devices before (left) and after (right) the blood collection procedure.
 | ||
| Fig. 1 Mitra device without blood (left) and after collection and drying of approximately 23.4 μL of the whole blood L-2 RM sample (right). | ||
Every Hemaxis DB 10 possesses four microfluidic channels (Fig. 2a) that are filled with blood (Fig. 2b) by capillarity. In this case, a droplet of blood was obtained at the tip of a micropipette, which was placed in contact with the inlet and aspirated into the system to simulate sampling from a fingertip. Then, to produce the DBSs it is only necessary to close the system and a reproducible amount of blood (10 μL) is deposited (see Fig. 2c) onto the standardized clinical filter paper, Whatman 903 in this case.30
 | ||
| Fig. 2 Hemaxis DB 10 device (a) before adding blood (whole blood L-2 RM); (b) with the capillaries filled with blood (approximately 10.0 μL); and (c) after depositing the blood samples onto the filter paper, thus producing the DBSs. | ||
Capitainer B also offers four sites for blood deposition: the red dashed areas (Fig. 3a). When blood is added to each site (e.g., after a finger prick for real patients or via a micropipette in this work, 30 μL), the microchannel above is filled with a constant blood volume (13.5 μL). After this, the thin film at the inlet dissolves and any extra blood is absorbed by a paper matrix. Then, the thin film at the outlet also dissolves and each portion of 13.5 μL of blood is transported by capillarity to the corresponding pre-perforated filter discs located on the other side of the device (see Fig. 3b and c). Such discs, which are glued to the device, can be detached using tweezers and analyzed (Fig. 3d). Such discs also make use of standardized clinical filter paper (Ahlstrom 222 filter paper).34,35
 | ||
| Fig. 3 Capitainer B device (a) before adding blood (obverse of the device); (b) filter paper discs without blood (reverse of the device); (c) DBSs formed after transferring the blood samples (whole blood L-2 RM); and (d) one DBS removed from the device. | ||
All the DBSs were left to dry at room temperature for at least 4 h before analysis.
2.4. Sample analysis
The CRMs whose analyses will be discussed in Section 3.2 were weighed using an external balance (Sartorius, Göttingen, Germany) inside Ni boats and then introduced into the Hydra IIC instrument and analyzed using the conditions shown in Table 1. The humidity content was taken into account for calculating the concentration of such samples, by introducing a sample portion in an oven at 95 °C for 4 hours. The amount of CRM used for analysis ranged between 1.0 and 16.5 mg, depending on the Hg content.Analysis of blood and urine reference samples was carried out in the same way, after producing DBSs and DUSs (see Section 2.3), without the need for weighing since the sample volume of every device is known in advance. For analysis of the blood sample with the lowest Hg content (whole blood L-1), four Mitra derived devices instead of just one were introduced into a quartz boat and analyzed together, thus improving the limit of detection (LOD).
For comparison, blood RMs were also analyzed directly (as liquids). 100 μL of each RM sample was measured per replicate, except for whole blood L-1, for which 200 μL were used.
In all cases, calibration was carried out using Hg aqueous standards introduced inside a quartz boat and following the program shown in Table 1.
3. Results and discussion
3.1. Hg analyzer parameters
Details on the operation of the Hg “analyzer” are provided in Section 2.1. It is a device that enables direct analysis of the sample, which is subjected to combustion in an O2 atmosphere, releasing Hg and other gases, most of which are removed in the catalyst furnace, while H2O vapors are removed by a membrane and an external dryer tube filled with Mg(ClO4)2 to avoid spectral interferences. Hg is preconcentrated in a gold trap and released after heating, to be measured by AAS at 253.7 nm.Four different temperatures can be modified: drying and pyrolysis in the combustion furnace, which operates between 50 °C and 1000 °C, the temperature of the catalyst furnace and the temperature of the amalgam furnace, and optimal values were investigated.
In practice, a temperature of 600 °C for the catalyst furnace was used, as this is recommended by the manufacturer for best operation. Concerning the amalgamator, it is necessary to use a temperature that is high enough to release Hg at a good rate in order to produce a well-defined peak profile, but that is not too high, in order to prevent any significant alteration of the characteristics (e.g., particle size) of the gold deposited in this furnace. Again, a value of 600 °C was found to be suitable for this step.
Since the idea was to construct the calibration curve with Hg aqueous standards while most of the samples were solids, the drying and pyrolysis steps were optimized for both conditions. The drying step had practically no influence on the solid samples; thus it was studied for the Hg aqueous standard. The usual volumes pipetted into the boats varied between 50 and 200 μL, and it was observed that a temperature of 150 °C for 120 s was sufficient to completely dry the solutions and generate Hg vapor. The use of lower temperatures, such as 100 °C, would require the use of longer times to dry such sample volumes. Similarly, the pyrolysis step had to remain at 600 °C for at least 120 s to guarantee the complete elimination of the solid matrixes. The effect of temperature was observed mainly for biological samples, which decomposed entirely (no ash residues left) at 600 °C, in addition to showing no significant memory effects. The use of lower temperatures results in the analytical signal increasing for each replicate measured.
Geological samples, of course, will always leave residues, which were easily cleaned with a swab. No evident benefit could be derived from the use of higher pyrolysis temperatures.
Although Hg may show different release mechanisms from each matrix, the use of gold trap preconcentration certainly helps in mitigating such effects, leading to similar Hg spectra regardless of the matrix. For instance, Fig. 4 compares the analytical signal of a Mitra derived DBS containing the RM whole blood L-2 (Hg concentration = 17.0 ± 3.4 μg L−1) with the one obtained using a Hg aqueous solution. Moreover, it can be stated that solid samples were always analyzed using nickel boats and the aqueous standards with a quartz boat, because the latter are always in an acidic (HCl) medium that reacts with nickel, degrading it. The sensitivity does not depend on the type of boat used, although quartz boats are usually purer, which positively influences the LODs, as will be discussed in Section 3.4.
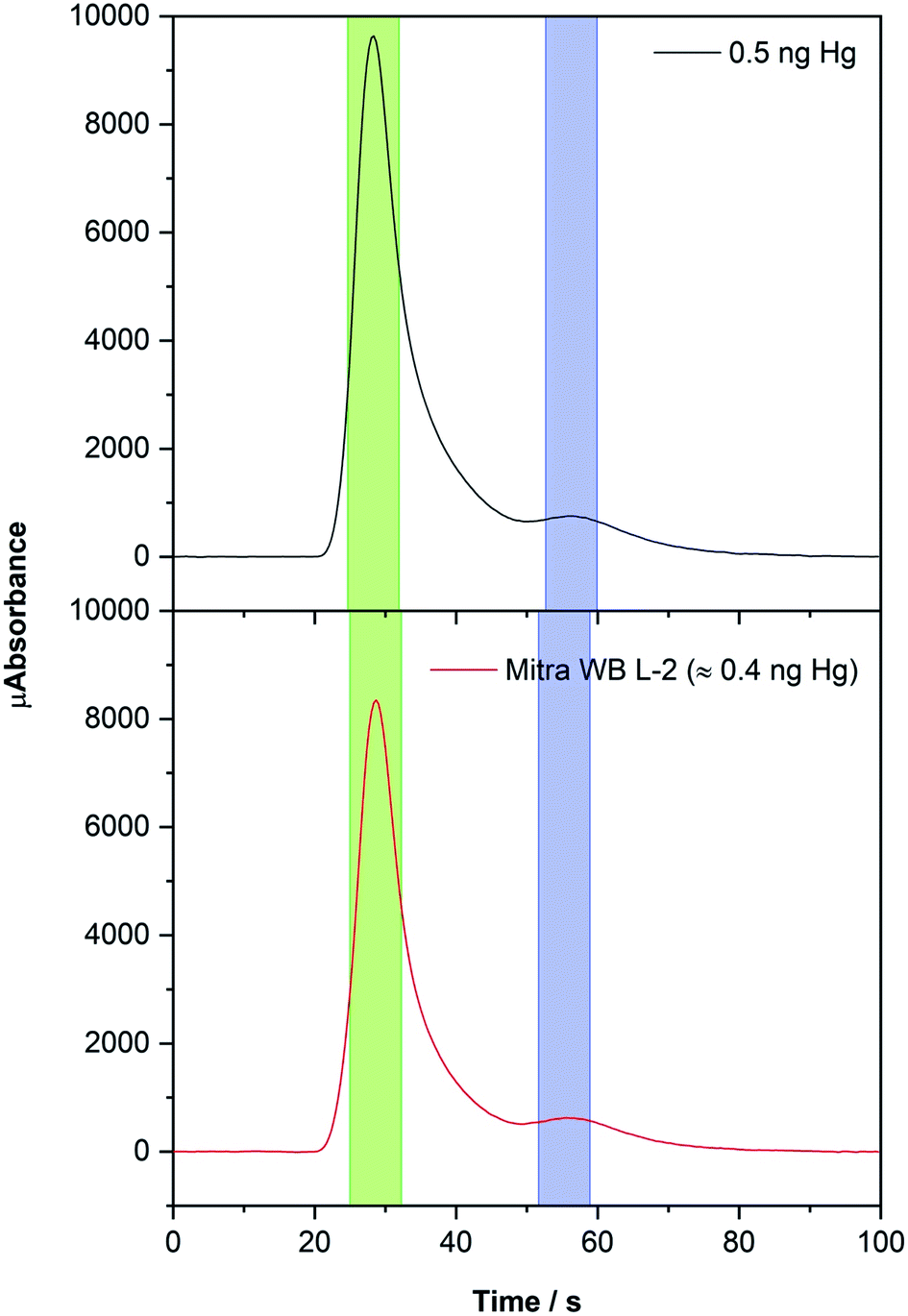 | ||
| Fig. 4 Hg time-resolved absorption spectra obtained from 100 μL of a 5 μg L−1 Hg standard solution (black line) using a quartz boat and from a Mitra derived DBS (red line, 23.4 μL of whole blood L-2 RM) using a nickel boat. The conditions listed in Table 1 were used. The green areas highlight the data values processed for higher sensitivity (longer cell) and the blue areas for lower sensitivity (shorter cell). | ||
The instrument is equipped with two different absorption cells with different lengths (one being 10 times longer) in tandem to increase the working concentration range, which thus leads to two peaks for each spectrum: for the longer cell, which obviously offers higher sensitivity, the peak maximum appears around 30 s, and for the shorter cell it appears at approximately 55 s (see Fig. 4). Each peak value is processed automatically using the instrument software (named Envoy) which sets the baseline and uses the absorbance values obtained in the vicinity of the peak maxima (7 seconds in total for each peak).
3.2. Evaluation of direct solid sample analysis
In order to evaluate the instrument response for the direct analysis of solid samples in general, several CRMs with different matrixes and different Hg levels were analyzed.The materials selected were: three geological (BCR 141R, NIST 2709 and NIST 1632d), three biological (NIST 1570a, NIST 1573a and NCS ZC 81002b), and one polymer (ERM-EC680) CRM (see Section 2.2 for more details). All the solid samples were directly weighed into the Ni boats and analyzed with the temperature program described in Table 1, while calibration was performed using Hg solutions.
It was observed that the response of the instrument in terms of sensitivity was very stable. In fact, a full calibration curve was constructed only once, using a blank and 7 standards with total Hg amounts between 0.05 and 10 ng. For analysis during the next 9 months, only one standard was measured besides the blank, and the sensitivity was adapted using this value. The RSD of the signal obtained for a 5 ng Hg standard (n = 8) during this period of time was 2.3%.
Fig. S1 (ESI†) shows the results obtained, highlighting the good agreement observed between the certified and experimental Hg concentrations, by means of a linear regression. Weighted least squares was used (Origin Pro 2019b, weight mode: instrumental) to avoid a strong influence of the sample with the higher Hg content, as well as to appropriately take into account all the sources of uncertainty, including the confidence intervals (C.I.) of the certified materials. With this approach, those values with lower uncertainty will show a higher weight in the fit.
The regression values show a slope near unity (95% C.I. 0.93093 ± 0.15568) and an intercept close to the origin (95% C.I. 0.0088 ± 0.0098), confirming that the method was suitable for direct solid sample analysis. The relative standard deviation (RSD) of the 7 samples varied between 2.1 and 8.7%, for loam soil and bituminous coal, respectively.
Sample ERM-EC680 contains significantly more Hg than the others and was analyzed using the values from the shorter and less sensitive absorption cell, to prove that this approach works as well. The value obtained was a bit low (experimental: 21.7 ± 1.1 mg kg−1, certified: 25.3 ± 1.0 mg kg−1), even though this is not so relevant for analysis of clinical samples, where such Hg values are not expected.
The agreement for the samples with lower contents is noteworthy (see Fig. S1†) and only in one case (NIST 1573a, experimental: 0.0460 ± 0.0014 mg kg−1, certified: 0.034 ± 0.04 mg kg−1), the confidence intervals did not overlap. It should be mentioned that some differences due to sample heterogeneity may occur, simply because the mass analyzed by means of the method proposed in this work is lower (see Section 2.4) than the minimum amount recommended by the CRM manufacturer and used to estimate the uncertainty of the CRMs (e.g., 150 mg for NIST 1573a and 500 mg for ERM-EC680).
Overall, it can be concluded that it is feasible to directly analyze all types of materials with a high Hg concentration range by simply calibrating with aqueous standards under the conditions used in this work, thus proving the lack of significant matrix effects.
3.3. Study of DBSs for Hg determination in blood
Each DBS device comes with the potential to absorb a nominal volume of blood, and such value is provided by the manufacturer as follows: Capitainer B: 13.5 ± 0.7 μL; Hemaxis DB 10: 10.0 ± 0.5 μL; and Mitra: 22.4 μL for real blood samples and 23.4 ± 1.0 μL for aqueous samples. The Capitainer B value is difficult to check (if the disc is detached for weighing it, then it may not absorb well when inserted back). Thus, measurements were carried out to test the values for the other devices.The Mitra volume was calculated by weighing recipients (n = 20) containing a blood RM before and after inserting the tip of the Mitra (thus leading to blood absorption). Assuming a 1.0 g mL−1 density, a volume of 24.1 ± 0.7 μL was obtained, where the uncertainty is expressed as the standard deviation. The same method was used for urine because, as will be explained in Section 3.5, this device will also be tested for such a sample. A value of 23.4 ± 0.9 μL was measured for urine, also assuming a 1.0 g mL−1 density.
Finally, five Hemaxis DB 10 devices, which produced 20 DBSs in total, were also evaluated by weighing them before and after absorbing the samples, and the volume of each DBS was estimated to be 10.0 ± 0.2 μL.
It is clear that the differences with the values provided by the manufacturers are minimal, and always within the uncertainty of the measurements. Thus, it seems that the manufacturers' values can be used in any lab without the need to do extra measurements and calculations, and we decided to use them herein as well for all further work: 13.5 μL for Capitainer B, 10.0 for Hemaxis DB and 23.4 μL for Mitra, as blood and urine reconstituted RMs are very similar to water in density.
Once the DBS volume values were established, the reproducibility of the three devices was evaluated. 10 DBSs for each device were produced as described in Section 2.3 and measured using the same conditions as for the previous solid samples. Since each device has a different volume, the instrument analytical signal was normalized (see Fig. 5).
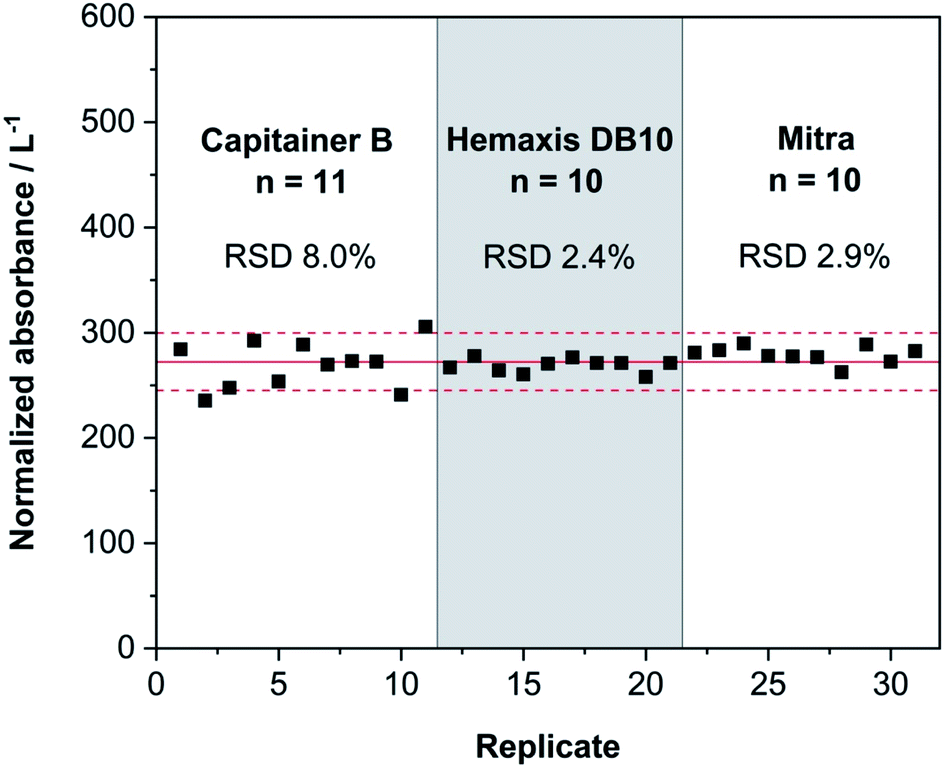 | ||
| Fig. 5 Results of consecutive measurements of DBSs produced with the whole blood L-2 RM using three different microsampling devices: Capitainer B, Hemaxis DB 10 and Mitra. The values were normalized according to the volume absorbed by each device, so they can be readily compared. The red line represents the normalized absorbance mean value from all the replicates and the dashed red lines represent 10% deviation from this mean value. | ||
Fig. 5 shows that the reproducibility of the three devices tested to produce DBSs is high. Almost all replicates lie within 10% of the mean value of normalized absorbance. Hemaxis DB 10 and Mitra imprecisions are below 3%, which is remarkable. As for Capitainer B (see Fig. 3), it shows higher data dispersion. Some DBS samples tore while they were collected due to the adhesive that attaches the DBSs on the system, which could explain the higher bias. Nonetheless, the values for Capitainer B are perfectly acceptable for clinical analyses. Overall, all the devices produce sufficient reproducibility for Hg determination at the trace level.
In any case, it is important to state that the method proposed analyzes whole DBS samples, such that no effects associated with the inhomogeneous distribution of the analyte within the DBS occur, which is not always the case for other solid sampling approaches.39–41
3.4. Figures of merit
After optimizing and evaluating the DBS analysis, the figures of merit were determined, and the results can be found in Table 2. The instrumental limits of detection (LOD, 3σ10 blanks/slope) and quantification (LOQ, 10σ10 blanks/slope) were calculated by analyzing empty Ni boats. For each DBS device, both the LOD and LOQ were calculated measuring the respective DBS blanks (n = 10) and considering their volumes.| Parameter | Values |
|---|---|
| a Four Mitra derived DBSs were analyzed at the same time to evaluate the LOD and LOQ. | |
| Calibration curve equation | μA = 463 + 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 880mHg (ng) 880mHg (ng) |
| Linear working range/ng | 0.05–10 |
| R 2 | 0.99999 |
| LOD/pg | 3.4 |
| LOQ/pg | 11 |
| Parameter | Microsampling devices | ||||
|---|---|---|---|---|---|
| Capitainer B | Hemaxis HB 10 | Mitra | Mitraquartz | Mitra4quartza | |
| LOD/μg L−1 | 3.2 | 2.6 | 2.5 | 1.2 | 0.32 |
| LOQ/μg L−1 | 11 | 8.7 | 8.3 | 4.1 | 1.1 |
The working linear range indicated in Table 2 is based on the use of only the longer absorption cell, appropriate for a low-level Hg concentration. It could be further expanded, between 1.0 and 100 ng with an R2 = 0.9976, by just selecting the signal provided by the shorter cell (see Fig. 4). Thus, although it should not be needed for analysis of clinical samples, the method would be capable of measuring a range of more than 3 orders of magnitude using atomic absorption.
In terms of absolute LOD values, a few picograms can be detected, showing remarkable sensitivity. In terms of relative values (concentration), the values obtained are somewhat less impressive, as values lower than 1 μg L−1 can be obtained using other approaches,9,10,13 although comparable values have been reported using direct analysis of blood via high-resolution continuum source graphite furnace AAS.5 This can all be explained considering the very small volume of the samples (lower than 25 μL) used herein for producing DBSs, which obviously results in higher relative LOD values. Nonetheless, the LOQs obtained for all the DBS devices are sufficiently low to detect significant exposure to Hg.
If, on the other hand, the goal were to determine basal levels (the average concentration worldwide is around 2 μg L−1 for people who do not eat fish6), these values would not be low enough. A good sample for checking this aspect is Seronorm whole blood L-1 (1.48 ± 0.30 μg L−1), so an alternative approach was investigated for its analysis. The Ni boats used for analysis typically show higher blank signals compared to quartz boats. Therefore, the possibility of introducing several DBSs into a quartz boat (Mitra4quartz) was considered. In this regard, the Mitra device was selected as it already contains more blood volume than the others and the simultaneous analysis of 4 devices should suffice.
By means of this approach, a significant improvement in terms of the LOD (approx. 10 times) was achieved, thus enabling the determination of Hg at low μg L−1 levels in DBSs. However, it should be emphasized that 4 Mitra devices are used for each replicate (approximately 94 μL), and their use would be justified only if an accurate determination at basal levels is needed.
3.5. Determination of Hg in blood and urine using DBSs
After optimizing the methods, three whole blood RM samples were analyzed with the DBS devices, L-1, L-2 and L-3, whose concentrations are listed in Table 3. As discussed in Section 3.4, determination of whole blood L-1 was only possible by simultaneously measuring the signal derived from the release of mercury from 4 replicate Mitra-based DBS samples introduced in one quartz boat. The whole blood RMs were also analyzed by pipetting them directly into the Ni boats, and their results are shown in Table 3.| Sample | Hg concentration/μg L−1 | ||||
|---|---|---|---|---|---|
| Reference | Directa | Mitra | Capitainer B | Hemaxis HB 10 | |
| a Direct analysis of the liquid samples. b The concentration was calculated analyzing 4 Mitra derived DBSs at the same time, corresponding to a total blood volume of approximately 94 μL. | |||||
| Whole blood L-1 | 1.48 ± 0.30 | 1.52 ± 0.77 | 1.61 ± 0.38b | — | — |
| Whole blood L-2 | 17.0 ± 3.4 | 17.2 ± 0.5 | 17.4 ± 2.3 | 18.8 ± 3.7 | 14.8 ± 1.1 |
| Whole blood L-3 | 25.8 ± 5.2 | 27.8 ± 6.0 | 24.6 ± 8.0 | 27.6 ± 3.0 | 25.5 ± 0.9 |
| Urine L-2 | 44.0 ± 8.9 | 47.0 ± 1.0 | 45.3 ± 3.0 | — | — |
As can be seen, a good agreement with the reference values was obtained in all the cases, demonstrating the potential of the technique for the direct determination of Hg in DBSs. Moreover, the RSDs obtained varied between 1.6 and 13%, which is suitable for a technique based on of fast sampling and direct analysis.
Finally, one advantage of the Mitra swab-shape is that it could collect other types of liquid samples just by immersion of its tip into the sample. As discussed in the introduction, urine is a fluid usually evaluated for assessing occupational exposure to inorganic Hg. Therefore, a urine RM sample L-2 was tested using the Mitra device and the results are shown in Table 3. An excellent agreement with the reference value was observed, with very good precision (RSD of 2.7%). This result evidenced the potential of the Mitra device as an alternative for analyzing small volumes of liquid samples as a solid sample.
It can finally be mentioned that Mitra derived samples containing whole blood L-2 were analyzed 7 months after they were prepared. They were stored in a closed lab bench drawer, protected from light, at room temperature. The result obtained was 19.5 ± 0.4 μg Hg L−1, which overlaps with the reference value, showing that the analyte remains stable in this type of sample, at least during this period of time. Therefore, the Mitra device is a suitable analytical tool to store constant amounts of liquid samples (as DMSs) for further analysis.
4. Conclusions
Current DBS microsampling devices show a very promising performance for Hg determination in whole blood, facilitating easy home collection. Using a technique based on direct solid sampling, combustion, Au amalgamation and AAS monitoring, fast analysis of DBSs is enabled, and accurate results for Hg can be obtained via external calibration with aqueous standards. The LODs can be decreased via summation of the response from the simultaneous combustion of several DBSs, such that low μg L−1 levels can be determined. Finally, the Mitra device also demonstrated its potential for urine analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the European Regional Development Fund for financial support through Interreg POCTEFA 176/16/DBS. The Aragon Government (Construyendo Europa desde Aragón) and project PGC2018-093753-B-I00 (MCIU/AEI//FEDER, UE) are also acknowledged. Márcia A. M. S. da Veiga received a grant from the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP grant#2017/20707-2). Raúl Garde acknowledges his predoctoral grant BES-2016-078971 (associated with project CTQ2015-64684-P) from the Ministerio español de Ciencia, Innovación y Universidades.References
- G. F. Nordberg, B. A. Fowler and M. Nordberg, Handbook on the toxicology of metals, Academic Press, London, 4th edn, 2015 Search PubMed.
- World Health Organization, Exposure to Hg: a major public health concern, WHO Document Production Services, Geneva, 2007, https://www.who.int/ipcs/features/Hg.pdf Search PubMed.
- World Health Organization, Guidance for identifying populations at risk from Hg exposure, UNEP Chemicals, Geneva, 2008, https://www.who.int/foodsafety/publications/chem/Hgexposure.pdf Search PubMed.
- W. McKelvey, B. Alex, C. Chernov, P. Hore, C. D. Palmer, A. J. Steuerwald, P. J. Parsons and S. E. Perlman, J. Urban Health, 2018, 95, 813–825 CrossRef PubMed.
- M. Aramendía, A. Guarda, D. Leite and M. Resano, J. Anal. At. Spectrom., 2017, 32, 2352–2359 RSC.
- G. Nordberg, D. Brune, L. Gerhardsson, P. Grandjean, O. Vesterberg and P. O. Wester, Sci. Total Environ., 1992, 120, 17–21 CrossRef CAS PubMed.
- A. Castaño, S. Pedraza-Díaz, A. I. Cañas, B. Pérez-Gómez, J. J. Ramos, M. Bartolomé, P. Pärt, E. P. Soto, M. Motas, C. Navarro, E. Calvo and M. Esteban, Sci. Total Environ., 2019, 670, 262–270 CrossRef PubMed.
- R. Y. A. Hassan, M. S. Kamel, H. N. A. Hassan and E. Khaled, J. Electroanal. Chem., 2015, 759, 101–106 CrossRef CAS.
- D. E. Nixon, M. F. Burritt and T. P. Moyer, Spectrochim. Acta, Part B, 1999, 54, 1141–1153 CrossRef.
- B. M. W. Fong, T. S. Siu, J. S. K. Lee and S. Tam, J. Anal. Toxicol., 2007, 31, 281–287 CrossRef CAS PubMed.
- S. J. Christopher, S. E. Long, M. S. Rearick and J. D. Fassett, Anal. Chem., 2001, 73, 2190–2199 CrossRef CAS PubMed.
- S. Q. Abad, P. Rodríguez-González, W. C. Davis and J. I. G. Aloso, Anal. Chem., 2017, 89, 6731–6739 CrossRef PubMed.
- R. Akramipour, M. R. Golpayegani, S. Gheini and N. Fattahi, Talanta, 2018, 186, 17–23 CrossRef CAS PubMed.
- R. Guthrie and A. Susi, Pediatrics, 1963, 32, 338–343 CAS.
- P. A. Demirev, Anal. Chem., 2013, 85, 779–789 CrossRef CAS PubMed.
- G. Nys, M. G. M. Kok, A.-C. Servais and M. Fillet, Trends Anal. Chem., 2017, 97, 326–332 CrossRef CAS.
- M. Resano, M. A. Belarra, E. García-Ruiz, M. Aramendía and L. Rello, Trends Anal. Chem., 2018, 99, 75–87 CrossRef CAS.
- T. W. McDade, S. Williams and J. J. Snodgrass, Demography, 2007, 44, 899–925 CrossRef PubMed.
- P. Bhatti, D. Kampa, B. H. Alexander, C. McClure, D. Ringer, M. M. Doody and A. J. Sigurdson, BMC Med. Res. Methodol., 2009, 9(76), 1–6 Search PubMed.
- P. M. M. De Kesel, S. Capiau, W. E. Lambert and C. P. Stove, Bioanalysis, 2014, 6, 1871–1874 CrossRef CAS PubMed.
- S. Velghe, L. Delahaye and C. P. Stove, J. Pharm. Biomed. Anal., 2019, 163, 188–196 CrossRef CAS PubMed.
- P. Denniff and N. Spooner, Anal. Chem., 2014, 86, 8489–8495 CrossRef CAS PubMed.
- M. Protti, R. Mandrioli and L. Mercolini, Anal. Chim. Acta, 2019, 1046, 32–47 CrossRef CAS PubMed.
- M. G. M. Kok and M. Fillet, J. Pharm. Biomed. Anal., 2018, 147, 288–296 CrossRef CAS PubMed.
- E. Bolea-Fernandez, K. Phan, L. Balcaen, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2016, 941, 1–9 CrossRef CAS PubMed.
- A. Cañabate, E. García-Ruiz, M. Resano and J. L. Todolí, J. Anal. At. Spectrom., 2017, 32, 78–87 RSC.
- Y. Anoshkina, M. Costas-Rodríguez and F. Vanhaecke, J. Anal. At. Spectrom., 2017, 32, 314–321 RSC.
- S. Capiau, E. Bolea-Fernandez, L. Balcaen, C. Van Der Straeten, A. G. Verstraete, F. Vanhaecke and C. P. Stove, Talanta, 2020, 208, 120055 CrossRef.
- Retrieved from https://www.neoteryx.com, last accessed 2019, September 26.
- L. A. Leuthold, O. Heudi, J. Déglon, M. Raccuglia, M. Augsburger, F. Picard, O. Kretz and A. Thomas, Anal. Chem., 2015, 87, 2068–2071 CrossRef CAS PubMed.
- R. Verplaetse and J. Henion, Anal. Chem., 2016, 88, 6789–6796 CrossRef CAS PubMed.
- Retrieved from http://hemaxis.com, last accessed 2019, September 26.
- Retrieved from https://capitainer.se/specification/, last accessed 2019, September 26.
- S. Velghe and C. P. Stove, Anal. Chem., 2018, 90, 12893–12899 CrossRef CAS PubMed.
- N. Spooner, A. Olatunji and K. Webbley, J. Pharm. Biomed. Anal., 2018, 149, 419–424 CrossRef CAS PubMed.
- G. Lenk, S. Sandkvist, A. Pohanka, G. Stemme, O. Beck and N. Roxhed, Bioanalysis, 2015, 7, 2085–2094 CrossRef CAS PubMed.
- E. Marguí, I. Queralt, E. García-Ruiz, E. García-González, L. Rello and M. Resano, Spectrochim. Acta, Part B, 2018, 139, 13–19 CrossRef.
- M. Aramendía, L. Rello, F. Vanhaecke and M. Resano, Anal. Chem., 2012, 84, 8682–8690 CrossRef PubMed.
- M. Resano, L. Rello, E. García-Ruiz and M. A. Belarra, J. Anal. At. Spectrom., 2007, 22, 1250–1259 RSC.
- L. Rello, A. C. Lapeña, M. Aramendía, M. A. Belarra and M. Resano, Spectrochim. Acta, Part B, 2013, 81, 11–19 CrossRef CAS.
- M. Aramendía, L. Rello, S. Bérail, A. Donnard, C. Pécheyran and M. Resano, J. Anal. At. Spectrom., 2015, 30, 296–309 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ja00348g |
| This journal is © The Royal Society of Chemistry 2020 |