
A simple single-stage extraction method for Mo separation from geological samples for isotopic analysis by MC-ICP-MS†
Lanping
Feng
a,
Lian
Zhou
 *a,
Wenfeng
Hu
a,
Wen
Zhang
a,
Baichan
Li
a,
Yongsheng
Liu
a,
Zhaochu
Hu
a and
Lu
Yang
b
*a,
Wenfeng
Hu
a,
Wen
Zhang
a,
Baichan
Li
a,
Yongsheng
Liu
a,
Zhaochu
Hu
a and
Lu
Yang
b
aState Key Laboratory of Geological Processes and Mineral Resources, School of Earth Sciences, China University of Geosciences, Wuhan 430074, China. E-mail: zhcug@163.com; Fax: +86-27-67885096; Tel: +86-27-67885096
bNational Research Council Canada, 1200 Montreal Rd., Ottawa, Ontario K1A 0R6, Canada
First published on 30th September 2019
Abstract
A new, simple and efficient separation method to isolate Mo from complex geological sample matrices for high precision Mo isotope ratio measurements is described. The proposed separation method benefits from the high selectivity and high capacity of the TRU resin, which affords straightforward separation of Mo from complex sample matrices and achieves high yield (∼100%), efficient separation and low blank with the use of only a single mineral acid, HCl (1.5 M for loading and 0.5 M for eluting), and small volumes. As opposed to the commonly used separation protocols, neither redox reagents (e.g., H2O2 and ascorbic acid) nor highly toxic HF is required for the proposed separation protocol, making this separation method straightforward and more environment friendly to operate. Mo isotope ratios were measured by multi-collector inductively coupled plasma mass-spectrometry (MC-ICP-MS) using the 97Mo–100Mo double spike in combination with standard-sample bracketing for mass bias correction. The results obtained for igneous rocks, manganese nodules, shales and seawater samples are in good agreement with previously reported values, confirming that the proposed separation protocol is suitable for complex sample matrices and will further promote its geological applications.
1. Introduction
Molybdenum (Mo) isotopes have been applied to track the evolution of geological processes, as Mo is a redox-sensitive trace metal.1–4 Due to advances in mass spectrometry techniques, mass-dependent Mo isotopic variations have been investigated in various geological samples, demonstrating significant fractionation associated with numerous geochemical processes, such as adsorption,5–7 mineral dissolution and precipitation.8,9 As a result, up to 2.17‰ per amu variation of the Mo isotope has been reported in different reservoirs, making the Mo isotope a promising tool for geological and environmental applications.The Mo isotopic composition is commonly analyzed by MC-ICP-MS10–12 which may be susceptible to matrix effects and polyatomic interferences; therefore purification of Mo from the sample matrix is required prior to its determination to achieve accurate results. Furthermore, for the accurate determination of Mo isotope ratios, the isotope fractionation due to non-quantitative recovery of Mo during column separation should be avoided.10,13 Although the addition of a double spike before column separation could correct this fractionation to some extent, the matrix effects are still present. Due to the complexity of the sample matrix and the relatively low Mo concentrations in geological samples, a high degree of purification is required for the high precision determination of Mo isotopic composition by MC-ICP-MS.
Numerous protocols have been successfully developed for the separation and purification of Mo from geological materials. The majority of the reported separation methods used ion exchange chromatography to purify Mo. For example, two-column ion-exchange separation has been widely used for Mo separation from geological samples, wherein an anion-exchange column was used to remove Zr and most other matrix elements, followed by a cation-exchange column to mainly separate Mo from Fe.1,10,14–16 In addition, two-step anion exchange column chromatography using an Eichrom AG1X8 (200–400 mesh) has also been successfully used for Mo (and W) separation.17,18 Although these two-column separation protocols may sustain a large sample load tolerance and achieve high purity of the Mo fraction, they are time consuming with tedious elution steps. Therefore, a single-column separation method based on anion resins was developed to shorten the separation procedure.19–21 Recently, Willbold et al.20 accomplished the purification of Mo from low Mo content samples on a single anion-exchange column by using redox agents (ascorbic acid and hydrogen peroxide) in elution. Although good separation can be obtained, these conventional ion-exchange techniques require a variety of eluents and relatively large elution volumes.
Attributed to its high selectivity for target elements, specific extraction chromatography is increasingly used for performing high precision isotope ratio measurements, such as TRU resin for Sn isotope analysis22 and DGA resin for Sr, Nd, Pb, Hf and Ca isotope analysis.23–25 Benefitting from its high selectivity, specific extraction chromatography is usually accomplished by single-column separation with use of a relatively small amount of resin and small volume of the eluent. A simple and efficient extraction method for Mo separation from geological samples has been developed, which was based on the BPHA (N-benzoyl-N-phenylhydroxylamine) extraction material.26,27 This separation protocol shows advantages in terms of high column efficiency and recovery, and low blank.26 This extraction material, however, is commercially unavailable which hinders its widespread application. In addition, the high selectivity of this protocol relies on the use of HF which is highly toxic, corrosive and dangerous to use.
A commercially available TRU resin was originally designed for separating radioactive nuclides from nuclear waste solutions28 and was further explored for the isotopic analysis of U, Nd and Sn in geological samples.22,29,30 For the first time, Burkhardt et al.31 used this resin to separate Mo from Ru after most matrix elements were removed using a cation resin column and an anion resin column for meteorite samples. Migeon et al.32 also used this resin for the first time to remove the residual matrix elements (principally U) after purification using an anion resin column for U-rich samples. However, in these previous studies, multiple columns were employed, and the use of the TRU resin was limited. Therefore, the selectivity of the TRU resin to Mo has not been not fully explored until now.
The purpose of this work was to develop a simple, fast and efficient single-column extraction protocol to quantitatively separate Mo from a suite of geological materials for mass-dependent Mo isotope ratio measurements. To achieve this goal, we have investigated the absorption behavior of Mo on TRU resin in various acid media (i.e., 0.5 M, 1 M, 1.5 M, 2 M, and 3 M HCl; 1 M and 2 M HNO3). Under optimized conditions, Mo is well retained on the TRU resin in 1.5 M hydrochloric acid whereas all the interference elements (e.g., Na, Mg, Al, K, Ca, Fe, Cr, Mn, Co, Ni, Cu, Zn, Ga, Rb, Sr, Zr, Ru, Sn and REE) are poorly retained, and Mo is easily stripped from the resin by the use of more diluted (i.e., 0.5 M) hydrochloric acid. Based on this selectivity, a simple and efficient single-column separation protocol was established, which was validated by the Mo isotopic composition measurements of various geological samples using the MC-ICP-MS technique.
2. Experimental
2.1. Reagents and materials
Reagent grade hydrofluoric, nitric and hydrochloric acids (SRC, China) were purified twice using a Savillex™ DST-1000 sub-boiling distillation system (Eden Prairie, MN, USA). High-purity de-ionized water (DI-water, 18.2 MΩ cm−1) was produced from a Milli-Q element system (Burlington, MA, USA) and used throughout this work. Eichrom TRU resin of particle size 100–150 μm was sourced from Eichrom Technologies Inc. (Darien, IL, USA). The dry resin was first immersed in 6 M HCl and then was washed with 6 M HCl and DI-water, alternately. After the cleaning procedure, the suspended particles were removed, and the resin was stored in DI-water before loading onto a column. NIST Mo SRM 3134 (lot: 130418) was obtained from the National Institute of Standards and Technology (Gaithersburg, MD, USA). 97Mo–100Mo double spike was prepared as a mixture from enriched 97Mo and 100Mo isotopes, which were purchased from Oak Ridge National Laboratory (ORNL, USA). The detailed double spike design and preparation procedure are provided in the ESI.† Seven geological reference materials BCR-2 (basalt), BHVO-2 (basalt), AGV-2 (andesite), W-2 (diabase), SGR-1 (shale), NOD-P-1 (manganese nodule), and NOD-A-1 (manganese nodule) were purchased from the United States Geological Survey (Reston, VA, USA). The Mo concentration of BCR-2 (USGS certified value 248 μg g−1) is significantly higher than the average crustal Mo concentrations (1–3 μg g−1), indicating that this geological reference material is likely to be contaminated with respect to Mo.16,33 Moreover, the Mo isotopic composition of BCR-2 was also considered to be heterogeneous.16 Thus, all these geological reference materials except BCR-2 were used as test samples for Mo isotope ratio measurements. BCR-2 was just used for testing the elution behavior of Mo and other matrix elements on the TRU resin for its unique high Mo content which is better to identify tails and multiple peaks. What is more, a previous study has suggested that BHVO-2 may also be contaminated, resulting in a heterogeneous Mo mass fraction.34 However, a large number of tests have proved that BHVO-2 has a homogeneous Mo isotopic composition.19,20,35 Therefore, BHVO-2 still can be used to verify the column separation and instrumental measurement method for Mo isotope measurements. Seawater collected from the surface waters of the South China Sea was measured for Mo isotope ratios to evaluate the applicability of the proposed separation method to seawater.2.2. Sample digestion
Sample powder (3–250 mg) containing ∼100 ng Mo was quantitatively weighed together with an appropriate amount of the 97Mo–100Mo double spike into 15 mL Savillex PFA beakers (except two nodules, which were spiked after the digestion due to their very high Mo contents of ∼600 ppm). Manganese nodules were digested in 15 mL Savillex vials with 3 mL mixed acid of HCl + HNO3 + HF (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1) following the method described by Berezhnaya and Dubinin.36 Other samples were digested in 4 mL concentrated HF–HNO3 (1:1) mixture at 120 °C for 24 hours, and a few drops of concentrated HClO4 were added for sedimentary rock (SGR-1) to break up the organic materials. After digestion and drying at 120 °C on a hotplate, the samples were treated with 4 mL of aqua regia at 120 °C for 2 days to dissolve any fluoride formed during the HF digestion procedure. After acid exchange or during the mid-stage of the heating procedure, sealed PFA beakers were placed in an ultrasonicator for 3–5 minutes during the dissolution period. Finally, all digested solutions were dried and redissolved in 4 mL 1.5 M HCl prior to chemical separation.
:2:1) following the method described by Berezhnaya and Dubinin.36 Other samples were digested in 4 mL concentrated HF–HNO3 (1:1) mixture at 120 °C for 24 hours, and a few drops of concentrated HClO4 were added for sedimentary rock (SGR-1) to break up the organic materials. After digestion and drying at 120 °C on a hotplate, the samples were treated with 4 mL of aqua regia at 120 °C for 2 days to dissolve any fluoride formed during the HF digestion procedure. After acid exchange or during the mid-stage of the heating procedure, sealed PFA beakers were placed in an ultrasonicator for 3–5 minutes during the dissolution period. Finally, all digested solutions were dried and redissolved in 4 mL 1.5 M HCl prior to chemical separation.
2.3 Chromatographic separation
Mo separation was carried out using 0.55 mL TRU resin in a custom-made quartz glass column (0.40 cm inner diameter, 4.5 cm high and 0.55 mL resin bed). Prior to use, the TRU resin was cleaned with 3 × 3 mL 1 M HNO3 and 3 × 3 mL 0.5 M HCl, alternatively. Then, the columns were preconditioned with 2 × 2 mL 1.5 M HCl (Table 1). The spiked sample solution (4 mL) was loaded onto the column. The TRU Spec resin color changed from white to bright yellow at this stage, reflecting the extraction of Fe(III). Each 4 × 4 mL 1.5 M HCl was added to completely rinse off matrix elements (e.g., Na, Mg, Al, K, Ca, Ti, Fe, Zn, Cu, Zr and Ru). During this procedure, the column turned white after rinsing with about 4 mL 1.5 M HCl. Finally, 3 × 2 mL 0.5 M HCl was added to quantitatively elute Mo. The purified sample solutions were dried and the residue was treated with 0.5 mL HNO3 twice to destroy organic matter introduced during the column separation. Finally, the solutions were evaporated to dryness and re-dissolved in 5% HNO3 (weight percent) to obtain 50 ng g−1 Mo solution prior to MC-ICP-MS analysis. The whole separation procedure takes about 4 hours to complete.| Step | Eluent | Vol. |
|---|---|---|
| Cleaning | 1 M HNO3 | 3 × 3 mL |
| 0.5 M HCl | 3 × 3 mL | |
| Conditioning | 1.5 M HCl | 2 × 2 mL |
| Loading | 1.5 M HCl | 4 mL |
| Rinsing | 1.5 M HCl | 4 × 4 mL |
| Elution of Mo | 0.5 M HCl | 3 × 2 mL |
2.4. ICP-MS determination of matrix elements
Element concentrations were determined using an Agilent 7700x ICP-MS instrument (Agilent Technologies, Tokyo, Japan). The nebuliser gas and make-up gas flow rates were optimised to obtain good signal intensities for Li, Y, Ce and Tl, while keeping the CeO+/Ce+ and Ce2+/Ce+ ratios below 1.2%. Drift corrections were carried out using In as an internal standard and by repeatedly analysing a quality control solution (QC) as a drift monitor over the duration of a run. The calibration of the instrument was performed using four ‘external’ standard solutions for trace element analysis (1, 10, 25 and 50 ng mL−1) and four ‘external’ standard solutions for major elements (10, 100, 500 and 1000 ng mL−1), which covered the range of concentrations in the investigated samples.2.5. Mo isotopic composition analysis
Mo isotope ratio measurements were carried out on a Neptune Plus (Thermo Scientific, Bremen, Germany) MC-ICP-MS at the State Key Laboratory of Geological Processes and Mineral Resources, China University of Geosciences, Wuhan. Sample and standard solutions were introduced into the plasma through an Aridus II desolvator with a 50 μL min−1 PFA nebulizer. A sample jet cone and a skimmer X cone combination was used to improve the sensitivity, resulting in a typical Mo intensity of 5–7 V for 98Mo in 50 ng g−1 solutions. Willbold et al.20 reported quite high (up to several 10−1 V) argide interference signals on masses 94, 96 and 98 with the use of the X cone. However, the signals of 40Ar214N+, 40Ar216O+ and 40Ar218O+ were monitored by measuring a 5% HNO3 blank solution and were found to be <1 mV on our instrument. Furthermore, the X cone was also employed for Mo isotope ratio measurements by other groups and it was found to be insignificant of argides.16,32 This may be due to the different yields of polyatomic ions from different instruments or under different parameter settings. Isobaric interferences from Zr and Ru were monitored by collecting 91Zr and 99Ru in L4 and H3, respectively, and were found to be negligible (<10−5 V) for all samples. Typical operating conditions are summarized in Table 2.| Neptune plus | |
| Rf power | ∼1200 W |
| Cooling gas flow rate | ∼16 L min−1 |
| Auxiliary gas flow rate | ∼1.0 L min−1 |
| Carrier gas flow rate | ∼1.0 L min−1 |
| Cones | Jet-sampler cone, X-skimmer cone |
| Cup configurations | L4 (91Zr), L2 (94Mo), L1 (95Mo), C (96Mo) |
| H1 (97Mo), H2 (98Mo), H3 (99Ru), H4 (100Mo) | |
| Mass resolution | Low-resolution |
| Sensitivity | 5–7 V for 98Mo at 50 ng g−1 Mo |
| Blocks and cycles | 3 blocks × 10 cycle |
| Integration time | 4.194 s × 1 |
| Peak center and baseline | At start |
| Data correction | Off-line correction |
|
|
| Aridus II | |
| Solution uptake | ∼50 μL min−1 |
| Sweep gas flow rate | ∼2.65 L min−1 |
| Nitrogen gas | ∼0.07 L min−1 |
| Spray chamber temperature | 110 °C |
| Desolvator temperature | 160 °C |
The samples were introduced in a sequence: SRM 3134std-sample-SRM 3134std using a CETAC ASX-112 FR autosampler. Between samples, 5% HNO3 was introduced for 2–3 minutes to reduce the 98Mo signal to below 1 mV, eliminating any potential cross-contamination. The intensities of all Mo and interference isotopes obtained from the blank solution (5% HNO3) were subtracted from those of all samples. Deconvolution of the raw double spike data was implemented by the iteration method.37,38 The nature compositions of samples and bracketing standards were obtained by subtracting the spike from the mixture through the iteration method, and the iteration terminated when the change of the corrected 98Mo/95Mo ratio is less than 10−6. Mo isotopic compositions are reported in standard delta (δ) notation relative to the mean value of the bracketing SRM 3134 calculated from eqn (1):
| δ98Mo = [(98Mo/95Mo)sample/(98Mo/95Mo)SRM 3134 − 1] × 1000 | (1) |
During the past decade, various reference materials have been utilized in many laboratories for Mo isotope ratio measurements, such as JMC mono Mo standard solution,10,11 CPI mono Mo standard solution,6,21 and NIST SRM 3134.16,20,35,39,40 Recently, NIST SRM 3134 (lot # 891307) has been increasingly utilized and accepted as the delta zero reference material.5,41–43 However, the batch (lot # 891307) of NIST SRM 3134 was sold out. Consequently, a new batch (lot # 130418) was purchased from the NIST and used as the bracketing standard throughout this study. An aliquot of NIST SRM 3134 (lot # 891307) was obtained from Song et al.44 and δ98/95Mo was measured against our NIST SRM 3134 (lot # 130418) in this study. A δ98Mo value (relative to NIST SRM 3134, lot # 130418) of 0.002 ± 0.040‰ (2SD, n = 20) was obtained for NIST SRM 3134 (lot # 891307), confirming no significant Mo isotopic shift between these two batches of NIST SRM 3134. Thus, the data reported in this study are inter-laboratory comparable with previous values.
The precision of Mo isotope ratio measurements was evaluated using the results obtained in the bracketing standards (NIST SRM 3134), assuming that the standard deviation (SD) of the sample measurements equals to the standard deviation of the bracketing standard measurements. The advantage of using bracketing instead of samples to estimate the measurement uncertainty is that there are more bracketing standards analyzed than the samples during a batch run. The standard deviation of δ98Mo was 0.082‰ (2SD) based on the results of 334 measurements of the bracketing standard during eight months of measurement (Fig. 1), indicating that the long-term repeatability of our instrument was less than 0.082‰.
 | ||
| Fig. 1 Mo isotopic compositions of the bracketing standard NIST SRM 3134 analyzed over a period of eight months. The horizontal line represents the mean δ98Mo value. The grey area represents twice the standard deviation of the δ98Mo value of NIST SRM 3134. | ||
3. Results and discussion
3.1 Extraction behavior of elements on the TRU resin
The TRU Spec resin comprises 13% octyl(phenyl)-N,N-diisobutylcarbamoylmethylphosphine oxide (CMPO) dissolved in 27% tri-n-butyl phosphate (TBP) and supported on the inert polymeric substrate (60 wt%) Amberchrom CG-71ms.28 It was originally designed for the separation of radioactive nuclides from nuclear waste solutions and was further explored for the isotopic analysis of U, Th and Nd in geological materials for the extraction of U, Th and light rare earth elements (LREEs) in dilute nitric acid medium.29,30,45,46 Tin can be selectively absorbed on the TRU resin in hydrochloride acid and this makes it ideal for Sn purification.22,47,48 This resin has been used previously for the separation of Mo, but it was used in combination with an ion exchange column; therefore, the selective adsorption conditions of Mo have not been not fully explored.31,32 In addition, only 70–90% Mo yield was obtained in previously reported separation protocols using the TRU resin.31,32 In this study, the elution behavior of Mo on the TRU resin was investigated with use of various concentrations of nitric and/or hydrochloride acid medium in order to establish a novel and simple Mo separation method.The elution behavior of Mo and matrix elements (including Na, Mg, Al, K, Ca, Fe, Cr, Mn, Co, Ni, Cu, Zn, Ga, Rb, Sr, Zr, Ru, Sn and REE) was investigated by using the geological reference material BCR-2 (50 mg) which was loaded on the TRU resin (0.55 mL resin bed volume used) using 1 M and 2 M nitric acid or 0.5 M, 1 M, 1.5 M, 2 M and 3 M hydrochloric acid. The basalt geological reference material BCR-2 was chosen not only because of its significantly high Mo content (248 ppm, USGS certificate) but also for its complex matrix which represents true geological samples rather than synthetic mixtures. Previous studies had shown that major matrix ions (including Zr and Ru) were not much retained in both dilute nitric acid (1 M (ref. 49) and 2 M (ref. 28 and 30)) and hydrochloric acid (0.5 M,22,47 1 M (ref. 50) and 3 M (ref. 51)) medium; thus, 1 M and 2 M nitric acid and 0.5–3 M hydrochloric acid were investigated in this study. It was observed that nitric acid medium was not suitable as Mo was stripped off from the column with matrix elements in the 1 M and 2 M HNO3 loading step (Fig. S2†). As shown in Fig. 2, semi-quantitative ICP-MS analyses of the collected fractions illustrate that the major matrix elements (Na, Mg, Al, K, Ca, Ti, etc.), transition metals (Cu, Co, Ni, V), REEs and other interference elements (e.g., Zr, Ru) were not much retained in all the tested 0.5–3.0 M hydrochloride acid solutions, consistent with the results observed in previous studies.22 It should be noted that each matrix element has a separate elution curve, which is represented by black curves in Fig. 2 (and in the following Fig. 3). It appears that the figures (e.g., Fig. 2b and e) have two black lines because these curves are divided into two groups with a small difference. Fe and Ga exhibit similar adsorption properties in dilute hydrochloric acid; they were eluted as early as other matrix elements in 0.5 M HCl (Fig. 2a). However, as the concentration of HCl increases, the adsorption capacity of Fe and Ga gradually increases, and only 60.9% and 66.2% of Fe and Ga were eluted when the HCl concentration increased to 3 M, respectively (Fig. 2e). It is worth noting that the Zn adsorption capacity of the TRU resin does not change monotonically with the concentration of hydrochloric acid (Fig. 2). Nevertheless, under all tested acidic conditions, the remaining Zn content was negligible after 16 mL elution. It is clear that the adsorption capacity of Mo increased as the HCl concentration increased, and almost no Mo was detected in the eluent (<0.01 ppb) when the HCl concentration increased to 1.5 M. This result indicates that the TRU resin absorbs Mo efficiently in 1.5–3 M HCl medium.
 | ||
| Fig. 2 Elution profiles of BCR-2 (50 mg) under different concentrations (0.5 M (a), 1 M (b), 1.5 M (c), 2 M (d) and 3 M (e)) of hydrochloric acid. The sample was loaded in 2 mL and eluted with another 14 × 1 mL selected eluent. When the acidity was up to 1.5 M, no significant Mo (<0.01 ppb) was found in the collected fractions. | ||
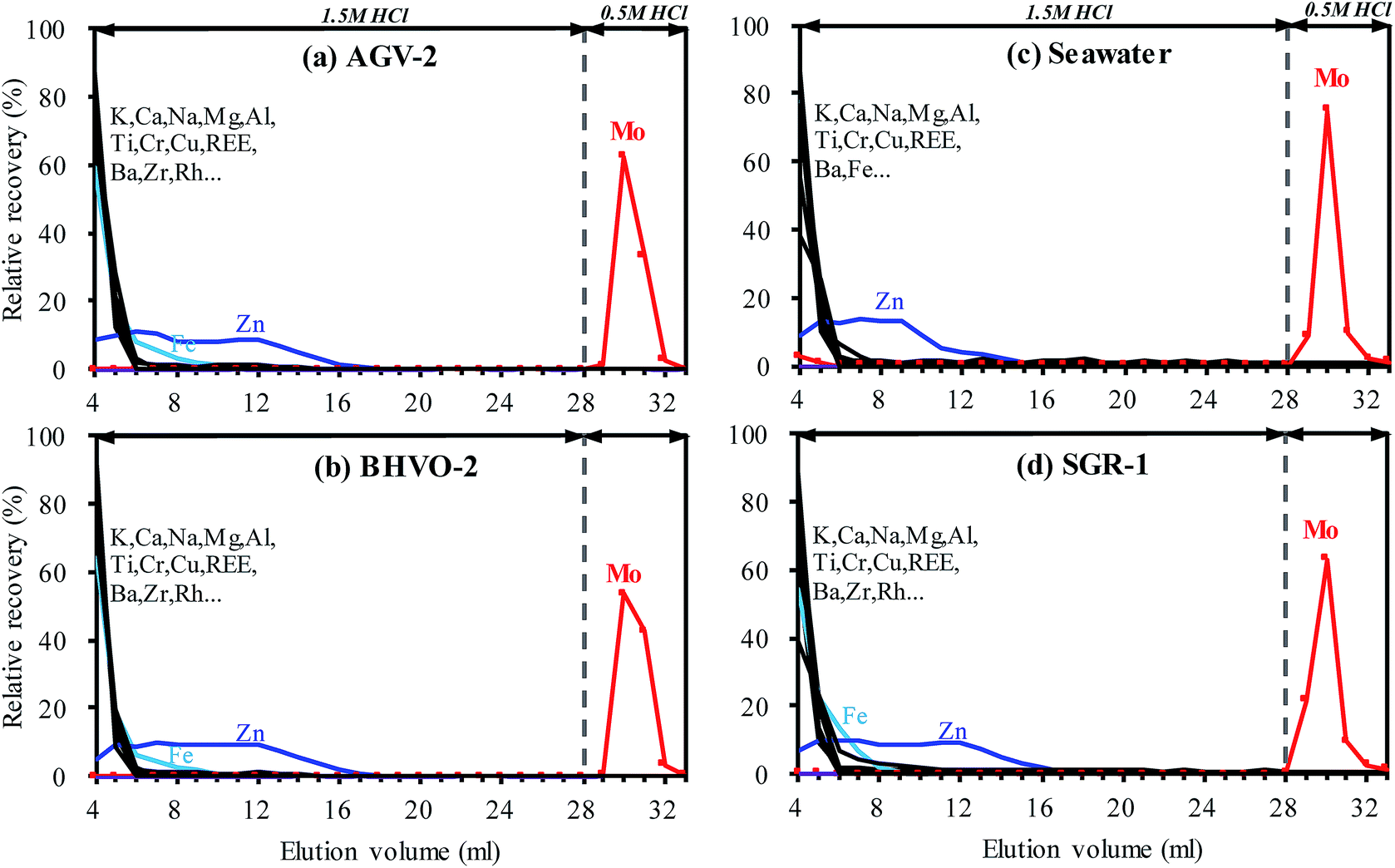 | ||
| Fig. 3 Elution profiles for AGV-2 (a), BHVO-2 (b), seawater (c) and SGR-1 (d) of the TRU resin with 0.55 mL resin bed. Each test sample solution contained ∼200 ng of Mo. Matrix elements were rinsed in 24 × 1 mL 1.5 mol L−1 HCl, while Mo was collected in 5 × 1 mL 0.5 mol L−1 HCl. | ||
3.2 Optimization of extraction chromatography
Based on the above results, it is evident that the TRU resin can be used as an ideal resin for Mo extraction. It is clearly that Mo can be strongly retained on the TRU resin in 1.5 M HCl medium whereas the majority of matrix elements (e.g., major elements, transition elements and REEs) were almost not adsorbed, while Zn, Fe and Ga were not much retained and can be removed with a small amount of the eluent, making this TRU resin potentially well-suited to the isolation of Mo from various geological samples. Importantly, Mo can be easily stripped from the TRU resin by using diluted 1 M HNO3 or 0.5 M HCl. In this study, 0.5 M HCl was employed as the eluent to obtain high-purity Mo since Sn is also not much retained in dilute nitric acid.22Consequently, the final protocol of Mo extraction chromatography was established simple steps of loading and rinsing in 1.5 M HCl and eluting in 0.5 M HCl, as presented in Table 1. This protocol was first tested by processing triplicate of a mono Mo standard solution. Each aliquot of Alfa Aesar Mo (600 ng Mo) was evaporated to dryness and then redissolved in 4 mL 1.5 M HCl. Then, these Mo standard solutions were loaded onto the TRU resin and rinsed with 4 × 4 mL 1.5 M HCl and eluted with another 3 × 2 mL 0.5 M HCl. All the sample loading, rinsing and elution fractions were collected and analyzed for Mo concentration by ICP-MS. Three independent experiments obtained a Mo yield of 101.5, 99.1 and 99.7%, respectively, with a mean value of 100.1 ± 2.5% (2SD, n = 3). These results confirm that the proposed protocol can achieve quantitative extraction of Mo from geological samples.
3.3 Adsorption capacity of the TRU resin
The maximum amount of Mo adsorbed per gram (so-called adsorption capacity) of the TRU resin was estimated following the method reported by Li et al.26 Briefly, the TRU resin column was loaded continuously (1 mL for each time) with a specific concentration (wMo, μg g−1) of the Mo standard solution until a detectable signal of Mo was observed in the collected fraction. The adsorption capacity (C, mg g−1) can be calculated from the following eqn (2):| C = wMoV/m | (2) |
3.4 Mo extraction from geological samples
High precision measurement of the Mo isotopic ratio is challenged by the isobaric interferences that arise from Ru (98Ru+ and 100Ru+) and the potential polyatomic interferences that arise from argide or oxynitride of Fe, Mn, Zn, Cu, Ni, Co and Cr (Table S2†). Although isobaric interferences that arise from Zr (92, 94, 96) do not interfere with the Mo isotopes involved in the double spike isotopes, efficient removal of Zr is required to obtain comprehensive Mo isotopic composition information. Clearly, elements such as Fe, Mn, Zn, Cu, Ni, Co, Cr and Zr are commonly abundant in natural geological samples, and most of the interferences originating from these elements could not be resolved instrumentally even if high resolution mode is utilized. Therefore, the abundance of matrix elements needs to be reduced to insignificant levels prior to analysis by mass spectrometry. Moreover, the matrix varies greatly in different types of geological samples, requiring the chemical purification method to tolerate different sample types.In this study, the proposed separation method was tested on four different geological reference materials AGV-2, BHVO-2, seawater and SGR-1. These samples cover several common types of geological samples, including igneous rock, sedimentary rock and water samples. As shown in Fig. 3, almost identical elution curves have been achieved for all sample matrices, indicating that Mo extraction has not been affected by any interfering matrix elements. Most of the matrix elements (e.g., K, Ca, Na, Mg, Al, Ba, Cu, V and Cr) were stripped from the TRU resin as soon as the sample was loaded in 4 mL 1.5 M HCl and reduced to negligible levels after rinsing with a small amount (i.e. 2–3 mL) of 1.5 M HCl. Whereas, more than 99% Zn was eluted after rinsing with 16 mL 1.5 M HCl.
Due to the high concentration of Fe and Ti in most geological samples, a rigorous chemical separation procedure is required to sufficiently remove these matrices prior to their measurements by MC-ICP-MS. However, it is difficult to effectively remove Fe and Ti by the commonly used anion-exchange technique through one column separation. Therefore, the Fe–Ti interferences were one of the major concerns for the separation of Mo by using anionic-exchange chromatography in a previous study.20 These troublesome interferences were usually overcome by using a special eluent e.g. 0.5 M HCl + 1 M HF,20 or further purified using a cation exchange column,1,15 making the separation procedure cumbersome. However, due to the weak adsorption of Fe and Ti on the TRU resin in 1.5 M HCl medium (Fig. 2c), the proposed separation protocol only needs a small amount of 1.5 M HCl to remove Fe–Ti, as shown in Fig. 3. The residual Fe and Ti were reduced to insignificant levels (e.g., [Fe]/[Mo] < 10 and [Ti]/[Mo] < 1, as discussed in the following section) after rinsing with 12 mL and 7 mL 1.5 M HCl, respectively. To ensure complete removal of Fe and Zn, an additional 8 mL of 1.5 M HCl was used for washing. Recoveries of Mo were found to be close to 100% for all samples (except seawater at 96.5%, Fig. 3c) in a total of 6 mL 0.5 M HCl elution fractions. Overall, quantitative recovery of Mo and complete elimination of the matrix elements were achieved for various matrices by employing the TRU resin.
3.5 Purity and recovery check
In order to assess the degree of purity and the recovery of the purified sample solutions, seven geological samples (BHVO-2, AGV-2, W-2a, SGR-1, NOD-P-1, NOD-A-1 and seawater) were separated following the proposed procedure (Table 1) and analyzed for element concentration by ICP-MS. The results show that most cation matrices were efficiently removed after the purification and yielded a very low ratio value to Mo (e.g., major elements (e.g., Na, Mg, Al, K and Ca, except Fe and Ti) < 10−2, transition elements (e.g., Cu, Zn, Ni, V and Cr) < 10−3, and REEs, Zr and Ru close to the 2% HNO3 blank level). Only traces of Ti and Fe were occasionally found in Mo isolates, and the [Ti]/[Mo] and [Fe]/[Mo] ratios were no more than 0.21 and 1.81, respectively.In order to assess the influence of matrix effects of the residual Fe and Ti in purified solutions on Mo isotope ratio measurements, 50 ppb Mo NIST SRM 3134 standard solutions were spiked with various amounts of Fe (Fe/Mo ranged from 0.5–10) or Ti (Ti/Mo ranged from 0.1–1), and the Mo isotope ratio was measured against a pure 50 ng g−1 Mo NIST SRM 3134 standard. According to the doping test (Fig. 4), the matrix effect on δ98Mo analyses is negligible (less than the instrument analysis precision of 0.08‰) even if the purified samples have a [Ti]/[Mo] ratio of up to 1 or a [Fe]/[Mo] ratio of up to 10. This result is well consistent with the observation reported by Liu et al.16 that the δ98Mo values were not sensitive to Fe when the [Fe]/[Mo] ratio was less than 30. Therefore, it can be concluded that the proposed separation protocol is well-suited for the separation of Mo from various sample matrices for high precision Mo isotope measurements.
 | ||
| Fig. 4 Assessment of influences of Fe and Ti on Mo isotopic ratio analysis by MC-ICP-MS. Range bars (2SD) were calculated based on at least three replicate measurements. The gray area represents typical instrumental analysis uncertainty on δ98Mo (0.08‰). | ||
Although the isotopic fractionation caused by incomplete Mo recovery during the column chemistry can be mitigated by the addition of the 97Mo–100Mo double-spike prior to chemical separation,11 quantitative recovery of Mo allows the use of other calibration methods (e.g., element doping method and standard sample bracketing method). One way to evaluate the Mo yield of the separation protocol is by comparing the Mo content between the unpurified and purified samples. ICP-MS analyses show that nearly quantitative recoveries of Mo (∼100%) for all the seven geological samples were obtained (detailed in Table 3), confirming the absence of Mo loss during column separation. This result further confirms that the proposed Mo extraction method is effective and not affected by the sample matrix.
| Reference material | Lithology | δ 98MoSRM 3134 | 2SD | N | Recoveryb (%) | Sources |
|---|---|---|---|---|---|---|
| a Number of independent digests of each geological reference material and each purified sample solution was measured 3 times by MC-ICP-MS. b Calculated from the Mo content ratio between the unpurified and purified samples. All the Mo contents were measured by the isotope dilution method using a 97Mo single spike, except the unpurified seawater sample for which the reference value reported by Zhao et al.35 was adopted. | ||||||
| BHVO-2 | Basalt | −0.03 | 0.05 | 12 | 102.0 ± 1.7 | This study |
| −0.05 | 0.09 | Zhao et al.35 | ||||
| −0.07 | 0.04 | Willbold et al.20 | ||||
| 0.00 | 0.07 | Siebert et al.52 | ||||
| −0.06 | 0.03 | Burkhardt et al.53 | ||||
| AGV-2 | Andesite | −0.12 | 0.08 | 7 | 102.1 ± 4.5 | This study |
| −0.14 | 0.05 | Zhao et al.35 | ||||
| −0.15 | 0.01 | Willbold et al.20 | ||||
| NOD-P-1 | Manganese nodule | −0.86 | 0.03 | 5 | 101.4 ± 1.0 | This study |
| −0.88 | 0.01 | Zhao et al.35 | ||||
| −0.88 | 0.15 | Barling et al.1 | ||||
| −0.83 | 0.10 | Asael et al.54 | ||||
| NODA-1 | Manganese nodule | −0.70 | 0.05 | 5 | 98.9 ± 6.7 | This study |
| −0.67 | 0.04 | Asael et al.54 | ||||
| −0.83 | 0.05 | Zhao et al.35 | ||||
| −1.20 | 0.15 | Barling et al.1 | ||||
| W-2a | Diabase | −0.03 | 0.06 | 6 | 98.3 ± 7.4 | This study |
| −0.04 | 0.05 | Wang et al.55 | ||||
| −0.05 | 0.06 | Burkhardt et al.53 | ||||
| −0.10 | 0.08 | Zhao et al.35 | ||||
| SGR-1a | Shale | 0.45 | 0.03 | 6 | 99.5 ± 2.1 | This study |
| 0.44 | 0.11 | Zhao et al.35 | ||||
| Seawater | Seawater | 2.04 | 0.03 | 9 | 97.5 ± 0.1 | This study |
| 2.04 | 0.06 | Zhao et al.35 | ||||
| 2.02 | 0.06 | Siebert et al.52 | ||||
| 2.04 | 0.07 | Pearce et al.19 | ||||
| 2.09 | 0.05 | Greber et al.39 | ||||
The potential column induced isotopic fractionation was further tested by checking the δ98Mo value of geological standards without using double-spike correction during column separation. This was carried out by adding the 97Mo–100Mo double-spike after the column separation procedure. The δ98Mo values obtained from seven geological standards spiked before and after the column separation procedure are compared in Fig. 5 (detailed in Table S3†). All data points fall on the 1:1 line, indicating that no isotopic fractionation had occurred as a result of the purification process and quantitative recovery of Mo was achieved.
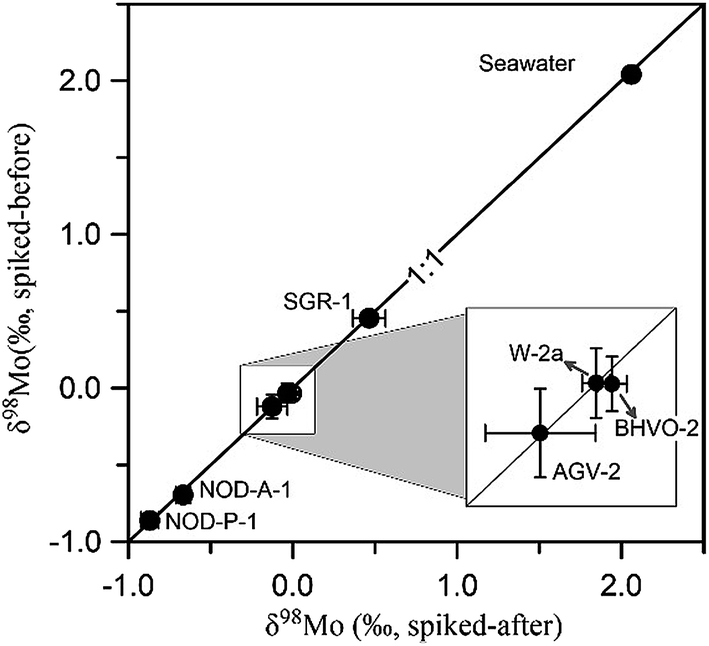 | ||
| Fig. 5 Comparison of Mo isotopic compositions of geological samples obtained from the results of the addition of the 97Mo–100Mo double spike before and after the column separation procedure. Range bars represent two standard deviations (2SD) of each measurement. | ||
3.5 Blanks and column reusability
The total procedural blank (including digestion and purification) was processed as samples and evaluated by monitoring the Mo contributions from reagents. The total procedural blank was measured three times with a mean value of 0.35 ± 0.25 ng (2SD). In addition, the column separation blank was also measured and only 0.03 ± 0.03 ng (2SD, n = 4) Mo was found, suggesting the digestion scheme produced the main contribution to the total blank Mo in our laboratory. This low column separation blank was mainly attributed to the use of a small amount of diluted high purity hydrochloric acid only.The stationary phase of the resin can be lost during repeated use with various harsh acids, thereby reducing the resin extraction capacity and impacting the sample separation efficiency. For instance, Li et al.26 reported that the capacity of the BPHA resin was reduced by ∼40% even with one time use for Mo purification. This reduction of resin capacity can be circumvented by replacing it with a new resin or supplementing the stationary phase,26 but the cost of sample purification will increase. The stability of the TRU resin has been evaluated by Horwitz et al.28 and insignificant impact on the adsorption of americium after rinsing with a large amount of water (600 mL) was found. This observation suggests that the TRU resin has good reusability due to its strong stability. In this study, the reusability of the TRU resin was evaluated by repeatedly processing Alfa Mo and BHVO-2 through a freshly prepared TRU resin, respectively, using the proposed separation scheme (Table 1). Eight separations of Alfa Aesar Mo solutions yielded a mean Mo recovery of 98.5 ± 4.4% (2SD, n = 8). In addition, the δ98Mo value of BHVO-2 obtained from the ninth separation is −0.02 ± 0.07 (2SD, n = 3), which is in good agreement with the recommend value (−0.05 ± 0.09 2SD, Table 3) reported by Zhao et al.35 The above results indicate that the TRU resin is reusable and can be used for Mo extraction at least nine times.
3.6 Precision and accuracy check
In order to demonstrate the validity of the separation procedure, seven international geological standards have been analyzed in replicate following the method outlined above. These geological standards vary greatly in matrices, including igneous rocks (W-2a, BHVO-2, AGV-2), sedimentary rocks of black shale (SGR-1), oceanic manganese nodules (NOD-P-1 and NOD-A-1), and seawater samples (Table 3). These samples were intentionally chosen to span a range of Mo concentrations and isotopic compositions, demanding the accurate and precise of Mo isotopic ratios for various geological samples. At least five replicates of each sample were digested and purified using the proposed separation procedure and each Mo fraction was measured three times by MC-ICP-MS (detailed in Table S3†). As shown in Fig. 6 and Table 3, δ98Mo of all geological references analyzed in this study is in good agreement with literature results. Previous studies suggested that NOD-A-1 has heterogeneous Mo isotopic compositions, with δ98Mo ranging from −0.67‰ to −1.20‰.1,35,54 A value of −0.70 ± 0.05‰ (2SD, n = 5) for NOD-A-1 was obtained in this study, in agreement with the value of −0.67 ± 0.04‰ (2SD) reported by Asael et al.54 The above results obtained in this study confirm the accuracy of the proposed column separation and measurement method. Moreover, as listed in Table 3, the measurement uncertainties for each sample ranged from 0.03 to 0.08‰ (2SD), indicating that geological samples can be analyzed accurately within the quoted precision of 0.08‰ (2SD). | ||
| Fig. 6 Comparison of the mean δ98MoSRM 3134 values obtained in this study with literature data, normalized to NIST SRM 3134. The filled symbols represent δ98MoSRM 3134 values obtained in this study, while the open symbols represent δ98MoSRM 3134 values from the literature. Range bars represent two standard deviations (2SD). | ||
4. Conclusions
A simple, fast and efficient column separation of Mo from geological sample matrices using the TRU resin is demonstrated, allowing precise and accurate Mo isotope ratio measurements in various geological sample matrices. The proposed method uses only one-column separation and exhibits excellent selectivity, high degree of purification and quantitative recovery of Mo from various geological sample matrices. The advantages of the proposed separation method include the following: (1) the entire separation procedure is very simple and time-saving (4 hours); (2) only diluted hydrochloric acid (1.5 M and 0.5 M) was used, making the method environmentally friendly; (3) extremely low column blank, low acid consumption and low memory effect of Mo on the TRU resin column were achieved. Based on the excellent selectivity for Mo, this separation protocol is expected to be suitable for the purification of ultra-low Mo content samples, such as carbonate samples, to further advance it applications in geological science.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank Dr Pansu Song for providing NIST SRM 3134 (lot # 891307). Constructive comments from two anonymous reviewers and efficient editorial handling of Harriet Brewerton are highly appreciated. This study was supported financially by the National Natural Science Foundation of China (Grants 41803014 and 41673013) and the Most Special Fund from the State Key Laboratory of Geological Processes and Mineral Resources, China University of Geosciences (MSFGPMR13).References
- J. Barling, G. L. Arnold and A. D. Anbar, Earth Planet. Sci. Lett., 2001, 193, 447–457 CrossRef CAS.
- C. Siebert, T. F. Nägler, F. von Blanckenburg and J. D. Kramers, Earth Planet. Sci. Lett., 2003, 211, 159–171 CrossRef CAS.
- J. Barling and A. D. Anbar, Earth Planet. Sci. Lett., 2004, 217, 315–329 CrossRef CAS.
- A. D. Anbar, Rev. Mineral. Geochem., 2004, 55, 429–454 CrossRef.
- E. K. King, S. S. Perakis and J. C. Pett-Ridge, Geochim. Cosmochim. Acta, 2018, 222, 584–598 CrossRef CAS.
- T. Goldberg, C. Archer, D. Vance and S. W. Poulton, Geochim. Cosmochim. Acta, 2009, 73, 6502–6516 CrossRef CAS.
- L. Wasylenki, B. Rolfe, C. Weeks, T. Spiro and A. Anbar, Geochim. Cosmochim. Acta, 2008, 72, 5997–6005 CrossRef CAS.
- A. R. Voegelin, T. F. Nägler, T. Pettke, N. Neubert, M. Steinmann, O. Pourret and I. M. Villa, Geochim. Cosmochim. Acta, 2012, 86, 150–165 CrossRef CAS.
- E. K. King, A. Thompson, O. A. Chadwick and J. C. Pett-Ridge, Chem. Geol., 2016, 445, 54–67 CrossRef CAS.
- A. D. Anbar, K. A. Knab and J. Barling, Anal. Chem., 2001, 73, 1425–1431 CrossRef CAS.
- C. Siebert, T. F. Nägler and J. D. Kramers, Geochem., Geophys., Geosyst., 2001, 2, 1032 CrossRef.
- D. Malinovsky, I. Rodushkin, D. C. Baxter, J. Ingri and B. Öhlander, Int. J. Mass Spectrom., 2005, 245, 94–107 CrossRef CAS.
- M. E. Wieser, J. R. De Laeter and M. D. Varner, Int. J. Mass Spectrom., 2007, 265, 40–48 CrossRef CAS.
- L. Zhou, T. J. Algeo, L. Feng, R. Zhu, Y. Pan, S. Gao, L. Zhao and Y. Wu, Chem. Geol., 2016, 428, 59–76 CrossRef CAS.
- A. J. Pietruszka, R. J. Walker and P. A. Candela, Chem. Geol., 2006, 225, 121–136 CrossRef CAS.
- J. Liu, H. Wen, Y. Zhang, H. Fan and C. Zhu, J. Anal. At. Spectrom., 2016, 31, 1287–1297 RSC.
- Y. Nagai and T. Yokoyama, J. Anal. At. Spectrom., 2016, 31, 948–960 RSC.
- Y. Nagai and T. Yokoyama, Anal. Chem., 2014, 86, 4856–4863 CrossRef CAS.
- C. R. Pearce, A. S. Cohen and I. J. Parkinson, Geostand. Geoanal. Res., 2009, 33, 219–229 CrossRef CAS.
- M. Willbold, K. Hibbert, Y.-J. Lai, H. Freymuth, R. C. Hin, C. Coath, F. Vils and T. Elliott, Geostand. Geoanal. Res., 2016, 40, 389–403 CAS.
- C. Archer and D. Vance, Nat. Geosci., 2008, 1, 597–600 CrossRef CAS.
- J. B. Creech, F. Moynier and N. Badullovich, Chem. Geol., 2017, 457, 61–67 CrossRef CAS.
- L. Feng, L. Zhou, L. Yang, W. Zhang, Q. Wang, S. Tong and Z. Hu, J. Anal. At. Spectrom., 2018, 33, 413–421 RSC.
- A. Retzmann, T. Zimmermann, D. Pröfrock, T. Prohaska and J. Irrgeher, Anal. Bioanal. Chem., 2017, 409, 5463–5480 CrossRef CAS.
- C. Li, X. Wang, J. Guo, Z. Chu and L.-J. Feng, J. Anal. At. Spectrom., 2016, 31, 1150–1159 RSC.
- J. Li, X.-R. Liang, L.-F. Zhong, X.-C. Wang, Z.-Y. Ren, S.-L. Sun, Z.-F. Zhang and J.-F. Xu, Geostand. Geoanal. Res., 2014, 38, 345–354 CrossRef CAS.
- N. Dauphas, L. Reisberg and B. Marty, Anal. Chem., 2001, 73, 2613–2616 CrossRef CAS.
- E. P. Horwitz, R. Chiarizia, M. L. Dietz, H. Diamond and D. M. Nelson, Anal. Chim. Acta, 1993, 281, 361–372 CrossRef CAS.
- S. Uchida, R. Garcia-Tenorio, K. Tagami and M. Garcia-Leon, J. Anal. At. Spectrom., 2000, 15, 889–892 RSC.
- C. Pin, D. Briot, C. Bassin and F. Poitrasson, Anal. Chim. Acta, 1994, 298, 209–217 CrossRef CAS.
- C. Burkhardt, T. Kleine, F. Oberli, A. Pack, B. Bourdon and R. Wieler, Earth Planet. Sci. Lett., 2011, 312, 390–400 CrossRef CAS.
- V. Migeon, B. Bourdon, E. Pili and C. Fitoussi, J. Anal. At. Spectrom., 2015, 30, 1988–1996 RSC.
- J. Li, X.-k. Zhu, S.-h. Tang and K. Zhang, Geostand. Geoanal. Res., 2016, 40, 405–415 CrossRef CAS.
- M. Willbold and T. Elliott, Chem. Geol., 2017, 449, 253–268 CrossRef CAS.
- P.-P. Zhao, J. Li, L. Zhang, Z.-B. Wang, D.-X. Kong, J.-L. Ma, G.-J. Wei and J.-F. Xu, Geostand. Geoanal. Res., 2016, 40, 217–226 CrossRef CAS.
- E. D. Berezhnaya and A. V. Dubinin, Geostand. Geoanal. Res., 2017, 41, 137–145 CrossRef CAS.
- L. Feng, L. Zhou, L. Yang, S. Tong, Z. Hu and S. Gao, J. Anal. At. Spectrom., 2015, 30, 2403–2411 RSC.
- W. Compston and V. M. Oversby, J. Geophys. Res., 1969, 74, 4338–4348 CrossRef CAS.
- N. D. Greber, C. Siebert, T. F. Nägler and T. Pettke, Geostand. Geoanal. Res., 2012, 36, 291–300 CrossRef CAS.
- E. K. Skierszkan, M. Amini and D. Weis, Anal. Bioanal. Chem., 2015, 407, 1925–1935 CrossRef CAS.
- T. F. Nägler, A. D. Anbar, C. Archer, T. Goldberg, G. W. Gordon, N. D. Greber, C. Siebert, Y. Sohrin and D. Vance, Geostand. Geoanal. Res., 2013, 38, 149–151 Search PubMed.
- H. Wen, J. Carignan, C. Cloquet, X. Zhu and Y. Zhang, J. Anal. At. Spectrom., 2010, 25, 716–721 RSC.
- T. Goldberg, G. Gordon, G. Izon, C. Archer, C. R. Pearce, J. McManus, A. D. Anbar and M. Rehkamper, J. Anal. At. Spectrom., 2013, 28, 724–735 RSC.
- P. Song, J. Wang, T. Ren, T. Zhou, Y. Zhou and S. Wang, Anal. Chem., 2017, 89, 9031–9038 CrossRef CAS PubMed.
- M. Ganio, K. Latruwe, D. Brems, P. Muchez, F. Vanhaecke and P. Degryse, J. Anal. At. Spectrom., 2012, 27, 1335–1341 RSC.
- C. Pin and S. Joannon, J. Anal. At. Spectrom., 2001, 16, 739–743 RSC.
- E. Yamazaki, S. Nakai, T. Yokoyama, S. Ishihara and H. F. Tan, Geochem. J., 2013, 47, 21–35 CrossRef CAS.
- E. Balliana, M. Aramendía, M. Resano, C. Barbante and F. Vanhaecke, Anal. Bioanal. Chem., 2013, 405, 2973–2986 CrossRef CAS.
- C. Pin and J. S. Zalduegui, Anal. Chim. Acta, 1997, 339, 79–89 CrossRef CAS.
- M. Haustein, C. Gillis and E. Pernicka, Archaeometry, 2010, 52, 816–832 CrossRef CAS.
- G. Brügmann, D. Berger and E. Pernicka, Geostand. Geoanal. Res., 2017, 41, 437–448 CrossRef.
- C. Siebert, J. C. Pett-Ridge, S. Opfergelt, R. A. Guicharnaud, A. N. Halliday and K. W. Burton, Geochim. Cosmochim. Acta, 2015, 162, 1–24 CrossRef CAS.
- C. Burkhardt, R. C. Hin, T. Kleine and B. Bourdon, Earth Planet. Sci. Lett., 2014, 391, 201–211 CrossRef CAS.
- D. Asael, F. L. H. Tissot, C. T. Reinhard, O. Rouxel, N. Dauphas, T. W. Lyons, E. Ponzevera, C. Liorzou and S. Chéron, Chem. Geol., 2013, 362, 193–210 CrossRef CAS.
- Z. Wang, J. Ma, J. Li, G. Wei, T. Zeng, L. Li, L. Zhang, W. Deng, L. Xie and Z. Liu, Geochim. Cosmochim. Acta, 2018, 226, 1–17 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ja00267g |
| This journal is © The Royal Society of Chemistry 2020 |