
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A green route to polyurethanes: oxidative carbonylation of industrially relevant aromatic diamines by CO2-based methyl formate†
Christine
Hussong
a,
Jens
Langanke
*ab and
Walter
Leitner
 *ac
*ac
aRWTH Aachen University, CAT Catalytic Center, Worringerweg 2, 52074 Aachen, Germany. E-mail: leitner@itmc.rwth-aachen.de
bCovestro Deutschland AG, Catalysis and Technology Incubation, Kaiser-Wilhelm-Allee 60, 51368 Leverkusen, Germany. E-mail: jens.langanke@covestro.com
cMax Planck Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45470 Mülheim a. d. Ruhr, Germany
First published on 28th August 2020
Abstract
The oxidative carbonylation of toluene-2,4-diamine (TDA) with methyl formate (MF), which can be produced from CO2, provides a possible route for the non-phosgene production of isocyanate precursors and enables a valuable utilization of the greenhouse gas. Extensive analysis of the product spectrum has provided detailed insight into the reaction network leading to the target product toluene-2,4-dicarbamate (TDC) and the most important side products. The most prominent one has been identified as methylene-bridged tetracarbamate 5, which is also an interesting precursor for applications in polyurethane chemistry. The side products are caused by three different reaction paths: N-formylation by MF, condensation with in situ formed formaldehyde, and N-methylation by in situ formed dimethyl carbonate (DMC). The influence of the catalyst on product distribution was evaluated for PdCl2/CuCl2 and a large number of heterogeneous Pd-catalysts. The oxidic support materials ZrO2, CeO2 and SiO2 were found to partially suppress the undesired side reactions leading to higher yields of TDC and tetracarbamate 5. The ratio of TDC to 5 was demonstrated to be affected significantly by the choice of the support. The synthetic protocol was extended to the synthesis of dicarbamates from 4,4′-methylenedianiline (MDA) and 2,4-diaminomesitylene (17). These results encourage further investigations into the design of selective catalysts for the production of isocyanate precursors from CO2 as a C1 source.
Introduction
The realization of the Green Chemistry principles depends primarily on the design criteria we are applying for the compounds and processes of the future: are they depleting or renewable, toxic or benign, persistent or readily degradable?1 Through strategic decisions in line with the goals of sustainability, benefits can be created which are multiplied in cascades. For example, the utilization of CO2 as a feedstock lowers waste whereby additionally it results in the avoidance of toxic phosgene, in a decrease of CO2 emissions, in a slowdown of the rise of CO2 levels, and finally in a mitigation of the global climate change.1 The research reported in this paper was motivated by this systems thinking, investigating the possibility to open a CO2-based pathway for the construction of the carbamate group as required in the production of polyurethane materials.The synthesis of aromatic dicarbamates is in the focus of current Green Chemistry research because this compound class provides a non-phosgene access to the corresponding diisocyanates, which are utilized in large quantities as industrially important building blocks in polyurethane chemistry.2N-Aryl carbamates can be synthesized, for example, by the reaction of aromatic amines with urea and alcohol3 or by the conversion of aromatic urea derivatives by means of alcohols or organic carbonates in the presence of PbO or sodium alcoholate etc.4 Aromatic amines can also be converted directly into the corresponding carbamates using organic carbonates, whereby organic lead and zinc salts or sodium alcoholates are capable of catalysing this reaction.5 In this context promising results were achieved utilizing Zn4O(OAc)6 in the transformation of toluene-2,4-diamine (TDA) with dimethyl carbonate (DMC).6 Another approach to N-aryl carbamates is the catalytic reductive carbonylation of nitro aromatics, whereupon the activity of the palladium/phenanthroline catalytic system is enhanced by addition of phosphoric acid.7 As a very atom efficient route comprising two C1 building blocks several research groups have investigated the oxidative carbonylation of aromatic amines by reacting them with carbon monoxide/methanol mixtures in the presence of various transition metal complexes.8
The use of the greenhouse gas carbon dioxide as a C1 building block is of particular interest in synthetic chemistry.9 In the last three years, several research groups have published their results on the synthesis of N-aryl carbamates through the direct reaction of aromatic amines with carbon dioxide using a wide variety of catalysts and reagents.10 However, due to the high thermodynamic stability of CO2 and the weak nucleophilicity of aromatic amines either stoichiometric quantities of organotin compounds or titanium alkoxides or extended reaction times are required.10 An approach to overcome this problem is the utilization of methyl formate (MF) in the oxidative carbonylation of aromatic amines for the synthesis of N-aryl carbamates.11–13 MF can be efficiently produced by CO2 hydrogenation integrated with the subsequent esterification of formic acid with methanol.14 Furthermore, MF can be catalytically decomposed to CO/MeOH in a controlled manner. The catalytic interconversion of CO2/formate/CO has been coined as the “formic acid triangle”.14a Thus, CO2-derived MF can be regarded as liquid, easily transportable and safe source for CO/methanol mixtures.11–15 The idea of using MF for the synthesis of aromatic carbamates was demonstrated previously only for the monofunctional model substrate aniline.11–13 Here, we present the first detailed investigations on the oxidative carbonylation of actual polyurethane precursors TDA, 4,4′-methylenedianiline (MDA) and 2,4-diaminomesitylene employing MF as CO2-based CO/MeOH source in combination with several Pd based catalysts.
Results and discussion
Oxidative carbonylation of toluene-2,4-diamine with methyl formate
Our research focused on the comprehensive study of the Pd-catalysed synthesis of TDC by the oxidative carbonylation of TDA using liquid MF and synthetic air (see Scheme 1).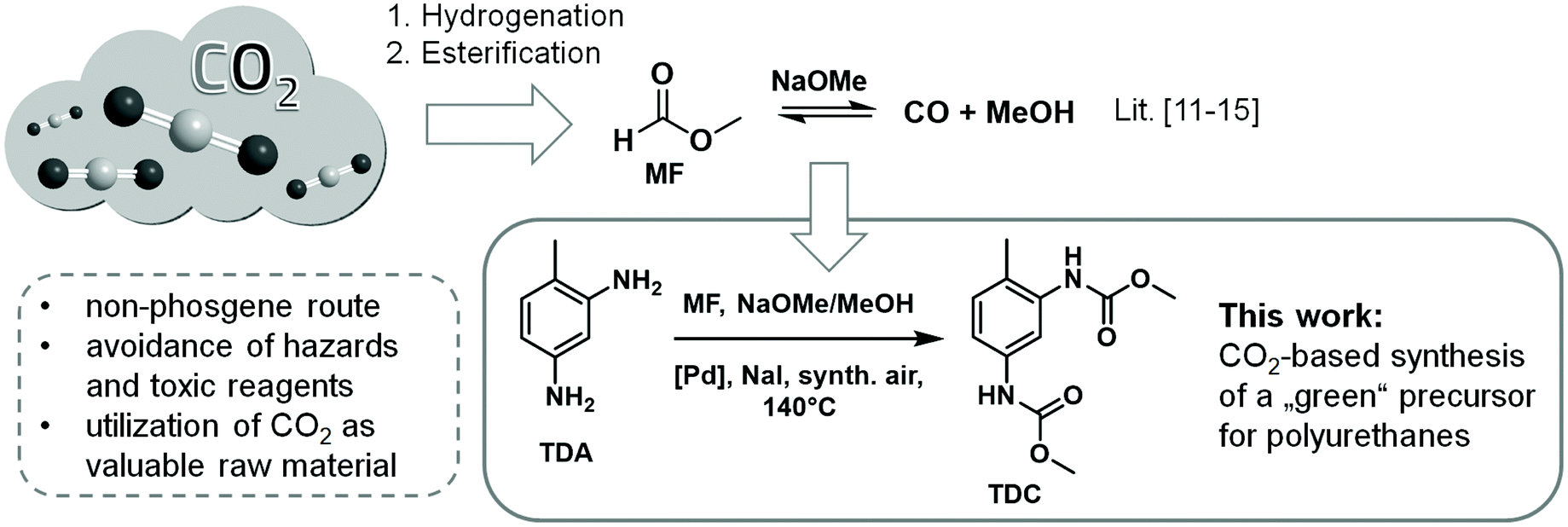 | ||
| Scheme 1 Synthesis of TDC by oxidative carbonylation of TDA with MF. | ||
The previously reported catalytic system Pd(10%)/C-NaI8a,12,13 was used as starting point for our investigations.11,16a MF and the methanolic sodium methanolate solution together with the catalyst system were pre-equilibrated in a stainless-steel reactor to generate the desired CO/methanol mixture.11–15 After heating to 140 °C for one hour, a constant CO pressure of 74 bar was formed and the reactor was pressurized with 29 bar of synthetic air. Using a HPLC pump the oxidative carbonylation was started by adding a methanolic TDA solution. The solvent and reagent MF was present in large excess (MF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TDA ca. 100:1) with low catalyst loading corresponding to 0.2 mol% total amount of Pd relative to TDA. After 4 h, the reaction was terminated by cooling the reactor to room temperature and the reaction mixture was analyzed by HPLC using an external calibration for the quantification of TDC. TDC was then isolated by column chromatography. While fair yields up to 39% of TDC could be obtained with this procedure, the performance of the catalyst system was found to be unstable under these conditions and a robust protocol could not be achieved.11,16a The complexity of the reaction network was identified as a major challenge, leading to a range of side products in varying composition. Hence, a crucial step in the development of the approach was the detailed analysis of the corresponding species and their reversible or irreversible formation in consecutive or parallel reaction pathways.
:TDA ca. 100:1) with low catalyst loading corresponding to 0.2 mol% total amount of Pd relative to TDA. After 4 h, the reaction was terminated by cooling the reactor to room temperature and the reaction mixture was analyzed by HPLC using an external calibration for the quantification of TDC. TDC was then isolated by column chromatography. While fair yields up to 39% of TDC could be obtained with this procedure, the performance of the catalyst system was found to be unstable under these conditions and a robust protocol could not be achieved.11,16a The complexity of the reaction network was identified as a major challenge, leading to a range of side products in varying composition. Hence, a crucial step in the development of the approach was the detailed analysis of the corresponding species and their reversible or irreversible formation in consecutive or parallel reaction pathways.
Unravelling the reaction network of oxidative carbonylation of TDA with MF
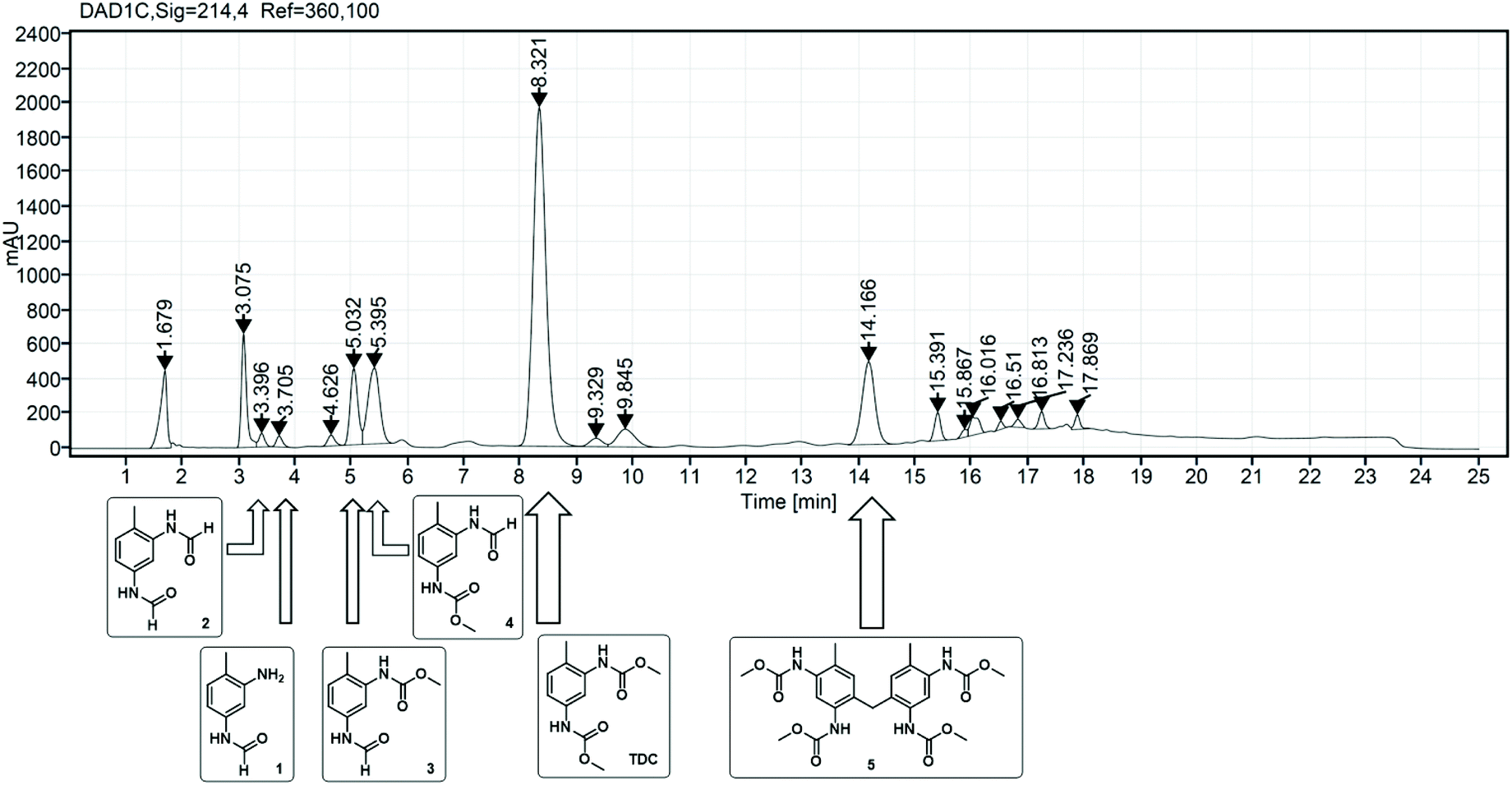 | ||
| Fig. 1 HPLC analysis of a typical reaction mixture of the oxidative carbonylation of TDA with MF: assignment of TDC and the identified side products 1–5 by UV detection at 214 nm. | ||
Formamides from N-formylation with MF
As already described in the literature, the non-catalytic N-formylation of aromatic amines with MF in a SN2t reaction is competing with the oxidative carbonylation, whereby formanilide (FAN) is formed in case of aniline as model substrate.11–13 It has not yet been completely clarified whether FAN is an intermediate which in the presence of oxygen further reacts palladium-catalysed with methanol forming the carbamate or whether FAN is only an unproductive side product.11,12 In the case of the industrially relevant substrate TDA, there are five different products possible from this reaction, whereby in addition to the formamide function, also an amine or carbamate function can be present.As part of the analysis of the product spectrum, we examined the reaction of TDA with MF in the presence of sodium methanolate, but in the absence of the Pd catalyst. In this way formation of formamide 1 (Rt = 3.7 min) and diformamide 2 (Rt = 3.3 min) was verified as shown in Fig. 2.17,18 For comparison, 2 was also synthesized according to literature from TDI and formic acid.18b The selective formation of the regioisomer 1 can be explained by the different reactivity of the two amino groups in TDA.19
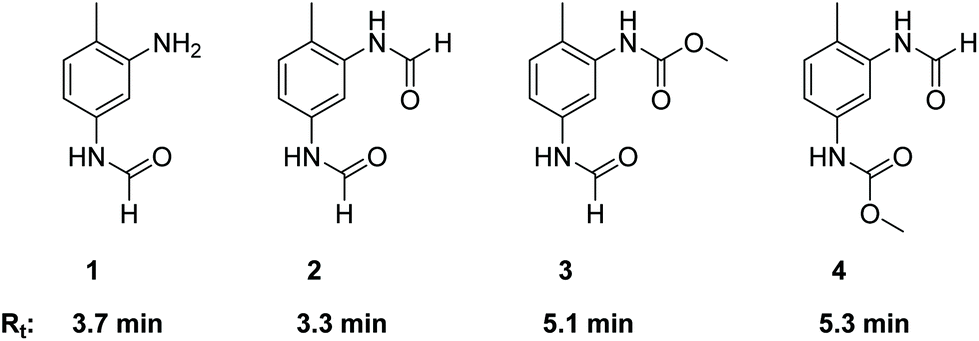 | ||
| Fig. 2 Structures and retention times (Rt) of formamide products resulting from the competing N-formylation during the oxidative carbonylation of TDA with MF. | ||
In order to investigate the role of N-formylation with regard to carbamate production, the oxidative carbonylation with MF was carried out using the mono-formamide 1 instead of TDA. From this reaction 2 (14%) and 3 (Rt = 5.1 min, 18%) were isolated by column chromatography together with 14% of TDC. The formation of TDC demonstrates that the formamide group has been converted into a carbamate unit under the conditions of the oxidative carbonylation. Nevertheless, the majority of the original formamide function remained unreacted. Furthermore, compounds 1–4 were detected by HPLC analysis in the reaction mixture upon injection of TDC under the conditions of the oxidative carbonylation instead of TDA, showing the possibility of exchanging the carbamate function for formamide groups by MF. These results indicate that formamide groups are less reactive than free amine groups regarding carbamate formation, constituting undesired side products for TDC production via oxidative carbonylation rather than intermediates as proposed in previous literature.11,12 However, the utilization of diformamide 2 for the direct non-phosgene TDI synthesis has been patented via a different route, making the formamides potentially useful side products in the context of carbamate production in general.20 Additionally, formamides are also key intermediates in the production of fragrances, pharmaceuticals, dyes and agrochemicals.18a Thus, the observed formation of 1 and 2 from TDA and MF under basic conditions suggests that the reaction of amines with “green” methyl formate additionally enables a sustainable route for the synthesis of industrially important formamides.18c
While the oxidative carbonylation of 1, as expected, resulted only in the formation of regioisomer 3, the reaction of TDA always led to 3 and 4 as demonstrated by isolating 4% of the pure regioisomer 4 (Rt = 5.3 min) by column chromatography. The NMR spectra of 1–4 were fully consistent with the cis/trans rotamers expected in solution.21–23 In the 1H NMR spectrum, the –NHC(O)H function shows a characteristic signal pattern due to the presence of cis- and trans-rotamers. The more stable cis-rotamer has a 3JH,H coupling constant of about 2 Hz, whereas the trans-rotamer appears with a 3JH,H coupling constant of about 11 Hz.21,22 Additionally, in the case of the cis-rotamer the protons ortho to the –NHC(O)H group are deshielded in comparison to the corresponding ortho-protons of the trans-rotamer.23
Methylene-bridged dimer from condensation with in situ formed formaldehyde
The side product with Rt = 14.2 min has been detected as the major side product in the HPLC analysis and is therefore of paramount importance. We succeeded in isolating this new compound in its pure form from the reaction mixture by column chromatography as yellow orange crystals. 1H NMR, 13C NMR and HR-MS analysis revealed the surprising result that this isolated compound can be characterized as the methylene-bridged tetracarbamate 5 (see Scheme 2). In other words, the most important side product of the oxidative carbonylation of TDA is “TDC-CH2-TDC” (5). This finding raised the question how the C1 bridge has been incorporated. A plausible proposal invokes partial Pd-catalysed oxidation of methanol to formaldehyde,24 which then reacts, possibly in situ, via electrophilic aromatic substitution and subsequent condensation with two molecules of TDA to give the tetraamine 13 as intermediate.25 Analogous transformations for the synthesis of 13 have been reported for example by conversion of TDA and methanol with a bio-catalyst (rat-liver S9) or by the direct reaction of TDA with formaldehyde.25,26 To verify the hypothesis, the methylene-bridged TDA dimer 13 was employed as substrate instead of TDA under the very same oxidative carbonylation conditions and led to formation of carbamate 5 (15%).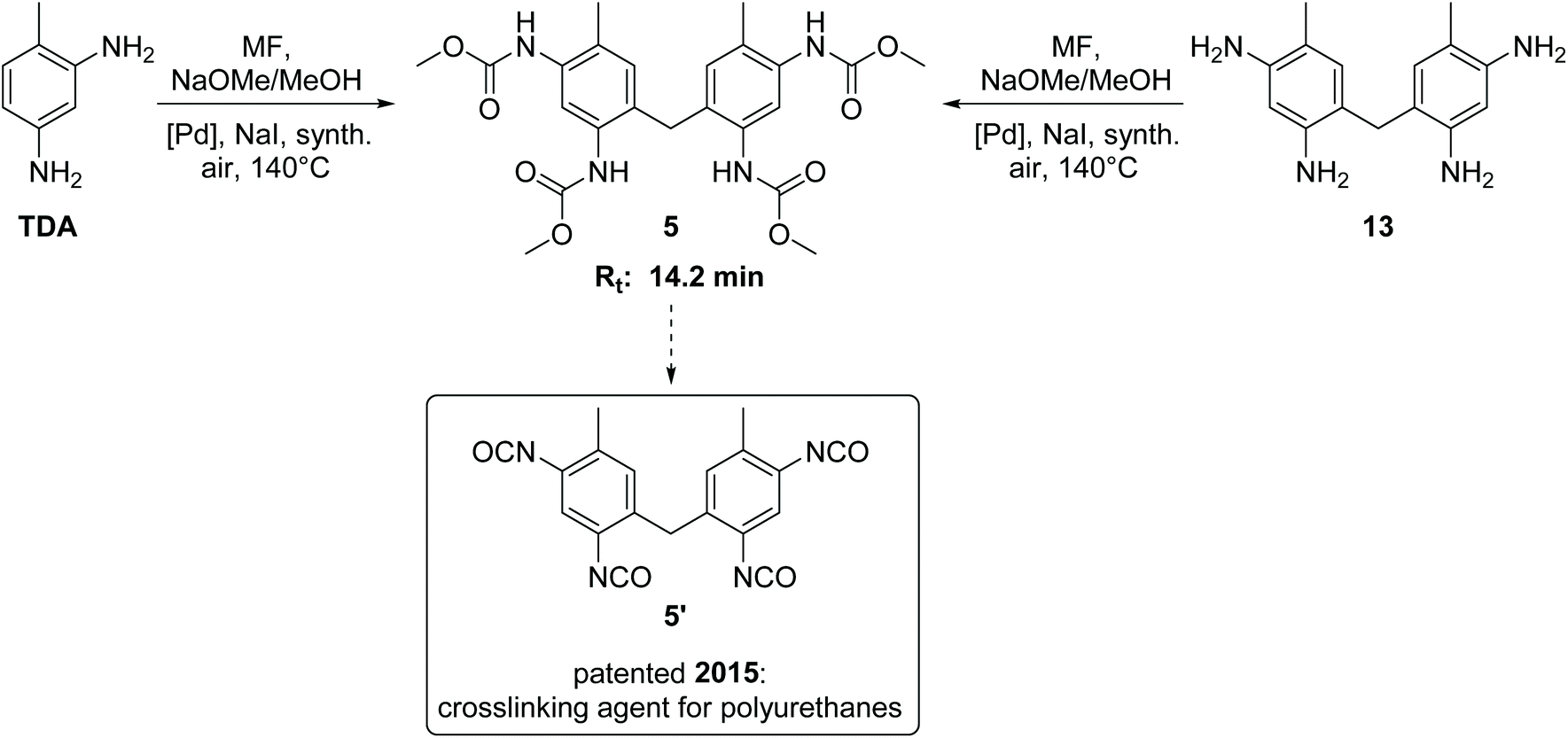 | ||
| Scheme 2 Synthesis, retention time (Rt) and application potential of the side product 5.11,27 | ||
The methylene-bridged TDC dimer 5 is a very interesting compound with high application potential. It can serve as a precursor for the synthesis of the tetraisocyanate 5′, which can be used as a highly reactive crosslinking agent in polyurethane chemistry.11,27 Contrary to the previously patented synthesis of 5′,27 our findings provide a non-phosgene, one pot route to this linker, making 5 a valuable co-product in this context encouraging further efforts to optimize its yield in addition to TDC.
Methylated products from N-methylation with in situ formed dimethyl carbonate
The side products with Rt ≈ 10 and Rt > 15 min (see Fig. 1) were identified as N-methylated products. Their origin can be associated with the presence of dimethyl carbonate (DMC) that is known to be formed under the conditions of oxidative carbonylation with MF.15 The presence of DMC in the reaction mixture was confirmed together with dimethyl oxalate (DMO) by GC-MS. Reaction of DMO with sodium methanolate can also lead to DMC formation.28 Furthermore, TDC is sensitive to methanolysis under thermal conditions leading to regeneration of the primary amine functions together also with DMC as coproduct.29As a reagent with dual reactivity, it is well described that DMC can not only convert amines into carbamates, but it can also cause N-methylations.30,31 In order to verify its potential for generating N-methylated side products, we examined the direct reactions of TDA, TDC, 1 and 2 with DMC. Thus, compounds 6–12 were isolated or at least identified in binary mixtures by 1H NMR, 13C NMR and HR-MS. Fig. 3 illustrates the corresponding structures and retention times (Rt) obtained by HPLC analysis. The presence of a free amino group in combination with an N-methylated carbamate or N-methylated formamide unit in compounds 8 and 10 reveal that the carbamate and formamide functions are more prone to N-methylation by DMC than the free amine.
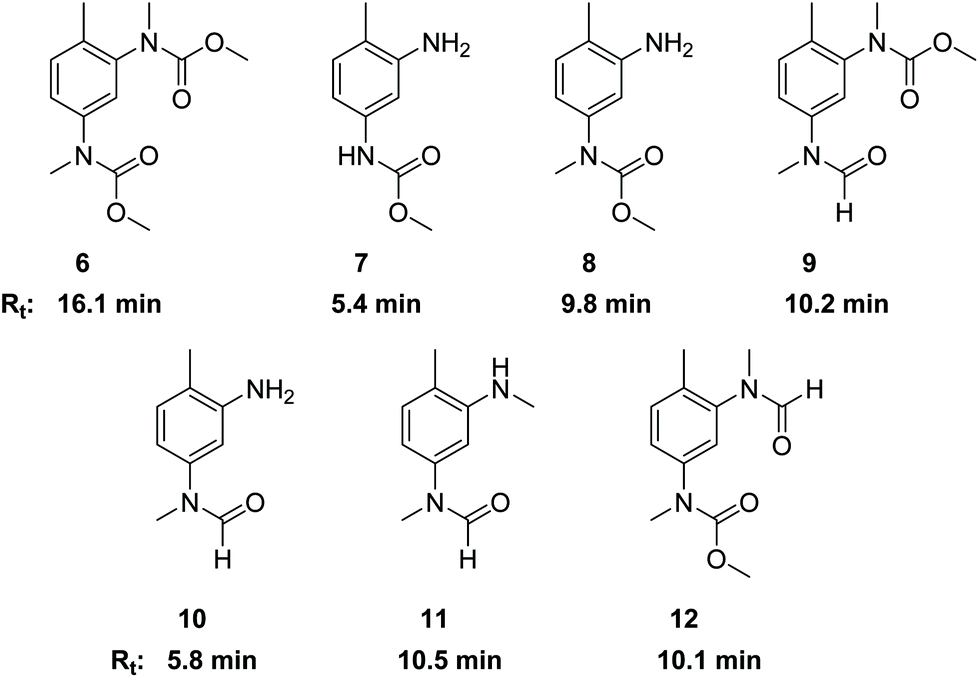 | ||
| Fig. 3 Structures and retention times (Rt) of compounds 6–12. | ||
Additionally, the tetraamine 13 was also reacted with DMC resulting in a complex mixture of compounds. The NMR-analysis clearly revealed both N-methylated amine and N-methylated carbamate functions. The corresponding retention times (Rt) were in the range between 16.5 and 18.7 min. Based on these results, we assign the areas in the HPLC analysis with Rt > 15 min mainly to N-methylated side products of the methylene bridged dimers.
Description of the full reaction network
In summary, the oxidative carbonylation of TDA with MF is characterised by a complex reaction network (see Fig. 4). The formation of side products can be divided into three main categories: first, N-formylation by the reaction with MF; second, the electrophilic aromatic substitution/condensation with formaldehyde resulting in methylene-bridged side products; and third the N-methylation by the reaction with DMC. Evaluating the side products with regard to potential benefits, they are of different significance. As described above, the formamide function can at least partly be transformed into a carbamate function under the conditions of the oxidative carbonylation, albeit at lower rates than the starting amine. Additionally, formamides are interesting industrial key intermediates and the diformamide 2 can be used as starting material for the non-phosgene TDI production.18,20 The dimeric side product 5 has an interesting application potential as precursor for a highly reactive crosslinking agent in polyurethane chemistry.27 Only the N-methylation is irreversible and leads to unproductive and undesired side products. Thus, the reaction of the starting materials and intermediates with DMC resulting in N-methylated compounds must be suppressed to improve the value generation of the process. This however, the formation of DMC can hardly be eliminated because, as mentioned above, it can be formed in three different ways. Consequently, the dual reactivity of DMC should be controlled by shifting it towards carbamate production, while simultaneously suppressing undesired N-methylations. This defines the central challenge for catalyst development in this field.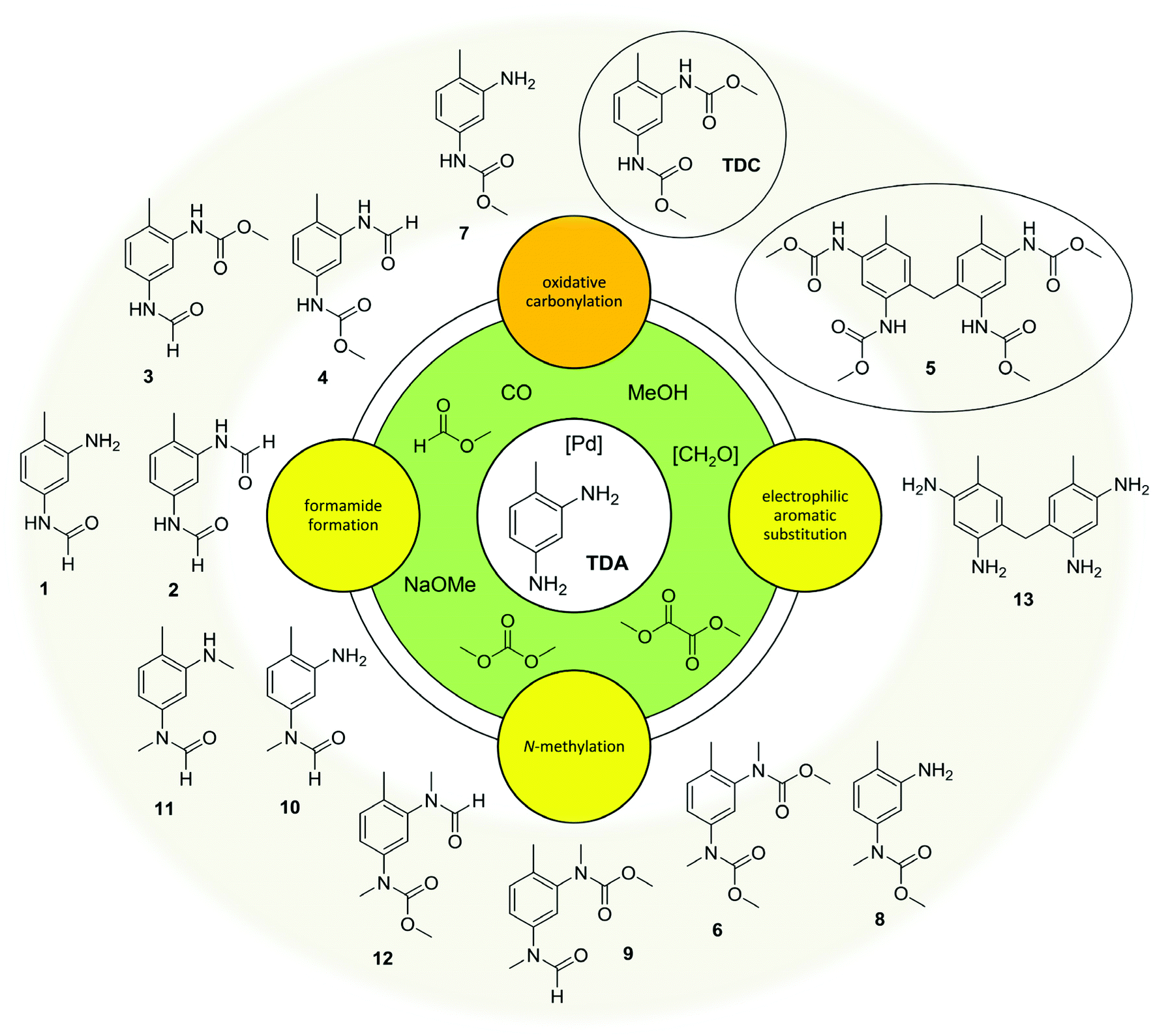 | ||
| Fig. 4 Oxidative carbonylation of TDA with MF: complex reaction network. | ||
Catalyst control of the reaction network
In order to evaluate to what extend the nature of the catalyst can be exploited to control the reaction network, eight heterogeneous palladium catalysts with 5% metal loadings on different supports were tested and compared to the homogeneous catalytic system PdCl2/CuCl2.8b In all experiments NaI was employed as promoter.8 Calibration of the HPLC method with standard solutions enabled quantification of TDC together with the potentially valuable side products 1–5 in the reaction mixture. The amount of N-methylated compounds was estimated based on the area percentage of the side products with Rt ≈ 10 min and Rt > 15 min in the chromatogram. Reactions were carried out for different time intervals to obtain insight into the development of conversion and selectivity to the individual products. All reactions were conducted at least twice to evaluate the robustness of the procedure and reproducibility of the catalyst performance. As proxy for catalyst efficiency, based on the total amount of Pd atoms the time dependence of an overall turnover number (TON) was used to define the turnover frequency (TOF). Preliminary results of a screening to identify suitable conditions were published previously.16The homogeneous system PdCl2/CuCl2 resulted in TDC yields reaching up to 38% and total yields of 1–5 + TDC of 53% in some experiments. However, the system exhibited inconsistent performance and large variations were observed between individual experiments. Consistent selectivities and TDC yields of 40–50% over independent experiments were achieved, however, using the oxidic supports ZrO2 and CeO2 exhibiting the highest TONs in the range of 190–200 with standard deviations of only 2–4%. For comparison, the averaged TONs of the other heterogeneous catalysts range between 143 and 184 with standard deviations of 9–16%. With the exception of TiO2, the formation of N-methylated products was lowest with the oxidic supports and maintained clearly below 10 area% in the HPLC analysis of the total product mixture for ZrO2, CeO2 and SiO2. This can be explained by the known activity of TiO2 to catalyse methylation reactions using DMC.31a In contrast, the support material CeO2 is known to suppress N-methylation when using Pd/CeO2 in the formation of carbamates from DMC.31 This is probably due to selective cleavage of DMC which is necessary for carbamate formation.32a The different behaviour of the two oxidic supports may be related to the high Lewis acidity of TiO2 as compared to the strong surface basicity of CeO2.32 Although N-methylation cannot be suppressed completely,31b this reactivity of Pd/CeO2 allows the catalyst to convert eventually formed DMC productively into carbamates also under the conditions of oxidative carbonylation. Therefore, the catalyst Pd/CeO2 was chosen for a detailed analysis of the reaction profile for the oxidative carbonylation of TDA (Fig. 5).
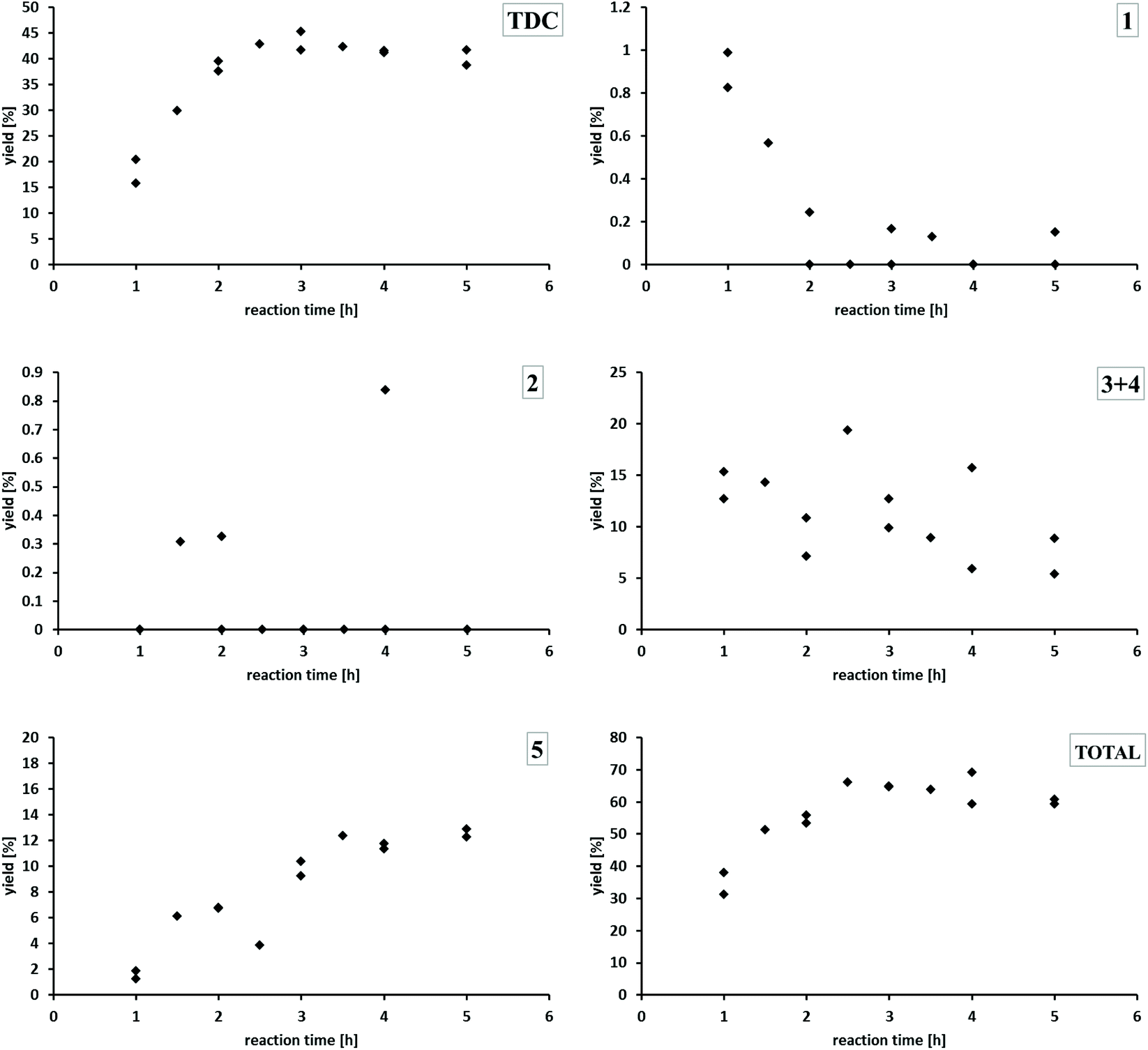 | ||
| Fig. 5 Yields of TDC, 1, 2, 3 + 4 and 5 in dependence of the reaction time together which the total yield of these products [50–83 bar CO; 3.7–5.9 vol% O2; 140 °C; MF (485 mmol); NaOMe (0.5 mmol); Pd(5%)/CeO2 (0.011 mmol Pd); NaI (0.6 mmol); TDA (5 mmol) in MeOH (5 mL)]. | ||
Using Pd/CeO2 under standard reaction conditions, the yield of TDC increases within the first two hours already up to 40%, reaching a plateau in the range of 39–45%. The mono-formylated TDA 1 is produced initially in very small amounts only and reacts further to disappear almost completely after 2 h. The concentration of the double formylated species 2 is never outside the detection limit. The concentration of the two regioisomers 3 and 4 containing both the formyl and carbamate function showed some variation, but a clear downward trend with reaction time can be noted. Generally, the concentration/time-profiles for species 1–4 are consistent with their formation as less productive intermediates for TDC formation. The yield of the methylene-bridged TDC dimer 5 increases over time reaching a nearly constant value of 13% after 3.5 hours. Comparing the concentration/time profiles of TDC and 5 indicates that they are formed in parallel pathways rather than 5 being a secondary product from TDC. The total yield of all potentially valuable compounds which could be quantified in the reaction mixture increases within the first three hours and then ranges between about 60 to 70%. At this stage, the product mixture is dominated by the two most valuable products TDC and 5 with 53% cumulated yield.
A notable correlation between the relative yields of TDC and 5 is observed for the different catalysts comparing the results with the highest total yield of these two target products (Fig. 6). The combined yields of TDC and 5 range between 48% and 53% in all these cases. As can be seen, higher yields of TDC correlate with significantly lower yields of 5, ranging from a TDC:5-ratio of nearly 9:1 for Pd/ZrO2 to 1.5:1 for Pd/C. As described before, the formation of TDC-CH2-TDC (5) can be explained as a result of the formation of formaldehyde as reactive C1 building block under the conditions of the oxidative carbonylation.24–26 The experimental result, that Pd(5%)/C gives the highest yield of 5 is in accordance with literature, since it has been suggested previously that during the Pd-catalysed decomposition of methanol carbonaceous overlayers, most likely elemental carbon, which are present during the reaction favour CH2O formation by hindering its dehydrogenation to CO.24a,f Furthermore, the equally high yields of 5 in the case of Pd(5%)/TiO2 and Pd(5%)/Al2O3 are also in line with literature, since both metal oxides have previously been used as support materials in the Pd-catalysed oxidation of methanol, whereby the formation of formaldehyde was observed both as an intermediate and as a product.24
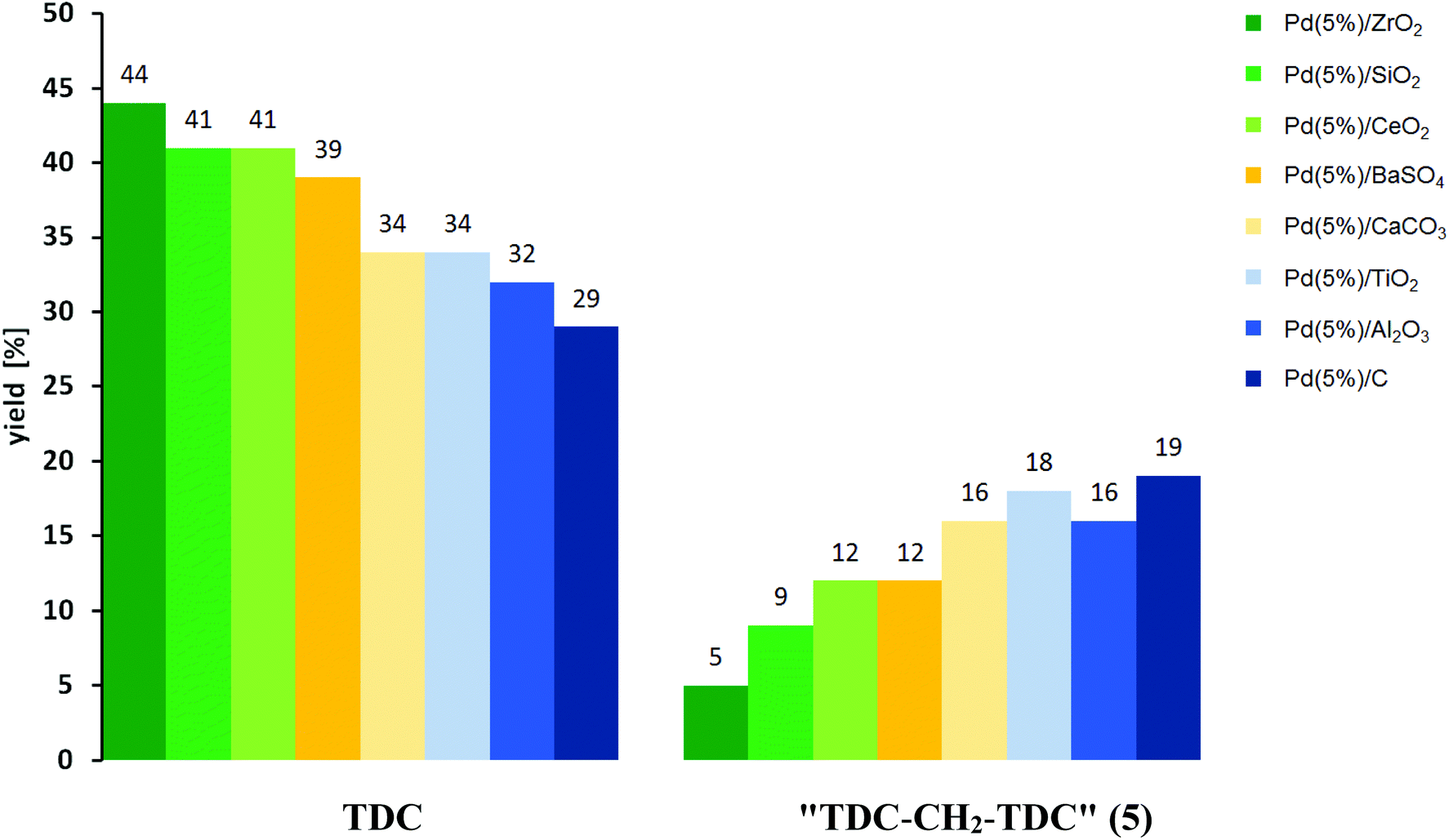 | ||
| Fig. 6 Influence of the support material on the palladium-catalysed oxidative carbonylation of TDA with MF. Reaction conditions: 485 mmol MF; 0.5 mmol NaOMe; 0.011 mmol Pd; 0.6 mmol NaI; 5 mmol TDA in 5 mL MeOH; 140 °C; reaction time 4 h. Quantified by HPLC-analysis using external standard solutions. | ||
Oxidative carbonylation of 4,4′-methylenedianiline and 2,4-diaminomesitylene
So far, the product analysis of the oxidative carbonylation of TDA with MF had provided detailed insights into the reaction network and based on these results the yield of the potentially valuable products could be significantly increased by choosing suitable catalysts minimizing some N-methylation as the most detrimental side reaction (compare Fig. 6). The method was next tested with industrially important 4,4′-methylenedianiline (MDA) and 2,4-diaminomesitylene. As catalyst, Pd(5%)/Al2O3 was applied because it is the cheapest material among those giving robust results with low methylation activity and because formation of the methylene bridged dimer is not relevant with both substrates.Subjecting MDA to the standard conditions of oxidative carbonylation revealed the formation of the desired dicarbamate MDC together with products from N-formylation in solution, as determined by HPLC analysis (see Scheme 3). Both MDC and 16 were isolated directly from the reaction mixture, whereas the two formamides 14 and 15 were identified by independent synthesis from MDA with MF in the presence of sodium methanolate. As illustrated in Fig. 7, the reactivity pattern of MDA is very similar to that of TDA. The dicarbamate MDC is formed rapidly in the beginning, the main side product 16 converts also to MDC more slowly over time. The total yield of both products reaches up to 40% with a maximum of 24% for MDC after 3.5 hours. A major limitation to reach higher yields is at least partly the fact that the ureas assumed as intermediates in carbamate formation12,31b are very poorly soluble in the methanol solvent leading to significant amounts of solid precipitates. Filtrating the reaction mixture resulted in 300 mg of solid material that consisted mainly of 1,3-bis(4-benzylphenyl)urea derivatives according to IR and NMR analysis.33
 | ||
| Scheme 3 Oxidative carbonylation of MDA with MF. | ||
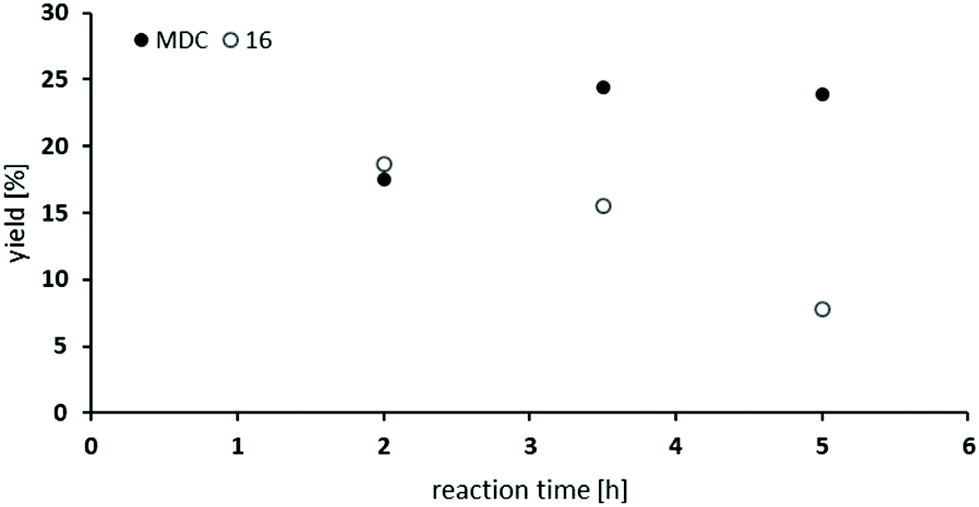 | ||
| Fig. 7 Oxidative carbonylation of MDA with MF: dependence of reaction time on the yield of MDC and 16 [69–72 bar CO; 3.6–4.5 vol% O2; MF (485 mmol); NaOMe (0.5 mmol); Pd(5%)/Al2O3 (0.011 mmol Pd); NaI (0.6 mmol); MDA (2.5 mmol) in MeOH (5 mL)]. | ||
The oxidative carbonylation of 2,4-diaminomesitylene (17) employing MF was studied using Pd(5%)/Al2O3 as well as Pd(5%)/SiO2 as catalysts (see Scheme 4). Both catalysts performed almost identical, and the yield of the dicarbamate 18 reached up to 30% after 5 h reaction time (Fig. 8). Again significant amounts of solid urea products were observed.12,31b,34 The mono-carbamate 19 was detected as intermediate. The formation of N-formamides was less pronounced, which may be reflecting steric hindrance. On the other hand, the presence of dimethylated amine 20 in the reaction mixture shows that N-methylation is not fully suppressed in this case.
 | ||
| Scheme 4 Oxidative carbonylation of 2,4,6-trimethyl-1,3-benzenediamine (17) with MF. | ||
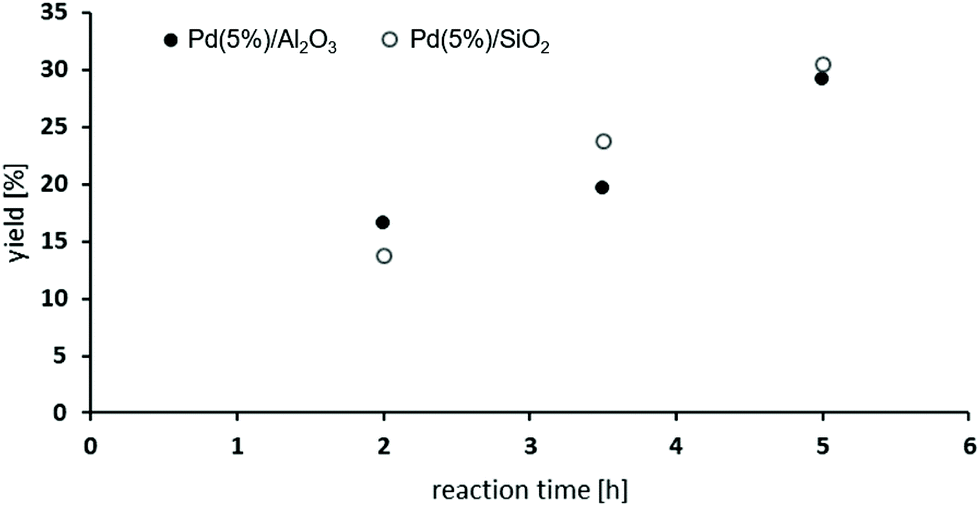 | ||
| Fig. 8 Oxidative carbonylation of 17 with MF: dependence of reaction time on the yield of 18 [65-81 bar CO; 4.2-5.2 vol% O2; 485 mmol MF; 0.5 mmol NaOMe; 0.011 mmol Pd; 0.6 mmol NaI; 5 mmol 17 in 5 mL MeOH]. | ||
Conclusions
The results of this study point out that the oxidative carbonylation of industrially relevant diamines utilizing CO2-based methyl formate (MF) as reagent indeed enables a green non-phosgene route to polyurethanes. In this regard the Pd-catalysed oxidative carbonylation of toluene-2,4-diamine (TDA) with MF has been studied in detail. Three different pathways causing side products have been found: Firstly, the nucleophilic substitution at the carbonyl group of MF (SN2t mechanism) without the participation of the Pd-catalyst or oxygen leading to formamides. Secondly, the electrophilic aromatic substitution/condensation with in situ formed formaldehyde resulting in the methylene-bridged tetracarbamate 5. Due to its own potential the dimer 5 is of particular interest for industrial application, making a valuable target product in the process, too. And thirdly, undesired N-methylations caused by the reaction with dimethyl carbonate (DMC) which is also produced during the oxidative carbonylation with MF.We tested PdCl2/CuCl2 and several heterogeneous Pd-catalysts with the result that the support materials ZrO2, CeO2 and SiO2 partially suppress undesired N-methylations. The combined yields of TDC and 5 reached up to ca. 50%, whereby higher yields of TDC correlate with lower yields of 5. Considering formamides as potential intermediates for further conversion to isocyanates as well, total yields of valuable products in the framework of polyurethane production reached up to 70%. The method of oxidative carbonylation with MF was extended to other diamines, whereby the solubility of urea intermediates in methanol was found to be a potential limitation.
The insights into the reaction mechanism resulting from the detailed side product and reaction network analysis and the findings concerning the influence of the support material of the catalyst on the reaction process in the case of TDC highlight the challenges and opportunities designing adequate catalysts for the synthesis of the industrially relevant diisocyanate precursors in commercially viable yields. This first study on oxidative carbonylation of industrially relevant substrate and product structures therefore is expected to contribute to unlocking the potential of CO2 as building block for polyurethane production following the Green Chemistry principles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding for the Carbon2Chem project by the German Federal Ministry of Education and Research (BMBF) under the grant number 03EK3042C. Open Access funding provided by the Max Planck Society.Notes and references
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- C. Six and F. Richter, Isocyanates, Organic, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, 2000 Search PubMed.
- L. Dou, X. Zhao, H. An, G. Wang and Y. Wang, Ind. Eng. Chem. Res., 2013, 52, 4408–4413 CrossRef CAS.
- (a) J. Wang, Q. Li, W. Dong, M. Kang, X. Wang and S. Peng, Appl. Catal., A, 2004, 261, 191–197 CrossRef CAS; (b) J. J. Gao, H. Q. Li and Y. Zhang, Chin. Chem. Lett., 2007, 18, 149–151 CrossRef CAS; (c) J. Gao, H. Li, Y. Zhang and W. Fei, Catal. Today, 2009, 148, 378–382 CrossRef CAS.
- (a) E. Reixach, N. Bonet, F. X. Rius-Ruiz, S. Wershofen and A. Vidal-Ferran, Ind. Eng. Chem. Res., 2010, 49, 6362–6366 CrossRef CAS; (b) A. Franzke, R. Baumann and M. Bock (BASF SE), US Pat8871965B2, 2014 Search PubMed; (c) S. Wang, G. Zhang, X. Ma and J. Gong, Ind. Eng. Chem. Res., 2007, 46, 6858–6864 CrossRef CAS; (d) P. Heitkämper, T. Schieb and S. Wershofen (Bayer AG), EU Pat0520273A2, 1992 Search PubMed.
- (a) S. Wershofen, S. Klein, A. Vidal-Ferran, E. Reixach and F. X. Rius-Ruiz (Bayer MaterialScience AG), Eur. Pat2230228A1, 2010 Search PubMed; (b) E. Reixach, R. M. Haak, S. Wershofen and A. Vidal-Ferran, Ind. Eng. Chem. Res., 2012, 51, 16165–16170 CrossRef CAS.
- (a) F. Ragaini, Dalton Trans., 2009, 6251–6266 RSC; (b) O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431–1455 RSC; (c) F. Ragaini, C. Cognolato, M. Gasperini and S. Cenini, Angew. Chem., Int. Ed., 2003, 42, 2886–2889 ( Angew. Chem. , 2003 , 115 , 2992–2995 ) CrossRef CAS PubMed.
- (a) P. Toochinda and S. S. C. Chuang, Ind. Eng. Chem. Res., 2004, 43, 1192–1199 CrossRef CAS; (b) B. Chen and S. S. C. Chuang, J. Mol. Catal. A: Chem., 2003, 195, 37–45 CrossRef CAS; (c) J. H. Park, T. S. Chang, Y. W. You, I. Heo, J. L. Lee, S. G. Han and K. Y. Jeong (Korea Research Institut of Chemical Technology), Int. PatWO107818A1, 2019 Search PubMed; (d) F. Shi, J. Peng and Y. Deng, J. Catal., 2003, 219, 372–375 CrossRef CAS; (e) F. Saliu, B. Putomatti and B. Rindone, Tetrahedron Lett., 2012, 53, 3590–3593 CrossRef CAS; (f) F.-M. Mei, L.-J. Chen and G.-X. Li, Appl. Organomet. Chem., 2010, 24, 86–91 CAS; (g) F. Shi and Y. Deng, Chem. Commun., 2001, 443–444 RSC; (h) Z.-H. Guan, H. Lei, M. Chen, Z.-H. Ren, Y. Bai and Y.-Y. Wang, Adv. Synth. Catal., 2012, 354, 489–496 CrossRef CAS; (i) A. Iturmendi, M. Iglesias, J. Munárriz, V. Polo, J. J. Pérez-Torrente and L. A. Oro, Chem. Commun., 2017, 53, 404–407 RSC.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- (a) D. Riemer, P. Hirapara and S. Das, ChemSusChem, 2016, 9, 1916–1920 CrossRef CAS PubMed; (b) N. Germain, I. Müller, M. Hanauer, R. A. Paciello, R. Baumann, O. Trapp and T. Schaub, ChemSusChem, 2016, 9, 1586–1590 CrossRef CAS PubMed; (c) T. Schaub, R. Baumann, R. Paciello and N. Germain (BASF SE), Int. PatWO177761A1, 2016 Search PubMed; (d) N. Germain, M. Hermsen, T. Schaub and O. Trapp, Appl. Organomet. Chem., 2017, 31, e3733 CrossRef; (e) Q. Zhang, H.-Y. Yuan, N. Fukaya, H. Yasuda and J.-C. Choi, ChemSusChem, 2017, 10, 1501–1508 CrossRef CAS PubMed; (f) Q. Zhang, H.-Y. Yuan, N. Fukaya, H. Yasuda and J.-C. Choi, Green Chem., 2017, 19, 5614–5624 RSC; (g) J.-C. Choi, N. Fukaya, Q. Zhang and H. Yasuda (National Institute of Advanced Industrial Science and Technology Jpn.), US Pat0185420A1, 2019 Search PubMed; (h) Q. Zhang, H.-Y. Yuan, N. Fukaya and J.-C. Choi, ACS Sustainable Chem. Eng., 2018, 6, 6675–6681 CrossRef CAS; (i) Y. Ren and S. A. L. Rousseaux, J. Org. Chem., 2018, 83, 913–920 CrossRef CAS PubMed; (j) M. Tamura, A. Miura, M. Honda, Y. Gu, Y. Nakagawa and K. Tomishige, ChemCatChem, 2018, 10, 4821–4825 CrossRef; (k) H.-Y. Yuan, Q. Zhang, N. Fukaya, X.-T. Lin, T. Fujitani and J.-C. Choi, Bull. Chem. Soc. Jpn., 2018, 91, 1481–1486 CrossRef CAS.
- W. Leitner, G. Franciò, M. Scott, C. Westhues, J. Langanke, M. Lansing, C. Hussong and E. Erdkamp, Chem. Ing. Tech., 2018, 90, 1504–1512 CrossRef CAS.
- M. S. Yalfani, G. Lolli, T. E. Müller, A. Wolf and L. Mleczko, ChemSusChem, 2015, 8, 443–447 CrossRef CAS PubMed.
- G. Lolli, A. Wolf, L. Mleczko and M. Yalfani (Bayer Technology Services), Int. PatWO158799A1, 2015 Search PubMed.
- (a) J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 ( Angew. Chem. , 2016 , 128 , 7416–7467 ) CrossRef CAS PubMed; (b) M. Scott, C. G. Westhues, T. Kaiser, J. C. Baums, A. Jupke, G. Franciò and W. Leitner, Green Chem., 2019, 21, 6307–6317 RSC; (c) A. Imberdis, G. Lefèvre and T. Cantat, Angew. Chem., Int. Ed., 2019, 58, 17215–17219 ( Angew. Chem. , 2019 , 131 , 17375–17379 ) CrossRef CAS PubMed; (d) J. J. Corral-Pérez, A. Bansode, C. S. Praveen, A. Kokalj, H. Reymond, A. Comas-Vives, J. VandeVondele, C. Copéret, P. R. von Rohr and A. Urakawa, J. Am. Chem. Soc., 2018, 140, 13884–13891 CrossRef PubMed; (e) J. J. Corral-Pérez, C. Copéret and A. Urakawa, J. Catal., 2019, 380, 153–160 CrossRef; (f) J. J. Corral-Pérez, A. Billings, D. Stoian and A. Urakawa, ChemCatChem, 2019, 11, 4725–4730 CrossRef.
- M. S. Yalfani, G. Lolli, A. Wolf, L. Mleczko, T. E. Müller and W. Leitner, Green Chem., 2013, 15, 1146–1149 RSC.
- (a) C. Hussong, J. Langanke and W. Leitner, Chem. Ing. Tech., 2020, 92 DOI:10.1002/cite.202000031; (b) We note that this averaged TOF does not reflect a mechanistic “catalytic activity” of Pd, as particle size and shape may vary with the support. It allows analysis of the catalysts on basis of effective usage of the noble metal, however.
- A. Albert, J. Chem. Soc., 1947, 244–250 RSC.
- (a) Z. Wu, Y. Zhai, W. Zhao, Z. Wei, H. Yu, S. Han and Y. Wei, Green Chem., 2020, 22, 737–741 RSC; (b) T. Nishikubo, E. Takehara and A. Kameyama, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3013–3020 CrossRef CAS; (c) A. Franzke, T. Mattke, J. Leschinski, R. Abdallah, M. Bock, R. Baumann and E. Ströfer (BASF SE), Int. PatWO067278A1, 2011 Search PubMed.
- T. Baba, A. Kobayashi, Y. Kawanami, K. Inazu, A. Ishikawa, T. Echizenn, K. Murai, S. Aso and M. Inomatac, Green Chem., 2005, 7, 159–165 RSC.
- (a) T. Mattke, E. Ströfer, J. Leschinski, R. Abdallah, A. Franzke and M. Bock (BASF SE), Eur. Pat2507206B1, 2014 Search PubMed; (b) R. W. Mason and J. L. Schuchardt (ARCO Chemical Technology, L.P.), Int. PatWO0224634A2, 2002 Search PubMed.
- R. J. Abraham, L. Griffiths and M. Perez, Magn. Reson. Chem., 2013, 51, 143–155 CrossRef CAS PubMed.
- M. Hosseini-Sarvari and H. Sharghi, J. Org. Chem., 2006, 71, 6652–6654 CrossRef CAS PubMed.
- R. Quintanilla-Licea, J. F. Colunga-Valladares, A. Caballero-Quintero, C. Rodríguez-Padilla, R. Tamez-Guerra, R. Gómez-Flores and N. Waksman, Molecules, 2002, 7, 662–673 CrossRef CAS.
- (a) M. Borasio, O. Rodríguez de la Fuente, G. Rupprechter and H.-J. Freund, J. Phys. Chem. B, 2005, 109, 17791–17794 CrossRef CAS PubMed; (b) N. Iwasa and N. Takezawa, Top. Catal., 2003, 22, 215–224 CrossRef CAS; (c) J. Lichtenberger, D. Lee and E. Iglesia, Phys. Chem. Chem. Phys., 2007, 9, 4902–4906 RSC; (d) N. Kruse, M. Rebholz, V. Matolin, G. K. Chuah and J. H. Block, Surf. Sci., 1990, 238, L457–L462 CrossRef CAS; (e) J. L. Davis and M. A. Barteau, Surf. Sci., 1990, 235, 235–248 CrossRef CAS; (f) G. Rupprechter, Catal. Today, 2007, 126, 3–17 CrossRef CAS; (g) G. Rupprechter, Adv. Catal., 2007, 51, 133–263 CAS; (h) R. Jiang, W. Guo, M. Li, D. Fu and H. Shan, J. Phys. Chem. C, 2009, 113, 4188–4197 CrossRef CAS; (i) S. Lee, B. Lee, F. Mehmood, S. Seifert, J. A. Libera, J. W. Elam, J. Greeley, P. Zapol, L. A. Curtiss, M. J. Pellin, P. C. Stair, R. E. Winans and S. Vajda, J. Phys. Chem. C, 2010, 114, 10342–10348 CrossRef CAS; (j) S. Sá, H. Silva, L. Brandão, J. M. Sousa and A. Mendes, Appl. Catal., B, 2010, 99, 43–57 CrossRef; (k) X.-K. Gu and W.-X. Li, J. Phys. Chem. C, 2010, 114, 21539–21547 CrossRef CAS; (l) R. W. McCabe and P. J. Mitchell, J. Catal., 1987, 103, 419–425 CrossRef CAS; (m) D. M. Pearson and R. M. Waymouth, Organometallics, 2009, 28, 3896–3900 CrossRef CAS; (n) R. Wojcieszak, E. M. Gaigneaux and P. Ruiz, ChemCatChem, 2012, 4, 72–75 CrossRef CAS; (o) R. Wojcieszak, E. M. Gaigneaux and P. Ruiz, ChemCatChem, 2013, 5, 339–348 CrossRef CAS; (p) R. Wojcieszak, M. N. Ghazzal, E. M. Gaigneaux and P. Ruiz, Catal. Sci. Technol., 2014, 4, 738–745 RSC; (q) R. Wojcieszak, A. Karelovic, E. M. Gaigneaux and P. Ruiz, Catal. Sci. Technol., 2014, 4, 3298–3305 RSC; (r) W. Tu and Y. H. Chin, ACS Catal., 2015, 5, 3375–3386 CrossRef CAS; (s) K. Czelej, K. Cwieka, J. C. Colmenares and K. J. Kurzydlowski, Langmuir, 2016, 32, 7493–7502 CrossRef CAS PubMed.
- M. L. Cunningham, H. B. Matthews and L. T. Burka, Chem. Res. Toxicol., 1990, 3, 157–161 Search PubMed.
- M. L. Cunningham, L. T. Burka and H. B. Matthews, Mutat. Res., 1990, 244, 273–277 CAS.
- H. Y. Choi, M. K. Yoo and J. H. Heo (Dongwoo Fine-Chem Co, Ltd.), Int. PatWO2015099285, 2015 Search PubMed.
- P. Foley (Celanese Corporation), US Patent4544507, 1985 Search PubMed.
- (a) X. Zhao, N. Wang, Y. Geng, H. An and Y. Wang, Ind. Eng. Chem. Res., 2011, 50, 13636–13641 CrossRef CAS; (b) S. Sun, N. Liang, H. An, X. Zhao, G. Wang and Y. Wang, Ind. Eng. Chem. Res., 2013, 52, 7684–7689 CrossRef CAS.
- (a) P. Tundo and V. Esposito, Green Chemical Reactions, Springer Verlag Dordrecht, 2008 CrossRef; (b) P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed; (c) P. Tundo, L. Rossi and A. Loris, J. Org. Chem., 2005, 70, 2219–2224 CrossRef CAS PubMed.
- (a) R. Juárez, P. Concepción, A. Corma, V. Fornés and H. García, Angew. Chem., Int. Ed., 2010, 49, 1286–1290 ( Angew. Chem. , 2010 , 122 , 1308–1312 ) CrossRef PubMed; (b) B. Puértolas, M. Rellán-Piñeiro, J. L. Núñez-Rico, A. P. Amrute, A. Vidal-Ferran, N. López, J. Pérez-Ramírez and S. Wershofen, ACS Catal., 2019, 9, 7708–7720 CrossRef.
- (a) S. Laursen, D. Combita, A. B. Hungría, M. Boronat and A. Corma, Angew. Chem., Int. Ed., 2012, 51, 4190–4193 ( Angew. Chem. , 2012 , 124 , 4266–4269 ) CrossRef CAS PubMed; (b) M. I. Zaki, M. A. Hasan and L. Pasupulety, Langmuir, 2001, 17, 768–774 CrossRef CAS; (c) M. I. Zaki, M. A. Hasan, F. A. Al-Sagheer and L. Pasupulety, Colloids Surf., A, 2001, 190, 261–274 CrossRef CAS; (d) L. Ferretto and A. Glisenti, Chem. Mater., 2003, 15, 1181–1188 CrossRef CAS; (e) D. Martin and D. Duprez, J. Phys. Chem., 1996, 100, 9429–9438 CrossRef CAS.
- J. Callison, F. Betzler, K. de Cuba, W. van der Borden, K. van der Velde, R. H. Carr, H. M. Senn, L. J. Farrugia, J. M. Winfield and D. Lennon, Ind. Eng. Chem. Res., 2012, 51, 11021–11030 CrossRef CAS.
- (a) F. Lortie, S. Boileau and L. Bouteiller, Chem. – Eur. J., 2003, 9, 3008–3014 CrossRef CAS PubMed; (b) C. Grundmann and H.-D. Frommeld, J. Org. Chem., 1966, 31, 157–162 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02412k |
| This journal is © The Royal Society of Chemistry 2020 |