
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-driven isomerization of cis,cis-muconic acid for the production of specialty and performance-advantaged cyclic biobased monomers†
Jack M.
Carraher‡
 ab,
Prerana
Carter‡
ab,
Radhika G.
Rao
ab,
Michael J.
Forrester
a,
Toni
Pfennig
ab,
Brent H.
Shanks
ab,
Eric W.
Cochran
a and
Jean-Philippe
Tessonnier
*ab
ab,
Prerana
Carter‡
ab,
Radhika G.
Rao
ab,
Michael J.
Forrester
a,
Toni
Pfennig
ab,
Brent H.
Shanks
ab,
Eric W.
Cochran
a and
Jean-Philippe
Tessonnier
*ab
aDepartment of Chemical and Biological Engineering, Iowa State University, Ames, IA 50011, USA. E-mail: tesso@iastate.edu
bCenter for Biorenewable Chemicals (CBiRC), Ames, IA 50011, USA
First published on 3rd September 2020
Abstract
The quest for green plastics calls for new routes to aromatic monomers using biomass as a feedstock. Suitable feedstock molecules and conversion pathways have already been identified for several commodity aromatics through retrosynthetic analysis. However, this approach suffers from some limitations as it targets a single molecule at a time. A more impactful approach would be to target bioprivileged molecules that are intermediates to an array of commodity and specialty chemicals along with novel compounds. Muconic acid (MA) has recently been identified as a bioprivileged intermediate as it gives access to valuable aliphatic and cyclic diacid monomers including terephthalic acid (TPA), 1,4-cyclohexanedicarboxylic acid (CHDA), and novel monounsaturated 1,4-cyclohexenedicarboxylic acids (CH1DA, CH2DA). However, accessing these cyclic monomers from MA requires to first isomerize biologically-produced cis,cis-MA to Diels–Alder active trans,trans-MA. A major impediment in this isomerization is the irreversible ring closing of MA to produce lactones. Herein, we demonstrate a green solvent-mediated isomerization using dimethyl sulfoxide and water. The mechanistic understanding achieved here elucidates the role of low concentrations of water in reducing the acidity of the system, thereby preventing the formation of lactones and improving the selectivity to trans,trans-MA from less than 5% to over 85%. Finally, a Diels–Alder reaction with trans,trans-MA is demonstrated with ethylene. The monounsaturated cyclic diacid obtained through this reaction (CH1DA) can be converted in a single step into TPA and CHDA, or can be directly copolymerized with adipic acid and hexamethylenediamine to tailor the thermal and mechanical properties of conventional Nylon 6,6.
Introduction
The polymer industry has become a key driver for the research and development of biorenewable chemicals due to end user appeal for green plastics.1–4 For instance, biobased terephthalic acid (TPA) is receiving significant attention due to its central role in the production of polyethylene terephthalate (PET), a commodity polyester broadly used in bottling and packaging.5–17 Other cyclic targets include specialty chemical 1,4-cyclohexanedicarboxylic acid (CHDA), which is gaining attention for tuning properties in polyesters while enhancing their sustainability.18,19 The market size for these cyclic building blocks is currently valued at $60 billion and is expected to further grow at an annual rate of 5%.20,21 Therefore, even a conservative 5% replacement by biobased drop-in chemicals represents tremendous opportunities for the emerging biorenewable chemical industry.Technologies developed to access cyclic molecules from renewable feedstocks have largely used retrosynthesis strategies. In the case of biobased TPA production, research has primarily focused on renewable p-xylene as a drop-in replacement for catalytic oxidation in the AMOCO process.6–8,13,22,23 An alternate route towards these cyclic diacids is through muconic acid (MA), an intermediate produced from either sugar or lignin using metabolically engineered yeasts and bacteria.24–28 The latter approach is particularly attractive as MA is a bioprivileged molecule with substantial potential for diversification to commodity and specialty chemicals, as well as novel molecules for enhanced end-use properties (Scheme 1).29–31 Previous work has already demonstrated the conversion of MA to an array of aliphatic commodity monomers including adipic acid and hexamethylenediamine,24,25,32–36 cyclic monomers such as ε-caprolactam,37,38 TPA and CHDA,5,19,39,40 and novel monounsaturated compounds such as 3-hexenedioic acid and 1,4-cyclohex-1/2-enedicarboxylic acid (CH1DA, CH2DA).19,41–44
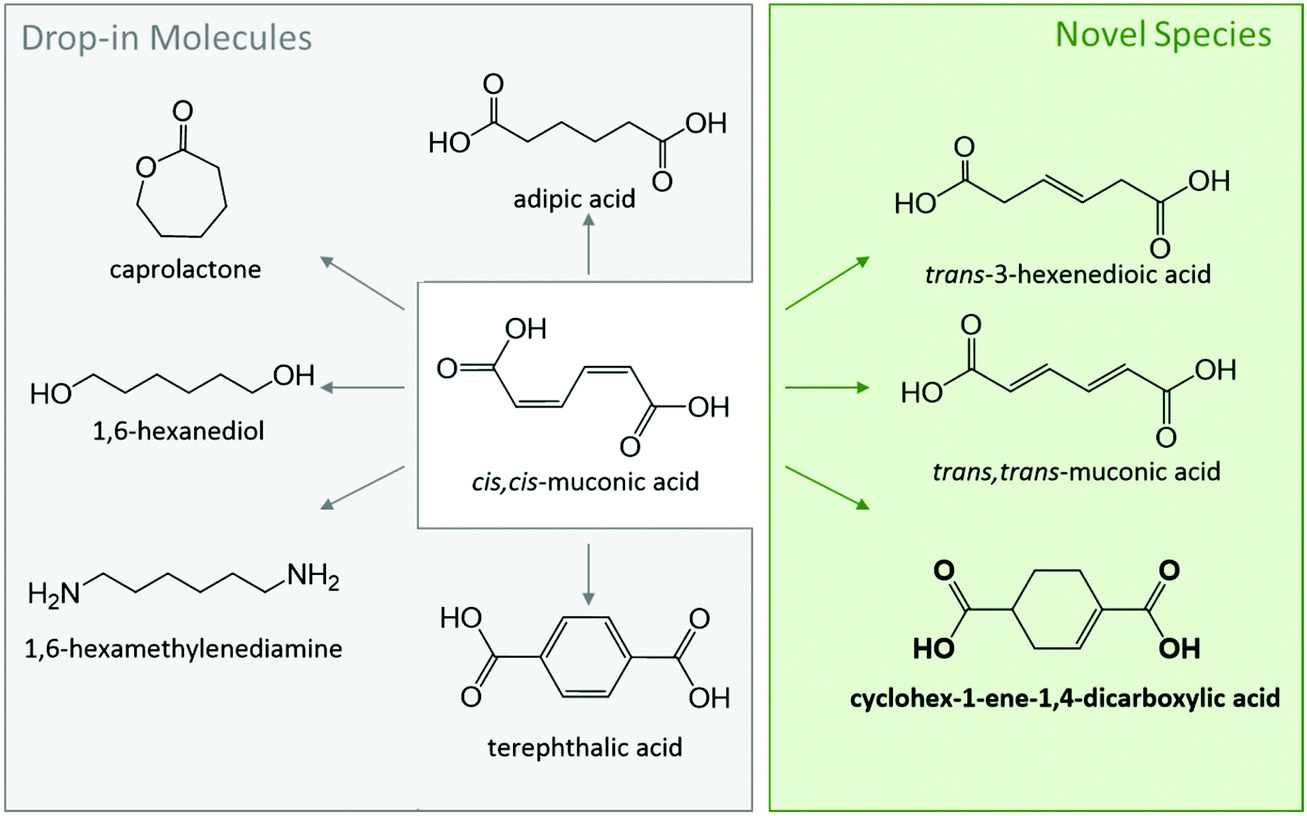 | ||
| Scheme 1 Bioprivileged molecule ccMA produced biologically from sugar or lignin can be diversified into various drop-in and novel species. | ||
Although the downstream production of cyclic molecules has experienced some significant advances,39,45 the initial isomerization of biologically-produced cis,cis-muconic acid (ccMA) to Diels–Alder active trans,trans-muconic acid (ttMA) remains a major bottleneck.46 Only a handful of MA isomerization technologies have been reported in the literature. Notable ones use catalysts such as Pd/C (methanol reflux) or iodine either under UV light or in solvents such as tetrahydrofuran, methanol, or acetonitrile.40,47–49 Various disadvantages, such as cost of noble metal catalysts, low selectivity, feed concentration limitations (e.g., 10 wt% MA for the iodine system), hinder them from reaching the commercial realm.5 The key obstacle in this seemingly simple isomerization is the spontaneous ring-closing of cis,cis- and cis,trans-MA (ctMA) to form the muconolactone (Mlac) and dilactone (Lac2). A previous report published by our group detailed the driving forces behind this lactonization when the isomerization is performed in water, and suggested several strategies to circumvent those driving forces.19
The present work investigates an organic solvent-mediated isomerization and demonstrates its clear potential to be scaled. It is shown here that the addition of water to the green aprotic solvent dimethyl sulfoxide (DMSO),50,51 enhances selectivity to ttMA (SttMA) by 13-fold at 20% conversion. A comprehensive mechanistic understanding of the role of water was achieved through a series of reactions with ctMA in DMSO/water solvent systems. As a proof of concept, ttMA was reacted with ethylene to produce the novel unsaturated molecule 1,4-cyclohex-1-enedicarboxylic acid (CH1DA); finally, a novel polyamide was formed through copolymerization with adipic acid and hexamethylenediamine. Overall, this study demonstrates a green, solvent-driven, scalable isomerization of MA in order to synthesize renewable cyclic diacids.
Results and discussion
While ccMA readily isomerizes to ctMA at room temperature,19,52 further isomerization to Diels–Alder active ttMA is hindered by the intramolecular lactonization between unsaturated carbon–carbon and carboxylic acid moieties. We previously showed that the aprotic solvent DMSO impedes this lactonization and allows for high SttMA (88%).19 To investigate this conversion on a more industrially relevant scale, a sample with a ccMA concentration on the order of 300 g L−1 (∼2 M concentration) was prepared in DMSO-d6. Despite 1H NMR spectra at such a high concentration being qualitative at best, heating a highly concentrated solution of ccMA at 100 °C overnight (t½ ∼ 2–4 hours) yielded primarily Mlac with no indication of the expected ttMA product. This result contrasted with our prior study,19 indicating an effect of reactant concentration on the isomerization reaction. Therefore, this system was investigated kinetically in order to develop optimization strategies for the isomerization to ttMA.Effect of muconic acid concentration in dry DMSO-d6
The experiments in this study used ctMA as the starting material, with concentrations ranging from 5 to 80 mM. Reactions were typically performed at 87 °C unless stated otherwise. The product selectivity at 20% and 50% conversion was determined as a function of initial ctMA concentration ([ctMA]0) as shown in Table 1 and Fig. 1. Maximum SttMA (19%) was observed at low conversion (ca. 20%) and the lowest [ctMA]0. SttMA gradually decreased from 19% to 3% as [ctMA]0 increased from 5 mM to 79 mM at 20% conversion. Additionally, SttMA at [ctMA]0 of 5 mM decreased from 19% to roughly 15% as conversion increased from 20% to 50%. Several new signals consistent with MA degradation and formation of unidentified products were observed for all [ctMA]0. This degradation was observed to occur more readily at elevated ctMA concentration. The significant drop in SttMA with increasing [ctMA]0 suggests an acid catalyzed pathway for lactonization analogous to the aqueous system.19 However, in this particular system, muconic acid is both the reactant and the catalyst as is shown in Scheme 2. | ||
| Fig. 1 Effect of [ctMA]0 on yields at 20% conversion (left axis) and the observed rate constant (right axis). 5–80 mM ctMA solutions in DMSO-d6 and heated to 87 °C. Kinetic traces and product distributions were obtained by 1H NMR monitoring with DMSO2 internal standard. | ||
 | ||
| Scheme 2 Bimolecular pathway for muconic acid catalyzed lactonization scheme. | ||
| [ctMA]0 (mM) |
k
obs×106![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (s−1) a (s−1) |
Half-life (days) | Selectivity (20% conversion) | Selectivity (50% conversion) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ccMA | ttMA | Mlac | Lac2 | ccMA | ttMA | Mlac | Lac2 | |||
| a k obs was determined at low conversion (ca. 20%) from eqn (1). Error in kobs was determined by least squares method to be ±1 × 10−7 s−1. | ||||||||||
| 5.5 | 2.6 | 3.1 | 0.8% | 19.0% | 35.0% | 37.3% | 0.4% | 14.6% | 26.0% | 31.0% |
| 29.0 | 1.9 | 4.2 | 6.3% | 12.0% | 51.5% | 25.0% | 1.6% | 6.4% | 34.8% | 34.2% |
| 45.0 | 1.8 | 4.4 | 6.5% | 6.3% | 55.0% | 26.0% | 1.8% | 4.0% | 37.6% | 35.6% |
| 79.0 | 2.1 | 3.8 | 7.5% | 3.0% | 47.5% | 15.0% | 1.6% | 3.0% | 36.4% | 32.4% |
Kinetic traces obtained for the consumption of ctMA are consistent with a first order rate equation (eqn (1)) for all [ctMA]t.
| ln([ctMA]t/[ctMA]0) = −kobs × t + const. | (1) |
Fig. 1 shows the observed rate constants (kobs) obtained from eqn (1) plotted against [ctMA]0. The trend for kobs with increasing [ctMA]0 is somewhat peculiar and initially unexpected as the isomerization of ccMA and ctMA to ttMA was shown to be unimolecular in an aqueous system.19 Therefore, an acid-catalyzed pathway that generates lactones would be expected to generate a linear plot with a y-intercept equal to the sum of all rate constants for unimolecular MA reactions and a slope corresponding to kacid-catalysed (M−1 s−1). The unimolecular reaction pathways would include isomerization, unimolecular lactonization, and unimolecular degradation (Scheme 3). However, kobs was found to initially decrease with increasing [ctMA]0 between 5 and 45 mM and then increase between 45 and 79 mM. The initial decrease at low [ctMA]0 suggested an equilibrium with a non-reactive species like an unreactive MA complex. Though kinetically observable, the non-reactive complex was not identifiable spectroscopically. At higher [ctMA]0 the acid catalyzed pathway shown in Scheme 2 appeared to be dominant. In addition to higher Mlac yields, elevated [ctMA]0 also resulted in significantly higher degradation to unidentified byproducts. Clearly, in the absence of water, this system is rather complex and will not offer high SttMA under industrially relevant conditions (i.e. high concentration). It was therefore decided to minimize efforts in this system and focus on approaches that take into consideration the effect of [ctMA]0: (i) an acid-catalyzed lactonization pathway and (ii) the potential for reversible formation of a non-reactive complex/es. The former is more important as it clearly dominates near the solubility limit. It was therefore concluded that decreasing the apparent acidity should improve SttMA.
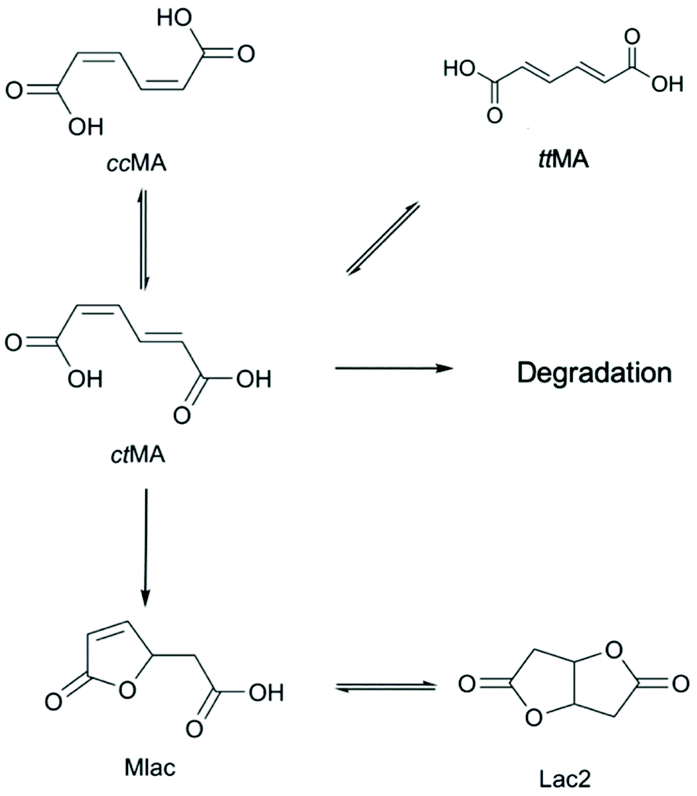 | ||
| Scheme 3 Unimolecular reaction pathways for ctMA. | ||
Effect of low water concentrations on muconic acid isomerization
The Dumesic group demonstrated that the addition of water to solid-acid catalyzed systems in DMSO and γ-valerolactone (GVL) can significantly decrease the activity of the catalyst.53 The decrease in catalyst activity could be attributed to a more widespread hydrogen bonding network near the active sites, which effectively decreases their pKa. This rationale was extended to our reaction system by introducing water to minimize the acid-catalyzed lactonization of ctMA. The experiments in this section were carried out using 48 mM ctMA in dry DMSO-d6 with varying concentrations of water ([H2O]). 1H NMR spectra of samples containing water (5 mM–744 mM) indicated that MA was in its fully protonated (ctMAH2) form under all conditions.At very low [H2O] the reactivity of ctMA was similar to that of the dry DMSO system at 20% conversion (compare 45 mM ctMA in Table 1 with 5 mM H2O in Table 2). SttMA at 96 mM H2O (H2O:MA ∼ 2:1) increased 13-fold to 81.5% (88% if conversion is considered as conv. = [ctMA]0 − ([ctMA]t + [ccMA]t)/[ctMA]0 due to the relatively rapid ccMA ↔ ctMA equilibrium). For [H2O] of 96 mM, high SttMA (82%) was maintained up to 50% conversion. Additionally, throughout this reaction the 1H NMR signal of H2O (3.34 ppm) gradually decreased with increase in conversion as shown in Fig. 2, until it was not observable after 52% conversion. The majority of the samples showed an increased SttMA with decreasing H2O signal, suggesting that an optimal ratio of H2O:MA exists for a highly selective production of ttMA (≥75%). This optimal range appears to be broad, varying from a H2O:MA ratio of 0.9:1 to 10:1.
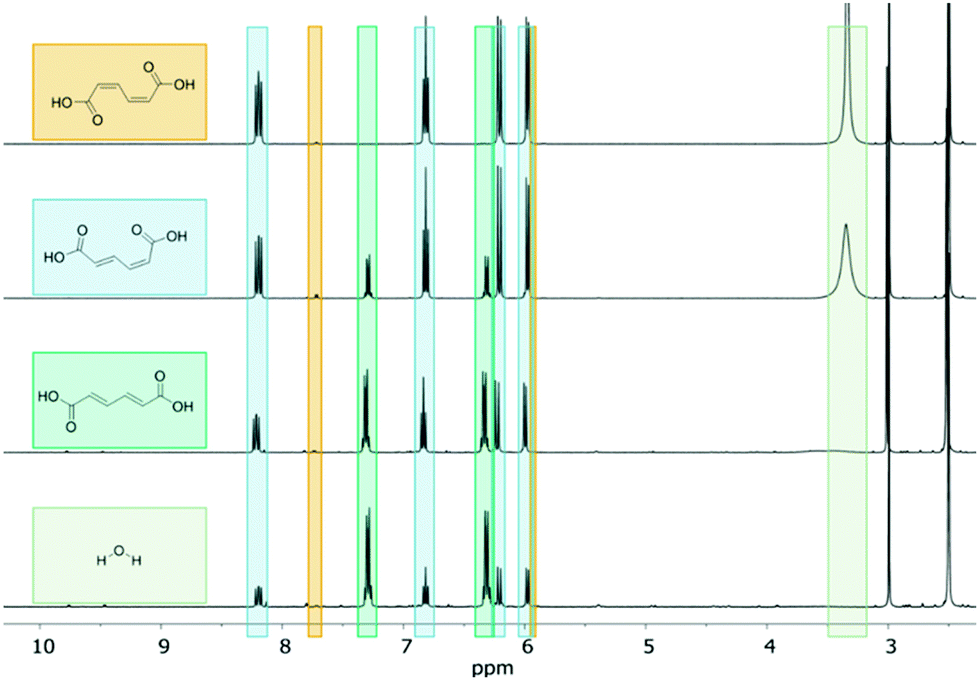 | ||
| Fig. 2 1H NMR (600 MHz, DMSO-d6) spectra of 48 mM ctMA + 96 mM H2O heated to 87 °C at 0 (top), 18.5, 52, and 72.5% (bottom) conversion. | ||
| [H2O]0 (mM) | 106 × kobsb (s−1) | Half-life (days) | Selectivity at 20% conversion | Selectivity at 50% Conversion | ||||
|---|---|---|---|---|---|---|---|---|
| ccMA | ttMA | Mlac | ccMA | ttMA | Mlac | |||
| a Selectivities at 20 and 50% conversion for 48 mM ctMA in DMSO-d6 with varying concentrations of water at 90 °C. b k obs acquired from fits to eqn (1) at 20% ctMA conversion. c 5 mM H2O sample also contains Lac2. | ||||||||
| 5.0 | 1.8 | 4.5 | 6.5% | 6.3% | 55.0%c | 1.8% | 4.0% | 37.6% |
| 37.7 | 1.4 | 5.9 | 7.5% | 70.0% | 3.5% | 1.8% | 69.8% | 2.8% |
| 55.6 | 1.4 | 5.9 | 7.5% | 75.0% | 3.0% | 1.8% | 71.2% | 2.8% |
| 70.1 | 1.3 | 6.2 | 7.5% | 77.5% | 4.0% | 1.9% | 79.0% | 2.7% |
| 74.6 | 1.5 | 5.4 | 7.5% | 76.0% | 3.0% | 1.8% | 78.2% | 2.6% |
| 96.1 | 1.5 | 5.5 | 7.5% | 81.5% | 3.0% | 1.8% | 82.0% | 2.6% |
| 115.8 | 1.7 | 4.7 | 7.5% | 80.0% | 2.5% | 1.8% | 70.0% | 2.4% |
| 157.4 | 1.6 | 5.0 | 7.5% | 80.0% | 2.5% | 1.8% | 72.0% | 2.2% |
| 322.0 | 1.6 | 5.0 | 7.5% | 75.0% | 4.5% | 2.1% | 83.7% | 2.5% |
| 478.0 | 1.6 | 5.0 | 7.5% | 75.0% | 4.5% | 2.4% | 85.7% | 2.4% |
| 744.0 | 1.6 | 5.0 | 7.0% | 70.0% | 3.0% | 2.4% | 79.0% | 2.4% |
Kinetically, the conversion of ctMA follows first order rate equations. As the water content was increased, there was an initial 20% drop in kobs between 5 mM and 38 mM, followed by a gradual increase (70–120 mM H2O), and a plateau from 120 mM to 744 mM. The initial decrease was expected under the assumption that an acid-catalyzed lactonization pathway exists in parallel to unimolecular isomerization, and that introduction of water to the system would decrease the apparent pKa. The sigmoidal shape observed in Fig. 3 could be indicative of a ctMA-2H2O complex that is roughly 20% more active for isomerization to ttMA than a ctMA-H2O complex. The plateau achieved at high [H2O] coupled with decreasing H2O signal and steady decline in SttMA above 160 mM H2O could also be indicative of H2O being a potential reactant in the degradation pathway.
 | ||
| Fig. 3 Effect of water content on yields at 20% conversion (left axis) and the observed rate constant (right axis). Microliter amounts of water were added to 600 μl of 48.0 mM ctMA solutions in DMSO-d6 and heated to 87 °C. Kinetic traces and product distributions were obtained by 1H NMR monitoring with DMSO2 internal standard. | ||
Probing the effect of muconic acid at low water concentration
To probe the effect of [ctMA]0 on the kinetics and yields at low water concentrations, these experiments were carried out with D2O:MA of (1.9–2.6):1 ([ctMA]0 ranging from 0.049–0.16 M). Under these conditions the water should interact primarily with the carboxyl moieties, lowering their apparent acidity and minimizing the acid-catalyzed lactonization shown in Scheme 2. This assumption is supported by NOESY NMR experiments showing proximity between the carboxyl moieties and H2O in the system (ESI 1†). However, it should be noted that the NOESY spectrum is not definitive proof for the absence of H2O association with internal carbon atoms.
The selectivity to cc- and ttMA at 20% conversion remained relatively constant between 50 and 160 mM ctMA (ca. 6 and 75–80%, respectively), but selectivity to Mlac increased 6-fold (Fig. 4). Additionally, kobs for ctMA consumption decreased with increasing [ctMA]0, similar to that observed under dry DMSO. However, unlike the trend observed in Fig. 1, the rate constants did not increase again at higher [ctMA]0. This observation supports the non-reactive complex hypothesis presented above.
 | ||
| Fig. 4 (a) Effect of [ctMA]0 with 2–2.6 equivalents of H2O (column 1) and D2O (columns 2–4) on yields at 20% conversion (left axis) and the observed rate constant (right axis) for ctMA solutions in DMSO-d6 heated to 87 °C. Kinetic traces and product distributions were obtained by 1H NMR monitoring with DMSO2 internal standard. (b) Comparison of Mlac yield in H2O and D2O. | ||
Effect of high water concentrations on muconic acid isomerization
D2O in concentrations ranging from 1.8 M to 16.0 M was added to a 50 mM ctMA solution resulting in a decrease in kobs for ctMA consumption from 2.1 × 10−6 s−1 to 1.0 × 10−6 s−1, as shown in Fig. 5. ttMA yields at 20% conversion remained relatively constant (15 ± 1%) up to 8.5 M D2O but were halved to 7.5% at 16 M D2O. This was accompanied by a slight decrease in equilibrium concentrations of ccMA and a 10-fold increase in Mlac yields (Fig. 5). Mlac yields rose steadily from 0.1% to 0.5% as [D2O] increased from 1.8 to 8.5 M, and then jumped to 5.5% at 16 M D2O. Rate constants obtained by initial rates analysis for Mlac formation (eqn (2)) were plotted as a function of [D2O]. The results suggested a ternary reaction in which water promotes lactonization through a push–pull type interaction, where one water molecule donates a proton to the δ carbon of the cis-carboxylic acid while another accepts the proton from the cis-carboxylic acid (Scheme 4). This observation is consistent with the large negative entropy of activation (ΔS‡ = −110 J (mol K)−1) obtained from simulations fitted to experimental data for the same reaction in an aqueous system.19 The rate constant for Mlac formation at 16 M D2O obtained from eqn (2) is consistent with the aqueous simulations (4 ± 2 × 10−7 s−1 at 16 M D2O and 3 × 10−7 s−1 at 55 M).19 This of course is only consistent if the reaction reaches kinetic saturation near 16 M D2O (i.e. D2O:ctMA = 320:1). The elementary rate constant for the ternary reaction shown in Scheme 4 was calculated to be 2 ± 1 × 10−9 M−2 s−1.| rateMlac = [Mlac]t/t → kMlac = rateMlac/{½([ctMA]0 − [ctMA]t)} | (2) |
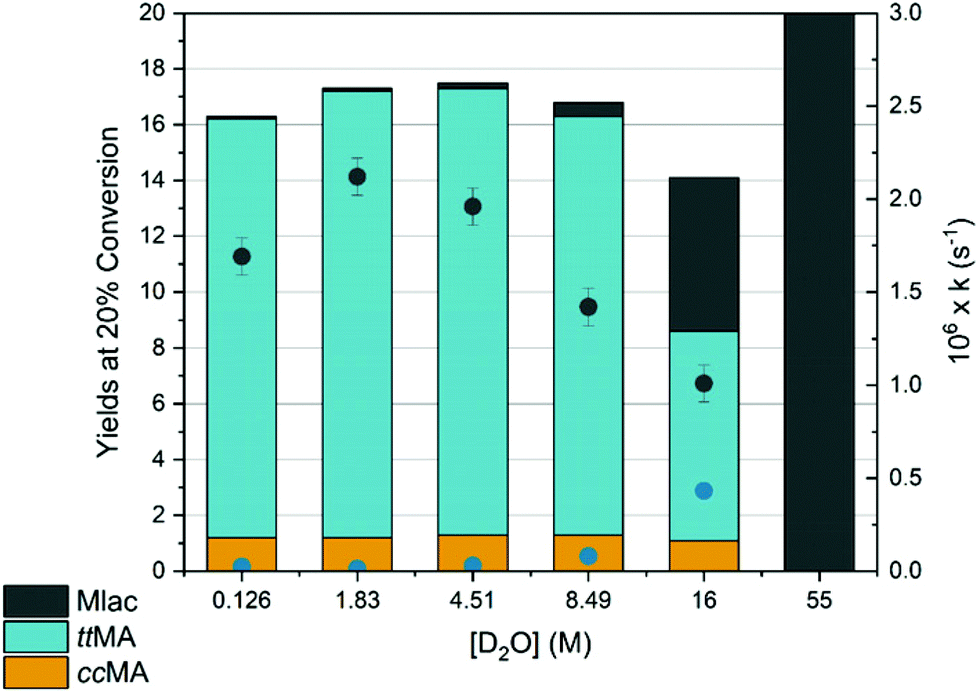 | ||
| Fig. 5 Effect of high water content on yields at 20% conversion (left axis). The observed rate constant for ctMA conversion (black ●, right axis) was obtained from eqn (1) and the observed rate constant for Mlac formation (blue ●, right axis) was estimated from eqn (2). ca. 50 mM ctMA solutions in DMSO-d6 + D2O were heated to 87 °C. Kinetic traces and product distributions were obtained by 1H NMR monitoring with DMSO2 internal standard. The column at 55 M D2O is from a previous experiment and the rate constant obtained is from simulations fit to experimental data in an aqueous system.19 | ||
 | ||
| Scheme 4 Ternary lactonization process with ctMAH2. | ||
In addition to the solvent driven lactonization, higher water content also showed a decrease in the rate constant for ttMA formation. The rate constant kttMA was obtained by the same method as kMlac shown in eqn (2). These rate constants were relatively unaffected up to 8.5 M D2O but decreased by nearly a factor of 5 from 8.5 to 16 M D2O. At 16 M D2O kttMA was roughly equivalent to kMlac whereas at 8.5 M D2O kttMA = 84 × kMlac. The increase in kMlac with [D2O] was expected for the reaction outlined in Scheme 4, however, the driving forces behind this significant decrease in kttMA were unclear. At 121 °C the ctMA to ttMA reaction was found to be reversible, vide infra. It is possible that water has an effect on the forward and/or reverse reaction(s). Therefore, the reactivity of 50 mM ttMA at 1.8 and 16 M D2O was tested, which confirmed reversibility at 87 °C. Comparison of kobs starting from both ctMA and ttMA yielded apparent equilibrium constants (Kapp) that vary with [D2O], Kapp = 7.2 and 1.5 at 1.78 M and 16 M D2O, respectively. The effect of water concentration on this equilibrium is exclusively a result of high water content slowing the forward reaction (ct → tt) as the reverse reaction (tt → ct) rate constant was the same at both 1.8 and 16 M D2O (ktt–ct = 3 ± 1 × 10−7 s−1).
The solvent driven lactonization at high water concentration in conjunction with the significant decrease in kttMA at 16 M D2O (neat D2O = 55 M) would help explain why ttMA is never observed in an aqueous system in the absence of catalyst like La3+.19
Effects of temperature & kinetic simulations
An investigation into the effects of temperature on the reaction system with ca. 2 equivalents of water was carried out. Product yields at 20% conversion and kobs for ctMA conversion are shown in Fig. 6. SttMA was relatively high for all three temperatures; 88, 80, and 88% at 60, 87, and 121 °C, respectively (91, 88, and 93% considering the ccMA ↔ ctMA equilibrium). The reaction at 121 °C was fast enough (t½ = 5.2 weeks, 5.2 days, and 1.7 hours at 60, 87, and 121 °C, respectively) that we were able to achieve equilibrium between ctMA and ttMA. While this is reasonable, it was surprising due to previous observations in which aged aqueous solutions of ttMA and ttMA in DMSO with no added water at 200 °C do not appear to yield ctMA. To probe the temperature effects further, kinetic simulations were run with KINSIM software in accordance with the simplified Scheme 4. Estimates for elementary rate constants were input based on the experimental values obtained from treatment of the data with either eqn (1) or (2). The rate constants were finely tuned until simulated kinetic traces matched experimental ones for all species, including the loss of signal due to degradation from ctMA. The equilibrium constants, rate constants, and activation parameters are shown in Table 3. While the simulations starting from ctMA fit well with the experimentally obtained kinetic traces, simulating the reverse reaction (e.g. starting from ttMA) required decreasing kcc–tt by a factor of 10. | ||
| Fig. 6 Experimental (■) and simulated (—) traces of ctMA isomerization at 2 equivalents of D2O at (a) 60 °C (b) 87 °C (c) 121 °C. | ||
a
| Reaction | Temperature (°C) | Activation parametersd | |||
|---|---|---|---|---|---|
| 60 | 87 | 121 | ΔH‡ kJ mol−1 | ΔS‡ J (mol K)−1 | |
| a All reactions were carried out in DMSO-d6 with 1.9–2.5 equivalents of D2O relative to 50 mM ctMA. b Equilibrium constants expressed with the subscript ‘isomer1–isomer2’ are for reversible isomerization and should not be confused with equilibrium constants with the subscript ‘isomer1,isomer2’ mentioned above as they represent K for formation of non-reactive complexes of the different isomers. c K ct–tt is an apparent equilibrium constant derived from kinetic information about the forward and reverse reactions obtained from fitting the simulation to experimental values and does not represent the thermodynamic equilibrium. d Activation parameters are calculated for the reactions outlined in Scheme 3 and are specific to the system at 50 mM ctMA with 2 equivalents of H2O. They do not take into consideration the equilibrium formation of potential non-reactive complexes. | |||||
| K cc–ct | 68 | 55 | 50 | — | — |
| K ct–tt , | 1.7 | 2.4 | 4.7 | — | — |
| k ct→tt | 2.5 × 10−7 | 1.2 × 10−6 | 1.4 × 10−4 | 110 ± 31 | −44 ± 85 |
| k tt→ct | 1.5 × 10−7 | 5.0 × 10−7 | 3.0 × 10−5 | 92 ± 28 | −103 ± 78 |
| k ct→Mlac | 4.0 × 10−9 | 3.1 × 10−8 | 6.0 × 10−6 | 128 ± 31 | −25 ± 86 |
| k ct→deg | 2.0 × 10−8 | 3.5 × 10−7 | 3.0 × 10−5 | 128 ± 14 | −11 ± 39 |
Perhaps non-reactive complexes dependent upon [MA] exist for each of the isomers (see next section on ccMA). The equilibria achieved for these unreactive complexes will likely have equilibrium constants that are dependent upon the specific isomer pair. That is to say Kcc,cc may be significantly different than Kct,ct, Ktt,tt, Kcc,ct, Kcc,tt, and Kct,tt. The focus of this investigation, however, is the unprecedented catalyst-free isomerization of ctMA to ttMA. Therefore, a more thorough study of these potential non-reactive complexes was not carried out at this time.
As a high SttMA was achieved with 2 equivalents of D2O at 121 °C (Fig. 6c), these conditions were selected to investigate the isomerization of ctMA under more industrially relevant conditions, namely at 70 and 300 g L−1 (ca. 0.5 and 2.0 M). In contrast to the experiment in dry DMSO-d6 that prompted this study (vide supra), the reaction with 2.0 M ctMA and 2 equivalents of D2O produced minute amounts of Mlac. However, the reaction quickly reached an equilibrium between ctMA and ttMA favoring ctMA (nearly 75:1 at 121 °C). This [ctMA]0 effect was not observed at 70 g L−1 (∼0.5 M), a concentration equivalent to the highest titers reported to date for biologically-produced ccMA. SttMA reached 78% at 50% conversion, which is similar to the selectivity achieved with a 48 mM solution (Table 2).
Isomerization from cis,cis-muconic acid
As was the case with ctMA, 1H NMR spectra obtained of solutions initially containing cc- and ttMA were consistent with the fully protonated form (MAH2). These experiments are outlined in Table 4. Kinetic traces for the isomerization of ccMA to ctMA were fitted to 1st order rate equations with R2 values ≥97%. Values for kobs decreased with increasing [ccMA]0 in the presence of 9 mM H2O. This is consistent with the potential formation of a non-reactive MA complex and the lack of an acid catalyzed pathway for ccMA.19 Addition of D2O to the system increased kobs implicating water as a non-innocent solvent consistent with the aqueous ΔS‡ = −88 J (mol K)−1 for cc- to ctMA isomerization of MAH2.19 This effect appears to reach saturation at high H2O:ccMA ratios. Unlike experiments in which ctMA was the initial isomer, no loss of signal was observed with reaction time, and selectivity to ctMA was 100%. This supports the above hypothesis that degradation of MA occurs from the ct-isomer.
| [ccMA]0 (mM) | [D2O] (M) | 107 × kobs (s−1) |
|---|---|---|
| a Isomerization was carried out at 22 ± 1 °C in DMSO-d6 and monitored by 1H NMR. Selectivity to ctMA was 100% and signal loss due to degradation was not observed. | ||
| 5 | 0.009 | 1.6 |
| 20 | 0.009 | 0.8 |
| 5 | 4.9 | 4.1 |
| 4 | 9.8 | 4.6 |
| 3 | 21.5 | 4.6 |
Isomerization from biobased cis,cis-muconate
The robustness of the isomerization process, specifically its tolerance to impurities, was investigated using a fermentation broth containing 70 g L−1 (∼0.5 M) of cis,cis-ammonium muconate (ESI†). The broth was first filtered over activated carbon to remove biogenic impurities and organic byproducts. ccMA was subsequently precipitated by acidification of the solution using 5 M sulfuric acid. The precipitate was recovered by filtration, rinsed with a minimum amount (10 ml) of 5 M sulfuric acid to prevent its redissolution, and dried overnight at room temperature. The biobased ccMA was then isomerized to ctMA using the procedure described previously. Further isomerization to ttMA was carried out in DMSO with 1.5 equivalents of H2O. The ttMA yield at 50% conversion reached 34.8%, which is on par albeit lower than the yield obtained for reagent-grade ccMA (39.5%), see Fig. 7. This difference can be at least partly attributed to residual sulfuric acid that catalyzed the lactonization of ccMA during the isomerization step to ctMA, as indicated by 1H NMR (ESI 2 and 3†). This issue could be easily addressed by improving the separation–purification process, which is beyond the scope of the present work. | ||
| Fig. 7 Product distribution and yields obtained at 50% conversion when the reaction was carried out with reagent-grade ccMA and biobased ccMA recovered from the fermentation broth. The reactions were performed with 48.0 mM ctMA solutions in DMSO-d6 and 2 equivalents of H2O and heated to 121 °C. Product distributions were obtained by 1H NMR monitoring with DMSO2 internal standard. | ||
Synthesis and copolymerization of the monounsaturated cyclic diacid CH1DA
Further conversion of Diels–Alder active ttMA to TPA and CHDA have been demonstrated in literature.19,54,55 Here, we show the preparation of novel monounsaturated cyclic diacids and their role on the physical and mechanical properties of Nylon-6,6 when copolymerized with adipic acid and hexamethylenediamine. ttMA underwent cycloaddition with ethylene in GVL to form cyclohex-1-ene-1,4-dicarboxylic acid (CH1DA) with >99% yield. Although cyclohex-2-ene-1,4-dicarboxylic acid (CH2DA) is the expected cycloadduct, CH2DA was not detected as it readily isomerizes in GVL likely due to the stabilization resulting from conjugation (Scheme 5). | ||
| Scheme 5 Reaction between ttMA and ethylene to produce mono-unsaturated cyclic diacid CH1DA. | ||
The synthesized CH1DA was further copolymerized with adipic acid and hexamethylenediamine to demonstrate the potential of this new monomer for tuning the properties of conventional Nylon. To maintain an equimolar ratio between the diacid and diamine, 10 to 25 mol% of adipic acid were replaced by CH1DA (Fig. 8). This range was selected taking into consideration the economic feasibility of using novel chemicals. Gel permeation chromatography (GPC), differential scanning calorimetry (DSC), and dynamic mechanical analysis (DMA) were used to characterize these polymers using Nylon-6,6 as a reference (ESI 4–6†). The corresponding results are summarized in Table 5. The addition of the cyclic diacid into Nylon's aliphatic backbone altered its crystallinity due to the defects introduced by CH1DA sequences. This change reduces the polymer's melting temperature from 261 to 233 °C, which facilitates its processing through blow molding. Moreover, introducing a cyclic molecule in the polymer backbone increased the rigidity of polymer chains, thereby increasing the storage modulus and glass transition temperatures. The addition of CH1DA allows control over Nylon-6,6 thermal properties without sacrificing its mechanical properties, as shown in Table 5. Overall, CH1DA offers a renewable alternative to commercially available products such as INVISTA's Dytek diamines with the additional benefit of providing an unsaturated bond. This unsaturation allows for further functionalization to insert properties such as hydrophobicity and flame retardance directly into Nylon's backbone.28
 | ||
| Fig. 8 Copolymerization of adipic acid (AA) and CH1DA with hexamethylene diamine. | ||
| Sample | M n [kDa] | PDI | T melt [°C] | G′ [GPa] | G′′ [MPa] | T g [°C] |
|---|---|---|---|---|---|---|
| a M n: number-average molecular weight with reference to poly(methyl methacrylate) standards; PDI: polydispersity index; G′, G′′: storage and loss moduli in the glassy plateau at 0 °C; Tg: glass transition temperature calculated from peak of tan(∂). | ||||||
| PA66 | 16.5 | 2.5 | 261.0 | 1.32 | 16.52 | 73.3 |
| CH1DA-10% | 15.8 | 2.1 | 250.2 | 1.65 | 17.39 | 75.8 |
| CH1DA-25% | 12.4 | 2.3 | 232.6 | 1.65 | 10.99 | 80.3 |
Conclusion
A novel solvent-driven, catalyst-free, isomerization of ctMA to ttMA was demonstrated and investigated mechanistically. The lactonization process that is dominant in aqueous media was overcome in solvent systems combining the polar aprotic solvent DMSO and water.19 In dry DMSO, MA generates a significant quantity of lactones through an acid-catalyzed pathway analogous to the aqueous system. This reaction is enhanced at higher [ctMA] due to MA acting as both the catalyst and reactant. The addition of 1 equivalent of water has the effect of reducing the apparent acidity of the system, thereby reducing lactonization. Consequently, the SttMA was increased by 13-fold.The production of ttMA is a relatively slow process, particularly at high [ctMA]. This is believed to be due to the formation of a non-reactive MA complex that slow the kinetics. The system is also limited by the reversibility of the system in DMSO/H2O mixtures, which was not previously observed in the aqueous system. Nevertheless, high yields of ttMA can be obtained at elevated temperatures in a relatively short period of time due to a shift in the ctMA to ttMA equilibrium constant favoring ttMA (nearly 5:1 at 121 °C). Although further increase in temperature would favor isomerization to ttMA, it needed to be balanced with the decomposition of DMSO at elevated temperatures.50
Continued addition of water sheds light on the role of solvent in both isomerization and lactonization for the aqueous system. Previous work supported by simulations and kinetic measurements indicated that the isomerization of ccMA to ctMA and the lactonization of ctMA to Mlac were unimolecular reactions. However, these reactions were both accompanied by relatively large entropies of activation (−88 and −110 J (mol K)−1, respectively).19 While the involvement of water in the isomerization of ccMA is still not definitively shown, it is strongly supported by the increase in kobs with water content and apparent saturation kinetics when D2O was in large excess of MA. Lactonization of ctMA, on the other hand, is strongly supported by the dependence of kMlac on [D2O]. Furthermore, this observed dependence on [D2O] indicates that this transformation is a ternary reaction in which water acts as both a proton donor and a proton acceptor (Scheme 2). In addition to the enhanced lactonization at high [D2O], kct→tt becomes slower while ktt→ct remains unchanged; resulting in a shift in the apparent equilibrium constant to favor ctMA over ttMA. Perhaps this offers an explanation as to why ttMA has never been observed to form in aqueous solutions without the presence of a catalyst.
Under optimal conditions (2 equivalents of water, 121 °C), ctMA is isomerized to ttMA with 88% selectivity, with Mlac and degradation products representing less than 3% of the mixture. The other species in solution are ctMA (in equilibrium with ttMA) and ccMA (in equilibrium with ctMA). In an industrial setting, ccMA and ctMA would be recovered and recycled, making it a scalable, green, and cost-efficient process.
This advancement in MA isomerization technology will not only allow for the development of biobased commodity chemicals such as TPA, but also for novel unsaturated cyclic molecules such as CH1DA. Incorporation of CH1DA in a polyamide backbone enabled tunable properties that could help with processability of polymers. Further modification of the alkene moiety in this diacid could additionally achieve targeted property enhancement in polyesters and polyamides.
Experimental
cis,cis-muconic acid (ccMA), trans,trans-muconic acid (ttMA), DMSO-d6 (99.96% D), γ-valerolactone (GVL), tetrahydrofuran (THF), hexamethylenediamine (HMDA), adipic acid (AA), tetramethylsilane and dimethyl sulfone (DMSO2) were purchased from Sigma-Aldrich. Ethylene gas was purchased from Airgas at 99.5% purity. cis,trans-Muconic acid (ctMA) was prepared by methods previously described in the literature.41 Extra-dry DMSO-d6 was refluxed over CaH2 and stored over sieves.For experiments in which strict water-free conditions were required, a mother solution containing ctMA and DMSO2 (internal standard) with dried DMSO-d6 was prepared in an inert atmosphere box. Experiments in which the effect of [MA] was investigated utilized 600 μL of the mother solution in J. Young tubes containing additional solid ctMA. ctMA concentrations were determined by NMR vs. DMSO2 internal standard in the mother solution. The tubes were sealed, placed in an Erlenmeyer flask with a thermometer, and heated in a laboratory oven. The experiments at 121 °C were carried out using an aluminum heating block equipped with a thermocouple. Samples were removed from the oven, cooled to room temperature, and analyzed by 1H NMR throughout the duration of the experiment. Experiments that investigated the effect of water were prepared in DMSO-d6 that had not been dried. Instead, aliquots of a mother solution containing ctMA and DMSO2 were added to J. Young tubes and H2O was added via syringe (<1 μL–100 μL). The water concentration was determined by 1H NMR signal relative to DMSO2 standard. High water concentration experiments (>1 M) utilized D2O to minimize interference with 1H NMR spectra. D2O was added to the mother solution with an electronic micropipette (>100 μL). Dilution of [ctMA] was adjusted by addition of solid ctMA and was again determined relative to [DMSO2] internal standard.
Cyclohex-1-ene-1,4-dicarboxylic acid (CH1DA) was produced through a Diels–Alder cycloaddition of ttMA with 500 psig ethylene in γ-valerolactone at 180 °C for 24 hours in a 50 ml pressurized reactor (Parr 4590 Series). The products in the liquid phase were filtered using a cellulose filter, washed repeatedly with water, and dried overnight in a drying oven. The dried product was then dissolved in DMSO-d6 and characterized using NMR using tetramethylsilane as internal standard.
To prepare the salt for polymerization, adipic acid and CH1DA were added in the molar ratio of x:(1 − x) with 0.75 ≤ x ≤ 1 and dissolved in methanol and THF, respectively. The molar equivalent of HMDA was then added to the resulting mixtures and heated to 40 °C. The precipitated salt was filtered, washed and dried overnight in a fume hood. Polymerization was carried out in a Parr reactor charged with the salt and 60 wt% water and pressurized with 100 psig N2. The mixture was heated to an internal temperature of 210 °C, held there for 80 minutes, followed by venting out the N2 and water. The sample was then allowed to polymerize at an internal temperature of 270 °C at atmospheric pressure for 1.5 hours and cooled to room temperature. Molecular weight of copolymers was obtained through gel permeation chromatography (GPC) using EcoSEC GPC system. Polymer samples of around 5 mg were dissolved in 1,1,1,3,3,3-hexafluoroisopropanol and compared to poly(methyl methacrylate) standards. Samples were then annealed in an oven for 6 hours at 150 °C. DSC was carried out on the copolymer using TA Q100. Dynamic mechanical analysis was performed using a TA ARES-G2 rheometer.
1H NMR spectra were collected with a Bruker AVIII600 spectrometer, and spectra were analyzed with MestReNova software. Data were plotted with OriginPro 9.1 software.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported in part by the National Science Foundation under grant number EEC-0813570 and the U.S. Department of Energy under grant number DE-EE0008492. The authors thank James Trettin for his help with synthesizing the polymers, Dustin Gansebom and Andrew Becker for their help with GPC analysis.References
- N. Hernández, R. C. Williams and E. W. Cochran, Org. Biomol. Chem., 2014, 12, 2834–2849 RSC.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef.
- R. P. Babu, K. O'Connor and R. Seeram, Prog. Biomater., 2013, 2, 8 CrossRef.
- T. Iwata, Angew. Chem., Int. Ed., 2015, 54, 3210–3215 Search PubMed.
- D. I. Collias, A. M. Harris, V. Nagpal, I. W. Cottrell and M. W. Schultheis, Ind. Biotechnol., 2014, 10, 91–105 CrossRef.
- C.-C. Chang, S. K. Green, C. L. Williams, P. J. Dauenhauer and W. Fan, Green Chem., 2014, 16, 585–588 RSC.
- H. J. Cho, L. Ren, V. Vattipalli, Y.-H. Yeh, N. Gould, B. Xu, R. J. Gorte, R. Lobo, P. J. Dauenhauer, M. Tsapatsis and W. Fan, ChemCatChem, 2017, 9, 398–402 CrossRef.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 Search PubMed.
- S. Bérard, C. Vallée and D. Delcroix, Ind. Eng. Chem. Res., 2015, 54, 7164–7168 CrossRef.
- T. Pfennig, R. L. Johnson and B. H. Shanks, Green Chem., 2017, 19, 3263–3271 RSC.
- T. Pfennig, A. Chemburkar, S. Cakolli, M. Neurock and B. H. Shanks, ACS Sustainable Chem. Eng., 2018, 6, 12855–12864 CrossRef.
- Y. Xiao, Q. Meng, X. Pan, C. Zhang, Z. Fu and C. Li, Green Chem., 2020, 22, 4341–4349 RSC.
- R. Y. Rohling, E. Uslamin, B. Zijlstra, I. C. Tranca, I. A. W. Filot, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2018, 8, 760–769 CrossRef.
- Y.-T. Cheng, J. Jae, J. Shi, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 1387–1390 CrossRef.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkóv, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- S. Thiyagarajan, H. C. Genuino, J. C. van der Waal, E. de Jong, B. M. Weckhuysen, J. van Haveren, P. C. A. Bruijnincx and D. S. van Es, Angew. Chem., Int. Ed., 2016, 55, 1368–1371 CrossRef.
- Y. Hu, Z. Zhao, Y. Liu, G. Li, A. Wang, Y. Cong, T. Zhang, F. Wang and N. Li, Angew. Chem., 2018, 130, 7017–7021 CrossRef.
- J. M. Carraher, T. Pfennig, R. G. Rao, B. H. Shanks and J.-P. Tessonnier, Green Chem., 2017, 19, 3042–3050 RSC.
- Purified Terephthalic Acid Market Size, Share | Industry Report, 2025, https://www.grandviewresearch.com/industry-analysis/purified-terephthalic-acid-market, (accessed June 3, 2020).
- Purified Terephthalic Acid (PTA) Market, https://www.marketsandmarkets.com/Market-Reports/purified-terephthalic-acid-market-150628286.html, (accessed June 3, 2020).
- T. Dai, C. Li, L. Li, Z. K. Zhao, B. Zhang, Y. Cong and A. Wang, Angew. Chem., Int. Ed., 2018, 57, 1808–1812 CrossRef.
- F. Wang and Z. Tong, ChemistrySelect, 2016, 1, 5538–5541 CrossRef.
- J. W. Frost and K. M. Draths, Synthesis of adipic acid from biomass-derived carbon sources, US5487987A, 1996.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 RSC.
- K. A. Curran, J. M. Leavitt, A. S. Karim and H. S. Alper, Metab. Eng., 2013, 15, 55–66 CrossRef CAS.
- M. Suástegui and Z. Shao, J. Ind. Microbiol. Biotechnol., 2016, 43, 1611–1624 CrossRef.
- M. Suastegui, J. E. Matthiesen, J. M. Carraher, N. Hernandez, N. Rodriguez Quiroz, A. Okerlund, E. W. Cochran, Z. Shao and J.-P. Tessonnier, Angew. Chem., Int. Ed., 2016, 55, 2368–2373 CrossRef CAS.
- B. H. Shanks and P. L. Keeling, Green Chem., 2017, 19, 3177–3185 RSC.
- B. H. Shanks and L. J. Broadbelt, ChemSusChem, 2019, 12, 2970–2975 CrossRef CAS.
- X. Zhou, Z. J. Brentzel, G. A. Kraus, P. L. Keeling, J. A. Dumesic, B. H. Shanks and L. J. Broadbelt, ACS Sustainable Chem. Eng., 2019, 7, 2414–2428 CAS.
- S. Capelli, D. Motta, C. Evangelisti, N. Dimitratos, L. Prati, C. Pirola and A. Villa, ChemCatChem, 2019, 11, 3075–3084 CrossRef CAS.
- A. Rosengart, S. Capelli, C. Pirola, A. Citterio, C. L. Bianchi, L. Prati and A. Villa, Chem. Eng. Trans., 2017, 57, 931–936 Search PubMed.
- W. Niu, K. M. Draths and J. W. Frost, Biotechnol. Prog., 2002, 18, 201–211 CrossRef CAS.
- C. Müller, et al., Method for producing hexamethylenediamine, WO2015086819A1, 2015 Search PubMed.
- D. R. Vardon, N. A. Rorrer, D. Salvachúa, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer, J. R. Dorgan and G. T. Beckham, Green Chem., 2016, 18, 3397–3413 RSC.
- R. Beerthuis, G. Rothenberg and N. R. Shiju, Green Chem., 2015, 17, 1341–1361 CAS.
- L. Coudray, V. Bui, J. W. Frost and D. Schweitzer, Process for Preparing Caprolactam and Polyamides Therefrom, US20130085255A1, 2013.
- R. Lu, F. Lu, J. Chen, W. Yu, Q. Huang, J. Zhang and J. Xu, Angew. Chem., Int. Ed., 2016, 55, 249–253 CrossRef CAS.
- J. W. Frost, A. Miermont, D. Schweitzer and V. Bui, Preparation of trans, trans muconic acid and trans, trans muconates, US8426639, 2013.
- J. E. Matthiesen, J. M. Carraher, M. Vasiliu, D. A. Dixon and J.-P. Tessonnier, ACS Sustainable Chem. Eng., 2016, 4, 3575–3585 CrossRef CAS.
- J. E. Matthiesen, M. Suástegui, Y. Wu, M. Viswanathan, Y. Qu, M. Cao, N. Rodriguez-Quiroz, A. Okerlund, G. Kraus, D. R. Raman, Z. Shao and J.-P. Tessonnier, ACS Sustainable Chem. Eng., 2016, 4, 7098–7109 CrossRef CAS.
- J.-P. Tessonnier, J. E. Matthiesen, T. Pfennig, B. H. Shanks and J. M. Carraher, Electrochemical isomerization of muconic acid, US10465043B2, 2016.
- J.-P. Tessonnier, J. M. Carraher, T. Pfennig and B. H. Shanks, Isomerization of muconic acid, US9957218B2, 2016.
- J. K. Ogunjobi, T. J. Farmer, C. R. McElroy, S. W. Breeden, D. J. Macquarrie, D. Thornthwaite and J. H. Clark, ACS Sustainable Chem. Eng., 2019, 7, 8183–8194 CrossRef CAS.
- I. Khalil, G. Quintens, T. Junkers and M. Dusselier, Green Chem., 2020, 22, 1517–1541 RSC.
- V. Bui, M. K. Lau, D. MacRae and D. Schweitzer, Methods for producing isomers of muconic acid and muconate salts, US8809583B2, 2014.
- J. W. Frost, A. Miermont, D. Schweitzer and B. Vu, Preparation of trans, trans muconic acid and trans, trans muconates, WO2010148049A2, 2010.
- A. E. Settle, L. Berstis, S. Zhang, N. A. Rorrer, H. Hu, R. M. Richards, G. T. Beckham, M. F. Crowley and D. R. Vardon, ChemSusChem, 2018, 11, 1768–1780 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- M. Martí, L. Molina, C. Alemán and E. Armelin, ACS Sustainable Chem. Eng., 2013, 1, 1609–1618 CrossRef.
- J. A. Elvidge, R. P. Linstead, P. Sims and B. A. Orkin, J. Chem. Soc., 1950, 2235 RSC.
- D. M. Alonso, J. M. R. Gallo, M. A. Mellmer, S. G. Wettstein and J. A. Dumesic, Catal. Sci. Technol., 2013, 3, 927–931 RSC.
- M. J. Burk, R. E. Osterhout and J. Sun, Semi-synthetic terephthalic acid via microorganisms that produce muconic acid, US9562241, 2014.
- V. Bui, J. W. Frost, D. A. Wicks, D. Schweitzer and A. Miermont, Cyclohexane 1,4 carboxylates, US8367859, 2013.
Footnotes |
| † Electronic supplementary information (ESI) available: NOESY NMR spectrum of MA in water, NMR spectra of biobased ccMA and ctMA, GPC, DSC, and DMA results of the synthesized polyamides, experimental method for the synthesis of biobased ccMA and 1,4-cyclohexanedicarboxylic acid. See DOI: 10.1039/d0gc02108c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |