
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical N-demethylation of tropane alkaloids†
Ali
Alipour Najmi
a,
Zhangping
Xiao
b,
Rainer
Bischoff
 a,
Frank J.
Dekker
b and
Hjalmar P.
Permentier
*a
a,
Frank J.
Dekker
b and
Hjalmar P.
Permentier
*a
aDepartment of Analytical Biochemistry, Groningen Research Institute of Pharmacy, University of Groningen, A Deusinglaan 1, 9713 AV Groningen, The Netherlands. E-mail: h.p.permentier@rug.nl
bChemical and Pharmaceutical Biology, Groningen Research Institute of Pharmacy, University of Groningen, A Deusinglaan 1, 9713 AV Groningen, The Netherlands
First published on 9th September 2020
Abstract
A practical, efficient, and selective electrochemical N-demethylation method of tropane alkaloids to their nortropane derivatives is described. Nortropanes, such as noratropine and norscopolamine, are important intermediates for the semi-synthesis of the medicines ipratropium or oxitropium bromide, respectively. Synthesis was performed in a simple home-made electrochemical batch cell using a porous glassy carbon electrode. The reaction proceeds at room temperature in one step in a mixture of ethanol or methanol and water. The method avoids hazardous oxidizing agents such as H2O2 or m-chloroperbenzoic acid (m-CPBA), toxic solvents such as chloroform, as well as metal-based catalysts. Various key parameters were investigated in electrochemical batch or flow cells, and the optimized conditions were used in batch and flow-cells at gram scale to synthesize noratropine in high yield and purity using a convenient liquid–liquid extraction method without any need for chromatographic purification. Mechanistic studies showed that the electrochemical N-demethylation proceeds by the formation of an iminium intermediate which is converted by water as the nucleophile. The optimized method was further applied to scopolamine, cocaine, benzatropine, homatropine and tropacocaine, showing that this is a generic way of N-demethylating tropane alkaloids to synthesize valuable precursors for pharmaceutical products.
Introduction
Naturally occurring tropane alkaloids, such as the anticholinergic agents atropine 1 and scopolamine 2, and the stimulant cocaine 3, are among the oldest drugs that have been used by humans. The important characteristic of these compounds is the presence of a tropane ring 4 including a tertiary N-methylamine group, in their chemical structure.1N-Demethylation of tropane alkaloids to their nortropane derivatives is a key step in the semi-synthesis of some important therapeutic agents.2,3 For example, noratropine 5 is an intermediate for the synthesis of the bronchodilator ipratropium bromide 6, which is in the WHO's list of essential medicines. Under the brand name of Combivent, ipratropium bromide had world-wide sales of $850 million and $950 million in 2008 and 2009, respectively, and is among the top 200 pharmaceutical products either by retail sales or prescriptions in the United States, in the last decade.4,5 Once 1 is demethylated to 5, 5 is alkylated with isopropyl bromide to give N-isopropyl-noratropine 7, which is quaternized with methyl bromide to give 6 (Fig. 1B), with the N-isopropyl and N-methyl substituents in axial and equatorial positions, respectively. In contrast, direct quaternization of 1 with isopropyl bromide leads to an isomeric mixture with the isopropyl group in both the axial and the equatorial positions.1,3 Pharmaceutical companies are currently using 7 for the preparation of 6.6 In a similar manner, norscopolamine 8 is used as an intermediate for manufacturing the bronchodilator oxitropium bromide 9 with N-ethyl and N-methyl substituents in the axial and equatorial positions, respectively, while the ethyl group is located in the equatorial position, if 8 is alkylated directly.1,7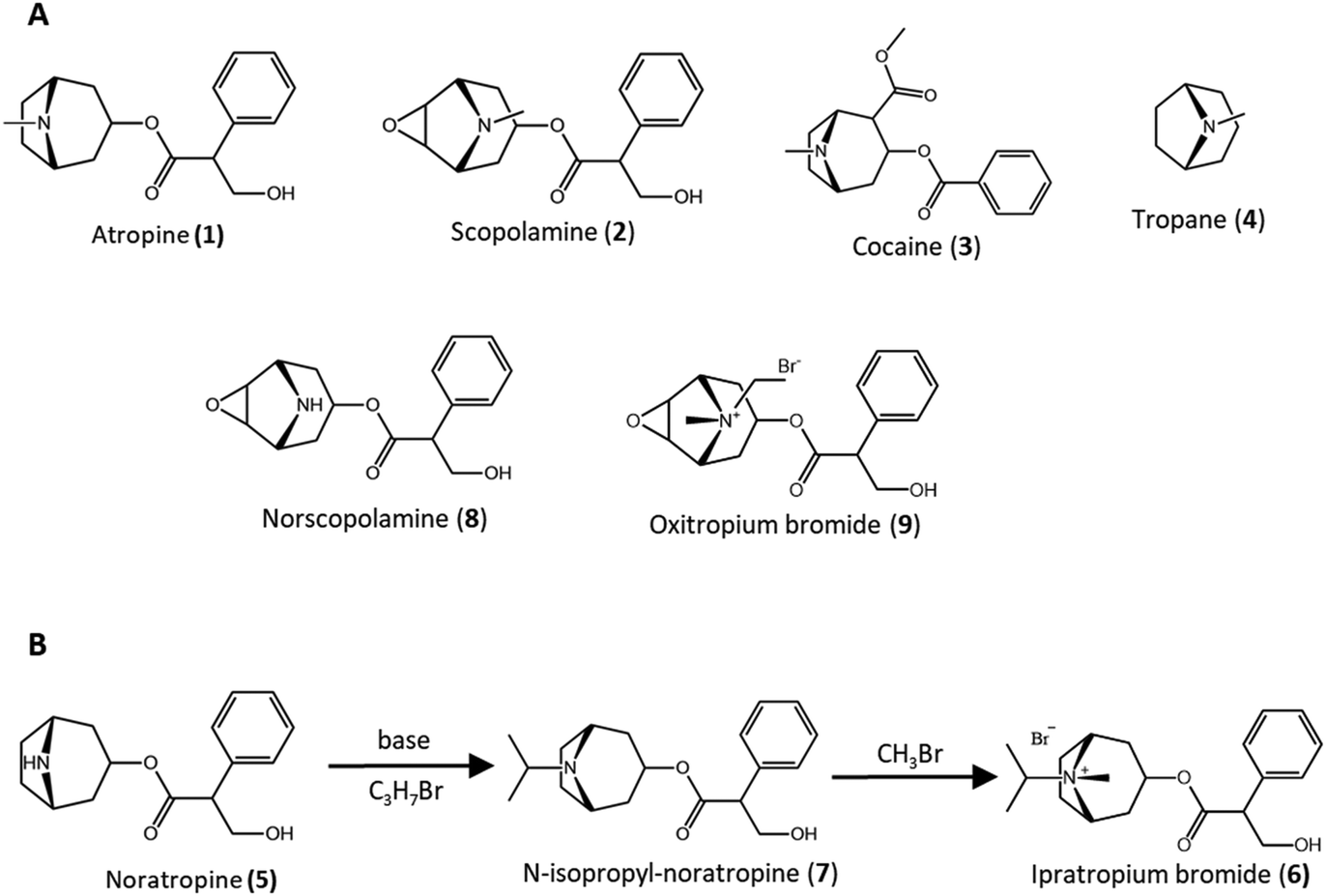 | ||
| Fig. 1 (A) Structure of some naturally occurring tropane alkaloids (1–3), tropane (4), norscopolamine (8), and the semi-synthetic bronchodilator oxitropium bromide (9); (B) synthesis of ipratropium bromide (6) from noratropine (5). | ||
Despite the importance of selective N-demethylation of tropane alkaloids, it has remained a challenging step for synthetic chemists.8 Different approaches have been reported for N-demethylation of the tertiary amine in atropine and scopolamine using agents such as 2,2,2-trichloroethyl chloroformate,9 α-chloroethyl chloroformate,10 KMnO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10,11 and photochemistry.12 Although these methods provide low to excellent yields (16–100%) of 5 and 8, they use toxic solvents and chemicals producing hazardous waste and by-products.
10,11 and photochemistry.12 Although these methods provide low to excellent yields (16–100%) of 5 and 8, they use toxic solvents and chemicals producing hazardous waste and by-products.
The original Polonovski reaction has proven to be effective for the N-demethylation of tertiary N-methylamines.13 Over the past decade, modifications of the reaction based on iron catalysts have been developed for the N-demethylation of tropane and opiate alkaloids.2,3,6,8,14–19 These reactions are based on a multi-step process, comprising the oxidation of the tertiary amine using m-chloroperbenzoic acid (m-CPBA) or H2O2 to the corresponding N-oxide, which is then isolated as the HCl salt. The N-oxide salt is N-demethylated with iron-based catalysts such as FeSO4·7H2O, FeCl2·4H2O, or Fe(NH4SO4)2,14,15 iron porphyrin complex,16,17 ferrocene,8 and iron powder2 which need to be separated from the reaction mixture by chelating with EDTA or TPPS (meso-tetra(4-sulfophenyl)porphyrin)14,15 or by filtration.16–18
Recent works have focused on using greener solvents such as ethanol and isopropanol, instead of chloroform, in the N-demethylation of N-oxides.3,6,18,20 In one approach, iron nanoparticles have been studied as catalyst for the N-dealkylation of various alkaloids reporting a yield of 85% for the synthesis of noratropine.18 However, chloroform is still in use for N-oxide formation in the first step of the reaction.18 Do Pham et al. studied the N-demethylation of atropine and scopolamine using Fe(III)–TAML (tetra-amidato macrocyclic ligand) as catalyst in ethanol without isolation of the N-oxide as the hydrochloride salt, reporting a yield of about 80% for noratropine and norscopolamine.3,6 However, this method uses 50 equivalents of H2O2, which must be deactivated at the end of the process by treatment with MnO2.3,6 A detailed comparison of these methods with respect to their advantages and disadvantages is provided in Table S1.†
Previous methods use either toxic organic solvents, require powerful oxidants or necessitate chromatographic purification or filtration to remove catalysts, all of which add to the overall cost of the procedure and increase the impact on the environment notably for large-scale production processes.2,3,6,8,14–19
Concerted N-demethylation/N-acylation strategies based on palladium catalysts have been reported for atropine and opiate alkaloids.21–24 Although these methods have been applied for the synthesis of the semi-synthetic opiates naltrexone22 and buprenorphine,23 the reaction is not selective for atropine, since the hydroxyl group is dehydrated, leading to the formation of apoatropine.21 In addition, application of palladium catalysts in commercial medicines leads to cost issues due to the need for palladium removal below the required level of 10 ppm.3
Organic electrosynthesis has several advantages over traditional methods, such as the limited use of hazardous chemicals, mild reaction conditions, a simple system design, scalability and notably sustainability.25–27 It is thus considered to be a “green” alternative to widely used organic synthesis methods.28,29 While electrochemistry has been applied for the oxidation of aliphatic amines,30–32 ferrocene-mediated oxidation of cyclohexylamines,33,34 and some phenethylamines35 as well as the synthesis of a wide-range of organic molecules (for a detailed list see a recent review36), there is no report regarding its application for the N-demethylation of bicyclic tertiary amines in general and tropane alkaloids in particular. Recently, Gul et al. reported optimized conditions for generating N-dealkylated lidocaine.37 In another study, electrochemical N-dealkylation of fesoterodine was reported.38 However, the reported concentrations (0.1–1 mM) are far lower than what is used in practical synthesis procedures, while only achieving less than 30% conversion to the N-dealkylated drugs.37,38
In order to overcome the challenges of the available organic synthesis methods, we developed a generic electrochemical N-demethylation strategy for tropane alkaloids that gives both good selectivity and yields in gram-scale synthesis. Product isolation is straightforward using liquid–liquid extraction. The reaction proceeds at room temperature in one step without the need for metal-based catalysts in an ethanol or methanol/water mixture. After having established the electrochemical synthesis reaction, we studied the mechanistic pathway showing that this reaction proceeds via the formation of an iminium intermediate that reacts with water to give nortropanes.
Methods
Reagents
Atropine, scopolamine·HBr, benzatropine·mesylate, tropane, clemastine fumarate, 4-diphenylmethoxy-1-methylpiperidine·HCl, sodium carbonate (Na2CO3), sodium sulfate (Na2SO4), ammonium hydroxide (NH4OH), and potassium cyanide (KCN) were purchased from Sigma-Aldrich. Ultra-pure HPLC grade acetonitrile (ACN) and dichloromethane (DCM) were purchased from Biosolve. Absolute ethanol and methanol were purchased from either Biosolve or VWR Chemicals. MP Ecochrom silica 32–63, 60 Å was used for column chromatography. Tropacocaine·HCl, cocaine, and homatropine·HBr were obtained from the Interfaculty Mass Spectrometry Center (IMSC), University of Groningen, Groningen, The Netherlands.Electrochemical methods
A thin-layer electrochemical flow-cell (μ-PrepCell 2.0, Antec-Scientific, Zoeterwoude, The Netherlands) was used to determine the optimal electrode material for the electrochemical N-dealkylation reaction. The μ-PrepCell has a 30 mm × 12 mm rectangular working electrode (glassy carbon, boron-doped diamond, TiO2, stainless steel, Pt or Au) and a counter electrode made from a conductive polyether ether ketone (PEEK) polymer. It has a reaction volume of about 20 μL. Reaction solutions were pumped into the flow-cell at flow-rates of 1–5 μL min−1 with a syringe pump (KD Scientific Inc., Holliston, MA, USA) using a glass syringe (Hamilton, Reno, NV, USA). The liquid from the outlet of the flow-cell was collected, and the fractions subjected to LC–MS analysis. Prior to use and between each experiment, the μ-PrepCell was flushed with the corresponding reaction mixture to assure stable conditions. Electrochemical synthesis was performed in a home-made two-electrode electrochemical cell using glassy carbon with 100 pores per inch (100 PPI; Goodfellow Cambridge Ltd, UK) as both anode and cathode. A glass test tube was used as reactor. Gram-scale electrochemical synthesis of noratropine in batch-cell was performed with a stack of GC electrodes, using four anode and four cathode electrodes (Fig. S1–S3†). Gram-scale electrochemical synthesis of noratropine was performed using an 8-channel flow-cell (in series) having 704 μL of total reactor volume (88 μL per channel) and 51 cm2 total surface area (106 mm × 3 mm open area per channel). A planar graphite electrode with dimension of 110 mm × 45 mm was used both as anode and cathode. Detailed information about the electrochemical flow-cell is reported elsewhere.39 Cyclic voltammetry (CV) experiments were carried out using a glassy carbon electrode (1.6 mm diameter, ALS Co.) as working electrode, a 100 PPI glassy carbon electrode as counter electrode, and an Ag/AgCl reference electrode. A 5 mM solution of compound in ethanol/water (2:1) including NaClO4 (0.1 M) as supporting electrolyte was used for CV experiments.
All electrochemical measurements were performed with an Autolab potentiostat (Metrohm AG, Herisau, Switzerland) using NOVA software (Metrohm AG) at room temperature under ambient atmosphere. Between each experiment, electrode surfaces were washed with water and ethanol and then dried under atmospheric conditions for both the μ-PrepCell and home-made cells.
1H and 13C NMR, LC–MS, and HRMS analysis
1H and 13C NMR spectra were recorded at 500 and 126 MHz, respectively, on a Bruker Avance 500 spectrometer. Chemical shifts were reported in ppm relative to the solvent. A C18 reversed-phase LC column (Hypersil gold, 100 × 2.1 mm, Thermo Scientific) was used for liquid-chromatography separation at a flow-rate of 300 μL min−1 (LC20-AD prominence, Shimadzu). Solvent A and B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. Mass spectrometry analysis was performed on a TSQ Quantum Ultra triple quadrupole mass spectrometer (Thermo Scientific) with electrospray ionization (ESI) in the positive mode. High resolution mass spectrometry analysis was performed on an Orbitrap Velos Pro (Thermo Scientific) with electrospray ionization (ESI) in positive mode.Liquid–liquid extraction (LLE)
:DCM (1:10) and extracted with 2 × 15 mL 1 M HCl. The aqueous extract was basified with 5 M aqueous NaOH and then extracted with 2 × 30 mL DCM.
:DCM (1:10) and extracted with 2 × 15 mL 1 M HCl. The aqueous extract was basified with 15% aqueous NH4OH and then extracted with 2 × 30 mL DCM.
The final organic extract was dried (Na2SO4), filtered and the solvent removed under vacuum to give the product. The yield was determined from the weight and 1HNMR of the isolated product.
Results and discussion
In initial experiments, an electrochemistry–liquid chromatography–mass spectrometry (EC–LC–MS) approach was used to investigate the feasibility of the electrochemical N-demethylation of atropine 1 using six commercially available flat electrodes with different materials but equal dimensions. The electrochemical reaction of atropine was investigated using an analytical electrochemical thin-layer flow-cell having a reactor volume of 20 μL. Alcohols (ethanol or methanol) were selected as solvents, since they are considered to be among the greenest solvents.20,40 As listed in Table 1, the electrochemical oxidation of atropine (oxidation potential of 0.95 V versus Ag/AgCl reference electrode) over carbon-based electrodes (GC and BDD) at constant current showed higher conversion to noratropine compared to noble-metal electrodes, while stainless steel and titanium oxide electrodes showed no N-demethylation or any other oxidation products at the applied conditions (Table 1; LC–MS chromatograms in Fig. S4†). Considering these results and the considerably lower price of glassy carbon electrodes compared to either BDD or noble-metal electrodes, a highly porous glassy carbon electrode (100 PPI grade) was selected and a home-made electrochemical batch-cell was fabricated to further optimize the reaction conditions (Fig. S2†).
As there is only one methyl group difference between atropine and noratropine, separation of these two compounds from any reaction mixture can be challenging. For this reason, it is important to have full conversion at the end of the reaction. Using only ethanol as solvent did not result in full conversion. Increasing the reaction time and decreasing the applied current either did not lead to complete conversion or resulted in overoxidation, a decreased yield and a complex reaction mixture of byproducts, as determined by LC–MS (Table 2, entries 1–5). Although increasing the applied current led to full conversion, analysis of the final reaction mixture by LC–MS showed a more complex reaction mixture including different overoxidized byproducts (see LC–MS chromatogram in Fig. S5†). We then considered whether addition of water to the reaction mixture would result in full conversion, as the electrochemical N-demethylation of atropine is similar to the Shono-type electrochemical oxidations, which have been applied for the activation and functionalization of C–H bonds adjacent to nitrogen atoms.41 It has been hypothesized that water or methanol may act as a nucleophile trapping the iminium intermediate produced during the electrochemical reaction.28,42,43 Increasing the water content of the reaction mixture led to full conversion and decreased the required time and charge for completion of the reaction (Table 2, entries 6–8), while giving a moderate yield of noratropine (44–67%). LC–MS analysis of the final reaction mixture still showed multiple byproducts (Table 2, entry 8). Reducing the applied current to 4 mA resulted in 82% isolated yield after silica-gel chromatography and a less complex final reaction mixture with only two minor byproducts (Table 2, entry 9; LC–MS chromatogram Fig. S6a†).
|
|
||||
|---|---|---|---|---|
| Entry | Current/time | Added water | Conversionb (%) | Yield (%) |
| a Absolute ethanol was used as solvent and the residual amount of water was not measured. b Determined by LC–MS. c Isolated yield (based on weight and 1HNMR) after silica-gel chromatography. d Isolated yield (based on weight and 1HNMR) after 3-step LLE. e Isolated yield (based on weight and 1HNMR) after 2-step-NaOH-LLE. f Isolated yield (based on weight and 1HNMR) after 2-step-NH4OH-LLE. | ||||
| 1 | 6 mA/3 h | 0a | 90 | 70b |
| 2 | 6 mA/5 h | 0a | 90 | 37b |
| 3 | 6 mA/7 h | 0a | 93 | 28b |
| 4 | 4 mA/6 h | 0a | 88 | 25b |
| 5 | 8 mA/3 h | 0a | 99 | 52b |
| 6 | 6 mA/5 h | 2.2 M (4% v/v) | 92 | 44b |
| 7 | 6 mA/5 h | 11 M (20% v/v) | 99 | 65b |
| 8 | 6 mA/3.5 h | 18.5 M (33% v/v) | 99 | 67b |
| 9 | 4 mA/4.5 h | 18.5 M (33% v/v) | 99 | 82c |
| 10 | 4 mA/4.5 h | 18.5 M (33% v/v) | 99 | 84d |
| 11 | 4 mA/4.5 h | 18.5 M (33% v/v) | 99 | 68e |
| 12 | 4 mA/4.5 h | 18.5 M (33% v/v) | 99 | 80f |

Considering the low complexity of the final reaction mixture, based on LC–MS analysis, we tested whether the desired product could be purified by liquid–liquid extraction (LLE). As waste production and solvent consumption at the final purification step of active pharmaceutical ingredients are responsible for more than half of the overall manufacturing expenses, there is a strong incentive to adopt other purification methods than chromatography.44,45 Do Pham et al. applied a three step LLE approach to isolate noratropine and norscopolamine from their final reaction mixture with high purity.3,6 Three different LLE techniques were evaluated and compared to chromatographic purification to isolate noratropine from the final reaction mixture obtained under the same reaction conditions showing that comparable yields to chromatographic purification can be obtained by three- or two-step LLE (Table 2, entries 10–12, LC–MS chromatogram of final reaction mixture Fig. S6b–d†). Another four replicates of the final reaction condition (Table 2, entries 9–12) were performed with another set of electrodes and reported in Table S2.†
In order to investigate the sensitivity of the electrochemical N-demethylation reaction to either solvent or supporting electrolyte, methanol was selected as an alternative solvent and four different supporting electrolytes were tested. As listed in Table 3, the electrochemical N-demethylation of atropine proceeded with good yield under all of these reaction conditions (LC–MS chromatogram of final reaction mixture Fig. S7†). However, using LiBr as supporting electrolyte required a longer reaction time to complete the conversion of atropine as compared to other supporting electrolytes.

Having studied different reaction conditions for the electrochemical N-demethylation of atropine, we investigated the generality of this approach by applying it to other tropane compounds (Table 4). We performed electrochemical N-demethylation of tropacocaine 10 in the home-made batch cell resulting in an isolated yield of 70% of nortropacocaine. It is noteworthy that performing the electrochemical reaction using its salt form (tropacocaine·HCl) did not lead to any conversion, possibly due to the acidic pH of the reaction solution with tropacocaine·HCl. Adding an equimolar of sodium carbonate to the reaction solution increased the pH of the solution to about 10 leading to 95% conversion (Table S3†). The facile N-demethylation reaction at basic pH is likely due to the fact that the lone–pair electrons of the tertiary amine group are more easily available for abstraction than under acidic conditions, when the amine is largely protonated.37,38,46,47 Besides tropane alkaloids, we have applied the electrochemical N-demethylation reaction to other examples of organic compounds having N-methylpiperidine or N-methylpyrrolidine in their chemical structures (Table S4†). Although the reaction proceeds with those compounds as well, the isolated yield of the secondary amine is lower than for the nortropanes, especially for N-methylpyrrolidine; the drug clemastine 14 was mostly converted to a product which was hydroxylated on the carbon adjacent to the nitrogen.
| Tropane alkaloids | Synthesized nortropanes | ||
|---|---|---|---|
| a 0.16 mmole. b 0.2 mmole. c Isolated yield after 2-step-NH4OH-LLE. d Isolated yield after silica-gel chromatography. e Electrochemical oxidation potential versus Ag/AgCl reference electrode (CV curves in Fig. S9†). f Faradaic efficiency (2-electron oxidation). | |||
| Scopolamine 2b |

|
83%c |

|
| 1.01 Ve | 4 mA/3 h | ||
| 74%f | |||
| Cocaine 3b |

|
60%d |

|
| 1.04 Ve | 4 mA/3 h | ||
| 53%f | |||
| Tropacocaine 10a |

|
73%c |

|
| 1.00 Ve | 4 mA/7 h | ||
| 22%f | |||
| Homatropine 11b |

|
63%c |

|
| 0.97 Ve | 4 mA/7 h | ||
| 24%f | |||
| Benzatropine 12a |

|
72%d |

|
| 0.91 Ve | 4 mA/12 h | ||
| 12%f | |||
Having studied the reaction conditions, the electrochemical N-demethylation of atropine was performed at the gram-scale using a stack of electrodes in a batch cell (Fig. S3†). As listed in Table 5, good yields of noratropine were obtained by a three-step LLE method after electrochemical reaction, with high purity as examined by 1H-NMR analysis (1H-NMR spectra of g-scale synthesis in ESI†), which can be used directly in subsequent steps for the synthesis of ipratropium bromide medicine.6
Flow synthesis is considered to be an interesting alternative to batch cell synthesis and electrochemical flow cells have been employed successfully for gram-scale synthesis.48 In order to make a comparison between the batch-cell and flow-cell synthesis of nortropanes in gram-scale, we used a newly designed electrochemical flow-cell,39 which has been applied to various electrochemical reactions.49,50 It is an 8-channel flow-cell, with a reactor volume of 88 μL per channel using graphite working and counter electrodes. The 8 channels of the flow-cell were used in series with the same solvent system as Table 5, entry 2. We performed a 2-gram scale electrosynthesis of noratropine. An isolated yield of 67% was obtained after three-step LLE, which is comparable to the batch-cell synthesis. A residence time of 42 min and 8 mA was found to be the optimal conditions. Increasing the applied current for the same residence time leads to lower yield and higher conversion to byproducts while decreasing the residence time at the same applied current did not lead to full conversion of atropine. It is noteworthy to mention that the ratio of the electrode surface area to the reactor volume of the batch-cell in gram-scale is calculated to be about 42 cm−1.51,52 This number is comparable to the surface-to-volume ratio of the flow-cell39 which is about 72 cm−1.
Since a considerable amount (80–90%) of non-aqueous waste generated from the manufacturing of active pharmaceutical ingredients (APIs) are solvents, the use of green solvents, such as ethanol, methanol and water, is a critical requirement.20 Besides, the application of heterogeneous catalysis plays a key role in developing new reaction processes to meet the target of green chemistry.53 The concept of catalyst immobilization and reuse, which has been recently defined as a new “key green chemistry research area” by big pharmaceutical companies,54 is addressed by our newly developed method using a cheap glassy carbon electrode for a prolonged period of time for different batches of the reaction in gram-scale. Finally, the purification of the synthesized noratropanes using a convenient LLE technique in high yields and purity is another advantage of the developed method avoiding a chromatographic purification step which is a growing demand in the pharmaceutical industry.45
Analysis of byproducts and mechanistic studies
Analysis of the final reaction mixture by LC–MS showed two main byproducts with m/z of 304.1 and 561.1 next to the produced noratropine (m/z of 290.1). Analysis of the aqueous extract in the first step of LLE, and the organic extract in the second step of LLE, showed a high intensity of these two byproducts compared to remaining noratropine, which confirms that the LLE method did not recover all of the products. The peak with m/z of 304.1 was therefore isolated using silica gel chromatography and characterized by 1H and 13C NMR and was confirmed to be N-formyl-noratropine which was also reported elsewhere as a byproduct of atropine demethylation.3,6 Interestingly, all of the other examined tropanes (compounds 10–12), except scopolamine, showed equivalent byproducts: analysis of the final reaction mixtures by LC–MS analysis showed for every compound a peak of M − 14 as the nortropane product, a peak of M + 14 as the N-formyl-nortropane product and a peak at [M] + [M − 14] − 5 as a probable dimer byproduct. In order to confirm the dimer structure, the electrochemical reaction was performed by tropane 4 as starting compound. As expected, a byproduct with m/z of 233.1 was produced. The byproduct was isolated and purified from the reaction mixture and its structure confirmed by 1H-NMR, 13C-NMR and HRMS as a quaternary amine, compound V-4 (Fig. 2). | ||
| Fig. 2 Proposed mechanism for the electrochemical N-demethylation of tropane alkaloids via the formation of an iminium intermediate reacting with water (route a), and predicted route for trapping the iminium intermediate with cyanide (route c). | ||
A basic understanding of the mechanistic pathways of the catalytic reactions facilitates the improvement of catalytic processes providing the opportunity for the rational design of the catalytic systems.53 The high dissociation energy of the C–N bond and the intrinsic stability of amines makes cleavage of the C–N bond challenging for synthetic chemists.55 Generally, two types of C–N bond cleavage by transition-metal catalysts (such as iron-based catalysts) have been studied broadly: (a) the oxidative addition of transition-metal catalysts to the C–N bond and, (2) the formation of intermediate imine or iminium species.56 The latter mechanism has also been proposed for the Shono-type electrochemical oxidation,57,58 which provides a route to activate and functionalize C–H bonds in the vicinity of a nitrogen atom. Shono-type oxidations proceed by the initial formation of a nitrogen-centered radical via direct electron transfer to the electrode, followed by a sequence of ET/PT/ET (electron/proton/electron transfer) steps leading to an iminium intermediate, which can be trapped with water or alcoholic solvents.28,42,43 Therefore, we hypothesized that the electrochemical N-demethylation of tropane alkaloids on glassy carbon electrodes may also proceed via the formation of an iminium intermediate, which subsequently reacts with water to form nortropanes, as depicted in Fig. 2 (route a).
C–N bond cleavage in the form of N-dealkylation, a common in vivo metabolic pathway for most of the amine-containing drugs catalyzed by Cytochrome P450 (CYP) enzymes, also proceeds via iminium intermediate formation59,60 which can be trapped by a nucleophile such as cyanide.61 In order to investigate the mechanism, we added an excess amount of potassium cyanide as a source of cyanide ions to trap the iminium intermediate (Fig. 2, route c). LC–MS analysis of the outlet of the cell (Fig. S8†) showed that, besides intact atropine at m/z = 290.1 (M) and noratropine at m/z = 276.1 (M − 14), a new compound at m/z = 315.1 (M + 25) was produced during the reaction. This compound (N-nitrilo-noratropine, VI-1, Fig. 2) can be formed by the addition of a CN− group (26 Da) to an iminium intermediate and abstraction of a hydrogen atom, supporting a reaction mechanism during which an iminium intermediate is generated by a two-electron oxidation reaction at the anode. N-Nitrilo-noratropine was synthesized in a home-made electrochemical cell and characterized by 1H and 13C NMR. Dimer formation during the electrochemical reaction can be explained by this mechanism as well, since the produced noratropine, a secondary amine, can act as a nucleophile and react with the iminium intermediate resulting in a tertiary diamine (compound IV, Fig. 2) which undergoes a two electron electrochemical oxidation reaction to form an amidinium structure. The produced formaldehyde from the removed methyl group was detected by LC–MS using a simple derivatization method, where acetylacetone reacts with formaldehyde in the presence of ammonium acetate to form the cyclized 3,5-diacetyl-1,4-dihydrolutidine product (m/z of 194.1),62,63 which can be easily detected by LC–MS analysis (Fig. S12†). In theory, only 2 F mol−1 electricity is required for a two-electron oxidation reaction.47 The electrochemical N-demethylation of atropine at gram scale was calculated to require 2.6 F mol−1.
Conclusions
We introduce an efficient, straightforward and green electrochemical N-demethylation strategy for tropane alkaloids based on oxidation on a glassy-carbon electrode. Using a commercially available flow cell (μ-PrepCell) and a home-made electrochemical batch cell, we screened a number of parameters. The optimized conditions were applied to a set of tropane alkaloids, with a focus on atropine and scopolamine. Compared to other iron-based catalytic methods, our electrochemical procedure proved to be generally applicable for producing nortropane derivatives in good to high yields with high purity at gram-scale without any need for chromatographic purification. The method is a convenient, one-step process using a water–ethanol/methanol co-solvent system avoiding multi-step processes, need for hazardous oxidizing agents such as H2O2 or m-CPBA, toxic solvents such as chloroform or metal-based catalysts. Other advantageous features of our approach comprise the use of an open-flask electrochemical reactor under ambient conditions and a low-cost porous glassy carbon electrode, which can be used for a prolonged period of time. We show that alternatively an electrochemical flow reactor can be used for gram-scale synthesis of nortropanes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the Open Technology Programme of Toegepaste en Technische Wetenschappen (TTW) with project number 15230 which is financed by the Netherlands Organisation for Scientific Research (NWO). The authors thank Prof. Dr Timothy Noël and Dr Gabriele Laudadio from the Micro Flow Chemistry & Synthetic Methodology group of the Eindhoven University of Technology, The Netherlands, for providing the electrochemical flow cell.References
- G. Grynkiewicz and M. Gadzikowska, Pharmacol. Rep., 2008, 60, 439–463 CAS.
- G. B. Kok, C. C. Pye, R. D. Singer and P. J. Scammells, J. Org. Chem., 2010, 75, 4806–4811 CrossRef CAS.
- D. D. Do Pham, G. F. Kelso, Y. Yang and M. T. W. Hearn, Green Chem., 2014, 16, 1399–1409 RSC.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- A. Fuentes, M. Pineda and K. Venkata, Pharmacy, 2018, 6, 43 CrossRef.
- D. D. Do Pham, G. F. Kelso, Y. Yang and M. T. W. Hearn, Green Chem., 2012, 14, 1189–1195 RSC.
- P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach: Third Edition, John Wiley & Sons, 2009 Search PubMed.
- G. B. Kok and P. J. Scammells, Bioorg. Med. Chem. Lett., 2010, 20, 4499–4502 CrossRef CAS.
- J. R. Pfister, J. Org. Chem., 1978, 43, 4373–4374 CAS.
- F. Balssa and Y. Bonnaire, J. Labelled Compd. Radiopharm., 2009, 52, 269–272 CrossRef CAS.
- M. J. Van Der Meer and H. K. L. Hundt, J. Pharm. Pharmacol., 1983, 35, 408–408 CrossRef CAS.
- J. A. Ripper, R. T. Tiekink and P. J. Scammells, Bioorg. Med. Chem. Lett., 2001, 11, 443–445 CrossRef CAS.
- D. Grierson, Org. React., 2004, 39, 85–295 Search PubMed.
- K. McCamley, J. A. Ripper, R. D. Singer and P. J. Scammells, J. Org. Chem., 2003, 68, 9847–9850 CrossRef CAS.
- S. Thavaneswaran and P. J. Scammells, Bioorg. Med. Chem. Lett., 2006, 16, 2868–2871 CrossRef CAS.
- Z. Dong and P. J. Scammells, J. Org. Chem., 2007, 72, 9881–9885 CrossRef CAS.
- G. Kok, T. D. Ashton and P. J. Scammells, Adv. Synth. Catal., 2009, 351, 283–286 CrossRef CAS.
- J. K. Awalt, R. Lam, B. Kellam, B. Graham, P. J. Scammells and R. D. Singer, Green Chem., 2017, 19, 2587–2594 RSC.
- J. K. Awalt, P. J. Scammells and R. D. Singer, ACS Sustainable Chem. Eng., 2018, 6, 10052–10057 CrossRef CAS.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- R. J. Carroll, H. Leisch, E. Scocchera, T. Hudlicky and D. P. Cox, Adv. Synth. Catal., 2008, 350, 2984–2992 CrossRef CAS.
- A. MacHara, D. P. Cox and T. Hudlicky, Adv. Synth. Catal., 2012, 354, 2713–2718 CrossRef CAS.
- A. MacHara, L. Werner, M. A. Endoma-Arias, D. P. Cox and T. Hudlicky, Adv. Synth. Catal., 2012, 354, 613–626 CrossRef CAS.
- B. Gutmann, P. Elsner, D. P. Cox, U. Weigl, D. M. Roberge and C. O. Kappe, ACS Sustainable Chem. Eng., 2016, 4, 6048–6061 CrossRef CAS.
- O. Hammerich and B. Speiser, Organic electrochemistry: revised and expanded, CRC Press, 2015 Search PubMed.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS.
- H. J. Schäfer, C. R. Chim., 2011, 14, 745–765 CrossRef.
- M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- K. K. Barnes and C. K. Mann, J. Org. Chem., 1967, 32, 1474–1479 CrossRef CAS.
- P. J. Smith and C. K. Mann, J. Org. Chem., 1969, 34, 1821–1826 CrossRef CAS.
- L. C. Portis, V. V. Bhat and C. K. Mann, J. Org. Chem., 1970, 35, 2175–2178 CAS.
- A. A. J. Torriero, M. J. A. Shiddiky, I. Burgar and A. M. Bond, Organometallics, 2013, 32, 5731–5739 CAS.
- A. A. J. Torriero, J. Morda and J. Saw, Organometallics, 2019, 38, 4280–4287 CAS.
- L. C. Portis, J. T. Klug and C. K. Mann, J. Org. Chem., 1974, 39, 3488–3494 Search PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CAS.
- T. Gul, R. Bischoff and H. P. Permentier, Electrochim. Acta, 2015, 171, 23–28 CAS.
- S. Torres, R. Brown, R. Szucs, J. M. Hawkins, G. Scrivens, A. Pettman, D. Kraus and M. R. Taylor, Org. Process Res. Dev., 2015, 19, 1596–1603 CrossRef CAS.
- G. Laudadio, W. de Smet, L. Struik, Y. Cao and T. Noël, J. Flow Chem., 2018, 8, 157–165 CrossRef.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- T. Shono, H. Hamaguchi and Y. Matsumura, J. Am. Chem. Soc., 1975, 97, 4264–4268 CAS.
- F. Wang, M. Rafiee and S. S. Stahl, Angew. Chem., Int. Ed., 2018, 57, 6686–6690 CrossRef CAS.
- K. J. Frankowski, R. Liu, G. L. Milligan, K. D. Moeller and J. Aubé, Angew. Chem., Int. Ed., 2015, 54, 10555–10558 CrossRef CAS.
- E. A. Peterson, B. Dillon, I. Raheem, P. Richardson, D. Richter, R. Schmidt and H. F. Sneddon, Green Chem., 2014, 16, 4060–4075 RSC.
- M. B. Hicks, W. Farrell, C. Aurigemma, L. Lehmann, L. Weisel, K. Nadeau, H. Lee, C. Moraff, M. Wong, Y. Huang and P. Ferguson, Green Chem., 2019, 21, 1816–1826 RSC.
- T. Shono, T. Toda and N. Oshino, Drug Metab. Dispos., 1981, 9, 481–482 CAS.
- A. A. Folgueiras-Amador, K. Philipps, S. Guilbaud, J. Poelakker and T. Wirth, Angew. Chem., Int. Ed., 2017, 56, 15446–15450 CrossRef CAS.
- D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573–4591 CrossRef CAS.
- G. Laudadio, A. D. A. Bartolomeu, L. M. H. M. Verwijlen, Y. Cao, K. T. De Oliveira and T. Noël, J. Am. Chem. Soc., 2019, 141, 11832–11836 CrossRef CAS.
- G. Laudadio, E. Barmpoutsis, C. Schotten, L. Struik, S. Govaerts, D. L. Browne and T. Noël, J. Am. Chem. Soc., 2019, 141, 5664–5668 CrossRef CAS.
- J. M. Friedrich, C. Ponce-de-León, G. W. Reade and F. C. Walsh, J. Electroanal. Chem., 2004, 561, 203–217 CrossRef CAS.
- J. Wang, Electrochim. Acta, 1981, 26, 1721–1726 CrossRef CAS.
- X. Liu, R. J. Madix and C. M. Friend, Chem. Soc. Rev., 2008, 37, 2243–2261 RSC.
- A. Steven, M. E. Kopach, F. Gallou, G. Moine, J. D. Hayler, S. Hughes, D. Entwistle, S. G. Koenig, M. C. Bryan, P. J. Dunn, F. J. Weiberth, F. Roschangar, M. R. Hickey and P. Richardson, Green Chem., 2018, 20, 5082–5103 RSC.
- K. Ouyang, W. Hao, W. X. Zhang and Z. Xi, Chem. Rev., 2015, 115, 12045–12090 CrossRef CAS.
- X. Jia, P. Li, Y. Shao, Y. Yuan, H. Ji, W. Hou, X. Liu and X. Zhang, Green Chem., 2017, 19, 5568–5574 CAS.
- T. Shono, Y. Matsumura, K. Tsubata, Y. Sugihara, S. Yamane, T. Kanazawa and T. Aoki, J. Am. Chem. Soc., 1982, 104, 6697–6703 CrossRef CAS.
- T. Shono, Y. Matsumura, K. Uchida, K. Tsubata and A. Makino, J. Org. Chem., 1984, 49, 300–304 CrossRef CAS.
- E. Nouri-Nigjeh, H. P. Permentier, R. Bischoff and A. P. Bruins, Anal. Chem., 2010, 82, 7625–7633 CrossRef CAS.
- J. W. Gorrod and G. Aislaitner, Eur. J. Drug Metab. Pharmacokinet., 1994, 19, 209–217 CrossRef CAS.
- A. A. Kadi, S. M. Amer, H. W. Darwish and M. W. Attwa, RSC Adv., 2017, 7, 36279–36287 RSC.
- W. J. Backe, Rapid Commun. Mass Spectrom., 2017, 31, 1047–1056 CrossRef CAS.
- S. Belman, Anal. Chim. Acta, 1963, 29, 120–126 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc00851f |
| This journal is © The Royal Society of Chemistry 2020 |