
A multicentre synergistic polyoxometalate-based metal–organic framework for one-step selective oxidative cleavage of β-O-4 lignin model compounds†
Hong-Rui
Tian‡
,
Yi-Wei
Liu‡
,
Zhong
Zhang
,
Shu-Mei
Liu
,
Tian-Yi
Dang
,
Xiao-Hui
Li
,
Xiu-Wei
Sun
,
Ying
Lu
 and
Shu-Xia
Liu
*
and
Shu-Xia
Liu
*
Key Laboratory of Polyoxometalate Science of the Ministry of Education, College of Chemistry, Northeast Normal University, Changchun, Jilin 130024, P. R. China. E-mail: liusx@nenu.edu.cn
First published on 29th November 2019
Abstract
A novel mixed-valence polyoxovanadate-based copper–organic framework, [CuI(bbi)]2{[CuI(bbi)]2VIV2VV8O26}·2H2O (NENU-MV-5, bbi = 1,1′-(1,4-butanediyl)bis(imidazole)), was facilely synthesized from routine reagents under mild hydrothermal conditions. Using NENU-MV-5 as a heterogeneous catalyst without any co-catalyst, one-step oxidative cleavage of β-O-4 lignin into phenols and aromatic acids with high catalytic activity and selectivity was realized under an oxygen atmosphere. No obvious decrease in activity was observed after five cycles, which indicates the excellent stability and sustainability of NENU-MV-5. The perfect catalytic performance of NENU-MV-5 can be attributed to the multi-site synergistic effect of the mixed-valence VV–O–VIV sites on polyoxovanadate for the oxidation of β-O-4 alcohol to β-O-4 ketone and the Cu(I) sites on the framework for the rapid cleavage of the Cα–Cβ bond of β-O-4 ketone. This system represented the first co-catalyst-free example for the one-step selective degradation of lignin catalyzed by a well-defined crystalline catalyst with definite composition and structure in a single solvent.
Introduction
Lignin, as a renewable biopolymer, is abundant in plants.1–3 Relatively stable C–O and C–C bonds link the primary units of lignin, in which the β-O-4 bonds account for ca. 50% of all linkages.4–6 Therefore, in order to obtain high value added aromatics, depolymerisation of lignin through the efficient catalytic cleavage of the β-O-4 bonds has attracted much attention from both industrial and academic researchers.7 The reported methods for the targeted cleavage of β-O-4 bonds in lignin include acid–base hydrolysis,8 pyrolysis,9,10 oxidation,11,12 and their combinations,13,14 among which the oxidation approach is considered as the most effective strategy owing to the well-retained premier functional groups (methoxyl, etc.) of the lignin polymer.15Homogeneous catalytic systems are widely used for the selective oxidative cleavage of lignin, and these catalysts can be divided into three categories: (i) transition metal (such as vanadium, cobalt, etc.) complex catalysts,16–18 which can realize the one-step conversion of lignin into small molecular aromatics; (ii) organocatalysts,14,19,20 in this system, nitroxyl-based co-catalysts (such as TEMPO, etc.; TEMPO = 2,2,6,6-tetramethylpiperidine-N-oxyl) are indispensable to complete the conversion of β-O-4 alcohol into β-O-4 ketone; and (iii) inorganic salt (VOSO4, CuCl, etc.) catalysts.15,21,22 A representative example is VOSO4/TEMPO, which can catalyze the oxidation of β-O-4 alcohol to β-O-4 ketone in acetonitrile, followed by the rapid Cα–Cβ bond cleavage of β-O-4 ketone catalysed by Cu(OAc)2/1,10-phenanthroline in methanol.21 Although these homogeneous catalytic systems are effective in catalysing the oxidative cleavage of β-O-4 lignin, they suffer from the difficulty in separation and recovery. Heterogeneous catalysis is an alternative strategy to resolve these problems. Recently, a Pd/CeO2 catalyst has been reported for the one-step oxidative cleavage of 2-phenoxy-1-phenylethanol using O2 as an oxidant in MeOH.23 Co-based composite catalysts also exhibited high performance for the oxidative cleavage of β-O-4 lignin with solid bases (K2CO3, NaOH, etc.) as co-catalysts.24,25 However, their ill-defined composition and structure prevented the further study of their catalytic mechanisms. More importantly, the requirement of two catalysts, two solvents, and usually the assistance of co-catalysts (such as nitroxyl species, acids, bases, etc.) in certain catalytic systems severely limit their practical application. Therefore, the development of efficient and sustainable heterogeneous catalysts with well-defined composition and structure but requiring no co-catalyst is highly desirable.
In this work, we synthesized a novel polyoxometalate-based metal–organic framework (POMOF),26,27 [CuI(bbi)]2{[CuI(bbi)]2VIV2VV8O26} (NENU-MV-5; bbi = 1,1′-(1,4-butanediyl)bis(imidazole)). Owing to the synergistic catalytic effect of multiple active sites in NENU-MV-5, it can catalyze the one-step oxidative cleavage of β-O-4 lignin using oxygen as an oxidant without any co-catalyst. In particular, the VV–O–VIV sites with mixed-valence (MV) vanadium act as the active centre for the oxidation of β-O-4 alcohol to β-O-4 ketone; then the obtained β-O-4 ketone is rapidly converted into target phenols and aromatic acids on Cu(I) sites through Cα–Cβ bond cleavage (Scheme 1). To the best of our knowledge, NENU-MV-5 is the first crystalline heterogeneous catalyst with well-defined composition and structure for the degradation of lignin.
 | ||
| Scheme 1 One-step oxidative cleavage of the β-O-4 bond by synergistic catalysis. | ||
Results and discussion
Composition and structural analysis of NENU-MV-5
Blue-violet strip NENU-MV-5 crystals were synthesized from sodium metavanadate, hydrazine hydrate, bbi and copper nitrate trihydrate under mild hydrothermal conditions. The molecular formula was determined as [CuI(bbi)]2{[CuI(bbi)]2VIV2VV8O26}·2H2O by single-crystal X-ray diffraction, elemental analyses and thermogravimetric analysis (TGA) (Fig. S1b†). NENU-MV-5 exhibited a 3D supramolecular structure (Fig. S4†) constructed by a 1D cationic chain [Cu(bbi)]+ (Fig. 1a) and a 2D anionic layer {[Cu(bbi)]2V10O26}2− (Fig. 1d) through electrostatic interaction. In this structure, Cu ions have two kinds of coordination modes. As shown in Fig. 1, Cu2 coordinates with two nitrogen atoms from two bbi molecules in a linear geometry to form the 1D chain (Fig. 1a) and Cu1 presents a four-coordinated structure with two nitrogen atoms from two bbi molecules and two terminal oxygen atoms from two {V10} clusters to construct the 2D layer (Fig. 1b and d). Bond valence sum (BVS) calculations28 demonstrated the +1 oxidation state of all Cu ions (Table S2†). [V10O26]4− exhibited a flat spherical structure with eight VO4 tetrahedra and two VO5 tetragonal pyramids linked by the vertex-sharing of O atoms (Fig. 1c and d). The BVS calculations revealed that the oxidation states of V ions (2V1, 2V2, 2V4, and 2V5) in VO4 are close to +5, and the oxidation states of V ions (V3 and V6) in VO5 are approximately close to +4 (Table S2†). X-ray photoelectron spectroscopy (XPS) was carried out to further confirm the oxidation states of Cu ions and V ions in NENU-MV-5. The Cu 2p3/2 peak at 932.2 eV in XPS demonstrates that the oxidation state of Cu ions in NENU-MV-5 is +1 (Fig. S3a†).29 Two legible peaks with electron binding energies of 517.15 eV and 516.2 eV can be assigned to V(V) 2p3/2 and V(IV) 2p3/2,30 respectively, and their peak area ratio is about 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S3b†). These results are consistent with the BVS calculation results and balance the skeleton charges of NENU-MV-5. It should be noted that reduced coordinatively unsaturated metal sites (CUSs) have been widely accepted that can adsorb O2 and activate O2 to produce active oxygen species through interacting with the lone pair electrons of O2, resulting in charge transfer from CUSs to O2.31,32 Remarkably, NENU-MV-5 combines two kinds of reduced CUSs (V(IV) and Cu(I)) in one compound, which is expected to be of great potential for O2 activation.
:1 (Fig. S3b†). These results are consistent with the BVS calculation results and balance the skeleton charges of NENU-MV-5. It should be noted that reduced coordinatively unsaturated metal sites (CUSs) have been widely accepted that can adsorb O2 and activate O2 to produce active oxygen species through interacting with the lone pair electrons of O2, resulting in charge transfer from CUSs to O2.31,32 Remarkably, NENU-MV-5 combines two kinds of reduced CUSs (V(IV) and Cu(I)) in one compound, which is expected to be of great potential for O2 activation.
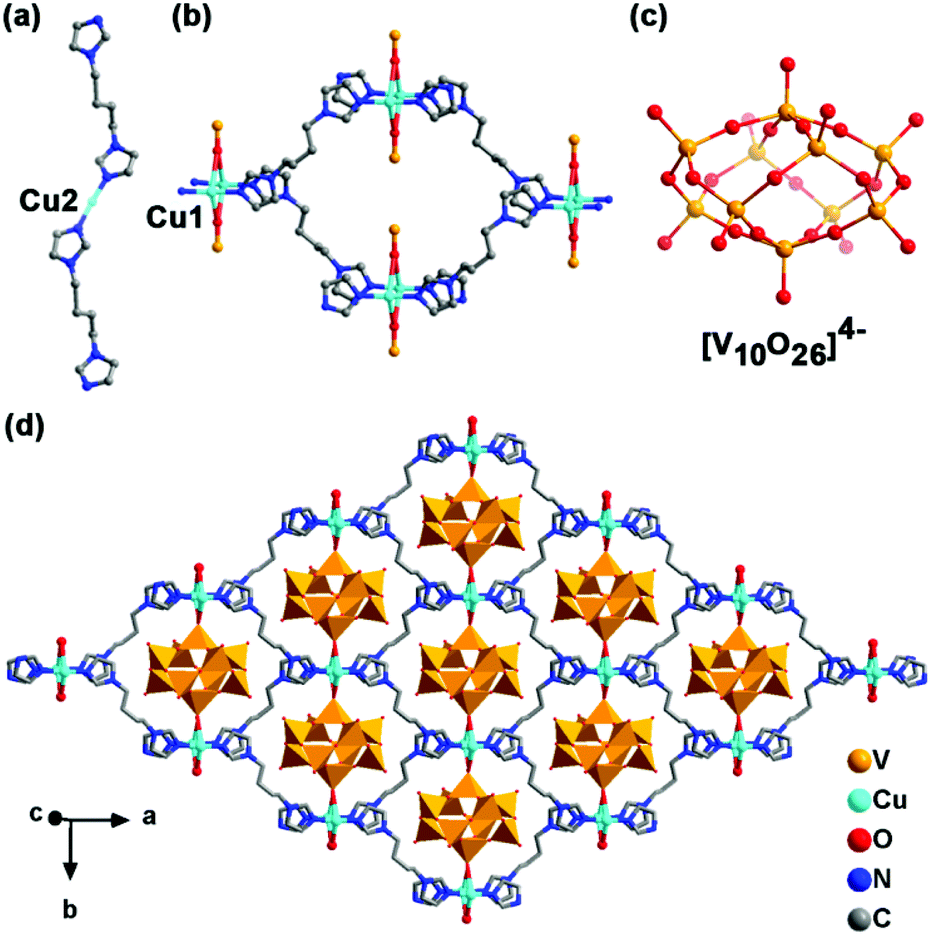 | ||
| Fig. 1 Coordination mode of Cu2 (a), Cu1 (b) and [V10O26]4− (c). (d) 2D layer of {[Cu(bbi)]2V10O26}2−. Hydrogen atoms and lattice water molecules are omitted for clarity. | ||
Catalytic performance of NENU-MV-5 for lignin model compounds
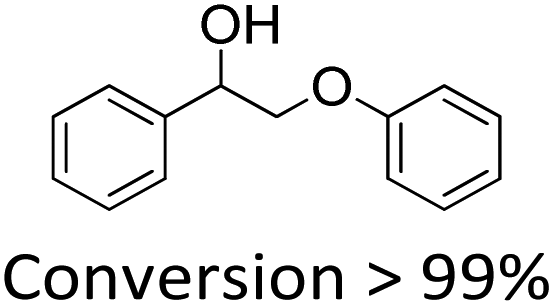
1-Phenyl-2-phenoxyethanol (1) is a widely used model compound for the study of lignin catalytic degradation. However, the oxidative cleavage of 1 usually involves two steps with two catalysts,14,33 the addition of a co-catalyst,24,25 and sometimes even two solvents.19,21 We found that NENU-MV-5 could catalyze the oxidative cleavage of 1 to aromatics in one step without any co-catalyst using only methanol (MeOH) as the sole solvent. The catalytic reaction was carried out at 100 °C under an O2 atmosphere (0.4 MPa). For a better dispersion in reaction media, NENU-MV-5 crystals were ground and the Brunauer–Emmett–Teller (BET) surface area of the used catalyst was 15 m2 g−1 (Fig. S5†). We monitored the reaction process by gas chromatography with flame ionization (GC-FID) and the products were identified by gas chromatography with mass spectrometry (GC-MS); more than 99% of compound 1 was converted within 12 hours, and the yields of phenol (1b), benzoic acid (1c) and methyl benzoate (1d) were 60%, 9.5% and 90%, respectively. Trace 2-phenoxy-1-phenylethanone (1a) was also detected with a yield of less than 1% (Table S3,† entry 1). It is noteworthy that the major products of the oxidative cleavage of β-O-4 lignin are usually 1b and/or 1c,21,34 whereas 1d (90%) was detected as the main product in our reaction system. Considering the presence of Lewis acid sites (Cu/V) on NENU-MV-5, we speculated that 1d was generated from the esterification reaction of the resultant 1c and MeOH solvent. In order to prove our conjecture, we replaced substrate 1 with commercially available benzoic acid (1c) under both N2 and O2 atmospheres keeping the other conditions the same, and a large amount of methyl benzoate (1d) was indeed detected (Table S4,† entries 1 and 2).The previously reported catalytic process for the selective oxidative cleavage of β-O-4 lignin usually requires two catalysts, specifically, first oxidizing lignin β-O-4 alcohol (1) into β-O-4 ketone (1a) with one catalyst, followed by the Cα–Cβ bond cleavage of 1a into phenol (1b) and benzoic acid (1c) with another.19,21 In contrast, in our work, NENU-MV-5 was used as the only catalyst and the obtained main products were 1b, 1c and 1d with only a trace amount of 1a being detected. This result indicated a cascade reaction process that 1a was degraded immediately after being generated from the oxidation of 1. To clarify the tandem catalytic process, we replaced 1 with stoichiometric intermediate 1a without changing the other reaction condition, and 1a was completely converted into 1b, 1c and 1d in less than 2 h (Table S3,† entry 3) whereas the conversion of 1 was only 30% (Table S3,† entry 4) at the same time. The obvious differences demonstrated that the oxidation of 1 to 1a was a rate-determining step for the degradation of 1 on NENU-MV-5. According to the related literature,35–38 we proposed a synergistic catalysis of multiple active sites to explain the one-step oxidative cleavage of 1 on NENU-MV-5. Firstly, MV-vanadium in VV–O–VIV of NENU-MV-5 catalysed the oxidation of the Cα–OH bond of 1 to 1a; subsequently, the Cα–Cβ bond of 1a was promptly cleaved to obtain 1b and 1c on Cu(I) sites.
To confirm the aforementioned assumptions, a range of control experiments were carried out using VOSO4, NaVO3, CuCl, Cu(NO3)2 and (Et4N)4(V10O26)·H2O containing V(IV), V(V), Cu(I), Cu(II) and MV-{V10} cluster as catalysts, respectively (Table 1). (Et4N)4(V10O26)·H2O (Fig. S6†) was synthesized according to the literature39 in which [V10O26]4− had the same structure as that in NENU-MV-5. These catalytic results showed that VOSO4 and (Et4N)4(V10O26)·H2O, both possessing V(IV), as catalysts gave only 1a in 44.6% and 96.7% yields, respectively (Table 1, entries 1 and 5). In contrast, V(V)-containing NaVO3 and V-free CuCl and Cu(NO3)2 as catalysts exhibited no catalytic activity (Table 1, entries 2–4). Considering the different oxidation states of V in these catalysts, we can deduce that V(IV) is the catalytically active site for the oxidation of 1 to 1a, but it is not active for the C–C bond cleavage of 1a to 1b and 1c. In contrast, copper(I/II) was inactive for the oxidation of β-O-4 alcohol to β-O-4 ketone; it was catalytically active for the cleavage of 1a to 1b and 1c (Table 1, entries 8 and 9). It was also observed that the catalytic activity of (Et4N)4(V10O26)·H2O involving MV-vanadium was distinctly higher than that of VOSO4, which might be caused by the collaborative effect of MV-vanadium in the {V10} cluster. The VV–O–VIV mode could promote electron transfer through intervalence charge transfer (IVCT) to expedite the catalytic reaction.40–42 For comparison, a physical mixture of VOSO4 and NaVO3 was used to catalyze the oxidative cleavage of 1; the conversion (merely 30%) was obviously lower than that of (Et4N)4(V10O26)·H2O (Table 1, entries 5 and 6), which could be ascribed to the separated V(IV) and V(V) lacking IVCT between them. Overall, the role of each active centre of NENU-MV-5 in the oxidative cleavage of 1 could be clarified; VV–O–VIV was effective for the catalytic oxidation of 1 to 1a and then Cu(I) catalysed the Cα–Cβ bond cleavage of 1a to 1b and 1c.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Conv. (%) | Yieldd (%) | |||
| 1a | 1b | 1c | 1d | |||
| n.d. = not detected.a Reaction conditions: Substrate (0.5 mmol), catalyst (0.1 mmol, based on V or Cu), MeOH (2 mL), O2 (0.4 MPa), 100 °C, 12 hours.b Catalyst (0.02 mmol VOSO4 + 0.08 mmol NaVO3).c 2 hours.d Yield was determined by GC using biphenyl as an internal standard. | ||||||
| 1 as the substrate | ||||||
| 1 | VOSO4 | 44.6 | 44.6 | n.d. | n.d. | n.d. |
| 2 | NaVO3 | <1 | n.d. | n.d. | n.d. | n.d. |
| 3 | CuCl | <1 | n.d. | n.d. | n.d. | n.d. |
| 4 | Cu(NO3)2 | <1 | n.d. | n.d. | n.d. | n.d. |
| 5 | (Et4N)4(V10O26)·H2O | 96.7 | 96.7 | n.d. | n.d. | n.d. |
| 6b | VOSO4 + NaVO3 | 30.0 | 30.0 | n.d. | n.d. | n.d. |
| 7 | NENU-MV-5 | >99 | <1 | 60.0 | 9.5 | 90.0 |
| 1a as the substrate | ||||||
| 8c | CuCl | 85.0 | — | 55.0 | 32.0 | 51.0 |
| 9c | Cu(NO3)2 | 65.0 | — | 51.5 | 15.4 | 71.0 |

Reaction mechanism of NENU-MV-5 for catalysing the conversion of β-O-4 lignin model compounds
The mechanism of NENU-MV-5 for the catalytic cleavage of lignin was proposed based on the experimental results. The catalytic oxidative cleavage of lignin model compound 1 with NENU-MV-5 involves two processes: firstly, VV–O–VIV catalyzes the Cα–OH bond oxidation of 1 to 1a, and then, the Cα–Cβ bond of 1a is cleaved on Cu(I). In both the processes, oxygen is necessary owing to the suppressive activities in the absence of O2 (Table S3,† entries 5 and 6). A radical reaction mechanism for the oxidative cleavage of lignin with oxygen as an oxidant has been reported, in which the main active species might be ˙O2−, HOO˙ and/or ˙OH.43 MeOH is an effective scavenger of ˙OH,44 and it is used as a solvent in the oxidative cleavage of 1; thus ˙OH would not be the active species. To confirm the active oxygen species involved in the oxidative cleavage of 1, an electric paramagnetic resonance (EPR) test was performed on NENU-MV-5 with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), which has been regarded as the most commonly used trapper for detecting spin-reactive species.45 The characteristic peak of the DMPO–˙O2− adduct in both MeOH (Fig. 2a) and acetonitrile (Fig. S7†) was observed as compared to the absence of NENU-MV-5,46,47 indicating that NENU-MV-5 could produce ˙O2− by interacting with dissolved oxygen in the solvent. Notably, the oxidative cleavage of 1 does not occur in acetonitrile (Table S3,† entry 2); thus ˙O2− may not be the active species of the oxidative cleavage of 1. According to the previous reports,48 ˙O2− alone is not effective in directly reacting with β-O-4 alcohol, while HOO˙ is able to abstract the Cα–H of β-O-4 alcohol to β-O-4 ketone. Therefore, it is reasonable to speculate that HOO˙ is the active species in which 1 is oxidized to 1a, as HOO˙ is acquired by the combination of ˙O2− with H+ from MeOH, which also explains the phenomenon that the oxidative cleavage of 1 could proceed in MeOH rather than in acetonitrile. In order to understand the activation of O2 by NENU-MV-5 to produce ˙O2−, the O2 adsorption/desorption test of NENU-MV-5 was performed. As shown in Fig. 2b, an obvious hysteresis in the O2 desorption isotherm indicated the presence of a strong interaction between NENU-MV-5 and oxygen.49 Generally, reduced CUSs could adsorb and activate O2. However, there are two kinds of reduced CUSs in NENU-MV-5, V(IV) and Cu(I). It is well known that coordinatively unsaturated Cu(I) can interact with oxygen to form a superoxocopper(II) or μ-peroxocopper(II) complex but not free ˙O2−,50–52 and our previous experimental results show that Cu(I/II) is not catalytically active for the oxidation of 1 to 1a (Table 1, entries 3 and 4). Hence, we deduce that ˙O2− is obtained from the O2 activated by V(IV). In addition, the BVS calculations demonstrate that the V(IV) atoms are positioned on the top and bottom of the MV-{V10} cluster (Fig. 1c), and the five-coordinated V(IV) geometry in the MV-{V10} cluster is similar to the vanadium centre configuration of the formation of [O]2− vacancy in polyoxovanadate.53 Oxygen vacancy is inclined to absorb O2 molecules and convert them into ˙O2−.54 These results illustrate that ˙O2− could be generated from the interaction of V(IV) with O2. The tetracoordinated V(V) in NENU-MV-5 can interact with 1-phenyl-2-phenoxyethanol (1) to form a five-membered ring to activate the Cα–H bond to promote the rapid oxidation of 1 to 1a, and this phenomenon has been confirmed by Hanson and co-workers.34 Accordingly, we proposed that the high catalytic performance of VV–O–VIV catalyses the oxidation of 1 to 1a, which could be attributed to the synergistic effect of pentacoordinated V(IV) for activating O2 to ˙O2− and tetracoordinated V(V) for activating the Cα–H bond of 1. The obtained ˙O2− combines with H+ from MeOH to give HOO˙, which abstracts the hydrogen atom of Cα to achieve the oxidation of 1 to 1a. | ||
| Fig. 2 (a) DMPO spin trapping EPR spectra for DMPO–˙O2− radical species in methanol. (b) O2 adsorption/desorption isotherms of NENE-MV-5, P0 = 0.4 MPa. | ||
Next, we investigated the reaction of Cu(I) sites in NENU-MV-5 catalysed Cα–Cβ bond cleavage of 1a to 1b and 1c using O2 as an oxidant. According to our previous experimental results, the obtained methyl benzoate (1d) in the oxidative cleavage of 1a is generated by the esterification of the resultant benzoic acid (1c) and solvent MeOH (Table S4,† entries 1 and 2); so we deduce that the Cα–Cβ bond of 1a is firstly cleaved. To confirm our conjecture, the oxidative cleavage of methyl benzoylformate (the intermediate of the preferential cleavage of the Cβ–O bond of 1a, Scheme S1†)23,24 by NENU-MV-5 was carried out under the same conditions as those for 1a and no 1c and 1d were detected (Scheme S2†), suggesting that the Cβ–O bond was not first broken but the Cα–Cβ bond was first broken in the oxidative cleavage of 1a. It is well known that coordinatively unsaturated Cu(I) can react with oxygen to produce Cu(II)–OO˙, which is an effective oxidant for the cleavage of the C–C bond.55 Hence, in the process of the oxidative cleavage of 1a catalyzed by Cu(I), we proposed that Cu(I) firstly interacted with O2 to give Cu(II)–OO˙, which catalysed the Cα–Cβ bond cleavage of 1a to 1c and phenol formate, and then the decarboxylation of the obtained phenol formate to phenol (1b) and CO2 occurred. The limewater test confirmed the production of CO2 in the reaction (Fig. S8†). Remarkably, the amount of CO2 (1.65 mmol) calculated from the obtained CaCO3 (165 mg, 1.65 mmol) collected with limewater (Scheme S3†) was obviously higher than the amount of CO2 (0.5 mmol) produced by the oxidative decarboxylation of 1 (Table S5†). Considering that phenol is easily oxidized by HOO˙ to CO2 and H2O (Scheme S4†),56 the generation of extra CO2 (1.15 mmol equivalent to 0.19 mmol phenol) which was comparable to the reduced phenol (0.2 mmol) indicated that part of the phenol produced by the oxidative cleavage of 1 was oxidized to CO2 and H2O.
Based on the experimental results and the above discussion, a possible mechanism was proposed for the catalytic reaction process (Scheme 2). Initially, MV-vanadium in VV–O–VIV of NENU-MV-5 oxidizes β-O-4 alcohol to β-O-4 ketone via synergistic catalytic action. In particular, pentacoordinated V(IV) and tetracoordinated V(V) of VV–O–VIV (a) interact with oxygen and β-O-4 alcohol, respectively (b). V(IV) adsorbs and activates oxygen to afford ˙O2− (c), and V(V) interacts with β-O-4 alcohol to form a stable five-membered ring to activate Cα–H (e). ˙O2− (c) combines with H+ from MeOH to give HOO˙ (d), which extracts hydrogen from Cα–H (e) and further forms the transition state f. Subsequently, the transition state f undergoes a single electron transfer to give intermediate β-O-4 ketone (g). Noticeably, the process of the oxidation of β-O-4 alcohol to β-O-4 ketone activates Cβ–H.23 Cu(I) (h) in NENU-MV-5 reacts with oxygen to give Cu(II)–OO˙ (i), where Cu(II) coordinates with the carbonyl oxygen of β-O-4 ketone and Cβ–H is oxidized by Cu(II)–OO˙ to the transition state j. Finally, j rearranges to k, which experiences the cleavage of the Cα–Cβ bond and decarboxylation to produce benzoic acid, phenol and CO2. Methyl benzoate is obtained by the esterification reaction of MeOH and benzoic acid under the catalysis of Lewis acids (Cu/V in NENU-MV-5).
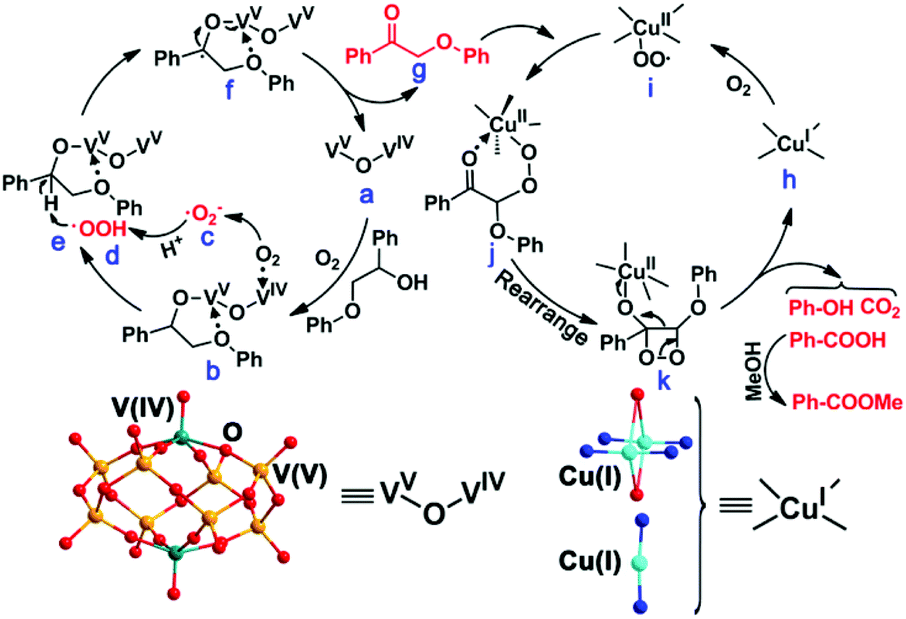 | ||
| Scheme 2 Proposed catalytic mechanism of NENU-MV-5. | ||
Oxidative cleavage of other lignin model compounds with NENU-MV-5
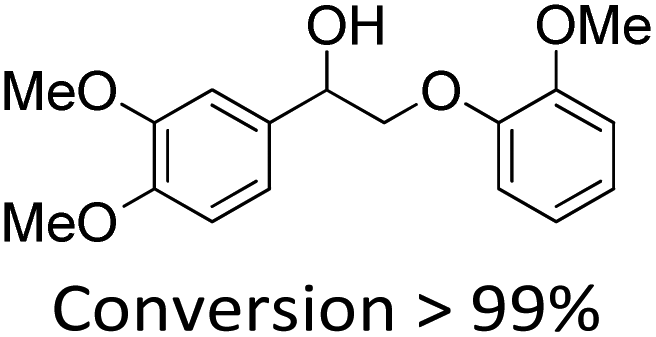
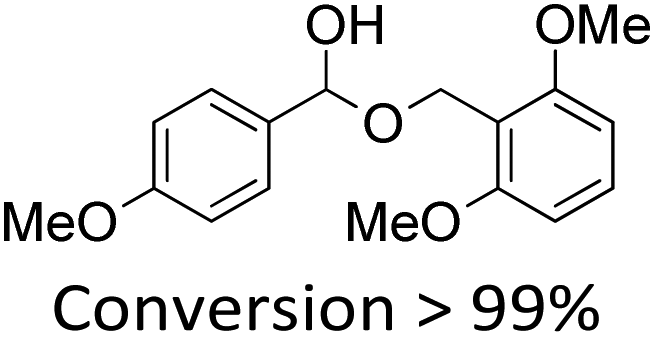
It is known that β-O-4 lignin model compounds with more functional groups, such as the methoxyl group, Cγ–OH, etc., are structurally more similar to native lignin.13,23,57 Hence, we investigated the catalytic behaviours of NENU-MV-5 towards the oxidative cleavage of a series of β-O-4 lignin model compounds with methoxyl and Cγ–OH groups. As shown in Table 2, NENU-MV-5 could catalyze the oxidative cleavage of lignin model compounds with different amounts of methoxyl groups to afford the corresponding value-added aromatics under the same conditions as 1 (Table 2, entries 2–7). Although the Cγ–OH group makes the oxidative cleavage of the Cα–Cβ bond more difficult,21 NENU-MV-5 can still catalyse the oxidative cleavage of the lignin model compounds with Cγ–OH to phenols, aromatic acids and esters in a good yield by appropriately increasing the reaction temperature (120 °C) and prolonging the reaction time (18 hours) (Table 2, entry 8). These results demonstrated that NENU-MV-5 can efficiently and selectively catalyze the β-O-4 lignin model compounds with characteristic functional groups (methoxyl and Cγ–OH groups, which are usually present in native lignin) to give aromatics, and indicated its potential to catalyse the oxidative cleavage of native lignin.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Model compounds | Product | Entry | Model compounds | Product | ||||
| Phenol | Acid | Ester | Phenol | Acid | Ester | ||||
| Yieldc (%) | Yieldc (%) | ||||||||
| a Reaction conditions: Substrate (0.5 mmol), NENU-MV-5 (0.01 mmol), MeOH (2 mL), O2 (0.4 MPa), 100 °C, 12 hours. b 120 °C, 18 hours. c Yield was determined by GC using biphenyl as an internal standard. | |||||||||
| 1 |
|
60.0 | 6.5 | 90.0 | 5 |
|
65.0 | 4.4 | 94.8 |
| 2 |

|
55.0 | 9.0 | 91.0 | 6 |
|
53.0 | 7.6 | 92.0 |
| 3 |
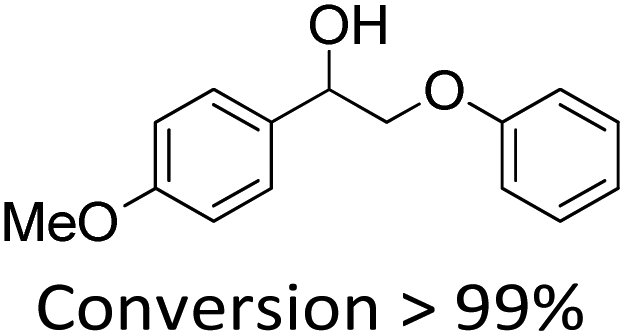
|
62.0 | 7.8 | 91.5 | 7 |

|
54.5 | 5.3 | 94.0 |
| 4 |
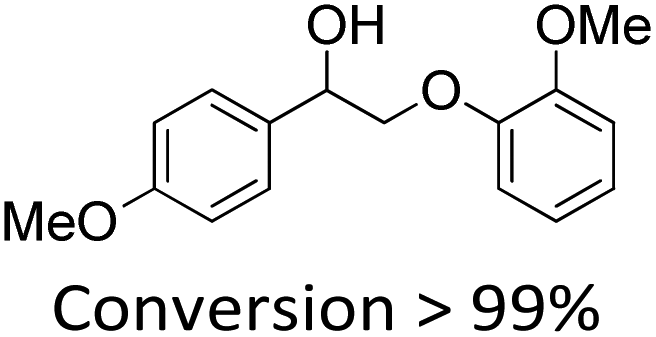
|
58.0 | 8.3 | 91.1 | 8b |

|
42.0 | 4.5 | 92.8 |
Recycling experiments
NENU-MV-5 exhibited excellent stability and recyclability. There was no significant decrease in activity after five cycles (the conversion of 1 was maintained at 99–96%). Moreover, the yields of phenol, benzoic acid and methyl benzoate almost remained constant during the five cycles (Fig. 3). Powder X-ray diffraction (PXRD) and Fourier-transform infrared (FTIR) spectra (Fig. 4a and b) of the recovered catalyst matched well with those of a fresh catalyst, indicating its structural integrity. In addition, there was no noticeable difference in XPS results, which demonstrates the well-maintained oxidation states of cuprous ions and vanadium ions in the catalyst before and after the reaction (Fig. 4c and d).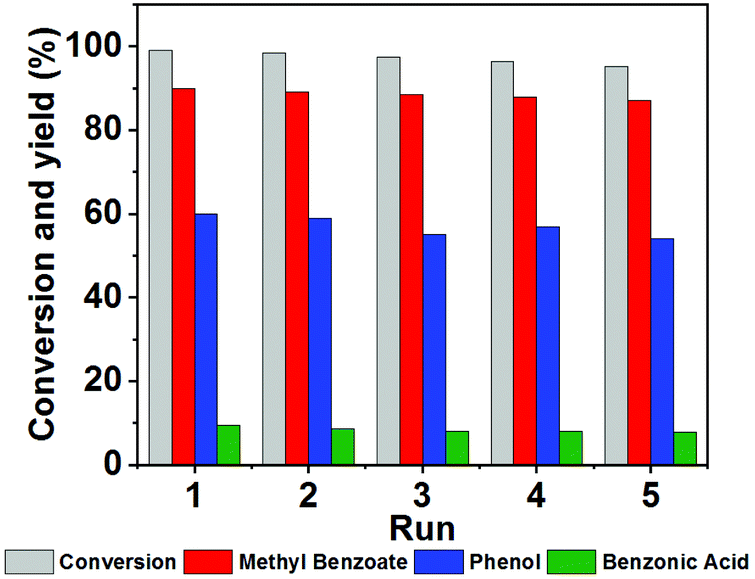 | ||
| Fig. 3 The recyclability of NENU-MV-5. Reaction conditions: Lignin model compound 1 (0.5 mmol), NENU-MV-5 (0.01 mmol), MeOH (2 mL), O2 (0.4 MPa), 100 °C, 12 hours, biphenyl as an internal standard. | ||
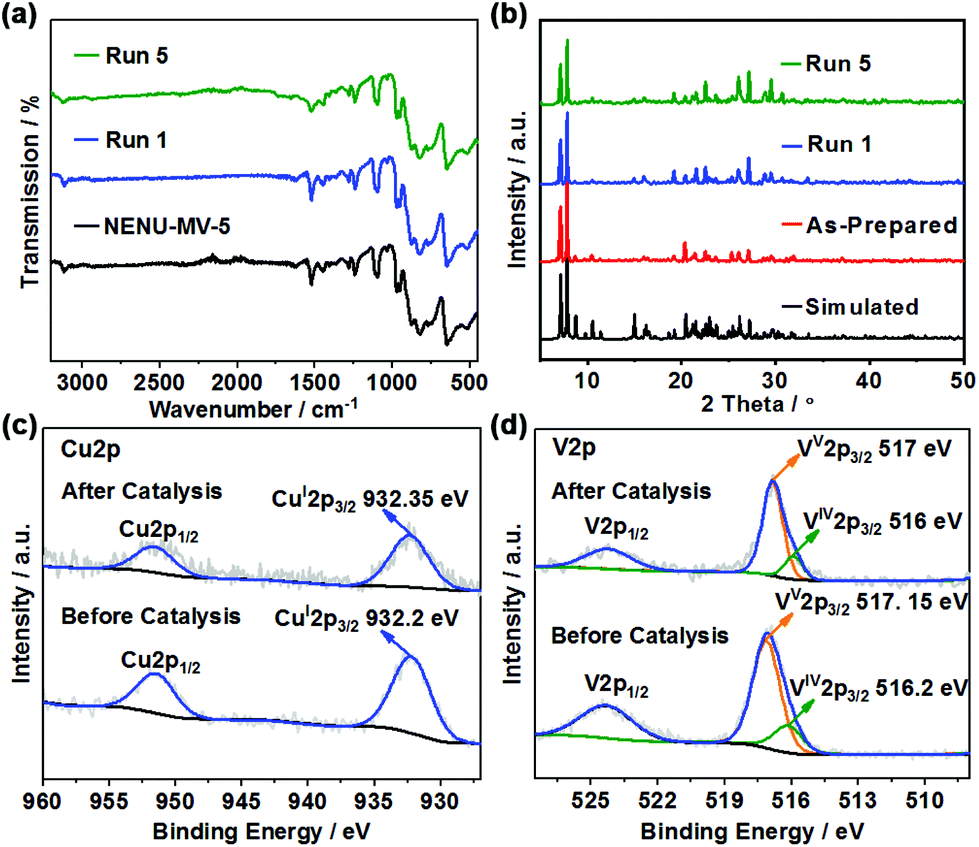 | ||
| Fig. 4 The FTIR spectra (a), PXRD patterns (b) and XPS results (oxidative states of cuprous ions (c) and vanadium ions (d)) of the recovered catalyst NENU-MV-5 after five catalytic reactions. | ||
Compared with the reported catalysts for the oxidative cleavage of β-O-4 lignin (Table S6†), NENU-MV-5 exhibited several advantages. First of all, as a heterogeneous catalyst, NENU-MV-5 showed comparable catalytic activity with homogeneous catalysts (Table S6,† entries 2 and 3).34,58 Secondly, the one-step degradation of lignin model compounds was realized in NENU-MV-5 by the synergy of multiple sites, whereas two steps with two catalysts were required in certain homogeneous catalytic systems (Table S6,† entries 9–11).14,19,20 Thirdly, NENU-MV-5 exhibited higher activity than palladium- and cobalt-based heterogeneous catalysts (Table S6,† entries 5–7),23–25 and does not require the assistance of any co-catalysts (such as K2CO3, NaOH, etc.). Hence, NENU-MV-5 can be considered as a sustainable and environmentally friendly catalyst for the oxidative cleavage of lignin into value-added aromatics.
Conclusions
In conclusion, we synthesized a MV-{V10} cluster-based Cu–MOF, NENU-MV-5, containing multiple catalytically active sites. Using NENU-MV-5 as a heterogeneous catalyst without any co-catalyst, β-O-4 lignin can be degraded into high value added aromatics in one step with high catalytic activity and recyclability. The catalytic mechanism was proposed and confirmed based on the experimental results. This work demonstrates that multiple active sites can be integrated into one material through constructing a polyoxometalate-based metal–organic framework, which provides an insight into designing multifunctional catalysts for organic transformations.Experimental
Materials and instrumentation
All of the reagents and solvents were of analytical grade, purchased from commercial sources and used without further purification. The ligand 1,1′-(1,4-butanediyl)bis(imidazole) (bbi) and lignin model compounds were synthesized according to the literature methods59,60 and characterized by 1H NMR (the synthesis and 1H NMR spectra are shown in the ESI†). Elemental analyses (C, H and N) were performed on a PerkinElmer 2400 CHN elemental analyzer. V and Cu were analyzed using a PLASMA-SPEC(I) ICP atomic emission spectrometer. FTIR spectra were collected (as KBr-pressed pellets) using a Nicolet 170SXFT-IR spectrophotometer in the range of 400–4000 cm−1. Thermogravimetric analysis (TGA) was conducted under a nitrogen atmosphere with a heating rate of 10 °C min−1 on an SDT 2960 Simultaneous DSC-TGA from TA Instruments up to 600 °C. Powder X-ray diffraction (PXRD) measurements were performed on a SmartLab instrument with Cu Kα (λ = 1.5418 Å) radiation and X-ray 40 kV/30 mA over the angular range 2θ 5°–50° at a scan rate of 10° min−1. Electron paramagnetic resonances (EPR) were obtained using an MS5000 EPR spectrometer. Nitrogen and oxygen adsorption/desorption measurements were performed by using a Hiden isochema IGA 100B instrument at 77 K and 298 K, respectively. X-ray photoelectron spectra (XPS) were collected using a Thermo ESCALAB 250 X-ray photoelectron spectrometer. 1H NMR spectra were recorded using a Bruker Avance 600 (1H: 600 MHz) system at ambient temperature. Data are reported as chemical shifts in ppm relative to TMS (0.00 ppm) for 1H. The following abbreviations are used to explain the multiplicities: s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. The conversion of lignin model compounds and the products were confirmed by gas chromatography (GC) using an Agilent 7800A instrument equipped with an HP-5 column. The products were identified by gas chromatography with mass spectrometry (GC-MS) on an Agilent 7890A-5975C instrument. The products were quantified by GC using a Shimadzu GC-2014 instrument equipped with an SH-Rtx-Wax column.Synthesis of NENU-MV-5
NaVO3 (122 mg, 1 mmol) and N2H4·H2O (80%, 19 μL, 0.32 mmol) were added into 10 mL of H2O. The resulting mixture was stirred for 1 hour at room temperature, and then bbi (95 mg, 0.5 mmol) and CuNO3·3H2O (121 mg, 0.5 mmol) were consecutively added. Then the mixture was stirred for another 1 hour at room temperature. The pH of the mixture was about 5.0. Then the mixture was placed in a 23 mL Teflon reactor and kept under autogenous pressure at 150 °C for 3 days. After this the mixture was cooled to 140 °C in 1 hour and kept at this temperature for 3 hours. Then the mixture was cooled to room temperature at a rate of 10 °C per hour, and blue-violet strip crystals were obtained (yield: 119 mg, 60% based on V). Elemental analysis for NENU-MV-5: C, 24.26%; H, 3.26%; N, 11.31%; V, 25.72%; Cu, 12.83%; found: C, 24.18%; H, 3.30%; N, 11.43%; V, 25.59%; Cu, 12.61%; FTIR (KBr, cm−1, Fig. S1a†): 3117(w), 1525(m), 1451(w), 1406(w), 1360(w), 1283(w), 1241(m), 1105(m), 966(s), 876(s), 824(s), 773(s), 654(s).Crystallography
Single-crystal X-ray diffraction (SXRD) data of NENU-MV-5 were collected on a Bruker diffractometer using Mo Kα radiation (λ = 0.71073 Å) at 295 K. The empirical absorption correction was based on equivalent reflections. The structure was solved by direct methods and refined by full-matrix least-squares fitting on F2 using the SHELXTL crystallographic software package. Non-hydrogen atoms were refined with anisotropic displacement parameters during the final cycles. All hydrogen atoms of the organic molecule were placed based on geometrical considerations and were included in the structure factor calculation. CCDC 1912680† contains the crystallographic data for NENU-MV-5. Crystal data and structure refinement are summarized in Table S1.†General procedure for the catalytic oxidation cleavage of lignin model compounds
0.5 mmol of lignin model compounds, 0.1 mmol of a catalyst (based on V or Cu), biphenyl (internal standard) and 2 mL of MeOH were added to a 10 mL autoclave reactor with an internal Teflon insert. Then, the reactor was charged with 0.4 MPa O2, heated to 100 °C and stirred for 12 h. The reactor was cooled to room temperature, the catalyst was centrifuged and the supernatant was diluted with MeOH. The conversion of lignin model compounds and the products were confirmed by gas chromatography-mass spectrometry (GC-MS).Recycling experiment of NENU-MV-5
The reusability of the catalyst NENU-MV-5 was tested using 2-phenoxy-1-phenylethanol (1) as the substrate. After the completion of the first catalytic reaction, the catalyst was recovered by centrifugation and then washed three times with MeOH. The recovered NENU-MV-5 was dried under air overnight and reused for the next run under the same conditions.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant No. 21872021, 21671033 and 21901135).References
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- Y. Shao, Q. Xia, L. Dong, X. Liu, X. Han, S. F. Parker, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, S. Yang and Y. Wang, Nat. Commun., 2017, 8, 16104 CrossRef PubMed.
- L. Dong, L. Lin, X. Han, X. Si, X. Liu, Y. Guo, F. Lu, S. Rudić, S. F. Parker, S. Yang and Y. Wang, Chem, 2019, 5, 1521–1536 CAS.
- Q. Meng, M. Hou, H. Liu, J. Song and B. Han, Nat. Commun., 2017, 8, 14190 CrossRef.
- Z. Hao, S. Li, J. Sun, S. Li and F. Zhang, Appl. Catal., B, 2018, 237, 366–372 CrossRef CAS.
- Y. Jing, Y. Guo, Q. Xia, X. Liu and Y. Wang, Chem, 2019, 5, 2520–2546 CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- A. K. Deepa and P. L. Dhepe, ACS Catal., 2015, 5, 365–379 CrossRef CAS.
- Y.-T. Cheng, J. Jae, J. Shi, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 1387–1390 CrossRef CAS.
- S. Chu, A. V. Subrahmanyam and G. W. Huber, Green Chem., 2013, 15, 125–136 RSC.
- S. Lotfi, D. C. Boffito and G. S. Patience, ChemSusChem, 2015, 8, 3424–3432 CrossRef CAS PubMed.
- J. Mottweiler, T. Rinesch, C. Besson, J. Buendia and C. Bolm, Green Chem., 2015, 17, 5001–5008 RSC.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249 CrossRef CAS.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS.
- B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. P. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794–804 CrossRef CAS.
- S. K. Hanson, R. Wu and L. A. P. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS.
- B. Biannic and J. J. Bozell, Org. Lett., 2013, 15, 2730–2733 CrossRef CAS PubMed.
- Y. Ma, Z. Du, J. Liu, F. Xia and J. Xu, Green Chem., 2015, 17, 4968–4973 RSC.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- M. Wang, J. Lu, X. Zhang, L. Li, H. Li, N. Luo and F. Wang, ACS Catal., 2016, 6, 6086–6090 CrossRef CAS.
- M. Wang, J. Lu, L. Li, H. Li, H. Liu and F. Wang, J. Catal., 2017, 348, 160–167 CrossRef CAS.
- W. Deng, H. Zhang, X. Wu, R. Li, Q. Zhang and Y. Wang, Green Chem., 2015, 17, 5009–5018 RSC.
- H. Luo, L. Wang, G. Li, S. Shang, Y. Lv, J. Niu and S. Gao, ACS Sustainable Chem. Eng., 2018, 6, 14188–14196 CrossRef CAS.
- S. Liu, L. Bai, A. P. van Muyden, Z. Huang, X. Cui, Z. Fei, X. Li, X. Hu and P. J. Dyson, Green Chem., 2019, 21, 1974–1981 RSC.
- S. Liu, L. Xie, B. Gao, C. Zhang, C. Sun, D. Li and Z. Su, Chem. Commun., 2005, 5023–5025 RSC.
- C.-Y. Sun, S.-X. Liu, D.-D. Liang, K.-Z. Shao, Y.-H. Ren and Z.-M. Su, J. Am. Chem. Soc., 2009, 131, 1883–1888 CrossRef CAS.
- I. Brown, J. Appl. Crystallogr., 1996, 29, 479–480 CrossRef CAS.
- P. Liu and E. J. M. Hensen, J. Am. Chem. Soc., 2013, 135, 14032–14035 CrossRef CAS.
- J. Dong, J. Hu, Y. Chi, Z. Lin, B. Zou, S. Yang, C. L. Hill and C. Hu, Angew. Chem., Int. Ed., 2017, 56, 4473–4477 CrossRef CAS PubMed.
- G. T. Babcock and M. Wikström, Nature, 1992, 356, 301–309 CrossRef CAS.
- M. P. Lanci, V. V. Smirnov, C. J. Cramer, E. V. Gauchenova, J. Sundermeyer and J. P. Roth, J. Am. Chem. Soc., 2007, 129, 14697–14709 CrossRef CAS PubMed.
- N. D. Patil, S. G. Yao, M. S. Meier, J. K. Mobley and M. Crocker, Org. Biomol. Chem., 2015, 13, 3243–3254 RSC.
- S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott and D. L. Thorn, Inorg. Chem., 2010, 49, 5611–5618 CrossRef CAS PubMed.
- B. Chen, X. Huang, B. Wang, Z. Lin, J. Hu, Y. Chi and C. Hu, Chem. – Eur. J., 2013, 19, 4408–4413 CrossRef CAS.
- S. Zavahir, Q. Xiao, S. Sarina, J. Zhao, S. Bottle, M. Wellard, J. Jia, L. Jing, Y. Huang, J. P. Blinco, H. Wu and H.-Y. Zhu, ACS Catal., 2016, 6, 3580–3588 CrossRef CAS.
- X. Ren, P. Wang, X. Han, G. Zhang, J. Gu, C. Ding, X. Zheng and F. Cao, ACS Sustainable Chem. Eng., 2017, 5, 6548–6556 CrossRef CAS.
- M. Wang, L. H. Li, J. M. Lu, H. J. Li, X. C. Zhang, H. F. Liu, N. C. Luo and F. Wang, Green Chem., 2017, 19, 702–706 RSC.
- A. Bino, S. Cohen and C. Heitner-Wirguin, Inorg. Chem., 1982, 21, 429–431 CrossRef CAS.
- I. Chakraborty, P. Baran, Y. Sanakis, A. Simopoulos, E. Fachini and R. G. Raptis, Inorg. Chem., 2008, 47, 11734–11737 CrossRef CAS.
- P. Yin, P. Wu, Z. Xiao, D. Li, E. Bitterlich, J. Zhang, P. Cheng, D. V. Vezenov, T. Liu and Y. Wei, Angew. Chem., Int. Ed., 2011, 50, 2521–2525 CrossRef CAS.
- S. Wang, Y. Liu, Z. Zhang, X. Li, H. Tian, T. Yan, X. Zhang, S. Liu, X. Sun, L. Xu, F. Luo and S. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 12786–12796 CrossRef CAS PubMed.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- M. D. Keywood, J. H. Kroll, V. Varutbangkul, R. Bahreini, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2004, 38, 3343–3350 CrossRef CAS PubMed.
- U. Stoin, A. I. Shames, I. Malka, I. Bar and Y. Sasson, ChemPhysChem, 2013, 14, 4158–4164 CrossRef CAS PubMed.
- S. Weng, B. Chen, L. Xie, Z. Zheng and P. Liu, J. Mater. Chem. A, 2013, 1, 3068–3075 RSC.
- C. Xu, H. Liu, D. Li, J.-H. Su and H.-L. Jiang, Chem. Sci., 2018, 9, 3152–3158 RSC.
- H. Li, F. Qin, Z. Yang, X. Cui, J. Wang and L. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef CAS PubMed.
- W. Zhang, D. Banerjee, J. Liu, H. T. Schaef, J. V. Crum, C. A. Fernandez, R. K. Kukkadapu, Z. Nie, S. K. Nune, R. K. Motkuri, K. W. Chapman, M. H. Engelhard, J. C. Hayes, K. L. Silvers, R. Krishna, B. P. McGrail, J. Liu and P. K. Thallapally, Adv. Mater., 2016, 28, 3572–3577 CrossRef CAS.
- K. D. Karlin, S. Kaderli and A. D. Zuberbühler, Acc. Chem. Res., 1997, 30, 139–147 CrossRef CAS.
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed.
- M. Zhao, X.-W. Zhang and C.-D. Wu, ACS Catal., 2017, 7, 6573–6580 CrossRef CAS.
- B. E. Petel, W. W. Brennessel and E. M. Matson, J. Am. Chem. Soc., 2018, 140, 8424–8428 CrossRef CAS PubMed.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS PubMed.
- A. S. K. Tsang, A. Kapat and F. Schoenebeck, J. Am. Chem. Soc., 2016, 138, 518–526 CrossRef CAS PubMed.
- X. Zhong, Y. Lu, F. Luo, Y. Liu, X. Li and S. Liu, Chem. – Eur. J., 2018, 24, 3045–3051 CrossRef CAS PubMed.
- Q. Mei, H. Liu, X. Shen, Q. Meng, H. Liu, J. Xiang and B. Han, Angew. Chem., Int. Ed., 2017, 56, 14868–14872 CrossRef CAS PubMed.
- W. Huo, W. Li, M. Zhang, W. Fan, H.-M. Chang and H. Jameel, Catal. Lett., 2014, 144, 1159–1163 CrossRef CAS.
- C. Li, L. Zhao, J. Li, X. Ding, S. Chen, Q. Zhang, Y. Yu and X. Jia, Chem. Commun., 2010, 46, 9016–9018 RSC.
- M. Dawange, M. V. Galkin and J. S. M. Samec, ChemCatChem, 2015, 7, 401–404 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1912680. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9gc03626a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |