
A novel bicomponent Co3S4/Co@C cocatalyst on CdS, accelerating charge separation for highly efficient photocatalytic hydrogen evolution†
Yunpeng
Liu
ab,
Bingxiong
Wang
a,
Qiao
Zhang
a,
Siyuan
Yang
 c,
Yuhang
Li
d,
Jianliang
Zuo
a,
Hongjuan
Wang
b and
Feng
Peng
*a
c,
Yuhang
Li
d,
Jianliang
Zuo
a,
Hongjuan
Wang
b and
Feng
Peng
*a
aSchool of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou, 510006, China. E-mail: fpeng@gzhu.edu.cn
bSchool of Chemistry and Chemical Engineering, Key Laboratory of Fuel Cell Technology of Guangdong Province, South China University of Technology, Guangzhou, 510640, China
cCollege of Materials and Energy, South China Agricultural University, Guangzhou, 510642, China
dSchool of Chemistry, Sun Yat-sen University, Guangzhou, 510275, China
First published on 25th November 2019
Abstract
In order to realize the dream of the practical application of hydrogen energy from solar energy and water, it is necessary to use efficient and earth-abundant cocatalysts for photocatalytic H2 evolution. Compared with a one-component cocatalyst, the multicomponent cocatalysts with a synergistic effect can effectively accelerate the separation of photogenerated electron–hole pairs in photocatalysis. Hence, a novel bicomponent Co3S4/Co@C cocatalyst on a CdS semiconductor photocatalyst is successfully synthesized by a one-step hydrothermal method. The Co3S4/Co-CdS photocatalyst obtained exhibits a highly efficient photocatalytic H2 evolution rate of 15.17 mmol h−1 g−1, which is 2.8 times higher than that of Pt-CdS. Furthermore, the quantum efficiency of Co3S4/Co-CdS reaches a maximum value of 16.80% at 475 nm. The significant improvement in the performance is because of the unique energy level structure of the Co3S4/Co-CdS photocatalyst, which has the maximum efficiency of charge separation and migration. This work not only presents a new protocol for constructing bicomponent cocatalysts on a semiconductor structure, but also proves that the bicomponent Co3S4/Co@C is a promising and low-cost cocatalyst.
Introduction
Hydrogen, which is a highly calorific, clean fuel, can be used to solve the energy crisis and environmental pollution which are caused by the use of traditional nonrenewable fossil fuels.1–3 Photocatalytic hydrogen production is regarded as a feasible and ideal approach to produce highly purified hydrogen.4 One-component photocatalyst exhibits low-efficiency H2 evolution, because of the high activation potentials required for H2 production and the rapid recombination of the photogenerated electron–hole pairs. Hence, the multi-component photocatalysts built of semiconductors and cocatalysts are usually designed to achieve efficient photocatalytic hydrogen production.5 Cocatalysts can provide the highly active sites for H2 and O2 evolution and promote the separation of electron–hole pairs, improving the photocatalytic performance.6The cocatalyst for H2- and O2-evolution can enhance both the reliability and activity of the photocatalysts in photocatalytic water splitting based on the following three main factors: (i) compared with a semiconductor, the cocatalyst has a lower overpotential for hydrogen evolution reaction (HER)7 and oxygen evolution reaction (OER),8,9 (ii) the cocatalyst can promote the separation and migration of photogenerated charge carriers at the semiconductor-cocatalyst interface,10,11 and (iii) the cocatalyst can suppress the photocorrosion and enhance the stability of photocatalyst.12 The HER system using sacrificial agents has been widely developed for efficient hydrogen production. However, the reported cocatalysts for HER are mostly noble metals, such as Pt, Au and Pd.13–15 Hence, exploring inexpensive cocatalysts is highly desirable. To date, cost-effective and earth-abundant transitional metals or transition metal sulfides, such as Co, Cu, CoxSy and MoS2,16–21 have been reported as excellent cocatalysts for catalyzing proton reduction in a photocatalytic reaction. Furthermore, the coupling of metal and a semiconductor results in a Schottky junction at the interface of metal and semiconductor, facilitating the carrier separation and migration, hence dramatically boosting the efficiency of photocatalytic reactions.22,23 For instance, Kuang et al. reported that CsPbBr3 coupled with Pd could significantly enhance the photoelectron consumption rate due to the Schottky junction.24 The Schottky barrier will prohibit photogenerated electrons from returning to the semiconductor from the metal, thus, accelerating the electron kinetics of the photocatalytic reaction.25 Compared with the semiconductor photocatalyst, the cocatalyst usually has better electrical conductivity, which is important for the capture and transfer of photogenerated electrons, thus accelerating the carrier separation and migration.26 In conclusion, the cocatalyst not only serves as an electron sink, but also provides earth-abundant and effective proton reduction sites.
The research on cocatalysts is mostly confined to one component, and the exploration of multicomponent cocatalysts is still at a preliminary stage. The synergistic effects induced by multicomponent cocatalysts, such as the efficient charge migration and enhanced photostability, have been reported over the years.27,28 The synergistic effect at the interface of multicomponent cocatalysts leads to the modification of the electronic structure of different components, which contributes to the electron transfer reaction.29 The large contact interface is critical to obtain the strong coupling effect of the different components.30 Xiong et al. reported that the Pd@Pt cocatalysts significantly enhanced the activity of TiO2 for photocatalytic water splitting, compared with Pt or Pd, because the multicomponent Pd@Pt cocatalysts prolonged the lifetime of photogenerated electrons.31 At present, the research on multicomponent cocatalysts focusses on precious metals. Hence, exploring low-cost and earth-abundant materials as photocatalytic multicomponent cocatalysts is still challenging research.
With these motivations, the bicomponent Co3S4/Co@C cocatalyst coupled with a CdS semiconductor was prepared successfully through a one-step hydrothermal method using Co@C as a precursor. The Co3S4/Co-CdS heterostructure is well suited for the efficient photocatalytic H2 evolution due to two primary factors. Firstly, the Co metal can easily accept the photogenerated electrons from the CdS semiconductor, and this one-way transfer prevents the electrons from recombining with holes in the CdS. Secondly, the stepwise down Fermi level structure of Co and Co3S4 stimulates the transfer of electrons from Co to Co3S4, further promoting the charge separation and migration. Hence, the maximal photocatalytic H2 evolution rate of Co3S4/Co-CdS (15.17 mmol h−1 g−1) is 2.8 times higher than that of Pt-CdS. As the electron sink, the bicomponent Co3S4/Co@C cocatalyst not only displays a much better catalytic performance than Pt, but also provides a strategy for the substitution of noble metal cocatalysts.
Experimental section
Preparation of samples
A portion of Co(NO3)2·6H2O (2.99 g), 1.92 g of p-phthalic acid and 2.72 g of triethylene diamine were added to 150 mL of N,N-dimethylformamide under strong agitation. The mixed solution was evaporated at 80 °C until the solvent was completely removed. Then the powders were heated at 80 °C in the oven for 2 h. The as-prepared dry powders were calcined at 900 °C for 1 h under a N2 atmosphere, with a heating rate of 1.5 °C min−1. The Co@C cocatalyst was finally obtained after cooling down to room temperature.32A portion of Cd(CH3COO)2·2H2O (CdAc2, 0.2665 g) and 0.15 g of thioacetamide (TAA) were put into a 100 ml Teflon-lined stainless-steel autoclave.33 Next, 1.44–11.52 mg of the as-obtained Co@C samples, corresponding to 1.0%–8.0% mass ratios of Co@C to CdS, were mixed with 50 ml of deionized water with high intensity ultrasonic mixing for 20 min. Then the mixtures were transferred into the Teflon-lined stainless-steel autoclave and heated at 180 °C for 24 h. The as-prepared samples were washed several times with deionized water and ethanol. After drying for 8 h, the Co3S4/Co-CdS was obtained. The prepared Co3S4/Co-CdS with different mass ratios of Co@C to CdS (1.0, 2.0, 4.0, 5.0, 6.0, and 8.0 wt%) were labelled as SCS1, SCS2, SCS4, SCS5, SCS6 and SCS8, respectively. Under the same experimental procedure, the CdS was prepared without adding Co@C cocatalysts, and the Co3S4/Co@C cocatalyst was synthesized using Co@C as a precursor without adding Cd(CH3COO)2. Carbon (C) was prepared by a method similar to that of Co@C without adding Co(NO3)2·6H2O. The Co3S4@C was also prepared by a method similar to that of Co@C, but the hydrothermal time was extended to 48 h when the metal Co was almost vulcanized into Co3S4.
For comparison, the mechanically mixed samples of CdS with Co@C, Co3S4/Co@C, Co3S4@C or C were prepared by a self-assembly method.34 Typically, 7.20 mg of Co@C, Co3S4/Co@C, Co3S4@C or C was mixed with 144 mg of CdS, and ground for 40 min. The prepared samples were denoted as Co-CdS, (Co3S4/Co)-CdS, Co3S4-CdS and C-CdS, respectively. The Pt-CdS was prepared by an in situ photo-deposition method using H2PtCl6 aqueous solution (0.01 M).
Material characterizations
The morphologies of the samples were acquired using field-emission scanning electron microscopy (FESEM) on a Philips FEI Quanta 200 FEG instrument. Transmission electron microscopy (TEM) images were obtained on Philips-FEI Talos F200S microscope. X-ray powder diffraction (XRD) patterns were acquired using a Rigaku D/Max 2550 X-ray diffractometer with a Cu Kα radiation source (λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS) was used to investigate the surface chemical states of the photocatalysts, which were obtained using a Thermo ESCALAB 250 with an Al Kα X-ray source. Ultraviolet photoelectron spectrometry (UPS) was performed using a VG Scienta R4000 analyzer. The UV-vis diffuse reflectance spectra were recorded on a Hitachi U-3010 spectrophotometer. Photoluminescence spectra (PL) were obtained using a Hitachi F-7000 fluorescence spectrophotometer with an excitation wavelength of 360 nm. The SPV spectra were obtained using an Au-Light CEL-SPS 1000 (China). The time-resolved photoluminescence (TRPL) spectra were recorded with an Edinburgh Instruments FLS920 with an excitation wavelength of 380 nm, and the decay curves were fitted by the decay curves of the ExpDec2 function software.Electrochemical measurements
Photoelectrochemical measurements were carried out on a Gamry, Reference 600+ electrochemical workstation with a three-electrode system, using platinum foil, Ag/AgCl and 0.35 M Na2S–0.25 M Na2SO3 aqueous solution as the counter electrode, reference electrode and electrolyte, respectively. The working electrodes were prepared as follows: 2 mg of catalysts and 30 μL of naphthol were dispersed in 200 μL of ethanol and 770 μL of deionized water, followed by ultrasonic treatment for 20 min. Then 10 μL of the catalyst ink was dropped on the glassy carbon electrode (2.5 mm in diameter). The electrocatalytic activities (HER and OER) were evaluated by linear sweep voltammetry (LSV) at a scan rate of 10 mV s−1, and the NaOH electrolyte was firstly purged with pure N2 for 1 h. Electrochemical impedance spectroscopy (EIS) were acquired under 10 mV amplitude in the frequency range of 0.1 Hz to 100 kHz in the dark. All of the potentials were calibrated to the RHE.Photoelectrochemical property measurements
Photoelectrochemical tests were also carried out on a Gamry Reference 600+ electrochemical workstation with the same three-electrode system, using 1 M NaOH aqueous solution as the electrolyte. Typically, the working electrodes were prepared as follows: 1 mg of the as-prepared samples and 20 μL of naphthol were dispersed in 180 μL of ethanol, followed by ultrasonic processing for 20 min. Then 15 μL of the previous solution was uniformly dispersed onto FTO glass with an effective area of 1 cm2. Finally, the FTO glass was dried at 60 °C for 1 h. The Mott–Schottky plots were obtained at 1000 Hz. The transient photocurrent responses were measured under the illumination of a CEL-HXF 300, 300 W Xe lamp with a 420 nm cut off filter and bias potential of 0 V vs. Ag/AgCl. Under visible-light illumination, EIS was also tested in the frequency range of 0.01 Hz to 10 kHz.Photocatalytic activity evaluations
The photocatalytic H2 evolution tests were performed in a gas-circulating CEL-SPH2N-D9 (Au-Light, China) photocatalytic H2 evolution system under vacuum with magnetic stirring. A portion (5 mg) of the as-prepared sample was added to 80 ml of 0.35 M Na2S–0.25 M Na2SO3 aqueous solution, followed by ultrasonic treatment for 10 min. The light source was CEL-HXF300, 300 W Xe lamp equipped with 420 nm cut off filter. Photocatalytic H2 production was determined by a GC7920-TFA (Au-Light, China) on-line gas chromatograph equipped with a thermal conductivity detector (TCD). The apparent quantum efficiency (AQE) under the same conditions of photocatalytic H2 evolution was carried out at various monochromatic lights, which were obtained using bandpass filters (420, 450, 475, 500, 520 and 550 nm). The AQE at different wavelengths was calculated according to the following equation: AQE (%) = (2 × number of evolved H2 molecules/number of incident photons) × 100.Computational methods
The work functions of the cocatalysts were evaluated using the density functional theory (DFT) calculations of the DMol3 software package. The correction, based on Grimme's method, was used for the dispersion interaction.35 The atomic cutoff setting was 4.8 Å for all the elements. The k-point was set to 4 × 4 × 1. Furthermore, the water solvent effect was regarded as a COSMO solvation model with a dielectric constant of 78.54.36 The electrons were spin-polarized in all calculations. Both optimization and band structure calculations were tightly converged.Results and discussion
The preparation process of the samples is shown in Fig. 1a. The Co3S4/Co-CdS photocatalyst was synthesized by a facile one-step hydrothermal method using Co@C as the precursor. The TAA not only reacted with CDAc2 to form CdS, but also vulcanized Co into Co3S4. The TEM and SEM images of Co@C in Fig. 1b show that the Co nanoparticles with sizes of 10–20 nm are coated by the graphitic carbon. The lattice spacing values of 0.20 and 0.33 nm were attributed to the (111) facet of Co and the (002) facet of graphitic carbon, respectively, as shown in Fig. 1c. The EDX element mappings (Fig. S1, ESI†) confirmed that the Co nanoparticles were dispersed homogeneously in the graphitic carbon. The CdS alone existed in the form of nanospheres (Fig. S2a–2c, ESI†), and the lattice spacing values of 0.34 and 0.32 nm corresponded to the (002) facet and the (101) facet of hexagonal phase CdS (Fig. 1d), respectively. When the Co@C cocatalysts were added into the synthetic process, the CdS still kept its the nanosphere structure and hexagonal phase, as shown in Fig. S2d (ESI)† and Fig. 1e. Further observation showed that partial Co nanoparticles were vulcanized into Co3S4 (Fig. 1e), therefore, the Co3S4/Co@C cocatalysts were successfully obtained during the hydrothermal process. The HR-TEM image shows that the lattice spacing values of SCS5 were 0.20, 0.24, 0.34, 0.32 and 0.25 nm, corresponding to the (111) facet of Co, (400) facet of Co3S4,16 (002) facet, (101) facet and (102) facet of CdS, respectively, further confirming that the Co3S4/Co-CdS photocatalyst had been successfully synthesized. In addition, the white frame shown in Fig. 1e indicated the compact interaction between Co and Co3S4, that was beneficial for the efficient interfacial charge transfer.34 The STEM image and EDX element mappings of SCS5 in Fig. 1f showed that the Co and Cd elements were distributed in agreement with the S elements, and CdS was combined with the Co3S4/Co@C cocatalyst.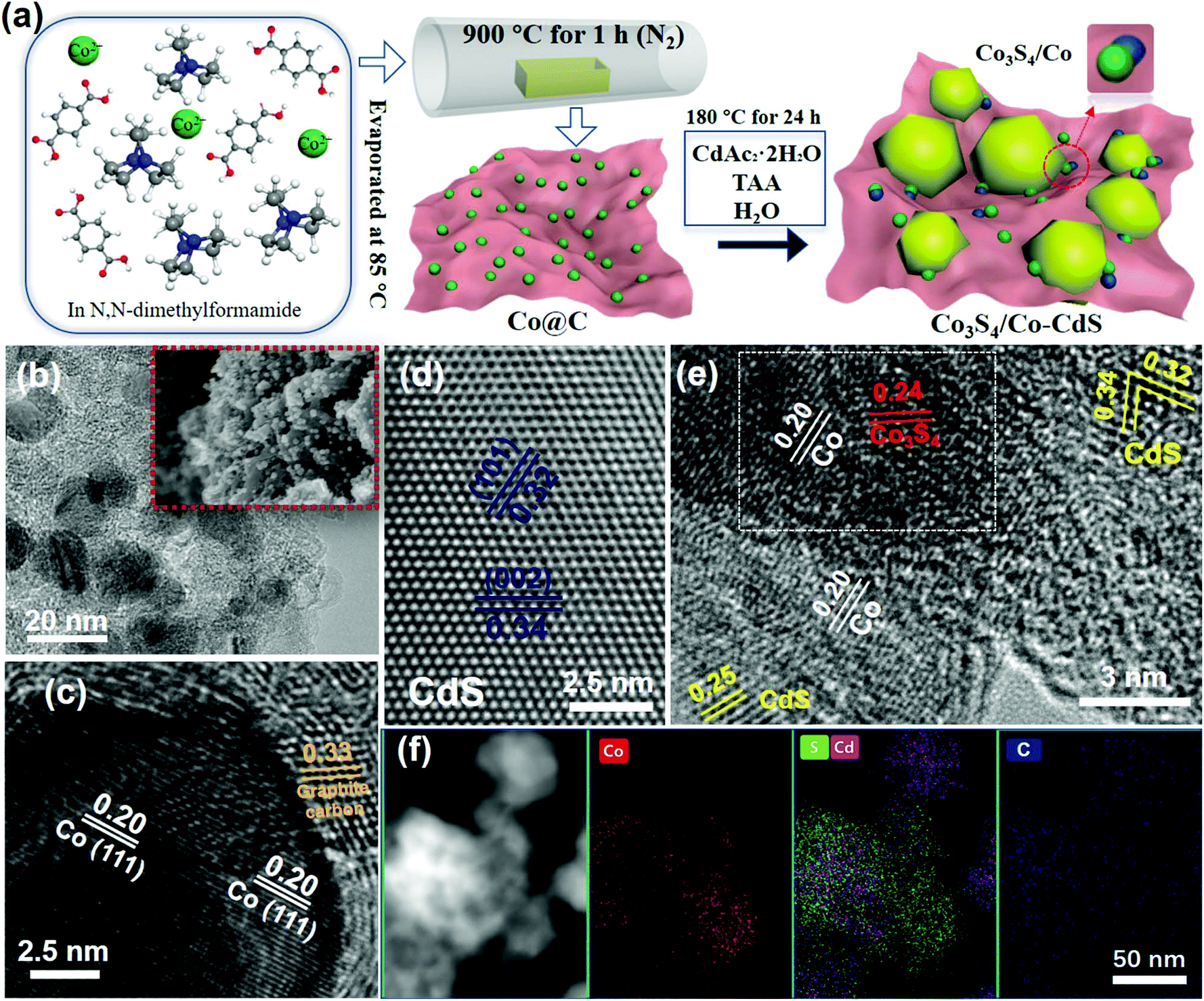 | ||
| Fig. 1 (a) Illustration of the synthesis of Co3S4/Co-CdS. (b) TEM and SEM (inset) and (c) HR-TEM images of Co@C. HR-TEM images of (d) CdS and (e) Co3S4/Co-CdS. (f) STEM image and the EDX element mapping of SCS5. | ||
The XRD was applied to investigate the crystal structure and phase, and the results are shown in Fig. 2a. The XRD patterns of all the as-prepared samples indicated the existence of a hexagonal wurtzite structure of CdS (JCPDS 41-1049), which was in agreement with the previous HR-TEM results. Furthermore, the unchanging XRD patterns indicated that the formation of the Co3S4/Co-CdS heterostructure did not affect the hexagonal wurtzite structure of the CdS. However, no intrinsic XRD peaks of Co, Co3S4 or graphitic carbon were observed even though the loading of the cocatalysts was up to 8%, which was probably caused by the high-dispersion of the Co3S4/Co@C cocatalyst. The XPS was used further to reveal the chemical valence status and composition of SCS5. The high-resolution XPS spectrum of Cd 3d (Fig. 2b) in the CdS shows two peaks at 405.4 eV and 412.1 eV, which were attributed to Cd 3d5/2 and Cd 3d3/2,37 respectively. The S 2p with double peaks (Fig. 2c) was ascribed to the typical metal–sulfur bonds in CdS.38,39 The XRD and XPS results confirmed that the CdS kept its original structure after coupling with Co3S4/Co@C. In the Co 2p high-resolution spectrum (Fig. 2d), a pair of peaks attributed to Co2+ ions at 781.89 eV for Co 2p3/2 and 796.70 eV for Co 2p1/2 were observed, and another doublet at 784.91 eV and 798.98 eV were ascribed to the Co 2p3/2 and Co 2p1/2 of the Co3+ ions,40,41 which indicated the presence of Co3S4. The peaks at 787.22 eV and 803.45 eV were satellite signals. Except for the peaks of Co2+ and Co3+ ions, the spin–orbit doublets belonging to Co0 at 787.22 eV for Co 2p3/2 and 803.45 eV for Co 2p1/2 were clearly observed,42 suggesting the presence of cobalt metal. These results combined with the results of the TEM analyses confirmed that the Co3S4/Co-CdS photocatalyst has been successfully synthesized.
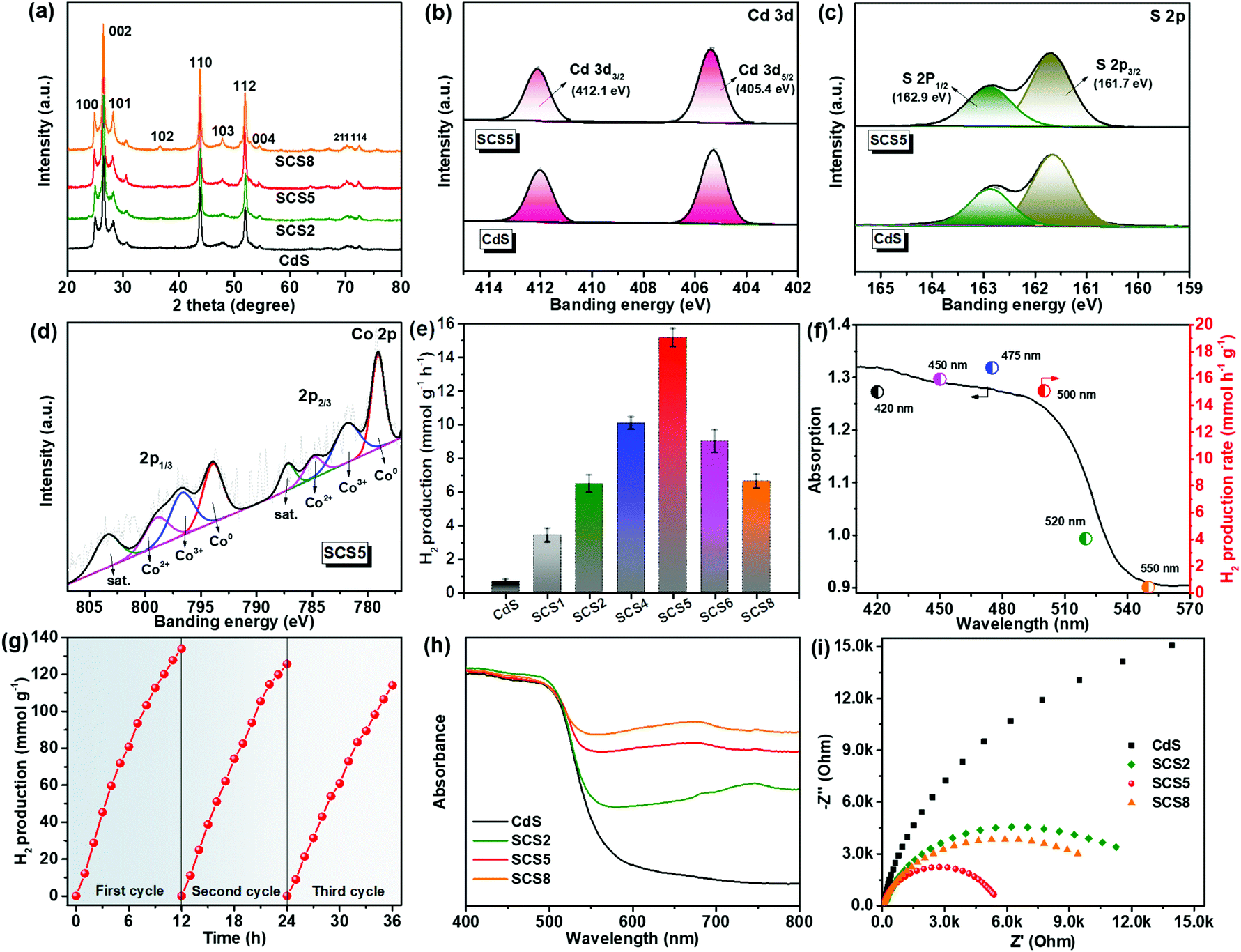 | ||
| Fig. 2 (a) XRD patterns of different samples. The XPS spectra of various elements: (b) Cd 3d, (c) S 2p, (d) Co 2p. (e) Photocatalytic H2 production rates, error bars represent standard deviation of three tests. (f) Apparent quantum efficiencies of SCS-5 based on different wavelengths and the corresponding UV-visible absorption spectrum. (g) Recyclability of hydrogen evolution tests of SCS-5. (h) UV-vis diffuse reflectance spectra. (i) Electrochemical impedance spectra in the frequency range of 0.01 to 10 kHz under visible light illumination. | ||
The photocatalytic H2 evolution of the samples were carried out under visible light irradiation (λ > 420 nm), as shown in Fig. 2e. The Co@C cocatalyst had no photocatalytic activity, and the pure CdS displays the low photocatalytic H2 evolution (0.65 mmol h−1 g−1). The H2 evolution was substantially enhanced, when the CdS was coupled with the Co3S4/Co@C cocatalyst. It is obvious that there was a volcano-type trend between the loading of the Co3S4/Co@C cocatalyst and the rate of H2 evolution. At the optimal loading of 5%, the SCS5 sample had the highest photocatalytic H2 evolution rate of 15.17 mmol h−1 g−1, which was 23.3 times higher than that of the CdS without the cocatalyst. Furthermore, the SCS5 exhibited a very efficient photocatalytic H2 evolution rate when compared with other similar composites (Table S1, ESI†). However, by further increasing the loading of the cocatalyst, a deterioration in the photocatalytic performance was observed, because of the covering of the surface-active sites on the CdS by excess cocatalyst. In addition, the determination of the AQE was carried out under different wavelengths (420, 450, 475, 500, 520 and 550 nm).43,44 The AQE of SCS5 reached a maximum value of 16.80% at 475 nm, achieving the high efficiency with noble metal-free photocatalysts (Fig. 2f). Furthermore, the recycling test for SCS5 showed a small loss of photoactivity after continuous illumination lasting for 36 h as shown in Fig. 2g. This stable H2 evolution for such a long illumination time suggests that CdS is stable and resistant to photocorrosion after coupling with the Co3S4/Co@C cocatalyst. The characterization results for SCS5 after the cycling tests are shown in Fig. S3 and (ESI).† No significant structural changes to SCS5 were observed in the SEM and TEM images after a long illumination. All the (101) facets of CdS, (111) facets of Co and (311) facets of Co3S4 were easily found. However, the peak intensities of CdS in the XRD patterns and XPS spectra decreased after the cycling tests. This may be caused by the slight photocorrosion of the CdS, leading to a small loss of photoactivity.
The UV-vis diffuse reflectance spectra (Fig. 2h) verified that the CdS exhibited a light absorption edge at about 550 nm. The Co3S4/Co-CdS photocatalysts showed the obvious improvement of visible light absorption, because of the black cocatalysts. Tauc plots (Fig. S5, ESI†) indicated that the Co3S4/Co@C cocatalysts had almost no influence on the band gap of the CdS. The positive tangential slopes in the Mott–Schottky plots confirmed the n-type semiconductor characteristics, as shown in Fig. S6 (ESI).† In addition, the smaller slopes in the Mott–Schottky plots of SCSx compared with those of CdS suggested a higher carrier density of SCSx due to the presence of the Co3S4/Co@C cocatalyst.45 The smallest slopes of SCS5 mean the highest carrier density, which favors the photoelectrochemical and photocatalytic performance.46 The EIS was performed under the visible-light illumination (>420 nm), as shown in Fig. 2i. Among all the samples, the SCS5 had the smallest hemicycle in the EIS Nyquist plots indicating the highest charge mobility. The EIS results confirmed that the Co3S4/Co-CdS photocatalysts were more favorable for separation and migration of the photogenerated charge carriers due to the excellent electrical conductivity of the Co3S4/Co@C cocatalyst, which was responsible for the superior photocatalytic performance of SCS5.
To investigate the origin of high photocatalytic activity on SCS5, the different Co@C and Co3S4/Co@C samples were inspected comprehensively. The XRD pattern of Co@C shows the typical diffraction peaks of metallic Co phases (Fig. 3a). After Co@C was vulcanized by TAA, the Co3S4 was successfully obtained with characteristic peaks at 2θ = 31.5° (311), 38.9° (400) and 55.1° (440). The TEM and HR-TEM analyses revealed that the metallic Co nanoparticles of Co3S4/Co@C retained their original structure when compared with that of Co@C (Fig. 3b), as shown in Fig. 3c. Furthermore, Co3S4 with the lattice spacings of 0.28 nm and 0.27 nm ascribed to (311) and (222) planes was adjacent to the Co nanoparticle, which indicated the intimate interface between Co and Co3S4 (Fig. 3d–e). These results demonstrated the coexistence of metallic Co and Co3S4 in Co3S4/Co@C, which was in agreement with the results shown in Fig. 1. The S 2p XPS peaks of the metal sulfide further confirmed the existence of Co3S4 in the Co3S4/Co@C cocatalyst (Fig. S7, ESI†), and the peaks at 168.5 eV and 169.6 eV were caused by the SO42− species from the excess TAA.47
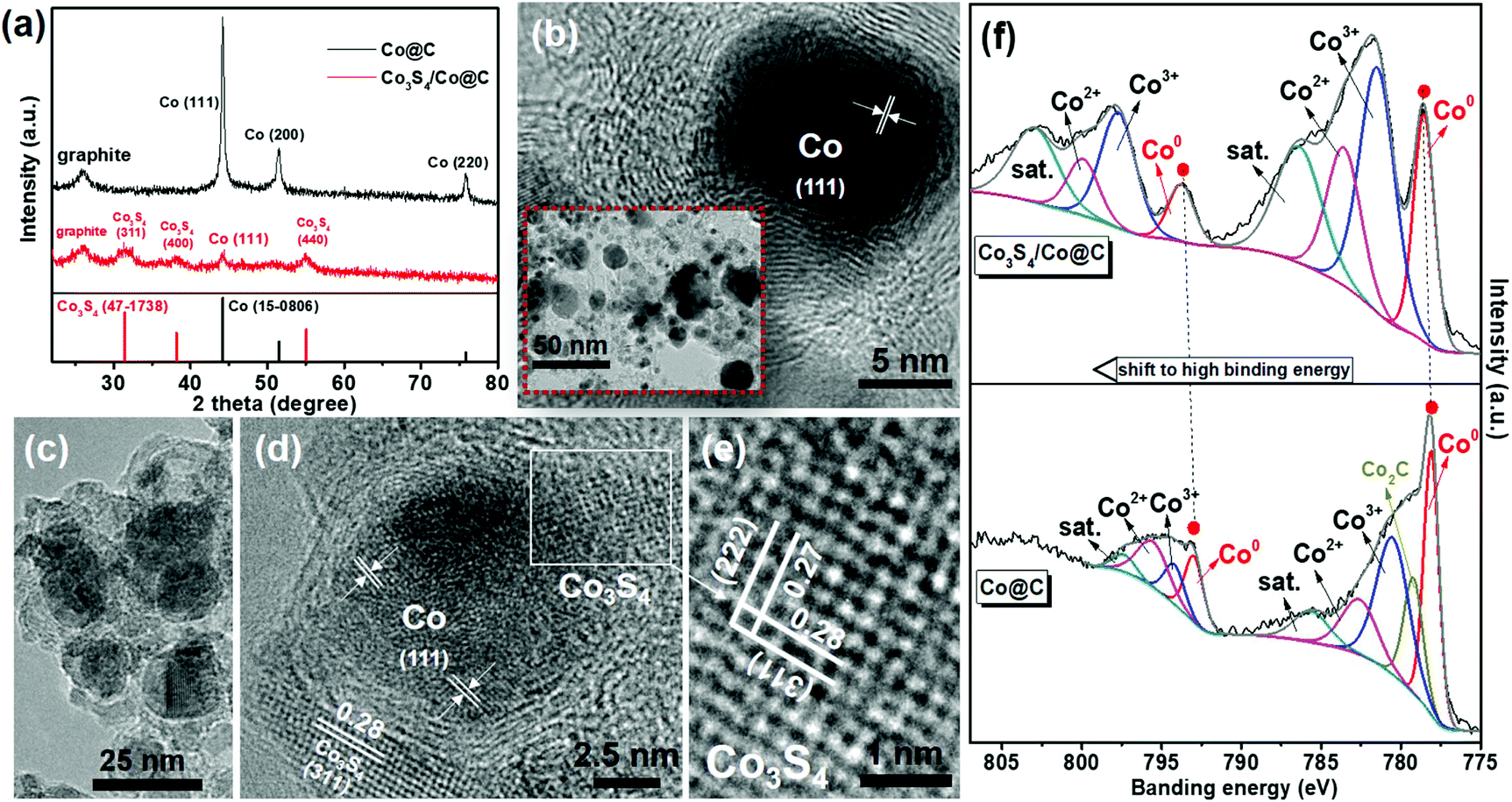 | ||
| Fig. 3 (a) XRD patterns. (b) TEM (inset) and HR-TEM images of Co@C. (c) TEM and (d and e) HR-TEM images of Co3S4/Co@C. (f) Co 2p XPS spectra of Co@C and Co3S4/Co@C. | ||
Most importantly, the XPS spectra shown in Fig. 3f directly demonstrate the electron redistribution at the Co3S4/Co interface. The formation of Co–S covalent bonds will decrease the electron density of Co species in the Co3S4/Co@C sample, accompanying the shift to higher binding energies for the Co 2p XPS peaks.32,48 This result proved the strong contact at the Co3S4/Co interface, which was beneficial for charge migration. In addition, compared to the C 1s XPS spectra of the Co@C and Co3S4/Co@C samples, there were no apparent changes for the graphitic-carbon structure,49 as shown in Fig. S8 (ESI).†
As shown in Fig. 4a, the EIS strongly correlate with the charge transfer resistance on the Co@C and Co3S4/Co@C cocatalysts. The semicircular diameter in the Nyquist plot of Co3S4/Co@C was much smaller than that of Co@C, and the HER activity of the Co3S4/Co@C was much higher than that of Co@C (inset in Fig. 4a).50 These results reveal that the Co3S4/Co@C cocatalyst has the faster charge transfer and kinetics for HER,51 which will boost the charge separation and migration in the photocatalyst. In general, the noble metal Pt is considered as a high efficiency cocatalyst for photocatalytic HER. However, the optimal sample of Pt-CdS (Fig. S9, ESI†) exhibited a lower photocatalytic H2 evolution rate (5.40 mmol h−1 g−1) than that of SCS5 under the same conditions. The designed Co3S4/Co-CdS photocatalyst exhibited 2.8 times higher activity than Pt-CdS. Thus, it is believed that the Co3S4/Co@C cocatalyst is an ideal substitute for Pt.
 | ||
| Fig. 4 (a) Polarization curves for the HER (inset) and electrochemical impedance spectra of Co@C and Co3S4/Co@C in the dark. (b) Photocatalytic H2 evolution, error bars represent the standard deviation of three tests. (c) Transient photocurrent responses. (d) PL spectra and electrochemical impedance spectra (inset) of CdS, C-CdS, Co-CdS, Co3S4-CdS and SCS5. (e) Surface photovoltage spectra, and (f) time-resolved PL spectra of CdS, Co-CdS and SCS5. | ||
Next, different control samples were synthesized to prove the excellent photocatalytic H2 evolution performance of Co3S4/Co-CdS, including the C-CdS (Fig. S10, ESI†), Co-CdS, and Co3S4-CdS (Fig. S11, ESI†) photocatalysts. Similarly, no intrinsic XRD peaks of Co and graphitic carbon were found in C-CdS, Co-CdS, and Co3S4-CdS (Fig. S12, ESI†), which were caused by the high-dispersion of Co3S4 and Co. The SCS5 sample also exhibited a stronger visible light absorption than that of C-CdS, Co-CdS, and Co3S4-CdS, as shown in Fig. S13 (ESI).† The photocatalytic H2 evolution rate of SCS5 was much higher than those of C-CdS, Co-CdS and Co3S4-CdS (Fig. 4b), which demonstrated that the high performance of Co3S4/Co@C cocatalyst was due to the synergistic effect of Co and Co3S4. Furthermore, the (Co3S4/Co)-CdS sample was also synthesized by mechanically mixing the Co3S4/Co@C cocatalyst and CdS. The TEM analyses and EDX element mapping shown in Fig. S14 (ESI)† proved the coexistence of Co3S4/Co and CdS. The mechanically mixed (Co3S4/Co)-CdS also showed a much higher photocatalytic activity than that of Co-CdS and Co3S4-CdS, because of the effect of Co3S4. However, SCS5 showed a higher photocatalytic activity (Fig. S15, ESI†) than that of (Co3S4/Co)-CdS, which was probably caused by the larger contact interface of the cocatalyst and the semiconductor.
The efficient interfacial charge separation and migration between Co3S4/Co and CdS were confirmed by the transient photocurrent responses, PL spectra, EIS, SPV and time-resolved PL spectra. The SCS5 exhibited a much higher photocurrent density than those of Co3S4-CdS, Co-CdS, C-CdS and CdS (Fig. 4c), indicating the effective separation and migration of photogenerated electron–hole pairs in the Co3S4/Co-CdS structure. The lowest emission intensity in the PL spectra (Fig. 4d) further confirmed the highest electron–hole separation efficiency on CdS was obtained using Co3S4/Co@C as cocatalyst. The EIS in the inset of Fig. 4d shows that SCS5, with the smallest hemicycle in the Nyquist plot, was more resistant to charge recombination, which indicated a fast charge transfer between the Co3S4/Co@C cocatalysts and CdS under illumination. All these characterizations indicated that SCS5 has the fastest rate of interfacial charge separation and migration, resulting in the highest photocatalytic H2 production rate. Furthermore, SPV was applied to investigate the photogenerated charge separation mechanism of the photocatalyst. The signal of the SPV spectrum depended on the type of semiconductor and direction of the migration of the photogenerated charge carriers.52 The positive SPV signal suggested a typical n-type semiconductor of CdS (Fig. 4e). The negative signal implied that photogenerated electrons were separated towards the cocatalysts in Co3S4-CdS, Co-CdS and SCS5.53 The weakest SPV signal on SCS5 revealed that the Co3S4/Co@C cocatalyst served as a most efficient electron acceptor, which would offset part of the positive photoelectric SPV signal.54 Meanwhile, the time-resolved PL spectra were used to obtain the charge carrier lifetimes of CdS coupling with different cocatalysts. As shown in Fig. 4f, the lifetime of the charge carrier on SCS5 (τav = 0.88 ns) was much shorter than those of Co-CdS (τav = 1.02 ns) and CdS (τav = 1.55 ns), signifying the effective charge migration from CdS to the Co3S4/Co@C cocatalyst.55,56 Photocatalyst coupling with a cocatalyst was supposed to have a shorter lifetime owing to the fast charge transfer from photocatalyst to cocatalyst and suppression of the electron–hole recombination. The decrease of a lifetime for the SCS5 sample revealed that the bicomponent Co3S4/Co was a more effective cocatalyst for charge transfer compared with the one-component Co cocatalyst. The previous results confirmed the efficient interfacial charge separation and transfer in the Co3S4/Co-CdS photocatalyst.
The work functions of the cocatalyst were confirmed by the DFT calculations, as shown in Fig. 5a and b. The work functions of Co and Co3S4 were 4.93 eV and 5.87 eV, respectively. The larger work function signifies the more positive Fermi level (vs. vacuum level).57 Hence, Co3S4 has a more positive Fermi level than that of Co, indicating that the photogenerated electrons tended to transfer to the Co3S4 from Co due to the different Fermi energy levels. The flat band energies from the Mott–Schottky plots vs. Ag/AgCl for CdS, Co-CdS and SCS5 were −1.04, −0.99 and −0.94 eV, respectively. Therefore, the Fermi levels vs. NHE (EAg/AgCl was about 0.197 eV vs. NHE, and the compensation potential was about 25 meV at room temperature) of CdS, Co-CdS and SCS5 were calculated to be −0.82, −0.77 and −0.72 eV, respectively. The more positive Fermi level of SCS5 was caused by the high working function of Co3S4. The conduction band (CB) position of CdS was generally more negative than the Fermi level by about −0.2 V,37,58 therefore, the CB position of CdS was calculated to be −1.02 eV. Furthermore, these results were also consistent with the UPS spectra. The UPS spectra in Fig. S16a (ESI)† indicated that the work functions of Co@C and Co3S4@C were 3.72 and 4.31 eV (vs. vacuum level), respectively. Therefore, the Fermi levels vs. NHE (NHE was −4.44 eV vs. vacuum level)59 of Co@C and Co3S4@C were calculated to be −0.72 and −0.13 eV, respectively, which further confirmed that the Co3S4 has a more positive Fermi level. Band gap structures of Co3S4/Co-CdS can be deduced from the previous analyses, as shown in Fig. S16b (ESI).† All the DFT calculations, Mott–Schottky plots and UPS proved that the most negative Fermi level of Co3S4 was in the Co3S4/Co-CdS photocatalyst, hence, the electrons were much easier to transfer to Co3S4, which had the active sites for photocatalytic hydrogen evolution.
 | ||
| Fig. 5 Fermi level calculated by DFT of (a) Co and (b) Co3S4. (c) Mott–Schottky plots of CdS, Co-CdS and SCS5. (d) Illustration of photogenerated carrier transfer between CdS and the Co3S4/Co@C cocatalyst. | ||
Based on the previous analyses, the carriers transfer mechanism on Co3S4/Co-CdS for photocatalytic H2 evolution is illustrated in Fig. 5d. Under the visible light irradiation, the photogenerated electrons were excited to the CB of CdS from the valence band (VB). Meanwhile, the holes in the VB were consumed by the sacrificial reagents on the surface of the CdS. As the conductive metal Co has a more negative Fermi level compared with the CB of CdS, the photogenerated electrons on CdS firstly transferred to the Co. In the Co3S4/Co@C cocatalyst, the Co3S4 has a more negative Fermi level than that of Co, leading to the transfer of electrons from Co to Co3S4. The electrons were captured by Co3S4 for the H2 evolution. This stepwise energy band structure notably stimulates the charge separation and efficient electron transfer between the CdS and Co3S4/Co@C cocatalyst, due to the strong confinement effect of the electrons in Co3S4/Co. The Co3S4/Co@C cocatalyst acts as the electron sink, which was beneficial for the rapid migration of the photogenerated electrons and the high efficiency photocatalytic H2 evolution reaction.
Conclusions
In summary, the bicomponent Co3S4/Co@C cocatalyst and CdS semiconductor were synthesized by a simple hydrothermal reaction using Co@C as a precursor. The novel Co3S4/Co-CdS heterostructure photocatalyst exhibited a high visible-light photocatalytic H2 evolution activity of 15.17 mmol h−1 g−1, with a high quantum efficiency of 16.80% at 475 nm. The Co3S4/Co@C cocatalyst served as a high efficiency electron sink, accelerating the charge separation and migration between the interface of the Co3S4/Co@C cocatalyst and CdS. Furthermore, owing to the stepwise down energy band structure caused by the different Fermi levels of Co and Co3S4, the photogenerated electrons on CdS rapidly transfer to the Co3S4via Co metal, which can efficiently inhibit the recombination of electrons and holes in CdS. This work not only presents an inexpensive earth-abundant Co3S4/Co@C cocatalyst to replace noble metal cocatalysts, but also uncovers the huge potential of multicomponent cocatalysts in renewable solar energy production.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (No. 21673080, 21802046, 51706231).Notes and references
- Y. Liu, Y. Li, S. Yang, Y. Lin, J. Zuo, H. Liang and F. Peng, ChemSusChem, 2018, 11, 2766–2775 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- Q. Zhao, J. Sun, S. Li, C. Huang, W. Yao, W. Chen, T. Zeng, Q. Wu and Q. Xu, ACS Catal., 2018, 8, 11863–11874 CrossRef CAS.
- Y. Miseki and K. Sayama, Adv. Energy Mater., 2018, 9, 1801294 CrossRef.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- G. Zhang, Z. A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- Y. Zhang, B. Tang, Z. Wu, H. Shi, Y. Zhang and G. Zhao, Green Chem., 2016, 18, 2424–2434 RSC.
- A. T. Garcia-Esparza, D. Cha, Y. Ou, J. Kubota, K. Domen and K. Takanabe, ChemSusChem, 2013, 6, 168–181 CrossRef CAS PubMed.
- C. Guo, P. He, R. Cui, Q. Shen, N. Yang and G. Zhao, Adv. Energy Mater., 2019, 9, 1900364 CrossRef.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- M. Tabata, K. Maeda, T. Ishihara, T. Minegishi, T. Takata and K. Domen, J. Phys. Chem. C, 2010, 114, 11215–11220 CrossRef CAS.
- R. Su, R. Tiruvalam, A. J. Logsdail, Q. He, C. A. Downing, M. T. Jensen, N. Dimitratos, L. Kesavan, P. P. Wells, R. Bechstein, H. H. Jensen, S. Wendt, C. R. Catlow, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2014, 8, 3490–3497 CrossRef CAS.
- M. Hojamberdiev, Y. Cai, J. J. M. Vequizo, M. M. Khan, R. Vargas, K. Yubuta, A. Yamakata, K. Teshima and M. Hasegawa, Green Chem., 2018, 20, 3845–3856 RSC.
- J. Yao, Y. Zheng, X. Jia, L. Duan, Q. Wu, C. Huang, W. An, Q. Xu and W. Yao, ACS Appl. Mater. Interfaces, 2019, 11, 25844–25853 CrossRef CAS.
- F. Zhang, H.-Q. Zhuang, J. Song, Y.-L. Men, Y.-X. Pan and S.-H. Yu, Appl. Catal., B, 2018, 226, 103–110 CrossRef CAS.
- G. Zhang, C. Huang and X. Wang, Small, 2015, 11, 1215–1221 CrossRef CAS.
- Z. Liu, Y. Huang, Q. Xiao and H. Zhu, Green Chem., 2016, 18, 817–825 RSC.
- X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS.
- Y. Zheng, J. Dong, C. Huang, L. Xia, Q. Wu, Q. Xu and W. Yao, Appl. Catal., B, 2020, 260, 118220 CrossRef CAS.
- J. Dong, Y. Shi, C. Huang, Q. Wu, T. Zeng and W. Yao, Appl. Catal., B, 2019, 243, 27–35 CrossRef CAS.
- J. D. Xiao, L. Han, J. Luo, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 1103–1107 CrossRef CAS.
- X. She, H. Xu, L. Li, Z. Mo, X. Zhu, Y. Yu, Y. Song, J. Wu, J. Qian, S. Yuan and H. Li, Appl. Catal., B, 2019, 245, 477–485 CrossRef CAS.
- Y.-F. Xu, M.-Z. Yang, H.-Y. Chen, J.-F. Liao, X.-D. Wang and D.-B. Kuang, ACS Appl. Energy Mater., 2018, 1, 5083–5089 CrossRef CAS.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- H. Husin, W.-N. Su, H.-M. Chen, C.-J. Pan, S.-H. Chang, J. Rick, W.-T. Chuang, H.-S. Sheu and B.-J. Hwang, Green Chem., 2011, 13, 1745–1754 RSC.
- Y. Ma, R. Chong, F. Zhang, Q. Xu, S. Shen, H. Han and C. Li, Phys. Chem. Chem. Phys., 2014, 16, 17734–17742 RSC.
- Q. Fu and X. Bao, Chem. Soc. Rev., 2017, 46, 1842–1874 RSC.
- A. Meng, L. Zhang, B. Cheng and J. Yu, Adv. Mater., 2019, 31, 1807660 CrossRef.
- M. Bowker, D. James, P. Stone, R. Bennett, N. Perkins, L. Millard, J. Greaves and A. Dickinson, J. Catal., 2003, 217, 427–433 CrossRef CAS.
- S. Bai, L. Yang, C. Wang, Y. Lin, J. Lu, J. Jiang and Y. Xiong, Angew Chem., Int. Ed., 2015, 54, 14810–14814 CrossRef CAS.
- Z.-H. Xue, H. Su, Q.-Y. Yu, B. Zhang, H.-H. Wang, X.-H. Li and J.-S. Chen, Adv. Energy Mater., 2017, 7, 1602355 CrossRef.
- X. Dai, M. Xie, S. Meng, X. Fu and S. Chen, Appl. Catal., B, 2014, 158–159, 382–390 CrossRef CAS.
- K. Zhang, J. Ran, B. Zhu, H. Ju, J. Yu, L. Song and S. Z. Qiao, Small, 2018, 14, e1801705 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- H. Guan, S. Zhang, X. Cai, Q. Gao, X. Yu, X. Zhou, F. Peng, Y. Fang and S. Yang, J. Mater. Chem. A, 2019, 7, 2560–2574 RSC.
- B. Chen, R. Li, G. Ma, X. Gou, Y. Zhu and Y. Xia, Nanoscale, 2015, 7, 20674–20684 RSC.
- G. Zhao, W. Zhou, Y. Sun, X. Wang, H. Liu, X. Meng, K. Chang and J. Ye, Appl. Catal., B, 2018, 226, 252–257 CrossRef CAS.
- W. Gu, L. Hu, W. Hong, X. Jia, J. Li and E. Wang, Chem. Sci., 2016, 7, 4167–4173 RSC.
- J. Du, T. Zhang, J. Xing and C. Xu, J. Mater. Chem. A, 2017, 5, 9210–9216 RSC.
- L. Liu, A. V. Puga, J. Cored, P. Concepción, V. Pérez-Dieste, H. García and A. Corma, Appl. Catal., B, 2018, 235, 186–196 CrossRef CAS.
- S. Chen, Y. Qi, T. Hisatomi, Q. Ding, T. Asai, Z. Li, S. S. Ma, F. Zhang, K. Domen and C. Li, Angew. Chem., Int. Ed., 2015, 54, 8498–8501 CrossRef CAS.
- G. Zhao, Y. Sun, W. Zhou, X. Wang, K. Chang, G. Liu, H. Liu, T. Kako and J. Ye, Adv. Mater., 2017, 29, 1703258 CrossRef.
- Y. Liu, Y. Li, F. Peng, Y. Lin, S. Yang, S. Zhang, H. Wang, Y. Cao and H. Yu, Appl. Catal., B, 2019, 241, 236–245 CrossRef CAS.
- K. Zhang, W. Zhou, L. Chi, X. Zhang, W. Hu, B. Jiang, K. Pan, G. Tian and Z. Jiang, ChemSusChem, 2016, 9, 2841–2848 CrossRef CAS.
- H. Wang and Z. Jin, Sustainable Energy Fuels, 2019, 3, 173–183 RSC.
- S. Liu, X. Zhang, G. Wang, Y. Zhang and H. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 34269–34278 CrossRef CAS PubMed.
- P. Chen, T.-Y. Xiao, Y.-H. Qian, S.-S. Li and S.-H. Yu, Adv. Mater., 2013, 25, 3192–3196 CrossRef CAS PubMed.
- X. Huang, Q. Shen, J. Liu, N. Yang and G. Zhao, Energy Environ. Sci., 2016, 9, 3161–3171 RSC.
- X. Xu, F. Nosheen and X. Wang, Chem. Mater., 2016, 28, 6313–6320 CrossRef CAS.
- R. Chen, F. Fan, T. Dittrich and C. Li, Chem. Soc. Rev., 2018, 47, 8238–8262 RSC.
- T. K. Townsend, N. D. Browning and F. E. Osterloh, Energy Environ. Sci., 2012, 5, 9543–9550 RSC.
- X. Yue, S. Yi, R. Wang, Z. Zhang and S. Qiu, Small, 2017, 13, 1603301 CrossRef.
- W. Tu, Y. Zhou, Q. Liu, S. Yan, S. Bao, X. Wang, M. Xiao and Z. Zou, Adv. Funct. Mater., 2013, 23, 1743–1749 CrossRef CAS.
- Z. Sun, H. Zheng, J. Li and P. Du, Energy Environ. Sci., 2015, 8, 2668–2676 RSC.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC.
- S. Trasatti, Pure Appl. Chem., 1986, 58, 955–966 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03323h |
| This journal is © The Royal Society of Chemistry 2020 |