
High fat diet incorporated with meat proteins changes biomarkers of lipid metabolism, antioxidant activities, and the serum metabolomic profile in Glrx1−/− mice†
Muhammad Ijaz
Ahmad
,
Muhammad
Umair Ijaz
,
Muzahir
Hussain
,
Iftikhar
Ali Khan
,
Noreen
Mehmood
,
Sultan Mehmood
Siddiqi
,
Congcong
Liu
,
Di
Zhao
,
Xinglian
Xu
,
Guanghong
Zhou
and
Chunbao
Li
 *
*
Key Laboratory of Meat Processing and Quality Control, MOE, Key Laboratory of Meat Processing, MARA, Jiangsu Collaborative Innovation Center of Meat Production and Processing, Quality and Safety Control, International Collaborative Laboratory of Animal Health and Food Safety, College of Food Science and Technology, Nanjing Agricultural University, 210095, Nanjing, China. E-mail: chunbao.li@njau.edu.cn; Fax: +86 25 84395679; Tel: +86 25 84395679
First published on 27th December 2019
Abstract
Red and processed meat consumption has been associated with oxidative stress, diabetes and non-alcoholic fatty liver disease (NAFLD). This study was aimed at exploring the effects of high-fat meat protein diets on potential metabolite biomarkers in Glrx1−/− mice, a well-documented mouse model to study NAFLD. Male Glrx1−/− mice were fed a control diet with 12% energy (kcal) from fat, a high-fat diet supplemented with casein (HFC) with 60% energy (kcal) from fat, and a high-fat diet supplemented with fish (HFF) or mutton proteins (HFM) for 12 weeks. The results of biochemical and histological analyses indicated that the intake of HFM increased hepatic total cholesterol, triglycerides, serum alanine transaminase and aspartate transaminase, and macro- and micro-vesicular lipid droplet accumulation, which were accompanied by altered gene expression associated with the lipid and cholesterol metabolism. HFF diet fed Glrx1−/− mice significantly ameliorated diet-induced NAFLD biomarkers compared to HFC and HFM diets. In addition, serum metabolome profiling identified metabolites specifically associated with lipid metabolism bile acid metabolism, sphingolipid and amino acid metabolism pathways. A HFM diet increased the abundance of LysoPC(15:0), LysoPC(16:0), LysoPC(20:1), LysoPE(18:2), LysoPE(22:0), LysoPE(20:6), O-arachidonoylglycidol, 12-ketodeoxycholic acid and sphinganine that are associated with NAFLD. The KEGG metabolic pathway of identified metabolites of high fat diets showed that the differential metabolites were associated with lipid metabolism, linoleic acid metabolism, amino acid metabolism, bile acid metabolism, sphingolipid metabolism, and glutathione metabolism pathways whereas HFF diet ameliorated NAFLD by modifying these pathways. These results provide potential metabolite biomarkers for NAFLD induced by HFM diet.
Introduction
Meat and their products do not only contain valuable nutrients for human health,1 but also contain saturated fatty acids, cholesterol, heme iron, sodium, and advanced glycation end products that may be harmful for patients with NAFLD.2 Indeed, the consumption of diets rich in red and processed meat has been associated with insulin resistance (IR), type 2 diabetes (T2D), oxidative stress and NAFLD.3,4 Several studies have reported that excessive consumption of refined carbohydrates, fats, in particular saturated fats, and protein from meat can cause NAFLD.5,6 Owing to the complex aetiology and pathogenesis of diabetes and obesity in relation to studying genetics in controls and patients, rodent models are powerful tools to investigate the molecular mechanisms of NAFLD pathogenesis.7 The intake of high fat diet has been associated with the progression and development of obesity, insulin resistance, oxidative stress and NAFLD,8,9 even after short periods of fat feeding.10 In a recent study, purified meat proteins showed different impacts from casein and soy proteins on gut microbiota, metabolites, and liver metabolism in young rats.11 Still, the pathophysiology of NAFLD and its progression induced by high fat meat protein diets remain unclear. In recent years, metabolomic profiling has provided new insights into the molecular signature of diseases associated with NAFLD. Multiple studies have identified key metabolites associated with NAFLD.12–18 Significant changes in key pathways involving the metabolism of bile acids,19 amino acids,14–18 steroids, hormones20,21 and fatty acids12,15,22 have been reported in subjects with NAFLD, making this an appealing target for future diagnostics.13 Indeed, the changes in these metabolites are likely to reflect specific pathways of liver injury related to NAFLD or advanced steatosis making them compelling biomarkers.Glrx1 is an abundant liver protein responsible for regulating oxidative stress and hepatic metabolism.23,24 Quantitative proteomic and metabolomic analyses showed that Glrx1 knockdown (KO) decreased the cellular level of glutathione (GSH) but increased reactive oxygen species (ROS) production. In contrast, the overexpression of Glrx1 has been shown to decrease cellular ROS levels.25 Our previous study showed that Glrx1−/− KO promoted inflammation and hepatic lipogenesis when fed processed meat proteins at a dose of 20% of protein.26 In a transgenic animal study, Glrx1 was observed to play an important role in regulating lipid and fatty acid metabolism, and Glrx1−/− mice that were fed a high-fat diet rich in cholesterol developed obesity, hyperlipidemia and NAFLD.24 Furthermore, Glrx1 is considered as a potential biomarker and key factor in the pathogenesis of chronic kidney disease and diabetes.27,28 In physiology, Glrx1 regulates nuclear factor-E2-related factor 2/Kelch-like ECH-associated protein1 (Nrf2/Keap1), nuclear factor κB (NF-kB), interleukin 1 beta (IL-1β) glutathionylation and sirtuin 1 activity.29–32 However, to the best of our knowledge, this study is the first on the key metabolomic signatures involved in NAFLD development caused by long-term high fat meat proteins in Glrx1−/− mice.
To assess the importance of Glrx1 in NAFLD mice, we generated a Glrx1 KO model by the deletion of Glrx1 gene using CRISPR cas9 technology, and studied the serum metabolomic profile to identify key metabolites and metabolic pathways in response to high-fat diets incorporated with casein (HFC), fish protein (HFF) or mutton protein (HFM). We also studied dyslipidemia and hepatic oxidative stress biomarkers in response to high-fat supplemented with meat protein diets. The LC-MS (G2-XS QTOF, Waters) platform was applied to analyze the serum metabolomic profiles.
Materials and methods
Diet preparation
Briefly, meat samples from longissimus dorsi muscles of domestic sheep, and dorsal white muscles of fish from bighead carp (Hypophthalmichthys nobilis) were obtained from a local meat company (Sushi, Jiangsu, China). Adipose and connective tissues were removed from meat samples, while bones, scales and fat were discarded from fish samples. All meat and fish samples were finely minced prior to protein extractions. Methylene chloride and methanol (V/V = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were used to remove fat. After de-fatting, the filtrates were collected, dried, ground, sieved and packed for further experiments. Casein was obtained from a commercial company (Shansong Biological Products Inc., Linyi, China). The diets were prepared according to a low fat diet (LFD) (D12450J, 10% kcal from lard New Brunswick, United States), or a high-fat diet (HFD) (D12492, 60% kcal from lard New Brunswick, United States) composition. The LFD and HFD diet formula compositions are shown in ESI S2 (Table 1†). The LFD contained protein (20%), lard (4.44%), soybean oil (5.55%), corn starch (31.05%), sucrose (34.51%), and maltodextrin10 (3.45%) while the HFD was comprised of protein (20%), lard (54.35%), soybean oil (5.55%), sucrose (6.70%), and maltodextrin10 (12.32%).
:1) were used to remove fat. After de-fatting, the filtrates were collected, dried, ground, sieved and packed for further experiments. Casein was obtained from a commercial company (Shansong Biological Products Inc., Linyi, China). The diets were prepared according to a low fat diet (LFD) (D12450J, 10% kcal from lard New Brunswick, United States), or a high-fat diet (HFD) (D12492, 60% kcal from lard New Brunswick, United States) composition. The LFD and HFD diet formula compositions are shown in ESI S2 (Table 1†). The LFD contained protein (20%), lard (4.44%), soybean oil (5.55%), corn starch (31.05%), sucrose (34.51%), and maltodextrin10 (3.45%) while the HFD was comprised of protein (20%), lard (54.35%), soybean oil (5.55%), sucrose (6.70%), and maltodextrin10 (12.32%).
Animals and experimental design
Transgenic Glrx1−/− male mice were generated by using CRISPR cas9 technology as previously described26,33 (also see ESI S1†).Acclimatization and feeding
Forty male C57BL/6J (6-weeks old) mice were obtained from Nanjing Biomedical Research Institute (NBRI) and housed in pairs in a specific pathogen free environment animal center (SYXK〈Jiangsu〉2011-0037). Following 1-week acclimation, mice were randomly assigned to the following dietary groups for 12-weeks: a low fat (LF) diet, a high fat (HF) diet containing casein protein, a high fat (HF) diet containing fish protein, or a HF diet containing mutton protein. After one week of adaption, mice were randomly assigned to four groups (n = 10 per group), and fed either a LFD, or HFD see ESI S2, Table 1.† For fish and mutton protein diets, casein was replaced with protein powders extracted from fish or mutton. Body weight and food intake were recorded weekly. All animal procedures were carried out in compliance with the guidelines for care and regulations of the Ethical Committee of Experimental Animal Centre of Nanjing Agricultural University and approved by the Experimental Animal Centre of Nanjing Agricultural University.Metabolic assays
The glucose tolerance test (GTT) was performed by intraperitoneal injection of glucose (2.0 g dextrose per kg) following overnight fasting prior to mice killing. Tail blood glucose levels were determined before and 15, 30, 45, 60, 90 and 120 min after glucose injection. Blood glucose was determined with a glucose meter (HemoCue, Angelholm, Sweden). The insulin tolerance test (ITT) was performed by intraperitoneal injection of insulin (0.75 U per kg body weight). After 4 h fasting, tail blood samples were collected before and 15, 30, 45 and 60 min after injection. Concentrations of insulin were measured by using the Hemo Cue glucose 201+ analyzer (HemoCue, Angelholm, Sweden). For glucose and insulin, the incremental area under the curve and decremental area under the curve were calculated by the trapezoidal method, respectively.Mice killing and tissue collection
The mice were sacrificed by cervical dislocation. Mice were deprived of food for 12 h before killing, and blood was collected and the samples were centrifuged at 12000g for 10 min. Serum was prepared and stored at −80 °C. Liver, colon, epididymal adipose tissues (EAT), perirenal fat and subcutaneous fat tissues were harvested, weighed and snap frozen in liquid nitrogen. All samples were stored at −80 °C until analysis.
Biochemical measurements
Serum total cholesterol (T-CHO), triglycerides (TG), high density lipoprotein (HDL), and low density lipoprotein (LDL) were determined with an automatic chemistry analyzer (Spotchem EZ, Nakagyoku, Kyoto, Japan) according to the manufacturer's instructions. Liver TG and T-CHO were determined using an automatic chemistry analyzer (Spotchem EZ, Nakagyoku, Kyoto, Japan). Serum alanine transaminase (ALT) and aspartate transaminase (AST) were determined using commercial kits (Abcam, Cambridge, MA) at 525 nm for ALT and 542 nm for AST using an automatic chemistry analyzer (Spotchem EZ, Nakagyoku, Kyoto, Japan).Hepatic antioxidant enzymes and inflammatory cytokines
Liver samples (200 mg) were homogenized in 2 mL of ice-cold physiological saline for 1 min. Following centrifugation at 5000g at 4 °C for 10 min, the protein content in the liver homogenate was assayed with a Bio-Rad protein assay (Hercules, CA). The antioxidant activities and oxidative status of glutathione peroxidase (GPx), catalase, superoxide dismutase (SOD), glutathione (GSH) and malondialdehyde (MDA) were measured using commercial kits (Cayman Chemical Co., Ann Arbor, MI) according to the manufacturer's instructions. Tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β) and interleukin 6 (IL-6) were determined with a multiplex detection kit (Bio-Rad, Hercules, CA, USA).Histological observations
For H&E staining, liver tissues were fixed in 10% formalin, paraffin embedded, and cut into 6 μm sections. For Oil Red O staining, liver tissues were cut into 8 μm cryosections, and stained with Oil red O for lipid analysis. Quantification of the droplet area, steatosis, denaturation, inflammation, and necrosis scores was performed by using Image Lab software (Version 7.01, Bio-Rad Laboratories, Hercules, CA) as previously described.34RT-qPCR
Liver samples (200 mg) were homogenized in 2 mL of ice-cold physiological saline for 1 min. Following centrifugation at 5000g at 4 °C for 10 min, the supernatants were aliquoted and RNA isolation was performed using the RNeasy mini kit (Qiagen, Hilden, Germany). The RNA concentration was determined by using a Nanodrop UV Visible spectrophotometer (ThermoFisher Scientific, Waltham, MA). RNA integrity and purification were determined by Gel Red (Biotium, Fremont, CA) stained agarose gel electrophoresis. Reverse transcription of RNA to cDNA was carried out with a Reverse Transcription kit (ThermoFisher Scientific, Waltham, MA). Real-time PCR was performed using gene-specific TaqMan primers (Invitrogen by ThermoFisher Scientific, Waltham, MA) listed in Table 1. Gene expression levels were measured by taking the comparative (2-ΔΔCT) method and followed by normalization to the reference gene β-actin.35| Gene | F | R |
|---|---|---|
| Srebf1 | CTCCAAGGTTTCGTCTGACG | TCCAGTGGCAAAGAAACACC |
| Scd1 | TGTCCTAAGGCCACCGGGTC | CGCATGCCTGTGATGCTCTGC |
| FASn | TGGGCCTGCTGTTCACA | TCCGATCCAGGTTTTTAAGTA |
| Cd36 | CTCCTGTGAACTCCTGTCCTT | AGCTGTCTGGCCAGTCAAC |
| ACC1 | ACATGGTCTGGGACTTCTGG | CAAGTTTTTGATGCCCTGGT |
| Srebf2 | GAGACCTGGGCAATGTGACT | GTTTACTGCGCAATCCCAAT |
| HMGCR | TCAAGCGTGACTTTGGGTCT | AGCGGAATAAGGCCTGTTGT |
| ACoX1 | GTCCACCGTGTATGCCTTCT | TCTGCAGATCGTTCATCTCG |
| Cyp7a1 | ACCGCTCCACCTGCCGTCAC | ACGGGCGACTCTAAGTGCTGC |
| Cyp27a1 | AAGAAGCAGACTGGAGGAGAAG | CAGGTTGTCACTCTCAGAACAGA |
| Cpt1a | GTGGCAAAGTTCAGTCACAA | TCCTCTTCTGGGCAACTGTC |
| Cyp7b1 | CCTCCGCTGCCATCAGTCAGT | TCGGCTGGGACTCGTGTTCA |
| β-Actin | TCCCTGTATGCCTCTGGTCGT | CCAGACGCAGGATGGCGTGA |
Western blotting
For the validation of Glrx1 KO mice, liver, colon and EAT (50 mg) were homogenized in RIPA buffer containing a protease inhibitor and a phosphatase inhibitor (ThermoFisher Scientific, Waltham, MA). The protein concentration was assayed using a Bio-Rad protein assay (Hercules, CA). The prepared samples were loaded onto 7–10% SDS-PAGE gels. Precision Plus Protein Dual Standards (Bio-Rad, Hercules, CA) marker was used on all gels. After separation by electrophoresis, proteins were transferred to an Immobilon-PSQ Membrane (EMD Millipore, Bedford, MA). Blots were blocked in 5% skimmed milk (Fisher Scientific, Waltham, MA) prepared in PBS with 1% Tween 20. For WB analysis the following primary antibodies were used to detect protein expression levels: Glrx (Abcam; Cambridge, UK) and GADPH (Cell Signaling, Beverly, MA). Membranes were incubated overnight at 4 °C with primary antibodies (1:1000) and were then incubated with the secondary antibodies (1:3000) for 4 h. The detection of protein bands and intensities was performed by using Image Quant LAS4000 (GE, Uppsala, Sweden) and Image J software (Version 1.4.3.67, Broken Symmetry Software, Madison, Wisconsin, USA), respectively.
Non-targeted metabolome analysis
000g at 4 °C for 5 min, the pellet was treated twice with the same process. The extracted serum samples were pooled and evaporated using the Thermo Speed VacSPD111 V (Thermo Fisher Scientific, Waltham, MA) for 4 h. The dried samples were subsequently reconstituted in 200 μL of 50% methanol. After centrifugation at 15000g for 5 min, the resulting supernatant was passed through a 0.22 μm Ministart RC 4 filter (Sigma-Aldrich, Shanghai, China). The samples were derivatized by trimethylsilylation by incubation in 10 μL of O methyl hydroxylamine hydrochloride (40 mg mL−1 in pyridine) at 40 °C for 30 min, followed by the addition of 90 μL of N, O-bis(trimethylsilyl)trifluoroacetamide containing 1% trimethylchlorosilane and incubation for 90 min. Finally, the derivatized samples were subjected to LC-MS analysis.
Results
Verification of the Glrx1−/− mice model
To verify the Glrx1−/− mice, we quantified Glrx1 gene expression in colon, liver and EAT of Glrx1−/− mice. In Glrx1−/− mice, the colon, liver and EAT exhibited significantly lower levels of Glrx1mRNA than that of β-actin (P < 0.05, Fig. 1A). Furthermore, the deletion was confirmed by western blotting analysis. Glrx1−/− mice had lower protein levels of Glrx1 than the values of GADPH (P < 0.05, Fig. 1B and C).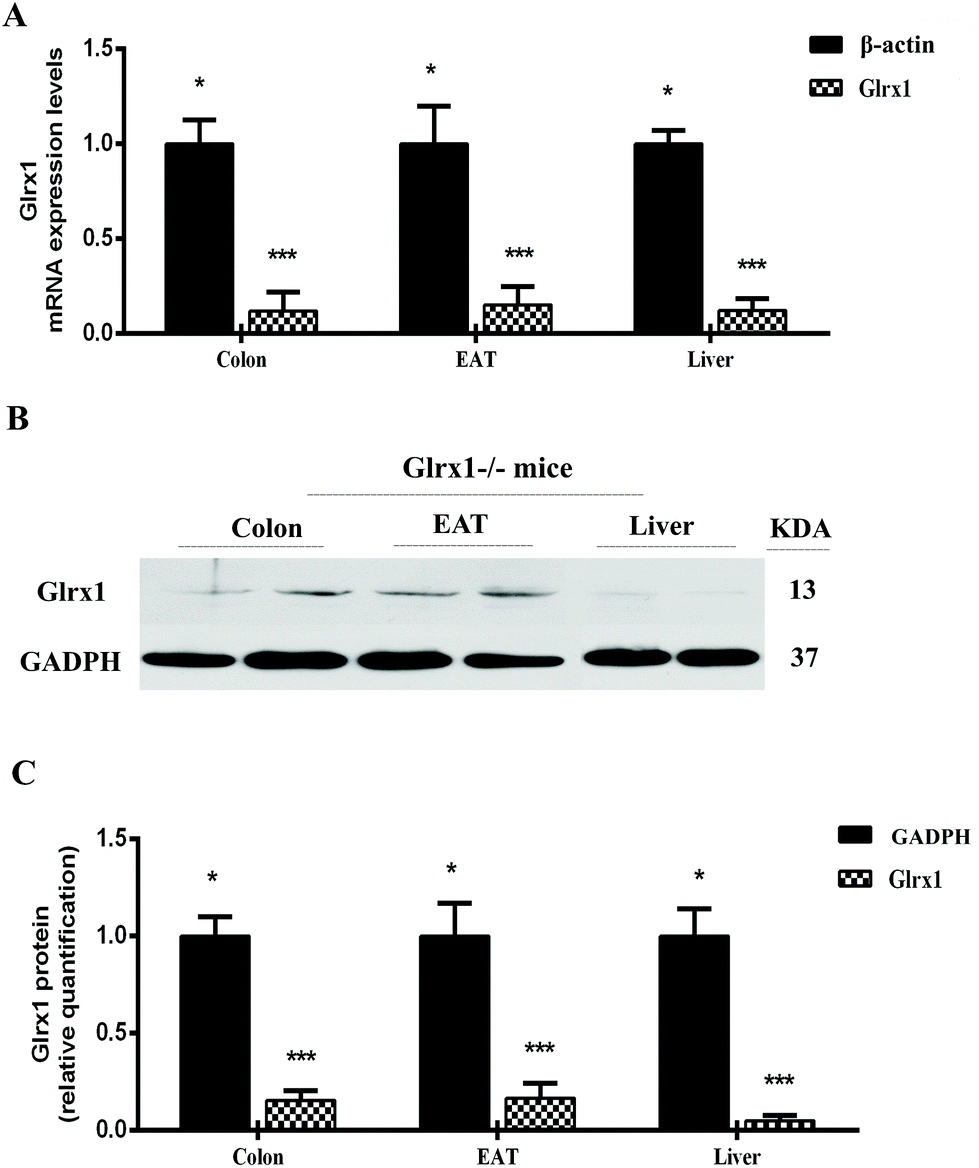 | ||
| Fig. 1 Validation of Glrx1 deletion. (A) Glrx1 mRNA in the colon, liver and epididymal adipose tissue (EAT) compared with β-actin in Glrx1−/− mice; (B) Glrx1 protein levels in the colon, liver and EAT. Representative western-blot of Glrx1 and GADPH. Data are presented as the means ± SD. * and *** indicate a significant difference according to Mann–Whitney test at P < 0.05 and P < 0.001, respectively. | ||
High-fat mutton protein induced obesity, insulin resistance, and hyperglycemia in Glrx1 deficient mice
The physical characteristics of Glrx1−/− mice fed a control or high fat diet are given in Table 2. Although Glrx1−/− mice fed a control diet showed a higher food intake compared with the high fat diet groups, the average daily caloric intake of high fat diet groups was significantly higher than that of the control group (P < 0.05). Also, high-fat diets induced a substantially higher body weight from the 3rd week to the 12th week compared with the control diet, in particular for the high fat mutton diet group (P < 0.05, Fig. 2A). However, the protein source in high-fat diets did not show significant differences in the growth performance of Glrx1−/− mice (P > 0.05). The GTT reveals that high-fat diets fed Glrx1−/− mice exhibited distinct glucose intolerance in that the areas under the curves were greater than that of the control group (P < 0.05, Fig. 2B). Glrx1−/− mice displayed elevated blood glucose levels at 30, 45, 60 and 90 min after the glucose injection in high-fat diet groups compared to that of the control diet group (P < 0.05) but there was no significant difference in the blood glucose levels at 0 and 15 min (P > 0.05). The high-fat fish protein diet group showed a slightly lower level of glucose intolerance than the high-fat mutton diet group (P < 0.05, Fig. 2C). The ITT indicates that HFC and HFM diet fed Glrx1−/− mice also showed significant insulin resistance (P < 0.05, Fig. 2D and E). There was no significant difference in insulin levels among the experimental groups at 0 min (P > 0.05). For the high-fat fish protein diet group, the serum insulin level decreased greatly within 30 min after insulin injection (P < 0.05, Fig. 2D), while it kept constant but was higher than the values of the control group at 45 min and 60 min (P < 0.05, Fig. 2D and E). This shows that fish protein diet may reduce high-fat-induced insulin resistance in Glrx1−/− mice.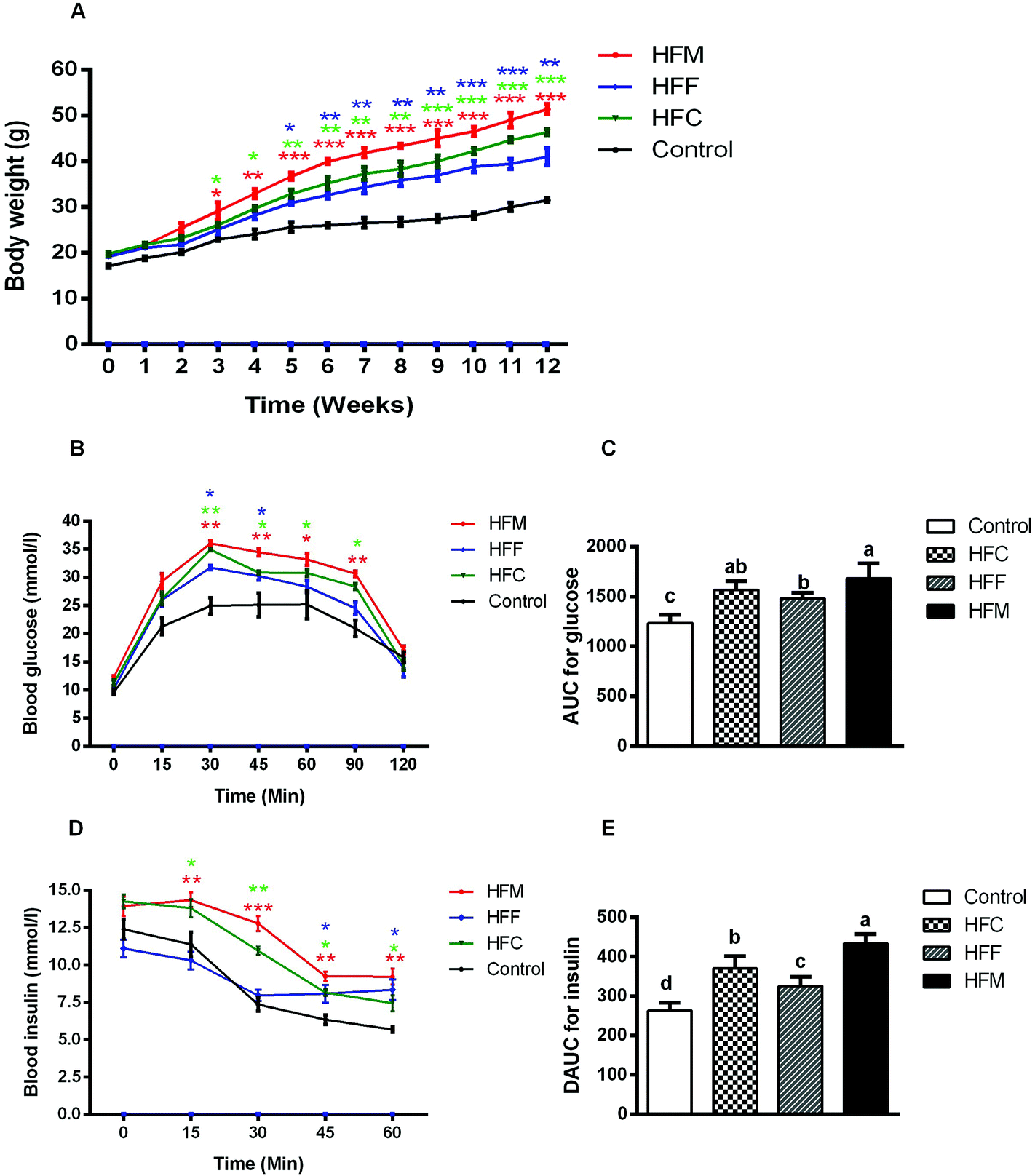 | ||
| Fig. 2 Physiological responses to different diets. (A) Body weight; (B) glucose response curve after administration of 2 mg glucose per g lean mass by gavage (n = 10 per group); (C) incremental area under the curve (IAUC) (n = 10 per group); (D) insulin response curve after administration of 0.75 units insulin per kg lean mass, n = 10 per group; (E) decremental area under the curve (DAUC) (n = 10 per group). The data represent group means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001 versus the control fed group. a,b,c,d Different letters indicate significant differences (P < 0.05). | ||
| Control | HFC | HFF | HFM | |
|---|---|---|---|---|
| Data are presented as means ± SD; a,b,c different subscripts indicate significant differences in measured variables (P < 0.05). | ||||
| Food consumption (g day−1) | 4.5 ± 0.71a | 3.4 ± 0.41a | 3.1 ± 0.52a | 3.9 ± 0.66a |
| Calorie intake (kcal day−1) | 13.8 ± 0.84c | 18.2 ± 0.98ab | 16.8 ± 0.63bc | 20.5 ± 0.77a |
| Body weight (g) | 30.86 ± 3.72c | 45.39 ± 4.22b | 40.85 ± 3.43b | 51.39 ± 5.14a |
| Liver weight (g) | 1.19 ± 0.10c | 2.25 ± 0.12ab | 1.84 ± 0.11bc | 2.90 ± 0.27a |
| Total fat weight (g) | 1.92 ± 0.28c | 5.35 ± 0.14b | 4.23 ± 0.23bc | 6.47 ± 0.39a |
| ALT (U L−1) | 9.92 ± 2.82c | 33.55 ± 2.20ab | 27.80 ± 6.37bc | 40.39 ± 4.82a |
| AST (U L−1) | 11.80 ± 1.41c | 32.37 ± 2.82b | 27.93 ± 3.14b | 42.58 ± 4.24a |
| LDL-C (mmol L−1) | 0.31 ± 0.05b | 0.66 ± 0.06ab | 0.50 ± 0.09ab | 0.81 ± 0.11a |
| HDL-C (mmol L−1) | 1.21 ± 0.11a | 0.85 ± 0.09ab | 1.04 ± 0.10ab | 0.69 ± 0.05b |
| TC (mmol L−1) | 2.39 ± 0.17c | 4.26 ± 0.22b | 3.86 ± 0.29b | 5.30 ± 0.31a |
| TG (mmol L−1) | 1.28 ± 0.14c | 1.91 ± 0.11ab | 1.67 ± 0.21bc | 2.36 ± 0.15a |
In addition, HFM diet fed Glrx1−/− mice had the highest weight gain, liver index and fat index (P < 0.05, Table 2). The intake of HFM induced obesity, insulin resistance, and higher fat deposition in Glrx1−/− mice. The HFM diet also induced the development of hyperlipidemia with higher levels of serum ALT, AST, LDL-C, TG and T-CHO, but a lower level of HDL-C (P < 0.05, Table 2). These results indicate that the dietary supplementation of mutton protein with a high-fat diet did not only induce hepatic lipid abnormalities, but also led to liver injury in Glrx1 deficient mice, which was further exacerbated by mutton protein diet.
HFM diet induced biochemical and histological markers of NAFLD in Glrx1 deficient mice
We evaluated the diet effects on the development of NAFLD by measuring the hepatic TG and TC contents. The intake of high-fat diets substantially increased hepatic TG and TC compared with the control (P < 0.05, Fig. 3A and B) and the HFM group showed higher values than the HFC and HFF groups (P < 0.05). H&E staining revealed higher fat accumulation with the development of more macro- and micro-vesicular lipid droplets in the liver of high-fat diet fed mice compared with the control mice. Lipid droplets in the HFF diet group appeared fewer in number than those in HFM and HFC groups (Fig. 3C). In addition, we performed oil Red O staining to assess the progression of hepatic steatosis. Significantly higher macrovesicular steatosis was seen in the livers of high fat diet fed mice, in particular for HFM-fed mice (Fig. 3D, ESI S2, Table 2†).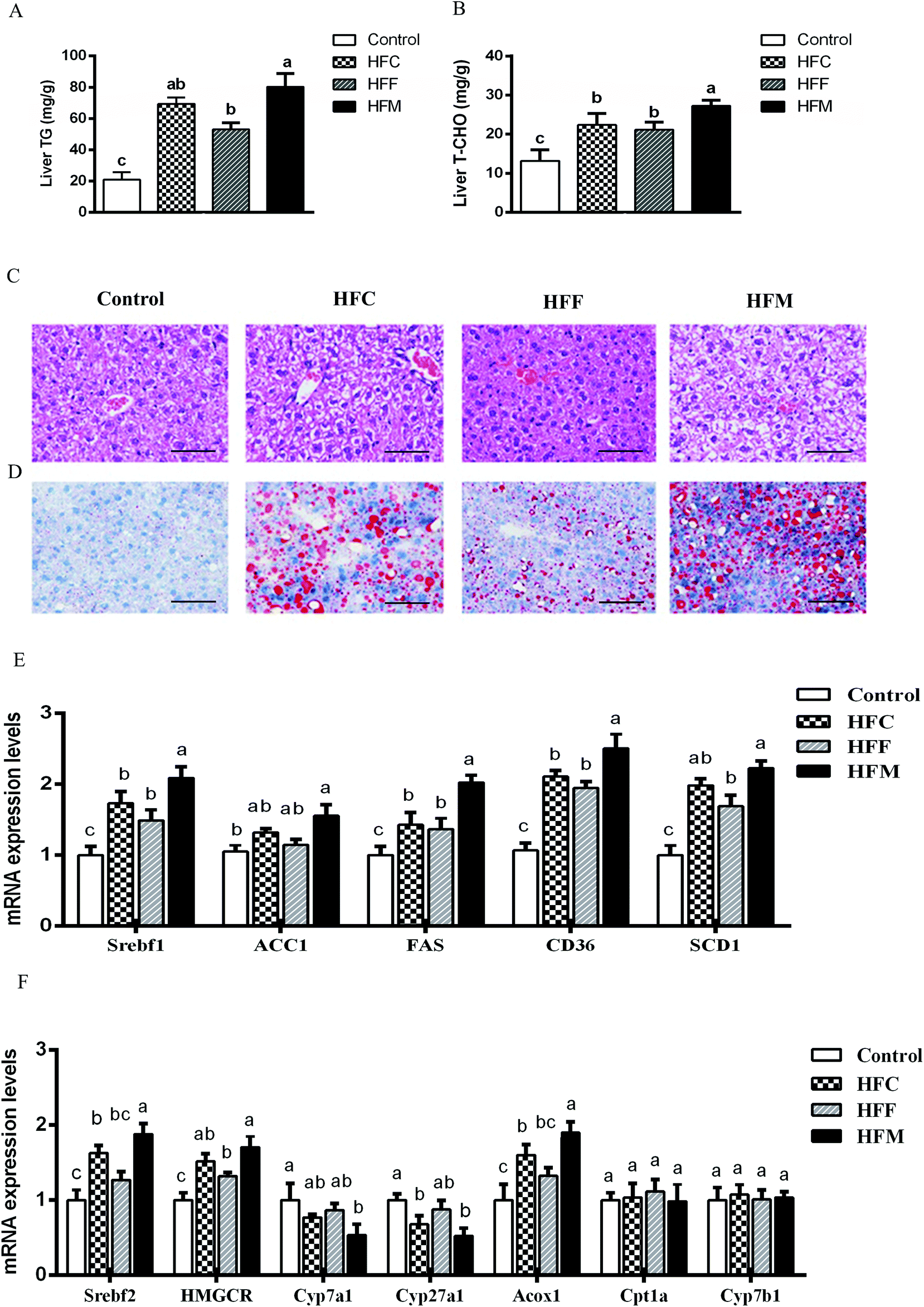 | ||
| Fig. 3 Glrx KO exacerbates diet-induced liver steatosis. (A) Triglyceride concentration in liver; (B) total cholesterol concentration in liver; (C) liver sections stained with H&E (scale bar: 100 μm); (D) liver sections stained with Oil Red O (scale bar: 100 μm) (original magnifications 400× for H&E and Oil red O staining); (E) mRNA levels of Srebfc1, ACC1, FAS, CD36, and SCD1 genes involving hepatic lipid metabolism; (F) mRNA levels of Srebfc2, HOMGCR, Cyp7a1, Cyp27a1, Acox1, Cpt1a, and Cyp7b1 genes involving hepatic cholesterol metabolism. Data are presented as the means ± SD (n = 10 mice per group). a,b,c Different letters indicate significant differences (P < 0.05). | ||
Glrx1 deficiency is associated with the hepatic dysregulation of lipid and cholesterol metabolism, and hepatic lipid metabolism is regulated by a delicate balance of anabolic and catabolic processes.36 In order to explore the mechanism involved in the lipid and cholesterol metabolism pathway, we quantified the mRNA levels of key lipogenesis enzymes and transcription factors being involved in cholesterol/bile acid biosynthesis and lipid metabolism. The intake of HFM diet markedly upregulated acetyl-CoA carboxylase (ACC1), sterol regulatory element-binding protein (Srebf1), fatty acid transporter (CD36), fatty acid synthase (FAS) and stearoyl-CoA desaturase-1 (SCD1) among diet groups (P < 0.05, Fig. 3E). In addition, the HFM diet also upregulated Srebf2, 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR), and peroxisomal acyl-coenzyme (ACoX1), but downregulated cholesterol 7 alpha-hydroxylase (Cyp7a1) and sterol 27-hydroxylase (Cyp27a1) (P < 0.05, Fig. 3F). Diets had no effect on the expression of genes involved in hepatic fatty acid oxidation, including carnitine palmitoyltransferase (Cpt1a) and 25-hydroxycholesterol 7-alpha-hydroxylase (Cyp7b1) in Glrx1−/− mice (P > 0.05). Thus hepatic lipid accumulation is unlikely to increase due to decreased lipid oxidation, but rather by de novo biosynthesis. These results indicate that Glrx1−/− mice are sensitive to HFM diet induced obesity, liver steatosis, and oxidative stress. Deficiency of this gene may lead mice to hyperlipidemia and hypercholesterolemia and impair fatty acid oxidation in a high-fat diet mode. Such an effect was aggravated by the inclusion of mutton protein in the diet. However, the inclusion of fish protein to high-fat diet showed a slightly protective effect against high fat diet induced hepatic dysregulation of lipid, cholesterol metabolism, and fatty acid oxidation.
Hepatic inflammation and antioxidant activities
NAFLD is associated with hepatic inflammation and excessive lipid accumulation.37 To assess the effects of Glrx1 deficiency on levels of inflammatory cytokines induced by high-fat diets, we measured the hepatic levels of TNF-α, IL-6 and IL-1β. Consistent with histological observations, the levels of hepatic TNF-α, IL-1β, and IL-6 were significantly increased by high-fat mutton diets compared with other diet groups (P < 0.05, Fig. 4A–C), and the HFC and HFM diets exhibited a stronger impact on these cytokines than the HFF diet (P < 0.05).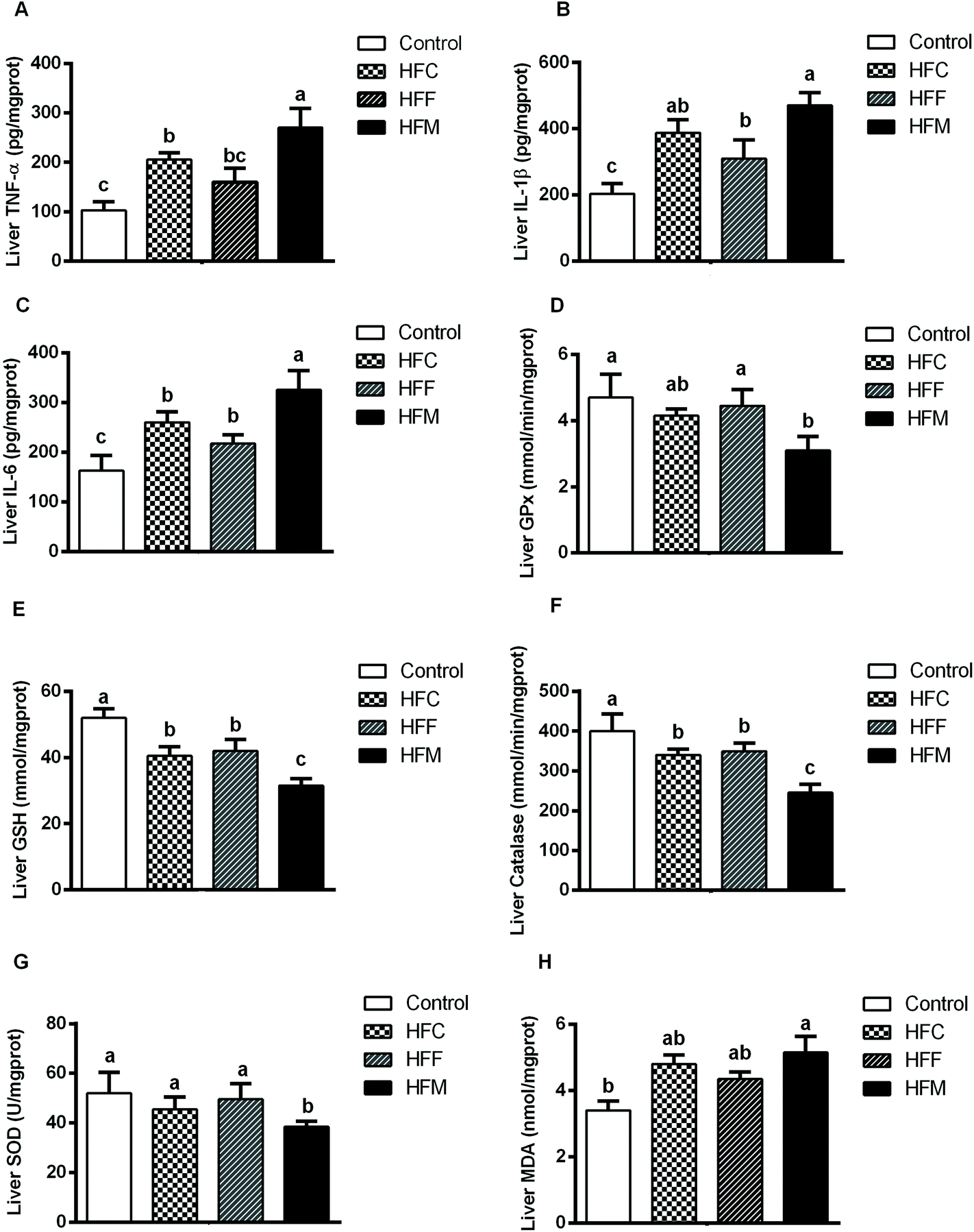 | ||
| Fig. 4 Effects of HFD on hepatic antioxidant capacity and pro-inflammatory cytokines. (A) TNF-α, tumor necrosis factor-α; (B) IL-1β, interleukin-1β; (C) IL-6, interleukin-6; (D) GPx, glutathione peroxidase; (E) GSH, glutathione; (F) CAT, catalase; (G) SOD, superoxide dismutase; (F) and MDA, malondialdehyde. Data are presented as the means ± SD (n = 10 mice per group). a,b,c Different letters indicate significant differences (P < 0.05). | ||
Glrx1 plays a key role in regulating oxidative stress by increasing the GSH level in the process known as protein de-glutathionylation.38,39 Furthermore, Glrx1 deficiency decreases antioxidant defense and increases ROS generation.25 To investigate the effect of dietary protein on the hepatic antioxidant capacity and redox status in Glrx1 deficient mice, we assessed hepatic oxidative stress by measuring the levels of different antioxidants. HFM and HFC diet fed Glrx1−/− mice had lower activities of GPx, GSH, catalase and SOD while the HFM diet group had higher hepatic MDA (P < 0.05, Fig. 4D–H).
Serum metabolomic profile
To gain insight into the metabolic mechanism, we investigated whether the intake of high-fat diets alters the key metabolic pathways involved in the development of NAFLD in Glrx1−/− mice. A total of 69 metabolites were identified, and the abundance of 37 metabolites was significantly different among diet groups. The majority of these metabolites were lysophosphatidylcholines (Lyso PC), lysophosphatidylethanolamines (Lyso PE), phosphatidylcholines (PC), phosphaethanolamines (PE), sphingolipids, eicosanoids, bile acids, amino acids, and their cofactor categories. The PCA plot showed that the first two principle components accounted for 73.1% of total variance and a good separation was observed among the diet groups indicating that these four groups have distinct metabolomic profiles (Fig. 5A). The top 15 metabolites that showed a consistent trend of alteration are listed in Fig. 5B. The HFM group had the highest concentrations of LysoPC(15:0), LysoPE(18:1), 12-ketodeoxycholic acid, sphinganine, O-arachidonoyl glycidol, Lyso PE (20:6), L-phenylalanine, 9-HODE, L-tyrosine and LysoPE(22:0). The HFF group had the highest concentrations of L-phenylalanine, L-tyrosine, creatine, glutathione and LysoPC(20:0) while the HFC group had the highest levels of Lyso PE (18:1), 12-ketodeoxycholic acid, sphinganine, 12(R)-HPETE, O-arachidonoyl glycidol, L-phenylalanine, 9-HODE and L-tyrosine compared with the control.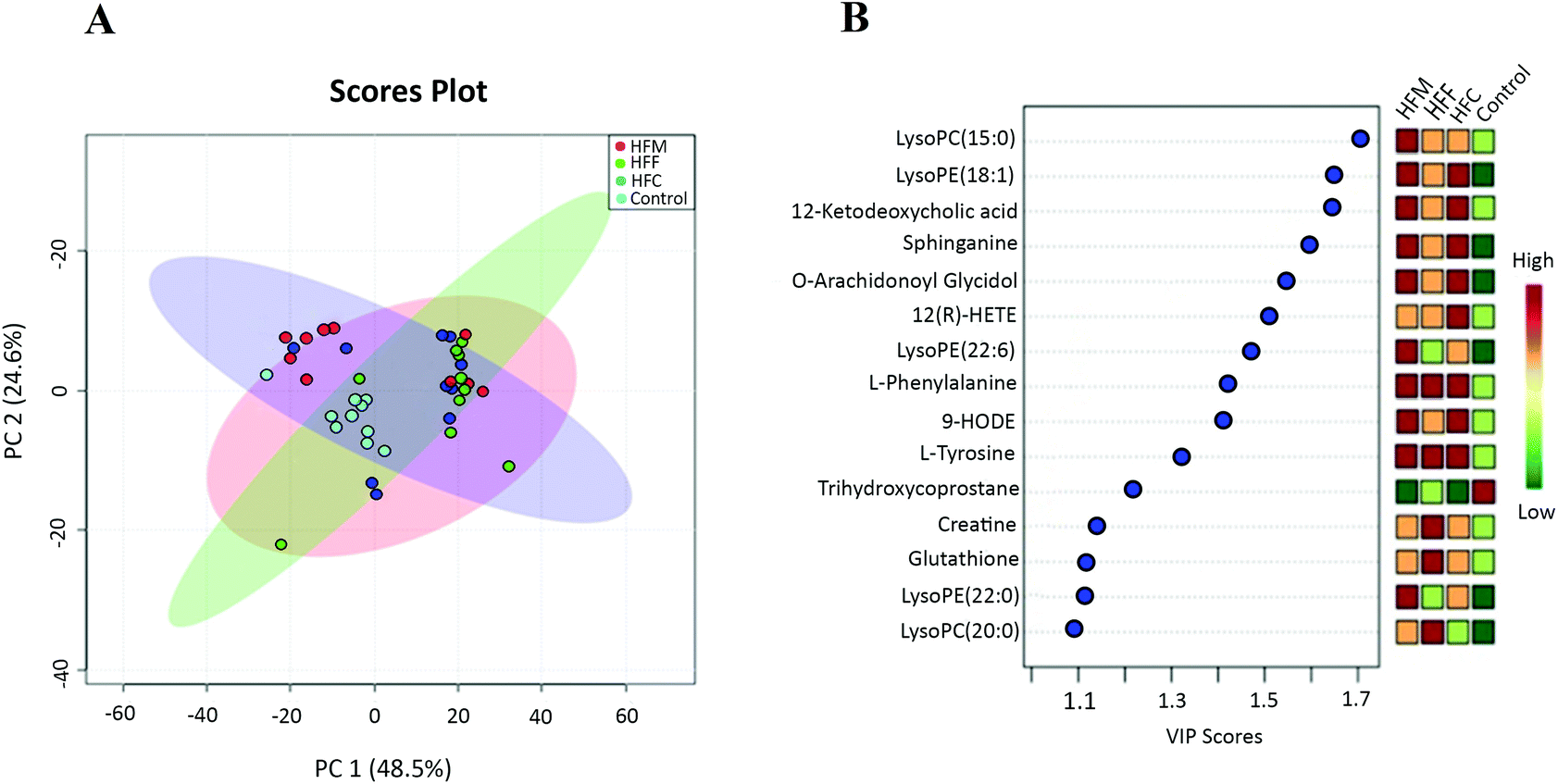 | ||
| Fig. 5 Serum metabolite profile of Glrx KO in response to different dietary proteins. (A) PCA score plot. Each point represents one biological sample; (B) the top 15 VIP scores of component 1. The left part lists significant difference of metabolites; the middle part shows the top 15 VIP scores; the right heatmap shows the concentration of metabolites. | ||
Furthermore, the OPLS-DA model was used to differentiate high fat diet groups from the control (Fig. 6A–C). The top 15 variables responsible for differences with VIP scores greater than 1 were selected. Compared to the control, the HFC group had higher concentrations of LysoPC(20:0), LysoPC(16:0), LysoPE(18:0), phenylethanolamine, 3-amino-2-naphthoic acid, 12-ketodeoxycholic acid, LysoPC(20:1), L-tyrosine, 12-hydroperoxy eicosatetraenoic acid (12(R)-HPETE), sphinganine, creatine, PE(18:0), O-arachidonoyl glycidol and 12-hydroxyoctadecaenoic acid (12-HODE) but a lower concentration of trihydroxycoprostane. The HFF group had higher concentrations of L-phenylalanine, L-tyrosine, phenylethanolamine, creatine, 12-ketodeoxycholic acid, PC(19:0), LysoPC(15:0), PC(22:1), PE(20:6), phytosphinganine, glutathione, 9-HODE, 12(R)-HPETE and LysoPE(18:1) but a lower concentration of trihydroxycoprostane. The HFM group had higher levels of LysoPC(15:0), LysoPC(16:0), LysoPC(17:0), LysoPC(20:0), LysoPC(20:6), LysoPE(18:1), L-phenylalanine, 9-HODE, O-arachidonoyl glycidol, L-tyrosine, 12-ketodeoxycholic acid, 1-heptadecanoyl-sn-glycero-3-phosphocholine, sphinganine and 13-HODE but a lower concentration of trihydroxycoprostane.
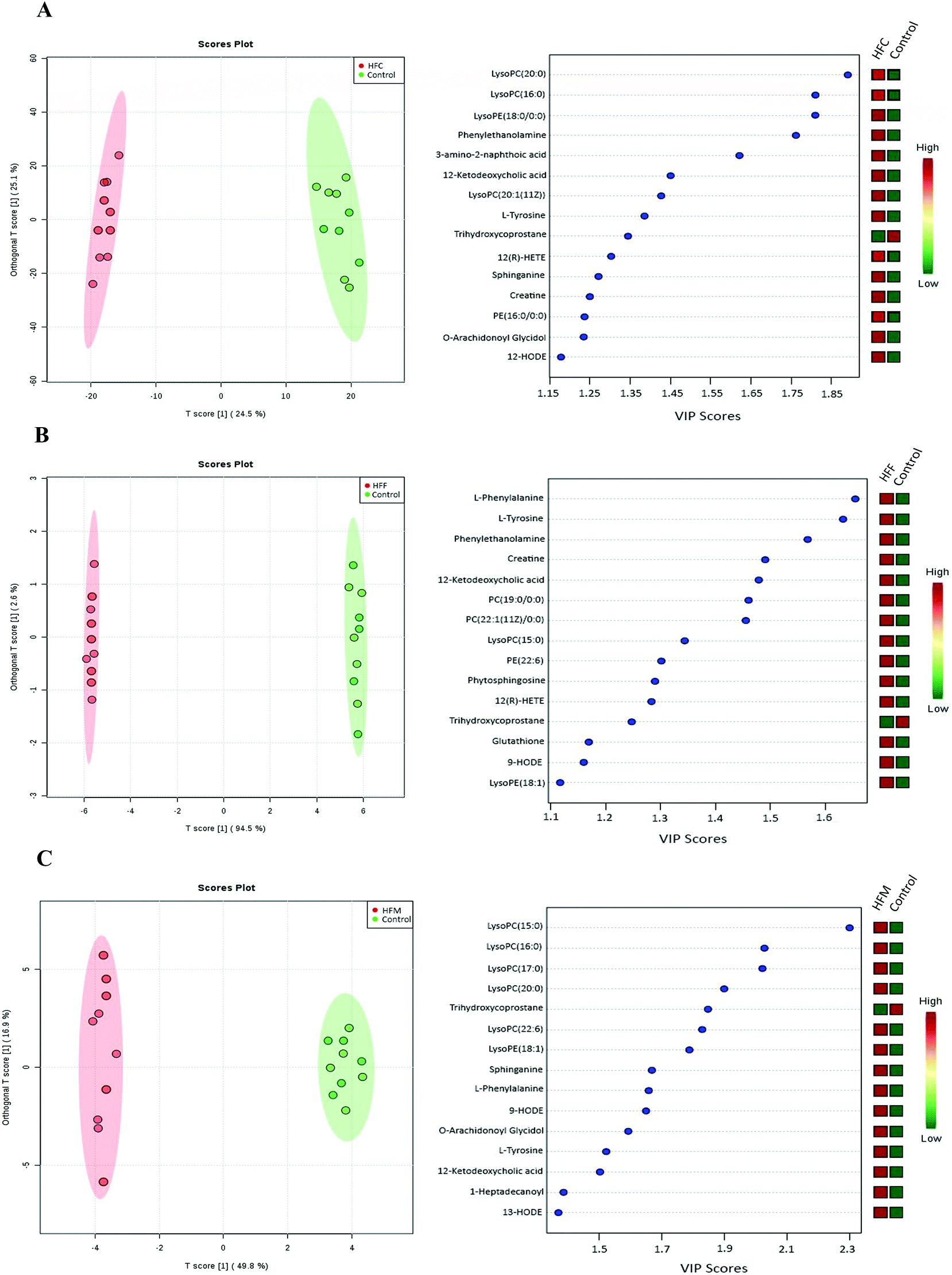 | ||
| Fig. 6 Pairwise comparisons between serum metabolite spectra obtained from the high fat casein (HFC), high fat fish (HFF) and high fat mutton (HFM) protein groups using OPLS analysis. Each figure has two parts: the left part is the OPLS score plot, the right part is the top 15 VIP scores. (A) HFC vs. control; (B) HFF vs. control; (C) HFM vs. control. | ||
In addition, we also applied the OPLS-DA model to differentiate the potential metabolites among high fat diet groups (Fig. 7A–C). The HFF group had lower concentrations of LysoPC(16:0), 1-monopalmitin, and LysoPE(18:2) but higher concentrations of creatine, glutathione, trihydroxycoprostane and rimexolone compared with the HFC group (Fig. 7A). Compared to HFF, the HFM had higher concentrations of 1-monopalmitin, LysoPC(16:0), and LysoPE(18:2) but lower concentrations of creatine, glutathione, and trihydroxycoprostane (Fig. 6B). Furthermore, the HFM had higher concentrations of LysoPC(16:0), PC(19:0), LysoPC(15:0), PE(18:1) and LysoPE(22:4) but a lower concentration of trihydroxycoprostane compared with the HFC group (Fig. 7C).
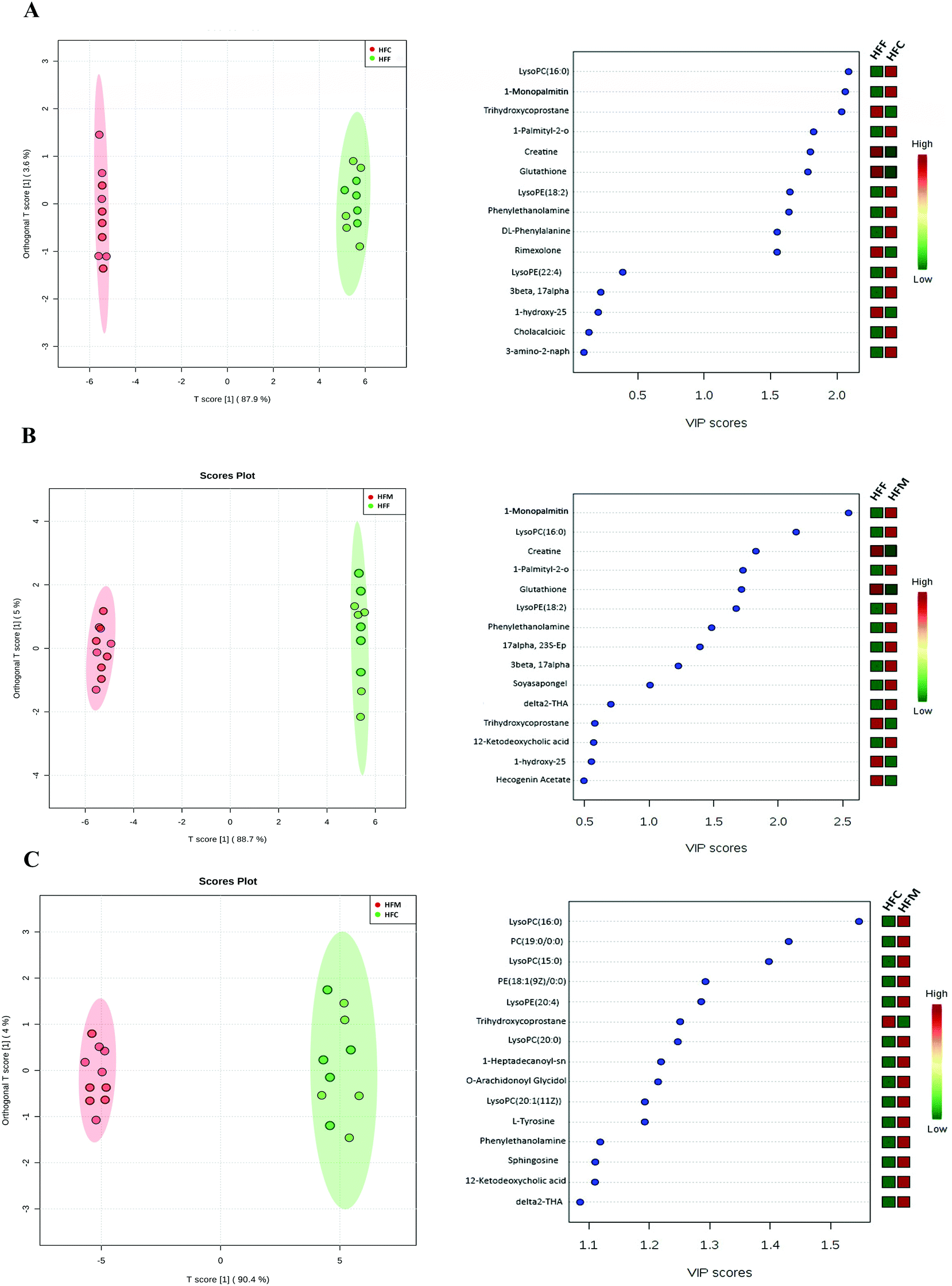 | ||
| Fig. 7 Pairwise comparisons between serum metabolite spectra obtained from the high fat casein (HFC), high fat fish (HFF) and high fat mutton (HFM) protein groups using OPLS analysis. Each figure has two parts: the left part is the OPLS score plot, the right part is the top 15 VIP scores. (A) HFF group vs. HFC protein group; (B) HFF group vs. HFM protein group; (C) HFC group vs. HFM protein group. | ||
Tables 3–6 summarize potentially differentiated compounds. We found that nine metabolic pathways were significantly changed by diets, involving glycerophospholipid metabolism, bile acid metabolism, steroid biosynthesis, purine metabolism, choline metabolism, sphingolipid metabolism, linoleic acid metabolism, phenylalanine metabolism, tyrosine metabolism, glycine and serine metabolism and glutathione metabolism. KEGG analysis showed that LysoPC(15:0), LysoPC(16:0), LysoPC(17:0), LysoPC(20:1), LysoPE(18:2), L-phenylalanine, L-tyrosine, sphinganine, DL-stearoylcarnitine, 9-HODE, 13-HODE, 12-ketodeoxycholic acid, creatine and glutathione may significantly affect glycerophospholipid metabolism, phenylalanine metabolism, tyrosine metabolism, mitochondrial beta-oxidation of long chain saturated fatty acids, linoleic acid metabolism, sphingolipid metabolism, bile acid metabolism, glycine and serine metabolism and glutathione metabolism. These pathways were predominantly affected by the high fat mutton diet compared with the other diet groups (P < 0.05, Tables 3–6).
| Putative I.D | [M/Z]+ | Formula | Identifier | HFC | HFF | HFM | Sub pathways |
|---|---|---|---|---|---|---|---|
| Fold change was calculated by dividing the mean of a metabolite in high-fat groups (HFC, HFF, HFM) by that in the control group. *p < 0.05, **p < 0.01, ***p < 0.001. | |||||||
| PC(19:0/0:0) | 538.3866 | C27H56NO7P | LMGP01050041 | 1.98** | 1.65** | 2.60*** | Glycerophospholipid metabolism |
| PC(18:1(9E)/0:0)[U] | 522.3562 | C26H52NO7P | None | 2.36** | 2.05** | 2.88*** | |
| PC(22:1(11Z)/0:0) | 577.4107 | C30H60NO7P | LMGP01050134 | 2.33** | 1.75** | 3.10*** | |
| LysoPC(15:0) | 482.3239 | C23H48NO7P | HMDB10381 | 3.20*** | 2.64** | 3.94*** | |
| LysoPC(16:0) | 496.3403 | C24H50NO7P | HMDB10382 | 2.15*** | 1.92** | 3.30*** | |
| LysoPC(17:0) | 509.3485 | C25H52NO7P | CAS50930-23-9 | 2.12** | 1.88** | 2.71*** | |
| LysoPC(20:0) | 552.4025 | C28H58NO7P | HMDB10390 | 2.34** | 1.64** | 2.50** | |
| LysoPC(20:1(11Z)) | 550.3864 | C28H56NO7P | HMDB10391 | 3.43*** | 3.33*** | 3.83*** | |
| LysoPC(22:6(4Z,7Z,10Z,13Z,16Z,19Z)) | 567.3324 | C30H50NO7P | HMDB10404 | 2.38** | 1.72** | 2.65*** | |
| PE(16:0/0:0) | 454.2930 | C21H44NO7P | None | 2.48** | 2.20** | 2.94*** | |
| PE(18:1(9Z)/0:0) | 480.3089 | C23H46NO7P | LMGP02050004 | 2.47*** | 2.40*** | 2.30*** | |
| PE(22:6(4Z,7Z,10Z,13Z,16Z,19Z)/0:0) | 526.2935 | C27H44NO7P | None | 4.55*** | 4.67*** | 4.42*** | |
| LysoPE(18:0/0:0) | 481.3168 | C23H48NO7P | HMDB11130 | 2.17** | 2.05** | 2.19** | |
| LysoPE(18:1(11Z)/0:0) | 479.3012 | C23H46NO7P | HMDB11505 | 2.24** | 1.67** | 2.88*** | |
| LysoPE(18:2(9Z,12Z)/0:0) | 478.2928 | C23H44NO7P | HMDB11507 | 2.87*** | 2.22** | 3.46*** | |
| LysoPE(22:0(7Z,10Z,13Z,16Z)/0:0) | 529.3168 | C27H48NO7P | HMDB11493 | 2.55** | 1.62** | 3.20*** | |
| LysoPE(20:4(8Z,11Z,14Z,17Z)/0:0) | 501.2860 | C25H44NO7P | HMDB11518 | 2.05** | 2.12** | 2.20** | |
| LysoPE(22:6(7Z,10Z,13Z,16Z)/0:0) | 567.3324 | C30H50NO7P | HMDB10404 | 2.35** | 2.42** | 3.12*** | |
| O-Arachidonoyl glycidol | 361.2733 | C23H36O3 | None | 3.17*** | 2.59** | 3.40*** | None |
| L-Phenylalanine | 165.0789 | C9H11NO2 | HMDB0612 | 2.53** | 1.87** | 2.84*** | Phenylalanine metabolism |
| L-Tyrosine | 181.0738 | C9H11NO3 | HMDB00158 | 2.45** | 1.34** | 2.60*** | Tyrosine metabolism |
| Phenylethanolamine | 120.0824 | C8H11NO | CSID975 | 2.11*** | 1.75** | 2.20*** | None |
| 3-Amino-2-naphthoic acid | 188.0745 | C11H9NO2 | None | 3.06*** | 3.02*** | 3.20*** | None |
| Sphinganine | 302.3049 | C18H39NO2 | CSID82609 | 3.35*** | 2.80** | 3.88*** | Sphingolipid metabolism |
| Phytosphingosine | 318.2999 | C18H39NO3 | CSID108921 | 2.52** | 2.30** | 3.10*** | |
| 3-Ketosphinganine | 282.2789 | C24H38O3 | CAS5130-29-0 | 2.69** | 2.60** | 2.99*** | |
| N,N-Dimethylsphingosine | 327.3131 | C20H41NO2 | HMDB13645 | 2.42** | 2.30** | 2.92*** | |
| 1-Heptadecanoyl-sn-glycero-3-phosphocholine | 510.3561 | C25H52NO7P | CAS50930-23-9 | 2.67** | 2.47** | 3.79*** | None |
| DL-Stearoylcarnitine | 438.3727 | C25H49NO4 | HMDB00848 | 2.56** | 2.40** | 2.97*** | Mitochondrial beta-oxidation of long chain saturated fatty acids |
| 9-HODE | 296.2351 | C18H32O3 | HMDB10223 | 3.77*** | 2.58** | 4.05*** | Linoleic acid metabolism |
| 13-HODE | 296.2351 | C18H32O3 | HMDB4667 | 3.25*** | 2.44** | 3.71*** | |
| 12(R)-HPETE | 303.2333 | C20H32O3 | CAS82337-46-0 | 2.28** | 2.12** | 3.30*** | None |
| 12-Ketodeoxycholic acid | 391.2842 | C24H38O4 | CAS5130-29-0 | 8.70*** | 6.30*** | 11*** | Bile acid metabolism |
| 3β-Hydroxy-5-cholenoic acid | 374.5471 | C24H38O3 | CSID83950 | 2.15** | 2.02** | 2.20** | |
| Trihydroxycoprostane | 443.3498 | C27H48O3 | CSID141053 | −13.34*** | −3.80** | −16.65*** | |
| Creatine | 132.0771 | C4H9N3O2 | CSID566 | 2.98*** | 3.55*** | 2.30** | Glycine and serine metabolism |
| Glutathione | 308.0912 | C10H17N3O6S | None | 2.77** | 3.67*** | 2.12** | Glutathione metabolism |
| Putative I.D | [M/Z]+ | Formula | Identifier | Fold change | Sub pathways |
|---|---|---|---|---|---|
| Fold change was calculated by dividing the mean of a metabolite in high-fat groups HFC by that in the HFF group. *p < 0.05, **p < 0.01, ***p < 0.001. | |||||
| PC(19:0/0:0) | 538.3866 | C27H56NO7P | LMGP01050041 | 0.75* | Glycerophospholipid metabolism |
| PC(18:1(9E)/0:0)[U] | 522.3562 | C26H52NO7P | None | 0.46* | |
| PC(22:1(11Z)/0:0) | 577.4107 | C30H60NO7P | LMGP01050134 | 0.23* | |
| LysoPC(15:0) | 482.3239 | C23H48NO7P | HMDB10381 | 0.60* | |
| LysoPC(20:0) | 552.4025 | C28H58NO7P | HMDB10390 | 0.34* | |
| LysoPC(20:1(11Z)) | 550.3864 | C28H56NO7P | HMDB10391 | 0.53* | |
| PE(16:0/0:0) | 454.2930 | C21H44NO7P | None | 1.24** | |
| PE(18:1(9Z)/0:0) | 480.3089 | C23H46NO7P | LMGP02050004 | 1.15** | |
| PE(22:6(4Z,7Z,10Z,13Z,16Z,19Z)/0:0) | 526.2935 | C27H44NO7P | None | 0.52* | |
| LysoPE(18:0/0:0) | 481.3168 | C23H48NO7P | HMDB11130 | 1.03** | |
| LysoPE(18:1(11Z)/0:0) | 479.3012 | C23H46NO7P | HMDB11505 | 1.84** | |
| LysoPE(18:2(9Z,12Z)/0:0) | 478.2928 | C23H44NO7P | HMDB11507 | 1.57** | |
| LysoPE(22:0(7Z,10Z,13Z,16Z)/0:0) | 529.3168 | C27H48NO7P | HMDB11493 | 1.05** | |
| LysoPE(20:4(8Z,11Z,14Z,17Z)/0:0) | 501.2860 | C25H44NO7P | HMDB11518 | 1.12** | |
| O-ArachidonoylGlycidol | 361.2733 | C23H36O3 | None | 0.67** | None |
| L-Phenylalanine | 165.0789 | C9H11NO2 | HMDB0612 | 0.23* | Phenylalanine metabolism |
| L-Tyrosine | 181.0738 | C9H11NO3 | HMDB00158 | 0.45* | Tyrosine metabolism |
| Phenylethanolamine | 120.0824 | C8H11NO | CSID975 | 1.21** | None |
| 3-Amino-2-naphthoic acid | 188.0745 | C11H9NO2 | None | 1.36** | None |
| Sphinganine | 302.3049 | C18H39NO2 | CSID82609 | 1.45** | Sphingolipid metabolism |
| Phytosphingosine | 318.2999 | C18H39NO3 | CSID108921 | 1.52** | |
| 1-Heptadecanoyl-sn-glycero-3-phosphocholine | 510.3561 | C25H52NO7P | CAS50930-23-9 | 1.50** | None |
| DL-Stearoylcarnitine | 438.3727 | C25H49NO4 | HMDB00848 | 1.36** | Mitochondrial beta-oxidation of long chain saturated fatty acids |
| 12(R)-HPETE | 303.2333 | C20H32O3 | CAS82337-46-0 | 1.08** | None |
| 12-Ketodeoxycholic acid | 391.2842 | C24H38O4 | CAS5130-29-0 | 2.82*** | Bile acid metabolism |
| 3β-Hydroxy-5-cholenoic acid | 374.5471 | C24H38O3 | CSID83950 | 0.95* | |
| Trihydroxycoprostane | 443.3498 | C27H48O3 | CSID141053 | −4.34*** | |
| Creatine | 132.0771 | C4H9N3O2 | CSID566 | −1.38** | Glycine and serine metabolism |
| Glutathione | 308.0912 | C10H17N3O6S | None | −1.27** | Glutathione metabolism |
| Putative I.D | [M/Z]+ | Formula | Identifier | Fold change | Sub pathways |
|---|---|---|---|---|---|
| Fold change was calculated by dividing the mean of a metabolite in high-fat groups HFM by that in the HFC group. *p < 0.05, **p < 0.01, ***p < 0.001. | |||||
| PC(19:0/0:0) | 538.3866 | C27H56NO7P | LMGP01050041 | 0.28* | Glycerophospholipid metabolism |
| PC(18:1(9E)/0:0)[U] | 522.3562 | C26H52NO7P | None | 0.15* | |
| LysoPC(15:0) | 482.3239 | C23H48NO7P | HMDB10381 | 0.41* | |
| LysoPC(16:0) | 496.3403 | C24H50NO7P | HMDB10382 | 1.43** | |
| LysoPC(20:0) | 552.4025 | C28H58NO7P | HMDB10390 | 0.08* | |
| LysoPC(20:1(11Z)) | 550.3864 | C28H56NO7P | HMDB10391 | 0.35** | |
| PE(16:0/0:0) | 454.2930 | C21H44NO7P | None | 0.42* | |
| PE(18:1(9Z)/0:0) | 480.3089 | C23H46NO7P | LMGP02050004 | 0.72* | |
| PE(22:6(4Z,7Z,10Z,13Z,16Z,19Z)/0:0) | 526.2935 | C27H44NO7P | None | 0.20* | |
| LysoPE(18:2(9Z,12Z)/0:0) | 478.2928 | C23H44NO7P | HMDB11507 | 1.34* | |
| LysoPE(22:0(7Z,10Z,13Z,16Z)/0:0) | 529.3168 | C27H48NO7P | HMDB11493 | 0.85* | |
| LysoPE(20:4(8Z,11Z,14Z,17Z)/0:0) | 501.2860 | C25H44NO7P | HMDB11518 | 0.88* | |
| O-Arachidonoyl glycidol | 361.2733 | C23H36O3 | None | 0.21* | None |
| L-Phenylalanine | 165.0789 | C9H11NO2 | HMDB0612 | 0.37* | Phenylalanine metabolism |
| L-Tyrosine | 181.0738 | C9H11NO3 | HMDB00158 | 0.25* | Tyrosine metabolism |
| Phenylethanolamine | 120.0824 | C8H11NO | CSID975 | 0.88* | None |
| 3-Amino-2-naphthoic acid | 188.0745 | C11H9NO2 | None | 0.62* | None |
| Sphinganine | 302.3049 | C18H39NO2 | CSID82609 | 1.25** | Sphingolipid metabolism |
| Phytosphingosine | 318.2999 | C18H39NO3 | CSID108921 | 1.32** | |
| 1-Heptadecanoyl-sn-glycero-3-phosphocholine | 510.3561 | C25H52NO7P | CAS50930-23-9 | 1.33** | None |
| DL-Stearoylcarnitine | 438.3727 | C25H49NO4 | HMDB00848 | 0.88* | Mitochondrial beta-oxidation of long chain saturated fatty acids |
| 12(R)-HPETE | 303.2333 | C20H32O3 | CAS82337-46-0 | 1.15** | None |
| 12-Ketodeoxycholic acid | 391.2842 | C24H38O4 | CAS5130-29-0 | 2.45** | Bile acid metabolism |
| 3β-Hydroxy-5-cholenoic acid | 374.5471 | C24H38O3 | CSID83950 | 1.15** | |
| Trihydroxycoprostane | 443.3498 | C27H48O3 | CSID141053 | −4.61*** | |
| Delta2-THA | 355.26 | C24H34O2 | HMDB-74388 | 3.53*** | None |
| 1-Palmityl-2-oleoyl-sn-glycerol | 609.34 | C37H72O4 | CSID4445455 | 2.36** | None |
| 1-Stearoyl-2-hydroxy-sn-glycero-3-phosphoethanolamine | 603.53 | C23H48NO7P | HMDB-69747-55-3 | 2.16** | None |
| 1-Monopalmitin | 510.36 | C19H38O4 | HMDB-24076 | 2.31** | None |
| Putative I.D | [M/Z]+ | Formula | Identifier | Fold change | Sub pathways |
|---|---|---|---|---|---|
| Fold change was calculated by dividing the mean of a metabolite in high-fat groups HFM by that in the HFF group. *p < 0.05, **p < 0.01, ***p < 0.001. | |||||
| LysoPC(20:0) | 552.4025 | C28H58NO7P | HMDB10390 | 0.76* | Glycerophospholipid metabolism |
| LysoPE(18:0/0:0) | 481.31 | C23H48NO7P | HMDB0011130 | 1.16** | |
| LysoPC(18:1(9Z)) | 521.34 | C26H52NO7P | HMDB0002815 | 2.04** | |
| MG(18:1(9Z)/0:0/0:0)[rac] | 357.30 | C21H40O4 | LMGL01010004 | 1.64** | |
| MG(22:6(4Z,7Z,10Z,13Z,16Z,19Z)/0:0/0:0) | 403.28 | C25H38O4 | HMDB0011587 | 0.67* | |
| 1-AG | 379.28 | C23H38O4 | HMDB0011578 | 1.73** | |
| O-Arachidonoyl glycidol | 361.2733 | C23H36O3 | None | 1.26** | None |
| L-Phenylalanine | 165.0789 | C9H11NO2 | HMDB0612 | 1.23** | Phenylalanine metabolism |
| L-Tyrosine | 181.0738 | C9H11NO3 | HMDB00158 | 1.55** | Tyrosine metabolism |
| Phytosphingosine | 318.2999 | C18H39NO3 | CSID108921 | 1.06** | Sphingolipid metabolism |
| 1-Heptadecanoyl-sn-glycero-3-phosphocholine | 510.3561 | C25H52NO7P | CAS50930-23-9 | 1.70** | None |
| DL-Stearoylcarnitine | 438.3727 | C25H49NO4 | HMDB00848 | 1.56** | Mitochondrial beta-oxidation of long chain saturated fatty acids |
| 12-Ketodeoxycholic acid | 391.2842 | C24H38O4 | CAS5130-29-0 | 4.22*** | Bile acid metabolism |
| Creatine | 132.0771 | C4H9N3O2 | CSID566 | −0.42* | Glycine and serine metabolism |
| Glutathione | 308.0912 | C10H17N3O6S | None | −0.78* | Glutathione metabolism |
| Delta2-THA | 355.26 | C24H34O2 | HMDB-74388 | 1.26** | None |
| 1-Palmityl-2-oleoyl-sn-glycerol | 594.52 | C37H70 | CSID4445455 | 3.30*** | None |
| 12(R)-HPETE | 336.23 | C20H32O4 | HMDB0004692 | 3.51** | None |
| 2-Linoleoyl glycerol | 354.52 | C21H38O4 | HMDB0011538 | 6.57** | None |
| 9,12,15-Octadecatrien-1-ol | 265.25 | C18H32O | HMDB-46167 | 1.52** | None |
Discussion
Excess consumption of red meat has been associated with the incidence of impaired glucose and insulin homeostasis, diabetes and NAFLD.3,4 In parallel, the prevalence of NAFLD is growing and has emerged as a leading cause for liver diseases.1 The accumulation of oxidized proteins is associated with multiple chronic diseases such as diabetes mellitus.5,6 Previous studies have shown that the intake of dietary proteins at a normal dose or high fat diets or western diets promoted inflammation, hyperlipidemia and NAFLD in Glrx1−/− mice.24,26 Also, Glrx1 plays a central role in the activation of p53, Nrf2/Keap1, IL-1β and associated signaling pathways at the cellular level in protection against oxidative stress.29–32 Despite extensive metabolomic research on the potential biomarkers of NAFLD, there has been no systemic approach on serum metabolic profiling of NAFLD induced by high-fat meat proteins in Glrx1−/− mice. The present study focused on the effect of high-fat meat protein diets on the serum biomarkers of NAFLD in Glrx1 deficient mice. Multivariate analysis of serum metabolites indicated that the high-fat mutton protein diet significantly shifted several regulatory pathways involved in oxidative stress, bile acid biosynthesis, amino acid metabolism, and fatty acid biosynthesis, however, fish protein diet alleviated high-fat induced NAFLD. Bile acids (BAs), amino acids, Lyso PC, Lyso PE, PC, PE and sphingolipid are well-known metabolites that are altered in NAFLD. All these compounds are involved in key hepatic metabolic pathways.40–42NAFLD is not only linked with distinctive changes in the plasma lipidomic profile but also with hepatic phospholipids and fatty acids. Fatty acids and phospholipids are the most abundant metabolites in liver and blood. A higher level of PC accumulation in the blood is directly correlated with the secretion of hepatic PC into blood induced by the high-fat diet.43 Likewise, LPC is an important signaling molecule involved in cell proliferation, invasion, and inflammation. Indeed, the high level of plasma LPCs has been associated with obesity and NAFLD.40,42 Several studies have reported that the intake of high fat diet significantly reduced the plasma concentrations of glycerophospholipids including PC and PE in human and rodent models.44,45 Still, the association between glycerophospholipids and obesity is not clear. Interestingly, the abundance of LysoPCs and LysoPEs, including LysoPC(15:0), LysoPC(16:0), LysoPC(17:0), LysoPC(20:0), LysoPC(20:6), LysoPE(18:1), LysoPE(18:2), LysoPE(22:0), LysoPE(22:6) and LysoPC(20:1), was increased in the HFM group compared with the control group. This is in agreement with previous studies in which higher PC levels were associated with diet induced NAFLD in mice.40–42
Oxidative stress is involved in the development of NAFLD. HETE and HODE, which are formed during enzymatic conversion by the action of lipoxygenases or by oxidation of non-enzymatic pathways, are important markers of oxidative stress.46 Lipoxygenase converts arachidonic acid to eicosanoids, which are key modulators of inflammatory pathways and associated with human NAFLD.47 Higher levels of 9-HODE and 13-HODE have been shown to be associated with NAFLD development.20 Likewise, endocannabinoids are also derivatives of arachidonic acid, and have been recently associated with NAFLD development.48 In the present study, the concentrations of 9-HODE, 13-HODE, 12(R)-HPETE and O-arachidonoylglycidol were strikingly upregulated in the high-fat meat protein diet group compared with the control.
The intake of high fat diet is associated with the accumulation of phospholipids, diacylglycerols, triacylglycerols, and free fatty acids in hepatocytes that ultimately leads to NAFLD. Hepatic lipogenesis is associated with the flow of fatty acids to the liver and depends on the dietary composition. Secondly, mono and polyunsaturated fatty acids are more prone to oxidation than saturated FAs and thus prevent accumulation of lipid fractions. Sphingolipid metabolites play a crucial role in inflammatory signaling,49 and chronic metabolic diseases, including obesity, T2DM and NAFLD. Chronic inflammatory diseases are characterized by a low but consistent activation of the immune system, and as a consequence, the body fails to give inflammatory responses to foreign stimuli.50 Most of these disease symptoms are associated with sphingolipid metabolism and inflammation. Sphingosine, dihydrosphingosine and phytosphingosine are potential and diagnostic biomarkers for the metabolomics of NAFLD.51 Likewise, studies have shown that fatty acids play a key role in the progression and development of NAFLD.12,15,22 Lipid-induced cell toxicity and apoptosis are associated with derivatives of fatty acids.15 In the present study, levels of sphingosine, phytosphingosine, 3-ketosphinganine and N,N-dimethylsphingosine were increased significantly in the HFM group compared with the control group and associated with the sphingolipid metabolism. This is in accordance with other studies on NAFLD patients.51,52
Previous studies indicated that changes in the amino acid metabolism were associated with NAFLD.14,15 Numerous studies showed higher levels of phenylalanine, tyrosine and glutamate in NAFLD.16,18 The irreversible metabolic conversion of phenylalanine to tyrosine mainly takes place in the liver where tyrosine is further metabolized, which impairs hepatic metabolism. An elevated tyrosine level has been detected in patients with NAFLD.16 In the present study, compared with the control group, mice fed with high fat diets had higher L-phenylalanine, L-tyrosine, phenylethanolamine, and 3-amino-2-naphthoic acid levels. Higher L-phenylalanine, L-tyrosine, and glutamate levels have been shown to be associated with high-fat-diet-induced oxidative stress, obesity, T2DM and NAFLD.16,53
In NAFLD patients, taurine and glycine-conjugated bile acids (BAs) and secondary BAs increased greatly.19,54 High-fat diets have also been shown to alter BA composition in rats.55 An increase in harmful BAs may contribute to repeated insults of inflammation that ultimately induce the progression of NASH.56 In this study, we observed that high-fat protein diets increased levels of 12-ketodeoxycholic acid and 3β-hydroxy-5-cholenoic acid in serum that involve bile acid metabolism. In addition, a HFM diet increased the level of serum ketodeoxycholic acid that plays a key role in dietary fat absorption and cholesterol homeostasis, which is in agreement with a pervious study that the intake of high fat diet increased the levels of primary and secondary bile acids.57
In summary, the present study identified key metabolites of glycerophospholipid metabolism, sphingolipid metabolism, phenylalanine metabolism, tyrosine metabolism, linoleic acid metabolism, bile acid metabolism, glycine and serine metabolism and glutathione metabolism pathways involved in NAFLD progression induced by a high-fat mutton protein diet. A high fat diet supplemented with fish meat protein significantly ameliorated diet-induced NAFLD serum metabolites, biomarkers associated with dyslipidemia and hepatic pro-inflammatory cytokines compared to HFC and HFM diets. These identified metabolites induced by high fat diets could be considered as possible metabolomic biomarkers to assess the development of NAFLD.
Author contributions
CL and MIA designed the experiment; MIA, MUI, MH, IAK, NM, and CCL performed the experiment; MIA analyzed the data; MIA and CL wrote the manuscript; DZ, XX and GZ gave critical comments.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by grants 31530054 (NSFC), CARS-35, and PADP.References
- L. Miele, V. Dall'armi, C. Cefalo, B. Nedovic, D. Arzani and R. Amore, A case control study on the effect of metabolic gene polymorphisms, nutrition, and their interaction on the risk of non-alcoholic fatty liver disease, Genes Nutr., 2014, 9, 383–391 CrossRef PubMed.
- W. H. Oddy, C. E. Herbison, P. Jacoby, G. L. Ambrosini, T. A. O'Sullivan and O. T. Ayonrinde, The Western dietary pattern is prospectively associated with nonalcoholic fatty liver disease in adolescence, Am. J. Gastroenterol., 2013, 108, 778–785 CrossRef CAS PubMed.
- S. Zelber-Sagi, D. Ivancovsky-Wajcman, N. F. Isakov, D. Orenstein, O. Shibolet and R. Kariv, High red and processed meat consumption is associated with non-alcoholic fatty liver disease and insulin resistance, J. Hepatol., 2018, 68, 1239–1246 CrossRef CAS PubMed.
- E. A. Hernandez, S. Kahl, A. Seelig, P. Begovatz, M. Irmler and Y. M. Kupriyanova, Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance, J. Clin. Invest., 2017, 127, 695–708 CrossRef PubMed.
- A. Bellavia, F. Stilling and A. Wolk, High red meat intake and all-cause cardiovascular and cancer mortality: is the risk modified by fruit and vegetable intake?, Am. J. Clin. Nutr., 2016, 104, 1137–1143 CrossRef CAS PubMed.
- R. Christensen, B. L. Heitmann, K. W. Andersen, O. H. Nielsen, S. B. Sorensen and M. Jawhar, Impact of red and processed meat and fiber intake on treatment outcomes among patients with chronic inflammatory diseases: protocol for a prospective cohort study of prognostic factors and personalized medicine, BMJ Open, 2018, 8, e018166 CrossRef PubMed.
- M. Den Boer, P. J. Voshol, F. Kuipers, L. M. Havekes and J. A. Romijn, Hepatic steatosis: a mediator of the metabolic syndrome, Lessons from animal models, Arterioscler., Thromb., Vasc. Biol., 2004, 24, 644–649 CrossRef CAS PubMed.
- K. Almind and C. R. Kahn, Genetic determinants of energy expenditure and insulin resistance in diet-induced obesity in mice, Diabetes, 2004, 53, 3274–3285 CrossRef CAS PubMed.
- M. Rossmeisl, J. S. Rim, R. A. Koza and L. P. Kozak, Variation in type 2 diabetes-related traits in mouse strains susceptible to diet induced obesity, Diabetes, 2003, 52, 1958–1966 CrossRef CAS PubMed.
- V. T. Samuel, Z. X. Liu and X. Qu, Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease, J. Biol. Chem., 2004, 279, 32345–32353 CrossRef CAS PubMed.
- Y. Zhu, X. Shi, X. Lin, K. Ye, X. Xu, C. Li and G. Zhou, Beef, chicken, and soy proteins in diets induce different gut microbiota and metabolites in rats, Front. Microbiol., 2017, 8, 1–11 Search PubMed.
- P. Puri, M. M. Wiest, O. Cheung, F. Mirshahi, C. Sargeant and H. K. Min, The plasma lipidomic signature of nonalcoholic steatohepatitis, Hepatology, 2009, 50, 1827–1838 CrossRef CAS PubMed.
- E. Alonso, D. Fernández-ramos and M. Varela-rey, Metabolomic identification of subtypes of nonalcoholic steatohepatitis, Gastroenterology, 2017, 152, 1449–1461 CrossRef PubMed.
- A. Mardinoglu, R. Agren and E. Kamp, Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease, Nat. Commun., 2014, 5, 3083–3083 CrossRef PubMed.
- J. Kaikkonen, P. Würtz and E. Suomela, Metabolic profiling of fatty liver in young and middle-aged adults: cross-sectional and prospective analyses of the Young Finns Study, Hepatology, 2017, 65, 491–500 CrossRef CAS PubMed.
- M. Kawanaka, K. Nishino, T. Oka, N. Urata, J. Nakamura and T. Suehiro, Tyrosine levels are associated with insulin resistance in patients with nonalcoholic fatty liver disease, Hepatic Med., 2015, 7, 29–35 CrossRef PubMed.
- R. Jin, S. Banton and V. Tran, Amino acid metabolism is altered in adolescents with nonalcoholic fatty liver disease an untargeted, high resolution metabolomics study, J. Pediatr., 2016, 172, 14–19 CrossRef CAS PubMed.
- M. Gaggini, F. Carli, C. Rosso, E. Buzzigoli, M. Marietti, V. D. Latta and A. Gastaldelli, Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance, Hepatology, 2018, 67, 145–158 CrossRef CAS PubMed.
- P. Puri, K. Daita and A. Joyce, The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids, Hepatology, 2017, 64, 170–181 Search PubMed.
- S. Polyzos, J. Kountouras and A. Tsatsoulis, Sex steroids and sex hormone-binding globulin in postmenopausal women with nonalcoholic fatty liver disease, Hormones, 2013, 12, 405–416 CrossRef PubMed.
- E. Koehler, J. Swain and S. Sanderson, Growth hormone, dehydroepiandrosterone and adiponectin levels in non-alcoholic steatohepatitis: an endocrine signature for advanced fibrosis in obese patients, Liver Int., 2012, 32, 279–286 CrossRef CAS PubMed.
- R. Loomba, O. Quehenberger and A. Armando, Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for noninvasive diagnosis of nonalcoholic steatohepatitis, J. Lipid Res., 2015, 56, 185–192 CrossRef CAS PubMed.
- M. Catta-Preta, L. Mendonca, J. Fraulob-Aquino and M. Aguila, A critical analysis of three quantitative methods of assessment of hepatic steatosis in liver biopsies, Virchows Arch., 2011, 459, 477–485 CrossRef PubMed.
- D. Shao, J. Han, X. Hou, J. Fry, J. Behring and F. Seta, Glutaredoxin-1 deficiency causes fatty liver and dyslipidemia by inhibiting sirtuin-1, Antioxid. Redox Signal., 2017, 27, 313–323 CrossRef CAS PubMed.
- F. Yang, M. Yi, Y. Liu, Q. Wang, Y. Hu and H. Deng, Glutaredoxin-1 silencing induces cell senescence via p53/p21/p16 signaling axis, J. Proteome Res., 2018, 17, 1091–1100 CrossRef CAS PubMed.
- A. M. Ijaz, I. M. Ijaz, M. Hussain, L. Congcong, X. Xu, C. Li and G. Zhou, Processed meat protein promoted inflammation and hepatic lipogenesis by upregulating Nrf2/Keap1 signaling pathway in Glrx-deficient mice, J. Agric. Food Chem., 2019, 67, 8794–8809 CrossRef PubMed.
- A. Levin, D. Nair, A. R. Qureshi, P. Bárány, O. Heimburger and B. Anderstam, Serum glutaredoxin activity as a marker of oxidative stress in chronic kidney disease: A pilot study, Nephron, 2018, 4, 249–256 CrossRef PubMed.
- D. Yatao, Z. Huihui, M. Sergio, H. Jesper, E. Neda and H. Arne, Plasma glutaredoxin activity in healthy subjects and patients with abnormal glucose levels or overt type 2 diabetes, Acta Diabetol., 2013, 2, 225–232 Search PubMed.
- A. N. Carvalho, C. Marques and R. C. Guedes, S-glutathionylation of Keap1: a new role for glutathione S-transferase pi in neuronal protection, FEBS Lett., 2016, 10, 1455–1466 CrossRef PubMed.
- N. L. Reynaert, A. Vliet, A. S. Guala, T. McGovern, M. Hristova and C. Pantano, Dynamic redox control of NF-kappa B through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13086–13091 CrossRef CAS.
- X. Zhang, P. Liu, C. Zhang, D. Chol, F. Zhao, H. Yu and H. R. Luo, Positive regulation of interleukin-1β bioactivity by physiological ROS-mediated cysteine S-glutathionylation, Cell Rep., 2017, 1, 224–235 CrossRef.
- N. Vall-Llaura, G. Reverter-Branchat, C. Vived, N. Weertman, M. J. Rodrıguez-Colman and E. Cabiscol, Reversible glutathionylation of Sir2 by monothiol glutaredoxins Grx3/4 regulates stress resistance, Free Radicals Biol. Med., 2016, 96, 45–56 CrossRef CAS.
- J. Joung, S. Konermann, J. Gootenberg, O. Abudayyeh, R. Platt and M. Brigham, Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening, Nat. Protoc., 2017, 12, 828–863 CrossRef CAS PubMed.
- M. Catta-Preta, L. Mendonca, J. Fraulob-Aquino and M. Aguila, A critical analysis of three quantitative methods of assessment of hepatic steatosis in liver biopsies, Virchows Arch., 2011, 459, 477–485 CrossRef PubMed.
- K. J. Livak and T. D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2-11CT method, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- Y. Kawano and D. E. Cohen, Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease, J. Gastroenterol., 2013, 48, 434–441 CrossRef CAS PubMed.
- E. Fabbrini, S. Sullivan and S. Klein, Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications, Hepatology, 2010, 51, 679–689 CrossRef CAS PubMed.
- C. Shelly and M. D. Lu, Glutathione synthesis, Biochim. Biophys. Acta, 2014, 5, 3143–3153 Search PubMed.
- S. I. Hashemy, C. Johansson, C. Berndt, C. H. Lillig and A. Holmgren, Oxidation and S nitrosylation of cysteines in human cytosolic and mitochondrial glutaredoxins: Effects on structure and activity, J. Biol. Chem., 2007, 19, 14428–14436 CrossRef PubMed.
- K. H. Pietiläinen, M. Sysi-Aho, A. Rissanen, T. Seppänen-Laakso and H. Yki-Järvinen, Acquired obesity is associated with changes in the serum lipidomic profile independent of genetic effects - a monozygotic twin study, PLoS One, 2007, 2, e218 CrossRef PubMed.
- H. Y. Kim, M. Kim, H. M. Park, J. Kim, E. J. Kim and C. H. Lee, Lysophospholipid profile in serum and liver by high-fat diet and tumor induction in obesity-resistant BALB/c mice, Nutrition, 2014, 30, 1433–1441 CrossRef CAS PubMed.
- H. Kim, J. H. Kim, S. Noh, H. J. Hur, M. J. Sung and J. Hwang, Metabolomic analysis of livers and serum from high-fat diet induced obese mice, J. Proteome Res., 2011, 102, 722–731 CrossRef PubMed.
- A. A. Noga and D. E. Vance, Insights into the requirement of phosphatidylcholine synthesis for liver functions in mice, J. Lipid Res., 2003, 44, 1998–2005 CrossRef CAS PubMed.
- B. Shirouchi, K. Nagao, N. Inoue, T. Ohkubo, H. Hibino and T. Yanagita, Effect of dietary omega 3 phosphatidylcholine on obesity-related disorders in obese Otsuka Long-Evans Tokushima fatty rats, J. Agric. Food Chem., 2007, 55, 7170–7176 CrossRef CAS PubMed.
- P. G. Rittes, Use of phosphatidylcholine for correction of localized fat deposits, Aesthetic Plast. Surg., 2003, 27, 315–318 CrossRef PubMed.
- D. Maciejewska, P. Ossowski, A. Drozd, K. Ryterska, D. Jamioł-Milc and M. Banaszczak, Metabolites of arachidonic acid and linoleic acid in early stages of non-alcoholic fatty liver disease-A pilot study, Prostaglandins Other Lipid Mediators, 2015, 121, 184–189 CrossRef CAS.
- V. Vangaveti, B. T. Baune and R. L. Kennedy, Hydroxyoctadecadienoic acids: novel regulators of macrophage differentiation and atherogenesis, Ther. Adv. Endocrinol. Metab., 2010, 1, 51–60 CrossRef CAS PubMed.
- G. Kunos and D. Osei-Hyiaman, Endocannabinoids and liver disease IV., Endocannabinoid involvement in obesity and hepatic steatosis, Liver Physiol., 2008, 5, G1101–G1114 Search PubMed.
- M. Maceyka and S. Spiegel, Sphingolipid metabolites in inflammatory disease, Nature, 2014, 510, 58–67 CrossRef CAS PubMed.
- R. Monteiro and I. Azevedo, Chronic inflammation in obesity and the metabolic syndrome, Mediat. Inflamm., 2010, 2010, 289645–289651 Search PubMed.
- S. Suárez-García, J. M. del Bas, A. Caimari, R. M. Escorihuela, L. Arola and M. Suárez, Impact of a cafeteria diet and daily physical training on the rat serum metabolome, PLoS One, 2017, 12, e0171970 CrossRef PubMed.
- S. Qi, S. Huang, X. Chen, Q. Huo, N. Xie and J. Xia, Liver tissue metabolic profiling and pathways of non-alcoholic steatohepatitis in rats, Hepatol. Res., 2017, 13, 1484–1493 CrossRef PubMed.
- S. C. Kalhan, L. Guo, J. Edmison, S. Dasarathy, A. J. McCullough and R. W. Hanson, Plasma metabolomic profile in nonalcoholic fatty liver disease, Metabolism, 2011, 60, 404–413 CrossRef CAS PubMed.
- B. C. Ferslew, G. Xie, C. K. Johnston, M. Su, P. W. Stewart and W. Jia, Altered bile acid metabolome in patients with nonalcoholic steatohepatitis, Dig. Dis Sci., 2015, 60, 3318–3328 CrossRef CAS PubMed.
- Y. Suzuki, R. Kaneko, M. Nomura, H. Naito, K. Kitamori and T. Nakajima, Simple and rapid quantitation of bile acids in rat serum and liver by UPLC-MS-MS: effect of high fat diet on glycine conjugates of rat bile acids, Nagoya J. Med. Sci., 2013, 75, 57–71 CAS.
- J. M. Ridlon, J. Alves, P. B. Hylemon and J. S. Bajaj, Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship, Gut Microbes, 2013, 4, 382–387 CrossRef PubMed.
- N. Jiao, S. S. Baker, A. Chapa-Rodriguez, W. Liu, C. A. Nugent and M. Tsompana, Suppressed hepatic bile acid signaling despite elevated production of primary and secondary bile acids in NAFLD, Gut, 2018, 67, 1881–1891 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fo02207d |
| This journal is © The Royal Society of Chemistry 2020 |