
Oleanolic acid enhances tight junctions and ameliorates inflammation in Salmonella typhimurium-induced diarrhea in mice via the TLR4/NF-κB and MAPK pathway†
Na
Dong‡
 ,
Chenyu
Xue‡
,
Lei
Zhang
,
Tingting
Zhang
,
Chensi
Wang
,
Chongpeng
Bi
and
Anshan
Shan
*
,
Chenyu
Xue‡
,
Lei
Zhang
,
Tingting
Zhang
,
Chensi
Wang
,
Chongpeng
Bi
and
Anshan
Shan
*
Institute of Animal Nutrition, Northeast Agricultural University, Harbin, 150030, P. R. China. E-mail: linda729@163.com; Tel: +860451 55190722
First published on 13th November 2019
Abstract
Salmonella typhimurium (S.T) is a common cause of acute, self-limiting food-borne diarrhea with severe intestinal inflammation and intestinal barrier damage. Oleanolic acid (OA), isolated from almost 2000 plant species, has been shown to have anti-inflammatory roles. The purpose of this study was to investigate the potential protective effects of OA on S.T-induced diarrhea and enteritis and to elucidate its anti-inflammatory mechanisms. A total of eighty BALB/c mice (4-week-old) were randomly divided into the control group (no S.T, no OA), the S.T group (S.T only), the S.T + OA group (S.T plus 100 mg kg−1 OA) and the OA group (100 mg kg−1 OA only). Compared with the S.T group, OA administration significantly reduced clinical symptoms and weight loss, and the severity of diarrhea and intestinal structural damage was significantly alleviated, which was confirmed by a decrease in the diarrhea index (DI) and jejunal histological damage. In addition, in the infected jejunum, OA maintained the expression and localization of occludin, claudin-1 and ZO-1 to protect the jejunal barrier, thereby maintaining the integrity of the gut barrier. Finally, OA treatment not only reduced the levels of COX-2 and iNOS but also inhibited the secretion of pro-inflammatory cytokines, such as IL-1β, IL-6 and TNF-α. Furthermore, western blotting results showed that OA treatment significantly inhibited IκB phosphorylation and degradation in intestinal tissues and the nuclear translocation of p65, and OA also decreased the level of TLR4 and the activation of the MAPK pathway. To summarise, OA can maintain the intestinal tight junction barrier and prevent diarrhea caused by S.T. as well as reduce intestinal inflammation through the NF-κB and MAPK signaling pathways.
Introduction
Salmonella typhimurium (S.T) is a Gram-negative bacterium of the family Enterobacteriaceae that is widely found in the intestines of humans and animals. It is widely distributed in nature and can cause serious harm to human and animal health by causing food poisoning and illness in humans and animals.1 It is one of the most common foodborne pathogens in the world. Over the past 100 years, there have been reports that S.T causes approximately 16 million cases of typhoid fever, 1.3 billion cases of gastroenteritis and 3 million deaths each year worldwide.2 The prevention and treatment of salmonellosis is extremely important due to its prevalence and high morbidity as well as the difficulty in applying preventive and control measures. S.T colonizes the small intestine and produces enterotoxins to destroy the intestinal barrier.3 Its infection is characterized by acute enterocolitis with inflammatory diarrhea. These disease symptoms are thought to be caused by an effective and rapid innate immune response to S.T by surgically binding pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharides (LPS) and flagellin.4 Due to the widespread harm caused by S.T in clinical and public health settings, considerable efforts have been made to improve the understanding of the pathogenic determinants of bacteria. Many in vitro studies using cell models are available, but these models do not represent the true natural environment present in the intestines of infected hosts. Many studies have used in vivo mouse models to study host–pathogen interactions and unravel the immune and cellular processes that occur during infection, which greatly expands our understanding of the pathogenesis of S.T-induced enterocolitis.5In recent years, many pathogens have shown resistance due to the abuse of antibiotics and biocides (i.e., disinfectants and preservatives) and heavy metal emissions.6 In addition, the Human Intestinal Microbiome Project indicates that early use of antibiotics not only permanently damages the gut microbiota but also causes many chronic health problems, such as type 2 diabetes, obesity and antibiotic-associated diarrhea.7 Looking for new biotherapeutics is critical to these new long-term chronic health problems. In recent decades, researchers have been studying plant-derived molecules derived from herbs, spices or essential oils that can be used for antibacterial and anti-inflammatory treatments.
Oleanolic acid (3β-hydroxy-12-en-28-oic acid, OA) is a pentacyclic triterpene that is ubiquitous throughout the plant kingdom. OA has been isolated from nearly 2000 plants,8 and the main sources of this compound include plants of the family Oleaceae.9 Many studies have shown that OA has a variety of biological activities, including anti-inflammatory, anti-tumor, anti-diabetic and neuroprotective effects.10,11 However, more information about the anti-enteritis mechanism of OA, especially in the protection of intestinal health, has not been elucidated. To investigate whether OA can prevent intestinal inflammation and diarrhea caused by harmful bacteria, we selected BALB/c mice infected with S.T to establish a mouse diarrhea model, and we further studied the protective effect of OA on the intestinal tract and its molecular mechanism. In this study, we found that OA not only protects the intestinal barrier but also inhibits the overexpression of intestinal pro-inflammatory factors caused by S.T infection. This finding reveals the potential of OA as an intestinal protectant.
Materials and methods
Chemicals and antibodies
Oleanolic acid (OA) (purity ≥98%) and phosphate-buffered solution (PBS) were obtained from Solarbio (Beijing, China); phenylmethanesulfonyl fluoride (PMSF), SDS-PAGE gel preparation kit and enhanced chemiluminescence (ECL) reagent were obtained from Beyotime (Shanghai, China); polyvinylidene fluoride (PVDF) membranes were obtained from Millipore (MA, USA); rabbit monoclonal primary antibodies against TLR4, MyD88, p-NFκB p65, NFκB p65, p-IκB-α, IκB-α, JNK, p-JNK, ERK, p-ERK, p38, and p-p38 were obtained from Cell Signaling Technology (MA, USA); rabbit monoclonal primary antibodies against ZO-1, occludin and claudin-1 were obtained from Abcam (Massachusetts, USA). Mouse monoclonal primary antibody against β-actin, iNOS, and COX-2 was obtained from Beyotime; the HRP-labeled goat anti-rabbit IgG (H + L) and HRP-labeled goat anti-mouse IgG (H + L) secondary antibody were obtained from Beyotime. ELISA kits of IL-1β, IL6 and TNF-α were purchased from Cusabio (Wuhan, China).Bacterial strains and growth conditions
S.T strain 14028 (ATCC) was provided from Northeast Agricultural University. The S.T strain frozen in glycerol medium was removed from the refrigerator. After being completely dissolved, the sterilization ring was sterilized by heating with an external flame of an alcohol lamp. After the sterilization ring was cooled, a small amount of bacterial liquid was inoculated onto a Luria–Bertani (LB) (Sigma-Aldrich) plate. The plates were put in a 37 °C bacterial incubator and cultured overnight. Monoclonal colonies were picked from LB plates and shaken overnight in 10 ml of LB medium at 37 °C and 180 rpm. On the next day, 100 μl of the bacterial solution in fresh LB medium was sampled, and the remaining culture was shaken for an additional 2 hours at 37 °C and 240 rpm until the OD600 was between 0.4 and 0.6 (logarithmic growth phase). The bacteria were then collected by centrifugation, and fresh LB was added.Ethical statement
Four-week-old male BALB/c mice (20 to 22 g) were provided by Liao Ning Chang Sheng Biotechnology Co., Ltd. Throughout the study, the mice were housed in an air-conditioned animal room with an indoor temperature of 23 ± 1 °C and a relative humidity of 40–60%; the mice received light from 8:00 to 20:00 daily. To adapt the mice to the laboratory environment, food and drinking water were freely supplied for 1 week before the start of the experiment. The protocols used in this experiment were approved by the Northeast Agricultural University Institutional Animal Care and Use Committee, and the ethical treatment of animals used in this study was approved by the Animal Welfare Committee protocol (#NEAU-[2013]-9) at Northeast Agricultural University (Harbin, China).Animals and experimental design
BALB/c mice (n = 80) were randomly divided into 4 groups (n = 20 per group) (Fig. 1): (1) the control group, (2) the S. typhimurium group (S.T), (3) the S. typhimurium + OA group (S.T + OA), and (4) the OA group (OA). The experiment was divided into two phases. The first phase was a pretreatment period of 10 days: mice of the OA and S.T + OA groups were administered OA (100 mg kg−1 day−1 p.o. according to the results of a preliminary test); the other groups were treated with a suspension by the same route as that of the OA group. The second phase was a three-day challenge period starting on day 8: mice in the S.T and S.T + OA groups were challenged with S.T (5.19 × 109 cfu mL−1 day−1 p.o.); the other groups were kept and treated with saline by the same route as that of the S.T group. During this time, fecal morphology and mouse body weight were recorded daily. The jejuna of mice in each treatment group were rapidly excised after sacrifice and placed in 4% paraformaldehyde for histological evaluation. The remaining jejunal tissue was rapidly frozen in liquid nitrogen and stored at −80 °C for biochemical assays and western blot analysis. | ||
| Fig. 1 Experimental design. | ||
Fecal examination
All mice were treated every morning after the start of the experiment and then placed in separate cages with the floor covered with filter paper and changed every hour for 6 hours. The amount, morphology and total amount of feces on the filter paper in the 6 hours were recorded. Three indicators were used to define the severity of diarrhea: loose stool incidence rate (LSIR), average loose stool grade (ALSG) and diarrhea index (DI). The LSIR is the ratio of the number of mouse loose stools to the total number of feces. The ALSG describes the extent of loose stools based on the average of the fecal diameter of each mouse on the filter paper. The loose fecal grade is divided into four grades according to the diameter of the feces on the filter paper: grade 1 (<1 cm), grade 2 (1–<2 cm), grade 3 (2–3 cm) and grade 4 (>3 cm). The DI is the result of LSIR multiplied by the ALSG.12H&E staining
The jejunal tissue of the same site was collected and fixed with 10% paraformaldehyde and embedded in paraffin. Sections were then stained with hematoxylin and eosin (H&E) to determine the severity of intestinal inflammation and the extent of mucosal damage and crypt damage. Images were captured at 100× and 200× magnification using a US Moticam 3000 photomicrography imaging system, and intestinal villus height and crypt depth were measured using a Motic Images Advanced 3.2 pathology image analysis system. Histopathological examination was performed by pathologists blinded to the study design.Cytokine assays
The jejunal segment (100 mg) was homogenized with PBS (w/v: 1/9) to evaluate proinflammatory factor levels. The TNF-α, IL-1β and IL-6 levels in jejunal samples were analyzed using an enzyme-linked immunosorbent assay (ELISA) kit (Cusabio, Wuhan, China) according to the manufacturer's instructions.Western blot analysis
The jejunum segment was homogenized. The total protein of jejunal tissue was treated and extracted using RIPA and PMSF. The lysate was treated on ice and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 minutes at 4 °C. The protein concentration was determined using a BCA Protein Assay Kit (Beyotime). Cellular protein extracts were separated by electrophoresis using a 12% SDS-polyacrylamide gel and electroblotted onto a polyvinylidene fluoride (PVDF) membrane. Afterward, the PVDF membrane was blocked with blocking solution (5% nonfat milk powder) for 2 hours at 37 °C. The membrane was incubated with the primary antibody of the protein of interest at 4 °C overnight and then washed 3 times with Tween 20/Tris buffered saline (TBST) for 10 minutes each.13 The membrane was incubated with the HRP-labeled secondary antibody corresponding to the primary antibody for 1 hour at 37 °C and washed with TBST as described above. Finally, samples were developed using a Super Enhanced Chemiluminescence (ECL) Plus Detection System (P0018, Beyotime).
000g for 30 minutes at 4 °C. The protein concentration was determined using a BCA Protein Assay Kit (Beyotime). Cellular protein extracts were separated by electrophoresis using a 12% SDS-polyacrylamide gel and electroblotted onto a polyvinylidene fluoride (PVDF) membrane. Afterward, the PVDF membrane was blocked with blocking solution (5% nonfat milk powder) for 2 hours at 37 °C. The membrane was incubated with the primary antibody of the protein of interest at 4 °C overnight and then washed 3 times with Tween 20/Tris buffered saline (TBST) for 10 minutes each.13 The membrane was incubated with the HRP-labeled secondary antibody corresponding to the primary antibody for 1 hour at 37 °C and washed with TBST as described above. Finally, samples were developed using a Super Enhanced Chemiluminescence (ECL) Plus Detection System (P0018, Beyotime).
Immunohistochemistry
The jejunal tissue wax pieces were sliced and dried. The sections were dewaxed in xylene and rehydrated by gradient alcohol to synthesize distilled water. The sections were placed in an antigen retrieval solution, and after heating for 10 minutes under low heat, they were naturally cooled and washed with PBS. Endogenous peroxidase activity was eliminated with 3% hydrogen peroxide solution for 15 minutes. Nonspecific staining was then blocked by incubation in 10% normal goat serum for 15 minutes. Next, sections were incubated with anti-ZO-1 primary antibody, anti-occludin primary antibody and anti-claudin-1 primary antibody overnight, followed by incubation with HRP-labeled secondary antibody for 30 minutes. The DAB chromogenic reagent was dripped onto the slice until the color was deepened to the correct hue. The sections were counterstained with Mayer's hematoxylin and then dehydrated and sealed. Finally, the sections were observed using the microscope, and pictures were taken at 400× magnification. The quantification of IHC was formalized into a parameter called mean optical density (MOD) detected using ImagePro Plus (Version 6.0, Rockville, MD, USA), which was obtained by dividing the optical density cumulative value of each point on the picture by the area of the target distribution.Statistical analysis
All data are expressed as mean ± standard deviation (SD). Data were analyzed using SPSS 20.0 software and the significance of the data between the groups was analyzed using one-way analysis of variance (ANOVA). Statistically, a P-value less than 0.05 was considered significant.Results
OA attenuated the severity and weight loss of mice with S.T-induced diarrhea
The body weight of the infected mice was significantly lower than that of those in the control group, and OA significantly attenuated this weight loss trend (P < 0.001) (Fig. 2A). In addition, we calculated the diarrhea index (DI) to estimate the degree of hazard caused by S.T based on the loose stool incidence rate (LSIR) and average loose stool grade (ALSG) (Fig. 2B). On day 1, the DI of the S.T induction group was significantly increased compared with that of the other groups (P < 0.01). In the next two days, the degree of diarrhea in the S.T-induced group was more severe than that in the control group (P < 0.001). At the same time, although the DI value of the OA-treated group was slightly increased, it remained at a low level, and there was no significant difference from the control group. The results indicate that OA has an antidiarrheal effect. | ||
| Fig. 2 Effect of OA on the development of the S.T-induced diarrhea model. (A) Mice were administered S.T orally for 3 days with or without OA pretreatment. Changes in body weights were recorded daily (n = 18). (B) Changes in DAI levels were evaluated everyday throughout the 9 days of the experimental period. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. the vehicle-treated control group; ##P < 0.01 and ###P < 0.001 vs. the S. Typhimurium-treated group. | ||
OA reduces histological changes in S.T-induced diarrhea mice
H&E stained jejunal tissue sections from control mice showed normal structure without histological changes. The S.T group showed disrupted jejunal structures, lamina propria edema, and intestinal epithelial capillary congestion, but no significant inflammatory cell infiltration was observed. The OA pretreatment significantly corrected structural deformation, relieved capillary congestion, and reduced edema (Fig. 3A). Compared with the control group, the height of jejunal villi and thickness of mucosa in the S.T group were significantly lower than those in the normal control group; however, the depth of the crypt significantly increased, and significant crypt hyperplasia occurred. By OA pretreatment, the villus height, crypt depth and mucosal thickness of the jejunum were restored to the control level (Fig. 3B, C and D).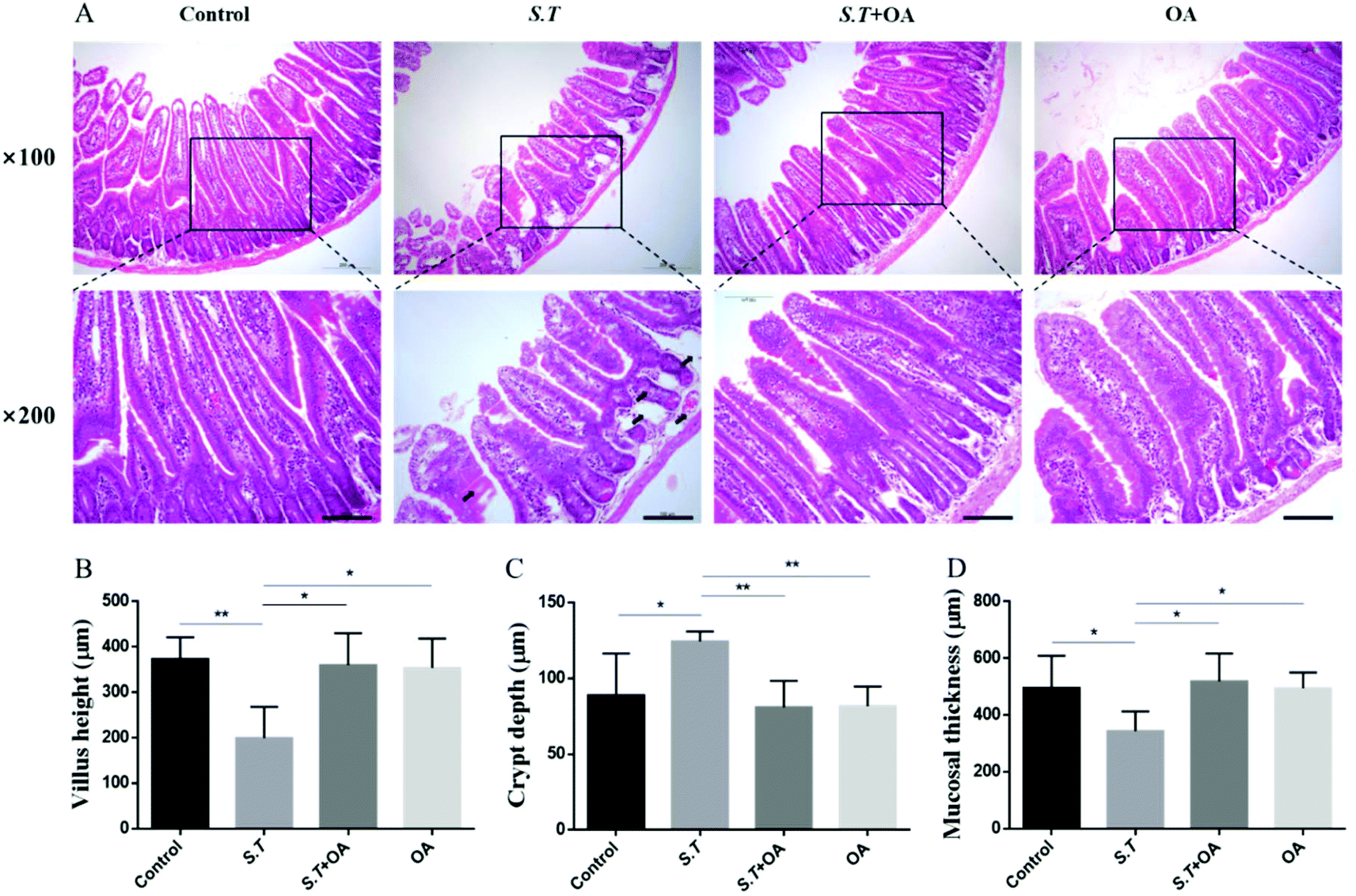 | ||
| Fig. 3 Effect of OA on the progression of S.T-induced damage. (A) Histological analysis of S.T-induced damage was performed by H&E staining. Original magnification, 100× and 200×. The bar is 100 μm. (B) Villus height. (C) Crypt depth. (D) Mucosal thickness. The values are the mean ± SD (n = 6) of three independent experiments. *P < 0.05 and **P < 0.01. | ||
OA enhances jejunal tight junction protein expression in S.T-administered mice
Tight junction (TJ) proteins play a key role in maintaining intestinal barrier integrity. Western blot analysis demonstrated that the jejunal expression of ZO-1, occludin, and claudin-1 decreased with the administration of S.T (Fig. 4). OA, however, attenuated the decreased expression of some of the TJ proteins that were detected. The protein expression levels of occludin, claudin-1 and ZO-1 in the S.T + OA group were higher than those in the S.T group. The expression and localization of occludin, claudin-1 and ZO-1 in the jejunum were observed by immunohistochemistry (Fig. 5). In control mice, occludin, claudin-1 and ZO-1 were detected throughout the basal layer of the intestinal mucosa and the outside of the intestinal villi. In the S.T challenge group, the expression of these TJ proteins was low or absent, indicating that the invasion of S.T severely impaired the localization and expression of these TJ proteins. However, specimens of the S.T + OA group showed localization and relatively complete expression of these TJ proteins. The IHC analysis results were quantified as mean optical density, and the results were consistent with the trend of western blotting results, indicating that OA effectively attenuated the decrease of TJ protein expression caused by S.T.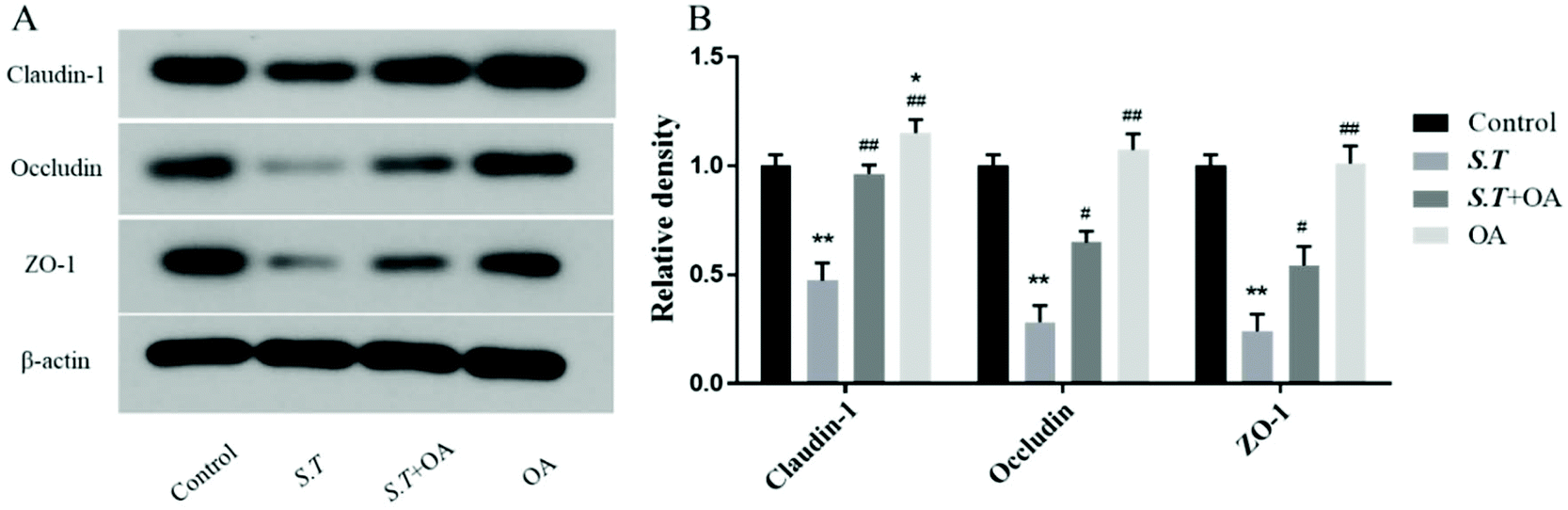 | ||
| Fig. 4 Effects of OA on the expression levels of the small intestine tight junction proteins ZO-1, claudin-1 and occludin in S.T-infected mice. (A) Western blot analysis of tight junction proteins from mice jejunum tissues. (B) Quantification of the protein levels; protein density values were normalized to the values from the control group. Data are presented as the mean ± SD, (n = 6). *P < 0.05 and **P < 0.01 vs. the control group; #P < 0.05 and ##P < 0.01 vs. the S.T-infected group. | ||
 | ||
| Fig. 5 (A) Immunohistochemistry for ZO-1, claudin-1 and occludin in each group (400×). Representative images of three independent tissue preparations are shown (n = 6). The bar is 50 μm. (B) Mean optical density (MOD) of IHC expression results. *P < 0.05 and **P < 0.01. | ||
OA inhibits S.T-induced proinflammatory cytokine production and the expression of COX-2 and iNOS
It has been reported that the overexpression of pro-inflammatory cytokines is an important part of the colonization mechanism of S.T.14 Therefore, we investigated the inhibitory effect of OA on the production of pro-inflammatory cytokines in S.T-induced jejunal tissue. As shown in Fig. 6A–C, S.T administration was found to induce TNF-α, IL-1β and IL-6 levels significantly, and OA reduced these increases. To further investigate the molecular mechanism by which OA inhibits diarrhea, we investigated the protein expression levels of pro-inflammatory enzymes, such as COX-2 and iNOS, in S.T-induced jejunal tissue from mice. As shown in Fig. 6D, S.T significantly induced the expression of COX-2 and iNOS in the mouse jejuna, and the expression of these proteins was significantly reduced by OA in mice infected with S.T.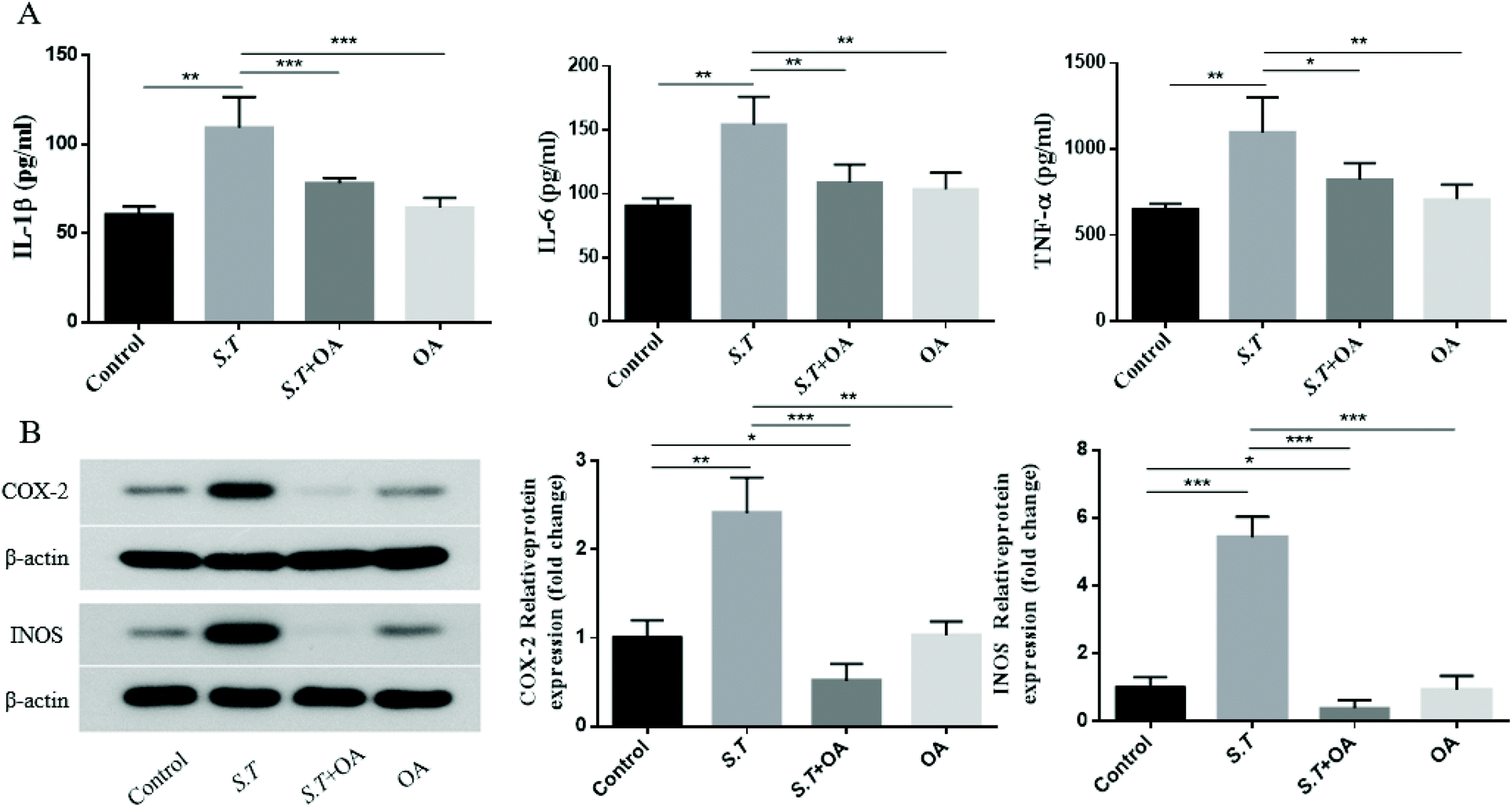 | ||
| Fig. 6 Effect of OA on the production of pro-inflammatory cytokines and the protein expression of iNOS and COX-2. (A) The production of pro-inflammatory cytokine levels was determined by EIA kits. (B) The expression levels of iNOS and COX-2 were determined by western blot analysis using specific antibodies. β-Actin was used as a loading control. The values are the mean ± SD (n = 6) of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
OA attenuates the activation of the NF-κB pathway and the expression of key proteins in the NF-κB pathway
TLR4 belongs to the family of innate immune receptors and is a conductive receptor for bacterial lipopolysaccharides in animal cells. TLR4 activates the downstream inflammatory pathway via the MYD88-dependent pathway.15 For example, TLR4 activates downstream NF-κB, which has a strong impact on mucosal inflammation and is an essential regulator in the immune environment. The key to NF-κB activation is the hydrolysis of inhibitory protein IκBs and the release of NF-κB dimers.16 Thus, we examined the effects of OA on the expression of TLR4 and MYD88 and the nuclear translocation of the p65 NF-κB subunit in jejunal tissues by western blot analysis. As shown in Fig. 7, the expression levels of TLR4 and MyD88 in the jejunum of mice infected with S.T significantly increased, and OA could effectively alleviate this condition. On the other hand, OA significantly inhibited the S.T-induced nuclear translocation of p65 in jejunal tissues. Furthermore, we determined that OA inhibits S.T-induced phosphorylation and degradation of IκB in mouse jejunal tissues. Taken together, these results indicate that OA inhibits the expression and activation of key proteins of the TLR4/NF-κB pathway, thereby inhibiting S.T-induced jejunal inflammation. | ||
| Fig. 7 Effect of OA on the nuclear translocation of NFκB and the protein expression of NFκB-dependent proteins in S.T-induced mouse jejunum. The values are the mean ± SD (n = 6) of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. the control group. #P < 0.05, ##P < 0.01 and ###P < 0.01 vs. the S.T group. | ||
OA inhibits MAPK activation in jejunal tissues
MAPK is an important method of regulating inflammation. Its activation has been shown to be associated with the development of intestinal inflammation. Therefore, we examined the phosphorylation of three important proteins in the MAPK pathway to confirm the activation of MAPK. As shown in Fig. 8, the phosphorylation levels of p38, ERK and JNK were quite significant in the S.T group. At the same time, as expected, oral OA significantly inhibited the activation of these three key proteins in the MAPK pathway. | ||
| Fig. 8 OA inhibits S.T-induced activation of key proteins of the MAPK pathway (p38, ERK and JNK). Data are presented as the means ± SD (n = 6). *P < 0.05 and **P < 0.01 vs. the control group; #P < 0.05 and ##P < 0.01 vs. the S.T group. | ||
Discussion
Infectious diarrhea is one of the most common diseases in the world; according to reports from the World Health Organization,17 infectious diarrhea is one of the five leading causes of death. S.T contamination of food sources is the most important cause of pathogenic diarrhea. Diarrhea, acute intestinal inflammation, and the presence of neutrophils in stool samples are important features of S.T infection.18 After entering the gastrointestinal tract, S.T invades the intestinal tract of the animal through the type 3 protein secretion system (T3SS), destroys the intestinal barrier and causes inflammation of the intestinal mucosa and then spreads in the host, causing severe intestinal inflammation. In addition, S.T faces dense microbiota in the gut. However, due to its unique virulence strategy, S.T can overcome the colonization resistance provided by the gut microbiota by inducing intestinal inflammation, and it can survive in a highly inflamed intestinal environment.14,19 Currently, clinical ST-induced diarrhea is typically treated with fluoroquinolones, such as ciprofloxacin, ampicillin, azithromycin or third-generation cephalosporins,20 but with the increasing resistance of S.T, it is necessary to find natural products that can be used long term without drug resistance and with good efficacy as a means of intestinal immune enhancement.OA is a pentacyclic triterpene compound that is ubiquitous throughout the plant kingdom. In nature, it exists in a variety of plants in the form of a free acid and glycosides. OA and its derivatives have several promising pharmacological activities, such as anti-inflammatory, antibacterial and antiviral activities.21 Based on various pharmacological effects of OA, we researched the anti-inflammatory roles of OA in the animal model of S.T-induced diarrhea. In the initial test, S.T (5.19 × 109 cfu mL−1 day−1) was able to cause mental fatigue and diarrhea in mice within 3 days and reduce the survival rate of mice within 5 days. Medium and high doses of OA (100 mg kg−1 day−1 and 150 mg kg−1 day−1, respectively) can significantly prevent diarrhea and death caused by S.T, but a low dose of OA (50 mg kg−1 day−1) had no impact. Based on these effects, OA (100 mg kg−1 day−1 p.o.) was used in our animal model to attenuate the development of S.T-induced diarrhea to evaluate the protective effect of OA on mice infected with S.T. We first examined the mice's body weights, diarrhea indexes and intestinal morphologies. From the initial results, OA effectively alleviated the weight loss and diarrhea index increase caused by S.T (Fig. 2). The small intestine is necessary for nutrient absorption, and its morphological integrity ensures its special function for water and other ions.22,23 Our results indicate that S.T infection can destroy the morphology of small intestinal mucosa, resulting in shortening of intestinal villi and increased crypt depth. Infected mice treated with OA showed increased villus height, decreased crypt depth and inhibited edema compared to infected mice, indicating that OA protects the mechanical barrier in the gut.
The structural basis of the intestinal mucosal mechanical barrier is the connection between intact intestinal epithelial cells and adjacent intestinal epithelial cells, which together form a selective barrier to the gut and regulate transepithelial cell transport of water and solutes. The junctions between epithelial cells are tight junctions (TJs), adhesion junctions, and gap junctions; tight junctions are the primary means of attachment between intestinal epithelial cells. TJs are a dynamic structure with a complex architecture. They consist of transmembrane barrier proteins (e.g., claudin, occludin and triterpenoids) and cytoplasmic scaffold proteins (e.g., ZO family proteins, cingulin and afadin).24 During infection, S.T will cross the intestinal epithelial barrier and rupture the tight junction of epithelial cells, which alters the basal permeability of the intestinal barrier.25 It has been reported that S.T infection of the intestinal epithelial cell model for more than 2 hours results in approximately 80% transepithelial electrical resistance loss and is capable of the specific dephosphorylation of occludin and degradation of ZO-1.26 In summary, the ability of S.T. to modulate the molecular composition of TJs helps it overcome the intestinal barrier and enter the circulation. Therefore, the regulation of TJ proteins is important for the treatment of diarrhea caused by S.T. Thus, we also analyzed the expression and localization of ZO-1, claudin-1 and occludin by western blot and immunohistochemical analysis. The results showed that OA treatment can restore S.T-induced TJ disruption, which indicates that OA may play an important role in maintaining TJ protein expression, thereby balancing barrier integrity and reducing intestinal damage.
Proinflammatory cytokines are key biomarkers released by intestinal immune cells after overactivation in the event of inflammation. Studies have shown that selective silencing or inhibition of the release of these inflammatory factors can reduce neutrophil migration and improve the development of intestinal inflammation.27 The inducible enzymes COX-2 and iNOS are known to be inflammatory proteins mainly expressed at the site of inflammation. COX-2 and iNOS are the main causes of increased PG levels (mainly PGE2) and NO production during inflammation. Excessive PGE2 and NO increase vasodilation and permeability, leading to intestinal mucosal congestion and edema.28 Under normal conditions, COX-2 will not be expressed in most cells, but it will be elevated under inflammatory conditions, thereby aggravating the stress response of inflammation.29 COX-2 can cause epithelial cell proliferation. It can also cause the secretion of Cl−1 in epithelial cells, which leads to the occurrence of diarrhea. Unlike COX-2, iNOS can catalyze the production of NO by L-arginine in vivo. The O2-interaction between NO and lamina propria produces peroxynitrite (ONOO−), causing nitrosylation of cellular nucleic acids, disrupting DNA structure, forming nitrosyl iron complexes, inducing protein kinase nitration, and ultimately generating damage to epithelial cells.30 Furthermore, in the high concentration state, NO induces the production of pro-inflammatory cytokines, such as TNF-α and IL-1β.31 The production of IL-1 and TNF-α can activate iNOS and promote the production of more NO by the body, thereby forming a positive feedback loop. Thus, the secretion of cytokines and the expression of NO itself are sustained, and the inflammatory reaction is more durable and more intense. A variety of detection strategies have demonstrated increased iNOS expression and NO production in human inflammatory bowel disease tissues. There is also evidence that the level of NO in iNOS is associated with disease activity in ulcerative colitis.32
The anti-inflammatory effects of triterpenoids have been widely reported. For example, ursolic acid (an isomer of oleanolic acid) has been shown to attenuate iNOS, COX-2 and TNF-α expression by downregulating NF-κB activation in an IFN-γ/LPS-stimulated mouse peritoneal macrophage model.33 Furthermore, in human umbilical vein endothelial cells (HUVEC), OA has been shown to inhibit the liposome-mediated release of high mobility group box 1 (HMGB1) and cell adhesion molecule (CAM) expression. It also has anti-inflammatory properties.34 HMGB1 is a protein that upregulates pro-inflammatory cytokines in several inflammatory diseases.35 Similarly, OA has been reported to attenuate LPS-induced proinflammatory responses by inhibiting NF-κB activation, thereby attenuating TNF-α expression in in vivo and in vitro studies.36 To verify the anti-inflammatory effect of OA, we first tested the expression of intestinal inflammatory factors in the mouse model of diarrhea. Consistent with our S.T-induced diarrhea experiment, S.T challenge induced the overexpression of intestinal inflammatory cytokines, while OA significantly inhibited the production of proinflammatory cytokines and the expression of COX-2 and iNOS in jejunal tissues, suggesting that the inhibitory effect of OA on enteritis was related to these proinflammatory mediators.
At the molecular level, the recognition of LPS, the major virulence component of S.T, is mainly mediated by TLR4.37 The TLR signal transduction pathway is derived from the intracellular TIR domain, which is conserved across all TLRs. There is early evidence that TLRs achieve dimerization and initiate signaling cascades by recognizing homologous ligands and, through a TIR-containing linker such as MyD88, activate a series of inflammatory pathways, such as the downstream NF-κB pathway.38 Natural plant-derived molecules have been found to exert anti-inflammatory effects by targeting the TLR4 activation pathway. Many recent studies have shown that the anti-inflammatory effects of natural plant compounds may occur through the MyD88 signaling pathway to inhibit the TLR4 pathway. For example, curcumin, 6-gingerol, 6-shogaol, 1-dehydro-10-glycanedionone, luteolin, quercetin, resveratrol, phenethyl caffeate, xanthohumol, genistein, berberine and sulforaphane are MyD88-dependent ways to reduce the activation of the TLR4 signaling pathway.39 Among them, it has been reported that in hepg2 cells, similar to the siRNA effect, celastrol (another pentacyclic triterpenoid) treatment significantly abolished the free fatty acid-induced TLR4-mediated signal cascade.40 Furthermore, cotreatment with TLR4 siRNA and celastrol was found to further attenuate the expression level of the downstream medium38 compared to each treatment alone. Similarly, in this study, OA inhibited S.T-induced expression of jejunal TLR and Myd88, suggesting that OA can effectively block the binding of S.T cell wall components to intestinal TLR, thereby preventing the activation of downstream inflammation-related pathways.
NF-κB and MAPK are two important signaling pathways that regulate inflammation in the downstream pathway of TLR4. Once activated, TLR4 induces and secretes large amounts of pro-inflammatory cytokines.41 There are five proteins in the mammalian NF-κB family, Rel A (p65), Rel B, c-Rel, p50/p105, and p52/p100; this family of proteins shares a Rel homeodomain (RHD) at the N-terminus, which results in it being classified as a family of NF-κB/Rel proteins.42 RHD is essential for dimerization and binding to κB homologous DNA. The κB inhibitor (inhibitor of κB and IκB) regulates NF-κB activity, and the IκB family consists of IκBα, β, ε, BCL-3, p100, and p105. The IκB protein masks the nuclear localization signal (NLS) of the classical NF-κB protein and retains it in the cytoplasm in an inactive state. The IκB kinase (IKK) promotes protein degradation by phosphorylating IκB, thus allowing classical NF-κB dimers to enter the nucleus from the cytoplasm and regulate target gene expression.43 It has been found that the effect of OA on NF-κB, OA treatment in vivo can inhibit DSS-induced activation of NF-κB and MAPK to improve colitis. In addition, OA inhibits the activation of NF-κB and the expression of pro-inflammatory cytokines in LPS-stimulated peritoneal macrophages in vitro.44 In this study, OA inhibits the p65 phosphorylation induced by S.T. and prevents degradation of the IκB-α protein, which potently inhibits NF-κB activation (Fig. 7). Thus, the above data indicate that OA can inhibit the TLR/MyD88/NF-κB pathway to regulate S.T-induced expression of jejunal inflammatory factors.
Apart from the NF-κB signaling pathway, signals that are generated by TLR are also transduced through the MAPK pathway to recruit pro-inflammatory cytokines and costimulatory molecules promoting inflammatory responses.45 MAPKs include ERK, p-38 and JNK, and they regulate various cellular response functions (cell proliferation, differentiation, development, transformation and apoptosis).46 After MAP kinase's activation, transcription factors in the cytoplasm or nucleus are in turn activated, triggering the expression of target genes, leading to biological responses, including pro-inflammatory mediators’ expression.47 Reports on the prevention of inflammation by plant extracts through MAPK have been reported in recent years. For example, oleuropein inhibits the IL-1β-induced expression of inflammatory mediators, such as NO, PGE2, COX-2 and iNOS, by inhibiting MAPK activation in human osteoarthritic chondrocytes;48 the three major compounds of red ginseng oil, linoleic acid, β-sitosterol, and stigmasterol, attenuate Aβ25–35-induced neuronal apoptosis and inflammation by regulating the MAPK/NF-κB pathway.49 We investigated the effects of OA on phosphorylation of ERK, JNK and P38 MAPK in jejunal tissues of infected mice; S.T induced activation of ERK, JNK and p38 MAPK. Importantly, OA pretreatment significantly inhibits the phosphorylation of P38, ERK and JNK, thus inhibiting the production of iNOS, COX-2 and pro-inflammatory cytokines, thereby exerting anti-inflammatory effects. Based on the detection results of key proteins in the TLR4/NF-κB and MAPK pathway, we plotted the mechanism of OA protecting intestinal immune barrier function, as shown in Fig. 9. By inhibiting the TLR4/myd88-dependent pathway, OA downregulated the activation of the NF-κB and MAPK pathways, thereby inhibiting the release of inflammatory factors. In addition, OA protects the expression and localization of tight junction proteins, such as ZO-1, which protects the intestinal barrier function.
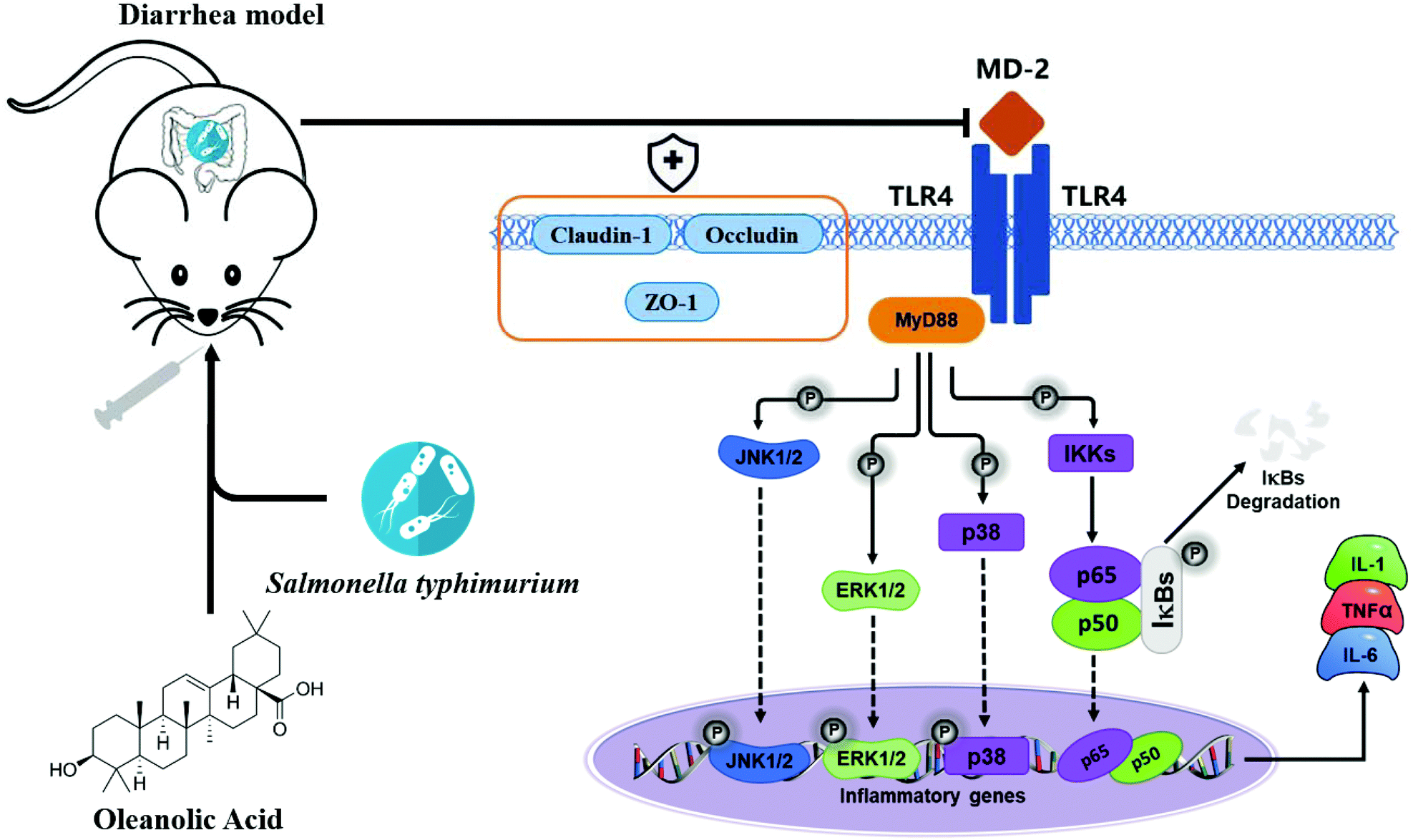 | ||
| Fig. 9 The diagram shows the mechanism by which OA attenuates intestinal inflammation and intestinal tight junction barrier damage through the TLR4/NF-κB and MAPK pathway. | ||
Conclusion
In conclusion, we established a mouse diarrhea model caused by Salmonella typhimurium and evaluated the protective effect of OA on the intestinal immunity and barrier in mice. The results of the in vivo tests demonstrate that OA increase the integrity of the intestinal tight junction barrier by maintaining the expression and localization of tight junction proteins in the gut. In addition, OA can effectively alleviate the development of intestinal inflammation. The molecular mechanism of its action occurs through the inhibition of TLR4 expression, inactivation of NF-κB, downregulation of NF-κB-associated pro-survival proteins, and inhibition of phosphorylation of key proteins in the MAPK pathway. Therefore, the results of this study confirm the ability of OA in increasing the tight junction integrity of the intestine, and clarify the anti-inflammatory mechanism of OA, providing clues for the role of OA in protecting intestinal health.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 31972580 and 31501914), the Natural Science Foundation of Heilongjiang Province (LH2019C023 and TD2019C001) and the China Agriculture Research System (CARS-35).References
- K. M. Lee, M. Runyon, T. J. Herrman, R. Phillips and J. Hsieh, Food Control, 2015, 47, 264–276 CrossRef.
- D. L. LaRock, A. Chaudhary and S. I. Miller, Nat. Rev. Microbiol., 2015, 13, 191 CrossRef CAS.
- P. Broz, M. B. Ohlson and D. M. Monack, in Gut Microbes, 2012 Search PubMed.
- K. Hallstrom and B. A. Mccormick, Front. Microbiol., 2011, 2, 88 Search PubMed.
- R. L. Santos, Front. Immunol., 2014, 5, 252 Search PubMed.
- A. C. Singer, H. Shaw, V. Rhodes and A. Hart, Front. Microbiol., 2016, 7, 1728 Search PubMed.
- J. E. Belizário and M. Napolitano, Front. Microbiol., 2015, 6, 1050 Search PubMed.
- E. O. Fukushima, H. Seki, K. Ohyama, E. Ono, N. Umemoto, M. Mizutani, K. Saito and T. Muranaka, Plant Cell Physiol., 2011, 52, 2050–2061 CrossRef CAS.
- J. Pollier and A. Goossens, Phytochemistry, 2012, 77, 10–15 CrossRef CAS.
- L. Caltana, D. Rutolo, M. L. Nieto and A. Brusco, Neurochem. Int., 2014, 79, 79–87 CrossRef CAS.
- S.-R. Yoo, S.-J. Jeong, N.-R. Lee, H.-K. Shin and C.-S. Seo, Pharmacogn. Mag., 2017, 13, 339 CAS.
- X. Han, Y. Pang, S. Liu, Z. Tan, S. Tang, C. Zhou, M. Wang and W. Xiao, Trop. J. Pharm. Res., 2014, 13, 1643–1651 CrossRef.
- J. Guo, H. Xing, M. Chen, W. Wang, H. Zhang and S. Xu, Sci. Total Environ., 2019, 663, 380–386 CrossRef CAS.
- B. Stecher, R. Robbiani, A. W. Walker, A. M. Westendorf, M. Barthel, M. Kremer, S. Chaffron, A. J. Macpherson, J. Buer and J. Parkhill, PLoS Biol., 2007, 5, 2177–2189 CrossRef CAS.
- C. R. H. Raetz and C. Whitfield, Annu. Rev. Biochem., 2002, 71, 635–700 CrossRef CAS.
- N. Kanarek, N. London, O. Schueler-Furman and Y. Ben-Neriah, Mol. Ther., 2010, 2, a000166 Search PubMed.
- World Health Organization, Fact Sheet, 2013 Search PubMed.
- M. Mellado-Ferreiro, V. Jarne-Betrán, M. Arteaga-Mazuelas and M. L. Abínzano-Guillén, in Anales del sistema sanitario de Navarra, 2016, vol. 39, pp. 139–141 Search PubMed.
- S. Y. Wotzka, B. D. Nguyen and W.-D. Hardt, Cell Host Microbe, 2017, 21, 443–454 CrossRef CAS.
- C. Lübbert and C. Lübbert, Expert Rev. Anti-Infect. Ther., 2016, 14, 193–206 CrossRef PubMed.
- S. Xiao, Z. Tian, Y. Wang, L. Si, L. Zhang and D. Zhou, Med. Res. Rev., 2018, 38, 951–976 CrossRef.
- A. Watson, C. Lipina, H. J. McArdle, P. M. Taylor and H. S. Hundal, Cell. Signalling, 2016, 28, 412–424 CrossRef CAS.
- S. Wang, X. Li, W. Wang, H. Zhang and S. Xu, Sci. Total Environ., 2019, 696, 134035 CrossRef CAS.
- K. Dokladny, M. N. Zuhl and P. L. Moseley, J. Appl. Physiol., 2016, 120, 692–701 CrossRef CAS.
- B. M. Schultz, C. A. Paduro, G. A. Salazar, F. J. Salazar-Echegarai, V. P. Sebastián, C. A. Riedel, A. M. Kalergis, M. Alvarez-Lobos and S. M. Bueno, Front. Immunol., 2017, 8, 191 Search PubMed.
- H. Köhler, T. Sakaguchi, B. P. Hurley, B. A. Kase, B. J. Kase, H. C. Reinecker and B. A. Mccormick, Am. J. Physiol.: Gastrointest. Liver Physiol., 2007, 293, G178 CrossRef PubMed.
- M. A. McGuckin, R. Eri, L. A. Simms, T. H. J. Florin and G. Radford-Smith, Inflamm. Bowel Dis., 2008, 15, 100–113 CrossRef.
- J. B. Kirsner, World J. Gastroenterol., 2001, 7, 175 CrossRef CAS.
- I. I. Singer, D. W. Kawka, S. Schloemann, T. Tessner, T. Riehl and W. F. Stenson, Gastroenterology, 1998, 115, 297–306 CrossRef CAS.
- Y.-Z. Wang, Y.-Q. Cao, J.-N. Wu, M. Chen and X.-Y. Cha, World J. Gastroenterol., 2005, 11, 46 CrossRef CAS.
- J. L. Wallace and M. J. S. Miller, Gastroenterology, 2000, 119, 512–520 CrossRef CAS.
- R. K. Cross and K. T. Wilson, Inflamm. Bowel Dis., 2003, 9, 179–189 CrossRef.
- D. S. Cha, J. S. Eun and H. Jeon, J. Ethnopharmacol., 2011, 134, 305–312 CrossRef.
- E.-J. Yang, W. Lee, S.-K. Ku, K.-S. Song and J.-S. Bae, Food Chem. Toxicol., 2012, 50, 1288–1294 CrossRef CAS.
- U. Andersson and K. J. Tracey, Annu. Rev. Immunol., 2011, 29, 139–162 CrossRef CAS.
- W. Lee, E.-J. Yang, S.-K. Ku, K.-S. Song and J.-S. Bae, Inflammation, 2013, 36, 94–102 CrossRef CAS.
- M. C. Royle, S. Tötemeyer, L. C. Alldridge, D. J. Maskell and C. E. Bryant, J. Immunol., 2003, 170, 5445–5454 CrossRef CAS.
- S. Mitchell, J. Vargas and A. Hoffmann, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2016, 8, 227–241 CAS.
- C.-Y. Chen, C.-L. Kao and C.-M. Liu, Int. J. Mol. Sci., 2018, 19, 2729 CrossRef.
- L. Han, B. Sun, C. Li, Y. Xie and L. Chen, Int. J. Mol. Med., 2018, 42, 2053–2061 CAS.
- J. Lai, Y. Liu, C. Liu, M. Qi, R. Liu, X. Zhu, Q. Zhou, Y. Chen, A. Guo and C. Hu, Inflammation, 2017, 40, 1–12 CrossRef CAS.
- S. C. Sun, Nat. Rev. Immunol., 2017, 17, 545 CrossRef CAS.
- B. Hoesel and J. A. Schmid, Mol. Cancer, 2013, 12, 86 CrossRef CAS.
- G.-D. Kang, S. Lim and D.-H. Kim, Int. Immunopharmacol., 2015, 29, 393–400 CrossRef CAS.
- M. K. Vidya, V. G. Kumar, V. Sejian, M. Bagath, G. Krishnan and R. Bhatta, Int. Rev. Immunol., 2018, 37, 20–36 CrossRef CAS.
- W. Zhang and H. T. Liu, Cell Res., 2002, 12, 9–18 CrossRef.
- B. Kaminska, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1754, 253–262 CrossRef CAS.
- Z. Feng, X. Li, J. Lin, W. Zheng, Z. Hu, J. Xuan, W. Ni and X. Pan, Food Funct., 2017, 8, 3737–3744 RSC.
- S. Lee, K. Youn and M. Jun, Food Funct., 2018, 9, 4122–4134 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fo01718f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |