
Coenzyme Q10 attenuates platelet integrin αIIbβ3 signaling and platelet hyper-reactivity in ApoE-deficient mice†
Fuli
Ya
 abc,
Xiaohong Ruby
Xu
d,
Zezhong
Tian
bce,
Reid C.
Gallant
d,
Fenglin
Song
f,
Yilin
Shi
abc,
Yinfan
Wu
abc,
Jianbo
Wan
g,
Yimin
Zhao
bce,
Reheman
Adili
h,
Wenhua
Ling
abc,
Heyu
Ni
dijkl and
Yan
Yang
*bce
abc,
Xiaohong Ruby
Xu
d,
Zezhong
Tian
bce,
Reid C.
Gallant
d,
Fenglin
Song
f,
Yilin
Shi
abc,
Yinfan
Wu
abc,
Jianbo
Wan
g,
Yimin
Zhao
bce,
Reheman
Adili
h,
Wenhua
Ling
abc,
Heyu
Ni
dijkl and
Yan
Yang
*bce
aDepartment of Nutrition, School of Public Health, Sun Yat-sen University (Northern Campus), Guangzhou, Guangdong Province 510080, China
bGuangdong Provincial Key Laboratory for Food, Nutrition and Health, Guangzhou, Guangdong Province 510080, China. E-mail: yangyan3@mail.sysu.edu.cn; Fax: +86-20-87330687; Tel: +86-20-87330687
cGuangdong Province Engineering Laboratory for Nutrition Translation, Guangzhou, Guangdong Province 510080, China
dDepartment of Laboratory Medicine, Keenan Research Centre for Biomedical Science, St Michael's Hospital, Unity Health Toronto, Toronto, Ontario, Canada M5B 1W8
eSchool of Public Health (Shenzhen), Sun Yat-sen University, Guangzhou, Guangdong Province 510006, China
fSchool of Food Science, Guangdong Pharmaceutical University, Guangzhou, Guangdong Province 510006, China
gState Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Taipa, Macau, 999078, China
hDepartment of Pharmacology, University of Michigan, Ann Arbor, MI 48109, USA
iCanadian Blood Services Centre for Innovation, Toronto, Ontario, Canada M5G 2 M1
jDepartment of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada M5S 1A1
kDepartment of Medicine, University of Toronto, Toronto, ON, Canada M5S 1A1
lDepartment of Physiology, University of Toronto, Toronto, ON, Canada M5S 1A1
First published on 26th October 2019
Abstract
Coenzyme Q10 (CoQ10) exists in a wide variety of foods and has promising cardiovascular benefits. However, its effects on platelets and integrin αIIbβ3 signaling during atherosclerosis have not been previously explored. Here, apolipoprotein E-deficient (ApoE−/−) mice were fed a standard diet, high-fat diet (HFD) or CoQ10-supplemented HFD for 12 weeks. We found that CoQ10 supplementation in ApoE−/− mice significantly alleviated formation of HFD-induced atherosclerotic lesions, and attenuated platelet hyper-aggregation and granule secretion, including CD62P, CD63 and CD40 ligand (CD40L) expression and platelet factor-4, β-thromboglobulin and activation normal T cell expressed and secreted (CCL5) release. CoQ10 supplementation decreased soluble fibrinogen and JON/A binding to αIIbβ3 on activated platelets, indicating that αIIbβ3-mediated inside-out signaling was attenuated. Additionally, CoQ10 down-regulated platelet αIIbβ3 outside-in signaling including decreasing phosphorylation of the β3 intracellular tail, cellular and sarcoma tyrosine-protein kinase (c-Src), and myosin light chain (MLC), and consistently attenuating platelet spreading and clot retraction. Importantly, platelet–monocyte aggregation that was primarily mediated by αIIbβ3 and can be blocked using an αIIbβ3-specific antagonist tirofiban was also markedly diminished by CoQ10. Thus, CoQ10 supplementation attenuates platelet hyper-reactivity via down-regulating both αIIbβ3 inside-out and outside-in signaling, which may play important preventive roles in atherothrombosis.
1. Introduction
Diet or dietary composition intervention is one of the key approaches to prevent cardiovascular diseases (CVDs).1–5 Coenzyme Q10 (CoQ10) is an effective natural antioxidant that is abundant in various foods as well as in human plasma.6,7 It has been well established that dietary CoQ10 supplements decrease the risk of CVDs.8 More recently, we also found that CoQ10 supplementation for 24 weeks improved lipid and glycemic profiles in dyslipidemic individuals.9 We and others previously demonstrated that CoQ10 inhibited the progress of atherosclerosis.10,11 Improvement of endothelial cell function and promotion of macrophage reverse cholesterol transport might partially contribute to the atheroprotective effects of CoQ10.10,12 Very recently, we have shown that CoQ10 up-regulates the platelet cAMP/PKA pathway and attenuates integrin αIIbβ3 signaling in vitro and in dyslipidemic human patients, as well as attenuates thrombus growth in a FeCl3-induced murine thrombosis model.13 However, the role of CoQ10 in platelet function, which is important for the initiation and development of atherosclerosis,14,15 particularly under proatherosclerotic conditions, has never been explored.Platelets are anucleate blood cells that not only play key roles in thrombosis and haemostasis, but also contribute to inflammation and atherosclerosis.15–20 For example, platelets can adhere to dysfunctional endothelium, release pro-inflammatory compounds, recruit circulating leukocytes, and thus promote the formation of atherosclerotic lesions.14,15,21 Therefore, enhanced platelet activation and aggregation contribute to the process of atherothrombosis, the major cause of CVDs such as heart attack and stroke, the leading cause of morbidity and mortality worldwide.22–25 Platelet integrin αIIbβ3 is the dominant platelet surface receptor; it mediates firm platelet adhesion and is indispensable for platelet aggregation.26–28 Platelet integrin αIIbβ3-mediated inside-out and outside-in signaling plays critical roles in the process of atherothrombosis.29,30 The integrin αIIbβ3 inside-out signalling process changes the conformation of the αIIbβ3 extracellular ligand binding domain from a low-affinity state to a high-affinity state for ligands such as fibrinogen (Fg), von Willebrand factor (VWF) and other ligands.16,31–34 Ligand binding to activated integrin αIIbβ3 subsequently initiates outside-in signaling, which induces a range of cellular events driving essential platelet processes including platelet spreading, platelet aggregation, granule secretion, and clot retraction.27,31,35
Recent evidence supports an important role of platelet integrin αIIbβ3-mediated signaling in the progression of atherogenesis via the initiation and modulation of inflammatory responses.36,37 ApoE−/− mice lacking the αIIb subunit of integrin αIIbβ3 showed a profound reduction in the burden of atherosclerotic lesions.37 Integrin αIIbβ3 is essential for the firm adhesion of platelets to dysfunctional endothelial cells and actively contributes to the recruitment of monocytes onto the intima during atherosclerotic plaque formation.37 Moreover, loss of platelet junctional adhesion molecule A (JAMA), a molecule that is associated with integrin αIIbβ3 in resting platelets and negatively regulates αIIbβ3 outside-in signaling,38,39 led to increased plaque formation, particularly in the early stages of atherosclerosis.36 Activated integrin αIIbβ3 ligand binding triggers a cascade of events that enhance expression of platelet adhesion receptors such as CD62P (P-selectin) and CD40 ligand (CD40L), and pro-inflammatory cytokine/chemokine release including platelet factor-4 (PF4) and activation normal T cell expressed and secreted (CCL5),40,41 which play crucial roles in atherogenesis.42 Given the protective roles of CoQ10 in atherosclerosis and CVDs, the current study aims to investigate whether the atheroprotective effects of CoQ10 involve the attenuation of platelet hyper-reactivity, and whether platelet integrin αIIbβ3 signaling is modulated during this process in an atherosclerosis model using ApoE−/− mice fed on a high-fat diet.
2. Materials and methods
2.1. Chemicals and reagents
CoQ10, CoQ9 and thrombin were purchased from Sigma-Aldrich (St Louis, MO, USA). Tirofiban was purchased from Selleck Chemical (Houston, Texas, USA). CytoPainter Phalloidin-ifluor 488, antibodies against Fg and fibronectin were purchased from abcam (Cambridge, UK). Antibodies against anti-phospho-integrin β3Tyr747, anti-phospho-SrcTyr418, anti-integrin αIIb (CD41), anti-phospho-MLCSer19, GAPDH and β-actin were obtained from Cell Signaling Technology (MA, USA). Soluble Fg was purchased from Cayman (Hamburg, Germany). The following antibodies were used for flow cytometry: phycoerythrin (PE)-conjugated anti-mouse CD62P, allophycocyanin (APC)-conjugated anti-mouse CD63, PE-conjugated anti-mouse CD40L, APC-conjugated anti-mouse αIIb, fluorescein isothiocyanate (FITC)-conjugated anti-mouse CD45, and PerCP-efluor 710-conjugated anti-mouse CD115 were obtained from eBioscience (San Diego, CA, USA). Soluble FITC-conjugated Fg was purchased from Thermo Fisher Scientific (Waltham, MA, USA) and PE-conjugated JON/A was purchased from Emfret Analytics (Eibelstadt, Germany).2.2. Animals and diets
Male homozygous ApoE−/− mice (C57BL/6 genetic background), aged 8 weeks, were obtained from Jackson Laboratories. Mice were housed under standard conditions with a 12 h light–dark cycle at 22–24 °C and free access to food and water. After 2 weeks of adaptation, the animals were randomly divided into three groups and fed either a standard normal AIN-93G diet (negative control (NC) group), a high-fat diet (HFD group; containing 0.15% cholesterol, 21% fat, and 19.5% casein; Medicience Ltd, Jiangsu, China) or a HFD supplemented with CoQ10 (1800 mg kg−1 diet) (CoQ10 group) for 12 weeks. The dose of CoQ10 was based on our previous study.10,43 The food consumption and body weight of mice were monitored weekly. All experiments were approved by the Animal Care and Use Committee of Sun Yat-sen University (permit number: SYXK [Yue] 2017-0080). At the end of the treatment period, murine blood was collected and tissue samples were stored at −80 °C until analysis.2.3. Quantification of atherosclerotic lesions
Atherosclerotic lesions in the aortic sinus were measured as described previously in ref. 36,44. Briefly, the heart and the whole aorta from ApoE−/− mice were excised after whole body in situ perfusion with 4% paraformaldehyde (PFA). Following an overnight fixation and adventitia removal, upper sections of hearts were embedded in an optimal cutting temperature (OCT) compound (Sakura Tissue-Tek, CA, USA) and frozen at −80 °C. For quantification of the maximum lesion area within the aortic sinus, frozen sections (8 μm) were obtained from the aortic sinus using Leica cryostats (Leica, Rijswijk, The Netherlands) and were stained with 0.5% Oil-red-O (Sigma-Aldrich, Inc. St Louis, MO) to measure the atherosclerotic lesion size. For plaque area quantification in the whole aorta, the aorta was longitudinally opened and stained with Oil-red-O to visualize lipid deposition. Digital micrographs were acquired using a Leica microscope (Leica Microsystems, Heidelberg, Germany), and the lesion areas were analyzed using Image-Pro Plus image analysis software (Media Cybernetics Corporation, USA). The plaque area was calculated as the percentage of the Oil-red-O+ stained area by dividing the area of the internal aortic lumen or the whole aorta.2.4. Blood collection and platelet preparation
Murine blood was collected following cardiac puncture of anesthetized ApoE−/− mice in a tube containing 3.8% acid citrate dextrose (ACD; 38 mM citric acid, 75 mM trisodium citrate, 100 mM dextrose). Platelet-rich plasma (PRP) was obtained by centrifugation at 300g for 7 min. Gel-filtered platelets were then prepared from the PRP using a Sepharose 2B column in piperazine-1,4-bisethanesulfonic acid (PIPES) buffer (PIPES 5 mM, NaCl 1.37 mM, KCl 4 mM, glucose 0.1%, pH 7.0), according to our previously described methods.1,2,45,46 After isolation of the PRP, the remaining sample was centrifuged at 2500g for 15 min at 4 °C to separate the platelet-poor plasma (PPP). Then, the PPP was further centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 5 min to remove the remaining cells and stored at −80 °C until further analysis for plasma inflammatory factors.
000g for 5 min to remove the remaining cells and stored at −80 °C until further analysis for plasma inflammatory factors.
2.5. Platelet aggregation assays
The concentration of gel-filtered platelets in PIPES was adjusted to 2.5 × 108 platelets per mL with the use of PIPES buffer.1,2,45,47,48 Platelet aggregation was performed using a Chronolog aggregometer (Chrono-Log Corp, Havertown, USA) in gel-filtered platelets at 37 °C with a sample stirring speed of 1000 rpm using our previously described methods.16,23,26 Platelet aggregation was stimulated by 0.1 U mL−1 thrombin in the presence of 1 mM Ca2+. The change in light transmission was monitored and recorded for 6 min.2.6. Detection of platelet surface expression of CD62P, CD63, and CD40L, as well as Fg and JON/A binding to platelets by flow cytometry
Gel-filtered platelets (1 × 106 platelets per mL) isolated from ApoE−/− mice were labeled with PE-conjugated anti-mouse CD62P, APC-conjugated anti-mouse CD63, PE-conjugated anti-mouse CD40L, FITC-conjugated Fg, and PE-conjugated JON/A antibodies for 20 min, respectively, followed by the addition of thrombin for 2 min in the presence of 1 mM Ca2+ to stimulate platelets. Samples were then fixed with 1% PFA, and analyzed by using a CytoFLEX flow cytometer (Beckman Coulter, CA, USA) within 30 min. The data were analyzed by using CytExpert 2.0 (Beckman Coulter, CA, USA).4,492.7. Measurement of platelet PF4, β-TG and CCL5 secretion
Murine gel-filtered platelets (1 × 108 platelets per mL) prepared from ApoE−/− mice were stimulated with 0.5 U mL−1 thrombin in the presence of 1 mM Ca2+, followed by centrifugation at 10000g for 5 min at 4 °C. The cell-free supernatant was transferred into another tube and the concentrations of PF4 (R&D Systems, MN, USA), β-thromboglobulin (β-TG; Abcam, Cambridge, UK) and CCL5 (eBioscience, CA, USA) were determined using commercial enzyme-linked immunosorbent assay (ELISA) kits.
2.8. Measurement of platelet spreading on immobilized Fg
Platelet spreading on immobilized Fg was carried out according to the previously described methods.50 Briefly, gel-filtered platelets were prepared from ApoE−/− mice after 12-week intervention. The Fg (100 μg mL−1) coated coverslips were blocked with 1% bovine serum albumin (BSA) for 60 min at 37 °C, followed by incubation with gel-filtered platelets for additional 60 min. Non-adherent platelets were then removed by washing with phosphate buffered saline (PBS) prior to fixing with 4% PFA. Platelets were permeabilized with 0.1% (v/v) TritonX-100, followed by staining with 0.1% (v/v) Alexa 488 conjugated-phalloidin for 60 min. Subsequently, the coverslips were washed with PBS, and adherent platelets were imaged with a 100× magnification oil immersion lens. The surface area of individual platelets was quantified using ImageJ software (NIH, USA).2.9. Clot retraction assays
Clot retraction assays were performed as previously described in ref. 50. Briefly, the pooled gel-filtered platelets (2 × 108 platelets per mL) from two mice were added to aggregometer tubes in the presence of 200 μg mL−1 Fg and 1 mM Ca2+. Clot retraction was initiated by adding 0.1 U mL−1 thrombin and monitored every 30 min for 60 min at 37 °C, and documented photographically using a Nikon camera. Images were analyzed using the NIH ImageJ software (NIH, MD, USA).2.10. Flow cytometry for platelet–monocyte aggregation
Platelet–monocyte aggregation assay was performed as described previously in ref. 36,51. Freshly isolated leukocytes (1 × 106 mL−1) and platelets (1 × 108 mL−1) were suspended in HBSS containing 5 mg mL−1 human albumin and 10 mM HEPES. The blood cells were then labeled with APC-conjugated anti-mouse CD41, FITC-conjugated anti-mouse CD45, and PerCP-efluor 710-conjugated anti-mouse CD115 for 20 min at room temperature in the dark. Platelets were then stimulated by using 1 U mL−1 thrombin for 2 min in the presence of 1 mM CaCl2 and MgCl2. In some experiments, platelet–monocyte interactions were analyzed after pretreatment of the platelets with tirofiban (1 μg mL−1). Platelet–monocyte aggregation was measured by using a CytoFLEX flow cytometer and the data were analyzed by using CytExpert 2.0.2.11. Measurement of plasma levels of PF4, CCL5, IFNγ, TNFα, IL-6, Fg, and fibronectin
The plasma levels of PF4 (R&D Systems), β-TG (Abcam), CCL5 (eBioscience), interferon γ (IFNγ; Abcam), tumor necrosis factor α (TNFα; Abcam), interleukin 6 (IL-6; Abcam), Fg (Abcam), and fibronectin (Abcam) were measured in ApoE−/− mice after 12-week CoQ10 supplementation using commercial ELISA kits according to the manufacturer's instructions.2,522.12. Measurement of plasma CoQ10 levels
The plasma CoQ10 concentrations in ApoE−/− mice were determined by high performance liquid chromatography (HPLC) with electrochemical detection (HPLC-EC), according to our previously described methods.10,53 In brief, the frozen plasma sample was thawed at room temperature, and then a 100 μL aliquot of the sample was placed in a 1.8 mL capped polypropylene tube containing 50 μL of internal standard solution (CoQ9). The sample was then mixed with 850 μL of cold 1-propanol. All tubes were vortex-mixed and centrifuged at 21000g for 10 min at 4 °C. The resulting supernatant was transferred to a glass autosampler vial. A 20 μL aliquot of 1-propanol extract from a vial was injected immediately into an automated HPLC. The analytical column was a Microsorb-MV reversed-phase C18 column (4.6 mm × 150 mm; 5 μm bead size; Varian, Lake Forest, CA, USA). The mobile phase for the isocratic elution was prepared as follows: sodium acetate trihydrate (6.8 g), 15 mL of glacial acetic acid, and 15 mL of 2-propanol were added to 695 mL of methanol and 275 mL of hexane, PH = 6.0. The mobile phase was filtered through a 0.2 μm nylon or analogous filter. The flow rate was 1 mL min−1.
2.13. Western blotting analysis
Protein lysates from platelets exposed for 60 min to immobilized Fg or BSA, or untreated gel-filtered platelets were lysed with lysis buffer (50 mM Tris, pH 7.4, 0.1% SDS, 150 mM NaCl, 1% nonidet P 40 (NP40), and 0.5% sodium deoxycholate) supplemented with protease and phosphatase inhibitors for 30 min, followed by centrifugation at 12000g for 15 min. The proteins were separated by SDS-PAGE. Western blotting analysis was performed using our previously described methods.2,45,54
2.14. Immunohistochemistry for CD68+ macrophages and CD4+ T cells in the aortic sinus atherosclerotic lesions
Aortic sinus sections were fixed in cold acetone for 10 min, washed in PBS, and blocked with 3% BSA for 30 min at room temperature. Sections were then incubated with primary antibodies including CD68 (Abcam, Cambridge, UK) for macrophages, and CD4 (Novus Biologicals, CO, USA) for CD4+ T cells overnight at 4 °C. This was followed by incubation with biotin-conjugated secondary antibodies, avidin–biotin complex, and a 3,3′-diaminobenzidine (DAB) substrate. Sections were counterstained with haematoxylin and images were acquired using a Leica microscope.44,55 Data were analyzed using Image-Pro Plus image analysis software.2.15. Quantitative reverse transcriptase real-time polymerase chain reaction (qRT-PCR)
After 12 weeks of intervention, total RNA was extracted56 from the aorta using a mirVanaTM miRNA isolation kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. After RNA isolation, 1 μg of total RNA was reverse transcribed to cDNA using a RT-PCR kit (TaKaRa, Dalian, Japan). Primer sequences for all genes (TNFα, IFNγ, and IL-6) are listed in Table S1.† The housekeeping gene β-actin was used as an internal control for quantification. Quantification was acquired on the ABI prism Vii7 Sequence Detection System (Applied Biosystems, CA, USA).2.16. Statistical analysis
GraphPad Prism version 5.01 software (GraphPad Inc., San Diego, CA, USA) was used for statistical analyses. Data are expressed as the means ± standard errors of the means (SEM). Differences between two groups were analyzed using unpaired t-tests and a one-way analysis of variance (ANOVA) followed by the Newman–Keuls test was used for multiple comparisons. P < 0.05 was considered to be statistically significant.3. Results
3.1. CoQ10 supplementation reduces atherosclerotic lesion formation in ApoE−/− mice
Cross-sectional or en face plaque extension was evaluated in the standard normal AIN-93G diet (NC), HFD and CoQ10-supplemented HFD ApoE−/− mice after 12 weeks of intervention. Oil-red-O staining showed that 12-week HFD feeding significantly increased the atherosclerotic lesion size in the aortic sinus compared with the NC group (Fig. 1A and B). The mean atherosclerotic lesion area in the aortic sinus of the CoQ10 group (17.48% ± 2.03%) was less than that of the HFD group (33.59% ± 4.59%) (Fig. 1A and B). Moreover, CoQ10 supplementation markedly decreased the HFD-enhanced lesion area in en face aortic plaque extension (Fig. 1C and D). Therefore, CoQ10 supplementation reduces atherosclerotic lesion formation in ApoE−/− mice.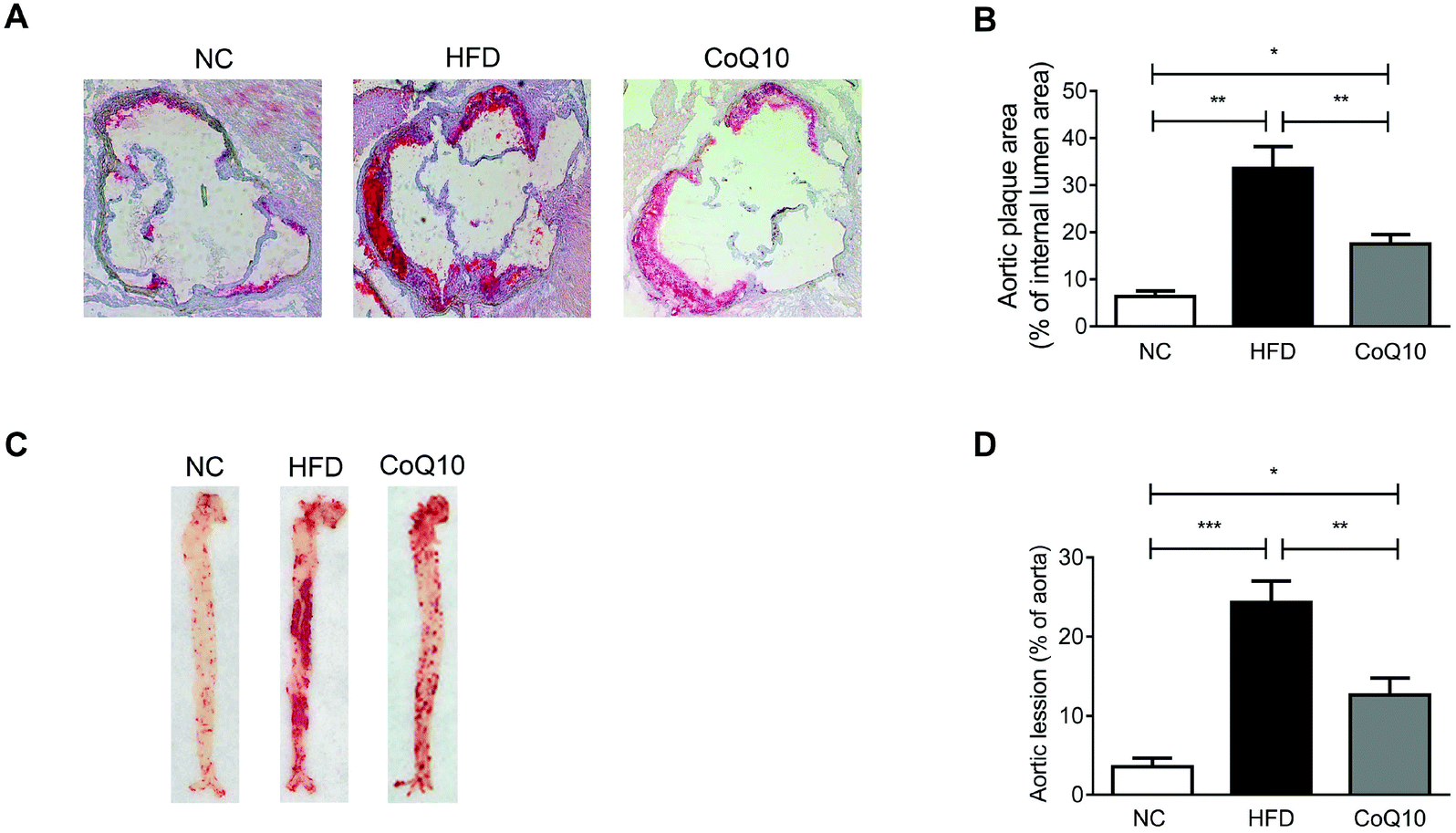 | ||
| Fig. 1 CoQ10 supplementation reduces atherosclerotic lesion formation in ApoE−/− mice. (A and B) Representative images of the atherosclerotic areas in Oil-red-O stained aortic sinus of NC, HFD or CoQ10-supppemented ApoE−/− mice (A) and quantification of the lesion area (B) are presented (n = 10). (C and D) Representative images of en face staining of the entire aorta are shown (C), and Oil-red-O-positive plaque surface is determined (D) (n = 10). Data are expressed as the means ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 as assessed by ANOVA followed by the Newman–Keuls test for multiple comparisons. NS, not significant difference. | ||
3.2. CoQ10 supplementation attenuates platelet aggregation and granule secretion
To assess whether CoQ10 was systemically absorbed following 12 weeks of supplementation, the plasma CoQ10 concentrations were determined in NC, HFD and CoQ10-supplemented ApoE−/− mice. We found that the plasma concentrations of CoQ10 in the NC group were comparative to that in the HFD group (Fig. 2A). CoQ10 supplementation resulted in a higher plasma CoQ10 concentrations by about 20-fold compared to mice fed on the HFD (Fig. 2A). To further determine whether CoQ10 supplementation acts on platelet function in relation to atherosclerosis in ApoE−/− mice, we examined platelet aggregation and granule secretion using gel-filtered platelets. We found that CoQ10 treatment significantly attenuated HFD-induced platelet aggregation to the level of the NC group in response to thrombin (Fig. 2B and C).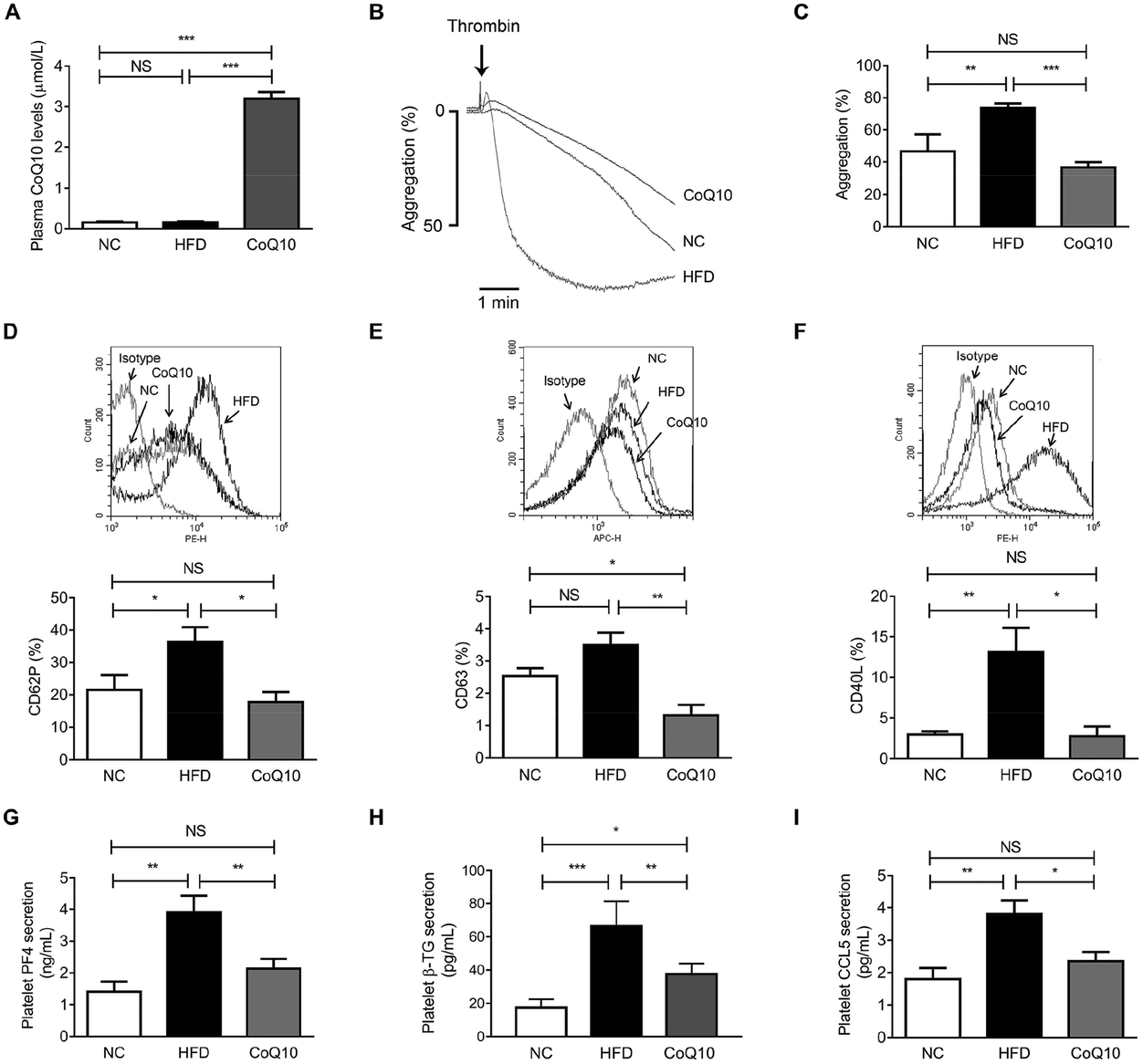 | ||
| Fig. 2 CoQ10 supplementation attenuates platelet aggregation and granule secretion in ApoE−/− mice. (A) Plasma CoQ10 concentrations are measured from ApoE−/− mice after 12 weeks of intervention (n = 10). (B and C) Gel-filtered platelet aggregation is stimulated by 0.1 U mL−1 thrombin (n = 10). Representative tracings (B) and summary data (C) are presented. (D) Surface expression of CD62P on gel-filtered platelets prepared from ApoE−/− mice is stimulated by 0.1 U mL−1 thrombin and analyzed by flow cytometry (n = 10). (E) Same as D, except platelet surface expression of CD63 stimulated by 1 U mL−1 thrombin is quantified (n = 4). (F) Same as D, except platelet surface expression of CD40L stimulated by 2 U mL−1 thrombin is quantified (n = 4). (G–I) Gel-filtered platelets prepared from ApoE−/− mice are stimulated by 0.1 U mL−1 thrombin for 5 min. The levels of PF4 (G), β-TG (H) and CCL5 (I) secretion from platelets are measured (n = 5). Data are expressed as the means ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 as assessed by ANOVA followed by the Newman–Keuls test for multiple comparisons. NS, not significant difference. | ||
When platelets are activated by agonists like thrombin, collagen and ADP, platelet granules containing abundant membrane-bound proteins are expressed substantively on the platelet surface and the soluble proteins (e.g., PF-4, β-TG, and CCL5) are secreted into the circulation, which promote platelet aggregation, and atherosclerosis.42 As shown by flow cytometric analysis, compared to the NC group, HFD feeding alone significantly increased thrombin-stimulated platelet surface expression of CD62P and CD40L in ApoE−/− mice, which were markedly inhibited by CoQ10 supplementation to the level of the NC group (Fig. 2D and F). We observed no significant increase in thrombin-stimulated platelet surface expression of CD63 in the HFD mice when compared to the level of NC mice, which was also significantly attenuated by CoQ10 supplementation (Fig. 2E). However, we did not observe any significant differences in platelet surface CD62P or CD63 expression in resting platelets among NC, HFD and CoQ10 mice (Fig. S1A and 1B†). Furthermore, 12-week CoQ10 supplementation markedly reduced thrombin-stimulated platelet secretion of PF4, β-TG and CCL5 in mice fed on the HFD (Fig. 2G–I). Additionally, HFD-increased plasma levels of PF4, β-TG and CCL5 were also favorably attenuated by CoQ10 (Fig. S2A–2C†). Thus, these data demonstrate that the dietary supplement of CoQ10 attenuates platelet aggregation and granule secretion in ApoE−/− mice.
3.3. CoQ10 supplementation inhibits platelet integrin αIIbβ3 inside-out signaling
Platelet αIIbβ3-mediated inside-out signaling results in the activated αIIbβ3 binding to its ligands (e.g., Fg, VWF, and fibronectin), which is central to platelet aggregation and granule secretion.29,31 To investigate whether CoQ10 supplementation affects the interactions between Fg and αIIbβ3 in ApoE−/− mice, soluble FITC-Fg binding to activated platelets was measured (a typical assay for platelet αIIbβ3-mediated inside-out signaling29,38,39). We found that 12-week CoQ10 supplementation significantly inhibited the increased Fg-binding caused by a HFD to thrombin-activated platelets (Fig. 3A and B). Moreover, CoQ10 supplementation in HFD mice inhibited JON/A binding to thrombin-activated platelets, an antibody specific for the activated conformation of αIIbβ3 in murine platelets (Fig. 3C and D). This effect was not due to decreased surface expression of integrin αIIb on platelets following CoQ10 supplementation in mice because comparable levels of αIIb were detected in HFD platelets by both flow cytometry (Fig. 3E) and western blotting (Fig. 3F). No significant difference was observed in the percentage of soluble FITC-Fg or JON/A binding to unstimulated platelets among NC, HFD and CoQ10 mice (Fig. S1C and 1D†). In addition, CoQ10 supplementation did not affect plasma levels (Fig. S3A and 3B†) nor platelet expression levels of Fg and fibronectin (Fig. S3C–3E†). Therefore, CoQ10 supplementation inhibits platelet αIIbβ3 integrin inside-out signaling.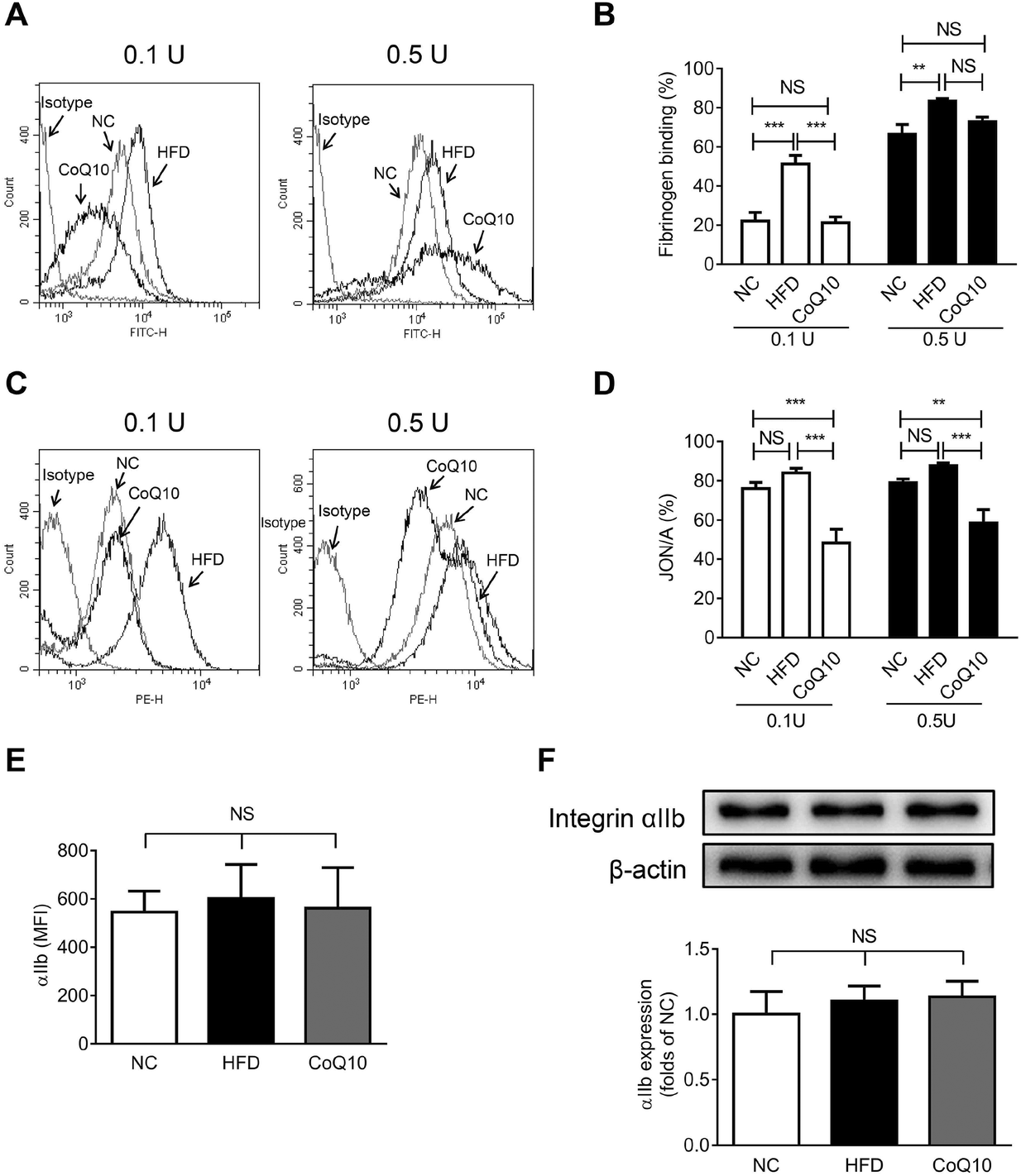 | ||
| Fig. 3 CoQ10 supplementation inhibits platelet integrin αIIbβ3 inside-out signaling in ApoE−/− mice. (A–D) Gel-filtered platelets are prepared from NC, HFD or CoQ10 mice. Soluble FITC-labeled Fg (A and B) or PE-labeled JON/A (C and D) binding to platelets that are stimulated by 0.1 U mL−1 or 0.5 U mL−1 thrombin is analyzed by flow cytometry (n = 10). (E) Surface expression of αIIb on resting gel-filtered platelets prepared from ApoE−/− mice is analyzed by flow cytometry using APC-labeled anti-mouse αIIb (n = 4). (F) Protein levels of αIIb on resting gel-filtered platelets prepared from ApoE−/− mice are analyzed by western blotting (n = 4). Data are expressed as the means ± SEM. **P < 0.01 and ***P < 0.001 as assessed by ANOVA followed by the Newman–Keuls test for multiple comparisons. NS, not significant difference. | ||
3.4. CoQ10 supplementation down-regulates platelet integrin αIIbβ3 outside-in signaling
Platelet spreading on immobilized Fg and clot retraction are primarily regulated by integrin αIIbβ3-dependent outside-in signaling.27 We further determined the role of CoQ10 supplementation in platelet spreading on immobilized Fg and clot retraction in ApoE−/− mice by using gel-filtered platelets. As shown in Fig. 4, platelets exposed to immobilized BSA remained discoid in NC, HDF and CoQ10 groups, while the platelets exposed to immobilized Fg spread and the surface area was significantly greater (Fig. 4A and B). Interestingly, ApoE−/− mice fed a HFD had a profoundly enhanced surface area of the spread platelets when exposed to immobilized Fg which was significantly decreased by CoQ10 supplementation (Fig. 4A and B). In addition, we found that CoQ10 treatment ameliorated the enhanced clot retraction caused by a HFD (Fig. 4C and D). Platelets from HFD mice were completely retracted in 60 min, whereas platelets from CoQ10 supplemented ApoE−/− mice started retracting at 30 min and reached about 40% by 60 min (Fig. 4C and D).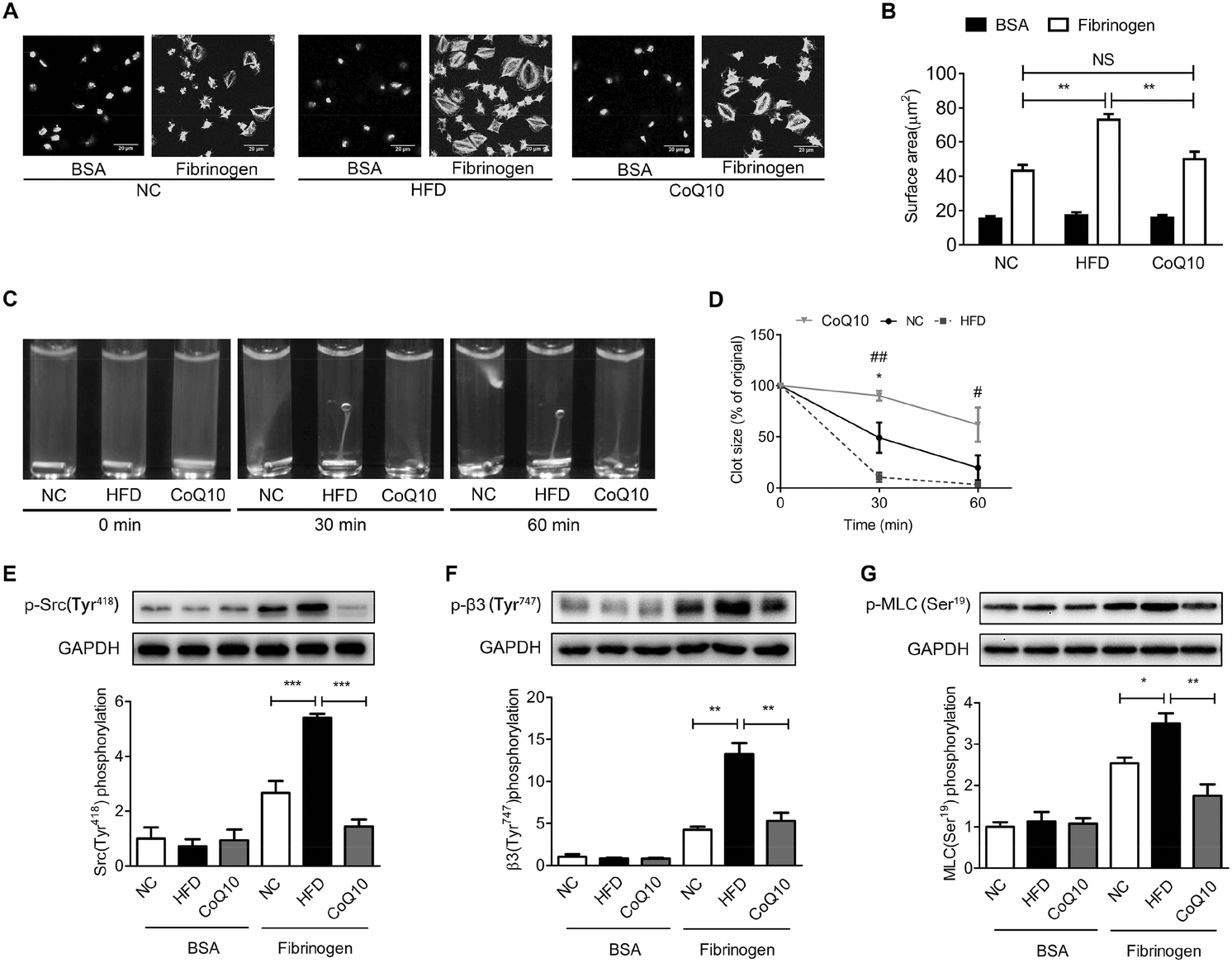 | ||
| Fig. 4 CoQ10 supplementation down-regulates platelet integrin αIIbβ3 outside-in signaling in ApoE−/− mice. (A and B) Representative images of platelets prepared from ApoE−/− mice after 60 min of spreading on immobilized Fg or BSA are presented (A), and the mean surface area of individual platelets is quantified (B) (n = 5). **P < 0.01; NS, not significant difference. (C and D) Assays of clot retraction are performed using gel-filtered platelets prepared from ApoE−/− mice. Representative photographs (C) and quantitation of clot retraction percentage (D) are given (n = 5). *P < 0.05: CoQ10 group versus NC group; #P < 0.05 and ##P < 0.01: CoQ10 group versus HFD group in panel D. (E–G) Protein lysates from NC, HFD or CoQ10 platelets exposed for 60 min to immobilized Fg or BSA are analyzed by western blotting using specific antibodies against phospho-c-Src (Tyr418) (E), or phospho-integrin β3 (Tyr747) (F), or phospho-MLC (Ser19) (G) (n = 4). *P < 0.05, **P < 0.01 and ***P < 0.001 in E–G. All data are expressed as the means ± SEM and assessed by ANOVA followed by the Newman–Keuls test for multiple comparisons. | ||
We have also further examined whether CoQ10 can directly affect platelet αIIbβ3 outside-in signaling. The effectors of integrin αIIbβ3 outside-in signaling include phosphorylation of a host of enzymes, adaptors, and cytoskeletal components such as the sarcoma tyrosine-protein kinase family, β3-integrin intracellular tail, etc.27 As shown in Fig. 4E and F, the phosphorylation of cellular and sarcoma tyrosine-protein kinase (c-Src) and the β-integrin tail were minimal in platelets exposed to BSA. However, this phosphorylation was markedly increased when platelets were exposed to Fg (Fig. 4E and 4F). CoQ10 supplementation significantly attenuated HFD-induced phosphorylation of c-Src (Fig. 4E) and the β-integrin tail (Fig. 4F) in platelets exposed to Fg. Myosin light chain (MLC) is the downstream effector of αIIbβ3 outside-in signaling. The phosphorylation of MLC on ser19 facilitates myosin interaction with actin filaments, which is essential for platelet shape change, spreading, granule secretion, and clot retraction.57 As shown in Fig. 4G, CoQ10 supplementation markedly down-regulated HFD-increased phosphorylation of MLC in platelets exposed to Fg. Therefore, our data demonstrate that the dietary supplement of CoQ10 down-regulates platelet αIIbβ3 outside-in signaling in ApoE−/− mice.
3.5. CoQ10 supplementation attenuates platelet–monocyte aggregation
It has been shown that platelet integrin αIIbβ3 outside-in signaling can directly mediate atherosclerosis through forming platelet–monocyte aggregates that promote monocyte activation and adhesion to the arterial endothelial cells, etc.36,37,39 To investigate whether CoQ10 supplementation can decrease platelet–monocyte interactions, activated platelets isolated from NC, HFD, and CoQ10-supplemented ApoE−/− mice were incubated with leukocytes. Indeed, HFD mice showed significantly enhanced platelet–monocyte aggregation, which was markedly decreased by CoQ10 supplementation (Fig. 5). Interestingly, similar to CoQ10 supplementation, tirofiban, a specific αIIbβ3 antagonist that can interfere with outside-in signaling by preventing Fg binding to αIIbβ3,36 also decreased the elevated platelet–monocyte interactions in mice fed a HFD. Importantly, in combination with tirofiban, CoQ10 did not exhibit an additive or synergistic effect on platelet–monocyte aggregation (Fig. 5), indicating that the inhibitory effect of CoQ10 on platelet–monocyte aggregation was mainly mediated by reducing the αIIbβ3 signaling.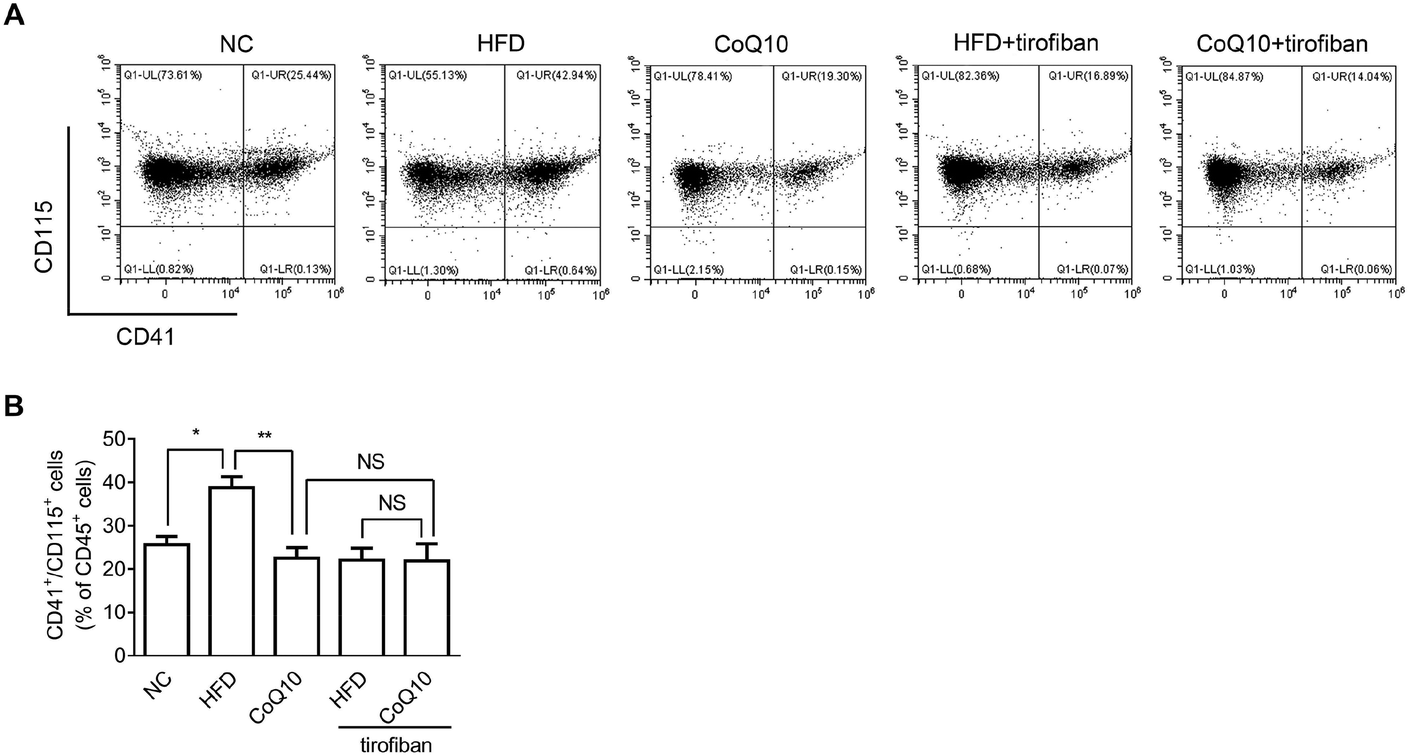 | ||
| Fig. 5 CoQ10 supplementation attenuates platelet–monocyte aggregation in ApoE−/− mice. Thrombin-activated platelets and leukocytes prepared from NC, HFD and CoQ10-supplemented ApoE−/− mice are incubated in the absence or presence of tirofiban (1 μg mL−1) and platelet–monocyte aggregates (CD41+/CD115+ cells) are quantified by flow cytometry as a percentage of all CD45+ cells (n = 5). (A) Representative images and (B) summary data are presented. Data are expressed as the means ± SEM. *P < 0.05 and **P < 0.01 assessed by ANOVA followed by the Newman–Keuls test for multiple comparisons. NS, not significant difference. | ||
4. Discussion
Hyper-reactive platelets possess a lower threshold for platelet aggregation and activation, which triggers vascular inflammation in both the early (i.e. atherosclerosis) and late (i.e. thrombosis) stages of atherothrombosis.1,15 Additionally, increased platelet reactivity is known to enhance the risk for CVDs.2,15,58,59 Utilizing an atherosclerosis model in ApoE−/− mice fed on a HFD, we identified that CoQ10 supplementation alleviated the progression of atherosclerosis. CoQ10 down-regulated platelet integrin αIIbβ3 signaling, leading to decreased platelet spreading, clot retraction, platelet aggregation, and pro-inflammatory granule factor secretion, as well as platelet–monocyte aggregation. Therefore, through attenuating integrin αIIbβ3 signaling and platelet hyper-reactivity, CoQ10 may be a promising agent for the prevention and/or slowing down the process of atherosclerosis and thus protecting against CVDs.Emerging evidence shows that platelet-mediated inflammatory responses represent an important step in the process of atherosclerosis.14,15,21 Platelet granule secretion is important in platelet-mediated inflammation and also contributes to atherosclerosis.42 We found that after 12 weeks, platelet surface expression of CD62P, CD40L and CD63 was enhanced in ApoE−/− mice fed on a HFD; these increases were attenuated in the CoQ10 treatment group. Platelet CD62P plays crucial roles in mediating monocyte rolling by interacting with monocyte P-selectin glycoprotein ligand-1 (PSGL-1), which accelerates the progression of atherosclerosis.60 Platelet CD40L is also a key mediator of platelet-induced inflammation in atherosclerosis by stimulating leukocyte activation and recruitment and activation of endothelial cells.61 We additionally demonstrated that 12-week CoQ10 supplementation decreased plasma levels and platelet release of PF4, β-TG and CCL5 in HFD mice. PF4 and β-TG are platelet-specific and are among the most abundant platelet proteins secreted following activation.59 They can enhance platelet aggregation, trigger recruitment of monocytes, promote monocyte activation and differentiation, etc.62 Circulating CCL5 levels are also considered to reflect platelet activation and secretion in vivo, although other cell types such as endothelial cells and macrophages can also secrete it.62 CCL5 is also an important mediator of inflammation in atherosclerosis because of its role in leukocyte recruitment and activation.63 In the present study, CoQ10 supplementation directly inhibited platelet CCL5 release following thrombin stimulation of gel-filtered platelets prepared from ApoE−/− mice fed on a HFD, indicating that decreased plasma CCL5 levels may have been partially driven by inhibition of platelet CCL5 secretion. To the best of our knowledge, this is the first study to identify a relationship between CoQ10 and platelet granule secretion in a model of atherosclerosis. Our current data indicate that the inhibitory effects of CoQ10 on platelet proinflammatory granule molecule secretion may at least partially contribute to its attenuation of atherogenesis.
Platelet integrin αIIbβ3 signaling plays vital roles in mediating platelet functions including platelet spreading, aggregation, granule release, etc.31,40,64 Ligand binding to activated integrin αIIbβ3 triggers a cascade of events that culminate in the increased surface expression of adhesion receptors and release of pro-inflammatory cytokines/chemokines.40 Engagement of αIIbβ3 on platelets upregulates CD40L and triggers CD40L-dependent matrix degradation by endothelial cells, which is critical for accelerating the development of atherosclerosis.65 Up-regulating integrin αIIbβ3 outside-in signaling can result in increased platelet release of PF-4 and CCL5.36 Our current study revealed that the dietary supplement of CoQ10 down-regulated αIIbβ3-mediated inside-out and outside-in signaling in ApoE−/− mice fed on a HFD, which may result in the attenuation of platelet aggregation and granule secretion. However, it is unclear how CoQ10 attenuates αIIbβ3 signaling during the process of atherosclerosis. It has been shown that αIIbβ3 activation can be mediated by multiple upstream pathways, including collagen-engaged GPVI signaling, calcium mobilization, cAMP/PKA signaling, etc.66,67 We recently demonstrated that CoQ10 attenuated platelet function predominantly by up-regulating cAMP/PKA signaling, leading to decreased platelet integrin αIIbβ3 outside-in signaling in vitro and in dyslipidemic human patients.13 Moreover, it has been reported that CoQ10 acts physiologically as an electron carrier in the mitochondria and an essential cofactor for ATP production.7 However, we recently showed that CoQ10 attenuated platelet function was independent of platelet cellular ATP synthesis or platelet mitochondrial metabolism, since CoQ10 treatment did not affect cellular ATP levels13 or alter the platelet mitochondrial membrane potential (Fig. S4†) in human platelets in vitro. Whether the cAMP/PKA signaling and platelet mitochondrial metabolism were modulated by CoQ10 supplementation and involved an attenuation of atherosclerosis in ApoE−/− mice fed on a HFD merit further investigation. Our current data indicate that through down-regulating platelet αIIbβ3 signaling, CoQ10 can effectively attenuate HFD-induced platelet hyperreactivity during the process of atherosclerosis in ApoE−/− mice.
It has been well established that the platelet–leukocyte interactions actively contribute to the platelet-induced chronic inflammatory processes, which facilitate the development of atherosclerotic lesions.68 Our results demonstrated that CoQ10 supplementation decreased HFD-enhanced platelet–monocyte aggregation in ApoE−/− mice. Platelet–monocyte interactions can be mediated by: platelet αIIbβ3-Fg-monocyte αVβ3, and platelet GPIbα-monocyte Mac-1, platelet P-selectin (CD62P)-monocyte PSGL-1, platelet CD40L-monocyte CD40, etc.69 We found that plasma and platelet expression levels of Fg (Fig. S3†) and platelet expression levels of αIIbβ3 (Fig. 3) and GPIbα (Fig. S5†) in ApoE−/− mice following feeding on a HFD were all unaffected after 12 weeks of CoQ10 supplementation. However, CoQ10 supplementation ameliorated the increased soluble FITC-Fg binding and JON/A monoclonal antibody binding to activated αIIbβ3 following HFD, indicating that CoQ10 reduced αIIbβ3-mediated inside-out signaling. CoQ10 also down-regulated αIIbβ3 outside-in signaling. These mechanisms might directly contribute to the inhibition of platelet–monocyte aggregation. This is further confirmed by our finding that interfering with Fg-αIIbβ3 interactions using a specific αIIbβ3 antagonist tirofiban, HFD-induced platelet–monocyte aggregation was markedly inhibited. This result was consistent with a report by Karshovska et al. who demonstrated a critical role of platelet integrin αIIbβ3 signaling in the modulation of platelet–monocyte interactions in atherosclerotic plaque development.36,38 Moreover, we also showed that CoQ10 supplementation did not further decrease the inhibitory effect of tirofiban, indicating that the attenuation of αIIbβ3 signaling by CoQ10 is necessary for its inhibitory role in platelet–monocyte interactions. Additionally, reducing platelet surface CD62P and CD40L expression by CoQ10 may also synergistically contribute to the decreased platelet–monocyte aggregation. Thus, our data indicate that by down-regulating platelet integrin αIIbβ3 signaling and attenuating granule secretion, CoQ10 supplementation can inhibit platelet–monocyte aggregates and attenuate the formation of atherosclerotic lesions.
Previous studies indicated that CoQ10 inhibited atherosclerosis, which may be partially attributed to the ability of CoQ10 to improve lipid profiles,8 where the increased circulating oxidized low-density lipoprotein (ox-LDL) can trigger platelet hyperreactivity.70 Interestingly, there have been some controversies regarding whether CoQ10 can improve lipid profiles.9,71,72 Several clinical studies did not observe CoQ10 supplementation could decrease serum LDL levels in patients with CVDs.71,72 Although our current study observed that CoQ10 supplementation decreased plasma levels of LDL in ApoE−/− mice fed on the HFD (Fig. S6†), the inhibitory effects of CoQ10 on platelet hyper-reactivity are unlikely caused by decreasing LDL, because our in vitro studies using human gel-filtered platelets, in the absence of lipoproteins, found that agonist-induced platelet aggregation was inhibited by CoQ10 treatment.13 Moreover, an attenuation of inflammatory responses by CoQ10 supplementation may also contribute to its alleviation of atherogenesis, since we observed that CoQ10 decreased HDF-enhanced CD68+ macrophage and CD4+ T cell infiltration (Fig. S7A–7D†), arterial mRNA expression of (Fig. S7E–7G†) and plasma levels of (Fig. S7H–7J†) pro-inflammatory factors including TNFα, IFNγ and IL-6. It was previously shown that up-regulation of platelet integrin αIIbβ3 outside-in signaling increased macrophage and T cell infiltration, and expression levels of pro-inflammatory factors in atherosclerotic lesions.36 Integrin αIIbβ3 antagonists can reduce circulating pro-inflammatory factors in patients with acute coronary syndromes.73 Although currently unknown, it is possible that CoQ10 down-regulated αIIbβ3-mediated signaling may partially contribute to the alleviation of inflammatory responses, which merits further investigation. It is conceivable that other platelet-independent anti-inflammatory mechanisms mediated by CoQ10 might be also involved in the process of atherosclerosis. Since CoQ10 could directly modulate the function of endothelial cells and macrophages,10,12,53 which may synergistically contribute to the attenuation of atherosclerotic lesion formation via decreased inflammatory responses.
5. Conclusions
In conclusion, our current study provides the first direct link between CoQ10 supplementation and platelet function during the process of atherosclerosis in ApoE−/− mice. We demonstrate that CoQ10 down-regulated platelet integrin αIIbβ3-mediated signaling, leading to decreased platelet hyper-reactivity (e.g., platelet aggregation, granule secretion, and platelet–monocyte aggregation). These effects of CoQ10 may profoundly contribute to the alleviation of atherosclerotic lesion formation. Since CoQ10 can also attenuate platelet hyper-reactivity in dyslipidemic human patients and inhibit thrombus formation in mice,13 through controlling aberrant platelet activation, the dietary supplement of CoQ10 may be a promising strategy for the protection against cardiovascular events (e.g., atherosclerosis and thrombosis).Author contributions
Fuli Ya designed the research, carried out experiments, analyzed the data and wrote the draft manuscript. Xiaohong Ruby Xu analyzed the data, revised and edited the draft manuscript. Reid C. Gallant revised and edited the draft manuscript. Zezhong Tian, Yilin Shi, Yinfan Wu, and Yimin Zhao performed some experiments. Heyu Ni analyzed the data, designed and edited the draft manuscript. Jianbo Wan, Reheman Adili and Wenhua Ling edited the draft manuscript. Yan Yang was the principal investigator who designed the research and analyzed the data, and had primary responsibility for the final content. All authors reviewed and approved the final manuscript.Abbreviations
| ApoE−/− | Apolipoprotein E-deficient |
| APC | Allophycocyanin |
| ACD | Acid citrate dextrose |
| β-TG | β-Thromboglobulin |
| BSA | Bovine serum albumin |
| CoQ10 | Coenzyme Q10 |
| CCL5 | Activation normal T cell expressed and secreted |
| CVDs | Cardiovascular diseases |
| CD40L | CD40 ligand |
| c-Src | Cellular and sarcoma tyrosine-protein kinase |
| DAB | Diaminobenzidine |
| ELISA | Enzyme-linked immunosorbent assay |
| Fg | Fibrinogen |
| FITC | Fluorescein isothiocyanate |
| HFD | High-fat diet |
| HPLC-EC | High performance liquid chromatography with electrochemical detection |
| IFNγ | Interferon γ |
| IL-6 | Interleukin 6 |
| JAMA | Junctional adhesion molecule A |
| MLC | Myosin light chain |
| NC | Negative control |
| OCT | Optimal cutting temperature |
| ox-LDL | Oxidized low-density lipoprotein |
| PF4 | Platelet factor-4 |
| PE | Phycoerythrin |
| PRP | Platelet-rich plasma |
| PPP | Platelet-poor plasma |
| PIPES | Piperazine-1,4-bisethanesulfonic acid |
| PFA | Paraformaldehyde |
| PSGL-1 | P-Selectin glycoprotein ligand-1 |
| qRT-PCR | Quantitative reverse transcriptase real-time polymerase chain reaction |
| SEM | Standard errors of the means |
| TNFα | Tumor necrosis factor α |
| VWF | von Willebrand factor |
Conflicts of interest
The authors have declared no conflicts of interest.Acknowledgements
This work was supported by the National Science Foundation of China (No. 81872617), the Shenzhen Science and Technology Innovation Commission (No. 201803073000433), the Guangzhou Science, Technology and Innovation Commission (No. 201804020045), Canadian Institutes of Health Research (project MOP 119540) and Canadian Institutes of Health Research (foundation 389035), Heart and Stroke Foundation of Canada (Ontario), and Canada Foundation for Innovation.References
- X. R. Xu, Y. Wang, R. Adili, L. Ju, C. M. Spring, J. W. Jin, H. Yang, M. A. D. Neves, P. Chen, Y. Yang, X. Lei, Y. Chen, R. C. Gallant, M. Xu, H. Zhang, J. Song, P. Ke, D. Zhang, N. Carrim, S. Y. Yu, G. Zhu, Y. M. She, T. Cyr, W. Fu, G. Liu, P. W. Connelly, M. L. Rand, K. Adeli, J. Freedman, J. E. Lee, P. Tso, P. Marchese, W. S. Davidson, S. P. Jackson, C. Zhu, Z. M. Ruggeri and H. Ni, Apolipoprotein A-IV binds alphaIIbbeta3 integrin and inhibits thrombosis, Nat. Commun., 2018, 9, 3608 CrossRef PubMed.
- F. Song, Y. Zhu, Z. Shi, J. Tian, X. Deng, J. Ren, M. C. Andrews, H. Ni, W. Ling and Y. Yang, Plant food anthocyanins inhibit platelet granule secretion in hypercholesterolaemia: Involving the signalling pathway of PI3K-Akt, Thromb. Haemostasis, 2014, 112, 981–991 CrossRef PubMed.
- Y. Dong, H. Cheng, Y. Liu, M. Xue and H. Liang, Red yeast rice ameliorates high-fat diet-induced atherosclerosis in Apoe(-/-) mice in association with improved inflammation and altered gut microbiota composition, Food Funct., 2019, 10, 3880–3889 RSC.
- Y. Yang, Z. Shi, A. Reheman, J. W. Jin, C. Li, Y. Wang, M. C. Andrews, P. Chen, G. Zhu, W. Ling and H. Ni, Plant food delphinidin-3-glucoside significantly inhibits platelet activation and thrombosis: novel protective roles against cardiovascular diseases, PLoS One, 2012, 7, e37323 CrossRef CAS PubMed.
- Y. Yao, Y. Chen, R. Adili, T. McKeown, P. Chen, G. Zhu, D. Li, W. Ling, H. Ni and Y. Yang, Plant-based Food Cyanidin-3-Glucoside Modulates Human Platelet Glycoprotein VI Signaling and Inhibits Platelet Activation and Thrombus Formation, J. Nutr., 2017, 147, 1917–1925 CrossRef CAS PubMed.
- A. Emami and B. Bazargani-Gilani, Effect of oral CoQ10 supplementation along with precooling strategy on cellular response to oxidative stress in elite swimmers, Food Funct., 2018, 9, 4384–4393 RSC.
- J. Kaikkonen, T. P. Tuomainen, K. Nyyssonen and J. T. Salonen, Coenzyme Q10: absorption, antioxidative properties, determinants, and plasma levels, Free Radical Res., 2002, 36, 389–397 CrossRef CAS PubMed.
- B. J. Lee, Y. F. Tseng, C. H. Yen and P. T. Lin, Effects of coenzyme Q10 supplementation (300 mg/day) on antioxidation and anti-inflammation in coronary artery disease patients during statins therapy: a randomized, placebo-controlled trial, Nutr. J., 2013, 12, 142 CrossRef PubMed.
- P. Zhang, C. Yang, H. Guo, J. Wang, S. Lin, H. Li, Y. Yang and W. Ling, Treatment of coenzyme Q10 for 24 weeks improves lipid and glycemic profile in dyslipidemic individuals, J. Clin. Lipidol., 2018, 12, 417–427 e5 CrossRef PubMed.
- D. Wang, X. Yan, M. Xia, Y. Yang, D. Li, X. Li, F. Song and W. Ling, Coenzyme Q10 promotes macrophage cholesterol efflux by regulation of the activator protein-1/miR-378/ATP-binding cassette transporter G1-signaling pathway, Arterioscler. Thromb. Vasc. Biol., 2014, 34, 1860–1870 CrossRef CAS.
- P. K. Witting, K. Pettersson, J. Letters and R. Stocker, Anti-atherogenic effect of coenzyme Q10 in apolipoprotein E gene knockout mice, Free Radical Biol. Med., 2000, 29, 295–305 CrossRef CAS.
- K. L. Tsai, L. H. Chen, S. H. Chiou, G. Y. Chiou, Y. C. Chen, H. Y. Chou, L. K. Chen, H. Y. Chen, T. H. Chiu, C. S. Tsai, H. C. Ou and C. L. Kao, Coenzyme Q10 suppresses oxLDL-induced endothelial oxidative injuries by the modulation of LOX-1-mediated ROS generation via the AMPK/PKC/NADPH oxidase signaling pathway, Mol. Nutr. Food Res., 2011, 55(Suppl 2), S227–S240 CrossRef CAS.
- F. Ya, X. R. Xu, Y. Shi, R. C. Gallant, F. Song, X. Zuo, Y. Zhao, Z. Tian, C. Zhang, X. Xu, W. Ling, H. Ni and Y. Yang, Coenzyme Q10 Upregulates Platelet cAMP/PKA Pathway and Attenuates Integrin alphaIIbbeta3 Signaling and Thrombus Growth, Mol. Nutr. Food Res., 2019, e1900662 CrossRef PubMed.
- A. J. Murphy, N. Bijl, L. Yvan-Charvet, C. B. Welch, N. Bhagwat, A. Reheman, Y. Wang, J. A. Shaw, R. L. Levine, H. Ni, A. R. Tall and N. Wang, Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis, Nat. Med., 2013, 19, 586–594 CrossRef CAS PubMed.
- M. Gawaz, H. Langer and A. E. May, Platelets in inflammation and atherogenesis, J. Clin. Invest., 2005, 115, 3378–3384 CrossRef CAS PubMed.
- A. Reheman, H. Yang, G. Zhu, W. Jin, F. He, C. M. Spring, X. Bai, P. L. Gross, J. Freedman and H. Ni, Plasma fibronectin depletion enhances platelet aggregation and thrombus formation in mice lacking fibrinogen and von Willebrand factor, Blood, 2009, 113, 1809–1817 CrossRef CAS PubMed.
- K. L. Quinn, M. Henriques, A. Tabuchi, B. Han, H. Yang, W. E. Cheng, S. Tole, H. Yu, A. Luo, E. Charbonney, E. Tullis, A. Lazarus, L. A. Robinson, H. Ni, B. R. Peterson, W. M. Kuebler, A. S. Slutsky and H. Zhang, Human neutrophil peptides mediate endothelial-monocyte interaction, foam cell formation, and platelet activation, Arterioscler. Thromb. Vasc. Biol., 2011, 31, 2070–2079 CrossRef CAS PubMed.
- J. Li, J. A. Sullivan and H. Ni, Pathophysiology of immune thrombocytopenia, Curr. Opin. Hematol., 2018, 25, 373–381 CrossRef CAS.
- C. Li, S. Piran, P. Chen, S. Lang, A. Zarpellon, J. W. Jin, G. Zhu, A. Reheman, D. E. van der Wal, E. K. Simpson, R. Ni, P. L. Gross, J. Ware, Z. M. Ruggeri, J. Freedman and H. Ni, The maternal immune response to fetal platelet GPIbalpha causes frequent miscarriage in mice that can be prevented by intravenous IgG and anti-FcRn therapies, J. Clin. Invest., 2011, 121, 4537–4547 CrossRef CAS PubMed.
- X. R. Xu, R. C. Gallant and H. Ni, Platelets, immune-mediated thrombocytopenias, and fetal hemorrhage, Thromb. Res., 2016, 141(Suppl 2), S76–S79 CrossRef CAS PubMed.
- X. R. Xu, D. Zhang, B. E. Oswald, N. Carrim, X. Wang, Y. Hou, Q. Zhang, C. Lavalle, T. McKeown, A. H. Marshall and H. Ni, Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond, Crit. Rev. Clin. Lab. Sci., 2016, 53, 409–430 CrossRef CAS.
- C. Weber and H. Noels, Atherosclerosis: current pathogenesis and therapeutic options, Nat. Med., 2011, 17, 1410–1422 CrossRef CAS PubMed.
- A. Cameron-Vendrig, A. Reheman, M. A. Siraj, X. R. Xu, Y. Wang, X. Lei, T. Afroze, E. Shikatani, O. El-Mounayri, H. Noyan, R. Weissleder, H. Ni and M. Husain, Glucagon-Like Peptide 1 Receptor Activation Attenuates Platelet Aggregation and Thrombosis, Diabetes, 2016, 65, 1714–1723 CrossRef CAS PubMed.
- A. Reheman, X. Xu, E. C. Reddy and H. Ni, Targeting activated platelets and fibrinolysis: hitting two birds with one stone, Circ. Res., 2014, 114, 1070–1073 CrossRef CAS.
- E. J. Benjamin, P. Muntner, A. Alonso, M. S. Bittencourt, C. W. Callaway, A. P. Carson, A. M. Chamberlain, A. R. Chang, S. Cheng, S. R. Das, F. N. Delling, L. Djousse, M. S. V. Elkind, J. F. Ferguson, M. Fornage, L. C. Jordan, S. S. Khan, B. M. Kissela, K. L. Knutson, T. W. Kwan, D. T. Lackland, T. T. Lewis, J. H. Lichtman, C. T. Longenecker, M. S. Loop, P. L. Lutsey, S. S. Martin, K. Matsushita, A. E. Moran, M. E. Mussolino, M. O'Flaherty, A. Pandey, A. M. Perak, W. D. Rosamond, G. A. Roth, U. K. A. Sampson, G. M. Satou, E. B. Schroeder, S. H. Shah, N. L. Spartano, A. Stokes, D. L. Tirschwell, C. W. Tsao, M. P. Turakhia, L. B. VanWagner, J. T. Wilkins, S. S. Wong, S. S. Virani and American Heart Association Council on, Epidemiology Prevention Statistics, Committee Stroke Statistics, Subcommittee, Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association, Circulation, 2019, 139, e56–e528 CrossRef.
- H. Yang, A. Reheman, P. Chen, G. Zhu, R. O. Hynes, J. Freedman, D. D. Wagner and H. Ni, Fibrinogen and von Willebrand factor-independent platelet aggregation in vitro and in vivo, J. Thromb. Haemostasis, 2006, 4, 2230–2237 CrossRef CAS.
- T. N. Durrant, M. T. van den Bosch and I. Hers, Integrin alpha(IIb)beta(3) outside-in signaling, Blood, 2017, 130, 1607–1619 CrossRef CAS PubMed.
- H. Ni and J. Freedman, Platelets in hemostasis and thrombosis: role of integrins and their ligands, Transfus. Apher. Sci., 2003, 28, 257–264 CrossRef.
- B. Estevez, B. Shen and X. Du, Targeting integrin and integrin signaling in treating thrombosis, Arterioscler. Thromb. Vasc. Biol., 2015, 35, 24–29 CrossRef CAS PubMed.
- B. Shen, X. Zhao, K. A. O'Brien, A. Stojanovic-Terpo, M. K. Delaney, K. Kim, J. Cho, S. C. Lam and X. Du, A directional switch of integrin signalling and a new anti-thrombotic strategy, Nature, 2013, 503, 131–135 CrossRef CAS.
- B. Shen, M. K. Delaney and X. Du, Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction, Curr. Opin. Cell Biol., 2012, 24, 600–606 CrossRef CAS PubMed.
- H. Ni, C. V. Denis, S. Subbarao, J. L. Degen, T. N. Sato, R. O. Hynes and D. D. Wagner, Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen, J. Clin. Invest., 2000, 106, 385–392 CrossRef CAS PubMed.
- E. Dunne, C. M. Spring, A. Reheman, W. Jin, M. C. Berndt, D. K. Newman, P. J. Newman, H. Ni and D. Kenny, Cadherin 6 has a functional role in platelet aggregation and thrombus formation, Arterioscler. Thromb. Vasc. Biol., 2012, 32, 1724–1731 CrossRef CAS PubMed.
- A. Reheman, S. Tasneem, H. Ni and C. P. Hayward, Mice with deleted multimerin 1 and alpha-synuclein genes have impaired platelet adhesion and impaired thrombus formation that is corrected by multimerin 1, Thromb. Res., 2010, 125, e177–e183 CrossRef CAS PubMed.
- H. Ni, J. M. Papalia, J. L. Degen and D. D. Wagner, Control of thrombus embolization and fibronectin internalization by integrin alpha IIb beta 3 engagement of the fibrinogen gamma chain, Blood, 2003, 102, 3609–3614 CrossRef CAS PubMed.
- E. Karshovska, Z. Zhao, X. Blanchet, M. M. Schmitt, K. Bidzhekov, O. Soehnlein, P. von Hundelshausen, N. J. Mattheij, J. M. Cosemans, R. T. Megens, T. A. Koeppel, A. Schober, T. M. Hackeng, C. Weber and R. R. Koenen, Hyperreactivity of junctional adhesion molecule A-deficient platelets accelerates atherosclerosis in hyperlipidemic mice, Circ. Res., 2015, 116, 587–599 CrossRef CAS PubMed.
- S. Massberg, K. Schurzinger, M. Lorenz, I. Konrad, C. Schulz, N. Plesnila, E. Kennerknecht, M. Rudelius, S. Sauer, S. Braun, E. Kremmer, N. R. Emambokus, J. Frampton and M. Gawaz, Platelet adhesion via glycoprotein IIb integrin is critical for atheroprogression and focal cerebral ischemia - An in vivo study in mice lacking glycoprotein IIb, Circulation, 2005, 112, 1180–1188 CrossRef CAS PubMed.
- M. U. Naik, J. L. Caplan and U. P. Naik, Junctional adhesion molecule-A suppresses platelet integrin alphaIIbbeta3 signaling by recruiting Csk to the integrin-c-Src complex, Blood, 2014, 123, 1393–1402 CrossRef CAS PubMed.
- M. U. Naik, T. J. Stalker, L. F. Brass and U. P. Naik, JAM-A protects from thrombosis by suppressing integrin alphaIIbbeta3-dependent outside-in signaling in platelets, Blood, 2012, 119, 3352–3360 CrossRef CAS PubMed.
- S. J. Shattil and P. J. Newman, Integrins: dynamic scaffolds for adhesion and signaling in platelets, Blood, 2004, 104, 1606–1615 CrossRef CAS PubMed.
- H. Yang, S. Lang, Z. Zhai, L. Li, W. H. A. Kahr, P. G. Chen, J. Brkic, C. M. Spring, M. J. Flick, J. L. Degen, J. Freedman and H. Ni, Fibrinogen is required for maintenance of platelet intracellular and cell-surface P-selectin expression, Blood, 2009, 114, 425–436 CrossRef CAS.
- P. von Hundelshausen and M. M. N. Schmitt, Platelets and their chemokines in atherosclerosis-clinical applications, Front. Physiol., 2014, 5, 294 Search PubMed.
- K. Chen, X. Chen, H. Xue, P. Zhang, W. Fang, X. Chen and W. Ling, Coenzyme Q10 attenuates high-fat diet-induced non-alcoholic fatty liver disease through activation of the AMPK pathway, Food Funct., 2019, 10, 814–823 RSC.
- W. Lin, W. Wang, D. Wang and W. Ling, Quercetin protects against atherosclerosis by inhibiting dendritic cell activation, Mol. Nutr. Food Res., 2017, 61, 1700031 CrossRef PubMed.
- F. Ya, J. Tian, Q. Li, L. Chen, J. Ren, Y. Zhao, J. Wan, W. Ling and Y. Yang, Cyanidin-3-O-beta-glucoside, a Natural Polyphenol, Exerts Proapoptotic Effects on Activated Platelets and Enhances Megakaryocytic Proplatelet Formation, J. Agric. Food Chem., 2018, 66, 10712–10720 CrossRef CAS PubMed.
- Z. Zhai, J. Wu, X. Xu, K. Ding, R. Ni, W. Hu, Z. Sun and H. Ni, Fibrinogen controls human platelet fibronectin internalization and cell-surface retention, J. Thromb. Haemostasis, 2007, 5, 1740–1746 CrossRef CAS PubMed.
- Y. Wang, E. Vachon, J. Zhang, V. Cherepanov, J. Kruger, J. Li, K. Saito, P. Shannon, N. Bottini, H. Huynh, H. Ni, H. Yang, C. McKerlie, S. Quaggin, Z. J. Zhao, P. A. Marsden, T. Mustelin, K. A. Siminovitch and G. P. Downey, Tyrosine phosphatase MEG2 modulates murine development and platelet and lymphocyte activation through secretory vesicle function, J. Exp. Med., 2005, 202, 1587–1597 CrossRef CAS PubMed.
- G. Zhu, Q. Zhang, E. C. Reddy, N. Carrim, Y. Chen, X. R. Xu, M. Xu, Y. Wang, Y. Hou, L. Ma, Y. Li, M. Rui, T. N. Petruzziello-Pellegrini, C. Lavalle, T. W. Stratton, X. Lei, R. Adili, P. Chen, C. Zhu, J. A. Wilkins, R. O. Hynes, J. Freedman and H. Ni, The integrin PSI domain has an endogenous thiol isomerase function and is a novel target for antiplatelet therapy, Blood, 2017, 129, 1840–1854 CrossRef CAS PubMed.
- Y. Yang, M. C. Andrews, Y. Hu, D. Wang, Y. Qin, Y. Zhu, H. Ni and W. Ling, Anthocyanin extract from black rice significantly ameliorates platelet hyperactivity and hypertriglyceridemia in dyslipidemic rats induced by high fat diets, J. Agric. Food Chem., 2011, 59, 6759–6764 CrossRef CAS PubMed.
- A. J. Unsworth, N. Kriek, A. P. Bye, K. Naran, T. Sage, G. D. Flora and J. M. Gibbins, PPAR gamma agonists negatively regulate alpha IIb beta 3 integrin outside-in signaling and platelet function through up-regulation of protein kinase A activity, J. Thromb. Haemostasis, 2017, 15, 356–369 CrossRef CAS PubMed.
- L. Ma, E. Simpson, J. Li, M. Xuan, M. Xu, L. Baker, Y. Shi, I. Yougbare, X. Wang, G. Zhu, P. Chen, G. J. Prud'homme, A. H. Lazarus, J. Freedman and H. Ni, CD8+ T cells are predominantly protective and required for effective steroid therapy in murine models of immune thrombocytopenia, Blood, 2015, 126, 247–256 CrossRef CAS PubMed.
- I. Yougbare, S. Lang, H. Yang, P. Chen, X. Zhao, W. S. Tai, D. Zdravic, B. Vadasz, C. Li, S. Piran, A. Marshall, G. Zhu, H. Tiller, M. K. Killie, S. Boyd, H. Leong-Poi, X. Y. Wen, B. Skogen, S. L. Adamson, J. Freedman and H. Ni, Maternal anti-platelet beta3 integrins impair angiogenesis and cause intracranial hemorrhage, J. Clin. Invest., 2015, 125, 1545–1556 CrossRef PubMed.
- X. Yan, T. Shen, X. Jiang, X. Tang, D. Wang, H. Li and W. Ling, Coenzyme Q10 consumption promotes ABCG1-mediated macrophage cholesterol efflux: a randomized, double-blind, placebo-controlled, cross-over study in healthy volunteers, Mol. Nutr. Food Res., 2015, 59, 1725–1734 CrossRef CAS PubMed.
- F. Ya, Q. Li, D. Wang, S. Xie, F. Song, R. C. Gallant, Z. Tian, J. Wan, W. Ling and Y. Yang, Cyanidin-3-o-beta-Glucoside Induces Megakaryocyte Apoptosis via PI3 K/Akt- and MAPKs-Mediated Inhibition of NF-kappaB Signalling, Thromb. Haemostasis, 2018, 118, 1215–1229 CrossRef PubMed.
- I. Yougbare, W. S. Tai, D. Zdravic, B. E. Oswald, S. Lang, G. Zhu, H. Leong-Poi, D. Qu, L. Yu, C. Dunk, J. Zhang, J. G. Sled, S. J. Lye, J. Brkic, C. Peng, P. Hoglund, B. A. Croy, S. L. Adamson, X. Y. Wen, D. J. Stewart, J. Freedman and H. Ni, Activated NK cells cause placental dysfunction and miscarriages in fetal alloimmune thrombocytopenia, Nat. Commun., 2017, 8, 224 CrossRef PubMed.
- M. Xu, J. Li, M. A. D. Neves, G. Zhu, N. Carrim, R. Yu, S. Gupta, J. Marshall, O. Rotstein, J. Peng, M. Hou, S. Kunishima, J. Ware, D. R. Branch, A. H. Lazarus, Z. M. Ruggeri, J. Freedman and H. Ni, GPIbalpha is required for platelet-mediated hepatic thrombopoietin generation, Blood, 2018, 132, 622–634 CrossRef CAS PubMed.
- A. Aburima, K. S. Wraith, Z. Raslan, R. Law, S. Magwenzi and K. M. Naseem, cAMP signaling regulates platelet myosin light chain (MLC) phosphorylation and shape change through targeting the RhoA-Rho kinase-MLC phosphatase signaling pathway, Blood, 2013, 122, 3533–3545 CrossRef CAS PubMed.
- M. Yang, B. C. Cooley, W. Li, Y. Chen, J. Vasquez-Vivar, N. O. Scoggins, S. J. Cameron, C. N. Morrell and R. L. Silverstein, Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions, Blood, 2017, 129, 2917–2927 CrossRef CAS PubMed.
- P. Blair and R. Flaumenhaft, Platelet alpha-granules: basic biology and clinical correlates, Blood Rev., 2009, 23, 177–189 CrossRef CAS PubMed.
- P. C. Burger and D. D. Wagner, Platelet P-selectin facilitates atherosclerotic lesion development, Blood, 2003, 101, 2661–2666 CrossRef CAS PubMed.
- N. Gerdes, T. Seijkens, D. Lievens, M. J. Kuijpers, H. Winkels, D. Projahn, H. Hartwig, L. Beckers, R. T. Megens, L. Boon, R. J. Noelle, O. Soehnlein, J. W. Heemskerk, C. Weber and E. Lutgens, Platelet CD40 Exacerbates Atherosclerosis by Transcellular Activation of Endothelial Cells and Leukocytes, Arterioscler. Thromb. Vasc. Biol., 2016, 36, 482–490 CrossRef CAS PubMed.
- P. von Hundelshausen and M. M. Schmitt, Platelets and their chemokines in atherosclerosis-clinical applications, Front. Physiol., 2014, 5, 294 Search PubMed.
- P. von Hundelshausen, K. S. Weber, Y. Huo, A. E. Proudfoot, P. J. Nelson, K. Ley and C. Weber, RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium, Circulation, 2001, 103, 1772–1777 CrossRef CAS PubMed.
- B. Nieswandt, D. Varga-Szabo and M. Elvers, Integrins in platelet activation, J. Thromb. Haemostasis, 2009, 7((Suppl 1), 206–209 CrossRef PubMed.
- A. E. May, T. Kalsch, S. Massberg, Y. Herouy, R. Schmidt and M. Gawaz, Engagement of glycoprotein IIb/IIIa (alpha(IIb)beta3) on platelets upregulates CD40L and triggers CD40L-dependent matrix degradation by endothelial cells, Circulation, 2002, 106, 2111–2117 CrossRef CAS PubMed.
- A. Smolenski, Novel roles of cAMP/cGMP-dependent signaling in platelets, J. Thromb. Haemostasis, 2012, 10, 167–176 CrossRef CAS PubMed.
- Z. Li, M. K. Delaney, K. A. O'Brien and X. Du, Signaling during platelet adhesion and activation, Arterioscler. Thromb. Vasc. Biol., 2010, 30, 2341–2349 CrossRef CAS PubMed.
- P. da Costa Martins, N. van den Berk, L. H. Ulfman, L. Koenderman, P. L. Hordijk and J. J. Zwaginga, Platelet-monocyte complexes support monocyte adhesion to endothelium by enhancing secondary tethering and cluster formation, Arterioscler. Thromb. Vasc. Biol., 2004, 24, 193–199 CrossRef CAS PubMed.
- X. R. Xu, N. Carrim, M. A. Neves, T. McKeown, T. W. Stratton, R. M. Coelho, X. Lei, P. Chen, J. Xu, X. Dai, B. X. Li and H. Ni, Platelets and platelet adhesion molecules: novel mechanisms of thrombosis and anti-thrombotic therapies, Thromb. J., 2016, 14, 29 CrossRef PubMed.
- E. A. Podrez, T. V. Byzova, M. Febbraio, R. G. Salomon, Y. Ma, M. Valiyaveettil, E. Poliakov, M. Sun, P. J. Finton, B. R. Curtis, J. Chen, R. Zhang, R. L. Silverstein and S. L. Hazen, Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype, Nat. Med., 2007, 13, 1086–1095 CrossRef CAS PubMed.
- N. Suksomboon, N. Poolsup and N. Juanak, Effects of coenzyme Q10 supplementation on metabolic profile in diabetes: a systematic review and meta-analysis, J. Clin. Pharm. Ther., 2015, 40, 413–418 CrossRef CAS PubMed.
- H. Mabuchi, A. Nohara, J. Kobayashi, M. A. Kawashiri, S. Katsuda, A. Inazu, J. Koizumi and Hokuriku Lipid Research Group, Effects of CoQ10 supplementation on plasma lipoprotein lipid, CoQ10 and liver and muscle enzyme levels in hypercholesterolemic patients treated with atorvastatin: a randomized double-blind study, Atherosclerosis, 2007, 195, e182–e189 CrossRef CAS PubMed.
- M. I. Furman, L. A. Krueger, M. D. Linden, M. L. Fox, S. P. Ball, M. R. Barnard, A. L. Frelinger 3rd and A. D. Michelson, GPIIb-IIIa antagonists reduce thromboinflammatory processes in patients with acute coronary syndromes undergoing percutaneous coronary intervention, J. Thromb. Haemostasis, 2005, 3, 312–320 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fo01686d |
| This journal is © The Royal Society of Chemistry 2020 |