
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical fate of quaternary ammonium compounds in river water†
Priya I.
Hora
 and
William A.
Arnold
*
and
William A.
Arnold
*
Department of Civil, Environmental, and Geo- Engineering, University of Minnesota – Twin Cities, 500 Pillsbury Drive SE, Minneapolis, Minnesota 55455, USA. E-mail: arnol032@umn.edu; Fax: +1-612-626-7750; Tel: +1-612-625-8582
First published on 17th April 2020
Abstract
Quaternary ammonium compounds (QACs) are not completely removed during wastewater treatment and are frequently detected in surface waters and sediments. The photochemical transformation of QACs has not been thoroughly investigated as a potential degradation pathway affecting their fate in the environment. Kinetic studies of common QACs with and without aromatic groups under simulated and natural sunlight conditions were performed with model sensitizers and dissolved organic matter to estimate photochemical half-lives in the aquatic environment. All QACs investigated react with hydroxyl radicals at diffusion-controlled rates (∼2.9 × 109 to 1.2 × 1010 M−1 s−1). Benzethonium reacted via direct photolysis (ΦBZT,outdoor = 1.7 × 10−2 (mol Ei−1)). Benzethonium also reacted with the triplet excited state model sensitizer 2-acetylnaphthalene, but evidence suggests this reaction pathway is unimportant in natural waters due to faster quenching of the triplet 2-acetylnapthalene by oxygen. Reactivity with singlet oxygen for the QACs was minimal. Overall, reactions with hydroxyl radicals will dominate over direct photolysis due to limited spectral overlap of sunlight emission and QAC absorbance. Photolysis half-lives are predicted to be 12 to 94 days, indicating slow abiotic degradation in surface water.
Environmental significanceQuaternary ammonium compounds (QACs) are extensively used worldwide. The photochemical fate of QACs in aquatic environments is not fully understood. The present study comparatively evaluates the contributions of direct and indirect photolysis on the transformation of five QACs in the environment. Through a series of sensitizer experiments in buffer and river water experiments under simulated and natural sunlight, this work reveals that QACs are susceptible to indirect photolysis by hydroxyl radicals, but direct photolysis is only important for benzethonium. This study aids in assessing the role of photolysis in the overall fate of QACs in surface waters. |
1. Introduction
Since the late 1930s, quaternary ammonium compounds (QACs) have been a widely used class of chemicals, chemical mixtures, and additives in a variety of industrial, agricultural, clinical, and consumer product applications.1–3 Some QACs are designated by the U.S. Environmental Protection Agency and the Organization for Economic Cooperation and Development as high production volume chemicals (≥1 million pounds per year imported or manufactured). An extensive primary use of QACs is as cationic surfactants.4,5 QACs are used in fabric softeners, antistatic agents, commercial disinfectants, sanitizers, antimicrobials, biocides, herbicides, pesticides, food preservatives, detergents, and phase transfer agents, and active ingredients in pharmaceuticals, cosmetics, and personal care products such as shampoos and antibacterial soaps.1,2,6,7 QACs are also used as lytic biocides, corrosion inhibitors, and clay stabilizers in hydraulic fracturing.6,8–10QACs contain at least one hydrophobic hydrocarbon chain bonded to a positively charged quaternary nitrogen atom and have other alkyl groups that tend to be mostly short-chain substituents such as methyl or benzyl functional groups.1,11 The central nitrogen is covalently bonded to four carbon-containing “R”-substituents (R4N+).6 There are three major classes of QACs: benzylalkyl dimethyl ammonium compounds (BACs), alkyltrimethyl ammonium compounds (ATMACs), and dialkyldimethyl ammonium compounds (DADMACs).
QACs are introduced into aquatic ecosystems through point source pollution and the discharge of effluents from wastewater treatment plants and are considered emerging pollutants.1,2,7,12–16 QACs have been detected in environmental compartments ranging from surface water to sediments and may be found in septic and sewage impacted areas.7,8,17–19 The worldwide average concentration of QACs in domestic wastewater, treated effluents, and surface water has been reported to be approximately 500 μg L−1, 50 μg L−1, and 40 μg L−1, respectively.3,13,20 The three QAC classes most frequently detected in the environment are: DADMACs with alkyl chain lengths from C8 to C18, ATMACs with alkyl chain lengths from C12 to C18, and BACs with alkyl chain lengths from C12 to C18.1 Approximately 75% of QACs used annually are released into wastewater treatment systems and BAC is the most frequently found QAC group worldwide in municipal wastewater effluents at concentrations ranging from 20 to 300 μg L−1.14,19,21–23 Although a large fraction of QACs are removed during the wastewater treatment process, QACs are still detected in aquatic environments, especially at higher concentrations in locations downstream of wastewater effluent discharge.9,13,17,18,20,24–26
Understanding the fate of QACs is important because an estimated 32.5% of QACs produced worldwide are released into the environment,3 and they retain their biocidal properties after application.3,27–29 QACs are known to be aerobically biodegradable, but the extent of biodegradation is variable.4,8,30–32 Based on laboratory studies, rates can vary from hours to months and depend on factors such as QAC structure, concentration, and microbial acclimation.33–36 Results from some of these studies suggest that biodegradation rates decrease with increasing hydrophobic alkyl chain length. Additionally, the data indicate that QACs with a benzyl group have decreased biodegradation.8 Moreover, while water-soluble, QACs have a strong tendency to sorb to organic matter in water bodies due to favorable hydrophobic and electrostatic interactions,7,17,18,37 which might reduce bioavailability and inhibit biodegradation. QACs have been detected in sediments at concentrations higher than in the aqueous phase,18–20,24,38 suggesting that QACs are environmentally persistent8 and their transport and fate are dependent upon association with organic matter in surface waters.38
Ecotoxicological effective concentrations (EC50) of QACs reported for various fish, algae, crustaceans, daphnia, rotifers, and bacterial, and protozoan species range from tens of μg L−1 to mg L−1.1,2,32,39,40 While QACs generally have not been considered acutely toxic to aquatic organisms based on reported surface water concentrations below many EC50 values, more research is needed.2,7,39 QACs might still pose cumulative chronic effects. Concerns about the detection of QACs in the environment stem from the possibility that prolonged exposure to sub-inhibitory QAC concentrations co-selects for antibiotic resistant bacteria.3,28,41–45
The photochemical behavior of QACs in natural environments is still not well understood. BACs are considered stable to direct photodegradation.6,46 Direct photolysis occurs when a contaminant undergoes transformation, such as breaking of bonds, resulting from directly absorbing light energy.47 Many QACs lack chromophoric functional groups or exhibit poor absorption of light in the visible spectrum; thus, direct photolysis processes may be limited in natural waters. Indirect photolysis, facilitated by QAC association with dissolved organic matter (DOM), could be an important and under-studied attenuation mechanism. Indirect photolysis occurs when a photosensitizer such as organic matter in natural waters absorbs light energy, becomes electronically excited, and subsequently reacts with a contaminant or produces transient photochemically produced reactive intermediates (PPRIs) that are energetically capable of transforming target contaminants.47 PPRIs, including reactive oxygen species (ROS), such as hydroxyl radicals (˙OH) and singlet oxygen (1O2), and non-ROS excited triplet states of DOM (3DOM*), react with contaminants in natural systems thus enhancing degradation and increasing phototransformation rates.47–50 QACs may sorb to DOM, which is ubiquitous in natural waters, and this interaction affects their distribution, transformation, and bioavailability. In aquatic systems, DOM may act as a sink for QACs and as a photosensitizer. For some hydrophobic compounds, attachment to DOM can expose the pollutant to heterogeneous microenvironments with high concentrations of PPRIs51 that accelerate degradation, or alternatively, this association could shield compounds from light, inhibiting reactions.
This study examines the photochemical fate of 5 QACs in surface waters and addresses gaps in knowledge on biocide degradation.6 By conducting experiments in buffered ultrapure water using sensitizers and radical quenchers and in Mississippi River water, the relative contributions of direct and indirect photolysis for each QAC are determined. Kinetic studies allow for calculation of second-order reaction rate constants in radical-mediated processes, estimation of quantum yields where relevant, and estimation of environmental half-lives in natural waters.
2. Materials and methods
2.1 Chemicals and reagents
The quaternary ammonium compounds used in this study were: benzyldimethyl-n-dodecylammonium chloride (C12-BAC, 98%, Alfa Aesar), benzyldimethyltetradecyl-ammonium chloride hydrate (C14-BAC, 99%, TCI America), benzethonium chloride (BZT, 99%, TCI America), didodecyldimethylammonium bromide (C12-DADMA, 99.9%, Santa Cruz Biotechnology), and didodecyltrimethyl ammonium bromide (C12-ATMA, 99%, Acros Organics). Structures are shown in the ESI Table S1,† and these compounds were selected because they represent high use molecules in different QAC classes. Experimental aqueous stocks, solutions, and buffers were prepared using ultrapure water (resistivity 18.2 MΩ cm, EMD Millipore Corp.). Hydrogen peroxide (H2O2, 30%, Fisher) was used as an ˙OH sensitizer, Rose Bengal (RB, dye content 95%, Sigma-Aldrich) and 2-acetylnaphthalene (2-AN, 99%, Alfa Aesar), were used as 1O2 sensitizers, para-chlorobenzoic acid (pCBA, 98%, Acros Organics) was used as an ˙OH probe, furfuryl alcohol (FFA, 98%, Sigma-Aldrich) was used as an 1O2 probe, HPLC grade 2-propanol (IPA, 99.9%, Fisher) was used as an ˙OH quencher, L-Histidine (His, 99%, Sigma Aldrich) was used as an 1O2 quencher, and sorbic acid (99%, Alfa Aesar) was used as a quenching agent for triplet excited states. p-Nitroacetophenone and pyridine (PNAP-PYR) were used as an actinometer.52,53 Tetraoctylammonium bromide (C8-TAA, 98%, Sigma Aldrich) and tetrapentylammonium bromide (C5-TAA, 99%, Sigma Aldrich) were used as internal standards for mass spectrometry analysis. Sodium phosphate monobasic monohydrate (98%, Fisher), sodium phosphate dibasic anhydrous (99%, J.T. Baker), ammonium acetate (97%, Macron), sodium hydroxide (98%, Macron), hydrochloric acid (35%, TraceMetal Grade, Fisher), o-phosphoric acid (85%, HPLC Grade, Fisher), glacial acetic acid (99%, ACS Grade, BDH) and formic acid (88%, ACS Grade, Fisher) were used to prepare buffers for experiments and high-pressure liquid chromatography (HPLC) analysis. HPLC grade methanol (99.9%, Fisher) and HPLC grade acetonitrile (99.9%, Fisher or J.T. Baker) were used as HPLC eluents. Optima LC/MS grade (Fisher Scientific) acetonitrile, water, formic acid, and ammonium acetate were used as eluents for mass spectrometry.2.2 River water sample collection and analysis
Mississippi River water (MRW) grab samples were collected in Minneapolis, MN in clean (soaked in Alcanox overnight and rinsed 3× with tap water, deionized water, and ultrapure water), combusted (5 hours at 550 °C) Pyrex glass media bottles at Saint Anthony Falls Laboratory (February 27, 2019) and the University of Minnesota Boathouse dock (June 5, 2018). Whole water samples were immediately vacuum filtered through a combusted 0.7 μm glass fiber filter followed by a sterile 0.2 μm Omnipore membrane filter (Millipore) and then stored at 4 °C in the dark prior to photolysis experiments.River water samples were characterized immediately before conducting photolysis experiments. Nitrate and nitrite were measured by ion chromatography using a Metrohm Compact ion chromatograph model 930. Calibration curves were generated with ACS grade sodium salts and detection limits were 0.1 mg L−1 as N for both NO3− and NO2−. Dissolved organic carbon (DOC), as non-purgeable organic carbon, and dissolved inorganic carbon (DIC) were measured with a Shimadzu TOC-L total organic carbon analyzer. Calibration curves were generated using potassium hydrogen phthalate (≥99.95%, Sigma-Aldrich) for DOC and anhydrous sodium carbonate (ACS grade, Fisher) and sodium bicarbonate (99.7–100.3%, Sigma-Aldrich) for DIC. River water pH was measured using a Thermo Scientific Orion pH probe (calibrated with pH 4, 7, and 10 standard solutions from Fisher). This information is tabulated in Table S2.†
2.3 Absorption spectra
Ultraviolet-visible absorbance of river water as well as aqueous QAC, probe, and actinometer solutions was measured with a Shimadzu UV-1601PC spectrophotometer using 1 cm quartz cuvettes. The light absorption properties of the chemicals are shown in Fig. S1.†2.4 Analytical methods
Concentrations of QACs with an aromatic group (C12-BAC, C14-BAC, and BZT), the actinometer, and probe compounds were determined by HPLC using an Agilent 1100 LC with a variable wavelength detector. HPLC methods are summarized in Table S3.† C12-DADMA and C12-ATMA were quantified on a Thermo Dionex UltiMate 3000 RSLCnano system equipped with a Thermo TSQ Vantage triple quadrupole tandem mass spectrometer (LC-MS/MS) in positive electrospray ionization mode at the University of Minnesota Masonic Cancer Center using the following parameters: heated electrospray ionization, spray voltage of 2.6 kV, vaporizer temperature of 250 °C, sheath gas pressure of 25 psi, aux gas pressure of 20 psi, capillary temperature of 300 °C, SRM scan width (m/z) of 0.1 and scan time (s) of 0.25. Separation was performed on a Waters XSelect CSH C18 (3.5 μm, 130 Å, 50 × 2.1 mm) column. Flow rate was maintained at 250 μL min−1, sample temperature was 10 °C, the column was kept at room temperature, and 4 μL was injected in the column. Isocratic methods (10 minutes in duration) of mobile phases A: 2 mM ammonium acetate with 0.1% formic acid in water, B: 0.1% formic acid in acetonitrile, and C: isopropanol were used (65% A, 20% B, and 15% C for C12-ATMA and 37% A, 33% B, and 30% C for C12-DADMA). Flow was diverted to waste from 0 to 4.5 minutes. For C12-ATMA the m/z values monitored were 228.26 and 60.15 and the retention time was 6.1 min, and for C12-DADMA they were 382.44, 214.28, and 5.6 min.2.5 Model sensitizer photochemical experiments
Experiments were performed to determine the reactivity of QACs with ˙OH using H2O2 and with 1O2 using RB and 2-AN. Duplicate experiments were run in sealed quartz test tubes in an Atlas Suntest CPS+ solar simulator equipped with a 1500 W xenon arc lamp and 290 nm cutoff filter, though it has previously been reported that there is some light transmission <290 nm,49 at a light intensity over 300–800 nm of 765 W m−2 for ˙OH experiments, at 350 W m−2 for 1O2 experiments with RB, and at 765 W m−2 for 1O2 experiments with 2-AN. The different light intensities were used to control the time over which the reaction occurred. During ˙OH experiments, QACs (C12 and C14-BAC, BZT) and pCBA were spiked from concentrated aqueous stock solutions prepared in unbuffered ultrapure water to achieve an initial concentration of approximately 10 μM in 6–8 mL of phosphate buffer (10 mM, pH 7.0) with 1 mM H2O2. Sub-samples of 200 or 500 μL were withdrawn at regular time intervals using combusted glass Pasteur pipettes and dispensed into 1.5 mL amber glass HPLC vials with crimp caps. Equivalent experiments with C12-DADMA and C12-ATMA had initial QAC concentrations of 1 μM and a pCBA concentration of 5 μM. Sub-samples of 900 μL were dispensed into screw top HPLC vials with 100 μL internal standard mix in acetonitrile (C8-TAA for C12-DADMA and C5-TAA for C12-ATMA) for LC-MS/MS analysis and 500 μL samples were withdrawn for pCBA analysis via HPLC. Control experiments to account for direct photochemical and non-photochemical losses were performed. Direct controls were run in buffer with the QAC and probe. Dark controls containing the QAC, sensitizer, and probe compound were wrapped in aluminum foil. A quenched control spiked with 1% IPA was also simultaneously run.Singlet oxygen experiments were conducted in a similar fashion with test tubes containing 8 mL of phosphate buffer (10 mM, pH 7.0), 1–2 μM RB (ground-state reduction potential, Eo (S/S−) = −0.54 VSHE; triplet energy, ET = 171 kJ mol−1; triplet state reduction potential, Eo* (3S*/S−) = 1.23 VSHE; 1O2 quantum yield ΦΔ = 0.75),54 40 μM FFA, and 10 μM BZT or C12-BAC. L-Histidine (20 mM) was used as an 1O2 quencher. To explore the role of 1O2 and triplet states, experiments with deoxygenated (i.e., nitrogen-sparged) solutions were carried out. Additional experiments were conducted for BZT with 16 μM 2-AN (ground-state reduction potential, Eo (S/S−) = −1.48 VSHE; triplet energy, ET = 249 kJ mol−1; triplet state reduction potential, Eo* (3S*/S−) = 1.10 VSHE; 1O2 quantum yield ΦΔ = 0.71).54 Deoxygenated solutions were prepared in an anaerobic glove bag (97% N2/3% H2, Pd catalyst, Coy Laboratory Products Inc.) by spiking aqueous stocks (in deoxygenated ultrapure water) of BZT, 2-AN, and FFA in deoxygenated pH7 10 mM phosphate buffer, which was sparged with N2 gas for 2 hours. The solution was placed into quartz test tubes and further bubbled with N2 outside of the glove bag for 5–10 minutes prior to being sealed with foil and rubber septa caps. The deoxygenated tubes were sampled sacrificially at 20 and 50 minutes. Another experiment with BZT in deoxygenated phosphate buffer was run with 1 mM sorbic acid to quench triplets.55,56 Sorbic acid was stirred overnight on a heated stir plate and then sonicated and degassed with application of vacuum for 10–20 minutes at 30–40 °C before being brought into the glove bag. Other solutions were prepared as above, and all sample solutions were bubbled with N2 outside the glove bag for 30 minutes prior to being dispensed into test tubes under a nitrogen blanket and sealed with foil and rubber septa caps. Tubes were sampled sacrificially after 10, 20, 30, 40, and 50 minutes.
Steady-state concentrations of hydroxyl radicals ([˙OH]ss) and singlet oxygen ([1O2]ss were calculated from the ratio of the measured pseudo-first-order rate constant (kobs), determined via linear regression, and the bimolecular reaction rate constant of pCBA with ˙OH (k˙OH,pCBA = 5 × 109 M−1 s−1)57 and FFA with 1O2 (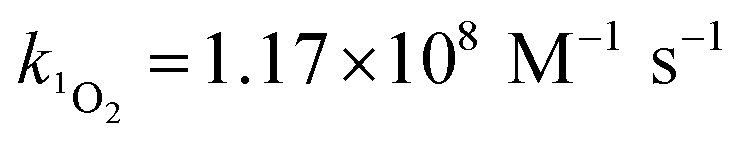 at 30 °C).58 Direct photolysis losses of the probes were negligible. Assuming a first-order kinetic dependence on each QAC and pCBA and that the reactions proceed independently, bimolecular reaction rate constants of QACs with ˙OH (k˙OH,QAC) were derived using competition kinetics according to eqn (1).
at 30 °C).58 Direct photolysis losses of the probes were negligible. Assuming a first-order kinetic dependence on each QAC and pCBA and that the reactions proceed independently, bimolecular reaction rate constants of QACs with ˙OH (k˙OH,QAC) were derived using competition kinetics according to eqn (1).
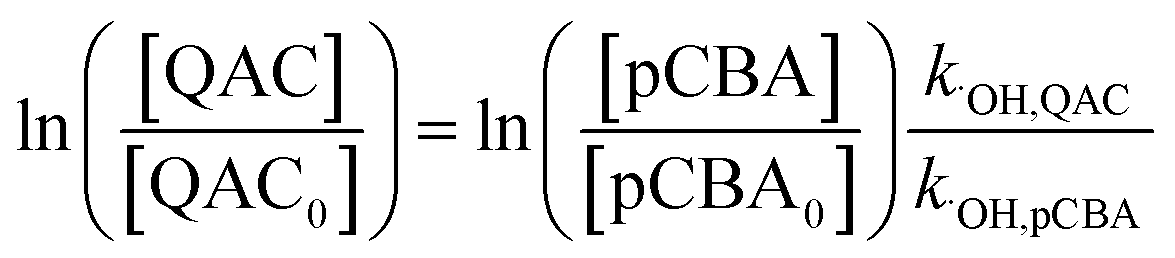 | (1) |
The rate constants for the respective QACs were obtained from linear regressions of logarithmic plots of normalized QAC versus pCBA concentrations in the same tube.
2.6 Simulated and outdoor photochemical experiments in river water
QAC photolysis experiments were performed on Mississippi River water and phosphate buffer under simulated and natural sunlight to assess the relative importance of direct photolysis and indirect photochemical transformation due to PPRIs. The radical-specific probe pCBA was used for the quantification of ˙OH steady-state concentrations in the same tube. Direct photolysis controls were performed for all QACs and pCBA in phosphate buffer solutions (10 mM, pH 7.0), and dark control experiments for QACs and pCBA in river water were performed in tubes wrapped with aluminum foil. River water solutions spiked with 1% IPA as a radical quencher were concurrently irradiated to evaluate the importance of indirect photolysis processes involving ˙OH. Time resolved samples of 200 or 500 μL were removed for HPLC analysis to determine reaction kinetics.For simulated sunlight experiments, C12 and C14-BAC and BZT were photolyzed in filtered Mississippi River water using a solar simulator. The light intensity was set to 765 W m−2, and samples were taken every 5 hours. All experiments were run in duplicate. For outdoor natural sunlight experiments, C12 and C14-BAC and BZT were photolyzed in filtered Mississippi River water collected on June 5, 2018 over a total of 26.8 hours from June 28, 2018 to July 6, 2018 (Table S4† summarizes the exposures) on the rooftop of the University of Minnesota Mechanical Engineering building (latitude: 44°58′30′′N, longitude: 93°14′1′′W, elevation: 860 ft, azimuth: 218°SW).
Aliquots of 6–8 mL initial volume reaction solutions in 10 mL quartz test tubes (13 mm o.d. and 11 mm i.d.) were sealed with pre-combusted aluminum foil squares and then rubber septa caps. The test tubes were irradiated at a 30° angle from the horizontal. Initial QAC and probe concentrations were nominally 10 μM, allowing for direct monitoring of reactant and probe compound concentrations and minimization of light screening.
The chemical actinometer PNAP-PYR was simultaneously irradiated during simulated and natural sunlight river water experiments to estimate the spectral irradiance of the light source, calculate direct photolysis quantum yields for QACs at low optical density, and plot long duration experimental data by dose via actinometer loss to compare laboratory and outdoor experiments. The PNAP-PYR actinometer was chosen because the half-life of the QACs was expected to be greater than 4 hours based on preliminary experiments. The concentrations of pyridine were set using relationships in Leifer52 such that the actinometer loss rate would occur over the same time scale as the QAC loss. The spectral irradiance of the solar simulator at wavelengths 276–400 nm was estimated using a bimolecular solution of 47.2 mM pyridine added to 10.7 μM p-nitroacetophenone for a desired half-life of 17 hours in river water experiments. For rooftop experiments, a 11.4 μM PNAP/52.3 mM pyridine solution was used. The molar absorptivity of PNAP (εPNAP) was calculated by linear regression of absorbance measurements of 3 analytical standards (concentrations 5, 10, and 15 μM) prepared in Milli-Q with trace acetonitrile.
The SMARTS 2.9.5 model59–61 was used to calculate the solar spectral irradiance (Fig. S1†) over the time course of outdoor experiments. Salient inputs are summarized in ESI Table S5.† Global tilted irradiances were calculated for each solar hour in an experimental day, summed, and divided by the total exposure hours for each day. The daily irradiances were then averaged for quantum yield calculations.
Direct photolysis quantum yields were calculated following the methods outlined in Dulin and Mill53 and Leifer52 with the updated PNAP-PYR quantum yield relationship published by Laszakovits et al.,62 summarized by eqn (2).
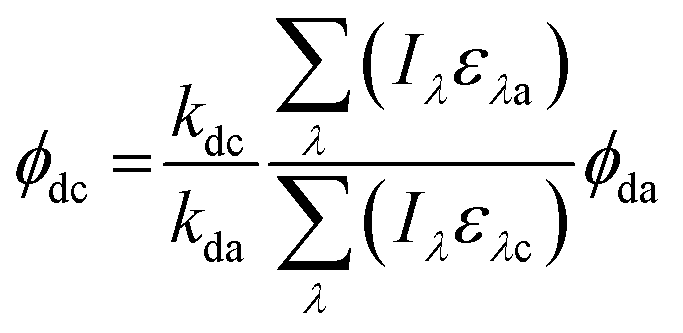 | (2) |
 = the slope of the logarithmic plot of direct photodegradation of the compound versus actinometer determined by linear regression. The wavelength range was 276 to 400 nm for the solar simulator and 280 to 400 nm for outdoor experiments.
= the slope of the logarithmic plot of direct photodegradation of the compound versus actinometer determined by linear regression. The wavelength range was 276 to 400 nm for the solar simulator and 280 to 400 nm for outdoor experiments.
2.7 Data analysis
Data points from replicate experiments were plotted individually. The slope of the regression lines and their standard error were used to calculate the required slopes/reaction rate constants and the associated 95% confidence intervals, respectively. Linear fits and generation of rate constants, standard errors of slopes, and 95% confidence intervals were performed with Origin, Version 2017 (OriginLab Corporation, Northampton, MA, USA).3. Results and discussion
3.1 Reactivity of QACs with ˙OH from H2O2 sensitizer experiments
Based on previous work with ionic liquids,63 ˙OH was hypothesized to be an important PPRI for the indirect photochemical transformation of QACs. Competition kinetics experiments were conducted with H2O2 and pCBA to determine bimolecular reaction rate constants between QACs and ˙OH (k˙OH,QAC). Fig. 1 shows the natural log of the ratio of concentration normalized by the initial concentration of QACs during a solar simulator irradiation experiment with H2O2 producing ˙OH. The C12-ATMA H2O2 40 minute data points (Fig. 1D) were treated as outliers due to anomalous instrumental measurements, indicated by open symbols, and excluded from the ensuing regression and analysis. For all the compounds studied, photolysis in the presence of ˙OH follows pseudo-first order kinetics (r2 ≥ 0.83). Dark controls dosed with H2O2 showed no significant change in QAC concentration. Quantification of steady-state ˙OH concentrations was done using pCBA (Fig. S2 and Table S6†).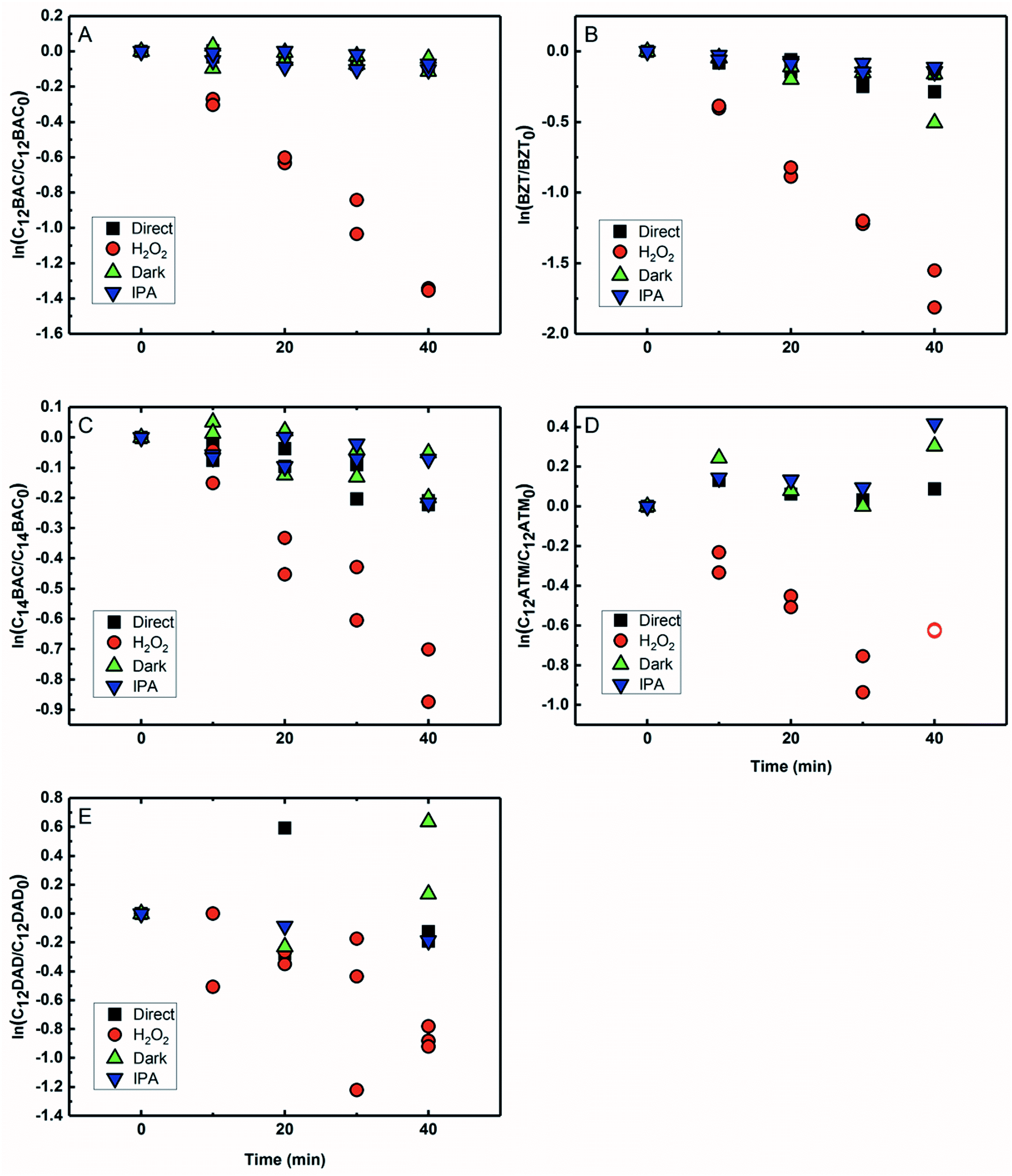 | ||
| Fig. 1 Phototransformation over time of: C12-BAC (A), BZT (B), C14-BAC (C), C12-ATMA (D), and C12-DADMA (E) in the presence of pCBA under simulated sunlight in phosphate buffer (direct, black squares), with 1 mM hydrogen peroxide (H2O2, red circles), H2O2 dark control (dark, green triangle), and H2O2 with 1% isopropanol (IPA, blue upside down triangle). Open symbols denote outliers. | ||
Fig. 2 shows the log–log plot of concentrations of the 5 QACs versus that of pCBA. Using the slopes of the linear regressions in Fig. 2 and the bimolecular reaction rate constant of pCBA with ˙OH, k˙OH,QAC values were calculated for the QACs using eqn (1) (Table 1).
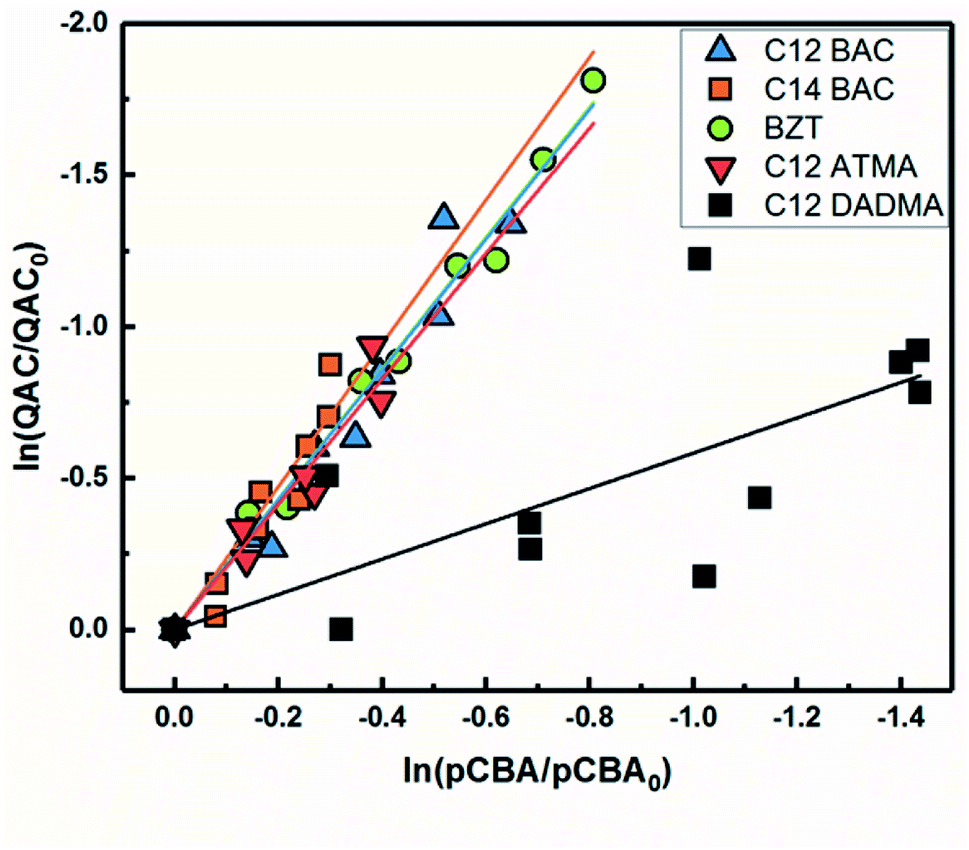 | ||
| Fig. 2 log–log concentrations of QACs versus para-chlorobenzoic acid from solar simulator irradiation experiments with hydrogen peroxide. Solid lines represent linear regressions. | ||
| QAC | Hydrogen peroxide | Solar simulator, river water | Outdoors, river water | ||
|---|---|---|---|---|---|
| k ˙OH,QAC (M−1 s−1) | [˙OH]ss (M) | k ˙OH,QAC (M−1 s−1) | [˙OH]ss (M) | k ˙OH,QAC (M−1 s−1) | |
| a Errors represent 95% confidence intervals associated with the regression slopes in Fig. 2. | |||||
| C12-BAC | (1.1 ± 0.1) × 1010 | (7.0 ± 0.2) × 10−16 | (1.1 ± 0.2) × 1010 | (3.8 ± 0.3) × 10−16 | (1.2 ± 0.1) × 1010 |
| C14-BAC | (1.2 ± 0.2) × 1010 | (6.7 ± 0.1) × 10−16 | (1.2 ± 0.3) × 1010 | (3.8 ± 0.3) × 10−16 | (1.2 ± 0.3) × 1010 |
| BZT | (1.08 ± 0.05) × 1010 | (6.1 ± 0.4) × 10−16 | (9.3 ± 1.3) × 109 | (3.9 ± 0.4) × 10−16 | (9.4 ± 3.2) × 109 |
| C12-ATMA | (1.03 ± 0.14) × 1010 | ||||
| C12-DADMA | (2.9 ± 1.0) × 109 | ||||
The results demonstrate that the QACs studied react with ˙OH at or near the diffusion-controlled limit (i.e., k˙OH,c = 5 × 109 − 1010 M−1 s−1), which is the case for many organic contaminants.57,64 The hydroxyl radical reacts unselectively with most organic compounds and reactions occur at different functional groups. Typically, ˙OH behaves as an electrophile and reacts with organic pollutants in two ways: (1) OH group addition to an aromatic ring or a double bond and (2) hydrogen atom abstraction from aliphatic carbon-hydrogen bonds.57,64 The structural differences of the studied QACs did not significantly affect the bimolecular rate constant, except the reaction between DADMA and ˙OH is about an order of magnitude lower than for the other compounds.
Similar to imidazolium cations,63 there was a slight trend seen for increasing k˙OH,QAC with the increasing side chain length of C12 and C14-BAC though the values are within error. For C12-DADMA, where reaction with ˙OH only occurs via H atom abstraction at C–H bonds in the side chains, rates were smaller than for BACs and BZT, and C12-ATMA. The exchange of a methyl group (ATMA) for a 12 carbon alkyl chain (DADMA) noticeably slows the reaction kinetics. The alkoxy groups in BZT have both inductive electron withdrawing and resonance electron-donating effects. The hydroxyl radical reacts rapidly with compounds containing aromatic rings with electron-donating substituents, and addition reactions still occur at appreciable rates in the presence of electron-withdrawing groups.64 The coincidence in rates for the BACs, BZT, and C12-ATMA suggests that H atom abstraction at C–H bonds in the side chain contributes to the overall reaction of BACs and BZT with ˙OH in addition to any reaction at the ring.
3.2 Assessing reactivity with other PPRIs
To further explore the role of specific PPRIs in QAC degradation in the presence of DOM and more definitively rule out additional pathways, sensitizer experiments were conducted to assess reactivity with 1O2 and triplet excited states. Because 1O2 is a selective electrophile preferentially reacting with electron-rich functional groups like sulfides, phenols, alkenes and aromatic compounds, anilines, furans, and other electron-rich heterocycles,58,65,66 it was hypothesized QACs would have limited interaction with this reactive oxygen species. Moreover, because DOM can be a microheterogeneous environment in which 1O2 potentially reaches higher steady-state concentrations51,67 and QACs will associate with DOM, enhanced reactivity might occur if in fact they do react with 1O2.Experiments with RB showed no reaction between C12-BAC (Fig. S3A†) and 1O2 (FFA was used to measure the bulk concentration of 1O2 − [1O2]ss = (3.9 ± 1.2) × 10−12 M) (Fig. S3B, Table S7†) under air saturated conditions, and there was no statistically significant difference between the pseudo-first-order rate constant for C12-BAC loss with RB and the histidine quenched and dark controls. No loss of C12-BAC was observed in the deoxygenated control as well, indicating no reactivity with triplet excited states of RB.
BZT exhibited possible reactivity with the triplet excited state of RB based on decay in the histidine quenched control, and BZT loss in the dark control suggested that BZT was also potentially reacting with RB (Fig. S4†). Additional control experiments (not shown) indicated that BZT interacts with RB under ambient, dark conditions and therefore a different 1O2 sensitizer, 2-AN, which is also a model 3DOM* sensitizer, was used.
The experiments with BZT and 2-AN illustrated minimal BZT loss due to 1O2 (Fig. S5A†), and there was no statistically significant difference between the 2-AN results and the dark control and the histidine control where 1O2 is quenched. A deoxygenated control experiment was also conducted. Interestingly, in the deoxygenated solution containing 2-AN, BZT exhibited an approximately 84% decrease after 50 minutes of irradiation. The addition of sorbic acid, which quenches triplet-excited states (with energy >200 kJ mol−1) of organic compounds, to the deoxygenated 2-AN solution slowed the reaction by 95%. These results indicate that BZT reacts with 32-AN*. FFA also reacted in the presence of 2-AN, but degassing the 2-AN solution resulted in a 23% decrease in the pseudo-first-order rate constant of FFA compared to FFA in an air-saturated solution (Fig. S5B†), indicating a role for both 1O2 and 32-AN*. Furthermore, minimal loss of FFA was observed in the deoxygenated solution upon addition of sorbic acid, reiterating that FFA is susceptible to degradation by organic triplets.68,69 Together, these results indicate that BZT does react with 32-AN*, but the reaction only occurs in the absence of oxygen, meaning quenching of the triplets by oxygen is faster than the reaction with BZT. Thus, BZT reaction with triplet excited state organic matter will likely be unimportant in environmental systems because dissolved oxygen in surface waters will efficiently scavenge triplets before they react with BZT.
3.3 Photochemical transformation of BACs in river water under simulated and natural sunlight
The transformation of BACs was then investigated in the presence of DOM in river water. Based on their chemical structures, no other processes besides reactivity with ˙OH would occur to transform ATMA and DADMA, and further experiments were not conducted in river water. Fig. 3A and B show the time course of C12-BAC and C14-BAC concentrations in phosphate buffer and Mississippi River water in the solar simulator. The tubes were irradiated for a total of 30 hours and this exposure was almost continuous with tubes being removed after 5 or 10 hour increments for sampling and then being returned to resume irradiation in the solar simulator. The loss in river water exhibits pseudo-first-order kinetics as attested to by logarithmic plots for QAC photolysis versus actinometer loss (Fig. S6†). To evaluate the oxidation of BACs by ˙OH, the probe pCBA was added to each MRW tube with BAC, allowing for competition kinetics and quantification of [˙OH]ss.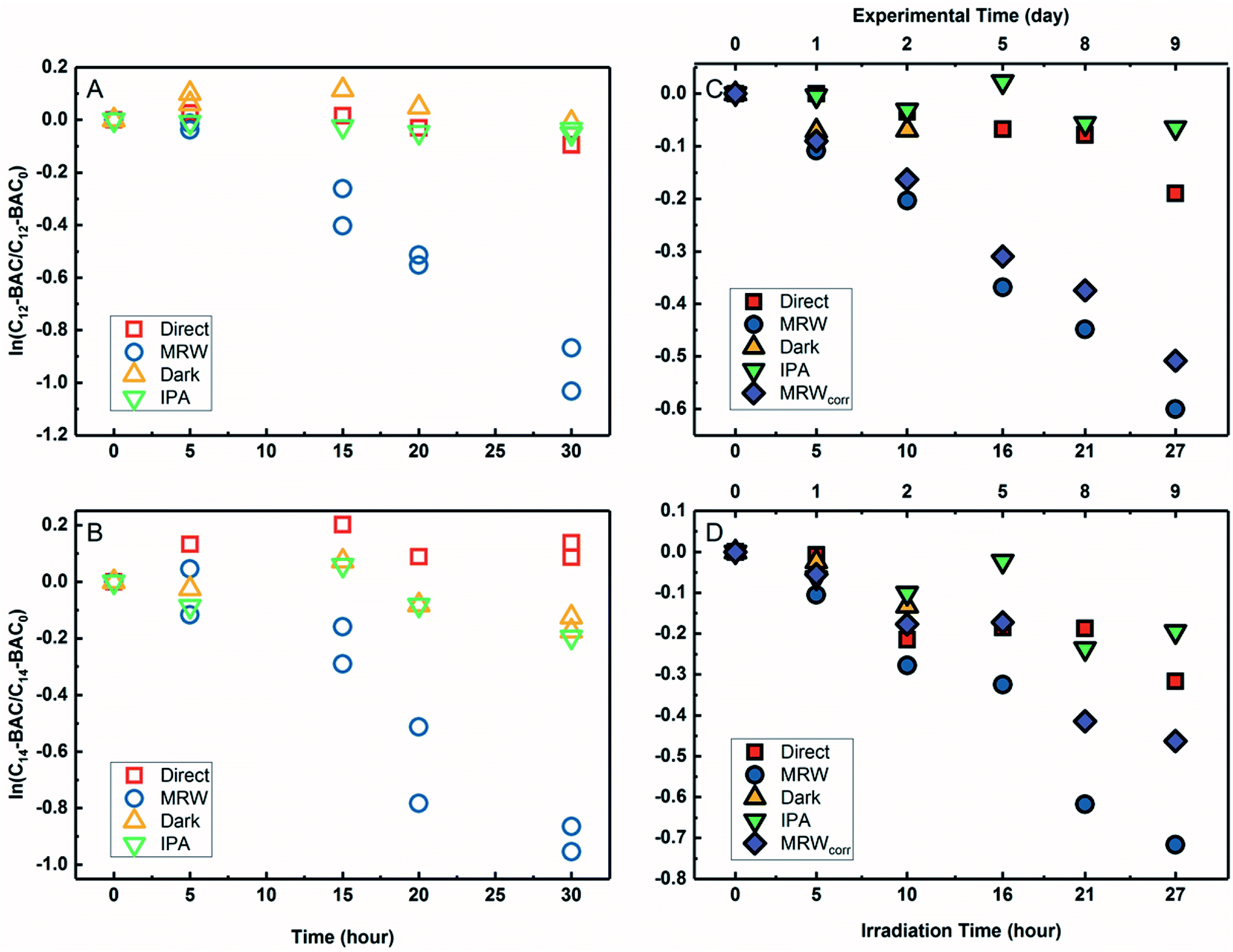 | ||
| Fig. 3 Photolysis of BACs with pCBA under simulated sunlight (C12: A, C14: B) or natural sunlight (C12: C, C14: D) in phosphate buffer (direct, red squares), Mississippi River water (MRW, blue circles), MRW dark control (yellow triangle), and MRW with 1% isopropanol (IPA, green upside down triangle). Indirect photochemical loss in river water is shown by purple diamonds (MRWcorr). Note the top x-axis for outdoor plots C and D showing the total duration of the experiment. | ||
For C12-BAC, 9% loss of the compound was seen in the direct photolysis control and 4% in the IPA quenched control. No apparent loss was exhibited in the dark control; in fact, a slight increase was observed possibly due to evaporation or variation in analytical reproducibility. The slopes for the direct (kdirect = (2 ± 1) × 10−3 h−1) and IPA quenched (kIPA = (1.7 ± 0.3) × 10−3 h−1) controls were within error. Estimating an indirect pseudo-first-order rate constant from the bimolecular reaction rate constant determined from H2O2 sensitizer experiments and the calculated [˙OH]ss from pCBA loss in MRW (Table 1) gives (2.7 ± 0.3) × 10−2 h−1, which is nearly identical to the overall rate constant for C12-BAC degradation in MRW ((2.9 ± 0.4) × 10−2 h−1 (Table S8†)), reaffirming that the loss seen in the MRW is due to the reaction with ˙OH.
For C14-BAC, a slight increase was observed in the direct control possibly from evaporation in the solar simulator or instrumental error in the measurements. The data in the direct, dark, and IPA quenched controls had consistent trends from 5 to 30 hours. While C14-BAC exhibited an average 14% and 18% decrease from the initial concentration in the dark and IPA quenched controls, respectively, the slopes (kdark = (3 ± 3) × 10−3 h−1 and kIPA = (5 ± 3) × 10−3 h−1) demonstrate negligible depletion in these controls compared to the loss in MRW. The overall rate constant for C14-BAC degradation in MRW was kMRW = (2.8 ± 0.6) × 10−2 h−1, which agrees with the estimated pseudo-first-order reaction rate constant for C14-BAC loss due to the reaction with ˙OH ((2.9 ± 0.4) × 10−2 h−1).
Photolysis proceeded faster in the presence of dissolved organic matter, indicating the importance of indirect photolysis (Table S8†). The addition of the radical inhibitor IPA reduced the photodegradation rates substantially in MRW for C12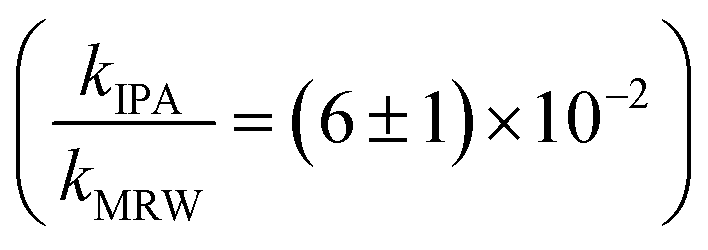 and C14-BAC
and C14-BAC 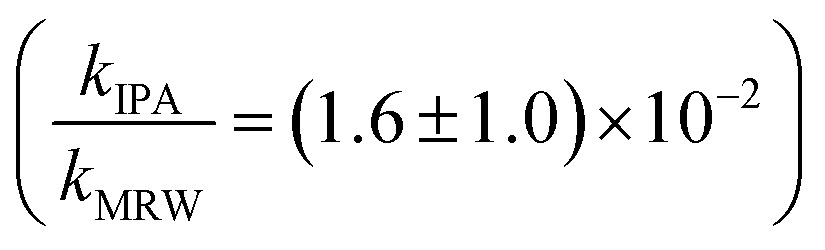 . The quencher results demonstrate that direct photochemical loss of BACs is likely to be relatively unimportant in natural systems and indicate that the ˙OH produced is responsible for BAC loss.
. The quencher results demonstrate that direct photochemical loss of BACs is likely to be relatively unimportant in natural systems and indicate that the ˙OH produced is responsible for BAC loss.
Analogous outdoor experiments were conducted in river water to verify the transferability of indoor photolysis results for the BACs. Fig. 3C and D show the time course of C12-BAC and C14-BAC concentrations in phosphate buffer and Mississippi River water in natural sunlight. Logarithmic plots of BAC versus actinometer loss (Fig. S7†) indicate that outdoor photolysis also follows pseudo-first-order kinetics. pCBA was again added to MRW tubes.
The overall rate constant for C12-BAC degradation in MRW in natural sunlight was (2.2 ± 0.1) × 10−2 h−1. Estimating an indirect pseudo-first-order rate constant from the bimolecular reaction rate constant determined from H2O2 sensitizer and solar simulator river water experiments and the calculated [˙OH]ss from pCBA loss in MRW (Table 1) gives (1.5 ± 0.1) × 10−2 h−1 and (1.5 ± 0.3) × 10−2 h−1, respectively. These estimates imply that 67 to 71% of C12-BAC loss in MRW in the outdoor experiment could be attributable to the reaction with ˙OH. IPA suppressed the reaction in river water substantially (kIPA/kMRW = (8 ± 8) × 10−2).
For C14-BAC, the overall rate constant in MRW was kMRW = (2.6 ± 0.4) × 10−2 h−1. The indirect pseudo-first-order rate constant estimates ((1.6 ± 0.3) × 10−2 h−1 from H2O2 sensitizer and (1.6 ± 0.4) × 10−2 h−1 from solar simulator river water experiments) suggest that 61 to 62% of C14-BAC loss in MRW in the outdoor experiment could be attributable to the reaction with ˙OH. IPA suppressed the reaction in river water, though not completely 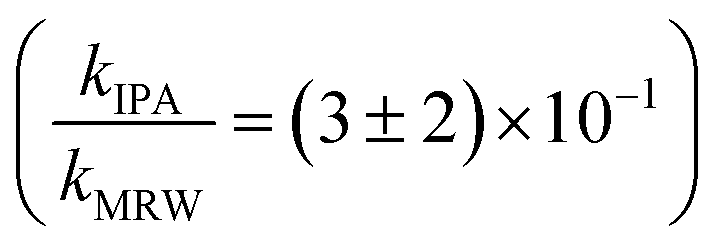 . At the end of the experiment, about an 18% decrease in C14-BAC concentration was seen in the quenched control and 27% in the direct control, though these losses are relatively minor compared to the losses in MRW over the same time period.
. At the end of the experiment, about an 18% decrease in C14-BAC concentration was seen in the quenched control and 27% in the direct control, though these losses are relatively minor compared to the losses in MRW over the same time period.
Because the overlap between the absorption spectra of the BACs and the solar spectrum is small, direct photochemical loss is slow and DOM might screen light affecting transformation rates. Additionally, BACs are hydrolytically stable. Though all C12-BAC control slopes are relatively rather small and associated with proportionate error (kdirect = (6 ± 2) × 10−3 h−1, kIPA = (2 ± 2) × 10−3 h−1, and kdark = (8 ± 9) × 10−3 h−1), a slight loss seems to be occurring in the MRW outdoors due to other abiotic and/or biotic mechanisms. The losses in C14-BAC are more pronounced. Slight thermal decay due to elevated temperatures outside, sorption to the walls of the test tube over the outdoor exposure period, and/or biodegradation could be occurring concomitantly with indirect photolysis to account for increased degradation in the controls.
Outdoor experiments took approximately 9 days while solar exposure time was a little over a day in total to account for weather and sunshine hours on the rooftop. While the river water was filter-sterilized prior to use in experiments and test tubes were capped, no special protocols were used to maintain sterile conditions for the entirety of the experiment. Therefore, it cannot be ruled out that the difference in predicted rate constants due to ˙OH and the observed rate constant is due to biodegradation because BACs are known to be susceptible to aerobic biodegradation.32–35,70
The outdoor river water (MRW) rates were thusly corrected for concentration changes in the controls by eqn (3).71 The indirect losses were plotted separately (MRWcorr in Fig. 3C and D), and the pseudo-first-order rate constants from MRWcorr were used to calculate bimolecular reaction rate constants with ˙OH in the outdoor experiments (see below).
| MRWcorr(t) = MRW(t)e(−kobs,control × t) | (3) |
3.4 BZT direct photolysis in river water under simulated and natural light
The degradation of BZT in MRW appeared to be a combination of direct and indirect photolysis (Fig. 4A) in the solar simulator. Correcting the BZT concentration measured in MRW to account for the loss due to direct photolysis using eqn (3) allowed for calculation of the indirect photolysis pseudo-first-order rate constant and for competition kinetics calculations. The indirect photolysis of BZT in river water is shown in Fig. 4 by the plotted MRWcorr data. The probe pCBA was used to evaluate the oxidation of BZT by ˙OH, by addition to each MRW tube with BZT, allowing for competition kinetics and quantification of [˙OH]ss.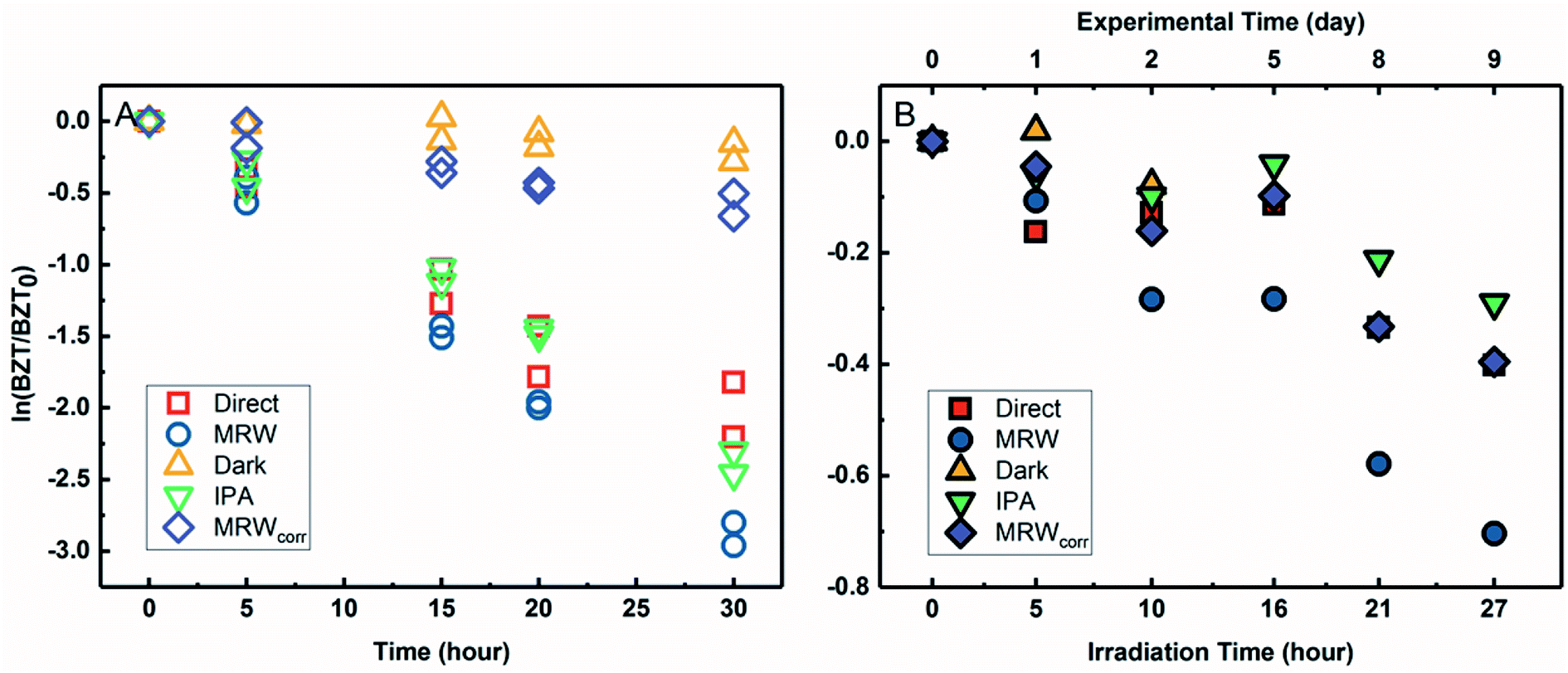 | ||
| Fig. 4 Photolysis of BZT with pCBA under simulated sunlight (A) or natural sunlight (B) in phosphate buffer (direct, red squares), Mississippi River water (MRW, blue circles), MRW dark control (yellow triangle), and MRW with 1% isopropanol (IPA, green upside down triangle). Indirect photochemical loss in river water is shown by purple diamonds (MRWcorr). Note the top x-axis for outdoor plot B showing the total duration of the experiment. | ||
The BZT direct photolysis quantum yield from solar simulator experiments was determined using eqn (2) and the photon fluence of the solar simulator determined from actinometry and the photon irradiance provided by the manufacturer (spectral flux, I0 = 5.4 Es L−1 s−1). Fig. S8† shows the logarithmic plot of BZT direct photolysis (from both the IPA quenched and direct controls) versus the PNAP actinometer. The slope of the line determined by linear regression (1.18 for IPA vs. PNAP and 1.11 for direct vs. PNAP) was used to calculate quantum yields. ΦBZT,indoor,IPA was determined to be 6.3 × 10−2 (mol Ei−1) while ΦBZT,indoor,direct was 5.9 × 10−2 (mol Ei−1). There is good agreement between the two estimates, demonstrating that the decrease of BZT seen in the IPA quenched control is due to direct photolysis and not any other process.
Outdoor experiments were carried out to compare data generated under simulated sunlight with those generated under natural sunlight and to verify the BZT quantum yield obtained in the laboratory because quantum yield calculations are sensitive to irradiance values. For BZT sunlight experiments (Fig. 4B), BZT loss exhibited pseudo-first-order kinetics in river water as seen in the logarithmic plots for QAC photolysis versus actinometer loss (Fig. S7†). As with the solar simulator experiment, pCBA was simultaneously irradiated in the MRW tubes.
The slopes of linear regressions for the BZT quenched and direct photolysis controls were identical within error due to overlapping confidence intervals. The data points were subsequently combined and fitted using a linear fit of concatenated data in Fig. S8,† which shows the logarithmic plot of BZT direct photolysis versus actinometer loss. The direct photolysis quantum yield, ΦBZT,outdoor, was determined to be 1.7 × 10−2 (mol Ei−1) using eqn (2), the slope of 0.21 from Fig. S8,† and the average global tilted irradiance over the timecourse of the rooftop exposures from SMARTS. The discrepancy in calculated quantum yields and half-lives can be explained by the fact that, as has been reported previously,49 though the Xe lamp has a filter that prevents the transmission of wavelengths below 290 nm, there is a bleed of wavelengths <290 nm, which introduces systematic error. This error is important for QACs like BZT which absorb weakly above 290 nm and thus direct photolysis rates are over-emphasized.72 In the terrestrial aquatic environment, direct photolysis occurs for chemicals that absorb photons at wavelengths ≥290 nm because the upper atmosphere screens out solar irradiance at shorter wavelengths. Despite errors introduced by the solar simulator and differences in quantum yields due to spectral overlap, direct photolysis of BZT would still occur in the environment, but the quantum yield determined in sunlight should be used in the calculation of predicted half-life.
3.5 Second-order rate constants with ˙OH in river water
Because pCBA was included in the river water experiments, competition kinetics can be used to determine the second order rate constant with ˙OH to test for any effect of the matrix or light source by comparing the results to those measured with H2O2. Fig. 5 shows the competitive oxidation of the QACs by ˙OH in MRW under simulated and natural sunlight corrected for losses not due to indirect photolysis. As with the ˙OH sensitizer experiments, BZT, C12-BAC, and C14-BAC react with ˙OH in the river water at or near diffusion controlled rates. Association with dissolved organic matter did not significantly influence the photochemical transformation rates of QACs with ˙OH based on the similarity of the values (within 10%) to those determined using H2O2 as a sensitizer.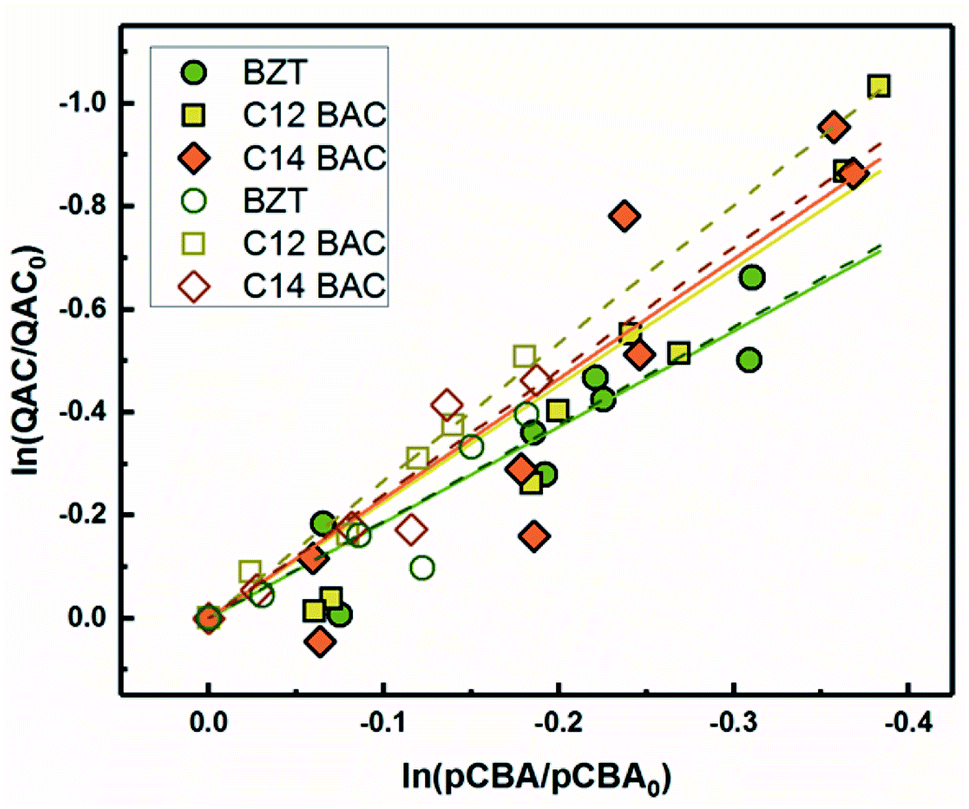 | ||
| Fig. 5 log-concentrations of QACs versus para-chlorobenzoic acid from solar simulator (solid symbols) and natural sunlight (open symbols) irradiation experiments in MRW. QAC concentrations are corrected to only reflect indirect photochemical loss due to ˙OH. Solid and dashed lines represent linear regressions for solar simulator and natural sunlight experiments, respectively. | ||
The outdoor experimental data overlay the indoor results in river water demonstrating that simulated sunlight experiments serve as good proxies for understanding the photochemical behavior of QACs in surface waters with respect to reaction with ˙OH. All calculated bimolecular reaction rate constants (Table 1) are within the 95% confidence intervals for the 3 sets of experiments serving as robust validation of the values. Due to the overlap of the indoor and outdoor results, we can be reasonably assured in our assumptions about the outdoor experimental data and that the indirect photolysis estimates are correct. Fig. 5 reaffirms that the photochemical fate of QACs in the environment will be highly dependent on ˙OH generation in natural waters.
3.6 Calculated half-lives in surface waters
The relative contributions of indirect and direct photolysis were evaluated by comparison of pseudo-first-order rate constants from experiments with river and ultrapure water under simulated and natural sunlight. For most of the QACs studied, direct photolysis under environmental conditions is either slow or nonexistent. Only BZT exhibited appreciable direct photochemical degradation. Direct photolysis half-lives for BZT were estimated to be 5.4–5.7 days under simulated sunlight and 20.6 days in natural sunlight using a direct photolysis rate constant calculated using the solar spectra modeled by SMARTS (Fig. S1†). While the reaction between QACs and ˙OH is diffusion controlled, QAC degradation via this process will be relatively slow in the environment. The bimolecular reaction rate constants with ˙OH were used to estimate half-lives of the tested QACs in sunlit surface waters using eqn (4) and assuming an [˙OH]ss of 1 × 10−16 M on average over 7 hours of daily sunshine.63 Maximum near-surface steady-state ˙OH concentrations range from 10−17 to 10−15 M in sunlit surface waters depending on the concentrations of photoproducing and scavenging species, latitude, and time of year.64,73,74 The half-lives of QACs in the photic zone presented in Table 2 reflect a lower bound (minimum persistence) because they are estimated based on conditions in which average maximum rates of ˙OH photoproduction occur in the photic zone of a water body at midday during the summertime.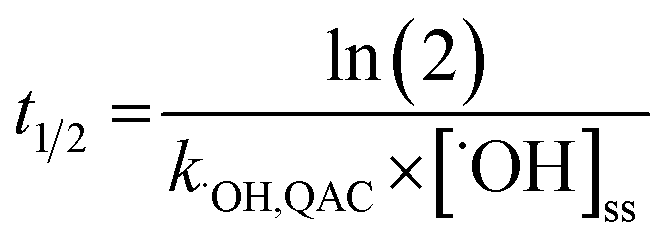 | (4) |
| QAC | t 1/2 (days) H2O2 | t 1/2 (days)c river water indoor | t 1/2 (days)c river water outdoor |
|---|---|---|---|
| a Errors represent 95% confidence intervals. b Calculated assuming 7 hours of sunshine per day. c BZT t1/2 including direct photolysis + ˙OH. | |||
| C12-BAC | 26 ± 2 | 24 ± 4 | 24 ± 2 |
| C14-BAC | 23 ± 3 | 24 ± 5 | 23 ± 6 |
| BZT | 26 ± 1 | 12 ± 1 | 12 ± 2 |
| ATMA | 27 ± 4 | ||
| DADMA | 94 ± 32 | ||
Because BZT is susceptible to both indirect and direct photolysis under relevant environmental conditions, the half-life of BZT in river water is estimated by extrapolating the experimental results for BZT and using the overall pseudo-first-order rate constant for photolytic loss (eqn (5)), where kc (ESI S3 eqn (1)†) is the direct photolysis pseudo-first-order rate constant of BZT in river water under natural sunlight.
| kobs,BZT = k˙OH,BZT × [˙OH]ss + kc | (5) |
While QACs demonstrate shorter lifetimes compared to ionic liquid cations,63 they still have half-lives on the order of weeks to months, highlighting their slow photochemical transformation in rivers and lakes, and their persistence in the aquatic environment. The results and half-life estimates were consistent for outdoor and indoor river water photolysis experiments as well as for H2O2 sensitizer experiments, reiterating the importance of ·OH over other PPRIs in the fate of QACs in surface waters.
4. Conclusions
The current study elucidates the photochemical fate of QACs in surface waters. For most of the QACs evaluated, indirect photolysis is more important than direct photolysis under simulated and natural sunlight. Through model sensitizer and quencher experiments, it is clear that all QACs studied will react predominantly with ˙OH produced in natural waters despite variations in structure, such as side-chain length and presence of aromatic rings. In waters with low steady-state ˙OH concentrations and deeper in the water column, the rates of reaction between QACs and ˙OH will be substantially slower and thus QAC lifetimes will be longer. The results of this study contribute to a more holistic and systematic evaluation of the fate of QACs in near-surface waters as well as the potential for their attenuation in the environment. Reported half-lives and rate constants can be used in future models to predict the fate of QACs in the environment, which could inform risk assessments and regulatory decisions on biocide usage given the growing public health concern over the proliferation of antibiotic resistant bacteria in the environment. Additionally, the hydroxyl radical reaction data could be applied to assessing the removal of QACs during advanced oxidation treatment.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We would like to acknowledge Dr Sarah Pati for help with LC-MS/MS measurements as well as Makenzie Pillsbury. Mass spectrometry was carried out in the Analytical Biochemistry Shared Resources of the Masonic Cancer Center, University of Minnesota, funded in part by Cancer Center Support Grant CA-77598. The authors thank Annika Heaps and Bryanna Lopez-Moran for assistance with laboratory work. This work was funded by the Minnesota Environmental and Natural Resources Trust fund (M.L. 2017, Chp. 96, Sec. 2, Subd. 04a) as recommended by the Legislative and Citizen Commission on Minnesota Resources.References
- C. Zhang, F. Cui, G. Zeng, M. Jiang, Z. Yang, Z. Yu, M. Zhu and L. Shen, Quaternary ammonium compounds (QACs): a review on occurrence, fate and toxicity in the environment, Sci. Total Environ., 2015, 518–519, 352–362 CrossRef CAS PubMed.
- G.-G. Ying, Fate, behavior and effects of surfactants and their degradation products in the environment, Environ. Int., 2006, 32, 417–431 CrossRef CAS PubMed.
- U. Tezel and S. G. Pavlostathis, Role of Quaternary Ammonium Compounds on Antimicrobial Resistance in the Environment in Antimicrobial Resistance in the Environment, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011, pp. 349–387 Search PubMed.
- M. T. García, E. Campos, J. Sanchez-Leal and I. Ribosa, Effect of the alkyl chain length on the anaerobic biodegradability and toxicity of quaternary ammonium based surfactants, Chemosphere, 1999, 38, 3473–3483 CrossRef.
- J. Hong, U. Tezel, D. Okutman Tas and S. G. Pavlostathis, Influence of quaternary ammonium compounds on the microbial reductive dechlorination of pentachloroaniline, Water Res., 2013, 47, 6780–6789 CrossRef CAS PubMed.
- G. A. Kahrilas, J. Blotevogel, P. S. Stewart and T. Borch, Biocides in hydraulic fracturing fluids: a critical review of their usage, mobility, degradation, and toxicity, Environ. Sci. Technol., 2015, 49, 16–32 CrossRef CAS PubMed.
- X. Li and B. J. Brownawell, Quaternary Ammonium Compounds in Urban Estuarine Sediment Environments – A Class of Contaminants in Need of Increased Attention?, Environ. Sci. Technol., 2010, 44, 7561–7568 CrossRef CAS PubMed.
- W. T. Stringfellow, J. K. Domen, M. K. Camarillo, W. L. Sandelin and S. Borglin, Physical, chemical, and biological characteristics of compounds used in hydraulic fracturing, J. Hazard. Mater., 2014, 275, 37–54 CrossRef CAS PubMed.
- A. Butkovskyi, H. Bruning, S. A. E. Kools, H. H. M. Rijnaarts and A. P. Van Wezel, Organic Pollutants in Shale Gas Flowback and Produced Waters: Identification, Potential Ecological Impact, and Implications for Treatment Strategies, Environ. Sci. Technol., 2017, 51, 4740–4754 CrossRef CAS PubMed.
- M. Elsner and K. Hoelzer, Quantitative Survey and Structural Classification of Hydraulic Fracturing Chemicals Reported in Unconventional Gas Production, Environ. Sci. Technol., 2016, 50, 3290–3314 CrossRef CAS PubMed.
- J. Yang, U. Tezel, K. Li and S. G. Pavlostathis, Prolonged exposure of mixed aerobic cultures to low temperature and benzalkonium chloride affect the rate and extent of nitrification, Bioresour. Technol., 2015, 179, 193–201 CrossRef CAS PubMed.
- A. C. Doherty, The distribution, fate and application as tracers of quaternary ammonium compounds (QACs) in sewage impacted estuaries, The Graduate School, Stony Brook University, Stony Brook, NY., 2013 Search PubMed.
- T. Ruan, S. Song, T. Wang, R. Liu, Y. Lin and G. Jiang, Identi fi cation and Composition of Emerging Quaternary Ammonium Compounds in Municipal Sewage Sludge in China, Environ. Sci. Technol., 2014, 48, 4289–4297 CrossRef CAS PubMed.
- E. Martínez-Carballo, C. González-Barreiro, A. Sitka, N. Kreuzinger, S. Scharf and O. Gans, Determination of selected quaternary ammonium compounds by liquid chromatography with mass spectrometry. Part II. Application to sediment and sludge samples in Austria, Environ. Pollut., 2007, 146, 543–547 CrossRef PubMed.
- A. C. Chiaia-Hernandez, M. Krauss and J. Hollender, Screening of lake sediments for emerging contaminants by liquid chromatography atmospheric pressure photoionization and electrospray ionization coupled to high resolution mass spectrometry, Environ. Sci. Technol., 2013, 47, 976–986 CrossRef CAS PubMed.
- M. Pateiro-Moure, M. Arias-Estévez and J. Simal-Gándara, Critical Review on the Environmental Fate of Quaternary Ammonium Herbicides in Soils Devoted to Vineyards, Environ. Sci. Technol., 2013, 47, 4984–4998 CrossRef CAS PubMed.
- X. Li, A. C. Doherty, B. Brownawell and P. A. Lara-Martin, Distribution and diagenetic fate of synthetic surfactants and their metabolites in sewage-impacted estuarine sediments, Environ. Pollut., 2018, 242, 209–218 CrossRef CAS.
- P. A. Lara-Martín, X. Li, R. F. Bopp and B. J. Brownawell, Occurrence of alkyltrimethylammonium compounds in urban estuarine sediments: Behentrimonium as a new emerging contaminant, Environ. Sci. Technol., 2010, 44, 7569–7575 CrossRef.
- S. G. Pati and W. A. Arnold, Comprehensive screening of quaternary ammonium surfactants and ionic liquids in wastewater effluents and lake sediments, Environ. Sci.: Processes Impacts, 2020, 22, 430–441 RSC.
- X. Li and B. J. Brownawell, Quaternary ammonium compounds in urban estuarine sediment environments – a class of contaminants in need of increased attention?, Environ. Sci. Technol., 2010, 44, 7561–7568 CrossRef CAS PubMed.
- M. Clara, S. Scharf, C. Scheffknecht and O. Gans, Occurrence of selected surfactants in untreated and treated sewage, Water Res., 2007, 41, 4339–4348 CrossRef CAS PubMed.
- E. Martínez-Carballo, A. Sitka, C. González-Barreiro, N. Kreuzinger, M. Fürhacker, S. Scharf and O. Gans, Determination of selected quaternary ammonium compounds by liquid chromatography with mass spectrometry. Part I. Application to surface, waste and indirect discharge water samples in Austria, Environ. Pollut., 2007, 145, 489–496 CrossRef PubMed.
- C. Zhang, U. Tezel, K. Li, D. Liu, R. Ren, J. Du and S. G. Pavlostathis, Evaluation and modeling of benzalkonium chloride inhibition and biodegradation in activated sludge, Water Res., 2011, 45, 1238–1246 CrossRef CAS.
- X. Li and B. J. Brownawell, Analysis of quaternary ammonium compounds in estuarine sediments by LC-ToF-MS: Very high positive mass defects of alkylamine ions as powerful diagnostic tools for identification and structural elucidation, Anal. Chem., 2009, 81, 7926–7935 CrossRef CAS PubMed.
- I. Ferrer and E. M. Thurman, Analysis of hydraulic fracturing additives by LC/Q-TOF-MS, Anal. Bioanal. Chem., 2015, 407, 6417–6428 CrossRef CAS PubMed.
- I. Ferrer and E. T. Furlong, Identification of alkyl dimethylbenzylammonium surfactants in water samples by solid-phase extraction followed by ion trap LC/MS and LC/MS/MS, Environ. Sci. Technol., 2001, 35, 2583–2588 CrossRef CAS PubMed.
- Antimicrobial Resistance in the Environment, ed. P. L. Keen and M. H. M. M. Montforts, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011 Search PubMed.
- M. Kim, M. R. Weigand, S. Oh, J. K. Hatt, R. Krishnan, U. Tezel, S. G. Pavlostathis and K. T. Konstantinidis, Widely used benzalkonium chloride disinfectants can promote antibiotic resistance, Appl. Environ. Microbiol., 2018, 84, e01201–18 CAS.
- M. C. Jennings, K. P. C. Minbiole and W. M. Wuest, Quaternary Ammonium Compounds: An Antimicrobial Mainstay and Platform for Innovation to Address Bacterial Resistance, ACS Infect. Dis., 2015, 1, 288–303 CrossRef CAS PubMed.
- H. Sütterlin, R. Alexy, A. Coker and K. Kümmerer, Mixtures of quaternary ammonium compounds and anionic organic compounds in the aquatic environment: Elimination and biodegradability in the closed bottle test monitored by LC-MS/MS, Chemosphere, 2008, 72, 479–484 CrossRef.
- M. T. García, E. Campos, J. Sánchez-Leal and I. Ribosa, Anaerobic degradation and toxicity of commercial cationic surfactants in anaerobic screening tests, Chemosphere, 2000, 41, 705–710 CrossRef.
- M. T. García, I. Ribosa, T. Guindulain, J. Sánchez-Leal and J. Vives-Rego, Fate and effect of monoalkyl quaternary ammonium surfactants in the aquatic environment, Environ. Pollut., 2001, 111, 169–175 CrossRef.
- U. Tezel, M. Tandukar, R. J. Martinez, P. A. Sobecky and S. G. Pavlostathis, Aerobic Biotransformation of n-Tetradecylbenzyldimethylammonium Chloride by an Enriched Pseudomonas spp. Community, Environ. Sci. Technol., 2012, 46, 8714–8722 CrossRef CAS PubMed.
- E. Ertekin, K. T. Konstantinidis and U. Tezel, A Rieske-Type Oxygenase of Pseudomonas sp. BIOMIG1 Converts Benzalkonium Chlorides to Benzyldimethyl Amine, Environ. Sci. Technol., 2017, 51, 175–181 CrossRef CAS PubMed.
- E. Ertekin, J. K. Hatt, K. T. Konstantinidis and U. Tezel, Similar Microbial Consortia and Genes Are Involved in the Biodegradation of Benzalkonium Chlorides in Different Environments, Environ. Sci. Technol., 2016, 50, 4304–4313 CrossRef CAS PubMed.
- Y. J. S. Lai, A. Ontiveros-Valencia, Z. E. Ilhan, Y. Zhou, E. Miranda, J. Maldonado, R. Krajmalnik-Brown and B. E. Rittmann, Enhancing biodegradation of C16-alkyl quaternary ammonium compounds using an oxygen-based membrane biofilm reactor, Water Res., 2017, 123, 825–833 CrossRef CAS PubMed.
- S. T. J. Droge and K. U. Goss, Ion-exchange affinity of organic cations to natural organic matter: influence of amine type and nonionic interactions at two different pHs, Environ. Sci. Technol., 2013, 47, 798–806 CrossRef CAS PubMed.
- B. J. Brownawell, H. Chen, J. M. Collier and J. C. Westall, Adsorption of organic cations to natural materials, Environ. Sci. Technol., 1990, 24, 1234–1241 CrossRef CAS.
- N. Kreuzinger, M. Fuerhacker, S. Scharf, M. Uhl, O. Gans and B. Grillitsch, Methodological approach towards the environmental significance of uncharacterized substances – quaternary ammonium compounds as an example, Desalination, 2007, 215, 209–222 CrossRef CAS.
- K. Jardak, P. Drogui and R. Daghrir, Surfactants in aquatic and terrestrial environment: occurrence, behavior, and treatment processes, Environ. Sci. Pollut. Res., 2016, 23, 3195–3216 CrossRef CAS PubMed.
- W. H. Gaze, N. Abdouslam, P. M. Hawkey and E. M. H. Wellington, Incidence of Class 1 Integrons in a Quaternary Ammonium Compound-Polluted Environment, Antimicrob. Agents Chemother., 2005, 49, 1802–1807 CrossRef CAS PubMed.
- M. R. Gillings, Integrons: past, present, and future, Microbiol. Mol. Biol. Rev., 2014, 78, 257–277 CrossRef PubMed.
- M. R. Gillings, M. P. Holley and H. W. Stokes, Evidence for dynamic exchange of qac gene cassettes between class 1 integrons and other integrons in freshwater biofilms, FEMS Microbiol. Lett., 2009, 296, 282–288 CrossRef CAS PubMed.
- M. R. Gillings, D. Xuejun, S. A. Hardwick, M. P. Holley and H. W. Stokes, Gene cassettes encoding resistance to quaternary ammonium compounds: a role in the origin of clinical class 1 integrons?, ISME J., 2009, 3, 209–215 CrossRef CAS PubMed.
- I. Mulder, J. Siemens, V. Sentek, W. Amelung, K. Smalla and S. Jechalke, Quaternary ammonium compounds in soil: implications for antibiotic resistance development, Rev. Environ. Sci. Bio/Technol., 2018, 17, 159–185 CrossRef CAS.
- United States Environmental Protection Agency Office of Prevention, Pesticides and Toxic Substances, Reregistration Eligibility Decision for Alkyl Dimethyl Benzyl Ammonium Chloride (ADBAC), Washington, DC, 2006 Search PubMed.
- M. E. Karpuzcu, A. J. McCabe and W. A. Arnold, Phototransformation of pesticides in prairie potholes: effect of dissolved organic matter in triplet-induced oxidation, Environ. Sci.: Processes Impacts, 2016, 18, 237–245 RSC.
- T. Zeng and W. A. Arnold, Pesticide Photolysis in Prairie Potholes: Probing Photosensitized Processes, Environ. Sci. Technol., 2013, 47, 6735–6745 CrossRef CAS PubMed.
- A. J. McCabe and W. A. Arnold, Reactivity of Triplet Excited States of Dissolved Natural Organic Matter in Stormflow from Mixed-Use Watersheds, Environ. Sci. Technol., 2017, 51, 9718–9728 CrossRef CAS PubMed.
- S. E. Page, W. A. Arnold and K. McNeill, Assessing the Contribution of Free Hydroxyl Radical in Organic Matter-Sensitized Photohydroxylation Reactions, Environ. Sci. Technol., 2011, 45, 2818–2825 CrossRef CAS PubMed.
- M. Grandbois, D. E. Latch and K. McNeill, Microheterogeneous Concentrations of Singlet Oxygen in Natural Organic Matter Isolate Solutions, Environ. Sci. Technol., 2008, 42, 9184–9190 CrossRef CAS.
- A. Leifer, The Kinetics of Environmental Aquatic Photochemistry: Theory and Practice, American Chemical Society, Washington, DC, 1988 Search PubMed.
- D. Dulin and T. Mill, Development and evaluation of sunlight actinometers, Environ. Sci. Technol., 1982, 16, 815–820 CrossRef CAS PubMed.
- K. McNeill and S. Canonica, Triplet state dissolved organic matter in aquatic photochemistry: reaction mechanisms, substrate scope, and photophysical properties, Environ. Sci.: Processes Impacts, 2016, 18, 1381–1399 RSC.
- J. E. Grebel, J. J. Pignatello and W. A. Mitch, Effect of halide ions and carbonates on organic contaminant degradation by hydroxyl radical-based advanced oxidation processes in saline waters, Environ. Sci. Technol., 2010, 44, 6822–6828 CrossRef CAS PubMed.
- J. E. Grebel, J. J. Pignatello and W. A. Mitch, Sorbic acid as a quantitative probe for the formation, scavenging and steady-state concentrations of the triplet-excited state of organic compounds, Water Res., 2011, 45, 6535–6544 CrossRef CAS PubMed.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (˙OH/˙O − in Aqueous Solution, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- E. Appiani, R. Ossola, D. E. Latch, P. R. Erickson and K. McNeill, Aqueous singlet oxygen reaction kinetics of furfuryl alcohol: effect of temperature, pH, and salt content, Environ. Sci.: Processes Impacts, 2017, 19, 507–516 RSC.
- NREL, SMARTS: Simple Model of the Atmospheric Radiative Transfer of Sunshine, https://www.nrel.gov/grid/solar-resource/smarts.html, accessed 6 September 2019 Search PubMed.
- C. A. Gueymard, Parameterized transmittance model for direct beam and circumsolar spectral irradiance, Sol. Energy, 2001, 71, 325–346 CrossRef CAS.
- C. Gueymard, SMARTS2, A Simple Model of the Atmospheric Radiative Transfer of Sunshine: Algorithms and Performance Assessment, Florida Solar Energy Center, 1995 Search PubMed.
- J. R. Laszakovits, S. M. Berg, B. G. Anderson, J. E. O'Brien, K. H. Wammer and C. M. Sharpless, p-Nitroanisole/Pyridine and p-Nitroacetophenone/Pyridine Actinometers Revisited: Quantum Yield in Comparison to Ferrioxalate, Environ. Sci. Technol. Lett., 2017, 4, 11–14 CrossRef CAS.
- S. G. Pati and W. A. Arnold, Photochemical Transformation of Four Ionic Liquid Cation Structures in Aqueous Solution, Environ. Sci. Technol., 2017, 51, 11780–11787 CrossRef CAS PubMed.
- R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental Organic Chemistry, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2002 Search PubMed.
- D. E. Latch, in Surface Water Photochemistry, The Royal Society of Chemistry, 2015, pp. 139–165 Search PubMed.
- W. A. Arnold and K. McNeill, Transformation of pharmaceuticals in the environment: Photolysis and other abiotic processes, in Comprehensive Analytical Chemistry, Elsevier, 2007, vol. 50, pp. 361–385 Search PubMed.
- F. L. Rosario-Ortiz and S. Canonica, Probe Compounds to Assess the Photochemical Activity of Dissolved Organic Matter, Environ. Sci. Technol., 2016, 50, 12532–12547 CrossRef CAS PubMed.
- M. Minella, L. Rapa, L. Carena, M. Pazzi, V. Maurino, C. Minero, M. Brigante and D. Vione, An experimental methodology to measure the reaction rate constants of processes sensitised by the triplet state of 4-carboxybenzophenone as a proxy of the triplet states of chromophoric dissolved organic matter, under steady-state irradiation conditions, Environ. Sci.: Processes Impacts, 2018, 20, 1007–1019 RSC.
- F. al Housari, D. Vione, S. Chiron and S. Barbati, Reactive photoinduced species in estuarine waters. Characterization of hydroxyl radical, singlet oxygen and dissolved organic matter triplet state in natural oxidation processes, Photochem. Photobiol. Sci., 2010, 9, 78–86 RSC.
- S. G. Pavlostathis, T. Konstantinidis, S. Oh, Z. Kurt, D. Tsementzi, M. R. Weigand, M. Kim, J. K. Hatt, M. Tandukar, S. G. Pavlostathis, J. C. Spain and K. T. Konstantinidis, Microbial Community Degradation of Widely Used Quaternary Ammonium Disinfectants, Appl. Environ. Microbiol., 2014, 80, 5892–5900 CrossRef PubMed.
- J. Wenk, U. von Gunten and S. Canonica, Effect of Dissolved Organic Matter on the Transformation of Contaminants Induced by Excited Triplet States and the Hydroxyl Radical, Environ. Sci. Technol., 2011, 45, 1334–1340 CrossRef CAS PubMed.
- M. W. Lam and S. A. Mabury, Photodegradation of the pharmaceuticals atorvastatin, carbamazepine, levofloxacin, and sulfamethoxazole in natural waters, Aquat. Sci., 2005, 67, 177–188 CrossRef CAS.
- P. L. Brezonik and J. Fulkerson-Brekken, Nitrate-Induced Photolysis in Natural Waters: Controls on Concentrations of Hydroxyl Radical Photo-Intermediates by Natural Scavenging Agents, Environ. Sci. Technol., 1998, 32, 3004–3010 CrossRef CAS.
- R. G. Zepp, J. Hoigne and H. Bader, Nitrate-induced photooxidation of trace organic chemicals in water, Environ. Sci. Technol., 1987, 21, 443–450 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0em00086h |
| This journal is © The Royal Society of Chemistry 2020 |