
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interactions of ferrous iron with clay mineral surfaces during sorption and subsequent oxidation†
Natacha
Van Groeningen
 a,
Laurel K.
ThomasArrigo
a,
James M.
Byrne
b,
Andreas
Kappler
b,
Iso
Christl
*a and
Ruben
Kretzschmar
a
a,
Laurel K.
ThomasArrigo
a,
James M.
Byrne
b,
Andreas
Kappler
b,
Iso
Christl
*a and
Ruben
Kretzschmar
a
aInstitute of Biogeochemistry and Pollutant Dynamics, Department of Environmental Systems Science, CHN, ETH Zürich, 8092 Zürich, Switzerland. E-mail: iso.christl@env.ethz.ch
bGeomicrobiology Group, Centre for Applied Geosciences (ZAG), University of Tübingen, Hölderlinstrasse 12, D-72074, Tübingen, Germany
First published on 6th May 2020
Abstract
In submerged soils and sediments, clay minerals are often exposed to anoxic waters containing ferrous iron (Fe2+). Here, we investigated the sorption of Fe2+ onto a synthetic montmorillonite (Syn-1) low in structural Fe (<0.05 mmol Fe per kg) under anoxic conditions and the effects of subsequent oxidation. Samples were prepared at two Fe-loadings (0.05 and 0.5 mol Fe added per kg clay) and equilibrated for 1 and 30 days under anoxic conditions (O2 < 0.1 ppm), followed by exposure to ambient air. Iron solid-phase speciation and mineral identity was analysed by 57Fe Mössbauer spectroscopy and synchrotron X-ray absorption spectroscopy (XAS). Mössbauer analyses showed that Fe(II) was partially oxidized (14–100% of total added Fe2+) upon sorption to Syn-1 under anoxic conditions. XAS results revealed that the added Fe2+ mainly formed precipitates (layered Fe minerals, Fe(III)-bearing clay minerals, ferrihydrite, and lepidocrocite) in different quantities depending on the Fe-loading. Exposing the suspensions to ambient air resulted in rapid and complete oxidation of sorbed Fe(II) and the formation of Fe(III)-phases (Fe(III)-bearing clay minerals, ferrihydrite, and lepidocrocite), demonstrating that the clay minerals were unable to protect ferrous Fe from oxidation, even when equilibrated 30 days under anoxic conditions prior to oxidation. Our findings clarify the role of clay minerals in the formation and stability of Fe-bearing solid phases during redox cycles in periodically anoxic environments.
Environmental significanceClay minerals are considered major sorbents in soils and sediments. However, their significance for the redox cycling of iron is still unclear. Under anoxic conditions, elevated concentrations of dissolved ferrous iron are observed. We demonstrate that at circumneutral to alkaline pH, surface-mediated oxidation of ferrous iron can occur under anoxic conditions in the presence of clays that are virtually free of structural iron. The retention of dissolved ferrous iron in the presence of clay minerals was evidenced to be mainly due to the formation of secondary Fe(II/III) phases. Subsequent aeration led to the complete oxidation of iron. In contrast to surface precipitation, adsorption of dissolved iron by layered aluminosilicates plays a minor role in iron retention and does not protect ferrous iron from oxidation. |
Introduction
Iron is ubiquitous in soils and sediments and of great importance in the biogeochemical cycling of metals.1–5 Its solubility in well-aerated soils is mainly controlled by the dissolution and precipitation of Fe(III)-(oxyhydr)oxides.6 During periods with anoxic conditions, Fe(III)-(oxyhydr)oxides serve as electron acceptors for dissimilatory metal-reducing bacteria leading to the release of aqueous Fe2+. Ferrous iron (Fe2+), being one of the most abundant redox-active species in anoxic soils, has a crucial role in controlling macronutrient and trace element cycles.7 The retention of Fe2+ in soils and sediments is, as for most divalent cations, governed by sorption processes (including adsorption as well as surface precipitation and polymerization)8 to minerals with a large surface area and specific sorption properties such as clay minerals, carbonates, and Fe(III)- and Mn(IV, III)-(oxyhydr)oxides. Under anoxic conditions, clay minerals are one of the most important sorbent phases as they are commonly found in high abundance in soils but are not susceptible to reductive dissolution. Aqueous Fe2+ can adsorb to clay mineral surfaces via cation exchange on permanently charged face surfaces, by the formation of surface complexes on edge surfaces, or by the formation of surface clusters and precipitates. Fe2+ is expected to effectively compete with some trace metal cations (e.g., Zn2+, Cd2+) for sorption sites on clay minerals,9,10 leading to mobilization of trace metals under anoxic conditions. On the other hand, immobilization of trace metals can occur through additional pathways such as incorporation into newly formed solids.Natural clay minerals often contain Fe(II) and Fe(III) in their crystal lattice11,12 and it has been shown that electrons can be transferred from ferrous iron to structural Fe(III).13–18 Iron-free clay minerals are generally considered to be redox-inactive within the natural redox potential of soils. However, Géhin et al.14 demonstrated that under strictly anoxic conditions Fe(II) was oxidized to Fe(III) on surfaces of clay minerals having very low iron contents. Potential electron acceptors in low-iron clay minerals could include trace Fe(III) impurities, other redox-active elements (e.g., Ti), or hydrogen.14 Following oxidation, the hydrolysis of Fe(III) may lead to the formation of oxyhydroxide clusters or precipitates on the clay mineral surface, as has been observed by many studies.12,15,19,20 However, uncertainties still prevail about the ability of Fe-free clay mineral to induce electron transfer from adsorbed Fe(II) to the clay mineral, leading to the formation of adsorbed Fe(III).19,21
When reduced soils are exposed to air, oxidation of sorbed Fe(II) by O2 is expected to occur, which may lead to the formation of polynuclear Fe(III)-oxyhydroxide clusters and surface precipitates. These oxidation products can effectively immobilize trace elements by co-precipitation.22–26 The nature of the surface precipitates formed by oxidation of ferrous iron depends on the reaction conditions (pH, rate of oxidation, suspension concentration, temperature, and concentration of impurities).27 However, if secondary Fe phases were formed first under anoxic conditions, the type of surface precipitates formed during oxidation could also be influenced by the solid phases formed in the absence of O2. It has been shown by Génin et al.28 that Fe(OH)2 oxidizes into chloride green rust (Cl-GR) and further oxidation of Cl-GR leads to the formation of lepidocrocite, goethite, or akaganeite, depending on the ratio of initial concentrations of Cl− and OH−. However, it has not been identified which Fe-bearing minerals form by oxidation of Fe(II) associated with clay mineral surfaces.
Since clay minerals are not susceptible to reductive dissolution, a large portion of the Fe pool in submerged soils is believed to be sorbed or incorporated into clay minerals.29 To date, few studies have been conducted on Fe2+ sorption on Fe-free clay minerals,14,21,30 which allows the determination of Fe2+ adsorption on clay minerals and the relevance of this process for Fe retention. Furthermore, the effects of subsequent oxidation on the speciation of Fe sorbed onto clay surfaces have not yet been determined. Therefore, the aims of this study were to investigate the interactions of Fe2+ with a low structural iron montmorillonite under anoxic conditions, and to characterize the Fe surface species and secondary phases formed during sorption. Additionally, we investigated the Fe species formed by oxidation of Fe sorbed on the clay mineral surface. The results will improve our understanding of the role of clay minerals in Fe redox cycling and phase (trans-)formation.
Experimental
Preparation of clay mineral
A synthetic montmorillonite (Barasym SSM-100), referred to as Syn-1 was purchased from the Source Clays Repository of the Clay Minerals Society (Purdue University, West Lafayette, IN). Syn-1 is very low in structural Fe and was selected for this study to minimize potential oxidation of adsorbed Fe2+ by structural Fe(III). Syn-1, was size fractionated (<2 μm), Ca saturated, and washed free of excess salt. The size-fractionated Ca-saturated Syn-1 was analyzed both as oriented and random oriented powder samples by X-ray diffraction (XRD, D8 Advance, Bruker). Diffractograms were recorded in Bragg–Brentano geometry from 3 to 80° 2θ with a step size of 0.02° 2θ and 4 s acquisition time per step, using Cu Kα radiation (λ = 1.5418 Å, 40 kV, and 40 mA). X-ray fluorescence analysis (XRF, XEPOS, Spectro) was performed for elemental analysis of the clay mineral. The measured diffractograms showed trace amounts of boehmite (γ-AlOOH) and total iron content was determined by XRF as 0.026 g kg−1. The specific surface area of Syn-1 was measured by multi-point N2-BET analysis (ASiQwin, Quantachrome) as 95 ± 2 m2 g−1.Anoxic ferrous iron sorption experiments
Batch sorption experiments were performed to quantify Fe2+ sorption on Ca-saturated Syn-1 as a function of pH (4–10) and CaCl2 concentration (0.1, 1.0, and 10 mM CaCl2) for two fixed ferrous iron concentrations (0.05 and 0.5 mM) at 25 ± 1 °C. The equilibration time was 24 h and the clay suspensions had a solid-to-solution ratio of 1 g L−1, giving Fe/clay ratios of 0.05 and 0.5 mol Fe per kg clay, respectively. Due to the high oxygen sensitivity of Fe2+, all batch sorption experiments were performed under anoxic conditions (O2 < 0.1 ppm). All solutions were prepared with O2- and CO2-free, doubly deionized (DDI) water (≥18.2 MΩ cm, Milli-Q, Millipore, Merck KGaA, Darmstadt, Germany), which was produced by boiling the water while purging it with pure nitrogen gas for at least 3 h outside the glovebox. This was done to prevent oxidation of dissolved Fe2+ and avoid the formation of carbonate solids at alkaline pH values contributing to the removal of Fe2+ from solution. All batch experiments were prepared in an anoxic glovebox from a stock clay suspension (25 g clay L−1), which was pre-equilibrated under a N2 atmosphere in the glovebox for 7 days. Before starting the sorption experiments, clay suspensions were equilibrated for 1 hour at pH 5 to remove CO2. Subsequently, aliquots of a Fe2+ stock solution (0.25 M made from a FeCl2 salt) were added to the clay suspension to obtain a total Fe2+ concentration of 0.05 and 0.5 mM. The suspension pH was adjusted by adding a pre-determined amount of CO2-free base (0.01 or 0.1 M NaOH) to reach the target pH value.The pH of the clay suspensions was measured for all samples at the end of the equilibration period. The samples were then passed through 0.22 μm nylon filters (VWR International) and the filtrates were acidified. Acidified samples were analyzed for Ca, Al, Si, Zr, V and Fe by inductively coupled plasma–optical emission spectrometry (ICP-OES, Vista MPX, Varian) (Ca, Al, Si) or mass spectrometry (ICP-MS, 8800 QQQ-ICP-MS, Agilent) (Fe, Zr, V).
Iron solid phase speciation
Two set of samples were prepared to investigate the solid-phase speciation of Fe and the effects of equilibration time (1 and 30 days), pH (7 and 8), Fe-loading (0.05 and 0.5 mol Fe per kg clay), and oxidation. The first group of samples were prepared for 57Fe Mössbauer spectroscopy and the second for XAS analysis. All batch sorption experiments were performed in 250 mL Nalgene® bottles and stirred at 800 rpm for 1 or 30 days under anoxic conditions. After equilibration, aliquots of the suspensions were sampled and filtered for analysis of solutes by ICP-OES or ICP-MS. For solid-phase analysis, 70 mL suspension was passed through a 0.22 μm cellulose nitrate filter (GE Healthcare Life Sciences) and solids were collected. The collected solids were then washed with deoxygenated CO2-free DDI water and subsequently dried in the dark in the glovebox. From the remaining suspensions, 50 mL was transferred into 50 mL polypropylene (PP) centrifuge tubes. These PP centrifuge tubes were then taken out of the glovebox and opened, in order to exchange the remaining headspace (∼5 mL) with ambient air, before shaking them end-over-end for 24 h. The solids of air-exposed suspensions were filtered, washed, and dried under ambient air.The specificity of Mössbauer spectroscopy to 57Fe was used to determine the oxidation states of Fe in the solid samples. Dried powders for Mössbauer analysis were loaded into Plexiglas holders (1 cm2) under anoxic conditions. The samples were placed in a closed-cycle He cryostat (SHI-850-I, Janis Research Co.) for measurement at 77 K. Spectra were collected with a constant acceleration drive system (WissEl, Wissenschaftliche Elektronik GmbH) in transmission mode with a 57Co/Rh source and calibrated against a 7 μm thick α-57Fe foil measured at room temperature. For two selected samples, the temperature was varied between 77 K and 15 K. All spectra were analyzed using the Recoil software31 by applying a Voigt-based fitting routine.32 The half-width at half maximum (HWHM) was fixed to a value of 0.130 mm s−1 for all samples.
X-ray absorption spectroscopy (XAS) at the Fe K-edge (7112 eV) was conducted at the SAMBA beamline of SOLEIL (Saint-Aubin, France). The dried solids were homogenized, pressed into 13 mm pellets and sealed between Kapton® tape. For transport to the synchrotron, samples were sealed in two layers of gas-tight-aluminum foil under N2 gas. Two samples were additionally prepared as oriented clay films for polarized-XAS measurement (α = 10°, 35°, 55°, and 80°). The oriented samples were prepared by collecting the clay on a cellulose filter (0.22 μm), separating the solid film from the filters, stacking the solid films, and sealing them between Kapton® tape. All measurements were conducted at 25 K to avoid beam damage and oxidation of oxygen-sensitive samples. Sample spectra were evaluated by linear combination fitting (LCF) to obtain information about the speciation of Fe and the formation of new Fe-bearing solid-phases. Shell-fit analyses of the extended X-ray absorption fine structure (EXAFS) spectra were also performed to obtain information about the short range local coordination environment of Fe. The nature of the backscattering atoms around Fe in the sorption samples was assessed using Morlet wavelet transforms (WT) of k3-weighted EXAFS data using the Fortran version of the HAMA software developed by Funke et al.33 Details on all measurements, data reduction, and analyses are provided as (ESI†).
Results and discussion
Sorption of ferrous iron to Syn-1
The pH-dependent sorption of Fe2+ to Syn-1 under anoxic conditions at different CaCl2 and Fe concentrations is shown in Fig. 1. At low pH (∼4), only small fractions of the total Fe2+ were sorbed to the clay, with the highest sorbed fraction in suspensions prepared with DDI water. This is consistent with non-specific adsorption of Fe2+ to cation exchange sites of the clay mineral and sorption competition with Ca2+. Between pH 4 and 7, sorption of Fe2+ increased strongly with pH and reached nearly 100% at circumneutral pH values. In the neutral to alkaline pH range, no clear sorption competition with Ca2+ was observed, pointing to inner-sphere surface complex formation of Fe2+ on clay mineral edge-surfaces and/or formation of surface clusters or precipitates. In all batch sorption experiments, the aqueous phase was thermodynamically undersaturated with respect to possible Fe-bearing solids at pH < 8 (see Table S1†), and hence, no secondary phases were expected to form by homogeneous nucleation and precipitation.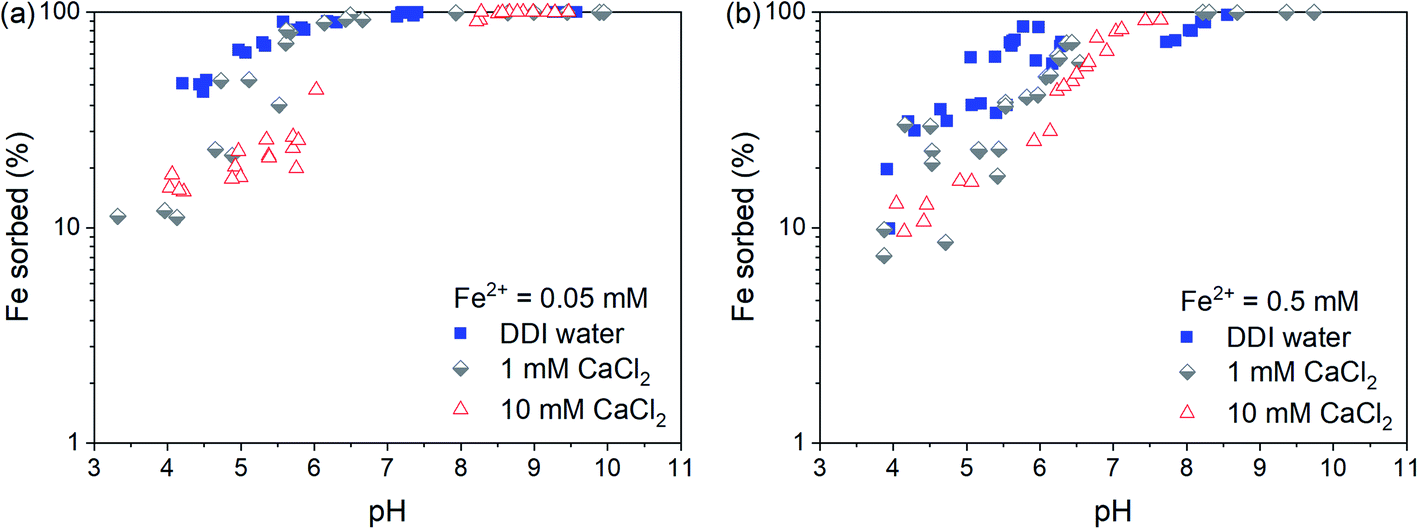 | ||
| Fig. 1 Percentage of Fe2+ sorbed on Syn-1 (∼1 g L−1) as a function of pH at different CaCl2 concentrations. Samples in DDI water had a Ca2+ concentration of ∼0.1 mM after 1 day equilibration. Total Fe2+ concentrations of (a) 0.05 and (b) 0.5 mM were added and equilibrated for 1 day with 1 g L−1 Syn-1. | ||
To investigate the sorption mechanism of Fe2+ to Syn-1 by XAS further, an additional set of samples was prepared at pH 7 or 8 under anoxic conditions containing 5 g L−1 clay, 0.25 or 2.5 mM Fe2+, and 50 mM CaCl2. The high CaCl2 concentration and pH values were selected to focus on sorption processes on edge surfaces, which are expected to dominate under these conditions. Furthermore, at pH < 8 the dissolved Fe2+ concentrations in the suspensions were always below saturation with respect to Fe(OH)2 (Table S1†).
In the XAS samples, the sorbed fraction of Fe2+ was higher at pH ∼8 than at pH ∼7 (Table S2†). The amount of sorbed Fe2+ also slightly increased with increasing equilibration time from day 1 to 30 (Table S2 and Fig. S3†), e.g., by 0.02 mol Fe per kg clay at pH 7. This observation is consistent with findings by Zhu et al.,34 who observed an increase in Fe2+ sorption to Syn-1 during 5 days at pH 7.5. Such increases of Fe2+ sorption suggest that slow sorption processes were taking place. Possible mechanisms explaining slow sorption kinetics include dissolution and precipitation reactions on surfaces.35–44
The derivative of the Fe K-edge XANES spectra of the solids from selected Fe2+ sorption samples are shown in Fig. 2, along with relevant Fe reference compounds for comparison. The position of the highest maximum in the derivatives of XANES spectra for the high Fe-loading anoxic sorption sample at 7125 eV suggests an Fe oxidation state of close to +2. However, the presence of a small shoulder at 7128 eV might suggest that a small fraction of Fe2+ was oxidized in these samples. For the low Fe-loading anoxic sample at pH ∼8, the position of the highest maximum is consistent with a valence state between +2 and +3, suggesting that sorbed Fe2+ was partially oxidized during sorption experiments. The low Fe-loading anoxic sample at pH 7 exhibited a maximum at 7127 eV, corresponding to a +3 oxidation state.
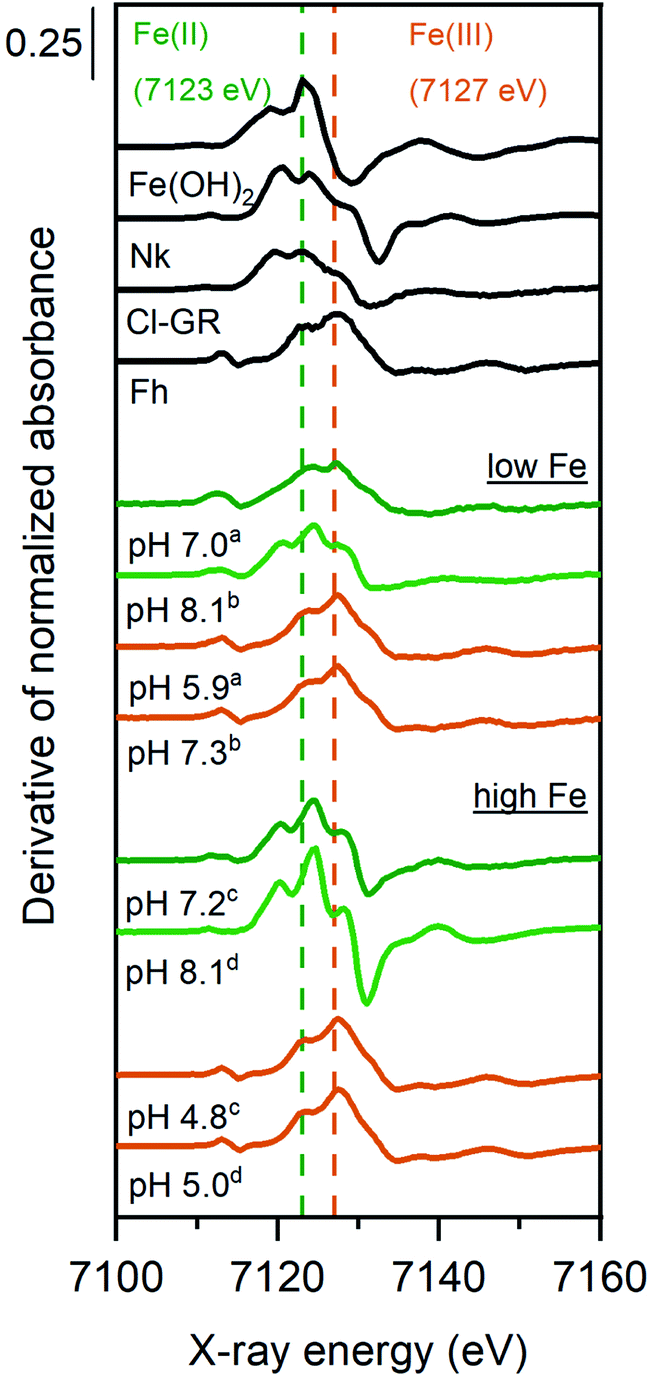 | ||
| Fig. 2 Derivative of normalized Fe K-edge XANES spectra of Fe2+ sorption samples in which Syn-1 (∼5 g L−1) was reacted with low (0.25 mM) or high (2.5 mM) Fe2+ concentrations at pH ∼7 or ∼8. Sorption samples were equilibrated for 1 day under anoxic conditions (green lines) and subsequently oxidized for 1 day (orange lines). Superscripts (a–d) indicate the corresponding pairs of anoxic and oxidized samples. Displayed pH values correspond to the pH measured at the end of the equilibration period for each sample. Selected reference spectra of solid phases containing Fe(II) and/or Fe(III) are shown for comparison (abbreviations: Nk = nikischerite, Cl-GR = chloride green rust, Fh = ferrihydrite). | ||
To quantify the oxidation states of sorbed Fe on Syn-1 under anoxic conditions with greater precision, Mössbauer spectroscopy was performed for selected solids (1 g L−1 clay experiments) from low and high Fe-loading samples (Fig. 3 and S8†). Fit parameters are detailed in Table S6.† Anoxic spectra (Fig. 3a and b) exhibited two doublets revealing the presence of ferric iron (Fe(III)) in addition to ferrous iron (Fe(II)). Accordingly, the spectra were fitted with one paramagnetic doublet with large quadrupole splitting (ΔEQ ∼2.68 mm s−1) assigned to Fe(II) and a second doublet with small quadrupole splitting (ΔEQ ∼0.81 mm s−1) consistent with Fe(III). At high Fe-loading, 14–28% of the sorbed iron was oxidized to Fe(III) under anoxic conditions and 52–100% for sorption experiments performed at low Fe-loading (Table S6†). The ferrous doublet had similar isomer shift (δ) and quadrupole splitting (ΔEQ) values as were reported for chloride green rust (δ = 1.27 mm s−1; ΔEQ = 2.68 mm s−1).45,46 The ferric doublet (ΔEQ ∼0.81 mm s−1; δ ∼0.46 mm s−1) was consistent with poorly crystallized or amorphous compounds such as ferric hydroxide Fe(OH)3 or ferrihydrite.47 Overall, the data clearly show that the added Fe2+ was partially oxidized upon sorption to Syn-1 clay under anoxic conditions. The relative abundance of Fe(III) was consistently higher at the low Fe-loading (Table S6†).
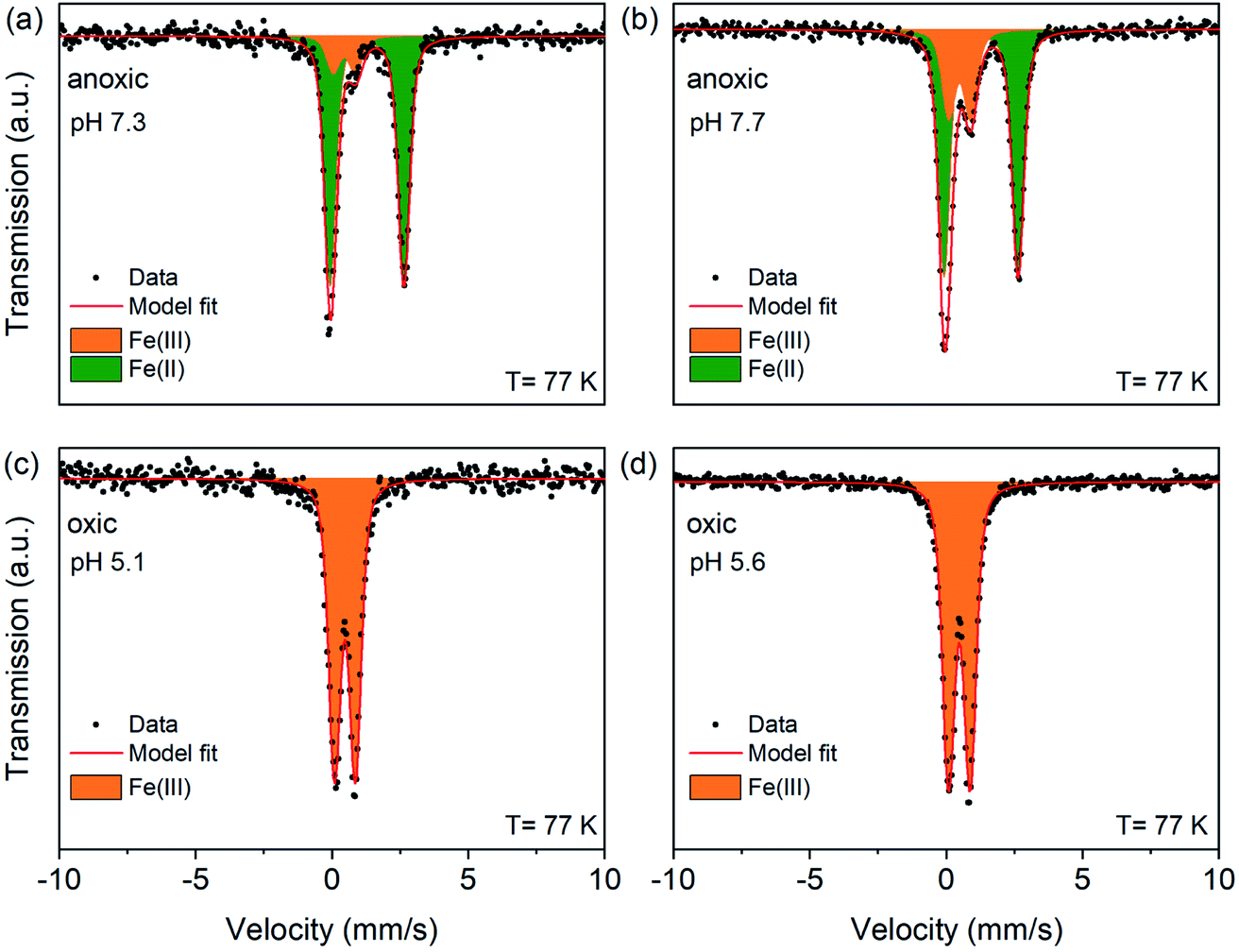 | ||
| Fig. 3 77 K Mössbauer spectra and fits of high Fe-loading solid samples. Syn-1 (1 g L−1) was reacted with 0.5 mM Fe2+ (enriched in 57Fe) at pH ∼7 or ∼8 under anoxic conditions during 1 day (a and b). Anoxic samples were subsequently exposed to air for 1 day (c and d). Displayed pH values correspond to the pH measured at the end of the equilibration period for each sample. In all graphs, symbols represent data and red lines the model fits. Corresponding fitting parameters are summarized in Table S6.† The Fe(II) doublet is represented as green area and the Fe(III) doublet as orange area. | ||
The presence of Fe(III) species in anoxic sorption samples confirms that even under anoxic conditions, Fe(II) can be oxidized to Fe(III) in the presence of clay minerals with very low structural Fe contents. The amount of structural Fe present in the Syn-1 (4.65 mmol Fe per kg) may have contributed to the oxidation of at maximum 0.93 and 9.3% of the added Fe2+ for high and low Fe-loading samples, respectively. Oxidation of Fe2+ by accidental exposure to O2 can be excluded as no Fe2+ oxidation was observed in Fe2+ control solutions without clay. An additional anoxic control experiment on Fe2+ sorption to Syn-1 was carried out in a glass bottle to ensure that no residual O2 sorbed to the walls of Nalgene® plastic bottles induced Fe2+ oxidation. The same results were obtained in experiments with glass and Nalgene® bottles, excluding residual sorbed O2 as a possible electron acceptor. Therefore, other reactive sites on the clay mineral surface must have induced electron transfer from Fe2+ to an electron acceptor. As only traces of Ti were present in the clay mineral (1.3 mmol Ti per kg) it can be excluded as main electron acceptor.
Géhin et al.14 suggested hydrogen to be a potential electron acceptor with the production of H2 that might be sorbed by the clay mineral and ensure the reversibility of the Fe(II) oxidation. In the study by Géhin et al.,14 the Fe2+ concentration added to a Fe-free clay (∼0.06 mmol Fe per g clay) was similar to the low Fe-loading sorption samples of this study. The partial pressure of H2 in both experiments was very low as they were performed in a 100% N2 atmosphere.14 Thermodynamic calculations demonstrate that at very low H2 partial pressures, H+ is a potential oxidant for Fe2+ at neutral to alkaline pH to form amorphous Fe(III)-hydroxides (Fig. S2a†). In the presence of more crystalline Fe(III) solids having lower iron solubilities such as e.g., lepidocrocite and goethite, H+ would represent an even more favorable oxidant for Fe2+ and might enable Fe2+ oxidation already at slightly acidic pH values (Fig. S2b†). The reduction of H+ would lead to the production of H2 and an increase of the partial pressure of H2. Concurrently, the Fe2+ activity is expected to decrease due to the formation of Fe(III) solid phases and progressing Fe2+ adsorption to mineral surfaces. Consequently, the ability of H+ to act as an oxidant for Fe2+ decreased during sample equilibration (see arrows in Fig. S2a†). This explains why the oxidation of Fe2+ was limited in our experiments. Accordingly, an increased percentage of Fe2+ oxidation with lower Fe-loading is expected, which is consistent with previous findings by Soltermann et al. and Géhin et al.14,30 confirming that the oxidation of Fe2+ is likely limited by the partial pressure of H2 and the Fe2+ activity in solution. In a control experiment without clay minerals present, Fe2+ oxidation was not observed. Based on this finding, we propose that clay minerals served as a catalyst for Fe2+ oxidation by H+ by facilitating the electron transfer via ligation of Fe2+ to protonated surface oxygen atoms. This catalytic effect of surfaces in electron transfer reactions may be important in various environmental settings. For instance, it has been shown that the reduction of organic pollutants by Fe2+ is enhanced when sorbed to mineral surfaces.48–50
To further investigate the possible solid-phase Fe species formed during the 1 and 30 days equilibration periods, linear combination fits (LCF) of Fe K-edge EXAFS spectra were performed (Fig. 4, Tables S11 and S12†). LCF analysis showed that EXAFS spectra of high Fe-loading anoxic samples were adequately fitted by including Fe(II)-hydroxide (β-Fe(OH)2) (6–13%), chloride green rust ([Fe32+Fe3+(OH)8]·[Cl·nH2O])45 (51–68%), and nikischerite (NaFe2+6Al3(SO4)2(OH)18(H2O)12)51 (19–40%). Nikischerite is a natural Fe(II)2Al(III)-layered double hydroxide (LDH) mineral with sulfate interlayer anions and is used here as a structural reference representing Fe(II)Al(III)-LDH phases in general.51 Fitting of the low Fe-loading anoxic samples (Fig. 4b) required the reference compound ferrihydrite, which is in line with the presence of Fe(III) as observed by both Mössbauer and XANES spectroscopy.
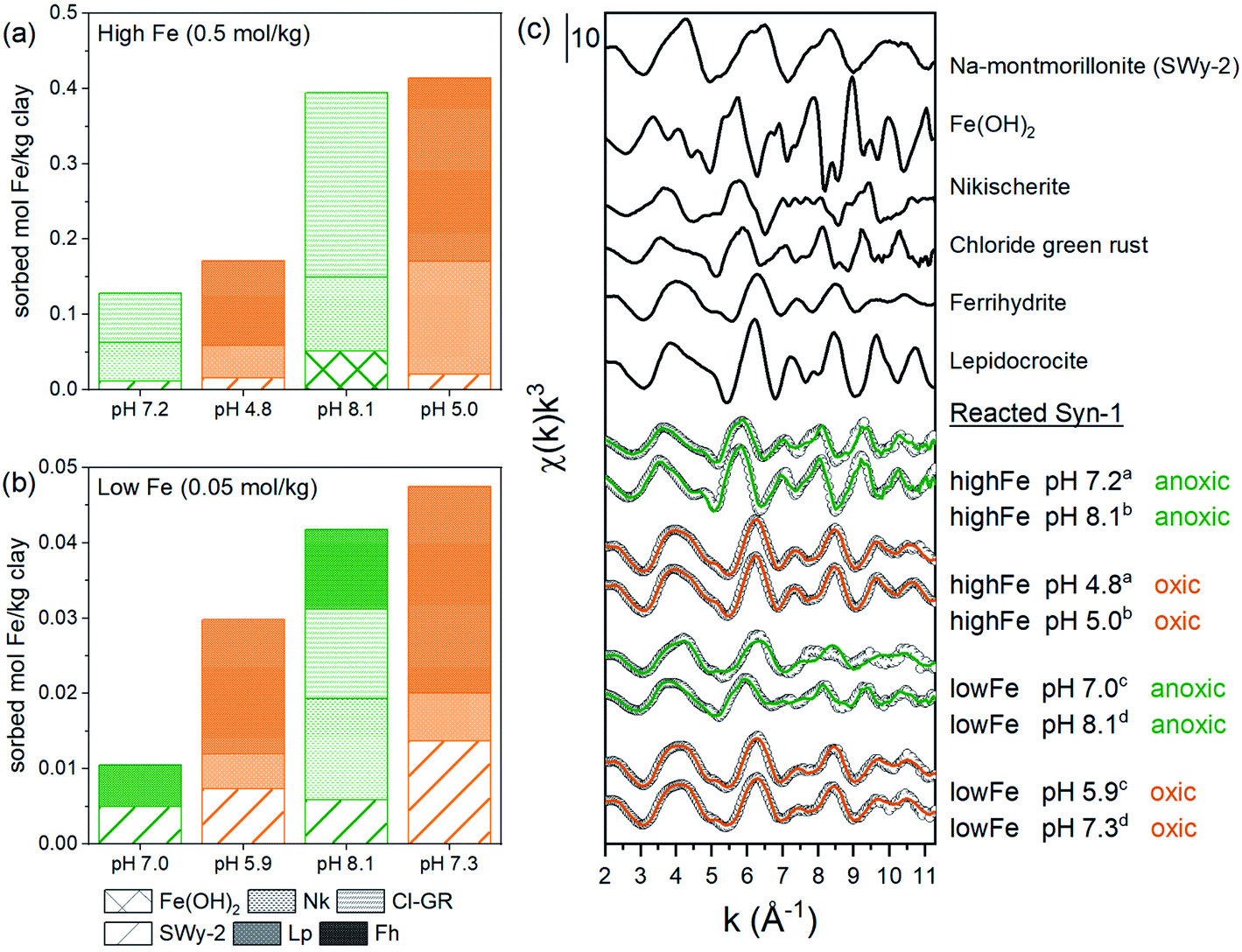 | ||
| Fig. 4 LCF fitting results of Fe K-edge EXAFS spectra of Fe2+ sorption samples equilibrated for 1 day under anoxic conditions (green lines) and subsequently oxidized for 1 day (orange lines) and relevant references: (a and b) absolute fractions of Fe references obtained by linear combination fitting of k3-weighted Fe K-edge EXAFS spectra for Fe2+ sorbed on Syn-1. High Fe load (a and c) and low (b and c) (0.25 mM) sorption samples. The fits were performed over a k-range of 2–11.3 Å−1 (kw = 3) with no fit constraints. The initial fit fractions (70.9–146.2%) were normalized to the sum equal to 100% and are reported in Tables S11 and S12† (abbreviations: Nk = nikischerite, Cl-GR = chloride green rust, Lp = lepidocrocite, Fh = ferrihydrite). (c) k3-weighted χ spectra of references (line): SWy-2, Fe(OH)2, nikischerite, chloride green rust, ferrihydrite and lepidocrocite are shown together with the k3-weighted χ spectra (open circles) of 1 day equilibrated samples with corresponding fit (green and orange colors correspond to anoxic and oxidized samples, respectively). Displayed pH values correspond to the pH measured at the end of the equilibration period for each sample. Superscripts (a–d) indicate the corresponding pairs of anoxic and oxidized samples. | ||
An improved fit (>10% decrease in R-factor, see Table S12†) was obtained for the low Fe-loading anoxic sorption sample equilibrated at pH ∼8 when Fe(II)-containing minerals, namely nikischerite and chloride green rust, were included. This is again consistent with the presence of Fe(II) species, as observed by Mössbauer spectroscopy. A comparison of the Fe oxidation states determined by Mössbauer spectroscopy for the 1 g L−1 batch experiments and by LCF of EXAFS spectra for 5 g L−1 batch samples is provided in Table 1. Both sets of batch experiments are in reasonable agreement and thus confirm that Fe2+ in anoxic Fe sorption samples can partially be oxidized in the presence of Syn-1 clay.
| High Fe load | Anoxic target pH | EXAFS | Mössbauer | ||
|---|---|---|---|---|---|
| Fe(II)a | Fe(III)a | Fe(II) | Fe(III) | ||
| a Assumption of a 3/1 Fe(II)/Fe(III) ratio as in stoichiometrically pure Cl-GR. b Note: parameter uncertainties are given in parentheses. | |||||
| Anoxic | 7 | 78 | 22 | 86 (1.3) | 14 (1.3) |
| Anoxic | 8 | 84 | 16 | 72 (0.6) | 28 (0.6) |
| Oxic | 7 | 0 | 100 | 0 | 100 |
| Oxic | 8 | 0 | 100 | 0 | 100 |
For the majority of the samples, the LCF significantly improved when an Fe(III)-containing clay mineral reference (SWy-2, containing 3 wt% Fe) was included as additional reference in the fits. Released Si from Syn-1 after 1 day equilibration between pH 6 and 10 amounted on average to ∼0.016 mmol Si per g clay compared to up to 8 times lower Si concentrations for anoxic sorption samples with Fe (see Fig. S6a and S7†). This provides additional evidence that the released Si from Syn-1 was removed from solution upon addition of Fe, either by adsorption or incorporation into newly formed Fe phases. It has been suggested by Soltermann et al.30 that Fe2+ can be taken up by the clay minerals, leading to the formation of Fe-bearing clay minerals.
In presence of elevated divalent metal (Me2+) concentrations (first-row transition series), it has been shown that Me(II)Al-LDH can form under alkaline conditions in the presence of Al-containing minerals.21,36,37,39,41–44,52–54 The formation of FeAl-LDH phases is favored over Fe(OH)2 in environments where Al3+ is present because FeAl-LDH phases are less soluble than Fe(OH)2 by two orders of magnitude.54 Yet, partial oxidation of Fe(OH)2 would lead to the formation of green rust, for which no information is available concerning its stability compared to FeAl-LDH.
The contributions of Fe(OH)2, Cl-GR, and nikischerite to the LCF of EXAFS spectra of high Fe-loading anoxic samples suggest that under our experimental conditions, Fe2+ was mainly sorbed through the formation of Fe2+-containing layered mineral phases. Furthermore, the inclusion of a reference spectrum for aqueous Fe2+ in the LCF did not improve the fits significantly, confirming that outer-sphere adsorption of Fe2+ to the clay surface was not an important sorption mechanism at neutral to weakly alkaline pH.
The goodness of fit in the LCF analysis of anoxic sorption samples was not fully satisfactory (R-factors > 5%, Tables S11 and S12†). Therefore, we further analyzed the data by shell-by-shell fitting of Fourier transformed (FT), k3-weighted EXAFS spectra (Fig. S11†), which is independent of reference spectra and may provide additional information about the average bonding environment of Fe in the samples.55 For all anoxic sorption samples, a first shell was observed at ∼1.5 Å (uncorrected for phase shift), representing the first oxygen shell surrounding the central Fe atoms. Further shells with R + ΔR between 2 and 3.5 Å were observed, which could be attributed to Fe–Fe, Fe–Al, or Fe–Si backscattering pairs.
For high Fe-loading anoxic samples (Tables 2 and S13†), the first shell was fitted with 4.1–5.4 O atoms at a distance of 2.10–2.12 Å, which is characteristic of an octahedral arrangement of O around Fe(II). The second shell at ∼2.8 Å (uncorrected for phase shift) can be fitted with Fe and Al or Si backscatterers.51,56,57 Due to the similar or even same crystallographic position of Fe2+, and Al3+ in LDH, it is difficult to unequivocally differentiate these two atoms. Additionally, constructive interferences from Si4+ atoms in greenalite,57 which are found at slightly larger crystallographic positions than Fe2+ and Al3+, could occur. Wavelet transformation (WT) of EXAFS spectra can complement the FT by resolving the k-dependence of the absorption signal, allowing better distinction between heavier and lighter backscattering atoms. The WT of the second coordination shell of the high Fe-loading anoxic samples and Fe references are shown in Fig. S13 and S14.† Unfortunately, WT did not allow for the distinction between Al and Fe path contribution to the second shell as both nikischerite and chloride green rust possessed a peak at k ∼6 Å−1. Therefore, three models with different single scattering paths for fitting the second shell in high Fe-loading anoxic samples were compared (Table S16 and Fig. S12†). Model 1 with only a second shell Fe–Fe path, Model 2 with Fe–Fe and Fe–Al paths, and Model 3 with Fe–Fe and Fe–Si paths, respectively. Model 1 returned a Fe–Fe distance of 3.20–3.21 Å, in agreement with crystallographic data of chloride green rust.56 Including an additional Fe–Al path (Model 2) resulted in a Fe–Al distance of 3.30 Å, which is significantly different from the Fe–Al distance in FeAl-LDH phases like nikischerite (3.12 Å).51 Similarly, including an additional Fe–Si path (Model 3) resulted in a fitted Fe–Si distance of 3.19 Å, which differs from the Fe–Si distance in greenalite (3.30 Å).57 Additionally, the inclusion of a Fe–Al or Fe–Si backscattering path to Model 1 did not decreased the reduced χ2-values by at least a factor two.55 Based on these results, Model 2 and 3 might not be suitable for the fitting of high Fe-loading anoxic samples. The aforementioned findings suggest that in high Fe-loading anoxic sorption samples mainly a LDH phase with Fe in the octahedral sheet formed. However, WT of EXAFS spectra of high Fe-load samples displayed as an intermediate between FeAl-LDH and Cl-GR, which might suggest that Al released from the clay minerals (see Fig. S6b†) was incorporated into the octahedral sheets of the newly formed Fe-phase. This is in agreement with the LCF fits of high Fe-loading anoxic sorption samples, which required both the nikischerite and Cl-GR reference. Therefore, it is likely that a mixed Fe(II)–Al(III)/Fe(III)-LDH phase formed under these conditions, as suggested by Starcher et al.44
In order to obtain information about the orientation of the newly formed secondary phases polarized EXAFS (P-EXAFS) spectra were recorded for the high Fe-loading sorption samples. In P-EXAFS neighbouring atoms along the electric field (or along polarization direction of the X-ray beam) are preferentially probed and atoms located in a plane perpendicular to this electric field direction are attenuated. Applying this method to clay mineral self-supporting films has the advantage of minimizing the contribution of cations from the tetrahedral sheets by orienting the layer ab plane to the electric field. Conversely, the contributions of cations from the octahedral sheet is extinguished in the perpendicular orientation of the electric field. The P-EXAFS spectra of the oriented high Fe-loading sorption sample recorded at α = 10°, 35°, 55° and 80° are presented in Fig. S15.† The presence of isosbestic points in the spectra provides evidence that the differences in the measured spectra were only due to orientation effects.58–61 The pronounced dependence upon orientation in the regions between the isosbestic points confirms the successful preparation of a clay film with preferred orientation.59–62 The angle dependence of the spectra also confirms the anisotropic formation of the secondary phase aligned with the clay mineral sheets for high Fe-loading anoxic sorption samples.
For the low Fe-loading anoxic samples, the first shell was fitted with 4.6–5.3 O atoms at a radial distance of 2.01–2.05 Å which is an intermediate distance of an octahedral arrangement of O atoms around Fe(II) and Fe(III).56,63 The fitting of a CN < 6 may suggest the presence of some tetrahedral Fe as found in ferrihydrite. This result is in agreement with the presence of Fh in the LCF of EXAFS spectra of this sample (Table S12†).
The FT of the EXAFS spectra of the low Fe-loading anoxic samples equilibrated at pH ∼8, showed a second peak at ∼2.75 Å (uncorrected for phase shift). This second shell was fitted with Fe located at a radial distance of 3.15–3.17 Å and a coordination number of 1.8–2.1. The fitted distance for the second shell corresponds with an intermediate distance of edge-sharing Fe atoms around Fe(II) and Fe(III) as in fougerite56 and lepidocrocite,64 respectively. This is consistent with the results obtained by LCF and Mössbauer spectroscopy.
The low Fe-loading anoxic sample equilibrated at pH ∼7 (Table 2) showed two overlapping small peak after the first shell, one at ∼2.3 Å and ∼3.1 Å (uncorrected for phase shift). Fitting of these two shells required a Fe–Al, Fe–Si and Fe–Fe backscattering path with distances of 2.84, 3.08 and 3.48 Å, respectively. Fitted distances for Fe–Al and Fe–Si paths are in good agreement with bond distances in phyllosilicates.65 The distance fitted for the Fe–Fe path at 3.48 Å is characteristic for double corner-sharing Fe as in ferrihydrite.63,66
| Fe–O | Fe–Fe | ΔE0e | Red. χ2f | R-factorg | |||||
|---|---|---|---|---|---|---|---|---|---|
| CNb | R (Å) | σ 2 (Å2) | CNb | R (Å) | σ 2 (Å2) | ||||
a The amplitude reduction factor S02, was set to 0.9 based on first shell optimization for all sorption samples (R-range 1.1–2.5 Å).
b Path degeneracy (coordination number).
c Mean half path length.
d Debye–Waller parameter. Debye–Waller parameter was fixed to 0.01 Å2 for Fe–Al and Fe–Si based on the σ2 obtained for Fe–Si and Fe–Al path by Soltermann et al.30σ2 (Fe) was fixed to 0.009 Å2 for Fe–Fe of low Fe-load sorption sample like was fitted for edge-sharing Fe–Fe for low Fe-load samples.
e Energy-shift parameter.
f Fit accuracy; reduced 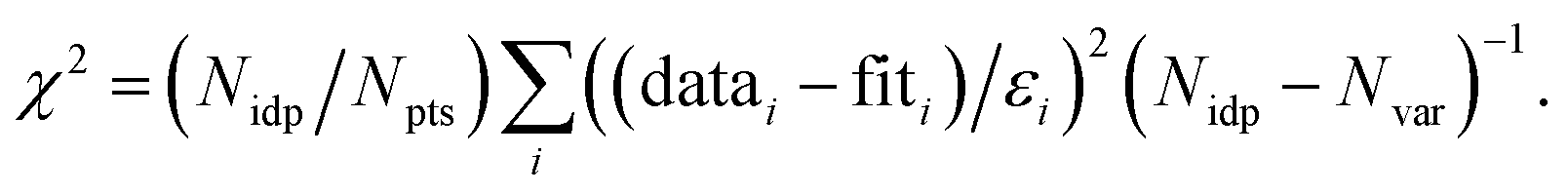 Nidp, Npts and Nvar are, respectively, the number of independent points in the model fit (11.2–13.7), the total number of data points (151–171), and the number of fit variables (6–10). εi is the uncertainty of the ith data point.
g
R-factor; Normalized sum of squared residuals Nidp, Npts and Nvar are, respectively, the number of independent points in the model fit (11.2–13.7), the total number of data points (151–171), and the number of fit variables (6–10). εi is the uncertainty of the ith data point.
g
R-factor; Normalized sum of squared residuals 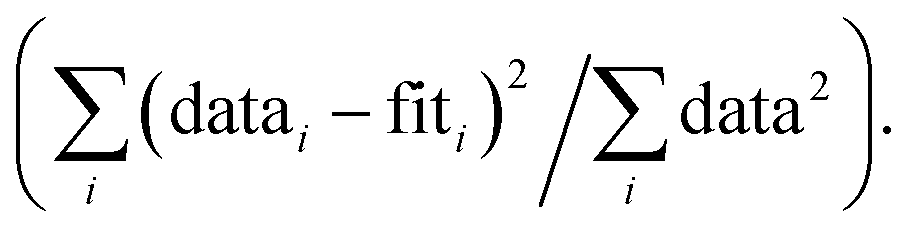 Note: The FT of k3-weighted EXAFS spectrum of the anoxic low Fe-loading samples at pH 8.1 was calculated over a k-range of 2–9.5 Å−1. The shell-fit analysis of k3-weighted Fe EXAFS spectrum of the anoxic low Fe-loading sample at pH 7.0 was performed in R-space R + ΔR-range of 1.1–4 Å. Note: The FT of k3-weighted EXAFS spectrum of the anoxic low Fe-loading samples at pH 8.1 was calculated over a k-range of 2–9.5 Å−1. The shell-fit analysis of k3-weighted Fe EXAFS spectrum of the anoxic low Fe-loading sample at pH 7.0 was performed in R-space R + ΔR-range of 1.1–4 Å.
|
|||||||||
| High Fe load | |||||||||
| pH 7.2 | 4.1 (0.8) | 2.10 (0.01) | 0.009 (0.003) | 2.6 (0.9) | 3.21 (0.01) | 0.006 (0.003) | 1.17 (1.79) | 110 | 0.0211 |
| pH 8.1 | 6.0 (0.5) | 2.12 (0.01) | 0.009 (0.001) | 4.5 (0.5) | 3.20 (0.01) | 0.007 (0.001) | −1.53 (0.7) | 235 | 0.0032 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||
| Low Fe load | |||||||||
| pH 8.1 | 4.6 (0.9) | 2.06 (0.02) | 0.012 (0.003) | 2.1 (0.4) | 3.17 (0.02) | 0.009 | 1.25 (1.81) | 14 | 0.0274 |
The WT of EXAFS spectra of low Fe-loading anoxic sample equilibrated at pH ∼7 (see Fig. S14c†) contained three maxima, one at k = 3, 7, and 10 Å−1 suggesting three different backscattering atoms contributing to the second shell. This result confirms that including the Fe–Fe, Fe–Al and Fe–Si backscattering paths in the model to fit low Fe-loading anoxic samples equilibrated at pH ∼7 is reasonable. Additionally, F-tests confirmed that the inclusion of Fe–Al and Fe–Si backscattering paths into the fit model significantly improved the goodness of the fit (S.I. Section 11†).
Previous studies have also investigated the sorption of Fe2+ to different clay minerals at various Fe-loadings.14,30 Collectively, the literature results suggested that depending on the Fe-loading, various Fe(II)- and Fe(III)-containing solids formed. At high Fe-loadings (0.53–2 mol Fe per kg) the formed secondary phases were predominantly Fe(II)-containing phases.19,21 At low Fe-loadings (0.002–0.08 mol Fe per kg) mainly Fe(III)-containing phases were observed.30,67 Our current study are in agreement with these results. The formation of an LDH phase with Fe in the octahedral sheet was observed for high Fe-loading anoxic sorption samples as shown by Jones et al.19 Contrary, in the study by Zhu et al.21 no Fe2+ oxidation was observed during sorption of Fe2+ onto Syn-1 and the formation of FeAl-LDH was suggested as the main solid phase forming. However, sorption experiments in the study by Zhu et al. were performed in an atmosphere with 5% H2 and 95% N2. Such elevated H2 concentration are expected to disable H+ to oxidize Fe2+ (cf. Fig. S2†) and to suppress the formation of green rust as the main secondary phase. In our low Fe-loading samples, over half of Fe(II) was oxidized to Fe(III), from which a large fraction was incorporated into newly formed clay minerals. This result shows that clay minerals can play an important role in the mobility of Fe2+ under anoxic conditions.
Effects of aeration of anoxic sorption samples on Fe speciation
To understand the effects of O2-exposure on the speciation of Fe in clay suspensions (Syn-1) that were anoxically equilibrated for 1 or 30 days with Fe2+, the anoxic suspensions were exposed to ambient air for 24 hours. Aeration resulted in a further increase in percent Fe sorption (see Fig. 4a, b and Table S2†), despite a drop in suspension pH (between 0.8 and 3.1 pH values) as a result of hydrolysis of Fe3+ (Table S3†). Due to the high O2 sensitivity of Fe2+ at near-neutral pH, rapid oxidation of adsorbed and dissolved Fe2+, hydrolysis of Fe3+, and precipitation of Fe(III)-(oxyhydr)oxides may occur. This results in the release of protons, explaining the observed decrease of suspension pH.After the equilibration of Fe2+ with Syn-1 under anoxic conditions, substantial amounts of aqueous Fe2+ still remained in solution (70–78% of added Fe at pH ∼7 and 7–16% at pH ∼8, see Table S2†). Additionally, some adsorbed Fe2+ may have been desorbed from clay surfaces as pH started to decrease. All dissolved and adsorbed Fe2+ is expected to rapidly oxidize upon exposure to O2 and form iron minerals such as lepidocrocite and/or ferrihydrite, depending on solution composition (e.g., Ca, Si).68 However, the remaining 22 to 30% (at pH ∼7) or 83 to 93% (pH ∼8) of the added Fe was sorbed to the clay, mostly by the formation of Fe(II) solid phases such as chloride green rust, FeAl-LDH, and/or Fe(III)-phyllosilicates, as was shown in the section above. The oxidation kinetics and resulting Fe species formed during aeration of such sorbed ferric Fe on clay minerals is far less understood.
Mössbauer spectroscopy showed that the 24 h aeration of anoxic Fe2+ sorption samples led to a complete oxidation of all added Fe, resulting in only a ferric doublet in the spectra of oxic samples (Fig. 3c and d). This finding shows that even 30 days equilibration under anoxic conditions, resulting in mainly the incorporation of Fe(II) into solid phases, did not prevent rapid oxidation to Fe(III). Additional Mössbauer spectra of two high Fe-loading oxic samples were measured at 45, 35, 25 and 15 K (Fig. S10†). The fitting results of the temperature-dependent Mössbauer spectra are given in Tables S7 and S8.† In general, the spectra displayed the characteristic effect of magnetic ordering with decreasing temperature. Ordered sextets had hyperfine parameters δ (between 0.49 and 0.50 mm s−1), ΔEQ (between −0.05 and −0.01 mm s−1) and hyperfine field (Bhf) (between 43.9 and 44.3 T) for the oxic samples at 15 K, which are in line with values reported for short range ordered Fe(III)-oxyhydroxides such as lepidocrocite and ferrihydrite.69 Additionally, temperature dependent Mössbauer spectra allow the determination of the blocking temperature, which is affected by crystallite size or interparticle interactions.70,71 We calculated the blocking temperature as the temperature at which the summed areas of the paramagnetic doublets and magnetic sextets (which includes poorly ordered and fully magnetically ordered sextets) of Mössbauer spectra are equal. Fig. S9† shows the relative abundance of doublets and sextets for the two oxic samples at pH 5.1 (pH 7.3 under anoxic conditions) and pH 5.6 (pH 8.4 under anoxic conditions). Both samples displayed a blocking temperature below the literature values for ferrihydrite and lepidocrocite (between 50 and 77 K).69,72 A blocking temperature of ∼32 K was calculated for the oxic sample pre-equilibrated at pH 7.3, compared to ∼42 K for the aerated suspension pre-equilibrated at pH 8.4. This indicates that oxidation of the anoxic sorption samples led to the formation of short range ordered Fe(III)-(oxyhydr)oxides, for which either precipitates had smaller crystallite size or show a lesser degree of interparticle interaction, potentially as a result of surface sorption effects, when formed at pH 7.3 than pH 8.4.
For all oxic sorption samples, the position of the Fe K-edge was at 7127 eV (Fig. 2), which corresponds to the oxidation state Fe(III) and is in line with Mössbauer data (Fig. 2). Linear combination fitting (LCF) results for Fe K-edge EXAFS spectra are presented in Fig. 4 (also Tables S11 and S12†). Oxidized sorption samples were well described (R-factor < 2%) with 3 reference compounds: ferrihydrite (45–60%), Fe(III)-containing phyllosilicates (24–36%), and lepidocrocite (13–19%). Essentially, all oxic sorption samples transformed into the same secondary phases regardless of their equilibration conditions (pH, Fe concentration) and time (1 or 30 days) under anoxic conditions (Tables S11 and 12†). Parts of the Fe(III)-containing phyllosilicates that have already formed under anoxic conditions were not affected by oxidation and remained as secondary phase after aeration. It was observed that the fraction of lepidocrocite formed was significantly lower for low Fe-loading samples compared to high Fe-loading samples.
The oxidation of aqueous ferrous iron at pH 7 and in a chloride background is known to lead to the formation of lepidocrocite, with crystallinity decreasing with decreasing pH.27 Below pH 4, oxidation of ferrous iron is expected to precipitate as ferrihydrite.27 As the pH of the oxic sorption samples remained above pH 4 and the concentration of Ca was elevated, the formation of lepidocrocite as the major Fe(III)-(oxyhydr)oxides is expected. However, the presence of dissolved silicate, released from the clay mineral, above 10 μmol L−1 has been shown to inhibit the formation of more crystalline iron oxides, such as lepidocrocite,73 and favor the formation of ferrihydrite.66,68 Therefore, the observation of ferrihydrite as the dominant secondary phase under oxic conditions in this study was more anticipated, as Si released from the clay mineral amounted to ∼80 μmol Si L−1 between pH 6 and 10 (Fig. S6a†). The observation of lepidocrocite as a second Fe(III)-(oxyhydr)oxides in the oxic samples can be explained by two mechanisms: (i) through the oxidation of green rust, which under certain conditions can be oxidized into lepidocrocite28 or (ii) through dissolved ferrous iron catalyzing the transformation of poorly-crystalline ferrihydrite to the more stable mineral lepidocrocite.74,75
A lower amount of lepidocrocite was formed in the low Fe-load samples, as direct oxidation of Fe2+ in presence of elevated Si concentrations (high Si/Fe ratio in solution) may mainly precipitate as ferrihydrite.73 However, only a small fraction of the sorbed Fe in the oxic samples was formed by direct oxidation of dissolved Fe2+ for low Fe-loading samples equilibrated at pH ∼8 (between 7 and 12%, see Fig. S4 and S5†) compared to samples equilibrated at pH ∼7 (between 60 and 65%, see Fig. S4 and S5†). Nevertheless both low Fe-loading samples resulted in the presence of similar solid phases. The higher fraction of lepidocrocite observed in the high Fe-loading oxic samples as compared to the low Fe-loading samples may be due to the oxidation of dissolved Fe2+ in the presence of a low Si/Fe ratio in solutions, which favors lepidocrocite formation over ferrihydrite or through the transformation of green rust into lepidocrocite.66,73 Because only a small fraction of the sorbed Fe (up to 20%, see Fig. S4 and S5†) was formed by direct oxidation of dissolved Fe2+ in high Fe-loading samples, lepidocrocite was primarily formed from the transformation of Fe-phases precipitated under anoxic conditions.
The LCF of EXAFS spectra of oxic samples significantly improved when an Fe(III)-containing clay mineral reference (SWy-2, representing Fe(III) in phyllosilicate structures) was included. Furthermore, measured Si in solution of oxic samples was 6–40 times lower than the Si released from Syn-1 after 1 day equilibration between pH 6 and 10 in absence of Fe (∼0.016 mmol Si per g clay) (Fig. S6a and S7†). Combined, this provides strong evidence for sorption and/or incorporation of Si into the Fe species formed upon aeration. Based on the LCF results, the amount of Fe sorbed by the formation of Fe-phyllosilicates (the fraction fitted by SWy-2) was calculated. Together with amount Si re-sorbed upon Fe2+ addition, this allowed to calculate the ratio of Fe to Si sorbed in the samples. The overall Fe/Si ratio of the newly formed Fe species was estimated to amount to 0.56–1.83 (Tables S4 and S5†). These values are in line with Fe/Si ratios one would expect for 1:1 Fe-phyllosilicate, like (Fe2+,Fe3+)3–2Si2O5OH and 2:1 Fe-phyllosilicate ((Fe2+,Fe3+)3–2Si4O10(OH)2).76
More detailed information about changes in local coordination environment of Fe upon aeration of the suspensions was obtained by shell-by-shell fitting of fourier transformed k3-weighted Fe K-edge EXAFS spectra (Fig. S11†). All oxic sorption samples showed a first shell at ∼1.5 Å (uncorrected for phase shift), representing the first oxygen shell surrounding the Fe atoms. A second shell was observed between 2 and 3.5 Å, which can be attributed to different Fe–Fe backscattering pairs. The first shell was fitted with 4.9–6.0 O atoms at a radial distance of 1.99–2.00 Å. This is consistent with an octahedral arrangement of the O atoms around a central Fe(III), as in lepidocrocite.63 The fitting of a CN smaller than 6 may suggest the presence of some tetrahedral Fe as found in ferrihydrite.77 This result is in agreement with the presence of Fh in the LCF of EXAFS spectra of oxic sample (Tables S11 and S12†). The second shell was composed of two peaks, one at ∼2.65 Å and a second peak at ∼3.11 Å (uncorrected for phase shift). These two peaks were successfully fitted with two Fe–Fe single backscattering paths. The first Fe backscattering path (Fe–Fe1) had a fitted distance of 3.07–3.09 Å with coordination numbers between 1.8–3.7, consistent with edge-sharing Fe octahedral.64 The second Fe backscattering path (Fe–Fe2) had a fitted distance of 3.42–3.45 Å with coordination numbers between 1.2–2.1, characteristic of double corner-sharing Fe octahedral.63,66 The fitted coordination numbers of edge-sharing Fe (Fe–Fe1) atoms increased with increasing Fe-loading, contrary to the fitted coordination numbers of corner-sharing Fe (Fe–Fe2) atoms (Tables S14 and S15†). These trends correspond well with the increasing fraction of lepidocrocite formed with increasing Fe-loading, as observed by LCF of EXAFS spectra of oxic sorption samples. Triangular multiple scattering (MS) paths Fe–O–O were not implemented in our fit model as including this path decreased the reduced χ2-values less than a factor two (see S.I. Section 15 for details†).55
Conclusions
Our results show that the formation of Fe-bearing secondary phases is the main sorption mechanism for Fe2+ uptake by clay minerals low in structural Fe under anoxic conditions. Clay minerals can enhance the formation of newly formed, layered Fe minerals such as green rusts, FeAl-LDH, and Fe-phyllosilicates, by providing nucleation sites and serving as a source of Al and Si. Furthermore, clay surfaces may act as catalysts for Fe2+ oxidation by hydrogen, especially at circumneutral to alkaline pH values and low H2 partial pressures. Therefore, the precipitation of Fe-bearing phases in presence of clays may be of importance in controlling Fe2+ solubility in anoxic groundwaters and soils.The nature of the secondary phases formed under anoxic conditions appeared to be controlled by Fe-loading, but also by other factors. Under the conditions used in our study (high Ca2+, no CO2), the formation of layered Fe minerals was favored. These conditions were chosen to ensure saturation of cation exchange sites with Ca2+ and exclude formation of iron carbonates, allowing us to identify the sorption mechanisms of Fe2+ to edge sites of the clay mineral. However, as submerged soils frequently contain elevated CO2 concentrations, the precipitation of carbonate green rust and also siderite (FeCO3) would also be expected in these environments.
After exposure of anoxic sorption samples to ambient air, sorbed Fe(II) was completely oxidized to Fe(III). Aeration led to a rapid transformation of sorbed Fe into ferrihydrite, lepidocrocite, and Fe(III)-bearing clay minerals, independent of the Fe-loading and the duration of the pre-equilibration under anoxic conditions. This demonstrates that clay minerals are unable to stabilize sorbed Fe(II) against oxidation. However, the presence of clay minerals favors the formation of Fe(III)-containing clays both under anoxic and oxic conditions. This process is expected to impact not only the solubility of iron in soils and sediments but also many redox transformations of organic and inorganic contaminants which can react with Fe-bearing clays.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank M. Fischer and K. Barmettler (Soil Chemistry Group, ETH Zurich) for assistance in the laboratory. We gratefully acknowledge M. Etique (University of Lorraine) for discussions on the synthesis of Fe(II) minerals and M. Siebecker (Texas Tech University) about Wavelet transformation. We acknowledge SOLEIL (proposal no. 20171097) for the provision of synchrotron radiation facilities and thank G. Landrot, E. Fonda and G. Alizon for their support during the synchrotron measurements. We are grateful to E. Elzinga (Rutgers University) for sharing with us the reference XAS spectra of Fe(OH)2 and nikischerite, T. Borch (Colorado State University) the XAS spectra of chloride green rust and A. Zitolo (SOLEIL) for the XAS spectra of aqueous Fe2+ and Fe3+. This research was funded by the Swiss National Science Foundation (grant no. 200021_156392).References
- P. Refait, L. Simon and J. M. R. Genin, Reduction of SeO42- anions and anoxic formation of iron(II)-iron(III) hydroxy selenate green rust, Environ. Sci. Technol., 2000, 34, 819–825 CrossRef CAS.
- V. Badaut, M. L. Schlegel, M. Descostes and G. Moutiers, In situ time-resolved X-ray near-edge absorption spectroscopy of selenite reduction by siderite, Environ. Sci. Technol., 2012, 46, 10820–10826 CrossRef CAS PubMed.
- J. Jonsson and D. M. Sherman, Sorption of As(III) and As(V) to siderite, green rust (fougerite) and magnetite: Implications for arsenic release in anoxic groundwaters, Chem. Geol., 2008, 255, 173–181 CrossRef CAS.
- S. Sengupta, P. K. Mukherjee, T. Pal and S. Shome, Nature and origin of arsenic carriers in shallow aquifer sediments of Bengal Delta, India, Environ. Geol., 2004, 45, 1071–1081 CrossRef CAS.
- T. Pal and P. K. Mukherjee, 'Orange sand' - A geological solution for arsenic pollution in Bengal delta, Curr. Sci., 2008, 94, 31–33 Search PubMed.
- W. L. Lindsay and A. P. Schwab, The chemistry of iron in soils and its availability to plants, J. Plant Nutr., 1982, 5, 821–840 CrossRef CAS.
- W. Stumm and B. Sulzberger, The cycling of iron in natural environments - Considerations based on laboratory studies of heterogeneous redox processes, Geochim. Cosmochim. Acta, 1992, 56, 3233–3257 CrossRef CAS.
- D. L. Sparks, Environmental soil chemistry, Academic Press, San Diego, 1995 Search PubMed.
- M. H. Bradbury and B. Baeyens, Modelling the sorption of Mn(II), Co(II), Ni(II), Zn(II), Cd(II), Eu(III), Am(III), Sn(IV),Th(IV), Np(V) and U(VI) on montmorillonite: Linear free energy relationships and estimates of surface binding constants for some selected heavy metals and actinides, Geochim. Cosmochim. Acta, 2005, 69, 875–892 CrossRef CAS.
- D. Soltermann, M. Marques Fernandes, B. Baeyens, J. Miehé-Brendlé and R. Dähn, Competitive Fe(II)–Zn(II) uptake on a synthetic montmorillonite, Environ. Sci. Technol., 2014, 48, 190–198 CrossRef CAS PubMed.
- J. B. Dixon, S. B. Weed and R. C. Dinauer, Minerals in soil environments, Soil Science Society of America, Madison, Wis., USA, 2nd edn, 1989 Search PubMed.
- F. Favre, J. W. Stucki and P. Boivin, Redox properties of structural Fe in ferruginous smectite. A discussion of the standard potential and its environmental implications, Clays Clay Miner., 2006, 54, 466–472 CrossRef CAS.
- A. Neumann, T. L. Olson and M. M. Scherer, Spectroscopic evidence for Fe(II)–Fe(III) electron transfer at clay mineral edge and basal sites, Environ. Sci. Technol., 2013, 47, 6969–6977 CrossRef CAS PubMed.
- A. Géhin, J.-M. Grenèche, C. Tournassat, J. Brendléd, D. G. Rancourt and L. Charlet, Reversible surface-sorption-induced electron-transfer oxidation of Fe(II) at reactive sites on a synthetic clay mineral, Geochim. Cosmochim. Acta, 2007, 71, 863–876 CrossRef.
- M. V. Schaefer, C. A. Gorski and M. M. Scherer, Spectroscopic evidence for interfacial Fe(II)–Fe(III) electron transfer in a clay mineral, Environ. Sci. Technol., 2011, 45, 540–545 CrossRef CAS PubMed.
- C. A. Gorski, M. Aeschbacher, D. Soltermann, A. Voegelin, B. Baeyens, M. Marques Fernandes, T. B. Hofstetter and M. Sander, Redox properties of structural Fe in clay minerals. 1. Electrochemical quantification of electron-donating and -accepting capacities of smectites, Environ. Sci. Technol., 2012, 46, 9360–9368 CrossRef CAS PubMed.
- C. A. Gorski, L. Klupfel, A. Voegelin, M. Sander and T. B. Hofstetter, Redox properties of structural Fe in clay minerals. 2. Electrochemical and spectroscopic characterization of electron transfer irreversibility in ferruginous smectite, SWa-1, Environ. Sci. Technol., 2012, 46, 9369–9377 CrossRef CAS PubMed.
- C. A. Gorski, L. E. Klupfel, A. Voegelin, M. Sander and T. B. Hofstetter, Redox properties of structural Fe in clay minerals: 3. Relationships between smectite redox and structural properties, Environ. Sci. Technol., 2013, 47, 13477–13485 CrossRef CAS PubMed.
- A. M. Jones, C. A. Murphy, T. D. Waite and R. N. Collins, Fe(II) interactions with smectites: Temporal changes in redox reactivity and the formation of green rust, Environ. Sci. Technol., 2017, 51, 12573–12582 CrossRef CAS PubMed.
- T. B. Hofstetter, R. P. Schwarzenbach and S. B. Haderlein, Reactivity of Fe(II) species associated with clay minerals, Environ. Sci. Technol., 2003, 37, 519–528 CrossRef CAS PubMed.
- Y. Zhu and E. J. Elzinga, Formation of layered Fe(II)-hydroxides during Fe(II) sorption onto clay and metal-oxide substrates, Environ. Sci. Technol., 2014, 48, 4937–4945 CrossRef CAS PubMed.
- T. Huynh, A. R. Tong, B. Singh and B. J. Kennedy, Cd-substituted goethites - A structural investigation by synchrotron X-ray diffraction, Clays Clay Miner., 2003, 51, 397–402 CrossRef CAS.
- L. N. Moyes, R. H. Parkman, J. M. Charnock, D. J. Vaughan, F. R. Livens, C. R. Hughes and A. Braithwaite, Uranium uptake from aqueous solution by interaction with goethite, lepidocrocite, muscovite, and mackinawite: An X-ray absorption spectroscopy study, Environ. Sci. Technol., 2000, 34, 1062–1068 CrossRef CAS.
- G. Ona-Nguema, G. Morin, F. Juillot, G. Calas and G. E. Brown, EXAFS analysis of arsenite adsorption onto two-line ferrihydrite, hematite, goethite, and lepidocrocite, Environ. Sci. Technol., 2005, 39, 9147–9155 CrossRef CAS PubMed.
- X. W. Xu, C. Chen, P. Wang, R. Kretzschmar and F. J. Zhao, Control of arsenic mobilization in paddy soils by manganese and iron oxides, Environ. Pollut., 2017, 231, 37–47 CrossRef CAS PubMed.
- K. Ehlert, C. Mikutta and R. Kretzschmar, Impact of birnessite on arsenic and iron speciation during microbial reduction of arsenic-bearing ferrihydrite, Environ. Sci. Technol., 2014, 48, 11320–11329 CrossRef CAS PubMed.
- U. Schwertmann and R. M. Cornell, Iron oxides in the laboratory:preparation and characterization, Wiley-VCH, Weinheim, New York, 2nd completely rev. and extended edn, 2000 Search PubMed.
- J. M. R. Génin, R. Aïssa, A. Géhin, M. Abdelmoula, O. Benali, V. Ernstsen, G. Ona-Nguema, C. Upadhyay and C. Ruby, Fougerite and FeII-III hydroxycarbonate green rust; ordering deprotonation and/or cation substitution; structure of hydrotalcite-like compounds and mythic ferrosic hydroxide Fe(OH)(2+x), Solid State Sci., 2005, 7, 545–572 CrossRef.
- J. W. Stucki, B. A. Goodman and U. Schwertmann, Iron in soils and clay minerals, D. Reidel, Dordrecht, 1988 Search PubMed.
- D. Soltermann, M. Marques Fernandes, B. Baeyens, R. Dähn, J. Miehé-Brendlé, B. Wehrli and M. H. Bradbury, Fe(II) sorption on a synthetic montmorillonite. A combined macroscopic and spectroscopic study, Environ. Sci. Technol., 2013, 47, 6978–6986 CrossRef CAS PubMed.
- K. Lagarec and D. G. Rancourt, Recoil − Mössbauer spectral analysis software for Windows., University of Ottawa, Ottawa, 1998 Search PubMed.
- D. G. Rancourt and J. Y. Ping, Voigt-based methods for arbitrary-shape static hyperfine parameter distributions in mossbauer-spectroscopy, Nucl. Instrum. Methods Phys. Res., Sect. B, 1991, 58, 85–97 CrossRef.
- H. Funke, A. C. Scheinost and M. Chukalina, Wavelet analysis of extended x-ray absorption fine structure data, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 094110 CrossRef.
- Y. Zhu, J. J. Liu, O. Goswami, A. A. Rouff and E. J. Elzinga, Effects of humic substances on Fe(II) sorption onto aluminum oxide and clay, Geochem. Trans., 2018, 19, 3 CrossRef PubMed.
- D. L. Sparks, Kinetics of Ionic Reactions in Clay-Minerals and Soils, Adv. Agron., 1985, 38, 231–266 CrossRef CAS.
- A. M. Scheidegger, G. M. Lamble and D. L. Sparks, Spectroscopic evidence for the formation of mixed-cation hydroxide phases upon metal sorption on clays and aluminum oxides, J. Colloid Interface Sci., 1997, 186, 118–128 CrossRef CAS PubMed.
- A. C. Scheinost, R. G. Ford and D. L. Sparks, The role of Al in the formation of secondary Ni precipitates on pyrophyllite, gibbsite, talc, and amorphous silica: A DRS study, Geochim. Cosmochim. Acta, 1999, 63, 3193–3203 CrossRef CAS.
- E. J. Elzinga and D. L. Sparks, Nickel sorption mechanisms in a pyrophyllite-montmorillonite mixture, J. Colloid Interface Sci., 1999, 213, 506–512 CrossRef CAS PubMed.
- A. M. Scheidegger, D. G. Strawn, G. M. Lamble and D. L. Sparks, The kinetics of mixed Ni-Al hydroxide formation on clay and aluminum oxide minerals: A time-resolved XAFS study, Geochim. Cosmochim. Acta, 1998, 62, 2233–2245 CrossRef CAS.
- A. M. Scheidegger, G. M. Lamble and D. L. Sparks, Investigation of Ni sorption on pyrophyllite: An XAFS study, Environ. Sci. Technol., 1996, 30, 548–554 CrossRef CAS.
- A. C. Scheinost and D. L. Sparks, Formation of layered single- and double-metal hydroxide precipitates at the mineral/water interface: A multiple-scattering XAFS analysis, J.Colloid Interface Sci., 2000, 223, 167–178 CrossRef CAS PubMed.
- W. Li, K. J. T. Livi, W. Q. Xu, M. G. Siebecker, Y. J. Wang, B. L. Phillips and D. L. Sparks, Formation of crystalline Zn-Al layered double hydroxide precipitates on gamma-alumina: The role of mineral dissolution, Environ. Sci. Technol., 2012, 46, 11670–11677 CrossRef CAS PubMed.
- A. N. Starcher, E. J. Elzinga and D. L. Sparks, Formation of a mixed Fe(II)-Zn-Al layered hydroxide: Effects of Zn co-sorption on Fe(II) layered hydroxide formation and kinetics, Chem. Geol., 2017, 464, 46–56 CrossRef CAS.
- A. N. Starcher, W. Li, R. K. Kukkadapu, E. J. Elzinga and D. L. Sparks, Fe(II) sorption on pyrophyllite: Effect of structural Fe(III) (impurity) in pyrophyllite on nature of layered double hydroxide (LDH) secondary mineral formation, Chem. Geol., 2016, 439, 152–160 CrossRef CAS.
- J. M. R. Génin, G. Bourrie, F. Trolard, M. Abdelmoula, A. Jaffrezic, P. Refait, V. Maitre, B. Humbert and A. Herbillon, Thermodynamic equilibria in aqueous suspensions of synthetic and natural Fe(II)-Fe(III) green rusts: Occurrences of the mineral in hydromorphic soils, Environ. Sci. Technol., 1998, 32, 1058–1068 CrossRef.
- P. Refait and J. M. R. Genin, Mechanisms of oxidation of Ni(II)-Fe(II) hydroxides in chloride-containing aqueous media: Role of the pyroaurite-type Ni-Fe hydroxychlorides, Clay Miner., 1997, 32, 597–613 CrossRef CAS.
- S. C. F. Auyeung, G. Denes, J. E. Greedan, D. R. Eaton and T. Birchall, A novel synthetic route to iron trihydroxide, Fe(OH)3 - characterization and magnetic-properties, Inorg. Chem., 1984, 23, 1513–1517 CrossRef CAS.
- J. Klausen, S. P. Trober, S. B. Haderlein and R. P. Schwarzenbach, Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions, Environ. Sci. Technol., 1995, 29, 2396–2404 CrossRef CAS PubMed.
- A. Neumann, T. B. Hofstetter, M. Skarpeli-Liati and R. P. Schwarzenbach, Reduction of polychlorinated ethanes and carbon tetrachloride by structural Fe(II) in smectites, Environ. Sci. Technol., 2009, 43, 4082–4089 CrossRef CAS PubMed.
- T. B. Hofstetter, A. Neumann and R. P. Schwarzenbach, Reduction of nitroaromatic compounds by Fe(II) species associated with iron-rich smectites, Environ. Sci. Technol., 2006, 40, 235–242 CrossRef CAS PubMed.
- D. M. C. Huminicki and F. C. Hawthorne, The crystal structure of nikischerite, Na Fe2+6 Al3(SO4)2 (OH)18 (H2O)12, a mineral of the shigaite group, Can. Mineral., 2003, 41, 79–82 CrossRef CAS.
- A. Voegelin, A. C. Scheinost, K. Buhlmann, K. Barmettler and R. Kretzschmar, Slow formation and dissolution of Zn precipitates in soil - A combined column-transport and XAFS study, Environ. Sci. Technol., 2002, 36, 3749–3754 CrossRef CAS PubMed.
- O. Jacquat, A. Voegelin, A. Villard, M. A. Marcus and R. Kretzschmar, Formation of Zn-rich phyllosilicate, Zn-layered double hydroxide and hydrozincite in contaminated calcareous soils, Geochim. Cosmochim. Acta, 2008, 72, 5037–5054 CrossRef CAS.
- L. Bhattacharya and E. J. Elzinga, A comparison of the solubility products of layered Me(II)-Al(III) hydroxides based on sorption studies with Ni(II), Zn(II), Co(II), Fe(II), and Mn(II), Soil Syst., 2018, 2, 20 CrossRef CAS.
- S. D. Kelly, B. Hesterdberg and B. Ravel, in Methods of Soil Analysis Part 5—Mineralogical Methods, SSSA Book Ser. 5.5. SSSA, ed. L. R. D. A. L. Ulery, Madison, WI, 2008, pp. 387–463, DOI:10.2136/sssabookser5.5.c14.
- F. Trolard, G. Bourrie, M. Abdelmoula, P. Refait and F. Feder, Fougerite, a new mineral of the pyroaurite-iowaite group: Description and crystal structure, Clays Clay Miner., 2007, 55, 323–334 CrossRef CAS.
- H. Shirozu and S. W. Bailey, Chlorite Polytypism .III. crystal structure of an orthohexagonal iron chlorite, Am. Mineral., 1965, 50, 868–885 CAS.
- R. Dähn, B. Baeyens and M. H. Bradbury, Investigation of the different binding edge sites for Zn on montmorillonite using P-EXAFS - The strong/weak site concept in the 2SPNE SC/CE sorption model, Geochim. Cosmochim. Acta, 2011, 75, 5154–5168 CrossRef.
- R. Dähn, A. M. Scheidegger, A. Manceau, M. L. Schlegel, B. Baeyens, M. H. Bradbury and M. Morales, Neoformation of Ni phyllosilicate upon Ni uptake on montmorillonite: A kinetics study by powder and polarized extended X-ray absorption fine structure spectroscopy, Geochim. Cosmochim. Acta, 2002, 66, 2335–2347 CrossRef.
- A. Manceau, D. Chateigner and W. P. Gates, Polarized EXAFS, distance-valence least-squares modeling (DVLS), and quantitative texture analysis approaches to the structural refinement of Garfield nontronite, Phys. Chem. Miner., 1998, 25, 347–365 CrossRef CAS.
- A. Manceau and M. L. Schlegel, Texture effect on polarized EXAFS amplitude, Phys. Chem. Miner., 2001, 28, 52–56 CrossRef CAS.
- A. Manceau, D. Bonnin, P. Kaiser and C. Fretigny, Polarized EXAFS of Biotite and Chlorite, Phys. Chem. Miner., 1988, 16, 180–185 CrossRef CAS.
- A. Manceau, Critical evaluation of the revised akdalaite model for ferrihydrite, Am. Mineral., 2011, 96, 521–533 CrossRef CAS.
- A. P. Zhukhlistov, Crystal structure of lepidocrocite FeO(OH) from the electron-diffractometry data, Crystallogr. Rep., 2001, 46, 730–733 CrossRef.
- L. L. Baker and D. G. Strawn, Fe K-edge XAFS spectra of phyllosilicates of varying crystallinity, Phys. Chem. Miner., 2012, 39, 675–684 CrossRef CAS.
- A. Voegelin, R. Kaegi, J. Frommer, D. Vantelon and S. J. Hug, Effect of phosphate, silicate, and Ca on Fe(III)-precipitates formed in aerated Fe(II)- and As(III)-containing water studied by X-ray absorption spectroscopy, Geochim. Cosmochim. Acta, 2010, 74, 164–186 CrossRef CAS.
- D. E. Latta, A. Neumann, W. A. P. J. Premaratne and M. M. Scherer, Fe(II)-Fe(III) electron transfer in a clay mineral with low Fe content, ACS Earth and Space Chem, 2017, 1, 197–208 CrossRef CAS.
- A. C. Senn, R. Kaegi, S. J. Hug, J. G. Hering, S. Mangold and A. Voegelin, Composition and structure of Fe(III)-precipitates formed by Fe(II) oxidation in water at near-neutral pH: Interdependent effects of phosphate, silicate and Ca, Geochim. Cosmochim. Acta, 2015, 162, 220–246 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The iron oxides : structure, properties, reactions, occurrences, and uses, Wiley-VCH, Weinheim, New York, 2nd, completely rev. and extended edn, 2003 Search PubMed.
- P. Gütlich, E. Bill and A. X. Trautwein, Mossbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications, Springer, 2011 Search PubMed.
- L. T. Kuhn, K. Lefmann, C. R. H. Bahl, S. N. Ancona, P. A. Lindgard, C. Frandsen, D. E. Madsen and S. Morup, Neutron study of magnetic excitations in 8-nm alpha-Fe2O3 nanoparticles, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 184406 CrossRef.
- C. M. Chen, R. Kukkadapu and D. L. Sparks, Influence of coprecipitated organic matter on Fe-(aq)(2+)-catalyzed transformation of ferrihydrite: implications for carbon dynamics, Environ. Sci. Technol., 2015, 49, 10927–10936 CrossRef CAS.
- A. M. Jones, R. N. Collins, J. Rose and T. D. Waite, The effect of silica and natural organic matter on the Fe(II)-catalysed transformation and reactivity of Fe(III) minerals, Geochim. Cosmochim. Acta, 2009, 73, 4409–4422 CrossRef CAS.
- C. M. Hansel, S. G. Benner and S. Fendorf, Competing Fe(II)-induced mineralization pathways of ferrihydrite, Environ. Sci. Technol., 2005, 39, 7147–7153 CrossRef CAS.
- C. M. Hansel, S. G. Benner, J. Neiss, A. Dohnalkova, R. K. Kukkadapu and S. Fendorf, Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow, Geochim. Cosmochim. Acta, 2003, 67, 2977–2992 CrossRef CAS.
- J. W. Gruner, The composition and structure of minnesotaite, a common iron silicare in iron formations, Am. Mineral., 1944, 29, 363–372 CAS.
- C. Mikutta, X-ray absorption spectroscopy study on the effect of hydroxybenzoic acids on the formation and structure of ferrihydrite, Geochim. Cosmochim. Acta, 2011, 75, 5122–5139 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. Calculation of saturation indices, protocol to ensure anoxic conditions, measured pH after equilibration, percentage of Fe sorbed by oxidation of dissolved ferrous iron, concentration analyses, concentration of Si release analysis, details on XAS data collection and reduction, additional Mössbauer spectra and XAS results. See DOI: 10.1039/d0em00063a |
| This journal is © The Royal Society of Chemistry 2020 |