
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooking and electronic cigarettes leading to large differences between indoor and outdoor particle composition and concentration measured by aerosol mass spectrometry†
Yuliya
Omelekhina
 a,
Axel
Eriksson
ab,
Francesco
Canonaco
c,
Andre S. H.
Prevot
c,
Patrik
Nilsson
a,
Christina
Isaxon
a,
Joakim
Pagels
a and
Aneta
Wierzbicka
*a
a,
Axel
Eriksson
ab,
Francesco
Canonaco
c,
Andre S. H.
Prevot
c,
Patrik
Nilsson
a,
Christina
Isaxon
a,
Joakim
Pagels
a and
Aneta
Wierzbicka
*a
aErgonomics and Aerosol Technology, Lund University, 221 00, Lund, Sweden. E-mail: aneta.wierzbicka@design.lth.se
bNuclear Physics Department, Lund University, 221 00, Lund, Sweden
cLaboratory of Atmospheric Chemistry, Paul Scherrer Institute, 5232 Villigen PSI, Switzerland
First published on 7th May 2020
Abstract
We spend about two thirds of our time in private homes where airborne particles of indoor and outdoor origins are present. The negative health effects of exposure to outdoor particles are known. The characteristics of indoor airborne particles, though, are not well understood. This study assesses the differences in chemical composition of PM1 (<1 μm) inside and outside of an occupied Swedish residence in real time with a High-Resolution Time-of-Flight Aerosol Mass Spectrometer (HR-ToF-AMS) and an Aethalometer. The chemical composition and concentration of particles indoors showed large differences compared to outdoors. The average indoor concentration was 15 μg m−3 and was higher than the outdoor 7 μg m−3. Organics dominated indoor particle composition (86% of the total mass) and originated from indoor sources (cooking, e-cigarette vaping). The average indoor to outdoor ratios were 5.5 for organic matter, 1.0 for black carbon, 0.6 for sulphate, 0.1 for nitrate, 0.2 for ammonium and 0.2 for chloride. The occupancy time accounted for 97% of the total measured period. Four factors were identified in the source apportionment of organic particle fraction by applying positive matrix factorization (PMF): two cooking factors, one e-cigarette factor and one outdoor contribution (OOA) organic factor penetrated from outside.
Environmental significanceThis study provides insight into differences in chemical characteristics of airborne particles PM1 (<1 μm) with the state-of-the-art techniques in real occupied apartment. The influence of active indoor and outdoor particle sources have been investigated. As in developed countries, we spend about 66% of our time in private homes, it is necessary to understand the exposure to airborne particles in indoor environments. Indoor concentrations were higher than outdoors due to contribution of the indoor sources, peak levels of organics indoors were reaching 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 μg m−3. Cooking activities, which belong to our everyday exposures, contributed to high levels of organic mass concentrations. Exposures indoors and their consequences require further investigations. 700 μg m−3. Cooking activities, which belong to our everyday exposures, contributed to high levels of organic mass concentrations. Exposures indoors and their consequences require further investigations.
|
Introduction
Epidemiological studies have linked human exposure to airborne fine particulate matter (<2.5 μm in diameter) to cardiopulmonary diseases and increased mortality.1–4 These studies are based on outdoor air measurements, however we spend the majority of our time indoors.5–7 Understanding the consequences of exposure to particulate matter in indoor environments is thus important, yet knowledge is sparse. In this paper, we focus on private homes, because in developed countries, this is where we spend about 66% of our time.5–7Airborne particle levels indoors can be affected by several processes, such as active indoor sources, occupant behavior, physicochemical processes indoors and infiltration of outdoor particles, which in turn depends on the outdoor particle characteristics and factors affecting infiltration such as type of building, its ventilation, airtightness, and airing practice.8–14 Recent studies show that some indoor sources emit ultrafine (<100 nm) and fine particles (<2.5 um) of a complex chemical nature. These sources include cooking,15–20 burning candles,21–23 burning incense,24,25 using cleaning products,26–30 and electronic cigarette vaping.31,32 The particle number and mass concentration indoors due to such activities can be orders of magnitude higher than outdoors.10,33–38
The majority of studies on the chemical composition of indoor particles use off-line techniques providing time-integrated values.33,39,40 This approach does not allow insight into time-resolved changes in particle chemistry. Yet knowledge of the chemical composition of indoor air and an understanding of the physicochemical processes occurring is important to assess potential health effects and develop effective control measures. Real-time aerosol mass spectrometry (AMS) enables time- and size-resolved assessment of chemical composition of airborne particles. AMS has been successfully used to study the transformations of outdoor particles when combined with positive matrix factorization techniques for their source apportionment.41–46 So far, these techniques have only been used in a few studies of specific indoor sources, but they have the potential to provide invaluable help for understanding the source contributions and chemical transformation of particles indoors. AMS has been used to investigate the chemical properties of such indoor sources as incense burning and cooking in laboratory chambers and experimental house.15,25,47–51 Incense burning in a laboratory chamber (30 L), studied with use of a High-Resolution Time-of-Flight Aerosol Mass Spectrometer (HR-ToF-AMS), emitted high particle mass concentrations PM1 (<1 μm in diameter) between 50 and 350 μg m−3, in which the chemical composition was dominated by organics.25 Three laboratory studies, using HR-ToF-AMS showed that particles emitted from different cooking and the heating of cooking oils are dominated by primary organic aerosols, while secondary aerosol production is negligible.49–51 A number of experiments were conducted in a research/test house to investigate the particle and gaseous emissions from the everyday activities, their removal indoors and chemical transformations with HR-ToF-AMS. The results showed that the cooking activities were the dominant source of submicron particle mass during the HOMEChem campaign. Cooking emitted the large amounts of organic aerosols (>100 μg m−3) and mass concentrations varied between the meal types. Organic cooking emissions mostly comprised from CxHy ion family, followed by CxHyOz+ representing the fatty acid composition of cooking oils. CxHyN+ and CxHyON+ contributed little to total particle mass (<1%).15
Particle concentrations indoors are always affected by particles of outdoor origin, which mix and undergo physicochemical transformations with both gas- and particle-phase pollutants generated by indoor sources. Various physicochemical aerosol transformations occur during outdoor-to-indoor transport. Penetration through the building envelope is size-dependent, and accumulation mode particles display the highest penetration probability.12 Some semi-volatile aerosol chemical species, such as ammonium nitrate and ammonium chloride, are susceptible to gas-to-particle partitioning, which depends on temperature, relative humidity, particle size, and the gas phase concentrations of each species as outdoor air is transported indoors.52–59 Thus, species' penetration into indoor environments can be reduced, depending on the factors listed above.
Outdoor-to-indoor aerosol transformations have been studied with offline techniques.50,52–54,60,61 Recently, an online high-resolution mass spectrometric technique has also been used for this purpose in indoor environments without any active indoor sources.55,62,63 Indoor to outdoor ratios (I/O) of the penetrated particle mass to indoor environments in winter ranged between 0.5–0.7 for organics, 0.3–0.5 for ammonium, 0.2 for nitrate, 0.5–0.6 for sulfate, and 0.6–0.9 for BC throughout the studies.55,63 Positive matrix factorization (PMF) was applied to the penetrated organic particle mass in a multi-use, mechanically ventilated university building. The following factors were identified: hydrocarbon-like (HOA), cooking (COA), and oxygenated (OOA) factors indoors.55 To the best of our knowledge, there is one study that includes co-existing active indoor sources while exploring the penetration of outdoor particles with an HR-ToF-AMS.64 Several indoor sources including the use of insect repellent devices and environment tobacco smoke and the influence of the outdoor penetration were studied in an office building with the Aerosol Time-of-Flight Mass Spectrometer (ATOFMS).64 To our knowledge, there are no published studies using HR-ToF-AMS to assess the chemical composition of particles in real residences that are actually occupied.
In this study, we performed simultaneous time- and size-resolved measurements of the chemical composition of particles inside and outside of an occupied Swedish residence. The overall aim was to investigate the chemical characteristics of PM1 inside and outside of the residence. The specific aims were to: (1) assess the differences in chemical particle characteristics between indoor and outdoor environments; (2) identify the origin of major contributors to indoor loadings; (3) investigate the chemical transformations of particles during outdoor-to-indoor transport and their contributions to indoor concentrations. The study is based on two and a half weeks of measurements and is part of a larger project “Airborne particles in our homes: cocktail effects, chemical composition, physical characteristics and toxicity” (Formas Dnr 942-2015-1029).
Experimental section
Measurement site
Simultaneous indoor and outdoor particle measurements were conducted from December 2016 to January 2017 in an occupied residence in Malmö, Sweden. The ground floor apartment with a total surface area of 117 m2 and volume of 322 m3, was naturally ventilated. It consisted from a kitchen, a dining room, two bathrooms, three bedrooms, and a living room (see Fig. S1†). A kitchen exhaust fan above the electric stove vented the cooking fumes to the outdoors.The residence is located in a two-story brick building (built in the 1930s) with an attic, surrounded by a garden that separates the building from the surrounding streets. A family of four lived in the apartment throughout the measurement campaign. The residents filled in logbooks recording their presence/absence from the home, daily activities, such as cooking, candle burning, using household products, vacuuming, sweeping, ironing, printing and opening windows.8 Occupancy time, defined as the time when at least one occupant was at home, accounted for 97% of the period, as measurements took place over a holiday period. The outdoor air pollution can be attributed to the regional background and local traffic from several major roadways within 100–550 m of the residence.
Sampling
Airborne particles were sampled from the apartment through two parallel stainless steel indoor and outdoor sampling lines. An inlet to indoor sampling line was situated in the living room, approximately 1.2 m above the floor and extending 0.5 m past the exterior of the building. The indoor sampling line led from living room through a window frame to the basement area, where the instruments were placed. The outdoor line was mounted outside of the window frame on the ground floor level of the apartment where it also led to the basement area, see Fig. 1. Each line was 4.1 m in length with a diameter of 4.5 mm. The indoor sampling line was heated with a heating wire and insulated to avoid condensation due to temperature differences. Average temperature (T) at the inlet in the living room was 24 (±0.4) °C, average T measured on the sampling line (under insulation) before entering the basement was 15.6 (±3.1) °C and average T in the basement area was 22 (±1.9) °C throughout the measurement period. An additional carrier flow (0.6 L min−1) was used to lower the residence time of indoor aerosols during sampling.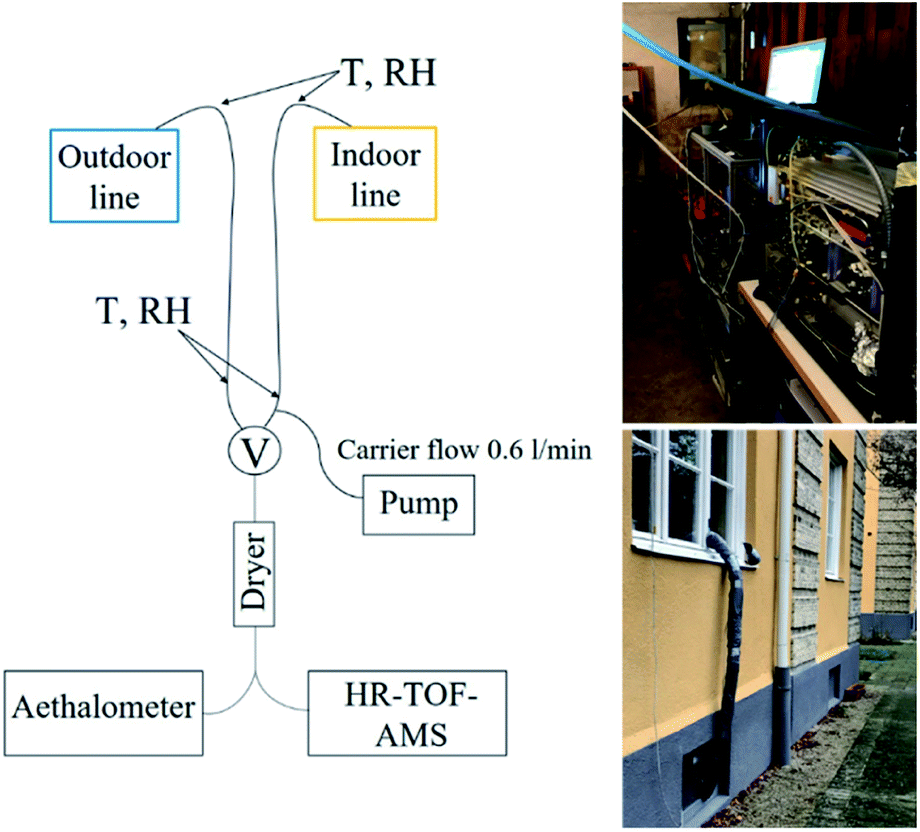 | ||
| Fig. 1 Schematics of the measurement setup. T = temperature, RH = relative humidity, V = switching valve switch, HR-ToF-AMS = High-Resolution Time-of-Flight Aerosol Mass Spectrometer. | ||
A switching valve alternated automatically between the indoor and outdoor lines with time intervals of 20 and 10 minutes, respectively. The aerosol sampled then passed through a Silica dryer (TOPAS, length 47 cm, RH < 30%) into the Time-of-flight Aerosol Mass Spectrometer (HR-ToF-AMS) and Aethalometer. The data acquired directly after valve switching (1.5 min) were excluded to avoid mixed indoor and outdoor air due to valve switching, data acquisition interval and residence time. Particle losses in the sampling line were calculated and ranged from 1 to 4% for 500 and 50 nm particles, respectively. However, the data has not been corrected for particle losses. The duration of the measurement campaign was one month, but for technical reasons, the data presented here covered a total of 16 days.
HR-ToF-AMS
The High Resolution Time-of-Flight Aerosol Mass Spectrometer (Aerodyne Research Inc., USA44) was used to measure mass concentrations and size-resolved mass distributions of organics, sulfate, nitrate, ammonium, and chloride in the size range 50–500 nm. The ToF-AMS was operated in mass spectrum (MS) and particle time-of-flight (PToF) modes with a time resolution of 30 seconds. Data were recorded in the V-mode. Ionization efficiency was calibrated using 300 nm (mobility) ammonium nitrate and ammonium sulfate particles. PToF sizing was calibrated with salt and polystyrene latex particles (PSL) in the size range 70–500 nm (vacuum aerodynamic diameter).43 CE efficiency of 1 was applied to both datasets.Aethalometer
(AE33, Magee65) was used to monitor black carbon (BC) mass concentration (at wavelength of 880 nm) inside and outside the residence.Other measurements
Indoor and outdoor temperatures, (T, °C), and relative humidity (RH, %) were recorded with a Testo sensors. The air exchange rate (AER) was measured on a separate occasion after the data measurement had finished. A tracer decay method was used. In short, N2O was released in the apartment and dispersed with use of two fans. G200 N2O monitor (Bedfont Scientific Limited) was used to measure the decay of N2O.Data processing
Indoor events were identified based on activity logbooks filled out by the occupants. A majority (76%) of the sources were identified based on the logbook. However, during data processing, some events not specified in the logbook were observed, and these were treated as “unknown” sources.Indoor to outdoor ratios (I/O) were calculated based on the average particle mass concentrations of given chemical components and were used to show differences in the chemical composition of particles.
The AMS analysis was performed using SQUIRREL v 1.6P and PIKA v 1.2P software written in IGOR Pro 6.37 (Wavemetrics, Inc., Portland, OR, USA).66
A Positive Matrix Factorization (PMF) was used to identify sources contributing to the mixture observed indoors and estimate the relative contributions to organic aerosol (OA) from AMS data. The PMF algorithm was applied using the bilinear model through a multilinear engine (ME-2). The PMF analysis of the organic aerosol matrix was performed on the HR-AMS data. For source apportionment we used graphic user interface SoFi 6.3 H45 Source Finder, Paul Scherer Institute, (PSI), which was written in the software package IGOR Pro 6.37.66
We explored four to seven factors with unconstrained runs in the PMF analysis. By increasing the number of more than four factors led to splitting of the factor profiles giving similar spectral pattern, but different mass concentrations. Thus, those factors did not present other particle sources. Additionally, we have tried to constrain cooking factor profiles (a-values 0.4–0.6). However, there was negligible changes to our solutions for four to seven factors and therefore we present the unconstrained result. A four-factor solution was chosen based on the dependence of Q/Qexp (Q/Qexp > 1.5) and the correlation of the obtained factor profiles and time series with reference spectra67 and the activity logbooks. The acceptable range of Q/Qexp ratio was between 1 and 5 based on the previous AMS studies.45 The residual did not decrease significantly with 5 or more factors compared to the 4 factor solution.
Sources identified by PMF, especially those classified as “unknown”, were scrutinized by further analyzing their mass spectra and comparing these to existing reference mass spectra (National Institute of Standards and Technology, NIST) and laboratory measurements, (see Fig. S2 in the ESI†). After the analysis, additional discussions with occupants helped to identify “unknown” indoor sources (i.e., those not logged to the activity logbooks).
Results and discussion
Chemical aerosol composition
Average particle mass concentrations over the whole measurement period were higher indoors in the studied apartment (15 μg m−3, ranging from 0.1 μg m−3 to 15700 μg m−3) than outdoors (7 μg m−3, ranging from 0.2 to 143 μg m−3). The AER in the apartment was 0.5 h−1. The differences between indoor and outdoor chemical particle mass compositions are presented in Fig. 2, based on the HR-ToF Aerosol Mass Spectrometer and Aethalometer datasets. The average particle mass concentration (μg m−3) for determined chemical species and their mass concentration fraction (%) are also presented in Fig. 2.
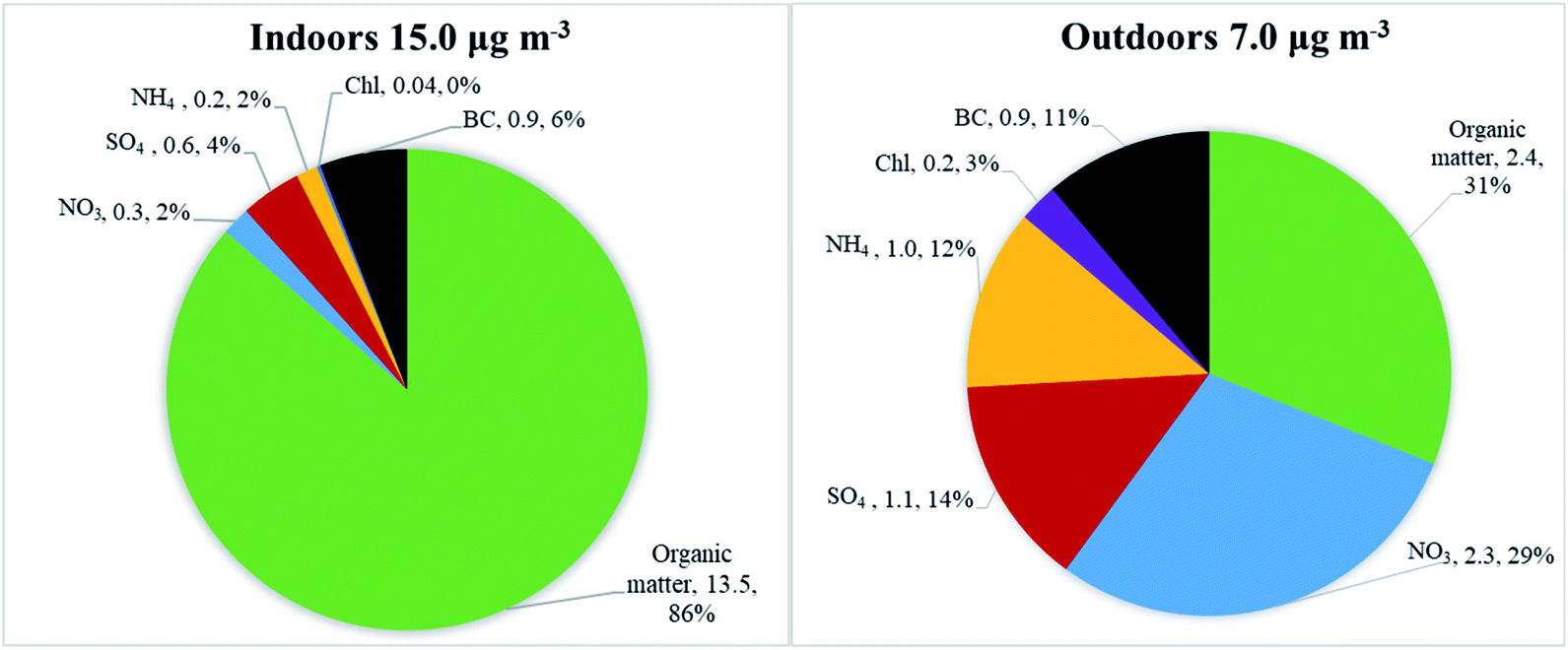 | ||
| Fig. 2 Differences between chemical composition of particles found indoors (left) and outdoors (right) given by fractions of average particle mass concentration during the whole measurement period. Pie chart description represents type of compound, average PM1 particle mass concentration (μg m−3), and mass concentration fraction (%). | ||
Indoors, the particles were dominated by organic matter (86% of the total mass, Fig. 2), followed by black carbon (6%), sulfate (4%) and nitrate (2%). The main contributors to the indoor particle levels were various types of cooking (frying, using the oven, deep-frying), e-cigarette vaping, and candle burning. These activities were identified based on the logbooks. The outdoor chemical composition was also dominated by organic matter (31%) and nitrate (29%), followed by BC (11%), sulfate (14%), ammonium (12%) and chloride (3%). The concentrations of sulfate, ammonium, nitrate, and chloride particle mass were lower indoors than outdoors. Sulfate and BC are non-volatile chemical species and are typically used to reflect penetration of outdoor particle mass upon outdoor-to-indoor transport as reported in previous studies.52,54–59 However, indoor levels of BC and sulfate in the apartment were also influenced by candle burning (based on information from logbooks and as seen in Fig. 3, discussed in detail under “Indoor–outdoor time series”). An enhanced decrease in ammonium nitrate and ammonium chloride mass concentrations upon outdoor-to-indoor transport, compared to sulfate and BC, can be explained by the chemical transformations of aerosols due to the temperature and relative humidity change (described in detail in section “Indoor–outdoor time series”).55,57,63,68,69
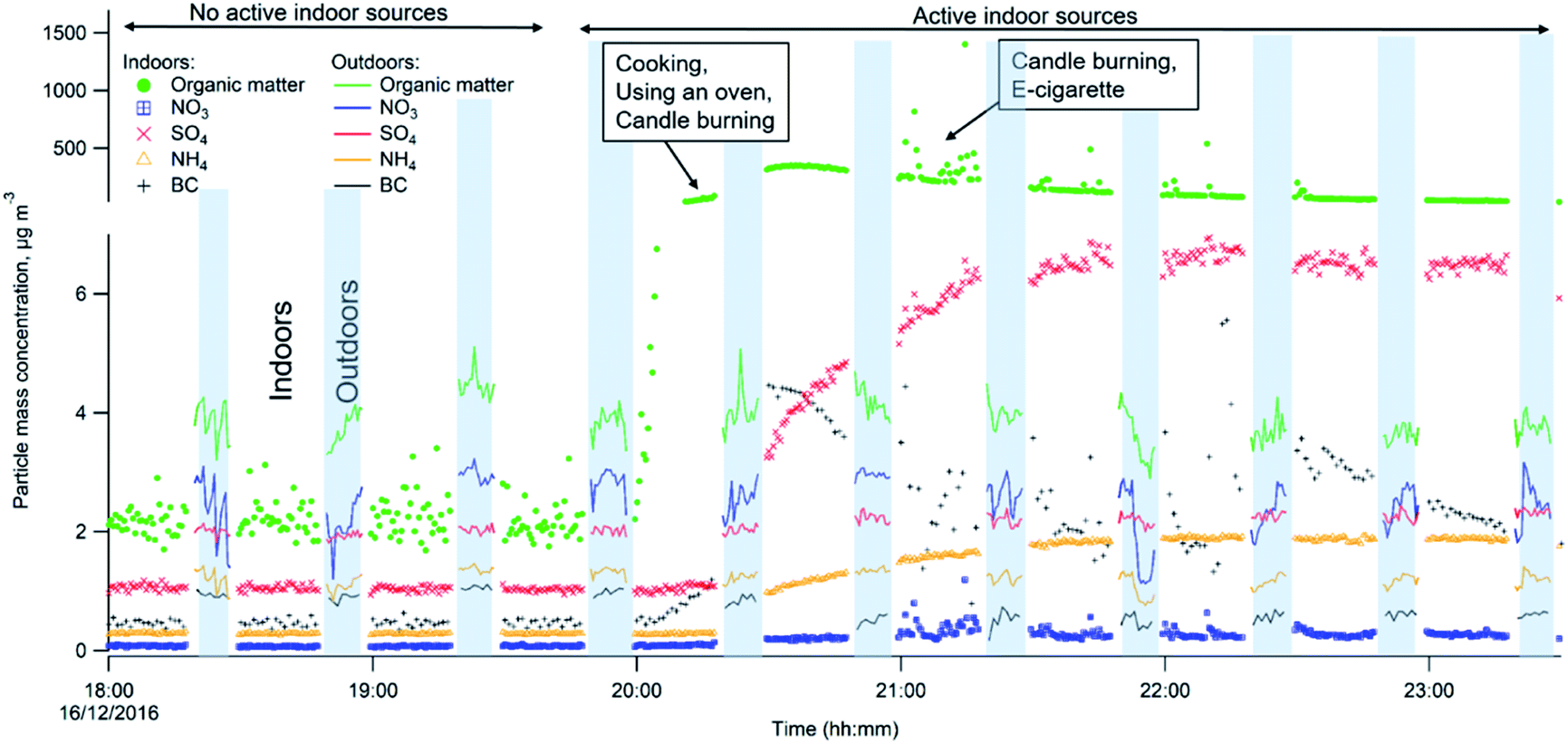 | ||
| Fig. 3 Particle mass concentrations of different chemical fractions during no active indoor sources (18:00–19:45), and with active indoor sources periods (19:45–24:00) inside and outside the residence during one evening. The changing pattern represents measurements with the switching valve alternating between indoors (starting position in this graph, no shading, 20 min) and outdoors (marked with shading, 10 min). Note the scale change on the Y-axis. | ||
The indoor to outdoor (I/O) ratio, calculated for the total measurement period, for organic matter was 5.5, followed by BC 1.0, sulfate 0.6, ammonium 0.2, nitrate 0.1, and chloride 0.2. Detailed concentration and summary statistics are presented in Table S1.† High I/O ratios of organic matter can be explained by the presence of the active indoor sources. The apartment was occupied 97% of the total measuring period as it was a winter holiday period. Influence of the outdoor pollution on indoor particle concentrations was small. Indoor mass concentrations were lower than outdoor concentrations only 23% of total measured time. We have also examined I/O ratios after we split the data into times with active indoor sources and times without any active sources (Table S2†). I/O ratio of organic matter during active indoor sources was 6.7, while during no active indoor sources it was 0.5. The latter represents penetration of organics from outdoors in case when there is no indoor source in operation. In general higher I/O ratios during active indoor sources compared to no active sources were observed for organics, BC and sulfate, which confirm contribution of indoor sources to indoor concentration of these species.
The I/O ratios obtained in this study, for the total measuring period, are comparable to those reported in earlier studies that did not include indoor sources55,63 for the majority of the species, with the exception for organics and BC. The I/O ratios measured in our study for organics and BC are higher, namely 5.0 vs. 0.5–0.7 and 1.0 vs. 0.6–0.9, respectively. The increased I/O ratios of organics and BC were due to the operation of indoor sources that were not present in the earlier studies. In previous studies, BC particle mass indoors was used as a tracer of the outdoor influence. However, in our study it is not possible to use the same approximation for quantifying BC concentration of outdoor origin since indoor sources (cooking, candle burning) were emitting BC on a daily basis. The I/O ratios of PM1 in winter in the earlier studies without indoor sources ranged between 0.2–0.3 for nitrate, 0.3–0.5 for ammonium, 0.5–0.6 for sulfate indoors with an AER between 1.9–3.1 h−1 in buildings with mechanical ventilation.55,63
Indoor–outdoor time series
Fig. 3 shows the differences in chemical composition and particle mass concentrations between indoor and outdoor air with and without active indoor sources during one evening, given as an example. The changing pattern (shown as different shades in Fig. 3) represents values when the automatic valve switched between indoors and outdoors.When there was no active indoors sources (18:00–19:45), the indoor particle mass concentrations of all measured chemical species were lower compared to outdoors (Fig. 3). This can be explained by particle losses during the outdoor–indoor penetration. It is clearly visible for organics, sulfate and BC, and is further influenced by loss due to phase changes, possibly driven by surface losses of the gas-phase in the case of semi-volatile ammonium nitrate aerosols. Penetrated particles from outdoors formed a background level indoors. A number of studies confirm that semi-volatile ammonium nitrate (NH4NO3) and ammonium chloride (NH4Cl) aerosols exist in equilibrium with gas phase ammonia, nitric acid, and hydrochloric acid. Thus, they are highly susceptible to changes in temperature, relative humidity and the gas phase concentrations of these compounds.52,54–58,68–72 During our measurements the average measured outdoor temperature Tout was 4.3 (±1.8 standard deviation, s.d.) and ranged from −8.8 to 9.7 °C. The average outdoor relative humidity RHout was 86.5 (±6)% and ranged from 58 to 100%. Indoors, average Tin was 24 (±0.4) °C and ranged from 20 to 26.1 °C. The average RHin was 41.4 (±3.7)% and ranged from 27 to 50%. We interpret that the rapid temperature change between cooler outdoor and warmer indoor environments led to a shift of the equilibrium of the nitrate and chloride with gas-phase species and volatilization reactions toward the gas phase; hence, there was an observed loss of particle phase ammonium nitrate and chloride indoors in Fig. 3 during the no active indoor sources period. Additional losses of ammonium nitrate indoors could result from sorption of the gas-phase to indoor surfaces as described earlier by Lunden et al.52
Indoor activities started to occur at 19:45 and indoor concentrations of organics, BC, ammonium and sulfate became higher indoors than outdoors. The indoor organic concentration reached maximum mass concentration of 351 μg m−3 as a result of the combined contribution of cooking with oven use, and candle burning. In comparison to our measurements, Brunekreef et al.73 showed similar maximum particle mass concentrations during cooking activities (200–300 μg m−3 in the particle size range of 100–300 nm). When candles were burning and e-cigarettes were vaping, adding to particles from cooking, organic mass concentrations reached 1405 μg m−3 in the three hours those sources were in operation (Fig. 3). Nguyen et al.31 found PM2.5 mass concentrations from e-cigarette vaping can vary between 15.5 and 37500 μg m−3 in vape shops. The emitted particle organic mass was mostly emitted from e-vaping as candles, during sooting conditions, emit predominantly particles with very low OC/EC ratios (<0.1),74 thus their contribution to OA is expected to be low. Candle burning was responsible for inorganic emissions such as BC, ammonium and sulfates. BC shows strong short-lived emission peaks, which ranged from 0.8 to 10.9 μg m−3 while the candle was burning (Fig. 3). Increased BC particle mass may be a result of the candle burning during sooting, as shown by Pagels et al.21 Sudden air movements (caused by people moving around in the residence or opening the entrance door) cause the candle flame to flicker and emit soot particles. The sulfate and ammonium concentrations gradually increased when the candle was burning and varied between 1.1–7.0 and 0.3–1.9 μg m−3, respectively, while outdoor sulfate and ammonium levels remained unchanged. The candle wick may have been infused with inorganic salts (e.g., sulfates, nitrates and phosphates) that control the consumption rate of the wick and prevent it from glowing after the flame has been extinguished.74 These particles may partly coagulate with the BC emissions.21 Danish EPA75 showed lower BC mass concentrations of 0.1–0.7 μg m−3 during candle burning under the sooting conditions. The Danish EPA75 found similar particle mass concentrations of ammonium 0.6–2.8 μg m−3 and sulfate 3.0–6.1 μg m−3, compared to our study.
Different cooking activities and candle burning were identified based on the information in the activity logbooks. However, occupants did not always log repeatedly occurring events such as e-cigarette vaping. Emissions of e-cigarette vapor were identified by fragmentation pattern of a compound emitted from the e-cigarette liquid (described in section Aerosol mass spectra), and confirmed by discussion with the occupants. Some activities such as toasting, vacuuming, and using cleaning spray, did not cause detectable changes in particle mass concentrations in the size range covered. Elevated mass concentrations during housekeeping activities have been shown to emit particles larger than 1 μm.14,38,76–78
Histograms of probability distribution comparing measured chemical species indoors and outdoors over the whole measurement period are presented in Fig. 4. One hour averaged data was used for the comparison. It can be seen that there are high particle mass concentrations of organics indoors due to frequently occurring indoor sources contributing with higher organic mass loading than outdoor concentrations. Outdoor organic mass concentrations are predominantly below 7 μg m−3. The distribution of particle mass concentrations of sulfate outdoors and indoors displays higher concentrations observed outdoors. Indoor concentrations represent mainly outdoor to indoor penetration, I/O ratio was 0.6. However, if sulfate indoors would originate only from outdoors we would see nearly identical distribution with lower concentrations. Some contribution at occasions when candles were burned (indoor sources) was observed, but sulfate concentrations indoors due to candle burning are low, which may explain similar distribution between indoor and outdoor at concentrations lower than 1 μg m−3. Substantially higher concentrations of nitrate, ammonium and chloride were measured outdoors compared to indoors. The larger shift in the probability distributions between indoor and outdoor in case of nitrate and ammonium can be due to losses during outdoor-to-indoor transport with some contribution from candles (indoor source) to the low concentrations measured indoors. In case of chloride there could be some contribution from cleaning products indoors at the low concentrations.
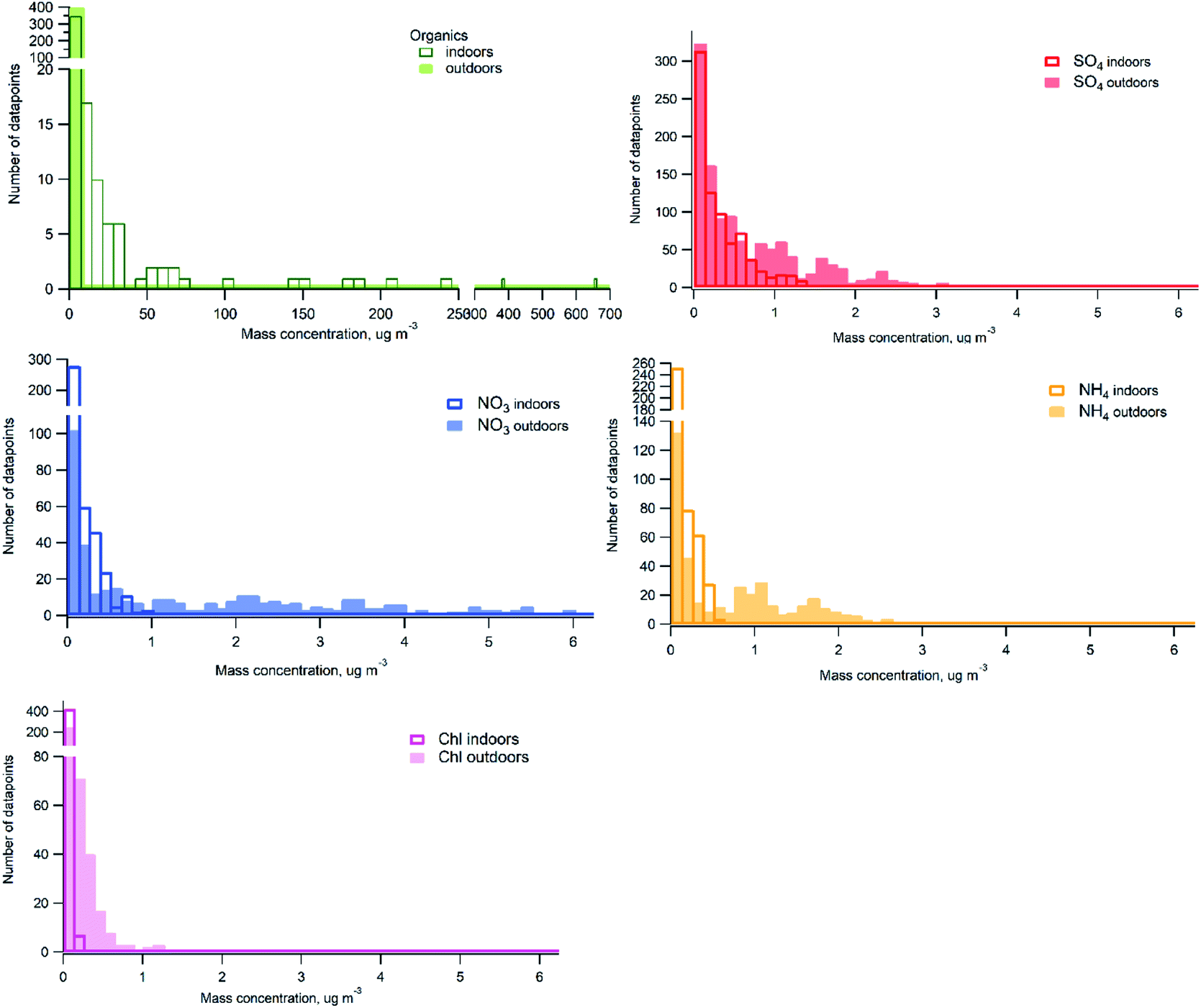 | ||
| Fig. 4 Probability distribution histograms of indoor and outdoor datasets (based on the 1 h-average data). | ||
In general, indoor sources have been shown to emit large amounts of submicrometer-size range particles, mainly of organic matter composition. The average particle mass concentration indoors was 15 μg m−3 throughout the measuring period. Particle mass concentration during periods with active indoor sources was 16.7 μg m−3 and during no active indoor sources 3.4 μg m−3 (Table S2†). The mass concentration values during entire measuring period during active indoor sources periods were below the recommended by WHO guidelines daily limit (25 μg m−3 within 24 hours) for outdoor PM2.5.79 However, measured peak concentrations in indoor air can reach as high concentrations as 351 μg m−3 (due to cooking or 15700 μg m−3 in close proximity to the place where e-cigarette was vaped (Table S1†)). The health impacts that are associated with short-term exposure to such high particle concentrations remain unknown. These exposures are likely repeated frequently, especially as many people cook on daily basis. Peak exposures go unnoticed when only 24 h averages are presented. It illustrates the importance of adequate ventilation to remove the pollutants accumulated indoors.
Aerosol mass spectra
Highly-resolved organic mass spectra (MS) of major families of the ion fragments, CxHy, CxHyO and CxHyOz, from cooking and vaping of the e-cigarettes are presented in Fig. 5. In Fig. 5A the average mass spectrum of all cooking activities is given. It was dominated by ion series of CnH2n−1+ (with mass to charge ratio m/z 41, 55, 69, 83…), CnH2n+1+ (m/z 43, 57, 71, 85…) and some oxygen-containing organics (m/z 29, 44, 60, 73). Measured spectra agree with results from earlier reported studies on emissions from cooking, which are briefly presented below.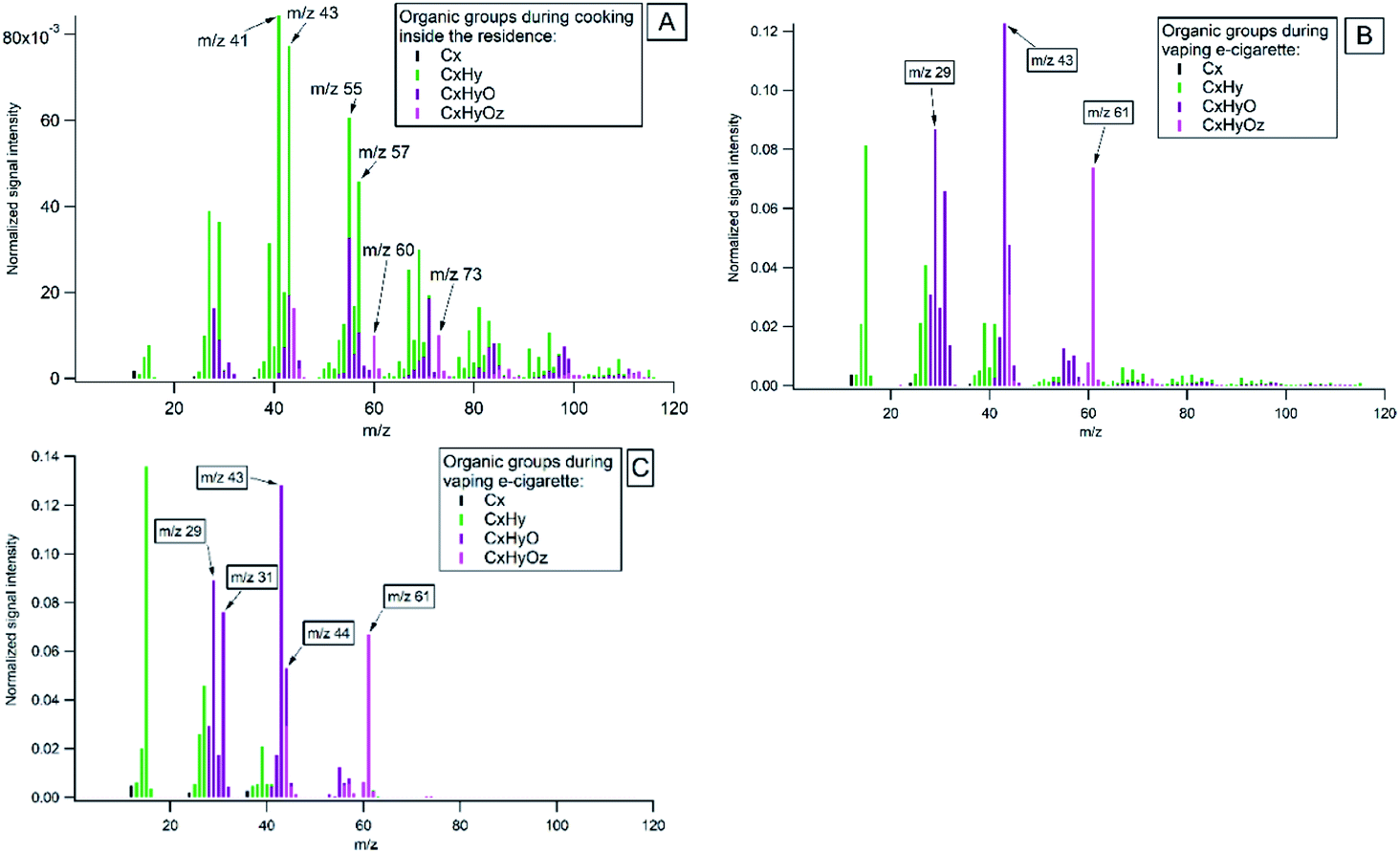 | ||
| Fig. 5 Average high-resolution organic mass spectral patterns during indoor activities inside the residence and reference laboratory spectra: (A) average of all cooking inside the residence; (B) vaping of e-cigarette inside the residence; (C) vaping of e-cigarette in the laboratory. | ||
It is known that during food treatment different processes and chemical reactions occur, such as denaturation of proteins, degradation of carbohydrates and fats at different temperatures. Studies have shown that frying and baking of foods of animal origin results in the formation of unbranched-chain saturated hydrocarbons and long-chain fatty acids47,49,80,81 with the most intensive ion peaks at C3H5 (m/z 41) and C4H7 (m/z 55).82 Hydrolysis of fats causes the formation of fatty acids with alcohol and carbonyl functional groups attached, and expressed by the CHO+ (m/z 29) ion. Duplissy et al.83 referred CHO+ ion as a product of thermal decomposition of cooking oils. Previous studies attributed peaks at m/z 60 (C2H4O2) and m/z 73 (C3H5O2) to fragmentation of sugars during cooking.47,80 Carbohydrates (e.g. starch, sugars and cellulose) undergo hydrolysis when heated with water. During the hydrolysis reaction, water reacts with the oxygen atom joining the sugar rings and breaks complex sugars down into single ring sugars.84 These monomer sugars may decompose to anhydrous sugars such as levoglucosan and similar molecules upon heating. The release of CO2 at m/z 44 was observed as a result of thermal decarboxylation of organic acids during cooking. It is used as an indicator of highly oxygenated organic aerosols.85 Our study confirmed that highly-resolved mass spectra of organic emissions from indoor activities showed different fragmentation patterns and can be used as reference spectra to identify different sources in indoor environments.
The organic mass spectrum of vaping the e-cigarettes (Fig. 5B) showed fragmentation peaks of glycerine (C3H8O3) at m/z 43 and m/z 61. Vegetable glycerine (VG) is one of the main constituent in e-cigarette, which is added as humectant for vapor production from e-liquid.86Fig. 5C shows a laboratory spectrum of vaping of e-cigarette, which is similar to the e-cigarette mass spectrum measured in the residence. The mass spectra obtained are comparable to the mass spectra of glycerine in the NIST database (NIST: glycerine, Fig. S2†).87 Another common main constituent of e-cigarette fluids is propylene glycol (PG). PG has a much higher vapour pressure than glycerine and rapidly evaporates in indoor air (NIST: propylene glycol).87
The differences between the average indoor and outdoor mass spectra's during the entire campaign are illustrated in Fig. S5.† Indoor mass spectra prominent peaks were at m/z's 41, 43, 55, 57, 60, 61, 71, 73 and represents combination of cooking, vaping of the e-cigarette. Outdoor spectra prominent peaks were at m/z's 41, 43, 44, 55, 57, 60, 71, 73 that represents formed oxygenated organic species, traffic, and biomass burning emissions.
PMF analysis
Indoor sources emit large amounts of PM1 predominately of organic matter origin, as seen in the results presented above. Thus, we focused on the characterization of the organic mass fraction by means of a Positive Matrix Factorization (PMF) analysis to identify and separate sources contributing to the indoor concentrations. PMF identified three primary factors: cooking OAI (COAI), cooking OAII (COAII), electronic cigarette OA (EOA); and one secondary factor: outdoor contribution (OOA) factor penetrated from outside.Fig. 6A shows the factors' profiles, and Fig. 6B shows the factors' time series. The mass spectra of the factor profiles allowed us to distinguish sources from each other by comparing obtained spectra with the reference spectra in an HR-AMS spectral database.67 PMF also enabled the identification of unknown sources such as e-cigarettes (by tracing the glycerine peak at m/z 61) and some cooking activities that were not logged in the activity logbook by the occupants. On average, the e-cigarette (EOA) was the major contributor (44%) to indoor loadings and resulted in particle mass concentrations of 6.8 μg m−3 (Fig. 7A). However, the contribution of EOA to observed concentrations may be overestimated because the placement of the inlet of the sampling line was in close proximity to where e-cigarette was vaped. This was only discovered after the measurements had been carried out, during discussions with the occupants about the identification of the peaks observed in the measured data. However, the data present the real concentrations measured in this specific apartment, and highlight the challenges of performing measurements in real occupied residences. E-cigarette vaping occurred fourteen times during the period measured by AMS.
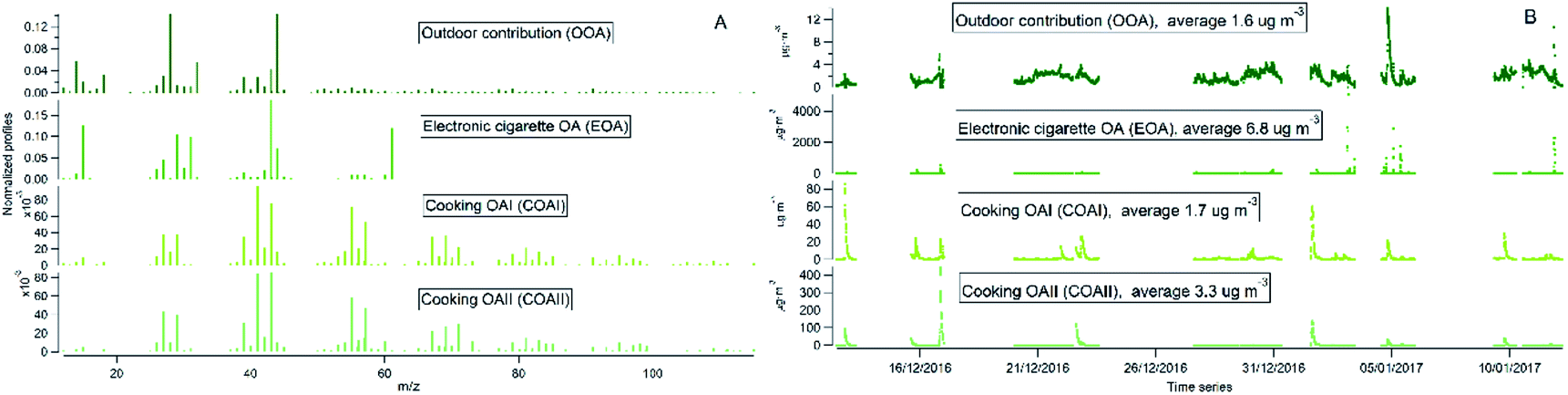 | ||
| Fig. 6 PMF analysis of high-resolution organic species indoors: (A) factors' profiles; (B) factors' time series. | ||
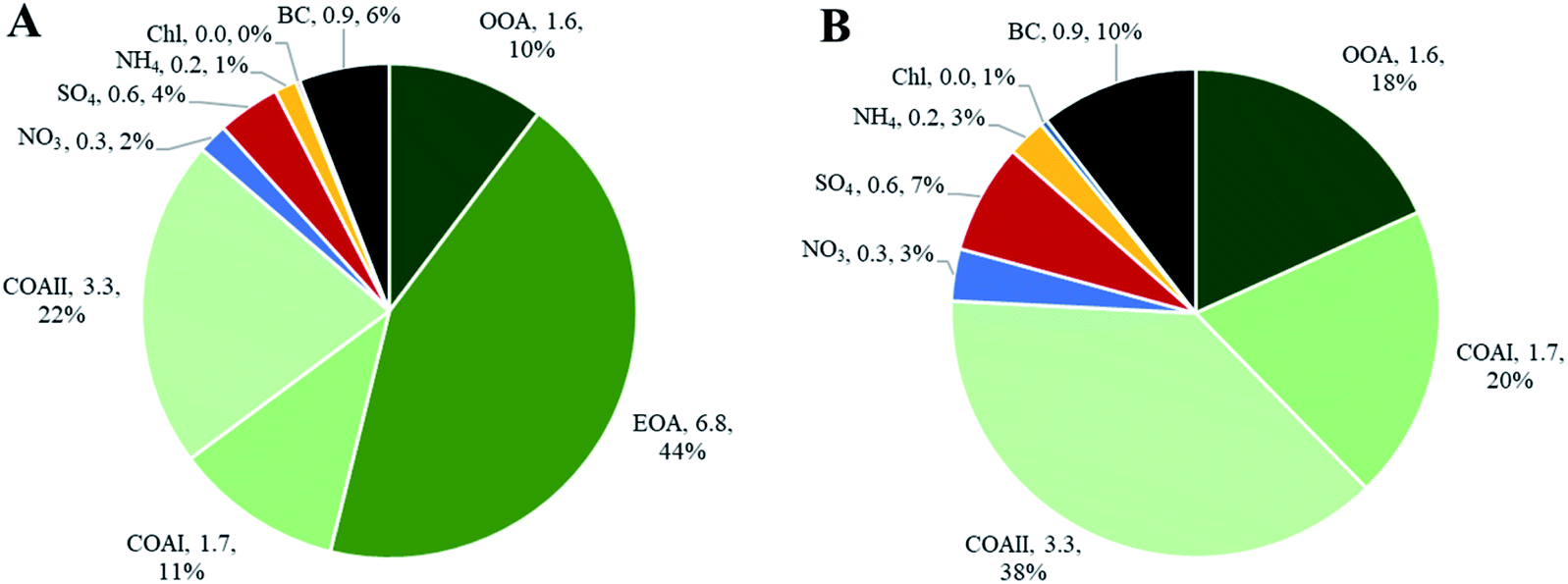 | ||
| Fig. 7 Average indoor particle mass concentration of chemical species including split of organics into contributing sources obtained from PMF analysis. Averages calculated on the basis of the whole measurement period: (A) particle mass concentrations of all species (in particle mass concentration, μg m−3; percentage, %); (B) particle mass concentrations of all species excluding e-cigarette factor (in particle mass concentration, μg m−3; percentage, %). Total average mass concentration indoors was 15 μg m−3. | ||
The second largest contributor indoors was cooking activities, which contributed to 33% of the total particle mass. Cooking events were frequent (n = 29). Of these, 10 were identified as cooking (in general without specification), 5 as baking, 9 as frying, 1 as deep frying according the activity logbooks. Two cooking factors, COAI and COAII, were retrieved during the PMF analysis. When comparing the PMF time series with activity logbooks of cooking events, it was not possible to identify a specific cooking activity (e.g. frying) as COAI or COAII. However, some of the cooking events (n = 8) were not logged by the participants and were retrieved from the PMF analysis. Both COA factor profiles had characteristic peaks at m/z's 41, 43, 55, 57, 60, 71 and 73. This is similar to results in previous studies.15,46,47,80,88–90 However, the difference between COAI and COAII factors was that the intensity of m/z's 43, 55, 57, 60, 71 and 73 of COAI was less pronounced compared to COAII (see Fig. S3†).
The peaks at m/z's 60 and 73 are commonly associated with wood burning in outdoor datasets (Biomass Burning Organic Aerosol PMF factor; BBOA) and have been linked to anhydrous sugars, such as levoglucosan, formed during low-temperature pyrolysis of cellulose in the biomass. Anhydrous sugars have also been detected during cooking91 with higher abundance in Hong Kong style cooking and lower abundance during meat cooking. Anhydrous sugars have been hypothesized to be emitted during high temperature cooking of vegetables.91 Vegetables are rich in carbohydrates that may decompose to single ring sugars through hydrolysis reactions and at higher temperature form anhydrous sugars. Thus, COAII may therefore represent high temperature cooking of vegetables and other food rich in carbohydrates, while COAI may represent low temperature cooking and cooking of food with low carbohydrate content. Logbooks in future studies should list both the cooking process and food type.
The signals at m/z's 43, 55, 57, 71 represent unbranched-chain saturated hydrocarbons and long-chain fatty acids formed during cooking. COA tracer ion ratios can help elucidate COA and its subcategories (i.e., specific cooking techniques and ingredients) in both indoor and outdoor source attribution efforts. The ratios of m/z 55:57 is often used to identify COA (e.g. separating COA from the commonly reported “hydrocarbon-like OA”, HOA typically attributed to traffic). Farmer et al.15 report 55:57 ratios of 2.26 and 1.80 for “oil splash” and “stir-fry”, respectively. We have found m/z 55:57 ratios of 1.33 and 1.25, for COAI and COAII factors, respectively. Interestingly, this puts values found in this study outside the range of literature values (1.5–4) reported by others15,55,80,92
The contribution of particle mass from outdoors reflected penetration of low volatile oxygenated organic species (1.6 μg m−3). The outdoor OA mass spectrum showed a peak at m/z 44 and was dominated by the CO2+ ion, which reflect the result of decomposition and fragmentation of oxygenated organic acids, as reported earlier.83,85 The retrieved outdoor contribution factor (OOA) from PMF analysis is similar to average outdoor mass spectra (Fig. S4†). It can be seen that not all the organic particle mass has penetrated inside (67%) due to size dependent penetration through the building envelope12 and evaporation of more volatile species (which include a lower fraction of m/z 44). It is important to note that e-cigarette is not a typical indoor source at home, the results reflect the habits of the occupants of this specific apartment (Fig. 7A). Thus, if we exclude e-cigarette as a source (Fig. 7B), the dominant indoor source would be cooking activities, which on average contributed with 5 μg m−3, or 58% out of the total mass (i.e. after deduction of e-cigarette contribution). In this case, the outdoor contribution would be 18%.
Probability distribution of individual sources contributions to measured organic particle mass concentration is presented in Fig. 8. Histograms were prepared on basis of 2 minutes averaged measurement data. It can be seen that EOA occurs frequently and contribute with varying mass concentrations of organics ranging from hundreds to thousands of μg m−3 (with maximum contributions about 4000 μg m−3). There is a difference between COAI and COAII contribution, COAII contributes with higher concentrations with majority of data points below 100 μg m−3, and some occasions with concentrations up to 350 μg m−3, whereas COAI contributes with concentrations lower than 65 μg m−3 and on majority of occasions with concentrations below 10 μg m−3. In contrast to all indoor sources, outdoor contribution OOA displays very low contribution below 10 μg m−3.
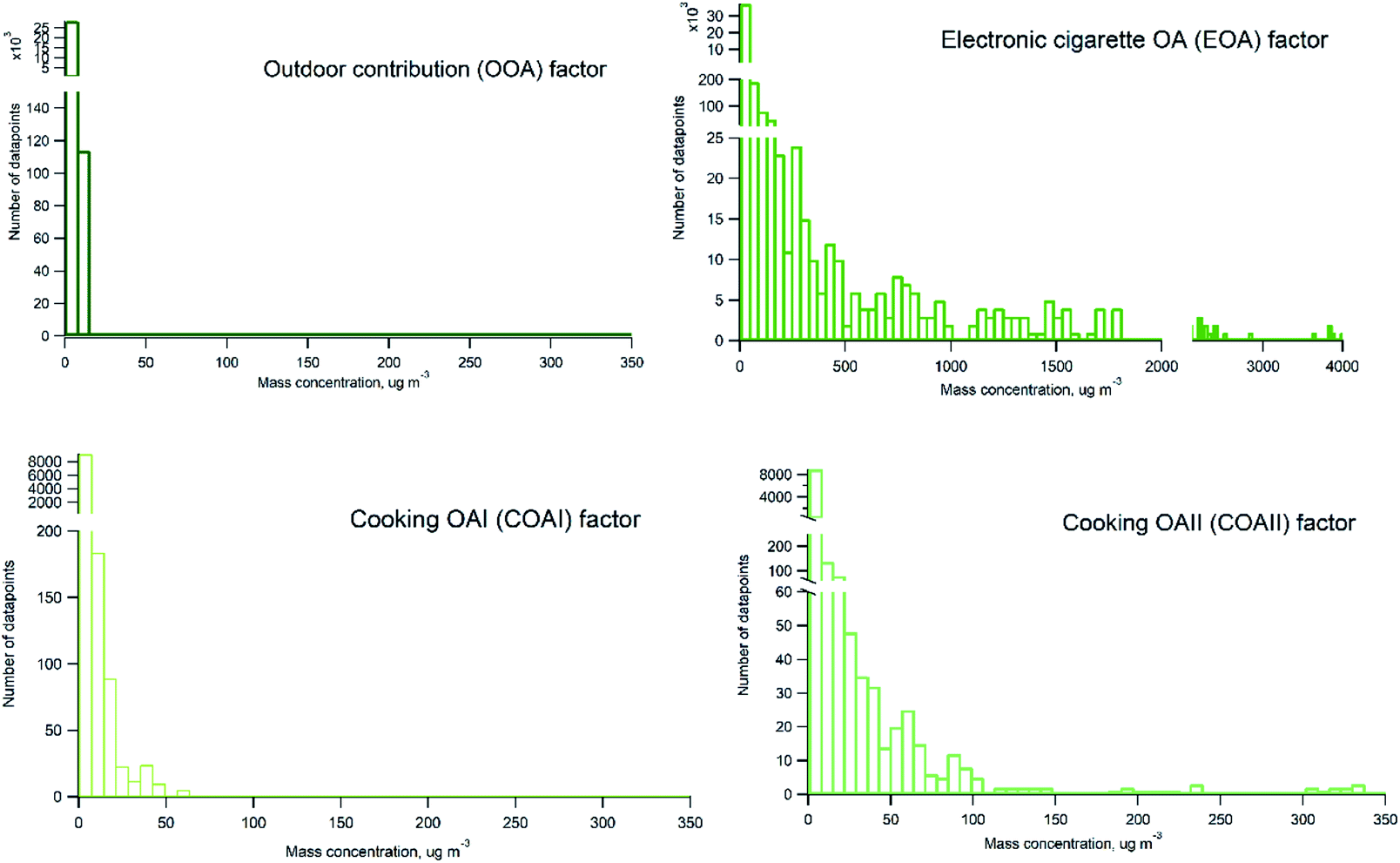 | ||
| Fig. 8 Probability distribution histograms of indoor PMF factors (based on the 2 min-average data). | ||
PMF source apportionment proved to be a useful tool for separating and identifying contributing sources to organic mass fractions. However, PMF was ineffective for retrieving the candle burning factor in both unconstrained and constrained runs. This is most likely because when candles burn, their mainly emit salts particles (phosphates and alkali nitrates) or BC depending on the burning conditions, as shown by Pagels et al.21 only small amounts of organics are emitted predominantly during extinguishing.
In real occupied indoor environments, such as the apartment in the study presented in this article, indoor sources of particles may occur at the same time or in a sequence of the activities. This results in particle mixtures from the indoor sources and the contribution of outdoor airborne particles, which are difficult to differentiate using traditional chemical methods. The results obtained from PMF source apportionment allowed us to estimate the contribution of specific sources to the concentrations measured in indoor air.
As an example, we present in Fig. 9 a series of pie charts of organic particle mass concentration from the retrieved PMF time series and inorganic mass during a sequence of indoor activities in one evening (the same as shown in Fig. 3). Fig. 9A represents PMF results of the separated factor time series from the specific evening. Each pie chart (B–D) represents a 10 min average concentration. The outdoor concentration during this time remained unchanged, as seen in Fig. 3.
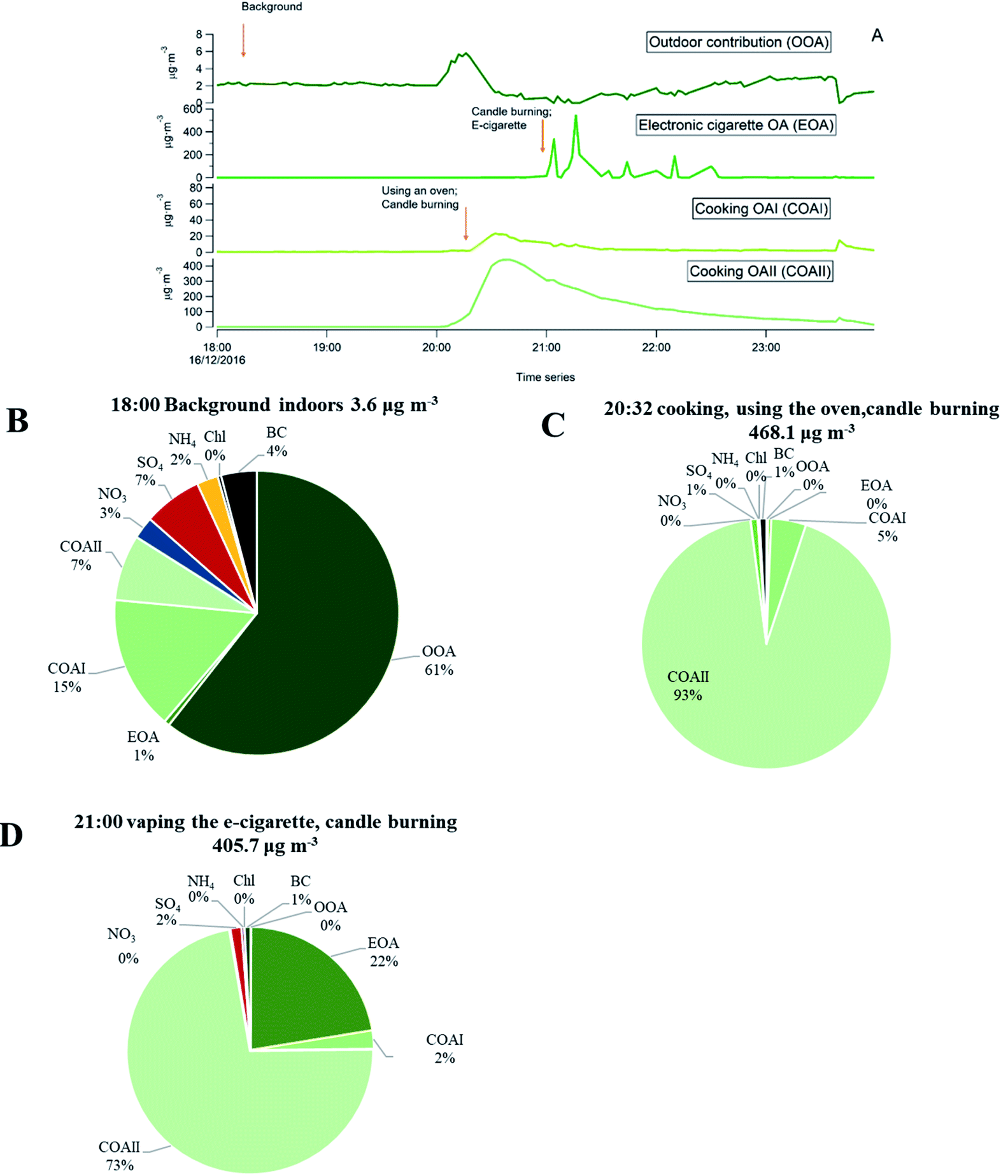 | ||
| Fig. 9 Differences in particle chemical composition indoors during the sequence of events during one evening – an example. OOA – outdoor contribution of organics; EOA electronic cigarette organics; COAI – cooking I organics; COAII – cooking II organics. (A) Time series plot of the different sources during this evening obtained from PMF. Arrows indicate start time of 10 min average from which each pie chart was made; (B) no active indoor source period (indoor background) (C) first peak concentration during this evening due to cooking with oven use and candle burning; (D) peak concentrations during candle burning and e-cigarette vaping (coinciding events) adding to earlier concentrations from cooking, oven use and candle burning. The pie chart description represents type of compound, and mass concentration fraction (%). | ||
As one can see from the Fig. 9B, when the apartment was vacant and no indoor sources were in operation, organics that penetrated from outdoors contributed to 61% of the particle mass indoors. Inorganics (NH4, SO4, BC and Chl), contributed to 16% of the indoor mass concentration. However, there was also a residual organic mass concentration (23%) from previous cooking events and vaping of the e-cigarette that have not been removed by the ventilation system of the apartment. When the occupants were cooking, burning candle and vaping electronic cigarette (Fig. 9C and D) the emissions were dominated by organic matter, followed by inorganic particle species. BC was emitted from candle burning in a sooting mode, sulfate and ammonium were emitted from candle wick. The described pie charts illustrate an example of how particle mass concentration of different chemical species changed during activity and non-activity periods indoors. We have observed such differences in chemical composition of particles with and without indoor activities during the entire measurement period.
Limitations of the study
The influence of the e-cigarette on particle mass concentrations may have been overestimated due to the close proximity of the sampling inlet to the place where e-cigarette was vaped. The inlet of the probe was placed in the living room to ensure some distance from the kitchen and to reduce the influence of strong indoor sources such as cooking. The location was also chosen for practical reasons: the preference of the occupants. However, it turned out that this was the place where e-cigarette was vaped, so the e-cigarette concentrations captured are “close to source” concentrations. Additionally it may be that e-cigarette vaping is not typically occurring source in indoor environments. The accumulation of the pollutants emitted by indoor sources may be enhanced indoors in winter season in Scandinavia as the ventilation is usually reduced (low AER) due to windows and doors kept closed.The mass concentrations presented in this study were assessed on the basis of AMS measurements that do not capture refractory components, and were measured in the size range 50–500 nm. Additionally, the length of the sampling line introduced some diffusional losses in sampling line, 1–4%; these losses have not been applied to the presented values. These factors contribute to underestimating the presented values of mass concentration.
Indoors, particles are likely liquid as majority of the mass were emitted from cooking93 and e-cigarette,94 thus they would have high CE. In this study, we have applied the collection efficiency (CE) of AMS as 1 to both indoor and outdoor datasets for consistency. However, CE has different values depending from particle composition size ranges and sampling locations.95
Reyes-Villegas et al.93 found the RIEOA value up to 3.06 and if we were to use that value it would decrease the cooking factor concentration by a factor of 2.2 in our datasets. Thus, further investigation of RIE with cooking experiments are needed. PMF is not effective for apportioning signal to sources, which contribute less than 5% of the mass concentration.96
Concentrations presented here were measured in one apartment with a specific air exchange rate, volume, and surface area available for particle deposition. Therefore when comparing to other studies, variations in these factors of the residence should always be considered.
Conclusions
This is the field-deployed study that assessed chemical characteristics of airborne particles PM1 (<1 μm) with the state-of-the-art High-Resolution Time-of-Flight Aerosol Mass Spectrometer (HR-ToF-AMS) and an Aethalometer inside and outside of an occupied Swedish residence. Chemical composition and concentration of particles indoors showed large differences in comparison to outdoors. Average indoor mass concentrations were twice higher compared to the outdoor loadings throughout the measuring period. Indoor mass concentrations were lower than outdoor concentrations only 23% of total measured time. Indoor active sources, namely, cooking (frying, using the oven, deep-frying) and e-cigarette vaping emitted large amounts of submicrometer-size range particles predominantly of organic matter origin (86% of the total mass). This has resulted in high indoor to outdoor (I/O) ratio for organic matter (5.5). The concentrations of sulfate, ammonium, nitrate and chloride particle mass were lower indoors than outdoors and reflected penetration of outdoor particle mass during outdoor-to-indoor transport. An enhanced decrease in ammonium nitrate and ammonium chloride mass concentrations upon outdoor-to-indoor transport, compared to sulfate and BC can be explained by the chemical transformations of aerosols. However, indoor levels of BC and sulfate in the apartment were also influenced by candle burning.Measured average particle mass concentration indoors of 15 μg m−3 was below the recommended by WHO guidelines daily limit (25 μg m−3 within 24 hours) for outdoor PM2.5.79 However, peak concentrations of organics in indoor air during cooking reached as high concentrations as 351 μg m−3. The cooking activities is our everyday activity and perhaps these exposures were likely repeated frequently. Peak exposures go unnoticed when only 24 h averages are presented. It illustrates the importance of adequate ventilation to remove the pollutants accumulated indoors.
A Positive Matrix Factorization (PMF) source apportionment analysis applied to organic particle fractions, allowed identification of contributing sources occurring simultaneously or as a sequence of events. Identified factors within organics were: two cooking factors (COAI, COAII), e-cigarette factor (EOA) and oxygenated outdoor factor (OOA). OOA contribution accounted on average to 10% of the total indoor particle organic mass, thus was not the main contributor indoors. PMF also allowed us to identify unknown indoor sources, such as cooking and e-cigarette vaping, by comparing the mass spectra obtained to the reference mass spectral database. The retrieved mass spectra of indoor sources can be of future use as reference spectra to enable the use of PMF for source apportionment in indoor environments.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This project was financed by the Swedish Research Council FORMAS (Project Dnr 942-2015-1029) and supported by EU COST Action CA16109.References
- M. Kampa and E. Castanas, Human health effects of air pollution, Environ. Pollut., 2008, 151(2), 362–367 CrossRef CAS PubMed.
- C. A. Pope III, R. T. Burnett, M. J. Thun, E. E. Calle, D. Krewski and K. Ito, et al., Lung Cancer, Cardiopulmonary Mortality, and Long-term Exposure to Fine Particulate Air Pollution, JAMA, J. Am. Med. Assoc., 2002, 287(9), 1132–1141 CrossRef PubMed.
- C. A. Pope and D. W. Dockery, Health Effects of Fine Particulate Air Pollution: Lines that Connect, J. Air Waste Manage. Assoc., 2006, 56(6), 709–742 CrossRef CAS PubMed.
- K. Katsouyanni, G. Touloumi, E. Samoli, A. Gryparis, A. Le Tertre and Y. Monopolis, et al., Confounding and Effect Modification in the Short-Term Effects of Ambient Particles on Total Mortality: Results from 29 European Cities within the APHEA2 Project, Epidemiology, 2001, 12(5), 521–531 CrossRef CAS PubMed.
- S. Brasche and W. Bischof, Daily time spent indoors in German homes – Baseline data for the assessment of indoor exposure of German occupants, Int. J. Hyg. Environ. Health, 2005, 208(4), 247–253 CrossRef PubMed.
- N. E. Klepeis, W. C. Nelson, W. R. Ott, J. P. Robinson, A. M. Tsang and P. Switzer, et al., The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants, J. Exposure Sci. Environ. Epidemiol., 2001, 11(3), 231–252 CrossRef CAS PubMed.
- J. A. Leech, W. C. Nelson, R. T. Burnett, S. Aaron and M. E. Raizenne, It's about time: a comparison of Canadian and American time–activity patterns, J. Exposure Sci. Environ. Epidemiol., 2002, 12(6), 427–432 CrossRef PubMed.
- A. Wierzbicka, M. Bohgard, J. H. Pagels, A. Dahl, J. Löndahl and T. Hussein, et al., Quantification of differences between occupancy and total monitoring periods for better assessment of exposure to particles in indoor environments, Atmos. Environ., 2015, 106, 419–428 CrossRef CAS.
- D. Rim, M. Green, L. Wallace, A. Persily and J.-I. Choi, Evolution of Ultrafine Particle Size Distributions Following Indoor Episodic Releases: Relative Importance of Coagulation, Deposition and Ventilation, Aerosol Sci. Technol., 2012, 46(5), 494–503 CrossRef CAS.
- C. He, L. Morawska and D. Gilbert, Particle deposition rates in residential houses, Atmos. Environ., 2005, 39(21), 3891–3899 CrossRef CAS.
- S. Bhangar, N. A. Mullen, S. V. Hering, N. M. Kreisberg and W. W. Nazaroff, Ultrafine particle concentrations and exposures in seven residences in northern California, Indoor Air, 2011, 21(2), 132–144 CrossRef CAS PubMed.
- W. Nazaroff, Indoor particle dynamics, Indoor Air, 2004, 14(s7), 175–183 CrossRef PubMed.
- L. Morawska, Indoor Environment. Airborne Particles and Settled Dust, WILEY-VCH Verlag GmbH & Co., 2003 Search PubMed.
- T. L. Thatcher and D. W. Layton, Deposition, resuspension, and penetration of particles within a residence, Atmos. Environ., 1995, 29(13), 1487–1497 CrossRef CAS.
- D. K. Farmer, M. E. Vance, J. P. D. Abbatt, A. Abeleira, M. R. Alves and C. Arata, et al., Overview of HOMEChem: House Observations of Microbial and Environmental Chemistry, Environ. Sci.: Processes Impacts, 2019, 21(8), 1280–1300 RSC.
- C. L. Wan MPW, G. N. Szeto, T. C. Chan and C. Y. H. Chao, Ultrafine particles and PM2.5 generated from cooking in homes, Atmos. Environ., 2011, 45(34), 6141–6148 CrossRef.
- University simulation of particulate matter formation during heating different cooking oils, ed. A. M. Torkmahalleh, U. Kadyrbayeva and A. Kadyrbayeva, AIChE Annual Meeting, 2016 Search PubMed.
- Y. Torkmahalleh MAZ, P. K. Hopke, A. Rossner and A. R. Ferro, Additive impacts on particle emissions from heating low emitting cooking oils, Atmos. Environ., 2013, 73, 194–198 CrossRef.
- T. Hussein, H. Korhonen, E. Herrmann, K. Hämeri, K. E. J. Lehtinen and M. Kulmala, Emission Rates Due to Indoor Activities: Indoor Aerosol Model Development, Evaluation, and Applications, Aerosol Sci. Technol., 2005, 39(11), 1111–1127 CrossRef CAS.
- T. Hussein TG, J. Ondracek, P. Dohanyosova, V. Zdimal, K. Hameri, M. Lazaridis, J. Smolik and M. Kulmala, Particle size characterization and emission rates during indoor activities in a house, Atmos. Environ., 2006, 40(23), 4285–4307 CrossRef.
- J. Pagels, A. Wierzbicka, E. Nilsson, C. Isaxon, A. Dahl and A. Gudmundsson, et al., Chemical composition and mass emission factors of candle smoke particles, J. Aerosol Sci., 2009, 40(3), 193–208 CrossRef CAS.
- L. Knight and C. Mendenhall, Candles and incense as potential sources of indoor air pollution: market analysis and literature review, National Risk Management Research Laboratory, 2001 Search PubMed.
- P. M. Fine, G. R. Cass and B. R. T. Simoneit, Characterization of fine particle emissions from burning church candles, Environ. Sci. Technol., 1999, 33(14), 2352–2362 CrossRef CAS.
- C. L. Wu, C. Y. H. Chao, G. N. Sze-To, M. P. Wan and T. C. Chan, Ultrafine Particle Emissions from Cigarette Smouldering, Incense Burning, Vacuum Cleaner Motor Operation and Cooking, Indoor Built Environ., 2011, 21(6), 782–796 CrossRef.
- Y. J. Li, J. W. T. Yeung, T. P. I. Leung, A. P. S. Lau and C. K. Chan, Characterization of Organic Particles from Incense Burning Using an Aerodyne High-Resolution Time-of-Flight Aerosol Mass Spectrometer, Aerosol Sci. Technol., 2012, 46(6), 654–665 CrossRef CAS.
- C. J. Weschler and N. Carslaw, Indoor Chemistry, Environ. Sci. Technol., 2018, 52(5), 2419–2428 CrossRef CAS PubMed.
- J. Schwarz, O. Makes, J. Ondracek, M. Cusack, N. Talbot and P. Vodicka, et al., Single Usage of a Kitchen Degreaser Can Alter Indoor Aerosol Composition for Days, Environ. Sci. Technol., 2017, 51(11), 5907–5912 CrossRef CAS PubMed.
- A. W. Nørgaard, J. D. Kudal, V. Kofoed-Sørensen, I. K. Koponen and P. Wolkoff, Ozone-initiated VOC and particle emissions from a cleaning agent and an air freshener: risk assessment of acute airway effects, Environ. Int., 2014, 68, 209–218 CrossRef PubMed.
- W. W. Nazaroff and C. J. Weschler, Cleaning products and air fresheners: exposure to primary and secondary air pollutants, Atmos. Environ., 2004, 38(18), 2841–2865 CrossRef CAS.
- B. C. Singer, H. Destaillats, A. T. Hodgson and W. W. Nazaroff, Cleaning products and air fresheners: emissions and resulting concentrations of glycol ethers and terpenoids, Indoor Air, 2006, 16(3), 179–191 CrossRef CAS PubMed.
- C. Nguyen, L. Li, C. A. Sen, E. Ronquillo and Y. Zhu, Fine and ultrafine particles concentrations in vape shops, 2019, (3), 721–988.
- M. Belka, F. Lizal, J. Jedelsky, M. Jicha and J. Pospisil, Measurement of an electronic cigarette aerosol size distribution during a puff, Epj Web Conf. 2017 Search PubMed.
- L. Morawska, A. Afshari, G. N. Bae, G. Buonanno, C. Y. H. Chao and O. Hänninen, et al., Indoor aerosols: from personal exposure to risk assessment, Indoor Air, 2013, 23(6), 462–487 CrossRef CAS PubMed.
- L. Morawska, G. A. Ayoko, G. N. Bae, G. Buonanno, C. Y. H. Chao and S. Clifford, et al., Airborne particles in indoor environment of homes, schools, offices and aged care facilities: the main routes of exposure, Environ. Int., 2017, 108, 75–83 CrossRef CAS PubMed.
- C. Isaxon, A. Gudmundsson, E. Z. Nordin, L. Lönnblad, A. Dahl and G. Wieslander, et al., Contribution of indoor-generated particles to residential exposure, Atmos. Environ., 2015, 106, 458–466 CrossRef CAS.
- G. Bekö, C. J. Weschler, A. Wierzbicka, D. G. Karottki, J. Toftum and S. Loft, et al., Ultrafine Particles: Exposure and Source Apportionment in 56 Danish Homes, Environ. Sci. Technol., 2013, 47(18), 10240–10248 Search PubMed.
- C. He, L. Morawska, J. Hitchins and D. Gilbert, Contribution from indoor sources to particle number and mass concentrations in residential houses, Atmos. Environ., 2004, 38(21), 3405–3415 CrossRef CAS.
- C. M. Long, H. H. Suh and P. Koutrakis, Characterization of Indoor Particle Sources Using Continuous Mass and Size Monitors, J. Air Waste Manage. Assoc., 2000, 50(7), 1236–1250 CrossRef CAS PubMed.
- C. P. Weisel, J. Zhang, B. J. Turpin, M. T. Morandi, S. Colome and T. H. Stock, et al., Relationship of Indoor, Outdoor and Personal Air (RIOPA) study: study design, methods and quality assurance/control results, J. Exposure Sci. Environ. Epidemiol., 2005, 15(2), 123–137 CrossRef CAS PubMed.
- B. J. Turpin, C. P. Weisel, M. Morandi, S. Colome, T. Stock and S. Eisenreich, et al., Relationships of indoor, outdoor, and personal air (RIOPA): part II. Analyses of concentrations of particulate matter species, Res. Rep. Health Eff. Inst., 2007, 130(2), 9–92 Search PubMed.
- J. D. Allan, J. L. Jimenez, P. I. Williams, M. R. Alfarra, K. N. Bower and J. T. Jayne, et al., Quantitative sampling using an Aerodyne aerosol mass spectrometer - 1. Techniques of data interpretation and error analysis, J. Geophys. Res. Atmos., 2003, 108(D3), 10 Search PubMed.
- J. T. Jayne, D. C. Leard, X. Zhang, P. Davidovits, K. A. Smith and C. E. Kolb, et al., Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles, Aerosol Sci. Technol., 2000, 33(1–2), 49–70 CrossRef CAS.
- M. R. Canagaratna, J. T. Jayne, J. L. Jimenez, J. D. Allan, M. R. Alfarra and Q. Zhang, et al., Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 2007, 26(2), 185–222 CrossRef CAS PubMed.
- P. F. DeCarlo, J. R. Kimmel, A. Trimborn, M. J. Northway, J. T. Jayne and A. C. Aiken, et al., Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 2006, 78(24), 8281–8289 CrossRef CAS PubMed.
- F. Canonaco, M. Crippa, J. G. Slowik, U. Baltensperger and A. S. H. Prévôt, SoFi, an IGOR-based interface for the efficient use of the generalized multilinear engine (ME-2) for the source apportionment: ME-2 application to aerosol mass spectrometer data, Atmos. Meas. Tech., 2013, 6(12), 3649–3661 CrossRef.
- M. Crippa, F. Canonaco, V. A. Lanz, M. Äijälä, J. D. Allan and S. Carbone, et al., Organic aerosol components derived from 25 AMS data sets across Europe using a consistent ME-2 based source apportionment approach, Atmos. Chem. Phys., 2014, 14(12), 6159–6176 CrossRef.
- L. Y. He, Y. Lin, X. F. Huang, S. Guo, L. Xue and Q. Su, et al., Characterization of high-resolution aerosol mass spectra of primary organic aerosol emissions from Chinese cooking and biomass burning, Atmos. Chem. Phys., 2010, 10(23), 11535–11543 CrossRef CAS.
- X. Ji, O. Le Bihan, O. Ramalho, C. Mandin, B. D'Anna and L. Martinon, et al., Characterization of particles emitted by incense burning in an experimental house, Indoor Air, 2010, 20(2), 147–158 CrossRef CAS PubMed.
- T. Liu, Z. Li, M. Chan and C. K. Chan, Formation of secondary organic aerosols from gas-phase emissions of heated cooking oils, Atmos. Chem. Phys., 2017, 17(12), 7333–7344 CrossRef CAS.
- F. Klein, U. Baltensperger, A. S. H. Prévôt and I. El Haddad, Quantification of the impact of cooking processes on indoor concentrations of volatile organic species and primary and secondary organic aerosols, Indoor Air, 2019, 29(6), 926–942 CrossRef CAS PubMed.
- F. Klein, N. J. Farren, C. Bozzetti, K. R. Daellenbach, D. Kilic and N. K. Kumar, et al., Indoor terpene emissions from cooking with herbs and pepper and their secondary organic aerosol production potential, Sci. Rep., 2016, 6, 36623 CrossRef CAS PubMed.
- M. M. Lunden, K. L. Revzan, M. L. Fischer, T. L. Thatcher, D. Littlejohn and S. V. Hering, et al., The transformation of outdoor ammonium nitrate aerosols in the indoor environment, Atmos. Environ., 2003, 37(39), 5633–5644 CrossRef CAS.
- R. B. Mosley, D. J. Greenwell, L. E. Sparks, Z. Guo, W. G. Tucker and R. Fortmann, et al., Penetration of Ambient Fine Particles into the Indoor Environment, Aerosol Sci. Technol., 2001, 34(1), 127–136 CrossRef CAS.
- M. Mozurkewich, The dissociation constant of ammonium nitrate and its dependence on temperature, relative humidity and particle size, Atmos. Environ. Part A Gen. Top., 1993, 27(2), 261–270 CrossRef.
- A. M. Johnson, M. S. Waring and P. F. DeCarlo, Real-time transformation of outdoor aerosol components upon transport indoors measured with aerosol mass spectrometry, Indoor Air, 2017, 27(1), 230–240 CrossRef CAS PubMed.
- L. M. Hildemann, A. G. Russell and G. R. Cass, Ammonia and nitric acid concentrations in equilibrium with atmospheric aerosols: experiment vs. theory, Atmos. Environ., 1984, 18(9), 1737–1750 CrossRef CAS.
- C. A. Pio and R. M. Harrison, Vapour pressure of ammonium chloride aerosol: effect of temperature and humidity, Atmos. Environ., 1987, 21(12), 2711–2715 CrossRef CAS.
- C. W. Spicer, D. V. Kenny, G. F. Ward and I. H. Billick, Transformations, Lifetimes, and Sources of NO2, HONO, and HNO3 in Indoor Environments, Air Waste, 1993, 43(11), 1479–1485 CrossRef CAS PubMed.
- S. V. Hering and E. Avol, Indoor-outdoor concentration ratios for fine particle mass and inorganic ions in twelve southern California homes, Second Colloquium on Particulate Air Pollution and Human Health, Proceedings of the Second Colloquium on Particulate Air Pollution and Human Health, Park City, Utah, 1996, pp. 3–52 Search PubMed.
- E. Diapouli, A. Chaloulakou and N. Spyrellis, Levels of ultrafine particles in different microenvironments — Implications to children exposure, Sci. Total Environ., 2007, 388(1), 128–136 CrossRef CAS PubMed.
- O. O. Hänninen, S. Alm, K. Katsouyanni, N. Künzli, M. Maroni and M. J. Nieuwenhuijsen, et al., The EXPOLIS study: implications for exposure research and environmental policy in Europe, J. Exposure Sci. Environ. Epidemiol., 2004, 14(6), 440–456 CrossRef PubMed.
- M. Avery Anita, M. S. Waring and P. F. DeCarlo, Seasonal variation in aerosol composition and concentration upon transport from the outdoor to indoor environment, Environ. Sci.: Processes Impacts, 2019, 21(3), 528–547 RSC.
- N. Talbot, L. Kubelova, O. Makes, J. Ondracek, M. Cusack and J. Schwarz, et al., Transformations of aerosol particles from an outdoor to indoor environment, Aerosol Air Qual. Res., 2017, 17(3), 653–665 CrossRef CAS.
- M. Dall'Osto, R. M. Harrison, E. Charpantidou, G. Loupa and S. Rapsomanikis, Characterisation of indoor airborne particles by using real-time aerosol mass spectrometry, Sci. Total Environ., 2007, 384(1), 120–133 CrossRef PubMed.
- L. Drinovec, G. Močnik, P. Zotter, A. S. H. Prévôt, C. Ruckstuhl and E. Coz, et al., The “dual-spot” Aethalometer: an improved measurement of aerosol black carbon with real-time loading compensation, Atmos. Meas. Tech., 2015, 8(5), 1965–1979 CrossRef CAS.
- 6.37 Igor Pro Search PubMed.
- HR AMS Database, available from: http://cires1.colorado.edu/jimenez-group/HRAMSsd/, 2018.
- A. W. Stelson and J. H. Seinfeld, Relative humidity and temperature dependence of the ammonium nitrate dissociation constant, Atmos. Environ., 1982, 16(5), 983–992 CrossRef CAS.
- N. Hodas and B. J. Turpin, Shifts in the Gas-Particle Partitioning of Ambient Organics with Transport into the Indoor Environment, Aerosol Sci. Technol., 2014, 48(3), 271–281 CrossRef CAS.
- J. Smolík, L. Mašková, N. Zíková, L. Ondráčková and J. Ondráček, Deposition of suspended fine particulate matter in a library, Heritage Sci., 2013, 1(1), 7 CrossRef.
- L. Mašková, J. Smolík and P. Vodička, Characterisation of particulate matter in different types of archives, Atmos. Environ., 2015, 107, 217–224 CrossRef.
- S. Hering and G. Cass, The Magnitude of Bias in the Measurement of PM25 Arising from Volatilization of Particulate Nitrate from Teflon Filters, J. Air Waste Manage. Assoc., 1999, 49(6), 725–733 CrossRef CAS PubMed.
- B. Brunekreef, N. A. Janssen, J. J. de Hartog, M. Oldenwening, K. Meliefste and G. Hoek, et al., Personal, indoor, and outdoor exposures to PM2.5 and its components for groups of cardiovascular patients in Amsterdam and Helsinki, Res. Rep. Health Eff. Inst., 2005, 127, 1–70 Search PubMed.
- F. Willhöft and R. Horn, Candles. Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- J. A. Hansen, P. B. Pedersen, T. N. Jensen, M. Poulsen, S. Thomann and O. Kilström, et al., Environmentally friendly candles with reduced particle emissions, The Danish Environmental Protection Agency, 2018 Search PubMed.
- E. Abt, H. H. Suh, P. Catalano and P. Koutrakis, Relative Contribution of Outdoor and Indoor Particle Sources to Indoor Concentrations, Environ. Sci. Technol., 2000, 34(17), 3579–3587 CrossRef CAS.
- A. R. Ferro, R. J. Kopperud and L. M. Hildemann, Elevated personal exposure to particulate matter from human activities in a residence, J. Exposure Sci. Environ. Epidemiol., 2004, 14(1), S34–S40 CrossRef CAS PubMed.
- C. Howard-Reed, L. A. Wallace and S. Emmerich, Deposition rates of fine and coarse particles in residential buildings: literature review and measurements in an occupied townhouse, National Institute of Standards and Technology, 2003 Search PubMed.
- Ambient (outdoor) air pollution, available from: https://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health Search PubMed.
- C. Mohr, J. A. Huffman, M. J. Cubison, A. C. Aiken, K. S. Docherty and J. R. Kimmel, et al., Characterization of Primary Organic Aerosol Emissions from Meat Cooking, Trash Burning, and Motor Vehicles with High-Resolution Aerosol Mass Spectrometry and Comparison with Ambient and Chamber Observations, Environ. Sci. Technol., 2009, 43(7), 2443–2449 CrossRef CAS PubMed.
- M. Dall'Osto, M. Paglione, S. Decesari, M. C. Facchini, C. O'Dowd and C. Plass-Duellmer, et al., On the Origin of AMS “Cooking Organic Aerosol” at a Rural Site, Environ. Sci. Technol., 2015, 49(24), 13964–13972 CrossRef PubMed.
- R. M. Silverstein, F. X. Webster and D. Kiemle, Spectrometric identification of organic compounds, John Wiley & Sons, Inc., 7th edn, 2005 Search PubMed.
- J. Duplissy, P. F. DeCarlo, J. Dommen, M. R. Alfarra, A. Metzger and I. Barmpadimos, et al., Relating hygroscopicity and composition of organic aerosol particulate matter, Atmos. Chem. Phys., 2011, 11(3), 1155–1165 CrossRef CAS.
- P. Barham, The science of cooking, Springer-Verlag Berlin Heidelberg, 1950 Search PubMed.
- N. L. Ng, M. R. Canagaratna, Q. Zhang, J. L. Jimenez, J. Tian and I. M. Ulbrich, et al., Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 2010, 10(10), 4625–4641 CrossRef CAS.
- J. Hartmann-Boyce, R. Begh and P. Aveyard, Electronic cigarettes for smoking cessation, BMJ [Br. Med. J.], 2018, 360, j5543 CrossRef PubMed.
- NIST database, available from: https://webbook.nist.gov/cgi/cbook.cgi?Name=glycerine%26Units=SI%26cMS=on 2018.
- J. Sun, Q. Zhang, M. R. Canagaratna, Y. Zhang, N. L. Ng and Y. Sun, et al., Highly time- and size-resolved characterization of submicron aerosol particles in Beijing using an Aerodyne Aerosol Mass Spectrometer, Atmos. Environ., 2010, 44(1), 131–140 CrossRef CAS.
- J. D. Allan, P. I. Williams, W. T. Morgan, C. L. Martin, M. J. Flynn and J. Lee, et al., Contributions from transport, solid fuel burning and cooking to primary organic aerosols in two UK cities, Atmos. Chem. Phys., 2010, 10(2), 647–668 CrossRef CAS.
- J. G. Slowik, A. Vlasenko, M. McGuire, G. J. Evans and J. P. D. Abbatt, Simultaneous factor analysis of organic particle and gas mass spectra: AMS and PTR-MS measurements at an urban site, Atmos. Chem. Phys., 2010, 10(4), 1969–1988 CrossRef CAS.
- X. Zhao, Q. Hu, X. Wang, X. Ding, Q. He and Z. Zhang, et al., Composition profiles of organic aerosols from Chinese residential cooking: case study in urban Guangzhou, south China, J. Atmos. Chem., 2015, 72(1), 1–18 CrossRef CAS.
- T. Liu, Z. Wang, X. Wang and C. K. Chan, Primary and secondary organic aerosol from heated cooking oil emissions, Atmos. Chem. Phys., 2018, 18, 11363–11374 CrossRef CAS.
- E. Reyes-Villegas, T. Bannan, M. Le Breton, A. Mehra, M. Priestley and C. Percival, et al., Online Chemical Characterization of Food-Cooking Organic Aerosols: Implications for Source Apportionment, Environ. Sci. Technol., 2018, 52(9), 5308–5318 CrossRef CAS PubMed.
- T. R. Sosnowski and M. Odziomek, Particle Size Dynamics: Toward a Better Understanding of Electronic Cigarette Aerosol Interactions With the Respiratory System, Front. Physiol., 2018, 9(853) Search PubMed.
- A. M. Middlebrook, R. Bahreini, J. L. Jimenez and M. R. Canagaratna, Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data, Aerosol Sci. Technol., 2012, 46(3), 258–271 CrossRef CAS.
- I. M. Ulbrich, M. R. Canagaratna, Q. Zhang, D. R. Worsnop and J. L. Jimenez, Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 2009, 9(9), 2891–2918 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0em00061b |
| This journal is © The Royal Society of Chemistry 2020 |