
Building a stable cationic molecule/electrode interface for highly efficient and durable CO2 reduction at an industrially relevant current†
Jianjun
Su
a,
Jun-Jie
Zhang
b,
Jiacheng
Chen
c,
Yun
Song
a,
Libei
Huang
a,
Minghui
Zhu
 *c,
Boris I.
Yakobson
b,
Ben Zhong
Tang
def and
Ruquan
Ye
*ag
*c,
Boris I.
Yakobson
b,
Ben Zhong
Tang
def and
Ruquan
Ye
*ag
aDepartment of Chemistry, State Key Laboratory of Marine Pollution, City University of Hong Kong, Hong Kong 999077, China. E-mail: ruquanye@cityu.edu.hk
bDepartment of Materials Science and Nano Engineering and Department of Chemistry, Rice University, 6100 Main Street, Houston, Texas 77005, USA
cState Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: minghuizhu@ecust.edu.cn
dDepartment of Chemistry, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction and Institute for Advanced Study, SCUT-HKUST Joint Research Institute, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong 999077, China
eHKUST-Shenzhen Research Institute, No. 9 Yuexing 1st Rd, South Area, Hi-tech Park, Nanshan, Shenzhen 518057, China
fCenter for Aggregation-Induced Emission, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Tianhe Qu, Guangzhou 510640, China
gCity University of Hong Kong Shenzhen Research Institute, Shenzhen, Guangdong 518057, China
First published on 8th December 2020
Abstract
Aggregation and leaching are two major obstacles to the synthesis of efficient and durable heterogeneous molecular catalysts. These problems are even more severe for charged molecules, not only resulting in unsatisfactory performance, but also leading to misleading evaluation of charged functionalities. In this work, methylation of cobalt(II) tetraamino phthalocyanine (CoTAPc) transforms its electron-donating amino groups into electron-withdrawing quaternary ammonium cations, which favor the formation of *COOH intermediate and the desorption of *CO; this is conducive to a 130% increase of the current density for the CO2 reduction reaction (CO2RR). However, the catalysts leach severely; consequently, the current density decays rapidly. To resolve this dilemma, we developed an in situ functionalization strategy by first covalently grafting CoTAPc onto carbon nanotubes via a diazo-reaction, followed by a complete methylation reaction. This is conducive to a 700% increase in CO partial current density compared to that of a physically mixed sample at −0.72 V vs. RHE with highly stable currents. In a flow cell, this covalently immobilized structure delivers an industrially relevant current density of 239 mA cm−2, CO selectivity of 95.6% at 590 mV overpotential and very low molecular loading of 0.069 mg cm−2. This work provides mechanistic insight and a design strategy for charged molecular catalysts for high-performance and stable heterogeneous electrolysis.
Broader contextHeterogenization of molecular catalysts can enhance the performance of the electrochemical CO2 reduction reaction (CO2RR) by facilitating the charge transfer. However, building a stable cationic molecular catalyst/electrode interface is challenging due to the improved solubility of charged molecular catalysts. This catalyst leaching issue will not only result in poor CO2RR performance, but also lead to a misleading evaluation of the charged functionalities and hamper the further development of charged electrocatalysts. In this work, we developed an in situ strategy for the formation of covalently immobilized quaternary ammonium-substituted cobalt phthalocyanine on carbon nanotubes. The covalent bonding significantly improves the stability, and the quaternary ammonium groups optimize the electronic structure towards CO2RR. A serendipitous CO2RR performance with a record high turnover frequency of 102 s−1 at −0.72 V vs. RHE is attained, which is equivalent to a 700% enhancement to that of the physically mixed samples. When tested in a flow cell at a very low molecular loading of 0.069 mg cm−2, the covalently immobilized structure enables an industrially relevant current density of 239 mA cm−2 and a CO selectivity of 95.6% at 590 mV overpotential. This work underscores the significance of a stable interface for charged molecules and has implications for the future development of stable heterogeneous electrocatalysts. |
Introduction
Electrochemical reduction of carbon dioxide (CO2) using renewable electricity is an effective process for the production of useful fuel and chemical commodities.1,2 This attractive process can not only mitigate global warming by fixing CO2, but also provide a pathway to store electricity as chemical energy.3,4 Many efforts have been devoted to efficiently achieve the electrochemical CO2 reduction reaction (CO2RR) with high activity, selectivity and stability at low overpotential.5,6 Among numerous examples, molecular complexes for the production of CO have attracted significant research attention, as CO is an important precursor for diverse commodity chemicals synthesized by the Fischer–Tropsch process.7,8 One advantage of molecular catalysts lies in their well-defined active sites and tunable properties, which will be conducive to understanding of structure–activity relationships and the rational design of high-performance catalysts.9,10Peripheral functionalization with different substituents has been proved to be an effective method to enhance the electrocatalytic performance of molecular catalysts.11,12 The functional groups can tune the electronic properties of the metal centers by inductive effects. For example, cobalt phthalocyanine with eight cyano substituents, CoPc(CN)8, shows higher activity and selectivity than CoPc by facilitating the formation of active Co(I) sites and CO desorption.13 In our previous work, electron-donating groups on cobalt tetraphenylporphyrin (CoTPP) were found to improve the CO2RR by increasing the electron density of the cobalt center and promoting the electrosorption of CO2.14 Another effect of peripheral functionalities is electrostatic interactions. It was suggested that cationic groups favor the formation of intermediates in the rate-determining step by electrostatic stabilization. Both enhancements have been observed on CoPc and CoTPP with quaternary ammonium cations compared to their unsubstituted forms.14,15
Heterogenization of molecular catalysts can achieve great improvement of current densities and stability by facilitating charge transfer.13,16,17 However, the existence of positive charge, such as pyridinium and quaternary ammonium groups, will increase the water solubility of the molecules, resulting in poor stability.18,19 The solubility issue can be mitigated by using an oppositely charged conductive substrate. For example, by using reduced graphene oxide (rGO) and nitrogen-doped reduced graphene oxide, both iron tetra-(4-N,N,N-trimethylanilinium)porphyrin (FeTMAP)20 and cobalt tetra-(4-N,N,N-trimethylanilinium)porphyrin (CoTMAP)21 can be immobilized through π–π stacking and electrostatic interactions. These strategies improve the durability of the catalyst; however, the turnover frequency (TOF) values are very small. We hypothesize that the strong electrostatic interactions between the molecular catalysts and rGO result in a sandwich structure and decrease the accessibility of CO2 to the metal center.22,23 In addition, the hydrothermal reaction for the catalyst synthesis can decompose the quaternary ammonium groups.21
To this end, there is a dilemma between the stability and activity of molecular catalysts in improving its CO2RR activity via charge engineering. Herein, combining experimental data and theoretical prediction, we first study the effects of cations on enhancing the CO2RR of molecular catalysts. Three model samples are used, namely CoPc, cobalt(II) tetraamino phthalocyanine (CoTAPc), and cobalt(II) tetra-(4-N,N,N-trimethylanilinium)phthalocyanine (CoTMAPc). As expected, CoTMAPc shows the highest CO2RR activity; however, the four quaternary ammonium groups significantly increase the water solubility, and the current densities decay quickly. To resolve this dilemma, we covalently immobilize CoTMAPc onto carbon nanotubes (CNTs) through a diazo-reaction between CoTAPc and CNTs followed by a methylation reaction. This strategy affords a high current density of 239 mA cm−2 and faradaic efficiency for CO (FECO) of 95.6% at −0.7 V vs. RHE with excellent stability in a flow cell configuration.
Results and discussion
Characterization and electrocatalytic performance of CoPc, CoTAPc and CoTMAPc catalysts
The cationically charged CoTMAPc was synthesized by methylation of CoTAPc with excess CH3I to transform the four amino groups into quaternary ammonium cations.24Fig. 1a shows the chemical structures and the photographs of the three molecules in water (left to right: CoPc, CoTAPc and CoTMAPc). The introduction of charged functional groups also greatly increases the water solubility (Fig. 1a). The transformation of the amino groups in CoTAPc into trimethylamino groups in CoTMAPc was confirmed by Fourier transform infrared (FT-IR) (Fig. S1, ESI†) and 1H nuclear magnetic resonance (1H NMR) (Fig. S2, ESI†) spectroscopy. The N–H stretching peaks at 3333 and 3208 cm−1 of CoTAPc disappeared in CoTMAPc.25 The prominent –CH3 stretching vibration in the –N+(CH3)3 group at 1484 cm−1 and the C–N bending vibration in –N+–(CH3)3 at 944 cm−1 appear in CoTMAPc.26 The 1H NMR spectrum of CoTMAPc also shows a strong signal at 3.18 ppm from the methyl proton.27 Raman spectra (Fig. S3, ESI†) of CoTAPc and the as-prepared CoTMAPc show a characteristic absorption peak at about 1000–1600 cm−1.28 In the UV-visible (UV-vis) spectra (Fig. S4, ESI†), the Q band absorbance peak of CoTMAPc is blue-shifted to 669.8 nm after methylation, which is attributed to the strongly electron withdrawing nature of the trimethylamino group.29 Therefore, the above characterization confirmed the successful synthesis of the positively charged CoTMAPc molecule.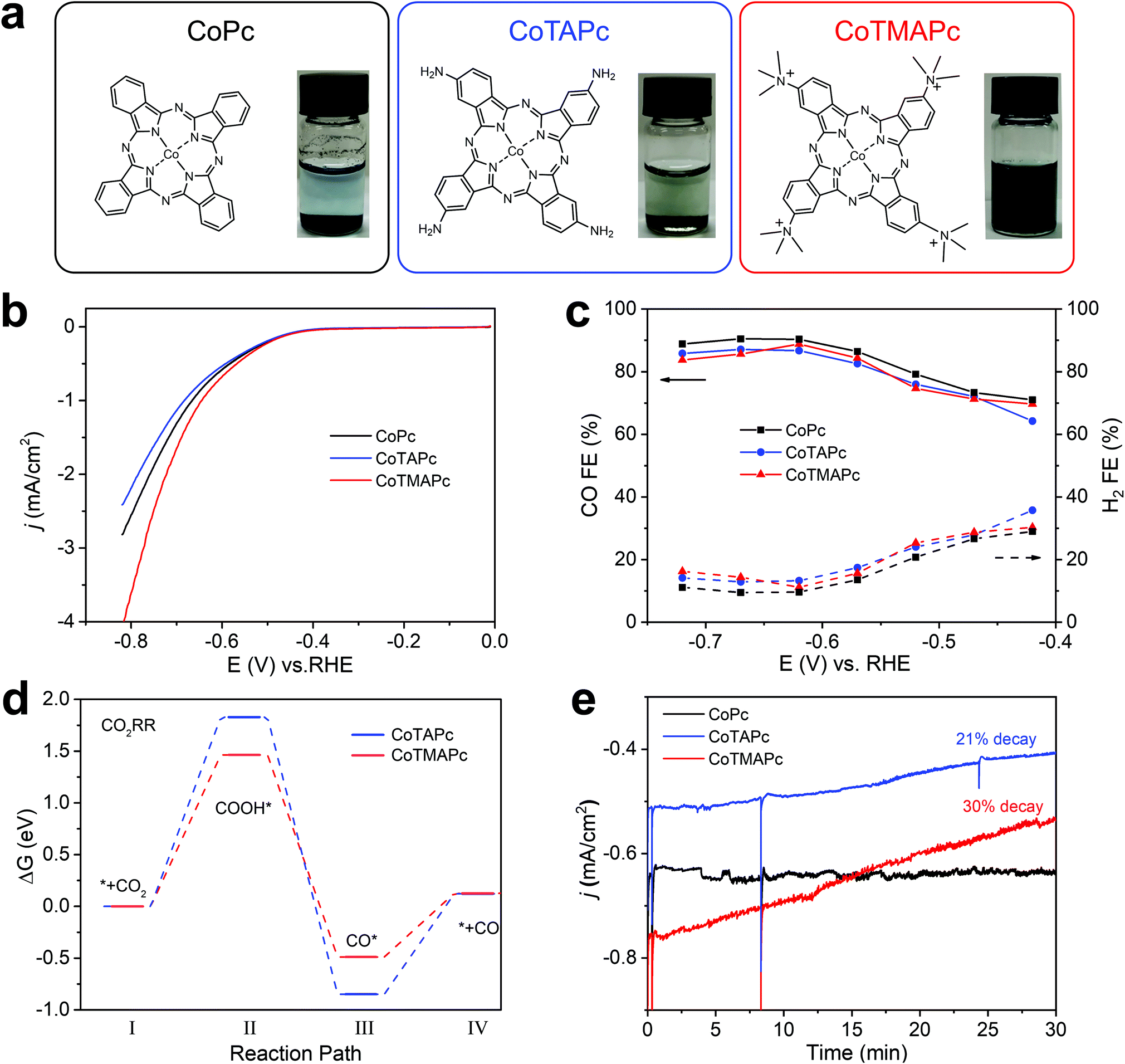 | ||
| Fig. 1 Electrocatalytic performance of CoPc, CoTAPc and CoTMAPc in CO2-saturated 0.5 M KHCO3. (a) Chemical structures and photographs of CoPc, CoTAPc and CoTMAPc in water (0.7 μmol mL−1). (b) Linear sweep voltammogram curves at a sweep rate of 10 mV s−1. (c) Faradaic efficiencies at different potentials. (d) CO2 free energy profiles for CO2RR to CO catalyzed by CoTAPc and CoTMAPc. (e) Long-term electrolysis at −0.62 V versus RHE. | ||
The CO2RR activities of the molecular catalysts were evaluated in a home-made three-compartment cell21,36 in CO2-saturated 0.5 M KHCO3 with a molecular loading of 7 × 10−9 mol cm−2. The working electrode was prepared by dispersing 4.5 μmol of molecular catalyst in 10 mL DMF with 200 μL Nafion and dropping 20 μL catalyst ink on carbon paper (0.5 inch diameter). Compared with CoPc, CoTAPc has a more negative redox potential in CO2 and N2 saturated electrolyte, while CoTMAPc has a more positive redox potential (Fig. S5, ESI†); this further demonstrates the electron-withdrawing effect of the peripheral cationic functional groups on the CoPc molecule. Linear sweep voltammetry (LSV) shows that all catalysts exhibited similar onset potentials at ∼−0.4 V vs RHE (Fig. 1b). The CO partial current densities (jCO) and turnover frequencies (TOFCO) exhibited similar trends (Fig. S6, ESI†). CoTMAPc shows much higher initial jCO (1.87 mA cm−2) and TOF (1.39 s−1) at −0.72 V than CoPc (jCO = 1.42 mA cm−2, TOF = 1.05 s−1) and CoTAPc (jCO = 1.19 mA cm−2, TOF = 0.88 s−1). As depicted in Fig. 1c, the FECO of these three molecules have roughly the same trend, which increases from 70% at their onset overpotentials to 85% at −0.57 V vs. RHE, then levels to ∼87% at potentials more negative than −0.57 V. The ∼120 mV dec−1 Tafel slopes (Fig. S7, ESI†) of all three molecules indicate a one-electron transfer rate-limiting step without a mass-transport limit.30,31
The variation of the Gibbs free energy of the reaction pathway was calculated to provide mechanistic insight into the CO2RR activity. The top and side views of CoTAPc and CoTMAPc and their intermediate steps of CO2RR are listed in Table S1 (ESI†). As shown in Fig. 1d, the change of the variation of the free energy, Δ(ΔG), for the rate-determining step (the conversion of CO2 to *COOH, where * indicates adsorbed species)30 is 1.46 eV on CoTMAPc, which is lower than CoTAPc by 0.37 eV. Moreover, the Δ(ΔG) for the CO desorption on CoTMAPc is only 0.62 eV, which is significantly lower than that of CoTAPc (0.97 eV). A diagram of the variations of the Gibbs free energy for the hydrogen evolution reaction (HER) processes of CoTAPc and CoTMAP is shown in Fig. S8 (ESI†). The Δ(ΔGH) of CoTMAPc was calculated to be 0.79 eV, which is higher than that of CoTAPc (0.77 eV), indicating that the HER reaction is more difficult for CoTMAPc. The computation results indicate that compared to the electron-donating amino groups, the electron-withdrawing quaternary ammonium groups on CoTMAPc favor the formation of *COOH and the desorption of CO, which collectively benefits the CO2RR to CO and is consistent with our experimental data.
However, the improved solubility of charged molecular catalysts results in very poor stability of CO2RR performance. During the stability measurement at −0.62 V in Fig. 1e, the CoPc shows a good stability in 30 min with almost no current decrease. However, the current densities of the CoTAPc and CoTMAPc samples decreased quickly by 20.9% and 30% after 30 min, respectively. This is because of the introduction of water-soluble amino and trimethylamino groups in CoTAPc and CoTMAPc, resulting in catalyst leaching during continuous electrolysis.
Synthesis and characterization of CoTMAPc@CNT
In order to solve the leaching problem of CoTMAPc molecules for CO2RR, we further developed a strategy to covalently immobilize CoTMAPc on CNTs. The CoTAPc was firstly anchored on the CNTs by a diazonium reaction, and the amino groups were then transformed into quaternary ammonium groups by a methylation reaction (Fig. 2a). To control the selectivity, there are two important steps in the synthesis. First, the ratio of isoamyl nitrite to CoTAPc was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 so that not all four amino groups of CoTAPc would be converted to diazonium. Second, the isoamyl nitrite was slowly added to the dispersed CoTAPc so that the amino groups were in great excess. Fig. 2b shows the change of the zeta potential of CoTAPc@CNT with CH3I equivalent in the methylation reaction. The zeta potential value of CoTAPc@CNT greatly increases from −22.8 mV to 20.2 mV when the CH3I equivalent increases from 6 to 10 and then levels off at ∼40 mV when the CH3I equivalent further increases from 20 to 50, which suggests complete methylation. The methylation can also be inferred from the XPS data. As shown in Fig. 2c, CoTMAPc@CNT has Co 2p3/2 and Co 2p1/2 signals at 781.1 eV and 796.1 eV, respectively, which are higher than those of CoTAPc@CNT at 780.0 eV and 795.2 eV. The variation is due to the transformation of electron-donating amino groups into electron-withdrawing quaternary ammonium groups, leading to a decrease in the electron density of Co. Similarly, the N 1s peaks (Fig. 2d) of the N atoms in phthalocyanine increase from 398.3 eV in CoTAPc@CNT to 398.4 eV in CoTMAPc@CNT.32 Moreover, the appearance of the N 1s peak at 401.9 eV in CoTMAPc@CNT further demonstrates the formation of quaternary ammonium.33 The N:Co atomic ratios of CoTAPc@CNT and CoTMAPc@CNT from XPS (Table S2, ESI†) are 11.06 and 11.00, respectively, which suggests that tethering and functionalization occurs in accordance with Fig. 2a. The UV-vis spectra (Fig. S9, ESI†) of CoTMAPc@CNT show an absorption peak at 684.2 nm, which slightly blue shifts compared to that of CoTAPc@CNT (687.8 nm). This trend agrees with the peak shift of molecular catalysts in Fig. S4 (ESI†). Additionally, the Q band absorbance peak of CoTMAPc@CNT (684.2 nm) was red-shifted compared to that of CoTMAPc (669.8 nm), while the Q band of CoTAPc@CNT (687.8 nm) was blue-shifted to that of CoTAPc (701.7 nm). This is because the grafted molecules lose an electron-donating NH2 group in CoTAPc@CNT and an electron-withdrawing N+(CH3)3 group in CoTMAPc@CNT. Fig. 2e presents the Raman spectra of pristine CNT, CoTAPc@CNT and CoTMAPc@CNT. CNT has two signature peaks at 1357 and 1582 cm−1, which correspond to the D and G bands. The CoTAPc@CNT and CoTMAPc@CNT exhibit some fine signals from molecular catalysts at 1000–1500 cm−1.34 The ratio of the D to G peak intensities (ID/IG) reflects the degree of disorder of carbon materials. The molecule-modified CNT samples have a higher ID/IG ratio (∼0.3) than pristine CNTs (0.27), which likely arises from disturbance of the surface lattice after the covalent grafting.35
:1 so that not all four amino groups of CoTAPc would be converted to diazonium. Second, the isoamyl nitrite was slowly added to the dispersed CoTAPc so that the amino groups were in great excess. Fig. 2b shows the change of the zeta potential of CoTAPc@CNT with CH3I equivalent in the methylation reaction. The zeta potential value of CoTAPc@CNT greatly increases from −22.8 mV to 20.2 mV when the CH3I equivalent increases from 6 to 10 and then levels off at ∼40 mV when the CH3I equivalent further increases from 20 to 50, which suggests complete methylation. The methylation can also be inferred from the XPS data. As shown in Fig. 2c, CoTMAPc@CNT has Co 2p3/2 and Co 2p1/2 signals at 781.1 eV and 796.1 eV, respectively, which are higher than those of CoTAPc@CNT at 780.0 eV and 795.2 eV. The variation is due to the transformation of electron-donating amino groups into electron-withdrawing quaternary ammonium groups, leading to a decrease in the electron density of Co. Similarly, the N 1s peaks (Fig. 2d) of the N atoms in phthalocyanine increase from 398.3 eV in CoTAPc@CNT to 398.4 eV in CoTMAPc@CNT.32 Moreover, the appearance of the N 1s peak at 401.9 eV in CoTMAPc@CNT further demonstrates the formation of quaternary ammonium.33 The N:Co atomic ratios of CoTAPc@CNT and CoTMAPc@CNT from XPS (Table S2, ESI†) are 11.06 and 11.00, respectively, which suggests that tethering and functionalization occurs in accordance with Fig. 2a. The UV-vis spectra (Fig. S9, ESI†) of CoTMAPc@CNT show an absorption peak at 684.2 nm, which slightly blue shifts compared to that of CoTAPc@CNT (687.8 nm). This trend agrees with the peak shift of molecular catalysts in Fig. S4 (ESI†). Additionally, the Q band absorbance peak of CoTMAPc@CNT (684.2 nm) was red-shifted compared to that of CoTMAPc (669.8 nm), while the Q band of CoTAPc@CNT (687.8 nm) was blue-shifted to that of CoTAPc (701.7 nm). This is because the grafted molecules lose an electron-donating NH2 group in CoTAPc@CNT and an electron-withdrawing N+(CH3)3 group in CoTMAPc@CNT. Fig. 2e presents the Raman spectra of pristine CNT, CoTAPc@CNT and CoTMAPc@CNT. CNT has two signature peaks at 1357 and 1582 cm−1, which correspond to the D and G bands. The CoTAPc@CNT and CoTMAPc@CNT exhibit some fine signals from molecular catalysts at 1000–1500 cm−1.34 The ratio of the D to G peak intensities (ID/IG) reflects the degree of disorder of carbon materials. The molecule-modified CNT samples have a higher ID/IG ratio (∼0.3) than pristine CNTs (0.27), which likely arises from disturbance of the surface lattice after the covalent grafting.35
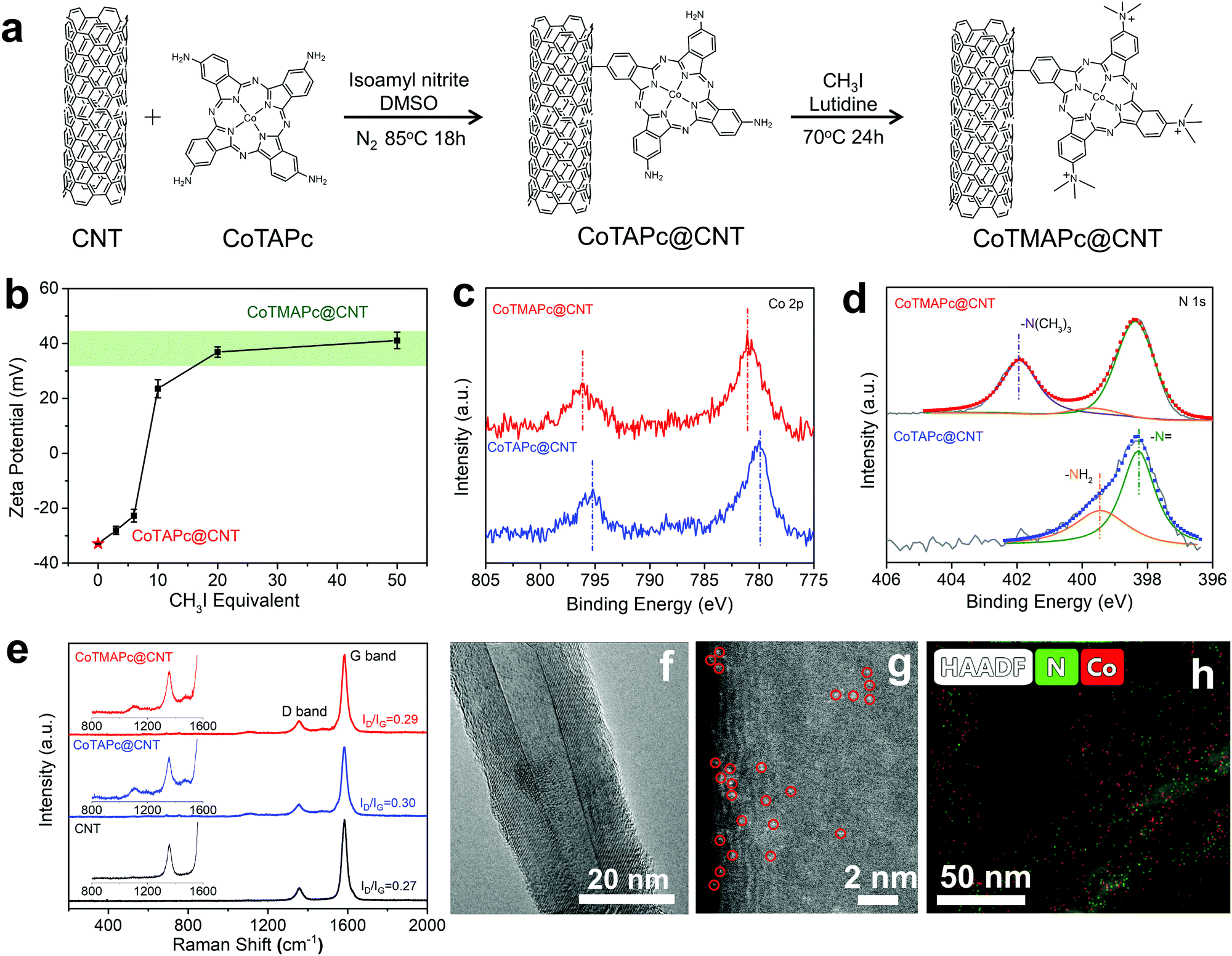 | ||
| Fig. 2 Strategy for heterogenizing CoTMAPc onto CNT. (a) Scheme for the synthesis of CoTMAPc@CNT. (b) Zeta potentials of CoTMAPc@CNT as a function of the CH3I equivalent. High-resolution XPS spectra of (c) Co 2p and (d) N 1s of CoTAPc@CNT and CoTMAPc@CNT. (e) Raman spectra of pristine CNT, CoTAPc@CNT and CoTMAPc@CNT. (f) A typical TEM image and (g) HADDF-STEM image of CoTMAPc@CNT. The circled bright spots highlight the dispersed Co centers. (h) N and Co EDS elemental mapping of CoTMAPc@CNT. | ||
Transmission electron microscopy (TEM) reveals that CoTAPc@CNT and CoTMAPc@CNT possess a similar smooth outer surface morphology to that of the pristine CNTs without obvious aggregation of molecular catalysts (Fig. 2f and Fig. S10, ESI†). The Z-contrast high-angle annular dark field high-resolution aberration-corrected scanning transmission electron microscopy (HADDF-STEM) image (Fig. 2g) of CoTMAPc@CNT shows the high dispersion of Co sites on the CNT surfaces, as indicated by the circled bright spots. The energy dispersive X-ray spectroscopy (EDS) maps (Fig. 2h) also show the uniform distribution of N and Co elements alongside the CNT. Inductively coupled plasma mass spectrometry (ICP-MS) was carried out to determine the Co contents in the as-prepared composite materials. The Co contents in CoTAPc@CNT and CoTMAPc@CNT were found to be approximately 0.55 wt%, corresponding to 5.7 wt% and 6.9 wt% of CoTAPc and CoTMAPc in the hybrid.
Electrocatalytic CO2RR performance of CoTMAPc@CNT
The CO2RR electrochemical performance of CoTAPc@CNT and CoTMAPc@CNT was tested with a total mass loading of 75 μg cm−2, corresponding to a molecule catalyst loading of ∼7 × 10−9 mol cm−2. We also took the physically mixed samples, CoTAPc/CNT and CoTMAPc/CNT, with the same molecule catalyst loading for comparison. We first estimated the number of electrochemically active sites by measuring the cyclic voltammograms (CVs) of the catalysts (Fig. S11a and c, ESI†). The surface-active Co sites of CoTAPc@CNT and CoTMAPc@CNT are 6.87 × 10−9 and 6.72 × 10−9 mol cm−2, which are very close to the theoretical content (7 × 10−9 mol cm−2), indicating that all the molecular catalysts are almost accessible and electrochemically active on the CNT surface. Moreover, after a 30 min chronoamperometric test, the numbers of electrochemically active sites of CoTMAPc@CNT and CoTAPc@CNT were 6.91 × 10−9 and 6.84 × 10−9 mol cm−2, respectively, which are almost the same as the values before the stability test (Fig. S11b and d, ESI†). However, for the physically mixed samples, the initial amounts of the electrochemically active species of CoTMAPc/CNT and CoTAPc/CNT were only 1.72 × 10−9 and 1.63 × 10−9 mol cm−2 (Fig. S12a and d, ESI†). These values were about four times smaller than the theoretical loading (7 × 10−9 mol cm−2). The low electrochemical active sites of the physically mixed samples were due to the molecular stacking in physical mixing composites and some initial leaching. Similar to the data in Fig. 1e, the current densities of CoTMAPc/CNT and CoTAPc/CNT also decreased after a 30 min stability test (Fig. S12b and e, ESI†), and the number of electrochemically active sites also reduced to 1.48 × 10−9 and 1.01 × 10−9 mol cm−2, respectively (Fig. S12c and f, ESI†). In order to quantify the Co leaching, we further conducted ICP analysis on the post-reaction electrolyte solution at 1 mg cm−2 catalyst loading. As shown in Table S3 (ESI†), no Co content was detected in the post-reaction electrolyte of CoTAPc@CNT and CoTMAPc@CNT. However, the CoTAPc/CNT and CoTMAPc/CNT showed 11.6% and 43.0% Co leaching after 30 min electrocatalyst, respectively, which is consistent with the current attenuation in Fig. S12b and e (ESI†). These results further proved the stability enhancement of the covalent bond. The stabilities of both grafted samples were further evaluated at different applied potentials (Fig. S13, ESI†). Notably, unlike the rapid attenuation of the physically mixed samples, both CoTMAPc@CNT and CoTAPc@CNT have a very stable current density at different potentials for 60 min. The long-term potentiostatic electrolysis of CoTMAPc@CNT was further undertaken at −0.62 V for 12 hours, as shown in Fig. 3e. The catalyst achieved a current density of 11.9 mA cm−2 and a FECO of 98% with negligible decay, which amounts to an excellent turnover number (TON) of 373000 over the 12 h of electrocatalysis. The excellent stability of CoTMAPc@CNT signifies the effective role of covalent bonds in stabilizing the molecular catalysts.
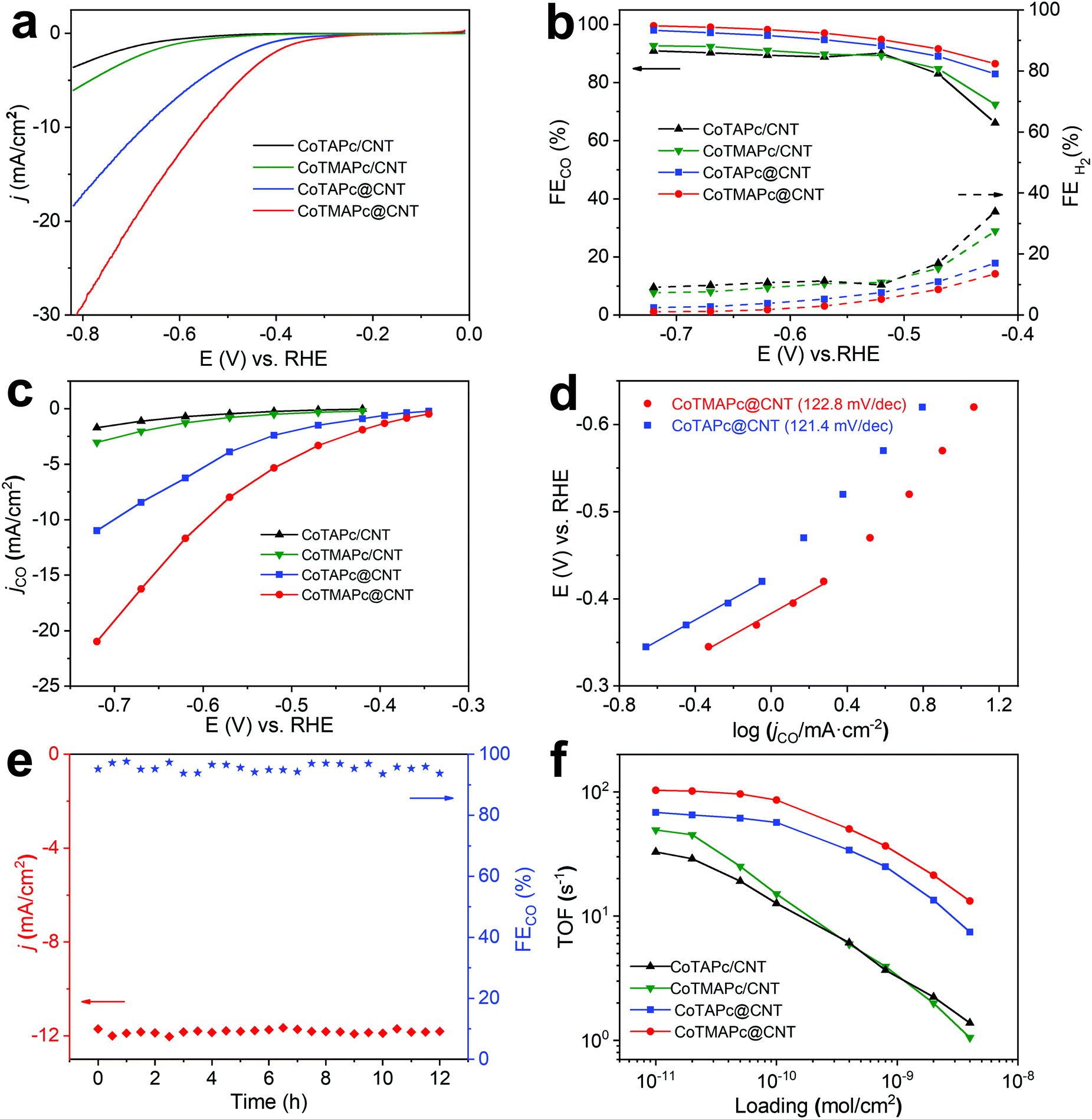 | ||
| Fig. 3 CO2RR activities of CoTAPc@CNT, CoTMAPc@CNT, CoTAPc/CNT and CoTMAPc/CNT in CO2-saturated 0.5 M KHCO3. (a) LSV curves acquired at a scan rate of 10 mV s−1. The catalyst loading is ∼7 × 10−9 mol cm−2. (b) FE of CO and H2. (c) CO partial current densities. (d) Tafel slopes of CoTAPc@CNT and CoTMAPc@CNT. (e) Long-term stability of CoTMAPc@CNT at −0.62 V vs. RHE and (f) TOFCO at −0.62 V with different catalyst loadings. | ||
As shown in the LSV curves (Fig. 3a), after covalent grafting on the CNT surface, CoTMAPc@CNT and CoTAPc@CNT present a low onset potential of −0.28 V and large total current densities of 30.7 mA cm−2 and 18.4 mA cm−2 at −0.82 V, respectively. All four samples exhibit similar trends of FECO, which increase from −0.42 to −0.52 V and remain stable at higher overpotentials (Fig. 3b). The FECO of CoTMAPc@CNT and CoTAPc@CNT could reach 99% and 98% at −0.72 V, while CoTMAPc/CNT and CoTAPc/CNT only attained 92% and 90% at the same potential. Static state measurements show that CoTMAPc@CNT and CoTAPc@CNT exhibit high jCO values of −21.0 mA cm−2 and −10.9 mA cm−2 at −0.72 V, almost 700% of those of CoTMAPc/CNT (−3.1 mA cm−2) and CoTAPc/CNT (−1.7 mA cm−2), respectively (Fig. 3c). The 1H NMR spectrum (Fig. S14, ESI†) shows no liquid products in the post-reaction electrolyte. Two control experiments were conducted: (1) carbon paper (CP) without deposited catalyst tested in CO2-saturated 0.5 M KHCO3, and (2) CoTMAPc@CNT with a molecular loading of 7 × 10−9 mol cm−2 tested in N2 saturated 0.5 M KHCO3. The CO partial current densities (jCO) at different potentials are shown in Table S4 (ESI†). The small jCO confirms that the CO production is negligible without CO2 or with CO2 but in the absence of catalyst. The reaction kinetics for the CO formation was analyzed by the Tafel slope (Fig. 3d). The Tafel slopes of 122.6 and 121.4 mV dec−1 for CoTMAPc@CNT and CoTAPc@CNT, respectively, indicate that there is no transport limitation and are consistent with the values for a rate-limiting single-electron transfer step.30
TOF is defined as the number of reduction products generated per electrocatalytic active site per unit time, representing the intrinsic activity of the catalyst.35 Here, the TOF values of different samples were calculated from the partial CO current density (Fig. S15, ESI†) with different molecular catalyst loadings (1 × 10−11 to 4 × 10−9 mol cm−2) at −0.62 V (Fig. 3f). At a loading of 1 × 10−11 mol cm−2, CoTMAPc@CNT exhibits a maximum TOFCO of 102.9 s−1, far greater than the 68.3 s−1 of CoTAPc@CNT, which indicates a higher CO2RR activity of CoTMAPc molecule. However, the physically mixed samples, CoTMAPc/CNT and CoTAPc/CNT, only show relatively low TOFCO values of 49.3 and 32.8 s−1, respectively. In general, the TOFs decrease at higher catalyst loadings because the current densities can be limited by mass transport. When the catalyst loading increases exponentially, the TOFCO values of CoTMAPc/CNT and CoTAPc/CNT decrease much more rapidly to 1.05 and 1.38 s−1 at a loading of 4 × 10−9 mol cm−2; this may result from aggregation of the molecular catalysts during electrode preparation and leaching during electrolysis.
The current density of CO2RR in H-type cells is limited by the slow diffusion and low solubility of CO2 in aqueous media, which can be resolved by using a flow cell design.8 Here, we further demonstrate that CoTAPc@CNT and CoTMAPc@CNT can deliver commercially relevant current densities in a flow cell configuration. The flow cell was assembled as shown in Fig. 4a. Both the CoTAPc@CNT and CoTMAPc@CNT composites showed much higher current densities than those collected in a customized three-compartment cell (Fig. S16, ESI†). CoTMAPc@CNT shows a high current density of 31.0 mA cm−2 and an FECO of 94.5% at a low overpotential of 290 mV (Fig. 4b) with negligible performance decay for 15 h (Fig. 4c). At −0.7 V vs. RHE (η = 590 mV), an outstanding current density of 239 mA cm−2 and FECO of 95.6% were achieved. Salt segregation on the cathode (gas channel) was observed at −0.6 V, which arises from the rapid consumption of water (CO2 + 2e− + H2O → CO + 2OH−) and leads to fluctuation of the current density. The excellent CO2RR activities of CoTMAPc@CNT are comparable to those of other reported catalysts for the conversion of CO2 to CO (Fig. 4d and Table S5, ESI†).8,37–41 For example, compared to other complexes containing CoPc, CoPc–CN/CNT and Ni single-atom catalysts (SAC), CoTMAPc@CNT, at a much lower molecular loading of 0.069 mg cm−2, delivers much higher current densities than other systems with smaller total cell voltages. Even in comparison to the performance of the precious metals Au and silver, the current density and selectivity of CoTMAPc@CNT still stand out.
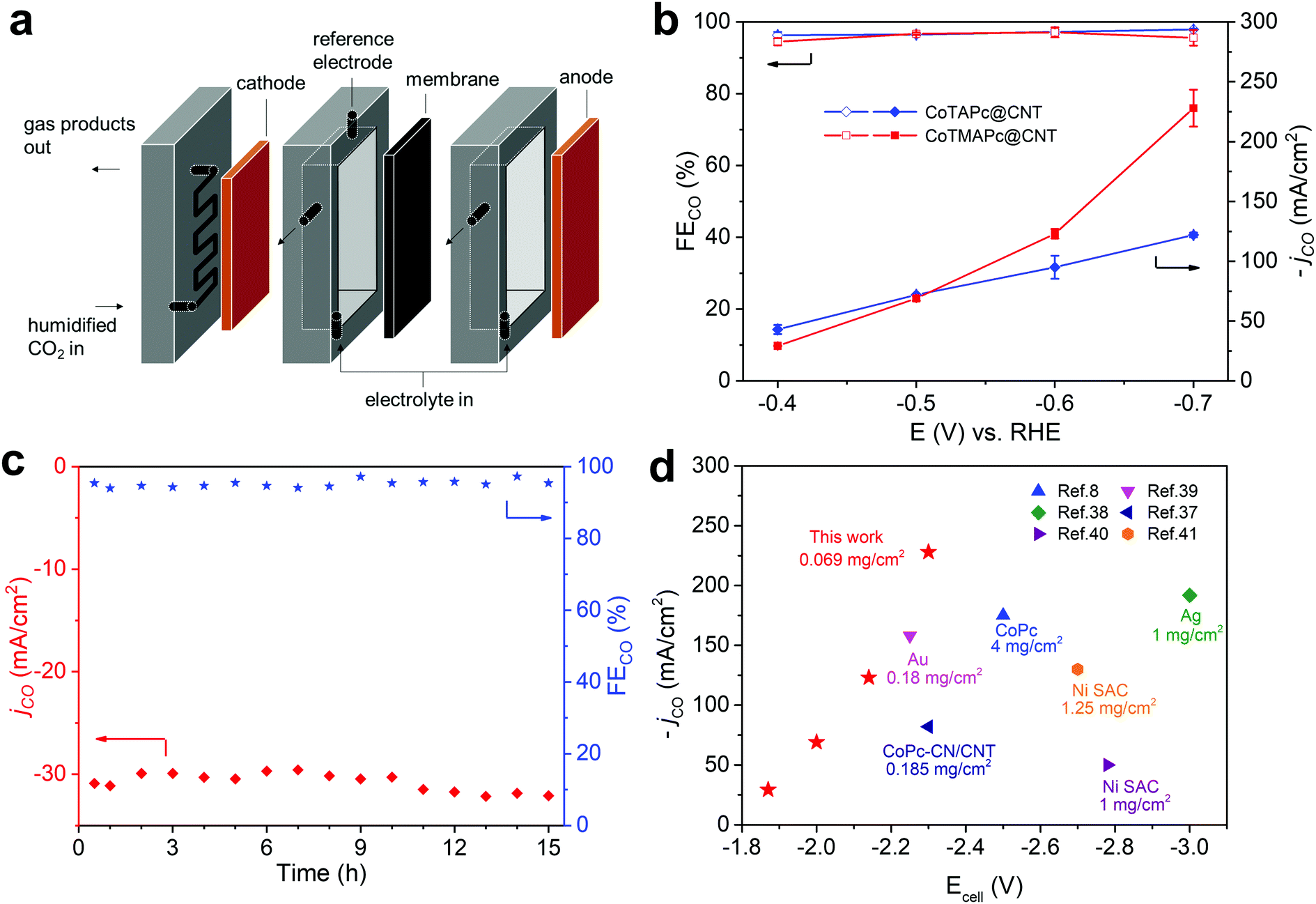 | ||
| Fig. 4 CO2RR performance in a flow cell. (a) Graphic illustration of the CO2 flow cell. (b) Current densities and selectivities of CO production at various potentials of CoTAPc@CNT and CoTMAPc@CNT. (c) Long-term electrolysis of CoTMAPc@CNT at −0.4 V vs. RHE. (d) Comparison of the CO2RR performance of CoTMAPc@CNT to literature data. The catalyst loadings are also listed for comparison. | ||
Conclusions
In conclusion, quaternary ammonium cation can improve the CO2RR activity of CoPc by decreasing the reaction barriers of intermediate formation and product desorption. However, the enhanced solubility of molecular catalysts due to the charged functional groups hampers their long-term electrolysis and underestimates their function. A strategy for in situ functionalization of the immobilized molecular complex on the CNT surface is developed by diazo-reaction and methylation reactions. With this design, our cationic catalyst CoTMAPc@CNT achieves a TOF of ∼100 s−1 at −0.62 V with negligible decay in current density. In a flow cell configuration, it delivers an industrially relevant current density of 239 mA cm−2 and a selectivity of 95.6% at a cell voltage of 2.3 V and a molecule loading of 0.069 mg cm−2. This study provides an approach for rational design of molecular catalysts for high-performance CO2 electroreduction and may have implications for other molecular catalysts in wide applications. It may also call for a revisit of the effects of charged functionalities in heterogeneous molecular electrochemistry.Experimental
Materials
All chemicals were used as received without any further treatment. Cobalt phthalocyanine (CoPc, 92%), potassium bicarbonate (KHCO3, 99.5%), iodomethane (CH3I), 2,6-lutidine (99%), isoamyl nitrite and dimethyl sulfoxide (DMSO) were purchased from J&K Scientific Ltd. Cobalt tetraaminophthalocyanine (CoTAPc, 95%) was purchased from PorphyChem SAS. Multiwalled carbon nanotubes (OD: 10–20 nm, L: 5–30 μm) were bought from XFNANO. N,N-Dimethylformamide (DMF), diethyl ether, and ethanol were purchased from LabScan. Deionized water (Millipore Milli-Q grade) with a resistivity of 18.2 MΩ cm was used in all experiments.Synthesis of CoTMAPc
CoTAPc (30 mg, 0.048 mmol) was dissolved in DMF (5 mL) and sonicated for 10 min. Then, 2,6-lutidine (100 μL) and iodomethane (100 μL) were added to the CoTAPc suspension and sonicated for another 10 min. Subsequently, the mixture was stirred at 70 °C for 24 h. After the reaction, the solution was poured into diethyl ether (25 mL) and the precipitate was centrifuged and repeatedly washed with diethyl ether and acetone. Finally, the green CoTMAPc product was vacuum dried at 60 °C.Synthesis of CoTAPc@CNT
CNT (100 mg) and CoTAPc (34.5 mg, 0.055 mmol) were added to DMSO (50 mL) solution and sonicated for 30 min. Isoamyl nitrite (7.4 μL, 0.055 mmol) was slowly added to the DMSO solution under magnetic stirring. Then, the solution was maintained at 85 °C for 18 h in N2 atmosphere. After the solution was cooled to room temperature, the product was collected by centrifugation and washed with DMF and water until the solvent became colorless. Finally, the CoTAPc@CNT composite was vacuum dried at 60 °C.Synthesis of CoTMAPc@CNT
CoTAPc@CNT (100 mg) was dispersed in DMF (50 mL). CH3I (1 mL) and 2,6-lutidine (1 mL) were added, and the mixture was stirred at 70 °C for 24 h. The final product was collected by centrifugation, washed with DMF (50 mL × 3) and DI water (50 mL × 3) and vacuum dried.Material characterization
The morphologies of the samples were characterized using transmission electron microscopy (TEM, Philips Technai 12) equipped with energy dispersive X-ray spectroscopy. Zeta-potentials were determined on a Dynamic Light Scattering Particle Size Analyzer (Malvem Zetasizer Nano ZS). Samples were dispersed in DI water and sonicated for 1 h before the test. ICP-atomic emission spectroscopy (ICP-OED) measurements were conducted on an Optima 8000 spectrometer. Samples were digested in hot concentrated HNO3 for 1 h and diluted to the desired concentrations. FT-IR spectra were taken on a PerkinElmer Spectrum 100 FT-IR spectrometer. UV-vis spectrophotometry was performed on a Shimadzu 1700 spectrophotometer in DMF solution with a concentration of 1 × 10−5 mol mL−1. Raman spectra were collected using a LabRAM HR800 laser confocal micro-Raman spectrometer with a laser wavelength of 514.5 nm. 1H NMR spectra were recorded on a Bruker 600 MHz ASCEND AVANCE III HD Nuclear Magnetic Resonance System (NMR-600). The X-ray photoelectron spectroscopy data were collected on a Thermo ESCALAB 250Xi spectrometer equipped with a monochromatic AlK radiation source (1486.6 eV, pass energy 20.0 eV). The data were calibrated with C 1s 284.8 eV. Scanning transmission electron microscopy was characterized on a double spherical-aberration-corrected FEI Themis Z microscope at 60 kV.Working electrode preparation
The carbon paper (Toray, TGP-H-060, Fuel Cell Store) was punched into discs with a diameter of 0.5 inches. For the preparation of the CoTMAPc@CNT and CoTAPc@CNT working electrodes, the catalyst ink was prepared by dispersing 5 mg of catalyst in 1 mL of DMF with 20 μL 5 wt% Nafion solution (Sigma Aldrich, Nafion 117, 5 wt%) and sonicated for 1 h. Then, 20 μL of the ink was drop-casted on the carbon paper and subsequently dried naturally overnight. The loading on the electrode was 75 μg cm−2 (or 7 × 10−9 mol cm−2 for molecular catalyst loading). For the preparation of CoPc, CoTAPc and CoTMAPc working electrodes, 4.5 μmol of molecular catalyst were dispersed in 10 mL DMF with 200 μL Nafion. After sonication, 20 μL catalyst ink was drop-casted on the carbon paper and dried naturally overnight. The catalyst loading was ∼7 × 10−9 mol cm−2. For the preparation of the physically mixed samples, CoTAPc/CNT and CoTMAPc/CNT, 4.5 μmol of molecular catalysts, 50 mg CNT and 200 μL Nafion were dissolved in DMF and sonicated. Then, 20 μL catalyst ink was dropped on the carbon paper and dried naturally overnight.Electrochemical measurements
The electrochemical performance was carried out in a customized three-compartment cell as previously reported.21,36 A platinum foil and Ag/AgCl leak-free reference (LF-2, Innovative Instrument Inc.) were used as the counter and reference electrodes, respectively. The working electrode was separated from the counter electrode by a Nafion-117 membrane (Fuel Cell Store). Before using, the leak-free reference was calibrated as reported.42 The reference electrode was calibrated with respect to the reversible hydrogen electrode. The calibrations were conducted in high-purity hydrogen-saturated 0.5 M H2SO4 electrolyte with a Pt wire as the working and counter electrode at a scan rate of 1 mV s−1. The average of the two potentials of each CV curve where the current crossed zero was taken to be the thermodynamic potential. All potentials in this study were converted to reversible hydrogen electrode (RHE) according to the Nernst equation (Evs.RHE = Evs.Ag/AgCl + 0.251 + 0.0592 × pH). 1.75 mL of 0.5 M KHCO3 solution electrolyte was added to the working and counter compartments, respectively. The cell was purged with high-purity CO2 gas (Linde, 99.999%, 10 sccm) for 10 min prior to and throughout the duration of all electrochemical measurements. The electrochemical measurements were controlled and recorded with a CHI 650E potentiostat. The automatic iR (95%) compensation was used. The pH values of CO2 and N2-saturated 0.5 M KHCO3 electrolyte were 7.25 and 8.36, which was detected by a pH meter (HI 2211, Hanna Instruments). Gas-phase products were quantified by an on-line gas chromatograph (Ruimin GC 2060, Shanghai) equipped with a methanizer, a Hayesep-D capillary column, a flame ionization detector (FID) for CO and a thermal conductivity detector (TCD) for H2. The CO2 flow rate was controlled at 10 sccm using a standard series mass flow controller (Alicat Scientific mc-50 sccm). Each run was 8 min long. GC was calibrated using standard mixture gas (Linde) and diluted with CO2 (Linde 99.999%).The flow cell contained a gas-diffusion layer (GDL), an anion exchange membrane (Fumasep FAA-3-PK-130), a nickel foam anode and a leak-free Ag/AgCl electrode. The working electrodes were prepared by drop-casting 200 μL of catalyst ink (5 mg mL−1) onto the microporous side of the GDL to reach a total mass loading of ∼1 mg cm−2, or a molecular loading of ∼0.069 mg cm−2. Prior to the electrochemical tests, the electrolytes (1 M KOH, 100 mL) were separately circulated in both the working and counter compartments using peristaltic pumps (Longer, BT100-2J) at flow rates of 5 mL min−1 and 100 mL min−1, respectively. The CO2 flow was kept constant at 50 sccm using a mass flow controller. A CHI650E potentiostat was employed to record the electrochemical responses. The electrolyte resistance between the reference electrode and working electrode was determined to be approximately 4 Ω using potential electrochemical impedance spectroscopy (PEIS) analysis and manually compensated. The gas products were analyzed by an online gas chromatograph.
Computation
The density functional theoretical (DFT) calculations were carried out with the Gaussian 16.43 The Becke exchange functional (B) and the Lee–Yang–Parr (LYP) correlation functional within a generalized gradient approximation (GGA) was used to describe the interaction between the ionic cores and electrons.44,45 The hybrid basic set was employed to optimize all structures and calculate Gibbs free energy. For H, C, N, and O atoms, the basis set b3lyp/6-311+g(d,p) was adopted here. For the metal atom Co, the outer and inner shell valence electrons are described by the effective pseudopotential double-ζ (LANL2DZ) basis set, separately from the core electrons, which were described by the LANL2 effective core potential (ECP). For the CO2RR, the gas-phase errors was corrected as previously reported.46 For the reaction steps involving transfer of a H+/e− pair, the free energy of the pair was set as half the free energy of gaseous H2 (H+ + e− ↔ 1/2H2).47Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Hong Kong Research Grant Council under Early Career Scheme (Project No. 21300620), Young Scientists Fund of the National Natural Science Foundation of China (Project No. 21905240), and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning and Shanghai Sailing Program (19YF1410600). Work at Rice (J. Z. and B. I. Y.) was supported by the National Science Foundation (CBET-1605848).References
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- F. P. García de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef.
- L. Yoon, S. Lee and K. Y. Wong, Chem, 2017, 3, 717–718 Search PubMed.
- H. Li, C. Qiu, S. Ren, Q. Dong, S. Zhang, F. Zhou, X. Liang, J. Wang, S. Li and M. Yu, Science, 2020, 367, 667–671 CrossRef CAS.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- J. Gu, C. S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS.
- D. H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O’Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- Y. Q. Zhang, J. Y. Chen, P. E. M. Siegbahn and R. Z. Liao, ACS Catal., 2020, 10, 6332–6345 CrossRef CAS.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef.
- M. Zhu, D. T. Yang, R. Ye, J. Zeng, N. Corbin and K. Manthiram, Catal. Sci. Technol., 2019, 9, 974–980 RSC.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS.
- K. Elouarzaki, V. Kannan, V. Jose, H. S. Sabharwal and J. M. Lee, Adv. Energy Mater., 2019, 9, 1900090 CrossRef.
- L. Xie, J. Tian, Y. Ouyang, X. Guo, W. Zhang, U. P. Apfel, W. Zhang and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 15844–15848 CrossRef CAS.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- J. Choi, J. Kim, P. Wagner, S. Gambhir, R. Jalili, S. Byun, S. Sayyar, Y. M. Lee, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Energy Environ. Sci., 2019, 12, 747–755 RSC.
- M. Zhu, C. Cao, J. Chen, Y. Sun, R. Ye, J. Xu and Y. F. Han, ACS Appl. Energy Mater., 2019, 2, 2435–2440 CrossRef CAS.
- J. Choi, P. Wagner, S. Gambhir, R. Jalili, D. R. Macfarlane, G. G. Wallace and D. L. Officer, ACS Energy Lett., 2019, 4, 666–672 CrossRef CAS.
- J. Choi, P. Wagner, R. Jalili, J. Kim, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Adv. Energy Mater., 2018, 8, 1801280 CrossRef.
- R. Honna, T. Tsukamoto, T. Shimada, T. Shiragami and S. Takagi, Clay Sci., 2015, 19, 53–58 CAS.
- X. Zhang, H. Liu, P. An, Y. Shi, J. Han, Z. Yang, C. Long, J. Guo, S. Zhao, K. Zhao, H. Yin, L. Zheng, B. Zhang, X. Liu, L. Zhang, G. Li and Z. Tang, Sci. Adv., 2020, 6, eaaz4824 CrossRef CAS.
- M. M. Bhuyan, H. Okabe, Y. Hidaka and K. Hara, J. Appl. Polym. Sci., 2018, 135, 45906 CrossRef.
- R. Asasutjarit, T. Theerachayanan, P. Kewsuwan, S. Veeranodha, A. Fuongfuchat and G. C. Ritthidej, AAPS PharmSciTech, 2015, 16, 1013–1024 CrossRef CAS.
- S. Liu, H. Bin Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S. T. Lee and Y. Li, Chem, 2017, 3, 652–664 CAS.
- M. Zhu, R. Ye, K. Jin, N. Lazouski and K. Manthiram, ACS Energy Lett., 2018, 3, 1381–1386 CrossRef CAS.
- J. S. Zeng, N. Corbin, K. Williams and K. Manthiram, ACS Catal., 2020, 10, 4326–4336 CrossRef CAS.
- N. Li, W. Lu, K. Pei and W. Chen, RSC Adv., 2015, 5, 9374–9380 RSC.
- C. Zhang and J. Zhao, Int. J. Electrochem. Sci., 2017, 12, 9161–9179 CrossRef CAS.
- J. Chen, J. Li, W. Liu, X. Ma, J. Xu, M. Zhu and Y. F. Han, Green Chem., 2019, 21, 6056–6061 RSC.
- M. Zhu, J. Chen, R. Guo, J. Xu, X. Fang and Y. F. Han, Appl. Catal., B, 2019, 251, 112–118 CrossRef CAS.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y. F. Han, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 CrossRef CAS.
- X. Lu, Y. Wu, X. Yuan, L. Huang, Z. Wu, J. Xuan, Y. Wang and H. Wang, ACS Energy Lett., 2018, 3, 2527–2532 CrossRef CAS.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H. R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, ACS Energy Lett., 2018, 3, 193–198 CrossRef CAS.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- R. Ye, Z. Peng, T. Wang, Y. Xu, J. Zhang, Y. Li, L. G. Nilewski, J. Lin and J. M. Tour, ACS Nano, 2015, 9, 9244–9251 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009, vol. 121, pp. 150–166 Search PubMed.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee02535f |
| This journal is © The Royal Society of Chemistry 2021 |