
Oriented proton-conductive nano-sponge-facilitated polymer electrolyte membranes†
Xin
Liu‡
 a,
Junfeng
Zhang‡
a,
Chenyang
Zheng
a,
Jiandang
Xue
a,
Tong
Huang
a,
Yan
Yin
*ab,
Yanzhou
Qin
a,
Kui
Jiao
a,
Qing
Du
a and
Michael D.
Guiver
*ac
a,
Junfeng
Zhang‡
a,
Chenyang
Zheng
a,
Jiandang
Xue
a,
Tong
Huang
a,
Yan
Yin
*ab,
Yanzhou
Qin
a,
Kui
Jiao
a,
Qing
Du
a and
Michael D.
Guiver
*ac
aState Key Laboratory of Engines, Tianjin University, Tianjin 300072, China. E-mail: yanyin@tju.edu.cn; guiver@tju.edu.cn
bState Key Laboratory of Separation Membranes and Membrane Processes, Tianjin Polytechnic University, Tianjin 300387, China
cCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China
First published on 17th December 2019
Abstract
Achieving high power output from proton exchange membrane fuel cells (PEMFCs) requires efficient proton transport in proton exchange membranes (PEMs). Since proton conductivity is closely related to membrane moisture content, operation at low relative humidity (RH) and elevated temperature has become a critical bottleneck for the practical application of PEMFCs due to severe PEM dehydration. While several strategies have sought to mitigate this, including external thermal and water management, coating of nano-cracked hydrophobic layers and optimization of membrane intrinsic water retention, only partial improvements have been realized. Here, using a membrane formulation of ferrocyano-coordinated poly(4-vinylpyridine) (CP4VP), phosphotungstic acid (PWA) and polysulfone (PSf), novel highly water-retentive PEMs are fabricated via a strong magnetic field. During magnetic-assisted membrane casting, CP4VP and PWA form a microporous Prussian blue analogue (PBA) framework with the new type of Fe–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–W bonding, which is paramagnetic and is thus simultaneously aligned in the through-plane (TP) direction of the membrane. The neutral PSf membrane component affords mechanical strength to the embedded TP-aligned conducting channels. This new type of microporous PBA framework is highly hydrophilic and proton conductive, with micropores of ∼5.4 Å diameter, which act as nano-sponges to absorb only more retentive non-freezable water, effective for proton conduction. These nano-sponges display efficient water absorption and retention at low RH and elevated temperatures, together with a much faster hydration process than the dehydration process. Furthermore, the TP-aligned PBA channels also enable faster water transport to promote PEM proton conduction beyond any previously reported water-retentive membrane. Consequently, the novel nano-sponge-like PEMs exhibit remarkable performance in both ex situ and in situ evaluations, especially at low RH and elevated temperature, largely prevailing over the commercial benchmark Nafion® 212.
N–W bonding, which is paramagnetic and is thus simultaneously aligned in the through-plane (TP) direction of the membrane. The neutral PSf membrane component affords mechanical strength to the embedded TP-aligned conducting channels. This new type of microporous PBA framework is highly hydrophilic and proton conductive, with micropores of ∼5.4 Å diameter, which act as nano-sponges to absorb only more retentive non-freezable water, effective for proton conduction. These nano-sponges display efficient water absorption and retention at low RH and elevated temperatures, together with a much faster hydration process than the dehydration process. Furthermore, the TP-aligned PBA channels also enable faster water transport to promote PEM proton conduction beyond any previously reported water-retentive membrane. Consequently, the novel nano-sponge-like PEMs exhibit remarkable performance in both ex situ and in situ evaluations, especially at low RH and elevated temperature, largely prevailing over the commercial benchmark Nafion® 212.
Broader contextOperational feasibility at low relative humidity and elevated temperature is one of the most pivotal issues to further effectuate the commercialization of proton exchange membrane fuel cells (PEMFCs). As the central component of PEMFC, current proton exchange membranes (PEMs) (such as state-of-the-art Nafion®) typically display severe membrane dehydration at low relative humidity and elevated temperature, causing a significantly decline in power output. Therefore, the regulation of water content in PEMs is of great importance. Current membrane humidification employs external thermal and water management systems, which increase manufacturing expenditure, cause parasitic power loss, and add both mass and size of PEMFCs, thereby restricting their application under practical variable environments. A major challenge at present is to develop alternative strategies other than external humidification, to intrinsically enhance water retention and proton transport in PEMs. A sponge is an example of an effective material allowing significant absorption and retention of water by strong capillary forces. Analogously, a sponge-like PEM with right-sized hydrophilic micropores may promote membrane hydration without external humidification, thereby providing high PEMFC performance in a hot and dry operational environment. |
Introduction
Maintaining sufficient hydration in proton exchange membranes (PEMs), especially at low relative humidity (RH) and elevated temperature, is indispensable to ensure productive power output in proton exchange membrane fuel cells (PEMFCs).1–3 For current PEMFCs, water is generally afforded and governed by external thermal and water management systems,4–6 thus complicating the manufacture and increasing the overall weight and volume, in addition to consuming some of the power generated by the PEMFC. Recently, a new strategy of coating PEM surfaces with nano-cracked hydrophobic layers was reported, whereby the PEMFC shows high power density at low RH and elevated temperature.7 While the hydrophobic barrier layer acts as a regulator to modulate water loss at elevated temperatures, the exterior nano-valve coating does not enhance the intrinsic conductivity of the PEM and may impede the rate of water sorption. To circumvent this, imparting intrinsic water retention within the PEM is an intuitively promising alternative.Several approaches to enhance the intrinsic water retention of PEMs have been exploited. Although membrane water retention is often improved, significant gains in both membrane proton conductivity and fuel cell output, as practical targets toward PEMFC application, remain elusive. The reasons for this vary according to each individual approach. The most widely used strategy is the fabrication of composite PEMs with hydrophilic inorganic fillers, such as silica,8,9 titania,9–12 zirconia,9,12 ceria,13,14 montmorillonite,15,16 halloysite,17,18 zeolite,19 palygorskite,20 cellulose21 and core–shell type or supporting type hybrid oxides.22–24 The incorporation of Pt-based catalytic filler is another approach.25 While these materials acquire or generate additional water within and around them, the majority of fillers are often proton nonconductive, so they may not exhibit the anticipated performance gains because this water is not closely associated with the proton-conducting components (PCs). Another approach incorporates water-sorptive polymeric microcapsules26–28 or hollow spheres29,30 into PEMs. However, these blend membrane modifications mainly increase the amount of freezable water (also known as free water), which is less retentive than non-freezable water (also known as bound water), especially at low RH and elevated temperature.31,32 Other approaches are polymer structure design33,34 or solvent assistance35,36 to facilitate increased water uptake, but such methods typically lead to a high proportion of freezable water in the enlarged hydrophilic domains of the PEMs, which are more prone to loss of water and proton conductivity at low RH and elevated temperature.
Apart from the focus on simply increasing water sorption in previous studies, further approaches for water retentive highly conductive PEMs may be promising to achieve progress on two other aspects. First, hydrophilic domains should overlap with proton conductive regions, thereby lessening non-participatory water for proton transport. This may be accomplished by avoiding the use of hydrophilic but nonconductive filler, or by the introduction of both hydrophilic and proton conductive fillers. Second, PEMs containing larger amounts of non-freezable water have much better water retention at low RH and elevated temperatures, which should improve proton conductivity and PEMFC performance. This could be achieved in PEMs with small hydrophilic domains having a size slightly larger than the diameter of a water molecule (2.8 Å), which would create a strong capillarity effect for water absorption and retention,37–39 and a shorter distance than that from the edge of the first hydration shell to PCs (6–6.5 Å),40–42 ensuring all water molecules are in close proximity and interact with the PCs (i.e. the more retentive non-freezable water). Prussian blue analogues (PBAs)43,44 have the potential to achieve this. PBAs are a family of metal–organic frameworks constructed by hexa-coordinated metal cations M1 and M2, which are bridged via cyano ligands (M1–CN–M2) in a face-centered cubic arrangement. With different species and valences of M1 and M2, the length of a semi cubic edge M1–CN–M2 will change slightly. The diameter of PBA micropores are in the range of ∼2.8 Å to 6 Å. PBAs can be broadly categorized into water-soluble types (containing additional alkaline metal ions) and water-insoluble types (not containing additional alkaline metal ions).45,46 The water-soluble ones are obviously impractical for improving the water retention of PEMs, while the water-insoluble ones exhibit very low water uptake due to the framework hydrophobicity, which prevent ingress of water. Besides, PBAs are not highly proton conductive due to their hydrophobicity and the absence of PCs. Even with optimization, proton conductivity at 20 °C is only ∼1 mS cm−1 at 100% RH, and it steeply declines at low RH.47 Thus, hydrophilic, but water-insoluble PBA proton conductors are needed.
Here, a new approach aimed at the improvement of PEM water retention is investigated. Phosphotungstic acid (PWA), a hydrophilic and strongly proton conductive material, is selected as the membrane filler. PWA, ferrocyano-coordinated poly(4-vinylpyridine) (CP4VP, a newly synthesized polymer, also both hydrophilic and proton conductive) and polysulfone (PSf, a tough matrix to afford good mechanical strength) are co-cast to fabricate composite membranes under a 35 tesla strong magnetic field. In our first communication, we reported the membrane formulation and magnetic assisted fabrication strategy.48 The preliminary primary findings of that study were the aligned proton channels, high PWA retention and prominent in situ membrane durability. After further in-depth experimental investigations, some important new findings are now reported in the present study. During membrane casting in a 35 tesla strong magnetic field, a water insoluble but highly hydrophilic and proton conductive Fe–CN–W PBA regular cubic framework is formed by PWA and CP4VP. The Fe–CN–W PBA framework is found to have ∼5.4 Å hydrophilic micropores, which act as a water-absorbing/retaining nano-sponge. The formation of a regular microporous hydrophilic PBA framework intrinsic to the aligned proton conductor channels and its water retention and fuel cell properties are the significant findings beyond our preliminary work, leading to a new water retention perspective and approach. Since the magnetically-induced formation of Fe–CN–W PBA becomes paramagnetic, it is simultaneously oriented by the magnetic field during membrane casting. Thus, the resulting composite membranes, in which through-plane (TP) aligned water/proton channels are embedded, have high TP-directional anisotropic proton conductivity, which is more advantageous for PEMFC configuration than in-plane (IP) proton conductivity. In addition, due to the water-insolubility of the PBA, this fabricating strategy concurrently circumvents the widespread problem of PWA leaching out from its composite PEMs,49,50 ensuring durable membrane properties. With these simultaneous concomitant effects, the PEMFCs based on these composite membranes exceed the power density and durability of commercial Nafion® 212 PEMFCs and previously reported water retentive PEMs.
Experimental
A list of the abbreviations is provided as Supplementary Note 1 (ESI†).Materials
Poly(4-vinylpyridine) (P4VP, Mw ∼ 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), PWA (99.9%) and 1,4,7,10,13-pentaoxacyclopentadecane (15-crown-5, 98%) were purchased from Sigma Aldrich, China. Sodium pentacyanoammineferroate(II) (SPCAF, 95%) was purchased from Tokyo Chemical Industry, Japan. PSf (Mw ∼ 60000) was obtained from Acros, China. Reagent grade methanol (99%), isopropanol (99%), dimethylformamide (DMF) (99%), benzene (99%) and aqueous HCl (1 M) were obtained from Aladdin, China. All the materials were used as received.
000), PWA (99.9%) and 1,4,7,10,13-pentaoxacyclopentadecane (15-crown-5, 98%) were purchased from Sigma Aldrich, China. Sodium pentacyanoammineferroate(II) (SPCAF, 95%) was purchased from Tokyo Chemical Industry, Japan. PSf (Mw ∼ 60000) was obtained from Acros, China. Reagent grade methanol (99%), isopropanol (99%), dimethylformamide (DMF) (99%), benzene (99%) and aqueous HCl (1 M) were obtained from Aladdin, China. All the materials were used as received.
Synthesis of CP4VP
A solution of SPCAF (10.9 g, 40 mmol) and 15-crown-5 (26.4 g, 120 mmol) in water (50 mL) was added dropwise to a magnetically stirred solution of P4VP (1.05 g, 10 mmol) in methanol (50 mL). The mixed liquid was sealed and stirred for 72 h at 40 °C. The solution was allowed to cool to room temperature, filtered through a syringe filter (0.45 μm polytetrafluoroethylene (PTFE)), mixed with 100 mL of 1 M HCl in an ice bath, and then very slowly decanted into isopropanol to precipitate CP4VP. The crude polymer product was re-dissolved in 100 mL of 1 M HCl in an ice bath, stirred for 1 h and reprecipitated in isopropanol. This procedure was carried out three times to obtain purified fully acidified polymer. After being dried at room temperature for 24 h in a vacuum oven, the resulting green-yellow colored CP4VP polymer was stored in a desiccator.Fabrication of composite membrane
For membrane fabrication, CP4VP and PWA (1:3 w/w, as the optimized ratio demonstrated in previous work48) were used as the combined PCs, and PSf was used as an inert non-conductive matrix to afford mechanical strength. A series of membrane materials were prepared containing various PC loadings of 0 wt%, 10 wt%, 20 wt%, 30 wt% and 40 wt% in the PSf matrix, respectively, with a total material amount of 0.4 g for each membrane. After the membrane materials were introduced into vials and dissolved in 5.0 g DMF and 1.0 g water, they were rotated on a test tube gyrator (Thermo Fisher Scientific, USA) for 12 h, filtered by syringe-driven filter (0.45 μm PTFE) and then degassed to remove bubbles. The obtained solutions were decanted into circular casting dishes (5 cm diameter), which were horizontally placed with a heater in the upright well of a 35 tesla water-cooled magnet to evaporate the solvent at 80 °C for 12 h. The membranes cast with magnetic field are named magnetic-cast membranes (MMs). Another comparative series of composite membranes were prepared with the identical membrane formula and casting conditions, but in the absence of the magnet. The membranes cast without magnetic field are named normal-cast membranes (NMs). MM-xPC and NM-xPC are used to refer the MM and NM with PC loading of x wt%, respectively. The cast membranes were then immersed in 1 M HCl for 24 h, and washed repeatedly by deionized water until the pH achieved 7.0. The thicknesses of all cast membranes were obtained in the range of 50–60 μm.
Characterization
The ultraviolet-visible (UV-vis) spectrum analyses were carried out using a Lambda 750 spectrophotometer (PerkinElmer, USA). X-ray photoelectron spectroscopy (XPS) tests were implemented by an Escalab 250 (Thermo Fisher Scientific, USA) instrument which employs a 150 W monochromatic Al Kα radiation. Fourier transform infrared spectra (FTIR) were conducted on a Nicolet iS10 system (Thermo Fisher Scientific, USA) in the range of 4000–700 cm−1. X-ray diffraction (XRD) patterns were collected by a D8 Advance X-ray diffractometer (Bruker, Germany) with a Cu Kα radiation generator at the scanning rate of 10° min−1. Positron annihilation lifetime spectra (PALS) were obtained by a “fast–fast” positron lifetime spectrometer (Ortec, USA), where the stacked membrane samples (total thickness >1 mm) were immobilized on both sides of the 20 μCi 22Na source until 1 × 106 counts were detected. The Brunauer–Emmett–Teller (BET) N2 physical adsorption–desorption curves and the pore size distribution of membrane samples were measured using a BELSORP-max instrument (MicrotracBEL, Japan). Theoretical calculations for the edge length of PBA cube were conducted by molecular dynamics simulation. The vibrating sample magnetometer (VSM) tests were implemented at 80 °C by a Squid-VSM system (Quantum Design, USA). Membranes were embedded in epoxy resin and solidified at 100 °C for 2 h, and then cut on an EMUC6 ultramicrotome (Leica, Germany) for section samples, which were observed by a Tecnai G2 F20 field emission transmission electron microscope (TEM) (FEI, USA) at an accelerating voltage of 200 kV. Tungsten, iron and sulfur elements were detected by an Enfinium 977 electron energy loss spectrometer (EELS) (Gatan, USA) using mapping technology, and the mapping time was 2 s. Cross-sectional membrane samples for S4800 field emission scanning electron microscope (SEM) (Hitachi, Japan) observation were freeze-fractured in liquid nitrogen and vacuum sputtered with a thin layer of platinum. The particle size of PWA dispersed in benzene was estimated by the dynamic light scattering (DLS) technique on a Zetasizer nano ZS90 apparatus (Malvern Instruments, UK). Thermogravimetric analysis (TGA) was conducted on a Q500 system (TA Instruments, USA) under nitrogen atmosphere with a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) measurements were carried out on a Q2000 instrument (TA Instruments, USA) under nitrogen atmosphere using a heating rate of 10 °C min−1. Dynamic vapor sorption (DVS) curves were recorded on a VTI-SA+ analyzer (TA Instruments, USA). Nuclear magnetic resonance (NMR) data were collected using an Avance 400 MHz wide-bore spectrometer (Bruker, Germany) with microimaging capability, where the deuterated water equilibrated membrane was blotted to remove any surface moisture and then quickly rolled and sealed in an NMR tube. The 2H spin–lattice relaxation time (T1) and self-diffusion coefficient (Dself) of deuterated water in the hydrated membrane samples were obtained using inversion-recovery sequences51,52 and pulsed-field gradient spin-echo method,53,54 respectively.Mechanical properties
Dumbbell-shaped samples were cut from membranes by a CP-25 sheet-punching machine (Creator, China). These samples were placed in a ZP (H) 32 chamber (Cincinnati Sub-Zero, USA) at 23 °C and 50% RH for 48 h before test, according to ASTM D882. The stress–strain curves were obtained by the tensile test on a Q800 system (TA Instruments, USA), at the speed of 5 mm min−1. The gauge length and the width of the samples were 30 mm and 5 mm, respectively.Water uptake
Membranes were equilibrated in liquid water for 1 h before the measurements of water uptake. Then membranes were immediately blotted to remove the surface water and weighed to obtain the wet mass. After being dried in a vacuum oven at 150 °C for 24 h, the membranes were weighed again to obtain the dry mass. The water uptake is calculated using the following equation: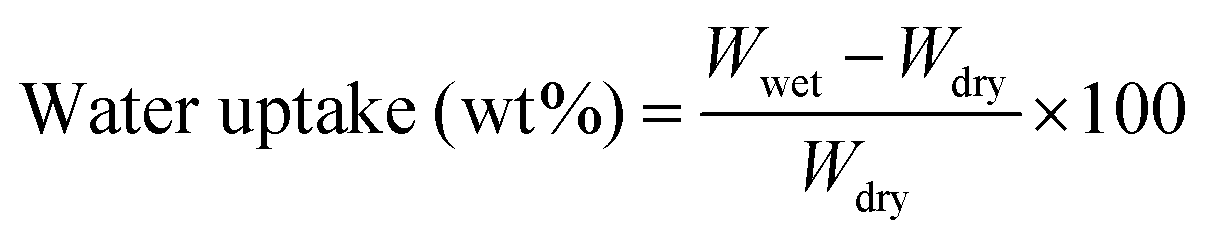 | (1) |
Proton conductivity
Membrane samples were equilibrated in liquid water or in a ZP (H) 32 chamber (Cincinnati Sub-Zero, USA) with controlled temperature and RH for 1 h. Then they were assembled in a custom built open-frame two-electrode clamp and further equilibrated for another 1 h. An autolab-PGSTAT302N electrochemical workstation (Metrohm, Switzerland) was employed for proton conductivity measurements using AC impedance technique. The membrane proton conductivity, σ, was calculated from: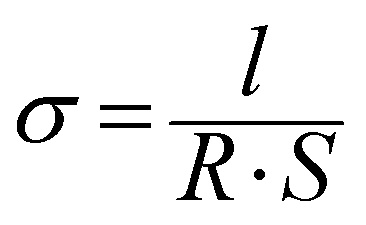 | (2) |
Membrane-electrode-assembly (MEA) preparation and single PEMFC test
Commercial Pt/C (60 wt% Pt, Johnson Matthey) was used as catalyst for both anode and cathode. The catalyst was ultrasonically dispersed in a binder (Nafion® D521 dispersion, Alfa Aesar, China) for 1 h, where the mass ratio of Nafion® to the catalyst was 20 wt%. The resultant dispersion was sprayed using an air gun (Iwata, Japan) onto carbon paper (Toray 250, Japan) to achieve 0.4 mg cm−2 catalyst loadings on both anode and cathode with an effective area of 4 cm2. MEAs were fabricated using the anode–membrane–cathode sandwich method by hot pressing under a pressure of 4 MPa at 120 °C for 3 min. Single cell PEMFC tests were conducted on a Fuel Cell Test System 850e (Scribner Associates, USA). Hydrogen and oxygen (or air) with controlled RH were supplied for anode and cathode at flow rates of 120 and 160 (or 450 for air) sccm, respectively. The polarization curves were recorded after the cells were activated at test temperature for 3 h.Results and discussion
Polymer synthesis, membrane fabrication and characterization
The synthesis mechanism of CP4VP is shown in Fig. 1. Detailed information for the synthesis and characterization of CP4VP have been elucidated elsewhere.48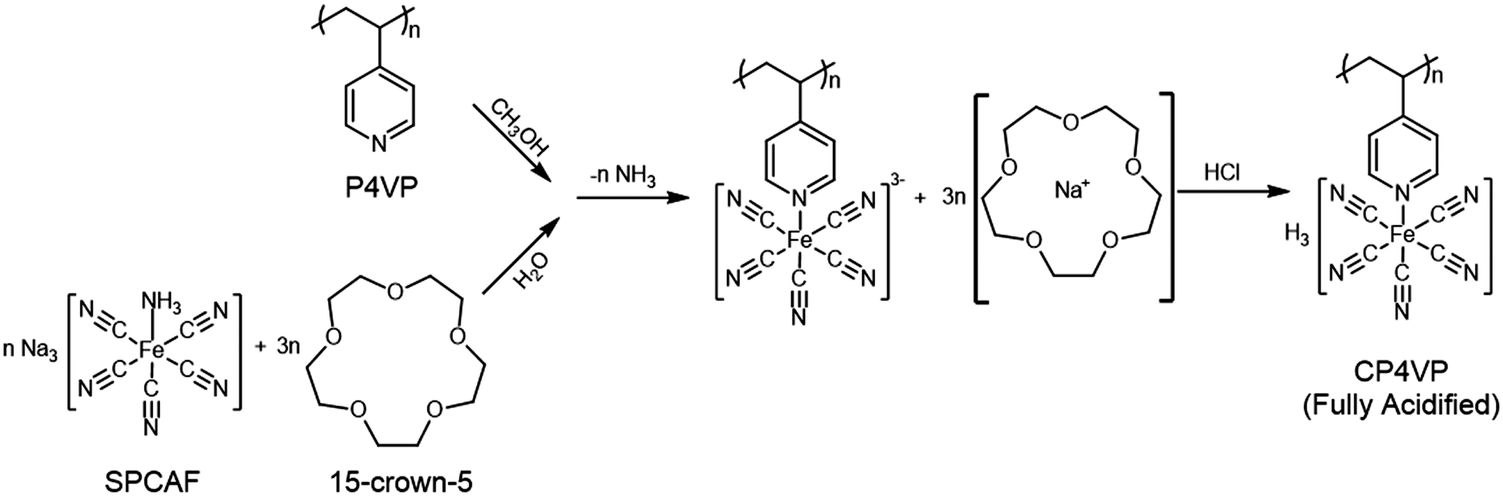 | ||
| Fig. 1 Synthesis of CP4VP from P4VP and SPCAF. After dissolution in water, a chelate is formed by the Na+ ion of SPCAF and the phase transfer catalyst 15-crown-5. Thus, SPCAF is transferred into the methanol phase to react with P4VP, forming the coordination polymer CP4VP. | ||
After membrane casting, different from NMs, MMs undergo a magnetically-induced interaction between CP4VP and PWA, and detailed characterization is provided in Supplementary Note 2 (ESI†), which includes UV-vis (Fig. S1, ESI†), XPS (Fig. S2, ESI†), FTIR (Fig. S3, ESI†) and related discussions. These results are in parallel with our previous work,48 where we explained the new interaction between CP4VP and PWA as the formation of ‘heteropoly blue’. In the present study, we have some significant new findings, which are as follows.
XRD is used to further study the interaction between CP4VP and PWA. The XRD curves of membrane components CP4VP, PWA and PSf are plotted in Fig. S4 (ESI†). CP4VP and PSf are amorphous, while PWA shows sharp crystalline peaks. Fig. 2a shows that both NM-0PC and MM-0PC have the same XRD curves as PSf, suggesting that magnetic assisted solution casting has no influence on the PSf matrix. NM-10PC and NM-20PC maintain amorphous features, but at a PC content of 30 wt% and above, NMs display some characteristic diffraction peaks consistent with bulk PWA, which is ascribed to PWA agglomeration, a problem often encountered in composite membranes with high filler loadings. In contrast, MMs with PC loadings have quite different behavior. All MMs show no peak corresponding with bulk PWA, regardless of PC loading, indicating that PWA agglomeration does not occur. Additional peaks appear with increasing trend when PC loading increases in MMs, correlating with PBA frameworks.55,56 Based on the previous evidence of the new magnetic field-induced chemical interaction between the N in the cyano ligand and the W in PWA, the PBA should be in the form of Fe–CN–W. This conclusion indicates that the N is connected to a particular W atom (i.e. a N–W covalent bond), which is different from the previous interpretation of electron delocalization on the formed heteropoly blue, hopping rapidly among all W atoms. This is the first time this type of PBA (Fe–CN–W) has been reported, and it possesses several interesting peculiarities. In the literature, W is always octa-coordinated with cyano ligands but not hexa-coordinated, and is connected with C (acts as M1 in the PBA structure template M1–CN–M2) but not N.57,58 Moreover, before the formation of the PBA framework, M1 and cyano ligand are in an exclusive coordination state, and M2 is in an individual cation state, but here the Fe(M1)-cyano ligand coordination in CP4VP is with an unusual pyridyl ligand and three additional active protons, and the W(M2) in PWA is in a chemically combined state. With such peculiarities, we conclude this PBA has numerous lacunary and dislocated lattices, as supported by the much wider and weaker XRD peaks (Fig. 2a) than normally seen.55,56 Based on the locations of these PBA peaks, together with their corresponding lattice planes labeled in Fig. 2a, a PBA lattice parameter of ∼10.8 Å is obtained by Bragg's law calculation, reflecting the length of 5.40 Å for a semi-cubic edge Fe–CN–W. This result verifies that the PBA framework in MMs creates abundant micropores with a size of 5.40 Å, just in the expectedly effective range of 2.8–6 Å for enhancing water retention.
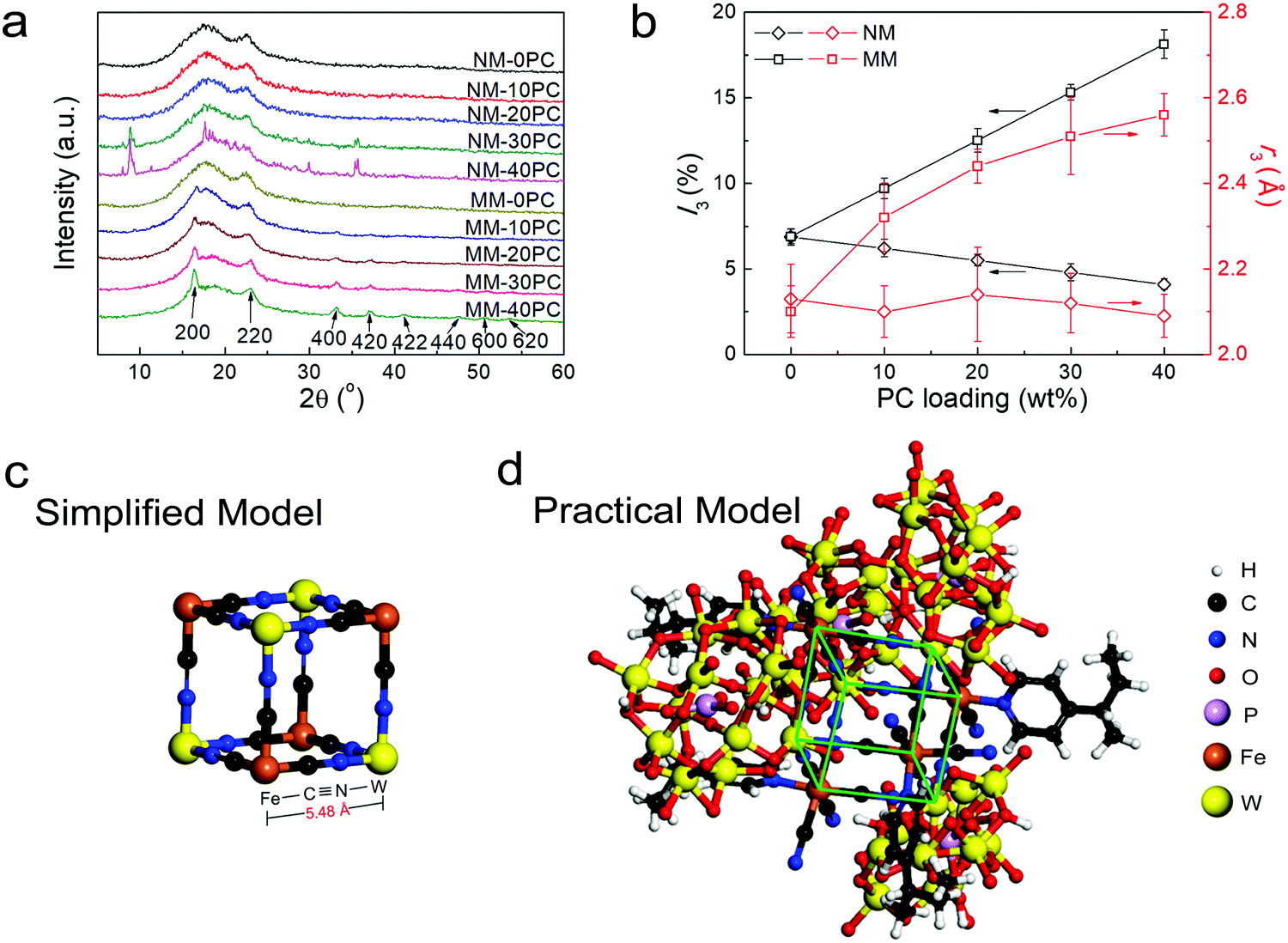 | ||
| Fig. 2 Evidence for the formation of PBA microporous frameworks in MMs. (a) XRD curves of NMs and MMs. (b) PALS measurements of NMs and MMs. (c) Simplified model for the minimum cubic unit of a Fe–CN–W PBA framework. (d) Practical model for the minimum cubic unit of a Fe–CN–W PBA framework. The good accordance between the estimated framework dimensions obtained by modeling and those obtained by experimental data provides strong evidence for the formation of Fe–CN–W PBA frameworks in MMs. | ||
While XRD analysis supports the formation of the PBA framework, the PALS technique is employed to further investigate the membrane microporosity. The results of I3 and r3, which reflect the intensities and average radii of the micropores in membrane samples, respectively, are plotted in Fig. 2b. For NMs, I3 shows a decreasing trend as PC loading increases, and a linear extrapolation to 100% PC reveals an I3 value of ∼0 for the PC, which suggests that inner pores are not formed without magnetic field in NMs. As a result, the obtained r3 is constant with various PC loading. MM-0PC and NM-0PC have similar data, suggesting that the magnetic field has no effect for the PSf matrix on the internal micropore structure. Different from NMs, the I3 values of MMs obviously increase with PC loading, which implies the micropores in MMs exist predominately in the hydrophilic domains. The r3 values in MMs also increase with PC loading, indicating that the pores in the Fe–CN–W PBA framework are larger than the pores in PSf. Based on the PALS data of MMs, an extrapolated r3 value of 2.70 Å, corresponding to the diameter of 5.40 Å, is obtained for the micropores in pure Fe–CN–W PBA framework, which is in good agreement with the lattice parameters estimated by XRD. Moreover, BET measurements further support this estimation for pore size (Fig. S5, ESI†). These results confirm the construction of micropores of a size suitable for water retention.
Theoretical calculations of the edge length of the minimum cubic unit of a Fe–CN–W PBA framework are also implemented. Fig. 2c shows a simplified cubic model containing only Fe, C, N and W atoms, which has an edge length of 5.48 Å. This value is in close agreement with the experimental value of 5.40 Å. Furthermore, a more accurate and practical model reflecting the actual structural assembly from CP4VP chains and PWA particles, is constructed in Fig. 2d (minimum cubic unit highlighted using green lines). In this practical model, the calculated value is 5.42 Å, which has a discrepancy of only 0.4% with the values obtained experimentally. The high consistency between simulation and experimental results provide strong evidence for the formation of microporous PBA in MMs.
Magnetism is another characteristic property for PBAs. With different species and valences of M1 and M2, PBAs may display various types of magnetism, such as diamagnetism, paramagnetism, ferromagnetism and antiferromagnetism.59–61 VSM measurements are used for determining magnetic transitions after the PBA formation in MMs, which verifies that the formed PBA is paramagnetic, and is simultaneously oriented by the magnetic field during membrane casting (Fig. S6, ESI†). Moreover, the driving force of the strong magnetic field appears to be central to the formation of PBA, by overcoming system entropy to attain a new system steady-state, different from that under normal conditions.62 Thus, it facilitates the transition from an originally diamagnetic membrane formulation to a paramagnetic microporous PBA framework. In fact, magnetic transitions and reactions induced by magnetic fields have also been found in other studies.63–66
Visual evidence for the isotropic and oriented structures for NMs and MMs, respectively, are provided by TEM. Since diamagnetic PSf is insensitive to magnetic field, NM-0PC and MM-0PC have a homogeneous appearance without obvious differences, as shown in Fig. S7 (ESI†). For NMs with increasing PC (Fig. 3a–d), ∼1.25 nm sized black dots are visible. Fig. 3a and b show they are randomly distributed, but with higher PC content of 30 wt% and above, agglomerates occur. The agglomeration coincides with the appearance of a bulk PWA peak in the XRD spectra. These black dots correlate with PWA particles, because the largest and heaviest element W in PWA permits the lowest electron penetration. This assertion is confirmed by DLS, where the size of PWA particles dispersed in benzene is estimated to be 1.24 nm (Fig. S8, ESI†). In contrast, Fig. 3e–h shows that MMs with PC exhibit directionally and evenly arranged PWA along the path of the magnetic field. As labeled on Fig. 3e and f, some dead-ends of the PWA chain appear in MM-10PC and MM-20PC, due to lower filler loading. Moreover, all MMs display halo-like areas around the PWA particles which are not found in NMs. Based on the preceding evidence for the formation of PBA framework (composed of CP4VP and PWA), these halo-like areas are deduced to be CP4VP domains. In the TEM images, these halo-like areas prevent PWA from agglomerating, even at high PC loading of >30 wt%, in good accordance with the XRD results. The brightest regions in the remainder of the TEM images are inferred to be PSf domains, because PSf has the lightest atoms. These inferences are verified by EELS mappings as shown in Fig. S9a–l (ESI†) (a–d for W, e–h for Fe and i–l for S, respectively, corresponding to the same areas of Fig. 3e–h). These results demonstrate that the CP4VP and PWA based paramagnetic PBA in MMs are synchronously oriented by the magnetic field during membrane casting, thus forming TP aligned hydrophilic and proton conductive nano-sponges, which are embedded in the PSf matrix. Cross-sectional SEM images of MMs are also shown in Fig. S10 (ESI†), which confirm the uniform and dense morphology of the samples.
 | ||
| Fig. 3 Cross-sectional TEM images of membranes. (a–d) NM-10PC, NM-20PC, NM-30PC and NM-40PC. (e–h) MM-10PC, MM-20PC, MM-30PC and MM-40PC. Compared with the isotropic NMs, MMs show aligned morphology along the TP direction. | ||
Properties of water in MMs
DSC tests are used to evaluate whether the microporous framework PBA structures in MMs enable high ratios of non-freezable water. Hydrated membranes equilibrated in liquid water at 40 °C are selected for the tests, the rationale being that equilibrium in liquid water provides the highest hydration levels, whereby there is more probability of the presence of less retentive freezable water. A moderate temperature of 40 °C allows the membranes to absorb considerable amounts of water, but is not expected to result in severe dehydration during sample manipulation. Thus, these conditions are appropriate to determine whether non-freezable water is dominant. In sharp contrast with Nafion® 212, the MMs show no evidence of melting peaks at 0 °C in Fig. 4a, indicating that even at the highest hydration levels, all water molecules closely interact with PC components and are highly retentive, which provides an important foundation for understanding the high water retention at low RH and elevated temperatures. Based on DSC integral calculation (Table S1, ESI†), the Nafion® 212 membrane equilibrated under the same conditions contains 55.0% of less retentive freezable water. Another evident distinction between the MMs and Nafion® 212 in Fig. 4a is the water evaporation/desorption process. The evaporation peaks of the MMs are around 150 °C, considerably higher than the boiling point of water, and the curve tails extend beyond 200 °C. In contrast, Nafion® 212 displays a much sharper water evaporation curve maximum slightly above 100 °C, and all water is desorbed before 140 °C. Thus, the retention of water in MMs is strikingly higher than that in Nafion® 212 at elevated temperatures.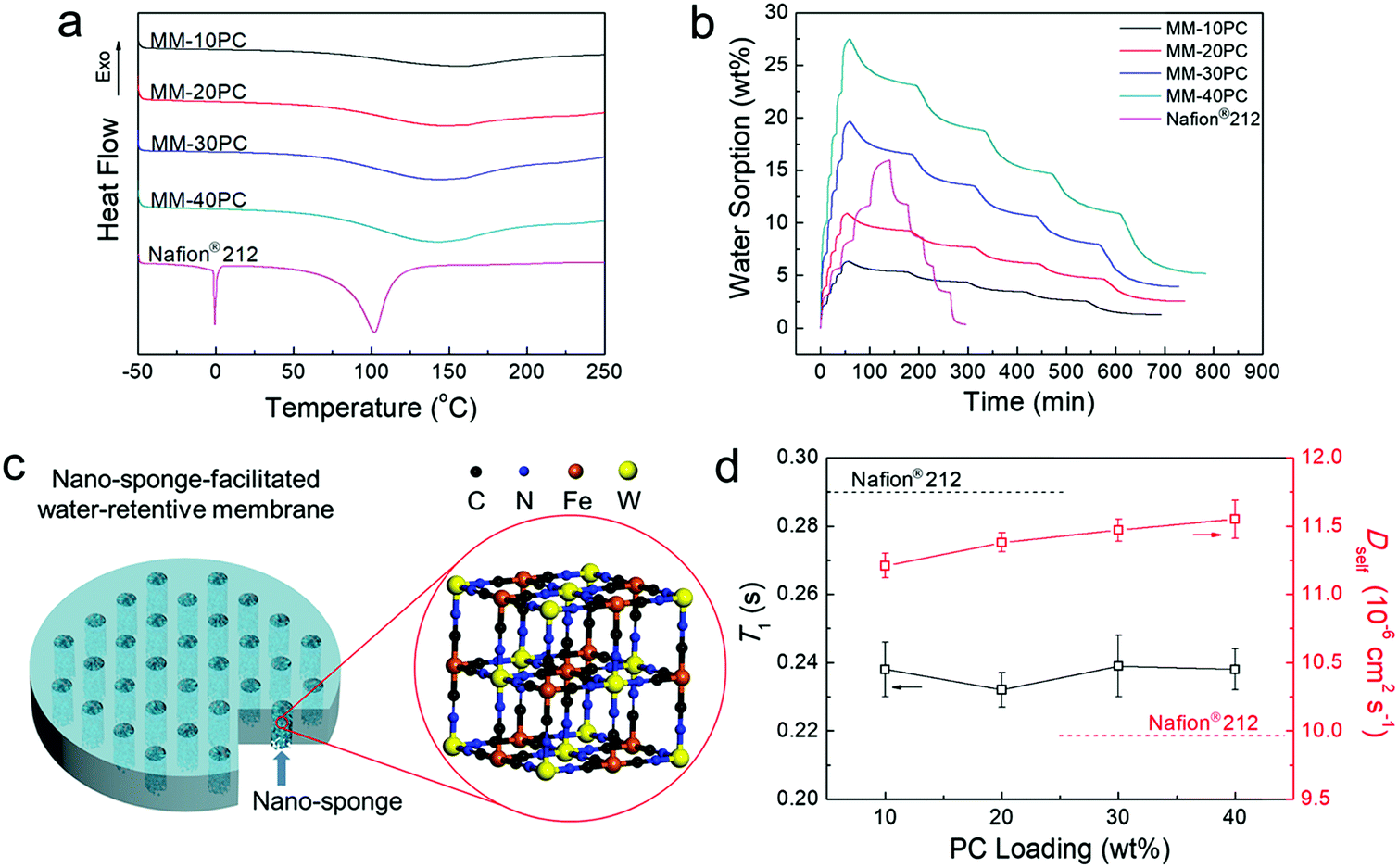 | ||
| Fig. 4 Properties of water in MMs and the diagram for nano-sponge-facilitated MM. (a) DSC curves of 40 °C liquid water equilibrated MMs and Nafion® 212. No freezable water is found in the MMs, and the water evaporation temperature is higher than that in Nafion® 212. (b) DVS curves of dehydrated MMs and Nafion® 212 at 40 °C. Compared with Nafion® 212, MMs have a much higher relative water sorption (Table S2, ESI†) at low RH, much faster sorption and much slower desorption rate (Table S3, ESI†) and much higher relative residual water (Table S4, ESI†). (c) Representation of the nano-sponge-facilitated MM. (d) NMR T1 and Dself of liquid deuterated water equilibrated MMs and Nafion® 212 at 40 °C. Since all water molecules in the MMs closely interact with the PCs, their rotational motions are slower than the water in Nafion® 212 (lower T1). However, the translational motions of the water molecules in MMs are faster because of the less tortuous TP aligned hydrophilic PBA channels (higher Dself). | ||
Water retention at low RH at 40 °C (the instrument operated at higher temperatures cannot support the entire RH range of 0–95%) on completely dehydrated membrane samples (150 °C under vacuum for 12 h) is further investigated by DVS. In Fig. 4b, MMs and Nafion® 212 display distinctly different behavior in their water sorption–desorption curves. First, MMs have moderate water uptakes in liquid water and relatively high water sorption at low RH (Table S2, ESI†). The MMs exhibit significant water sorption during the 0–20% RH sequence, which exceeds that of subsequent RH sequences, but Nafion® 212 has the highest water sorption in the 60% to 95% RH range. Second, Fig. 4b shows that the initial slopes of MMs for the sorption process are much steeper than those for the desorption process, implying that much shorter equilibrium times are needed for sorption than desorption. Table S3 (ESI†) shows that for each RH stage, the time ratios for water desorption/sorption equilibration in the MMs are approximately in the range of 11–13. In contrast, Nafion® 212 membrane displays a much more uniform and regular profile for both the initial slopes (Fig. 4b) and the equilibrium times (Table S3, ESI†) for both sorption and desorption. Third, after the sorption/desorption DVS test, the MMs still retain significant water that is not removed at 0% RH, which amounts to approximately 19–23% of that at 95% RH (Table S4, ESI†). To return to the initial completely dehydrated state before the DVS test, the MMs must be dried at over 150 °C under vacuum. In contrast, Nafion® 212 only retains a trace of water after the DVS test, and can be readily dehydrated at 60 °C in air.
The combined results of DSC and DVS indicate outstanding water retention of MMs at low RH and elevated temperature, which derives from the high hydrophilicity of the microporous PBA structure. The microporous PBA structure is composed of highly hydrophilic and proton conductive PCs (CP4VP and PWA), but it is unlike a normal PBA framework, which has analogously sized micropores that are hydrophobic. Consequently, water is readily sorbed in the PBA, and all the water is in a non-freezable form beneficial for proton transport. The hydrophilic micropore size of 5.4 Å is important to achieve a high degree of water retention. Being somewhat larger than the diameter of a water molecule (2.8 Å), it affords an effective capillarity effect for water absorption–retention.37–39 Concurrently, the generation of freezable water located outside the first hydration shell (6–6.5 Å) of the PC groups40–42 is averted, thus driving all the water molecules to interact closely with PC groups. Thus, non-freezable water is shielded from significant loss at low RH and elevated temperature. As far as we are aware, this is the first study utilizing hydrophilic micropores of a defined size to realize high water absorption and retention in PEMs at low RH at elevated temperature. Here, the hydrophilic PBA micropores with 5.4 Å size have an effectual capillary effect and act like a “sponge”. Fig. 4c is a representation of a water-retentive MM with oriented nano-sponges. In summary, equipped with these hydrophilic nano-sponges, the present MMs have the following characteristics: (a) absorption of considerable amounts of water at low RH, (b) efficient retention of water at elevated temperatures, (c) deployment of all water in sites effective for fast proton transport, and (d) a much faster hydration process than the dehydration process. Thus, the present PBA MMs intrinsically overcome some of the deficiencies in previous approaches for enhancing both internal and external water retention.
The TP orientation of the hydrophilic channels has additional advantages related to water transport. Since the strong interaction between water and PCs will obviously restrain the motion of water molecules, non-freezable water is much less mobile than freezable water, in both translational and rotational modes. Therefore, the two types of mobility of the water molecules in MMs should be mitigated, since non-freezable water is dominant in MMs compared with Nafion® 212, which contains a significant amount of freezable water.
Further analysis of the molecular mobility of water is conducted by NMR spectroscopy implemented on MM samples equilibrated in deuterated water at 40 °C. The T1s of deuterated water (reflecting rotational motion, not closely related to water transport) in MMs are lower than that of Nafion® 212 (Fig. 4d), indicating the rotational motion of the water in MMs are slower, in accordance with our expectation. In contrast, the Dself of deuterated water (reflecting translational motion, closely related to water transport) in MMs are higher than that of Nafion® 212 (Fig. 4d), indicating that the translational movement of water in MMs is faster. The promotion of self-diffusion of deuterated water in MMs compared with Nafion® 212 (∼15% improvement) is probably related to the TP aligned hydrophilic domains, where the water translational movement is less restricted than in the unoriented and tortuous hydrophilic domains of Nafion® 212. Furthermore, with increased PC loading, Dself exhibits a slightly rising trend, which we ascribe to the wider and better-connected hydrophilic domains with fewer dead-ends at higher PC loading, as observed in the TEM images (Fig. 3). Compared with Nafion® 212, although the increase in Dself (water transport) is not as marked as the results of DSC and DVS (water absorption and retention), it is an additional factor contributing to the overall improvement in the water environment in MMs. This result reveals that the aligned hydrophilic domains have not only the ostensible directional advantage of facilitating shorter translational distance, but also a hidden intrinsic benefit of enabling faster water transport.
Proton conductivity of MMs
The proton conductivities of MMs show conspicuous anisotropic behavior related to the TP-aligned proton channels in Fig. S11a (ESI†). Besides, the strong stability of proton conductivity shown in Fig. S11b (ESI†) indicates no CP4VP and PWA leach out of the MMs, due to the formation of water-insoluble PBA. Such strong PWA retention in MMs represents a considerable advance in fabrication strategy beyond typical PEM systems based on polymer–heteropoly acid interactions.67,68 While these results are similar with our previous findings,48 here we emphatically demonstrate the influence of the water-retentive microporous PBA framework on proton conductivity. In the following investigations, we focus chiefly on the TP conductivity because it is more related to the membrane configuration in PEMFC.The importance of the water-retentive PBA structure on the proton conductivities becomes more apparent in MMs at low RH and elevated temperature. The TP proton conductivities of MMs at 80 °C decline with lower RH (Fig. 5a), but the rate of decline is considerably less than commonly seen for the majority of PEMs.69,70 Even at 20% RH, the TP proton conductivity of MM-40PC is about 0.02 S cm−1. In contrast, Nafion® 212 exhibits a much sharper decline, with conductivities slightly higher than MM-20PC at RH > 80%, but ∼25% lower than MM-10PC at 20% RH. Here, it should be noted that the TP proton conductivity of Nafion® 212 is about 3 times that of MM-10PC in liquid water (Fig. S11a, ESI†), so from liquid water to 20% RH, MM-10PC has ∼4 times relative proton conductivity improvement over Nafion® 212. This phenomenon is the result of the strong water retention of MMs at low RH demonstrated in the above section, which promotes efficient proton transport. At a higher temperature of 120 °C (Fig. 5b), the TP proton conductivities of MMs are higher and decline moderately with RH as before, but Nafion® 212 shows a much steeper decline than at 80 °C, indicating that the benefit achieved by water retention on proton conductivity is even more marked.
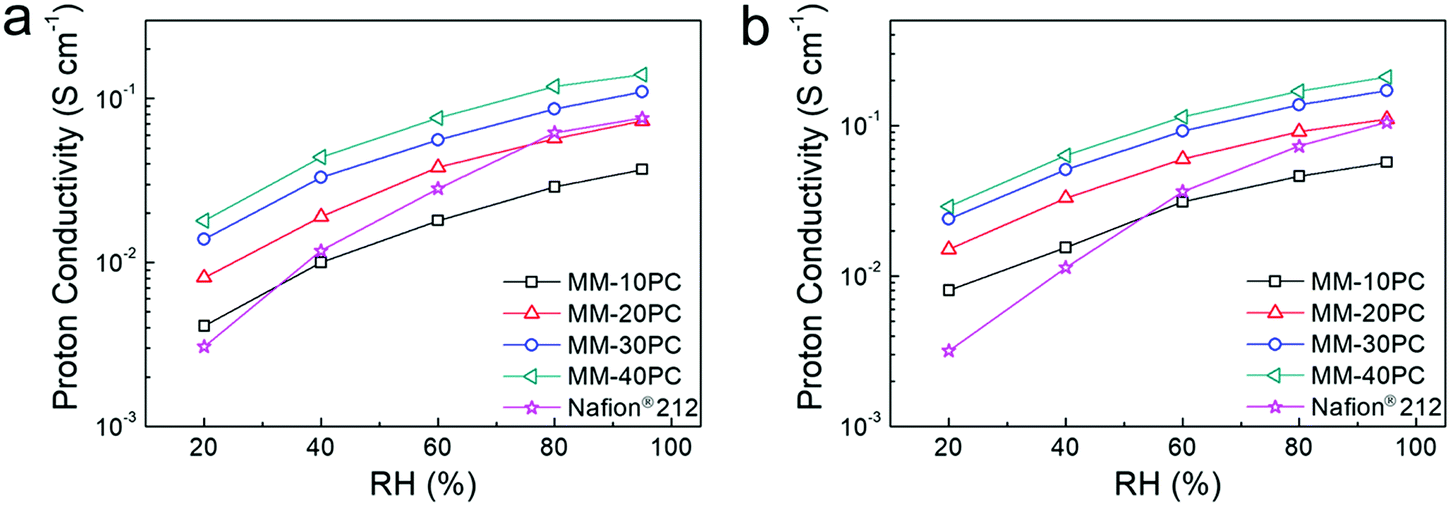 | ||
| Fig. 5 Comparative TP proton conductivities of MMs and Nafion® 212 at various RH at (a) 80 °C and (b) 120 °C. The water-retentive MMs show relatively moderate reductions in proton conductivity with RH, and the conductivities at 120 °C are universally higher than those at 80 °C. In contrast, Nafion® 212 displays a much steeper decline in proton conductivity at low RH, which is attributed to membrane dehydration. | ||
Comparative Arrhenius-type plots show the TP proton conductivity response at 80% RH (Fig. 6a) and at 40% RH (Fig. 6b) at different temperatures for MMs and Nafion® 212. Both plots for MMs show linearly increasing slopes of TP proton conductivities with temperature. In contrast, the slopes for Nafion® 212 exhibit inflections at 80 °C at 80% RH, and 60 °C at 40% RH, with negligible increases in conductivity thereafter. The efficacy of water retention in the sponge-like PBA structure of MMs is more apparent at 40% RH when compared to Nafion® 212. The perfluorocarbon hydrophobic structure of Nafion® 212 incurs severe dehydration at low RH and elevated temperature. In addition, molecular relaxation of Nafion® 212 could be another reason for the Vogel–Tamman–Fulcher type of conductivity increase.71,72 Nevertheless, the water-retentive and stiff PBA framework without segmental/phase motion in MMs may circumvent these two defects and maintain Arrhenius type increases in proton conductivity with temperature. The activation energies (Eas) at 80% RH and at 40% RH calculated from the conductivity slopes are listed in Table S5 (ESI†). The Eas of MMs at both 80% RH and 40% RH are significantly lower than those of Nafion® 212 below the inflection temperatures, which is attributed to the more effectual proton transport in the less tortuous TP orientated proton channels in MMs. It is noteworthy that MM-40PC (40 wt% PBA component) has a high TP proton conductivity of ∼0.049 S cm−1 at 20 °C and 80% RH. Extrapolating to 100 wt% PBA would give a conductivity of around 2.5 times that, which is over two orders of magnitude higher than any reported value of PBA, suggesting their merit in the design of new classes of PEMs.
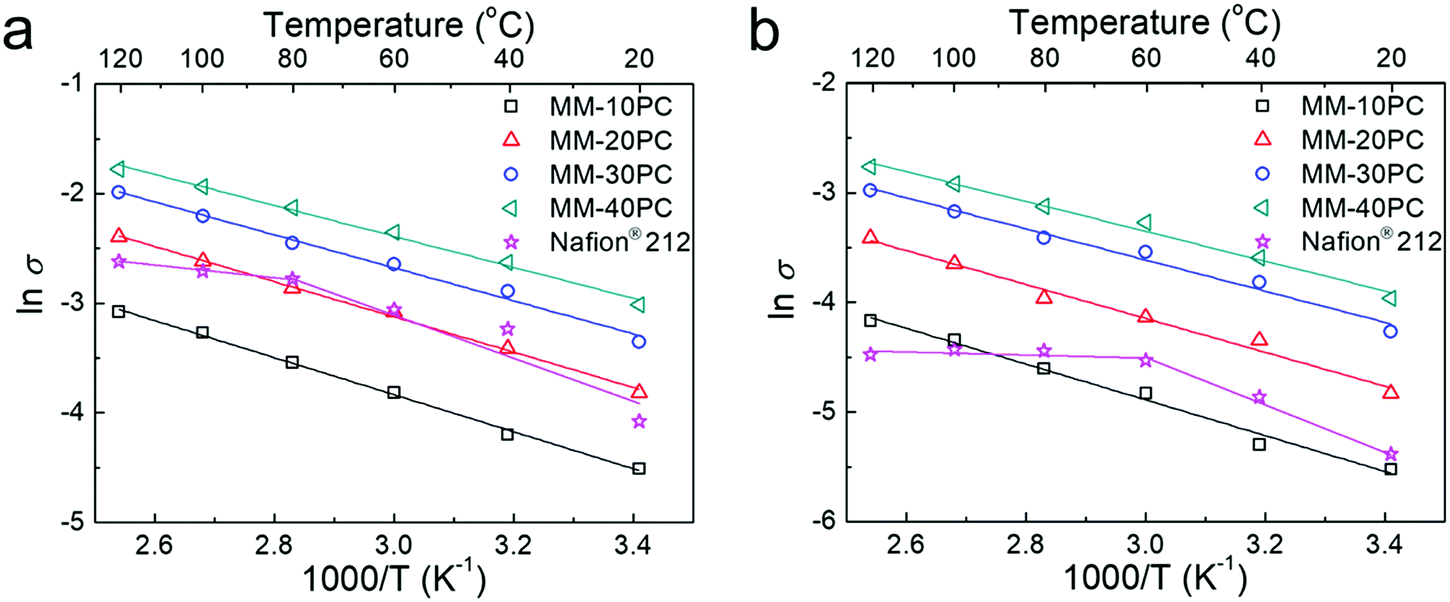 | ||
| Fig. 6 Comparative Arrhenius-type plots of TP proton conductivity of MMs and Nafion® 212 at various temperature at (a) 80% RH and (b) 40% RH. MMs show linearly increasing slopes of TP proton conductivities with temperature, while the slopes for Nafion® 212 exhibit inflections at 80 °C at 80% RH, and 60 °C at 40% RH, indicating that membrane dehydration occurs. | ||
Single PEMFC performance
Fig. 7 compares the polarization curves and derived power densities of PEMFCs (H2/O2 feed gases) using MMs and Nafion® 212 under different conditions, which clearly illustrates the performance improvements from TP orientation and water retention. The PEMFC assembled with MM-40PC has a maximum power density of 975 mW cm−2 when operated at 80 °C and 80% RH (Fig. 7a and c), which is ∼180% that of Nafion® 212. Under harsher conditions of 40% RH at 120 °C (Fig. 7b and d), the PEMFC using MM-40PC maintains a good maximum power density of 538 mW cm−2, in contrast with Nafion® 212, which shows a steep decline to 119 mW cm−2 due to membrane dehydration and consequential drop in proton conductivity. Associated data for PEMFC performance based on the other MMs are listed in Table S6 (ESI†). When the operation conditions change from 80% RH/80 °C to the harsher condition of 40% RH/120 °C, about ∼60% of the relative maximum power density is retained, much higher than that of Nafion® 212 (∼22%).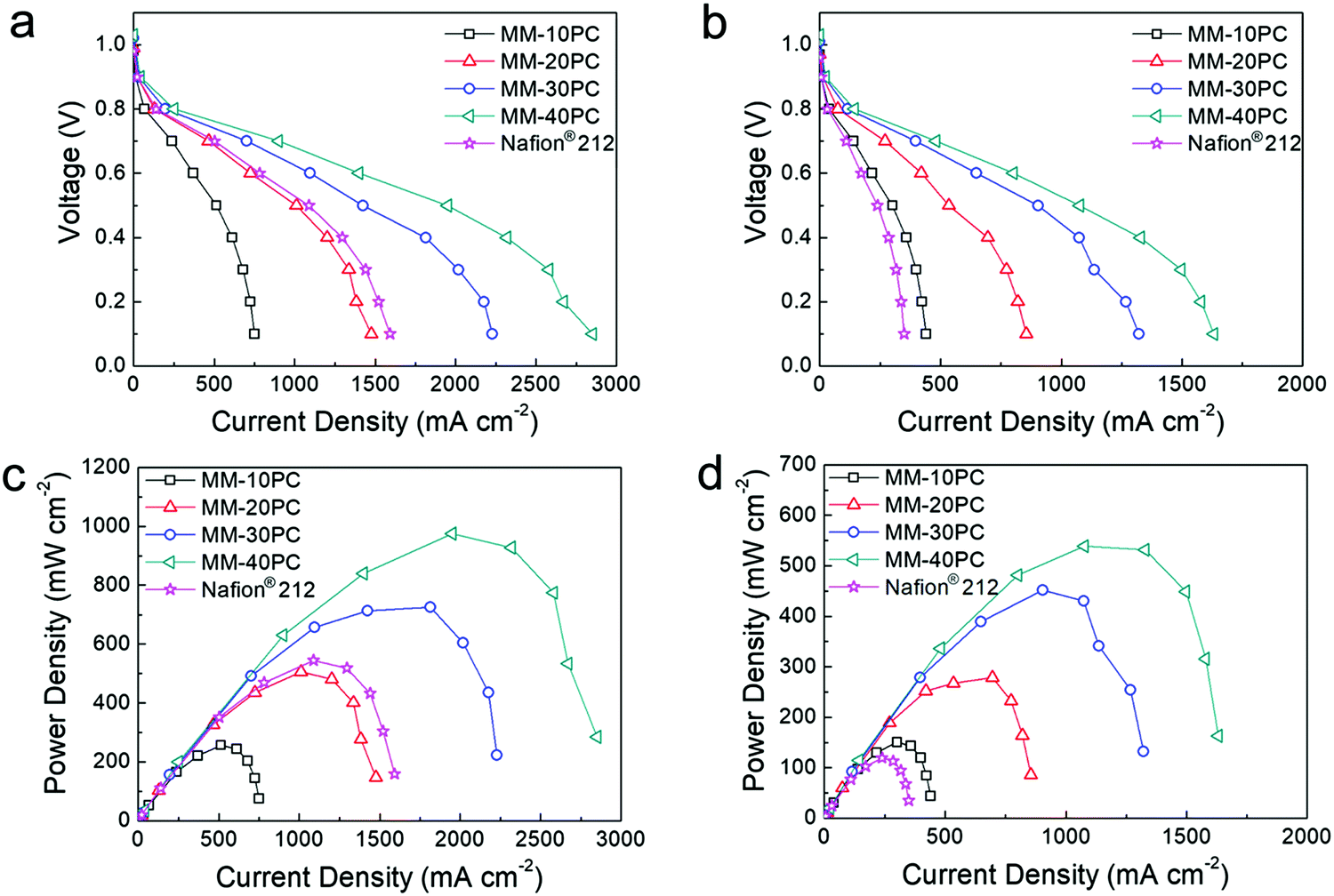 | ||
| Fig. 7 Comparative H2/O2 fuel cell performance of MMs and Nafion® 212 based PEMFCs. Polarization curves (a) at 80% RH/80 °C and (b) at 40% RH/120 °C. Power density curves (c) at 80% RH/80 °C and (d) at 40% RH/120 °C. MM-30PC and MM-40PC exhibit better performances than Nafion® 212 at 80% RH and 80 °C. Under more dehydrating conditions of 40% RH and 120 °C, all the MMs outperform Nafion® 212. The Pt/C loadings on anode and cathode are both 0.4 mg cm−2 (Pt loading 0.24 mg cm−2), and H2 and O2 are supplied at the flow rates of 120 and 160 sccm, respectively (standard atmospheric pressure without back pressure). | ||
For the PEMFC test using H2/air, the air is normally supplied at a higher flow rate than O2 to account for the lower density of oxygen in air. In preliminary experiments at 80% RH and 80 °C, we found that 450 sccm is an appropriate air flow rate to achieve ∼75% the PEMFC power output of that using 160 sccm O2. In Fig. S12a and c (ESI†), MMs and Nafion® 212 PEMFCs exhibit no evident relative performance differences, with both having ∼75% current and power densities of those in Fig. 7a and c, as assembled using the same gas diffusion and catalyst systems. However, at operating conditions of 40% RH and 120 °C, there is significant differentiation between the PEMFC performance based on MMs and Nafion® 212 (Fig. S12b and d, ESI†). MM-based PEMFCs still display identical performance ratios as they do in H2/O2 operation (∼75% of those in Fig. 7b and d), but the Nafion® 212 based PEMFC presents much lower relative remaining power (∼49% of that in Fig. 7b and d), probably owing to additional system dehydration caused by the high flow rate of dry and hot air. Such results demonstrate that the water-retentive MMs possess an additional performance advantage in high flow H2/air feed PEMFC operation at low RH and elevated temperatures.
Constant voltage durability tests are carried out at 0.7 V over a forty-day period to estimate the PEMFC longevity, where H2 and O2 are selected as the supplied gases since they are more aggressive conditions and critical to check PEMFC degradation than H2 and air. Fig. 8 shows that, under the two operation conditions of 80% RH/80 °C (Fig. 8a) and 40% RH/120 °C (Fig. 8b), MM based PEMFCs exhibit durable current density; MM-10PC, MM-20PC and MM-30PC show almost no reduction in current density, while MM-40PC has slight declines of 3.8% (80% RH/80 °C) and 4.5% (40% RH/120 °C) after 40 d, respectively. This decline is most likely related to the comparatively poorer mechanical properties of MM-40PC (Fig. S13, ESI†). TGA measurements further confirm that no structure decomposition of MMs occurs below 300 °C (Fig. S14, ESI†). This robust in situ durability shows considerable potential to achieve the U.S. Department of Energy (DOE) proposed technical target of PEMFC operation for 500 h at 120 °C without external humidification.73–75 In contrast, the Nafion® 212 based PEMFCs display much inferior durability, with decrements of ∼39.2% (80% RH/80 °C) and ∼55.4% (40% RH/120 °C) occurring within 20 d. From these advantages in power density and durability, the MMs fabricated here appear to be highly desirable for PEMFC application.
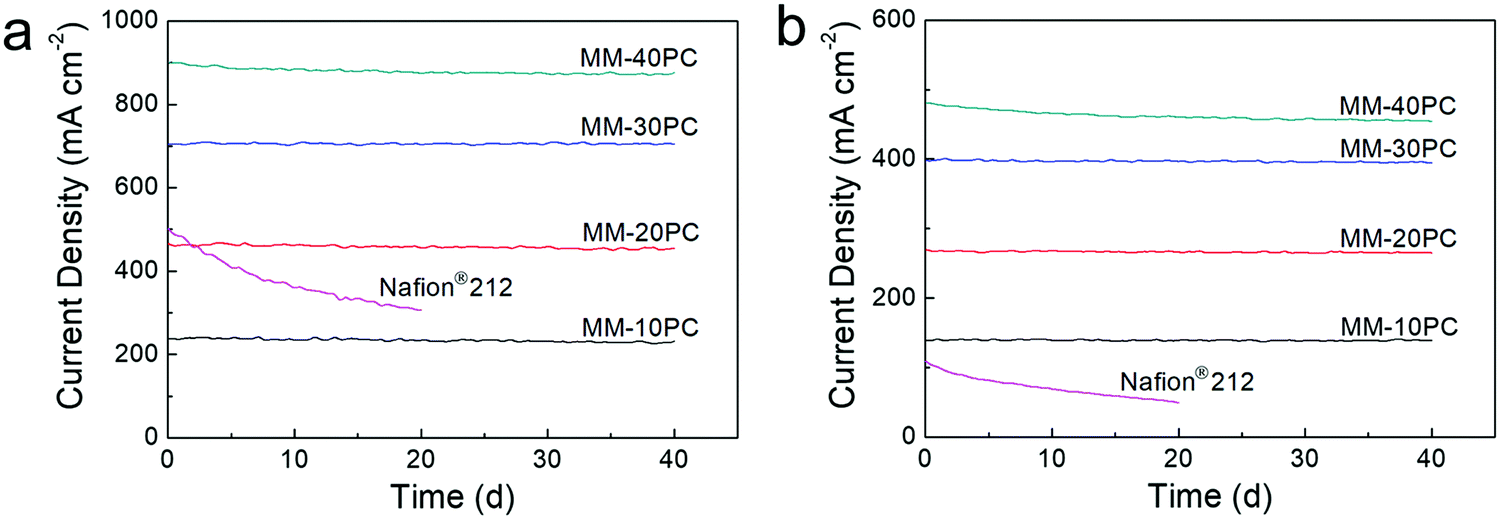 | ||
| Fig. 8 Constant voltage durability tests (0.7 V) of PEMFCs based on MMs and Nafion® 212. (a) 80% RH/80 °C. (b) 40% RH/120 °C. The MMs display robust in situ durability compared with Nafion® 212, showing considerable potential for long-term PEMFC application at low RH and elevated temperature, making the technical target of PEMFC operation for 500 h at 120 °C without external humidification accessible, set by the U.S. DOE. The Pt/C loadings on anode and cathode are both 0.4 mg cm−2 (Pt loading 0.24 mg cm−2), and H2 and O2 are supplied at the flow rates of 120 and 160 sccm, respectively (standard atmospheric pressure without back pressure). | ||
Conclusions
This is the first study to design and exploit hydrophilic micropores of a size that ensures all absorbed water molecules in PEM are located at the first hydration shell of PCs (i.e. the non-freezable water which strongly interacts with the PCs). Such approach effectively avoids the presence of less retentive freezable water, thus realizing high water absorption and retention in PEM at low RH conditions and elevated temperature. This size design approach is accomplished via the magnetically induced construction of a Fe–CN–W PBA framework from two hydrophilic proton conductive components, CP4VP and PWA, during membrane casting. The sponge-like framework contains abundant ∼5.4 Å sized hydrophilic micropores within the hydrophilic PBA domains. A strong water capillary effect in these right-sized hydrophilic micropores enable efficient water absorption, strong water retention, absence of freezable water and fast water and proton transport. Apart from its role in constructing the framework, the magnetic field induces paramagnetic properties, allowing the proton conducting Fe–CN–W PBA to simultaneously orientate in the TP direction during membrane casting. The resulting TP aligned hydrophilic and proton-conducting channels in MMs facilitate high TP proton conductivity and PEMFC output, even under conditions of low RH and elevated temperature. Additional details concerning the formation of PBA under magnetic field and the influence by polymer matrix and solvent during casting are provided in Supplementary Note 3 and 4 (ESI†). To clearly show the advantages of the nano-sponge membrane over previously reported water-retentive PEMs, a detailed comparison is listed in Table S7 (ESI†).
Beyond our initial work and interpretation of MMs with TP-orientated conducting structures reported previously,48 here we focus more on the aspects related with PBA structure and water retention. The important new discoveries are as follows: (a) construction of hydrophilic micropores with designed size (i.e. the Fe–CN–W PBA framework) in PEM, (b) strong water–PC interaction and 100% non-freezable water ratio associated with these hydrophilic PBA micropores, (c) high water absorption and retention obtained by a strong capillary effect in the hydrophilic PBA micropores, (d) faster water transport by less tortuous TP aligned PBA channels, (e) highly efficient proton transport via the synergistic aligned proton-conducting channels and strong water absorption, retention and transport, (f) much higher H2/O2 fuel cell performance and in situ durability than the industry standard Nafion® 212 membrane, especially at low RH and elevated temperature, coming close to an important technical target of the U.S. DOE and (g) additional water-retentive properties and higher power output in H2/air fuel cell operation with high air flow rate at low RH and elevated temperature operating conditions. The absence of freezable water may also be beneficial for PEMFC operations under ‘cold-start’ conditions.
Author contributions
M. D. G. conceived the study. M. D. G. and X. L. designed the experiments. M. D. G., Y. Y., J. Z. and X. L. wrote the manuscript. X. L., J. X., C. Z. and T. H. carried out the experiments and collected the data. X. L. and C. Z. implemented the molecular dynamics simulation work and prepared the data graphs. M. D. G., Y. Y., J. Z., X. L. and C. Z. contributed to the sketches. M. D. G., Y. Y., J. Z., Y. Q., K. J., Q. D. and X. L. discussed the results. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (21875161). The authors also thank the National Key Technology R&D Program (2018YFB0105601), the Natural Science Foundation of Tianjin (17JCZDJC31000), State Key Laboratory of Engines, and State Key Laboratory of Separation Membranes and Membrane Processes (M1-201704) for financial support.References
- H. Zhang and P. K. Shen, Chem. Soc. Rev., 2012, 41, 2382–2394 RSC.
- R. Devanathan, Energy Environ. Sci., 2008, 1, 101–119 RSC.
- Y. Hu, X. Li, L. Yan and B. Yue, Fuel Cells, 2017, 17, 3–17 CrossRef CAS.
- Z. Liu, J. Shen, H. Pei, Z. Tu, J. Wang, Z. Wan and W. Liu, Int. J. Energy Res., 2015, 39, 504–515 CrossRef CAS.
- T.-F. Cao, H. Lin, L. Chen, Y.-L. He and W.-Q. Tao, Appl. Energy, 2013, 112, 1115–1125 CrossRef CAS.
- S. Deabate, G. Gebel, P. Huguet, A. Morin and G. Pourcelly, Energy Environ. Sci., 2012, 5, 8824–8847 RSC.
- C. H. Park, S. Y. Lee, D. S. Hwang, D. W. Shin, D. H. Cho, K. H. Lee, T. W. Kim, T. W. Kim, M. Lee, D. S. Kim, C. M. Doherty, A. W. Thornton, A. J. Hill, M. D. Guiver and Y. M. Lee, Nature, 2016, 532, 480–483 CrossRef PubMed.
- Z. Jie, T. Haolin and P. Mu, J. Membr. Sci., 2008, 312, 41–47 CrossRef.
- N. H. Jalani, K. Dunn and R. Datta, Electrochim. Acta, 2005, 51, 553–560 CrossRef CAS.
- B. R. Matos, E. I. Santiago, J. F. Q. Rey, A. S. Ferlauto, E. Traversa, M. Linardi and F. C. Fonseca, J. Power Sources, 2011, 196, 1061–1068 CrossRef CAS.
- M. Lei, Y. J. Wang, C. Liang, K. Huang, C. X. Ye, W. J. Wang, S. F. Jin, R. Zhang, D. Y. Fan, H. J. Yang and Y. G. Wang, J. Power Sources, 2014, 246, 762–766 CrossRef CAS.
- G. Mohammadi, M. Jahanshahi and A. Rahimpour, Int. J. Hydrogen Energy, 2013, 38, 9387–9394 CrossRef CAS.
- Z. Wang, H. Tang, H. Zhang, M. Lei, R. Chen, P. Xiao and M. Pan, J. Membr. Sci., 2012, 421–422, 201–210 CrossRef CAS.
- K. Ketpang, K. Lee and S. Shanmugam, ACS Appl. Mater. Interfaces, 2014, 6, 16734–16744 CrossRef CAS PubMed.
- D. Xing, G. He, Z. Hou, P. Ming and S. Song, Int. J. Hydrogen Energy, 2011, 36, 2177–2183 CrossRef CAS.
- T. Fu, Z. Cui, S. Zhong, Y. Shi, C. Zhao, G. Zhang, K. Shao, H. Na and W. Xing, J. Power Sources, 2008, 185, 32–39 CrossRef CAS.
- J. Zhu, N. Guo, Y. Zhang, L. Yu and J. Liu, J. Membr. Sci., 2014, 465, 91–99 CrossRef CAS.
- X. Liu, S. He, G. Song, H. Jia, Z. Shi, S. Liu, L. Zhang, J. Lin and S. Nazarenko, J. Membr. Sci., 2016, 504, 206–219 CrossRef CAS.
- Y. Devrim and A. Albostan, Int. J. Hydrogen Energy, 2015, 40, 15328–15335 CrossRef CAS.
- F. Xu, S. Mu and M. Pan, J. Membr. Sci., 2011, 377, 134–140 CrossRef CAS.
- X. Xu, R. Li, C. Tang, H. Wang, X. Zhuang, Y. Liu, W. Kang and L. Shi, Carbohydr. Polym., 2018, 184, 299–306 CrossRef CAS PubMed.
- V. Di Noto, N. Boaretto, E. Negro, G. A. Giffin, S. Lavina and S. Polizzi, Int. J. Hydrogen Energy, 2012, 37, 6199–6214 CrossRef CAS.
- V. Di Noto, M. Piga, E. Negro, G. A. Giffin, S. Polizzi and T. A. Zawodzinski, RSC Adv., 2013, 3, 18960 RSC.
- C. Bi, H. Zhang, Y. Zhang, X. Zhu, Y. Ma, H. Dai and S. Xiao, J. Power Sources, 2008, 184, 197–203 CrossRef CAS.
- Y. Zhang, H. Zhang, C. Bi and X. Zhu, Electrochim. Acta, 2008, 53, 4096–4103 CrossRef CAS.
- J. Wang, H. Zhang, X. Yang, S. Jiang, W. Lv, Z. Jiang and S. Z. Qiao, Adv. Funct. Mater., 2011, 21, 971–978 CrossRef CAS.
- J. Wang, Z. Zhang, X. Yue, L. Nie, G. He, H. Wu and Z. Jiang, J. Mater. Chem. A, 2013, 1, 2267–2277 RSC.
- G. He, Y. Li, Z. Li, L. Nie, H. Wu, X. Yang, Y. Zhao and Z. Jiang, J. Power Sources, 2014, 248, 951–961 CrossRef CAS.
- E. Yan, J. Wang, Z. Jiang, H. Feng, L. Nie, T. Xu, X. Yang and X. Zhang, J. Mater. Chem. A, 2013, 1, 11762 RSC.
- H. Pu, D. Wang and Z. Yang, J. Membr. Sci., 2010, 360, 123–129 CrossRef CAS.
- B. Gupta, O. Haas and G. Scherer, J. Appl. Polym. Sci., 1995, 57, 855–862 CrossRef CAS.
- S. Feng and G. A. Voth, J. Phys. Chem. B, 2011, 115, 5903–5912 CrossRef CAS PubMed.
- A. S. Badami, A. Roy, H.-S. Lee, Y. Li and J. E. McGrath, J. Membr. Sci., 2009, 328, 156–164 CrossRef CAS.
- Z. Bai, M. Durstock and T. Dang, J. Membr. Sci., 2006, 281, 508–516 CrossRef CAS.
- X. Liu, S. He, Z. Shi, L. Zhang and J. Lin, J. Membr. Sci., 2015, 492, 48–57 CrossRef CAS.
- X. Liu, S. He, S. Liu, H. Jia, L. Chen, B. Zhang, L. Zhang and J. Lin, J. Membr. Sci., 2017, 523, 163–172 CrossRef CAS.
- Y. Huo and H. Zeng, Acc. Chem. Res., 2016, 49, 922–930 CrossRef CAS PubMed.
- K. Murata, K. Mitsuoka, T. Hirai, T. Walz, P. Agre, J. B. Heymann, A. Engel and Y. Fujiyoshi, Nature, 2000, 407, 599 CrossRef CAS PubMed.
- H. Omidian, J. G. Rocca and K. Park, J. Controlled Release, 2005, 102, 3–12 CrossRef CAS PubMed.
- J. Savage, Y.-L. S. Tse and G. A. Voth, J. Phys. Chem. C, 2014, 118, 17436–17445 CrossRef CAS.
- C. Chen, Y. L. Tse, G. E. Lindberg, C. Knight and G. A. Voth, J. Am. Chem. Soc., 2016, 138, 991–1000 CrossRef CAS PubMed.
- S. J. Paddison, Annu. Rev. Mater. Res., 2003, 33, 289–319 CrossRef CAS.
- Y. You, X.-L. Wu, Y.-X. Yin and Y.-G. Guo, Energy Environ. Sci., 2014, 7, 1643–1647 RSC.
- X. Xie, M. Ye, C. Liu, P.-C. Hsu, C. S. Criddle and Y. Cui, Energy Environ. Sci., 2015, 8, 546–551 RSC.
- F. S. Hegner, J. R. Galan-Mascaros and N. Lopez, Inorg. Chem., 2016, 55, 12851–12862 CrossRef CAS PubMed.
- A. K. Vipin, B. Hu and B. Fugetsu, J. Hazard. Mater., 2013, 258–259, 93–101 CrossRef CAS PubMed.
- S.-I. Ohkoshi, K. Nakagawa, K. Tomono, K. Imoto, Y. Tsunobuchi and H. Tokoro, J. Am. Chem. Soc., 2010, 132, 6620–6621 CrossRef CAS PubMed.
- X. Liu, Y. Li, J. Xue, W. Zhu, J. Zhang, Y. Yin, Y. Qin, K. Jiao, Q. Du, B. Cheng, X. Zhuang, J. Li and M. D. Guiver, Nat. Commun., 2019, 10, 842 CrossRef PubMed.
- D. Xu, G. Zhang, N. Zhang, H. Li, Y. Zhang, K. Shao, M. Han, C. M. Lew and H. Na, J. Mater. Chem., 2010, 20, 9239–9245 RSC.
- Y. Chen and H. Kim, J. Power Sources, 2009, 190, 311–317 CrossRef CAS.
- M. Neeman, K. A. Jarrett, L. O. Sillerud and J. P. Freyer, Cancer Res., 1991, 51, 4072–4079 CAS.
- T. A. Zawodzinski, C. Derouin, S. Radzinski, R. J. Sherman, V. T. Smith, T. E. Springer and S. Gottesfeld, J. Electrochem. Soc., 1993, 140, 1041–1047 CrossRef CAS.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- J. Lin, P.-H. Wu, R. Wycisk, P. N. Pintauro and Z. Shi, Macromolecules, 2008, 41, 4284–4289 CrossRef CAS.
- J. Nai, B. Y. Guan, L. Yu and X. W. Lou, Sci. Adv., 2017, 3, e1700732 CrossRef PubMed.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2005, 127, 6506–6507 CrossRef CAS PubMed.
- D. Pinkowicz, R. Podgajny, B. Nowicka, S. Chorazy, M. Reczyński and B. Sieklucka, Inorg. Chem. Front., 2015, 2, 10–27 RSC.
- B. Nowicka, T. Korzeniak, O. Stefańczyk, D. Pinkowicz, S. Chorąży, R. Podgajny and B. Sieklucka, Coord. Chem. Rev., 2012, 256, 1946–1971 CrossRef CAS.
- E. S. Koumousi, R. Jeon Ie, Q. Gao, P. Dechambenoit, D. N. Woodruff, P. Merzeau, L. Buisson, X. Jia, D. Li, F. Volatron, C. Mathoniere and R. Clerac, J. Am. Chem. Soc., 2014, 136, 15461–15464 CrossRef CAS PubMed.
- L. Samain, F. Grandjean, G. J. Long, P. Martinetto, P. Bordet and D. Strivay, J. Phys. Chem. C, 2013, 117, 9693–9712 CrossRef CAS.
- M. Okubo, D. Asakura, Y. Mizuno, T. Kudo, H. Zhou, A. Okazawa, N. Kojima, K. Ikedo, T. Mizokawa and I. Honma, Angew. Chem., Int. Ed., 2011, 50, 6269–6273 CrossRef CAS PubMed.
- Z. Yang, J. Wei, P. Bonville and M. P. Pileni, J. Am. Chem. Soc., 2015, 137, 4487–4493 CrossRef CAS PubMed.
- S.-K. Chen, B.-W. Jou and Y.-T. Tsai, Chin. J. Phys., 2018, 56, 303–307 CrossRef CAS.
- K. Mukai and T. Inoue, Carbon, 2017, 123, 645–650 CrossRef CAS.
- H. Li, M. M. Sadiq, K. Suzuki, P. Falcaro, A. J. Hill and M. R. Hill, Chem. Mater., 2017, 29, 6186–6190 CrossRef CAS.
- M. Krajewski, Nanoscale, 2017, 9, 16511–16545 RSC.
- L. Xu, H. Han, M. Liu, J. Xu, H. Ni, H. Zhang, D. Xu and Z. Wang, RSC Adv., 2015, 5, 83320–83330 RSC.
- A. R. Motz, M.-C. Kuo, J. L. Horan, R. Yadav, S. Seifert, T. P. Pandey, S. Galioto, Y. Yang, N. V. Dale, S. J. Hamrock and A. M. Herring, Energy Environ. Sci., 2018, 11, 1499–1509 RSC.
- D. W. Shin, M. D. Guiver and Y. M. Lee, Chem. Rev., 2017, 117, 4759–4805 CrossRef CAS.
- C. H. Park, C. H. Lee, M. D. Guiver and Y. M. Lee, Prog. Polym. Sci., 2011, 36, 1443–1498 CrossRef CAS.
- V. Di Noto, S. Lavina, E. Negro, M. Vittadello, F. Conti, M. Piga and G. Pace, J. Power Sources, 2009, 187, 57–66 CrossRef CAS.
- V. Di Noto, R. Gliubizzi, E. Negro and G. Pace, J. Phys. Chem. B, 2006, 110, 24972–24986 CrossRef CAS PubMed.
- C. Houchins, G. Kleen, J. Spendelow, J. Kopasz, D. Peterson, N. Garland, D. Ho, J. Marcinkoski, K. Martin, R. Tyler and D. Papageorgopoulos, Membranes, 2012, 2, 855–878 CrossRef CAS.
- S. Martin, P. L. Garcia-Ybarra and J. L. Castillo, Appl. Energy, 2017, 205, 1012–1020 CrossRef CAS.
- S. Y. Lee, D. W. Shin, C. Wang, K. H. Lee, M. D. Guiver and Y. M. Lee, Electrochem. Commun., 2013, 31, 120–124 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee03301g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |