
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revealing electrolyte oxidation via carbonate dehydrogenation on Ni-based oxides in Li-ion batteries by in situ Fourier transform infrared spectroscopy†
Yirui
Zhang‡
 *a,
Yu
Katayama‡
bc,
Ryoichi
Tatara
b,
Livia
Giordano
ab,
Yang
Yu
d,
Dimitrios
Fraggedakis
e,
Jame Guangwen
Sun
a,
Filippo
Maglia
f,
Roland
Jung
f,
Martin Z.
Bazant
eg and
Yang
Shao-Horn
*abd
*a,
Yu
Katayama‡
bc,
Ryoichi
Tatara
b,
Livia
Giordano
ab,
Yang
Yu
d,
Dimitrios
Fraggedakis
e,
Jame Guangwen
Sun
a,
Filippo
Maglia
f,
Roland
Jung
f,
Martin Z.
Bazant
eg and
Yang
Shao-Horn
*abd
aDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: yirui@mit.edu; shaohorn@mit.edu
bResearch Laboratory of Electronics, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cDepartment of Applied Chemistry, Graduate School of Sciences and Technology for Innovation, Yamaguchi University, Ube 755-8611, Japan
dDepartment of Material Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
eDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
fBMW Group, Petuelring 130, 80788 München, Germany
gDepartment of Mathematics, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 18th November 2019
Abstract
Understanding (electro-)chemical reactions at the electrode–electrolyte interface (EEI) is crucial to promote the cycle life of lithium-ion batteries. In this study, we developed an in situ Fourier-transform infrared spectroscopy (FT-IR) method, which provided unprecedented information on the oxidation of carbonate solvents via dehydrogenation on LiNixMnyCo1−x−yO2 (NMC). While ethylene carbonate (EC) was stable against oxidation on Pt up to 4.8 VLi, unique evidence for dehydrogenation of EC on LiNi0.8Co0.1Mn0.1O2 (NMC811) at voltages as low as 3.8 VLi was revealed by in situ FT-IR measurements, which was supported by density functional theory (DFT) results. Unique dehydrogenated species from EC were observed on NMC811 surface, including dehydrogenated EC anchored on oxides, vinylene carbonate (VC) and dehydrogenated oligomers which could diffuse away from the surface. Similar dehydrogenation on NMC811 was noted for EMC-based and LP57 (1 M LiPF6 in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 EC/EMC) electrolytes. In contrast, no dehydrogenation was found for NMC111 or surface-modified NMC by coatings such as Al2O3. In addition, while the dehydrogenation of solvents was observed in 1 M electrolytes with different anions, they were not observed on NMC811 in the concentrated electrolyte (EC/EMC with 3.1 M LiPF6), indicating lithium coordination could suppress dehydrogenation. Dehydrogenation of carbonates on NMC811 accompanied with rapid growth of interfacial impedance with increasing voltage revealed by electrochemical impedance spectroscopy (EIS), while the electrode–electrolyte combinations without dehydrogenation did not show significant impedance growth. Therefore, minimizing carbonate dehydrogenation on the NMC surface by tuning electrode reactivity and electrolyte reactivity is critical to develop high-energy Li-ion batteries with long cycle life.
:7 EC/EMC) electrolytes. In contrast, no dehydrogenation was found for NMC111 or surface-modified NMC by coatings such as Al2O3. In addition, while the dehydrogenation of solvents was observed in 1 M electrolytes with different anions, they were not observed on NMC811 in the concentrated electrolyte (EC/EMC with 3.1 M LiPF6), indicating lithium coordination could suppress dehydrogenation. Dehydrogenation of carbonates on NMC811 accompanied with rapid growth of interfacial impedance with increasing voltage revealed by electrochemical impedance spectroscopy (EIS), while the electrode–electrolyte combinations without dehydrogenation did not show significant impedance growth. Therefore, minimizing carbonate dehydrogenation on the NMC surface by tuning electrode reactivity and electrolyte reactivity is critical to develop high-energy Li-ion batteries with long cycle life.
Broader contextStability of the electrode–electrolyte interface (EEI) in Li-ion batteries is critical for high energy density and long cycle life. It is well known that carbonate-based electrolytes are reduced on negative electrode materials to form solid electrolyte interphase (SEI), which is electronically insulating but lithium-ion conducting to enable reversible lithium intercalation, but their possible (electro)chemical reactions on positive electrode materials are poorly understood. Ni-Rich LiNixMnyCo1−x−yO2 (NMC) materials provide enhanced energy densities but at the expense of shorter cycle life, and recent computational studies have shown that carbonate solvent molecules can be chemically oxidized or dehydrogenated on NMC, especially with increasing Ni content. In this study, we developed an in situ FT-IR method and revealed dehydrogenation pathways of carbonate solvents in the electrolyte (EMC/EC with 1 M LiPF6 or LiClO4) on composite LiNi0.8Mn0.1Co0.1O2 (NMC811) electrodes in situ at voltages as low as 3.8 VLi, which can be suppressed by decreasing surface oxygen reactivity of Ni-based oxides through coating or decreasing carbonate activity by using concentrated electrolytes (EMC/EC with 3.1 M LiPF6). These findings highlight the importance of in situ studies to capture reaction intermediates and suggest design strategies for more stable high-energy positive electrode materials. |
1. Introduction
Understanding the (electro-)chemical reactions at the electrified interface between positive electrodes and electrolyte is crucial to develop Li-ion batteries with long cycle life and safety.1–4 Layered lithium metal oxides are the most common positive electrode materials, among which LiNixMnyCo1−x−yO2 (NMC) provides high capacities and potentially lower costs for use in electric vehicles.5–9 While increasing Ni content in NMC from LiNi1/3Co1/3Mn1/3O2 (NMC111), LiNi0.6Co0.2Mn0.2O2 (NMC622) to LiNi0.8Co0.1Mn0.1O2 (NMC811) can greatly increase initial discharge capacities,5,6,10 the capacity retention decreases during cycling,10,11 which is accompanied by earlier onset for gas (O2 and CO2) evolution.10,12 Although the mechanistic details of electrolyte reactivity and reaction pathways on Ni-rich positive electrodes13–15 are not well understood, modifying electrode surfaces by ceramic coatings such as Al2O3,16–20 creating Ni-poor surfaces through concentration gradients21–24 and introducing additives in the carbonate electrolytes25–30 can greatly increase the capacity retention of Ni-rich electrodes, such as NMC811.Multiple reaction mechanisms on carbonate electrolyte oxidation have been proposed, including nucleophilic attack reactions between oxides and carbonate molecules,31–33 electrophilic attack,34,35 and dehydrogenation reactions, including EC dissociation by breaking C–H bond,36,37 and dissociation with oxygen vacancy formation.34 However, recent density functional theory (DFT) results13 show that EC dissociation on layered Ni-rich oxides is more energetically favorable than the other processes reported. The enhanced oxide-electrolyte reactivity for Ni-rich oxides10,12,38,39 can be attributed to having more metal–oxygen covalency or more oxygen p states pinned at the Fermi level,13,40,41 where there is greater driving force for the surface oxygen to dissociate or oxidatively dehydrogenate more carbonate solvents such as EC to form surface protic species (C3O3H4 → *C3O3H3+ + *H+) and reduce transition metal ions.13,42 For example, DFT calculations have shown that the dissociation energetics of EC (∼−2.6 eV) on NMC811 is thermodynamically favorable.13 Such proposed dehydrogenation of carbonate solvents is supported by ex situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and Raman spectroscopy, where EC dissociation on charged NMC is more visible with increasing Ni content in NMC or increasing lithium de-intercalation in NMC811.13 The protic species produced by dehydrogenation such as surface hydroxyl groups, can further react with LiPF6 salt, forming lithium nickel oxyfluorides,14 PF3O,43 LixPFyOz14 and HF species,14 validated by online electrochemical mass spectrometry and X-ray photoelectron spectroscopy (XPS).14,44,45
Although ex situ techniques have provided information on oxidative products from the electrolyte that remain on oxide surfaces, soluble species and dynamic changes of surface reaction intermediates formed as a function of potential are not captured.46In situ FT-IR measurements would be ideal to probe the reactivity between organic electrolytes and the oxide surface because it is highly sensitive to covalent bonds such as C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.47 While there are a few in situ FT-IR techniques previously reported in understanding the reduction48–50 or oxidation51,52 stability of organic electrolytes in lithium-ion batteries,48–51 it is not straightforward to apply these methods to study oxide powders, especially Ni-rich NMCs used in practical Li-ion batteries. For example, Kanamura et al.52 have developed a method for thin-film electrodes such as LiFePO453 and LiCoO254,55 sputtered on a gold plate and placed on the IR window with a micrometer, where the whole cell is flooded with electrolyte.52–55 These authors have observed carboxylate groups from propylene carbonate above 4.0 VLi during cyclic voltammogram of the LiCoO2 thin-film electrodes,54 while the exact oxidation products and the mechanisms were not well understood. Unfortunately, this method with thin film setups is not applicable to various composite electrodes of oxide powders due to the difficulty in synthesizing thin film electrodes for Li-containing Ni-rich NMCs.56 Moreover, Aurbach et al. have used a technique that purges the gaseous products from an electrochemical cell to an optical IR cell, which can detect gaseous species such as CO2 and CO from ionic liquid-based electrolyte with LiNi0.8Co0.15Al0.05O2 electrodes,57 but since it cannot detect interfacial species or solution phase species, the mechanistic details are not revealed. Other techniques including in situ DRIFTS58 or surfaced-enhanced Raman spectroscopy (SERS)59 have also been applied to the study of EEI formation for Li-rich oxides (Li1.2Ni0.2Mn0.6O2) and Lewis acid or Lewis base initiated EC decompositions have been proposed. However, in situ studies and direct evidence for interface reaction mechanisms on Ni-rich high-covalent NMCs are still missing.
O bonds.47 While there are a few in situ FT-IR techniques previously reported in understanding the reduction48–50 or oxidation51,52 stability of organic electrolytes in lithium-ion batteries,48–51 it is not straightforward to apply these methods to study oxide powders, especially Ni-rich NMCs used in practical Li-ion batteries. For example, Kanamura et al.52 have developed a method for thin-film electrodes such as LiFePO453 and LiCoO254,55 sputtered on a gold plate and placed on the IR window with a micrometer, where the whole cell is flooded with electrolyte.52–55 These authors have observed carboxylate groups from propylene carbonate above 4.0 VLi during cyclic voltammogram of the LiCoO2 thin-film electrodes,54 while the exact oxidation products and the mechanisms were not well understood. Unfortunately, this method with thin film setups is not applicable to various composite electrodes of oxide powders due to the difficulty in synthesizing thin film electrodes for Li-containing Ni-rich NMCs.56 Moreover, Aurbach et al. have used a technique that purges the gaseous products from an electrochemical cell to an optical IR cell, which can detect gaseous species such as CO2 and CO from ionic liquid-based electrolyte with LiNi0.8Co0.15Al0.05O2 electrodes,57 but since it cannot detect interfacial species or solution phase species, the mechanistic details are not revealed. Other techniques including in situ DRIFTS58 or surfaced-enhanced Raman spectroscopy (SERS)59 have also been applied to the study of EEI formation for Li-rich oxides (Li1.2Ni0.2Mn0.6O2) and Lewis acid or Lewis base initiated EC decompositions have been proposed. However, in situ studies and direct evidence for interface reaction mechanisms on Ni-rich high-covalent NMCs are still missing.
In this study, we have designed a new in situ FT-IR setup to examine the parasitic reactions between carbonate electrolytes and composite NMC powder electrodes used in practical Li-ion batteries as a function of voltage (or time) during galvanostatic (or potentiostatic) measurements. We first study electrolytes containing LiPF6 in EC using in situ FT-IR spectra, to examine its oxidation stability upon linear voltage sweep without NMC oxides, and their oxidation on NMC811 during charging, respectively. The spectra assignments are validated by DFT simulated spectra. We then study LiPF6 in EMC, and LP57 electrolyte (1 M LiPF6 in 3:7 EC/EMC), and examine their reactivity with NMC811 and NMC111 as a function of voltage. Next, we correlate the oxidation of different electrolytes by carbonate dehydrogenation on NMC811 and NMC111 with charge transfer impedance measured from electrochemical impedance spectroscopy (EIS). Finally, we propose the pathways and mechanisms for carbonate-based electrolyte decomposition on NMC that account for impedance growth and capacity fading. Through the in situ spectra on NMC surfaces, we track the interfacial reactions and understand the electrolyte decomposition mechanism on composite NMC powder surfaces in practical battery operations during galvanostatic charging, and interpret the strategies for EEI design by tuning electrode reactivity through coating materials, and tuning electrolyte activity by a concentrated electrolyte (EMC/EC with 3.1 M LiPF6).
2. Results and discussion
In situ FT-IR measurements of NMC811-mediated oxidation of EC upon charging
While EC with 1.5 M LiPF6 was stable against (electro)chemical oxidation up to 4.8 VLi on Pt, species derived from dehydrogenation of EC, including dehydrogenated EC (de-H EC, one hydrogen removed), vinylene carbonate (VC, two hydrogens removed), and oligomers with EC-like rings, were detected in situ upon charging NMC811 at voltages as low as 3.8 VLi. FT-IR spectra were first collected in situ at selected voltages with Pt only as the positive electrode, as the voltage of Li/Pt cells was linearly swept from open circuit voltage (OCV) at 3.1 VLi to 4.8 VLi (Fig. S1b, ESI†). Since the CO stretching region gives the strongest signal and is the most sensitive to molecule structure changes, the focus is to probe this CO stretching region. Two pronounced peaks in the CO region (1900 to 1700 cm−1) were revealed (Fig. S1c, ESI†), corresponding to CO stretching in EC (at ∼1800 cm−1) and Li+-coordinated EC (∼1773 cm−1),14,49 respectively. This assignment is in agreement with attenuated total reflection (ATR) measurements of 1.5 M LiPF6 in EC (Fig. S1a, ESI†) and DFT-simulated spectra (Fig. 1a). Voltage-dependent difference spectra obtained by subtracting each spectrum (in Fig. S1c, ESI†) by the spectrum at OCV revealed no new peaks appearing upon charging to 4.8 VLi (Fig. S1d, ESI†), indicating no significant electrolyte oxidation. This result is consistent with previous theoretical60–62 and experimental results63–65 for linear sweep voltammograms showing that carbonate electrolytes are stable against oxidation below 5 VLi on inert metals.
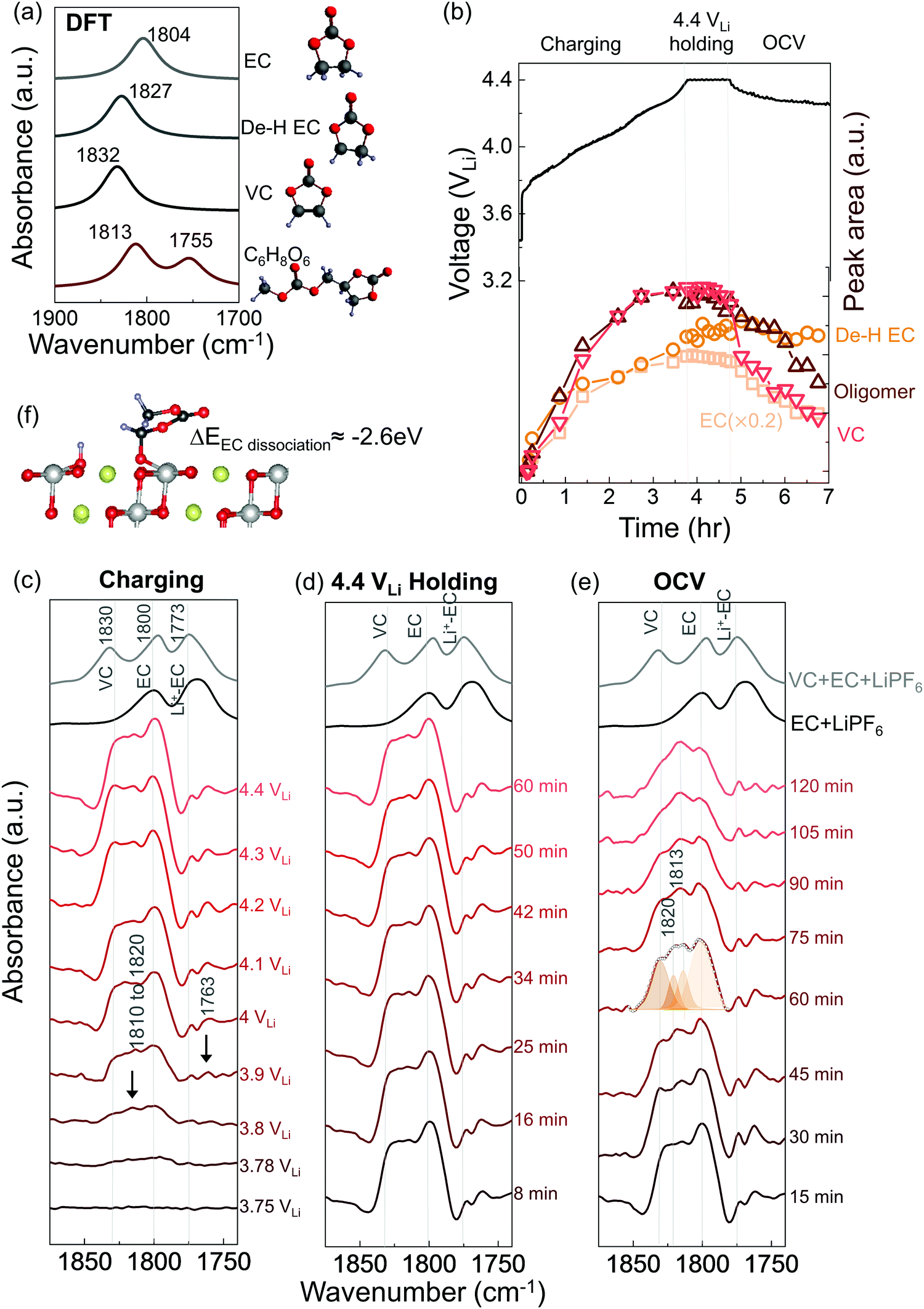 | ||
| Fig. 1
In situ FT-IR measurements on NMC811 surface in EC with 1.5 M LiPF6. (a) DFT-simulated spectra of EC, de-H EC, VC and oligomer C6H8O6. (b) Voltage profile (black) of NMC811 galvanostatically charged to 4.4 VLi, potentiostatically holding at 4.4 VLi, and resting at OCV, in 1.5 M LiPF6/EC electrolyte, with the in situ FT-IR cell, at a current of 27.5 mA g−1; deconvoluted peak areas (red) of EC and its decomposed products at ∼1813 cm−1 (de-H EC), ∼1820 cm−1 (oligomers with EC-like rings) and 1830 cm−1 (VC), during charging, potentiostatic holding and OCV. During the OCV, peak intensities of VC and oligomers decreased dramatically while de-H EC stayed almost the same in amount. (c) In situ FT-IR difference spectra (CO stretching region, in red) on NMC811 surface during galvanostatic charging to 4.4 VLi, (d) potentiostatic holding at 4.4 VLi, (e) resting at OCV in EC with 1.5 M LiPF6 electrolyte, and ATR spectra for 1.5 M LiPF6 in EC and 1.5 M LiPF6 in EC/VC (5:1) electrolyte solution (in black). (f) Diagram of EC dehydrogenation on LiNiO2 (similar to NMC811) and its energetics. | ||
Unfortunately, upon charging NMC811 from OCV to 4.4 VLi (Fig. 1b), significant changes were observed in the difference spectra (Fig. 1c) obtained from IR spectra collected in situ (Fig. S2, ESI†), indicative of oxidative instability. Broad intensities in the wavenumber range from 1850 to 1750 cm−1 were found to grow significantly from 3.8 VLi to 4.4 VLi, where individual peaks between 1830 and 1800 cm−1 became resolved at 3.8 VLi and grew in intensity. In addition, two small, sharp peaks at ∼1773 and ∼1763 cm−1 became visible upon charging from 3.9 VLi to 4.4 VLi. Upon further oxidation from 4.4 VLi to 4.8 VLi (Fig. S3, ESI†), the difference spectra remained unchanged. The peak at ∼1830 cm−1 that appeared at 3.8 V (Fig. 1c) could be assigned to VC, having two hydrogens removed from EC to form a CC bond in the ring, which assignment was supported by experimental ATR spectrum of VC-containing 1.5 M LiPF6 in EC (Fig. 1c), and calculated spectrum of VC by DFT (Fig. 1a). The peaks at ∼1800 cm−1 and ∼1773 cm−1 came from the CO stretching in EC and Li+–EC, respectively, and the peak intensity of EC grew with increasing voltage, possibly because bulk EC from electrolyte were attracted to surface during the adsorption and oxidation of EC on surface which could create a concentration gradient of EC in the electrolyte. The increased intensity ratio of VC to EC from 3.8 VLi to 4.8 VLi (Fig. S3c, ESI†) indicate more VC formed on the surface with increasing voltage. In addition, the peaks in the broad feature between 1820 to 1810 cm−1 and the peak at ∼1763 cm−1 (black arrow in Fig. 1c) can be attributed to small oligomers with EC-like rings, which could come from EC ring opening and polymerization.66–68 This assignment is supported by the DFT spectra (Fig. S3d, ESI† and Fig. 1a) of oligomers including C6H8O6 (computed 1813 and 1755 cm−1), C9H14O8 (computed 1819 and 1757 cm−1), and C7H10O6 (computed 1813 and 1750 cm−1). Further support came from time-dependent intensity measurements at OCV in Fig. 1e, which will be discussed below. Moreover, the feature around 1813 cm−1 (convoluted between 1820 to 1810 cm−1, black arrow in Fig. 1c) that appeared the earliest at ∼3.8 VLi could also contain CO stretching of dehydrogenated EC (de-H EC), having one hydrogen removed from EC and whose formation was energetically favorable with driving force around 2.6 eV (Fig. 1f).13,14 This peak position of de-H EC fell between EC and VC, which is in agreement with DFT (Fig. 1a), and the assignment was further supported by time-dependent intensity measurements at OCV in Fig. 1e to be discussed later. The deconvoluted peak areas of VC and oligomers were found to grow comparably with increasing voltage, indicating that they were gradually generated from EC and/or de-H EC, while the peak area of de-H EC grew less with increasing voltage (Fig. 1b), which might due to its consumption to further generate VC and oligomers. In summary, the appearance and growth of new peak features at ∼1830, ∼1820 to 1810 cm−1, and ∼1763 cm−1 provided direct evidence of electrolyte oxidation via EC dehydrogenation on charged NMC811 starting at voltages as low as ∼3.8 VLi, where EC could dissociate on electrode surface to remove one hydrogen and generate de-H EC, further remove another hydrogen to form VC, or combine with another EC to form oligomers.
While no significant changes were noted for the CO stretching region (1900 to 1700 cm−1) in the difference spectra during voltage holding at 4.4 VLi (Fig. 1d), most peaks were reduced in intensity with increasing time upon OCV following the 4.4 VLi voltage hold (Fig. 1b and e). These potentiostatic and open circuit voltage measurements were conducted followed by charging to 4.4 VLi to examine whether the species could stick on surface or could diffuse or dissolve away from surface. The spectrum at OCV (after 60 min) could be splitted into four features, VC at ∼1830 cm−1, de-H EC at ∼1813 cm−1, and oligomers at ∼1820 cm−1 (and 1763 cm−1) and EC at 1800 cm−1. The peaks of VC, oligomers and EC were reduced rapidly with increasing time at OCV, indicating these species diffused away from charged NMC811 into the electrolyte. Of significance to note that VC intensity was reduced faster than oligomers and EC, indicative of greater diffusivity or solubility, which is in agreement with the fact that VC was not detected in the ex situ DRIFT spectra.14 In contrast, the peak intensity of de-H EC (∼1813 cm−1) did not change significantly with increasing time (Fig. 1b and e), and it became the most dominant feature after resting for 60 minutes, suggesting de-H EC was anchored on the oxide surface, presumably by a C–Osurface bond formed during EC dehydrogenation on surface oxygen of NMC81113 (Fig. 1f), which is in agreement with the observation of this species in ex situ DRIFT spectra of charged NMC811 electrodes dried and removed from electrochemical cells.14 Next we discuss NMC811-mediated oxidation of EC in LP57 (1 M LiPF6 in EC/EMC) – a commonly used electrolyte for Li-ion batteries.
While LP57 (1 M LiPF6 in EC/EMC) was stable against oxidation on Pt, in situ FT-IR spectra revealed that EC was dehydrogenated to form de-H EC, vinylene carbonate (VC), and oligomers during galvanostatic charging similar to those discussed earlier for 1.5 M LiPF6 in EC. ATR measurements of bulk LP57 had four features (Fig. S5a, ESI†), including EC (∼1800 cm−1), Li+-coordinated EC (∼1773 cm−1), EMC (∼1750 cm−1) and Li+-coordinated EMC (∼1718 cm−1). On Pt, no new species appeared in the difference spectra during linearly voltage sweeping from OCV (∼3 VLi) to 4.8 VLi (Fig. S4, ESI†), indicative that the electrolyte was stable up to 4.8 VLi, which is in consistent with having both 1.5 M LiPF6 in EC (Fig. S1, ESI†) and 1 M LiPF6 in EMC (Fig. S5, ESI†) being stable on Pt. In contrast, spectra collected on NMC811 from OCV (3 VLi) to 4.8 VLi (Fig. S6, ESI†) was subtracted from the OCV spectrum to yield difference spectra (Fig. 2b), which revealed new peaks of EC-derived dehydrogenation species at voltages greater than 3.8 VLi, including de-H EC (∼1813 cm−1, greater than 3.9 VLi), VC (∼1830 cm−1, greater than 4.1 VLi), and oligomers with EC-like rings (∼1820 and ∼1763 cm−1, greater than 4.1 VLi) similar to Fig. 1. Changes in the difference spectra unique to the dehydrogenation of EMC during galvanostatic charging of NMC811 was not observed, which can be interpreted by the convolution of de-H EMC (discussed below in Fig. 3) with bulk EMC and with EC-derived oligomers, and/or having EC preferentially adsorbed on the NMC811 surface than linear alkyl carbonate EMC69,70 due to its higher dielectric constant.61 Experimental sum frequency generation results show that EC occupies over 90 mole% on lithium metal oxide (LiCoO2) surfaces in mixed carbonate solutions such as EC + DEC or EC + DMC, much higher than that in the bulk solution (43 mole%).70 After charging, we also discharged the cell and the difference spectra (Fig. S7, ESI†) showed that upon discharging from 4.8 VLi to 2 VLi, VC peak (1830 cm−1) and oligomer peaks (1820 and 1763 cm−1) gradually decreased in intensity, which agrees with the results during OCV (Fig. 1e) that VC and oligomers could diffuse or dissolve away. De-H EC still anchored on the surface, which is also in agreement with the results at OCV. Another interesting finding is that Li+–EC (1773 cm−1) peaks gradually grew larger, which was not observed during OCV, and this was probably because during discharge, Li+ migrated from bulk electrolyte to oxide surface to be intercalated, making more EC to become coordinated. As EC was found to oxidize by dehydrogenation upon charging at voltages as low as 3.8 VLi, we further examine and discuss NMC811-mediated oxidation of 1 M LiPF6 in EMC electrolyte to better understand the different reactivity of EC and EMC.
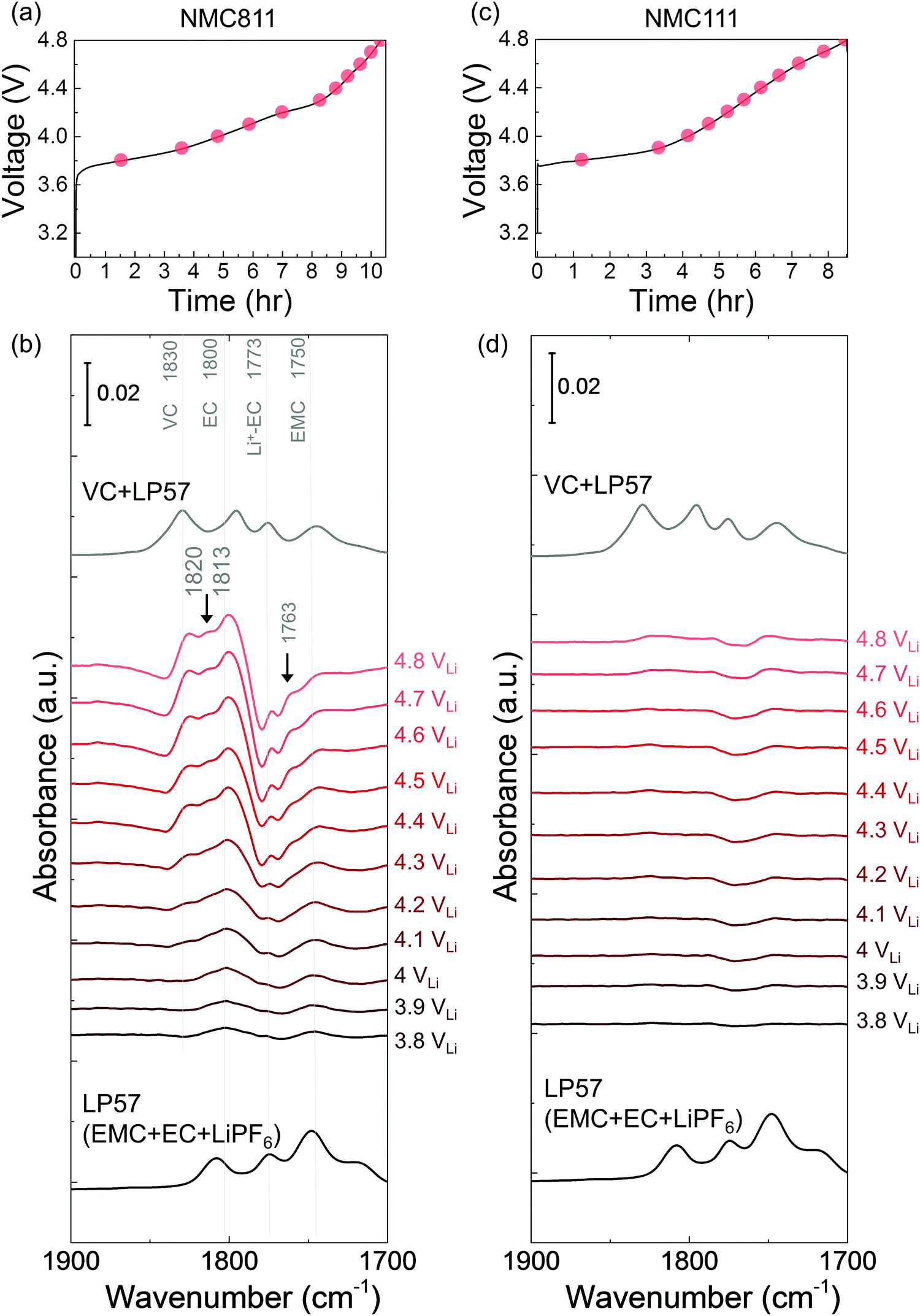 | ||
| Fig. 2
In situ FT-IR measurements on different NMC surfaces in LP57 upon first cycle charging to 4.8 VLi. Voltage profile during (a) NMC811, (c) NMC111 charged in LP57. In situ FT-IR cell was galvanostatically charged at 27.5 mA g−1. In situ FT-IR difference spectra in CO stretching region (in red) on (b) NMC811 and (d) NMC111 surface during charging in LP57 electrolyte, and ATR spectra (in black) for LP57 and LP57/VC solutions (1:1). On NMC811 surface, de-H EC (1813 cm−1, marked in arrow), VC (∼1830 cm−1), and oligomers with EC-like rings (∼1820 and ∼1763 cm−1, marked in arrows) arose in the spectra upon charging, while no obvious peaks appeared for NMC111 during first cycle charging. | ||
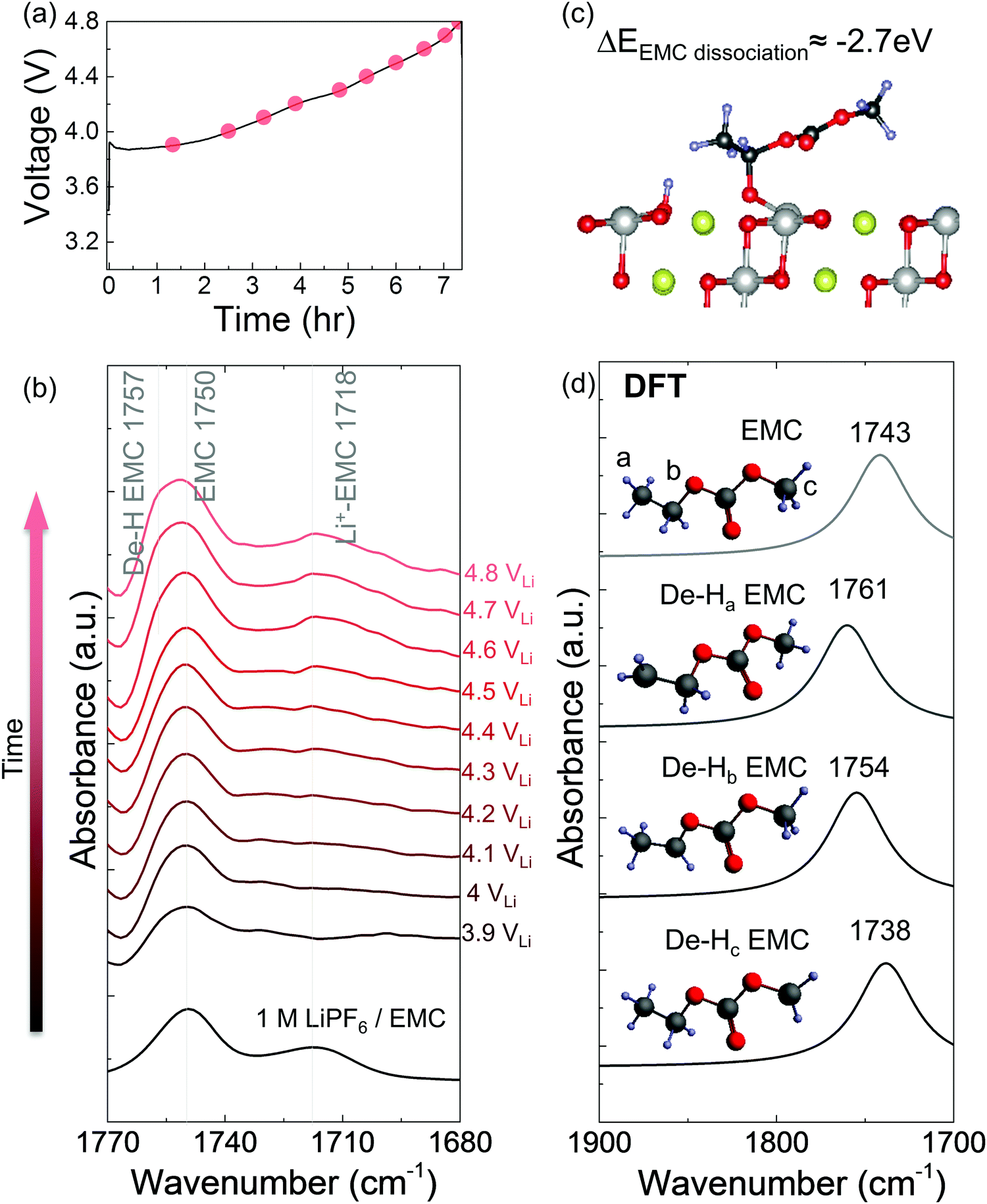 | ||
| Fig. 3
In situ FT-IR measurements on NMC811 surface in 1 M LiPF6/EMC. (a) Voltage profile during NMC811 charging in 1 M LiPF6/EMC, with voltage up to 4.8 VLi. In situ FT-IR cell was galvanostatically charged at 27.5 mA g−1. (b) In situ FT-IR difference spectra (CO stretching region) on NMC811 surface during charging in 1 M LiPF6/EMC electrolyte. (c) Diagram and energetics of EMC dehydrogenation on LiNiO2. (d) DFT computed spectra for EMC and de-H EMCs in the CO stretching region. | ||
In situ FT-IR measurements of NMC811-mediated oxidation of EMC upon charging
While EMC with 1 M LiPF6 was stable against oxidation to 4.8 VLi on Pt, in situ FT-IR spectra revealed that EMC became dehydrogenated (de-H EMC) on NMC811 with increasing voltage during galvanostatic charging (Fig. 3a). Ex situ ATR spectrum of pristine 1 M LiPF6 in EMC revealed two features of CO stretching at ∼1750 and ∼1718 cm−1 (Fig. 3b and Fig. S5a, ESI†), which can be assigned to EMC and Li+-coordinated EMC, respectively.14,71 During linearly voltage sweeping on Pt from OCV (3.1 VLi) to 4.8 VLi (Fig. S5b, ESI†), difference spectra (Fig. S5d, ESI†) obtained from subtracting spectra collected at different voltages (Fig. S5c, ESI†) by that at OCV show the growth of peaks corresponding to CO stretching of EMC at ∼1750 cm−1 and Li+-EMC at ∼1718 cm−1 with increasing voltage. Neither new peaks nor peak shifts was observed, indicating no visible electrolyte oxidation to 4.8 VLi, which is consistent with former DFT60–62 and experimental results63–65 of electrolyte (electro)chemical stability to ∼5 VLi. In contrast, the difference spectra collected on NMC811 during galvanostatic charging from OCV to 4.8 VLi (Fig. 3a) revealed that the peak of EMC at ∼1750 cm−1 shifted towards higher wavenumbers with increasing voltage (Fig. 3b), which could be assigned to de-Hb EMC, indicative of EMC oxidation by EMC dehydrogenation. This assignment, having one b-site hydrogen from carbon removed and a C–Osurface bond formed on the oxide in Fig. 3c, is supported by DFT computed spectra (Fig. 3d). As both EC and EMC (in 1.5 M LiPF6 in EC, 1 M in EMC and 1 M in EC/EMC mixture – LP57) were found to oxidize by dehydrogenation on NMC811 upon charging, we further examine the oxidation of LP57 by replacing NMC811 with NMC111, NMC622, and Al2O3-coated NMC622 upon charging to 4.8 VLi.
In situ FT-IR measurements of NMC111- and NMC622-mediated oxidation of LP57 upon charging
The dehydrogenation tendency of EC in LP57 on Ni-based oxide electrode decreased as decreasing metal-oxygen covalency13,72 or moving the Fermi level away from the oxygen p band center as predicted by DFT.13,73 Unlike NMC811, where EC was dehydrogenated upon charging to voltages as low as 3.8 VLi in the first cycle, no change was observed upon charging NMC111 to 4.8 VLi in the first cycle and subsequent two cycles. No new peaks were found for difference spectra collected for NMC111 upon the first galvanostatic charging from OCV to 4.8 VLi (Fig. 2c and d), indicative of no obvious solvent oxidation upon first cycle charging for NMC111, which is in agreement with recent ex situ FT-IR studies.14 In the subsequent third cycle, still no new peaks were found for difference spectra collected for NMC111 upon galvanostatic charging from OCV to 4.8 VLi (Fig. S9, ESI†). The difference in the dehydrogenation of EC or oxidation of LP57 between NMC811 and NMC111 observed in the in situ FT-IR measurements provides further experimental evidence to support previous DFT results, where the driving force for EC dehydrogenation on NMC811 with greater metal-oxygen covalency and more oxygen 2p states pinned at the Fermi level is much greater than that on NMC111.14,15,74,75While NMC622 did not show any evidence for carbonate dehydrogenation during the 1st cycle (Fig. S10, ESI†), dehydrogenated EC products including de-H EC (∼1813 cm−1, marked in black arrow), VC (∼1830 cm−1), and oligomers with EC-like rings (∼1820 and ∼1763 cm−1, marked in black arrows) were detected in the FT-IR difference spectra obtained upon the third charge (Fig. 4a and b). In contrast, the dehydrogenation of LP57 was not detected for NMC622 coated with Al2O3 in the 3rd charging (Fig. 4c and d). This difference in the dehydrogenation of LP57 between coated and uncoated NMC622 demonstrates the effect of surface “inert” coating layers76,77 such as Al2O3,78,79 ZrO2,80 HfO281 on eliminating electrolyte oxidation. Oxides such as Al2O3 were less thermodynamically favorable for dissociative adsorption of EC, and lowered the driving force to dehydrogenate carbonates,13,73,82,83 which is in agreement with former studies that coatings the electrode surfaces with Al2O3,78,79,83 ZrO280 or HfO2,81 or creating a Ni-poor surface to decrease M–O covalency21–23 can improve cycling performance. Coated Ni-rich NMC electrodes with greater capacity retention77–79,83 can be attributed to enhanced stability against dehydrogenation compared to electrodes without coatings. Next, we further examine the influence of carbonate solvent activity on the oxide-mediated electrolyte oxidation by changing electrolyte salt concentrations and salt anions.
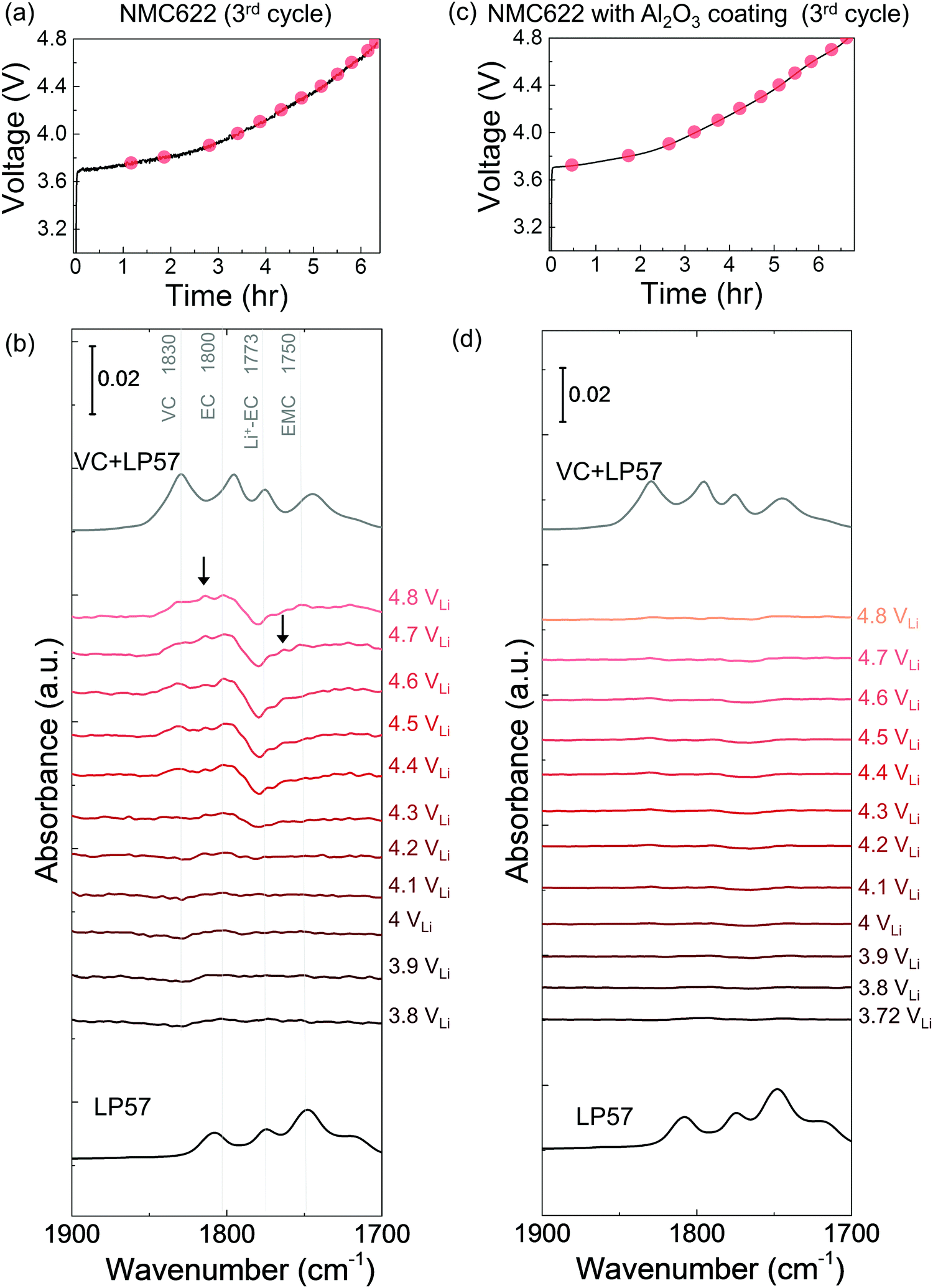 | ||
| Fig. 4
In situ FT-IR measurements on NMC622 surfaces with and without surface coating upon the 3rd cycle charging to 4.8 VLi in LP57. Voltage profile during charging of (a) NMC622, (c) NMC622 with Al2O3 coating. In situ FT-IR cell was galvanostatically charged at 27.5 mA g−1. In situ FT-IR difference spectra in CO stretching region (in red) on (b) NMC622, (d) NMC622 with Al2O3 coating electrodes during the 3rd cycle charging in LP57 electrolyte, and ATR spectra (in black) for LP57 solutions and LP57/VC solutions (1:1). On uncoated NMC622 surface, de-H EC (1813 cm−1, marked in arrow), VC (∼1830 cm−1), and oligomers with EC-like rings (1820 and 1763 cm−1, marked in arrows) arose in the spectra upon charging, while no obvious peaks appeared for coated NMC622 even during 3rd cycle charging. | ||
In situ FT-IR measurements of NMC811-mediated oxidation of concentrated and non-PF6− containing electrolytes on NMC811 upon charging
Increasing the salt concentration in the electrolyte was found to reduce NMC811-mediated dehydrogenation of EC upon charging. Ex situ ATR spectra (Fig. S13, ESI†) of EC/EMC with 1 M, 2 M, and 3.1 M LiPF6 revealed that increasing the salt concentration resulted in more Li+-coordinated solvent molecules (Li+–EC/EMC) but fewer free EC/EMC solvent molecules (low carbonate activity), in agreement with previous work.84–87 NMC811 charged in the concentrated electrolyte (3.1 M LiPF6 in EC/EMC), which contained fewer free EC or free EMC (molar fraction of free solvent in general concentrated electrolyte with dissociative salt is reported to be less than 10%),88–92 did not show dehydrogenation or oligomerization products from EC or EMC in the FT-IR difference spectra (Fig. 5a) upon charging to 4.8 VLi, unlike in LP57 (with 1 M LiPF6) where EC became dehydrogenated as discussed before (Fig. 2b). The overpotential in Fig. 5a could come from lower ionic conductivity and viscous nature of the concentrated electrolyte. Increasing the Li+ salt concentration in the electrolyte decreased the carbonate activity (Fig. S13, ESI†) in the electrolyte and enhanced the solvent stability against oxidative dehydrogenation on NMC811, which could potentially lead to stable cycling and longer cycle life for NMC811. This observation can interpret recent findings that increasing the LiBF493–95 or LiPF687,96,97 concentration can increase capacity retention of Ni-rich NMC,93–95 and enhance the cycle life96,98 since the dehydrogenation is suppressed by the decrease of free EC or EMC solvent activity in the concentrated electrolytes.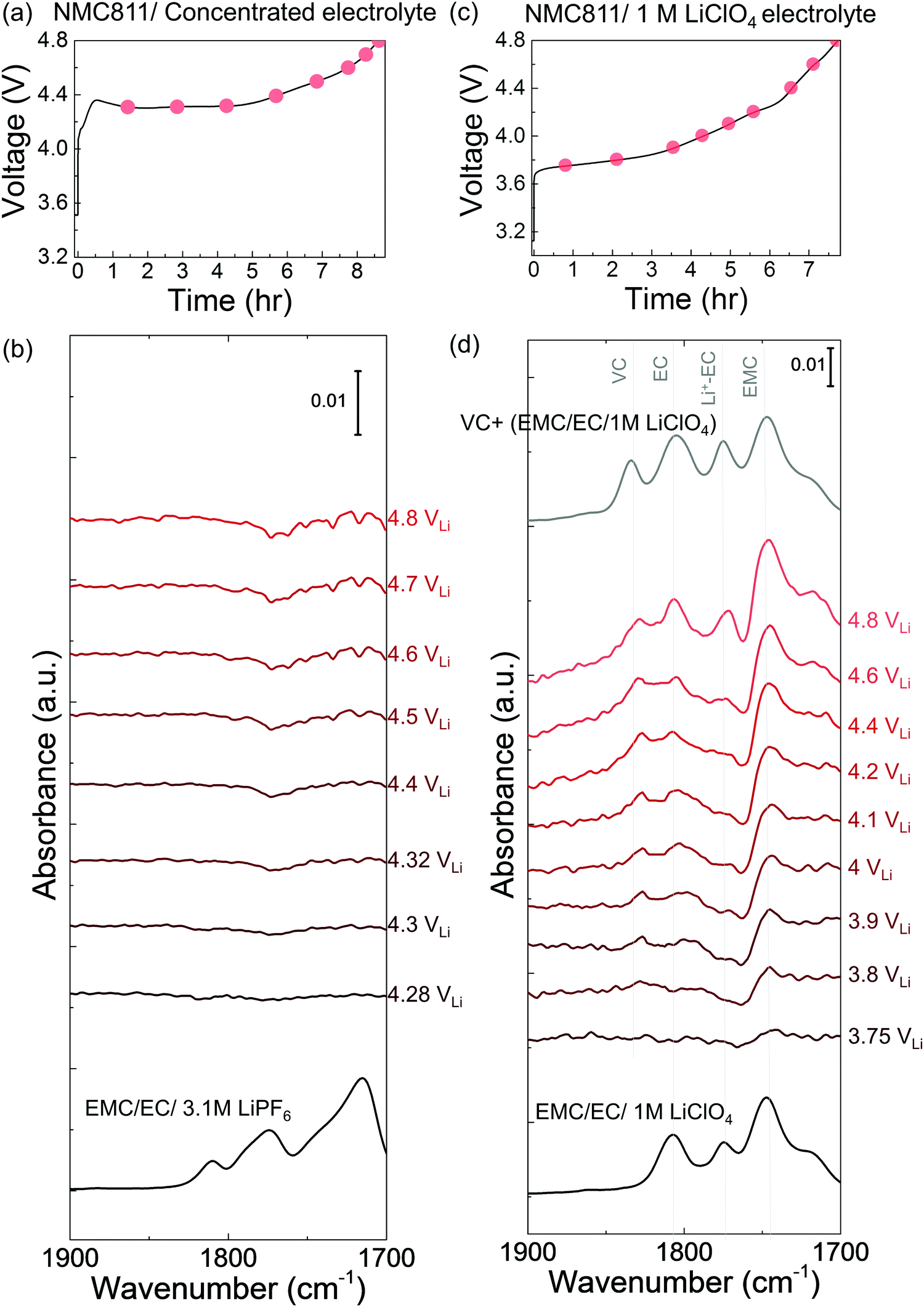 | ||
| Fig. 5
In situ FT-IR measurements on NMC811 surfaces upon charging to 4.8 VLi in concentrated (3.1 M) and in 1 M electrolyte. Voltage profile during charging of NMC811 (a) in concentrated electrolyte (EMC/EC with 3.1 M LiPF6), and (c) in EMC/EC with 1 M LiClO4. In situ FT-IR cell was galvanostatically charged at 27.5 mA g−1. In situ FT-IR difference spectra in CO stretching region (in red) on NMC811 during charging (b) in concentrated electrolyte (EMC/EC with 3.1 M LiPF6), and (d) in EMC/EC with 1 M LiClO4, and the ATR spectra (in black) for bulk electrolyte solutions and EMC/EC/LiClO4:VC =10:1 solution. | ||
Salts with smaller dissociation constants (TFSI− ∼ PF6− > AsF6− > ClO4−)88,99,100 in 1 M electrolytes have higher carbonate solvent activities. The smaller dissociativity of ClO4− in solvent indicates more cation–anion pairs, accordingly less Li+-coordinated EC (/EMC) compared to PF6−.101 With the different salt (1 M LiClO4 in 3:7 EC:EMC) but the same concentration (1 M) as LP57, NMC811 also showed dehydrogenation products from EC during the first charging in EMC/EC/1 M LiClO4. VC (∼1830 cm−1) appeared from 3.8 VLi, and the intensity increased as it was charged to 4.8 VLi (Fig. 5d), similar to LP57 (with 1 M LiPF6) case. De-H EC (∼1813 cm−1) and oligomer (∼1820 cm−1) features were convoluted between the VC (∼1830 cm−1) and EC (∼1800 cm−1) features, and they were less observable than in LP57 (Fig. 2b), possibly because the transformation to VC is faster or greater in LiClO4 due to the less dissociative LiClO4 salt87,88,99,100 and more free solvents in the electrolyte, giving rise to more dominant VC features. DFT calculations show that the solvent molecules are stabilized with Li+ interaction, while free solvents have higher energy levels of highest occupied molecular orbital (HOMO) and decreased stability against oxidation,88,102,103 where here we also show the higher tendency towards dehydrogenation with the less dissociative LiClO4 salt and less Li+-coordinated solvents. Furthermore, the kinetic barrier for H transfer would also be different between free carbonate and Li+ coordinated carbonate, giving lower energetic barrier to free carbonate to be decomposed on LiMO2 surface.75,87 The observation of VC in both EMC/EC with 1 M LiClO4 and LP57 confirmed the universal dehydrogenation of EC in 1 M electrolytes, which had higher carbonate activity or lower oxidative stability103 than concentrated electrolytes (3.1 M).
Correlating impedance growth with carbonate solvent dehydrogenation
While in situ FT-IR spectra revealed the dehydrogenation of solvents on NMC in situ as a function of voltage/time, we employed EIS (Fig. S16–S22, ESI†) to further reveal that the charge transfer and EEI impedance on NMC811 grew much more significantly than on NMC111 with increasing voltage, which could be attributable to dehydrogenated products on NMC811 surfaces (Fig. 2a and 6). NMC111, NMC622 with surface coating, and NMC811 in the concentrated electrolyte (3.1 M LiPF6 in EC/EMC) exhibit no significant impedance growth in contrast to NMC811 in LP57 (Fig. 6).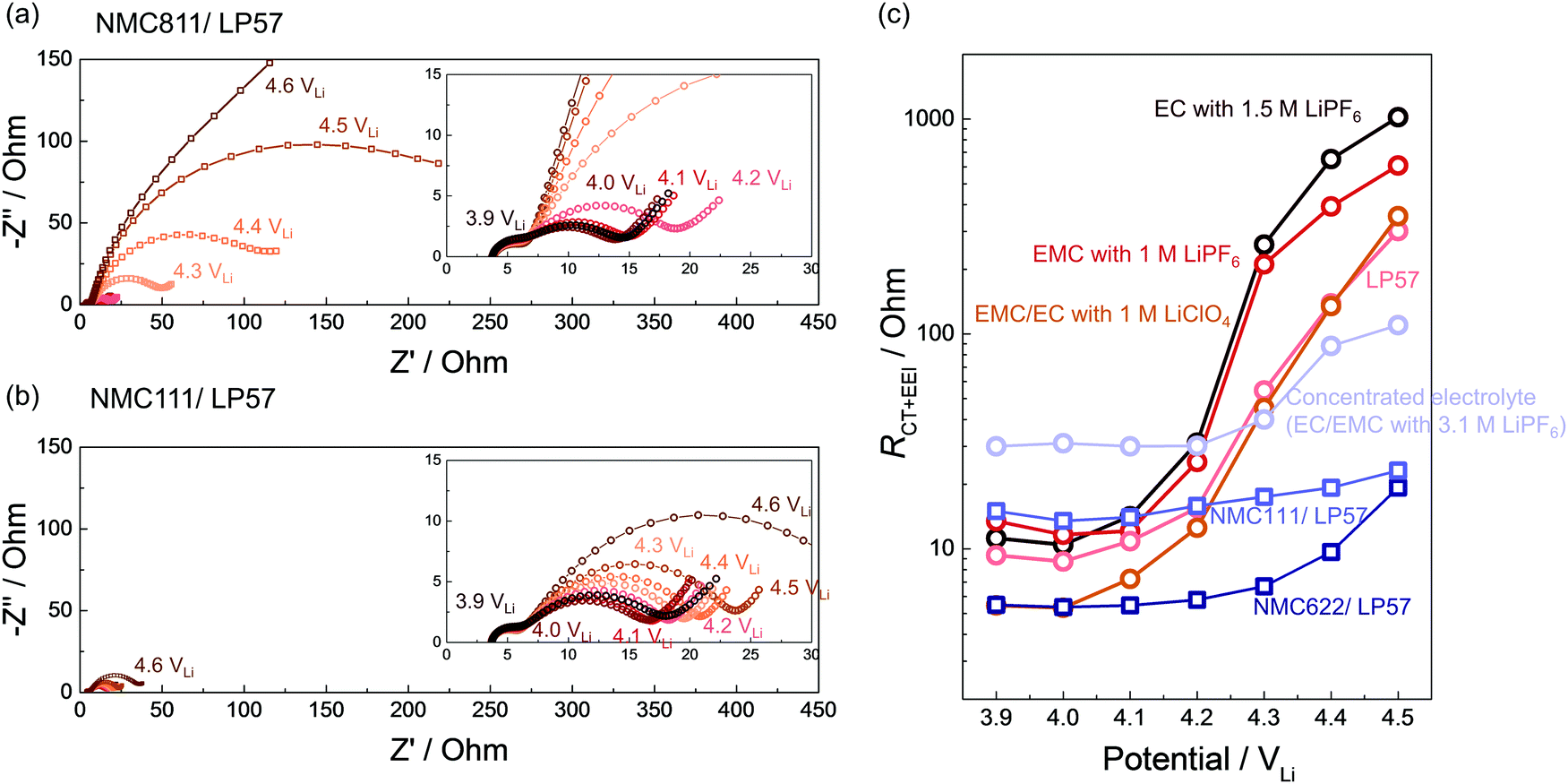 | ||
| Fig. 6 EIS measurement on NMC composite electrode during the first charging. Nyquist plot at different voltages for (a) NMC811 and (b) NMC111 charged in LP57. Inset figures show the enlarged Nyquist plot. (c) Charge transfer and EEI resistances (RCT+EEI) at different voltages measured by EIS, for NMC811 charged in EC with 1.5 M LiPF6, EMC with 1 M LiPF6, LP57, EMC/EC with 1 M LiClO4 and the concentrated electrolyte (3.1 M LiPF6 in EC/EMC), and for NMC111 and NMC622 charged in LP57. Those curves in (c) without specifying the electrode correspond to NMC811 electrode. | ||
EIS on NMC811 positive electrode collected at 3.9 VLi, 4 VLi and up to 4.6 VLi during the 1st cycle charging in LP57 showed huge growth in the low-frequency semicircle (right-hand side semicircle in Fig. 6a), which can be attributed to the charge transfer and EEI resistance (RCT+EEI, low-frequency resistance)104 (Fig. 6c), growing from ∼10 to over 300 Ohm during charging. The RCT+EEI impedance at 4.6 VLi was beyond the detected amplitude in this frequency range (down to 100 mHz) because it was almost at the end of charge. Similarly, for NMC811 in other non-concentrated electrolytes including EC with 1.5 M LiPF6, EMC with 1 M LiPF6, and EMC/EC with 1 M LiClO4, RCT+EEI all grew dramatically from ∼10 Ohm to ∼400 Ohm (Fig. 6c). The difference in RCT+EEI between EC with LiPF6, EMC with LiPF6, and LP57 at each voltage could come from the different dielectric constants105 and physical properties such as ionic conductivity and viscosity (Table 1) which could affect the solvation and de-solvation processes during charge transfer. This great increase of RCT+EEI upon NMC811 charging in all four 1 M or 1.5 M electrolytes can be correlated with the dehydrogenation and/or oligomerization products detected by in situ FT-IR reported above for 1 M or 1.5 M electrolyte cases (Fig. 1c, 2b, 3b and 5d), indicating that these organic products likely formed a resistive layer on NMC811 which passivated the surface and impeded charge transfer. The comparable impedance growth and similar dehydrogenation reactions between the PF6−-based electrolyte and ClO4−-based electrolyte noted for NMC811 further supports the hypothesis that solvent (carbonate) decomposition contributed most to the impedance growth, rather than commonly-perceived salt decomposition. Although LiPF6 salt could be attacked by surface protic species to form metal fluorides (MF)14,55,106 and resistive LiF,32 our result of a non-PF6− electrolyte revealed that even without severe salt decomposition, solvent dehydrogenation itself could account for great impedance growth on NMC811 in the ClO4−-based electrolyte.
| Ionic conductivity [mS cm−1] | Viscosity [mPa s] | |
|---|---|---|
| 1 M LiPF6 in EC:EMC = 3:7wt (LP57) |
8.88 | 3.00 |
| 1.5 M LiPF6 in EC | 7.19 | 11.0 |
| 1 M LiPF6 in EMC | 4.47 | 1.70 |
In contrast, NMC811 in the concentrated electrolyte did not exhibit as large impedance growth and RCT+EEI remained around 30–100 Ohm (Fig. 6c), which could be also be correlated with the more stable interface and lack of dehydrogenation from FT-IR result (Fig. 5a). This observation further supports the role of undesirable carbonate dehydrogenation in passivating the surface and increasing interfacial impedance, suggesting that lowering free solvent activity could facilitate stable cycling of NMC811. For NMC111 and NMC622, RCT+EEI stayed less than 50 Ohm through all the voltages (Fig. 6b and c) during the first cycle, which matched well with in situ FT-IR spectra that there was no profound solvent dehydrogenation in the first charging. The more stable interface and lower impedance growth is also in agreement with the better cycling stability of NMC111 and NMC622 than NMC811.11,107
Mechanistic discussion on electrolyte oxidation on Ni-rich NMC
We propose detailed mechanisms of electrolyte oxidative decomposition on layered Ni-rich metal oxide positive electrodes (Fig. 7) based on the species detected by in situ FT-IR experiments and their correlated contribution to EEI impedance growth and capacity loss. EMC molecules can dissociate b-site hydrogen (Fig. 3d) and adsorb on oxide surface through a C–Osurface bond, revealed by a blueshifted shoulder at ∼1757 cm−1 in Fig. 3b. The reaction is energetically favorable, which is closer to the dissociation on LiNiO2 surface with ΔEEMCdissociation = −2.7 eV calculated from DFT (Fig. 3c). EC molecules also dehydrogenate and form de-H EC that bonds with surface oxygen, revealed by the feature at ∼1813 cm−1 (Fig. 1). EC dissociation is also energetically favorable, with ΔEECdissociation close to −2.6 eV calculated from DFT (Fig. 1f). The driving force becomes even greater during delithiation and decreasing of x in LixMO2.14,15 EC can also further dehydrogenate a second hydrogen on the other carbon and form VC, which corresponds to the peak at ∼1830 cm−1 (Fig. 1c and 2b). Another pathway is that de-H EC could be further oxidized to open the ring, or form oligomers with EC-like rings such as C6H8O6, C9H14O8 and C7H10O6, revealed by the features at ∼1820 and ∼1763 cm−1 (Fig. 1c). The solvent dehydrogenation products may also be eventually oxidized to CO2 or CO.107 Organic products from carbonate decomposition can form a passivating layer on the oxide surface, through which Li+ needs to migrate to reach the oxides and undergo charge transfer,32 and therefore RCT+EEI shows great impedance growth during charging of NMC811 measured by EIS (Fig. 6c). The dehydrogenation of EC and EMC also generates protic species on the surface,13,107,108 which can further trigger reactions106 with the widely used LiPF6 salt to form HF14,108 and less-fluorine-coordinated species such as LixPFyOz,14,78 transition metal fluorides (MF)32 and PF3O.109,110 Salt decomposition might also contribute to EEI impedance, but our comparable results between LP57 (EMC/EC with 1 M LiPF6) and EMC/EC with 1 M LiClO4 show that solvent decomposition plays a much larger role. Combining in situ FT-IR spectra and EIS reveals that when there is undesirable dehydrogenation of solvents, there is great interfacial impedance growth at higher voltage, eventually leading to the loss of capacity when NMC811 is cycled to high voltages.107 On the NMC111 surface, which is less reactive, solvent is more stable against dehydrogenation, and the undesirable reactions to form passivating layers barely happen, leading to lower impedance and better capacity retention. The reactivity of NMC622 is between that of NMC811 and NMC111, so it did not yield dehydrogenated EC visibly during the first cycle, but gradually caused dehydrogenation in following cycles (such as the third cycle analyzed above).
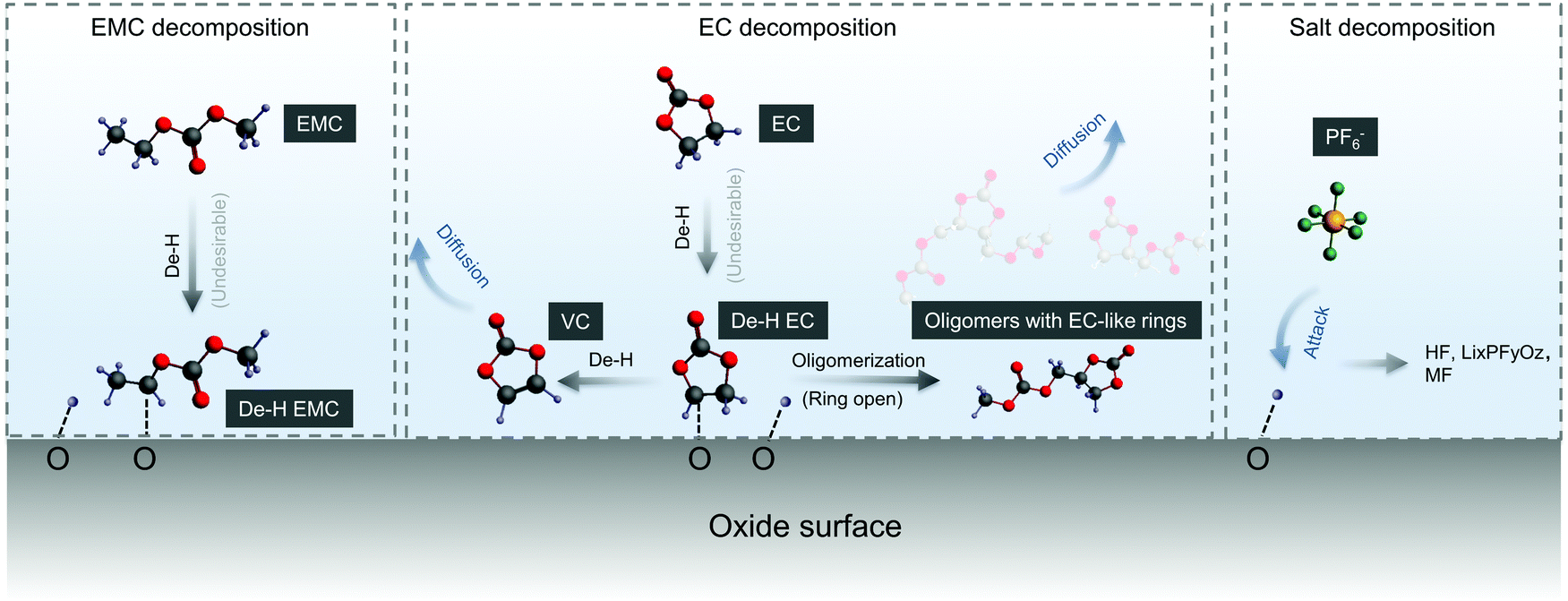 | ||
| Fig. 7 Proposed mechanism and pathways of electrolyte decompositions on NMC811. Electrolyte decompositions include solvent (EMC and EC) decomposition and coupled salt (PF6−) decomposition. Solvent decomposition happened by dehydrogenation first, and de-H EC could be further decomposed by removing another hydrogen or by oligomerization. The protic species on surface coming from dehydrogenation could further attack PF6− and lead to coupled salt decomposition.14 The decomposed species (mainly solvent decomposed species) would form a resistive layer at the EEI and lead to great impedance growth, eventually resulting in capacity loss for NMC811. | ||
The understanding of the electrolyte decomposition pathway suggests strategies to design a more stable interface, based on the principle of eliminating undesirable solvent dehydrogenation reactions that can be triggered followed by oligomerization and salt decomposition. This can be achieved by tuning the surface chemistry of electrode materials, e.g. using inert surface coating, such as ceramic thin films, which reduce the driving force for solvent dissociation. Although NMC622 yielded EC dehydrogenation after 3 cycles, NMC622 with Al2O3 coating did not cause visible solvent decomposition, which implies a more stable interface. This explains the mechanism for surface coatings such as Al2O3,78,79,83 HfO281 or ZrO280 to increase cycling stability and capacity retention. Another strategy to design a stable interface is to tune the solvent activity, e.g. by decreasing the number of free solvent molecules, which are less oxidatively stable than the molecules in the solvation shells.84,85,97 While NMC811 charged in non-concentrated electrolytes (1 M or 1.5 M), all showed undesirable solvent dehydrogenation and/or oligomerization, no obvious decomposition products were observed in concentrated electrolyte containing fewer free solvent molecules and the interface impedance didn’t grow as much as 1 M or 1.5 M cases, which could promote the cycling stability of NMC811.
Other approaches towards suppressing the undesirable solvent decomposition include adding additives in the electrolytes,111,112 with the idea of sacrificing additives to be oxidized rather than solvents. Interfacial stability can also be promoted by new types of solvents that are less likely to dissociate on oxides and new types of salts that are more stable and less likely to be attacked by protons. Exploring such strategies, including various electrolyte additives, will be the subject of our future work. The method of in situ FT-IR is also applicable to study the SEI formation on anodes, which is also within our plan to extend this method.
3. Conclusions
In this study, we developed an FT-IR method, which allowed the in situ studies on the reactivity of the electrolyte on NMC surfaces as a function of voltage. While ethylene carbonate (EC) remained stable against (electro)chemical oxidation on Pt up to 4.8 VLi, we found unique evidence for dehydrogenation of EC on LiNi0.8Co0.1Mn0.1O2 (NMC811) surface at voltages as low as 3.8 VLi. Three unique dehydrogenated species from EC were observed on the NMC811 surface, which included dehydrogenated EC (de-H EC, anchored on oxides), vinylene carbonate (VC), and dehydrogenated oligomers with EC-like rings, while the latter two can diffuse away from the NMC811 surface into the electrolyte. These observations indicate that electrolyte oxidation on NMC811 might not generate a protective film unlike the solid electrolyte interface (SEI) formed by EC reduction on graphite. Similar dehydrogenation was observed for EMC-based and LP57 electrolytes on NMC811. In contrast, no dehydrogenation was found for NMC111 or modified NMC surface by coatings such as Al2O3. Such oxidative dehydrogenation tendencies on the oxide chemistry is in good agreement with the driving force for EC dehydrogenation on surface oxygen of NMC predicted by recent density functional theory (DFT) calculations, which increased with lower Fermi level into the oxygen p band of oxides associated with greater Ni and less lithium in NMC. Interestingly, the dehydrogenation of EC and EMC was observed in different anions in the 1 M electrolytes, but not observed in the concentrated electrolyte (EC/EMC with 3.1 M LiPF6) on NMC811, indicating that the dehydrogenation of carbonates can be suppressed by increasing lithium coordination with solvents. Dehydrogenation of carbonates (EC and EMC) on the NMC811 surface was found to accompany with rapid growth of interfacial impedance with increasing charging voltage measured from EIS. In contrast, those electrode–electrolyte combinations without dehydrogenation were found to have no significant impedance growth. Therefore, it is crucial to minimize carbonate dehydrogenation on the NMC surface by tuning electrode reactivity and electrolyte reactivity for improving the cycle life and high energy of lithium-ion batteries.4. Experimental procedures
Experimental methods
:15 mass ratio, using a planetary centrifugal mixer (THINKY AR-100). The obtained slurry was drop-casted onto glassy fiber substrates (Whatman 934-AH, 10 mm in diameter) and dried at 100 °C. Then the composite composites were compressed at 0.5 T cm−2 using a hydraulic press to improve electrical conductivity. Finally the electrodes were completely dried at 120 °C under vacuum for 24 h. The active material loading was ∼6.5 mg cm−2, but it is noted here that the effective loading is smaller because some particles could permeate into the glassy fiber and not all of the loading is on the top surface.
The positive electrode for EIS experiments was composed of 85 wt% of NMC, carbon black (5 wt% KS6 and 2 wt% Super P, both from Timcal), and poly(vinylidene fluoride) (8 wt%, PVDF, Kynar). The mesh reference electrode was also composed of 80 wt% of Li4Ti5O12 (Itasco, >99.5%), 10 wt% of acetylene black (C-55, Chevron) and 10 wt% of PVDF. These materials were mixed together and thoroughly agitated in NMP. The obtained slurry was applied with a blade applicator onto aluminum foil (for NMC, 16 μm thickness) or 316 stainless-steel mesh (for Li4Ti5O12, 325 × 325 mesh, opening size 0.0017′′), and resulting sheet/mesh were dried at 70 °C. Next, each electrode was punched with a 1/2 inch diameter (1.27 cm ∅) for NMC and 18 mm diameter for Li4Ti5O12 mesh reference. The NMC composite were compressed at 6.3 T cm−2 using a hydraulic press. All of the electrodes were further dried in vacuum at 120 °C prior to cell assembling. The active material loading was ∼2.7 mg cm−2 for NMC and ∼1 mg cm−2 for Li4Ti5O12 respectively. Particle size of NMC111, NMC622 and NMC811 was examined with a scanning electron microscope (JEOL 5910, with secondary electron detector at accelerating voltage of 15 kV) and shown in Fig. S23 (ESI†) (d = 5–10 μm).
In this study, the electrolytes include 1 M LiPF6 in a 3:7 wt:wt ethylene carbonate (EC):ethyl methyl carbonate (EMC) (LP57, battery-grade, BASF), 1.5 M LiPF6 (>99.99%, battery-grade, Aldrich) in EC (battery-grade, BASF), 1 M LiPF6 in EMC (battery-grade, BASF), and concentrated electrolyte with 3.1 M LiPF6 in EMC/EC. For LiPF6/EC electrolyte, the salt concentration was 1.5 M rather than 1 M so that the electrolyte was in liquid phase at room temperature. The electrolytes (except for LP57) were prepared by simple mixing of LiPF6 and EC or EMC in the glove box. The residual water content in the electrolytes was measured by Karl Fischer titration and was less than 20 ppm.
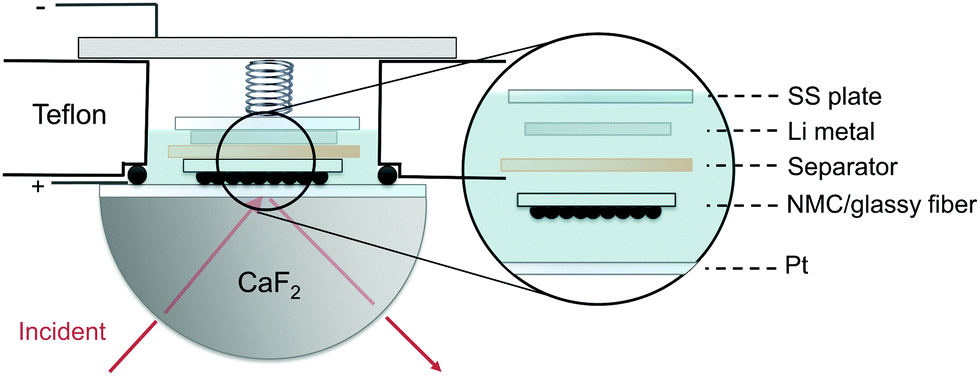 | ||
| Fig. 8 Schematic of in situ FT-IR spectro-electrochemical cell, where composite NMC drop-casted on glassy fiber substrate is the positive electrode with NMC particles faced towards the prism. | ||
After assembly, the cell was first rested for 6 hours. Next, galvanostatic charge were performed using BCS-COM (Biologic) and VMP3 (Biologic). Electrochemical behavior of the electrodes in the in situ FT-IR spectro-electrochemical cell was confirmed by galvanostatic measurements above (27.5 mA g−1). The actual or effective loading which could be charged might be smaller than measured: during electrode preparation (dropcasting) some particles could permeate through the glassy fiber substrate and stay on the other side of the substrate, and they may not be charged actually. Fig. S24 (ESI†) summarizes the charging time to reach 4.8 VLi during the first cycle for different electrode–electrolyte combinations in this paper, which shows comparable SOCs (8.2 hours on average with same charging rate at 27.5 mA g−1, variance within 20%, so the SOC reaches around 82%). For the measurements without oxides, sputtered Pt was the positive electrode and linear sweep voltammetry was performed.
At the same time during charging or linear voltage sweeping, in situ FT-IR measurements were performed on a Tensor II (Bruker) FT-IR equipped with deuterated triglycine sulfate (DTGS) detector inside an argon-filled glovebox. The FT-IR spectra were acquired in the single-reflection mode using an attenuated total reflection (ATR) accessory (Pike Vee-Max II, Pike Technologies) at an incident angle of 50 degrees. The spectral resolution was 4 cm−1 and the scan velocity was 1.6 kHz. Each spectrum was measured by superimposing 32 interferograms. All spectra were presented in the form of absorbance according to log(I0/I1), where I0 and I1 are the spectrum of background (blank Pt surface without electrolytes) and in situ spectrum of the sample, respectively.
For ex situ measurements on pristine electrolytes, the spectra were acquired in the ATR mode using a germanium (Ge) prism (Pier optics) at an incident angle of 50 degrees. Spectral settings were the same as in situ measurements. Although we assign the observed species based on DFT and solution ATR spectra, other species such as semicarbonates cannot be excluded, because there could be other features in the convoluted bands.
Computational methods
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 4) surface, modeled by a five-layer thick slab, as it has the lowest surface energy non-polar surface,125 and exposes the Li intercalation channels. The slabs were separated by at least 13 Å of vacuum and a dipole correction was applied to eliminate spurious interactions across the periodic boundary in the direction perpendicular to the surface. The coordinates of the adsorbate and the two upmost surface layers were allowed to fully relax, while the three bottom layers were fixed at the bulk positions.
4) surface, modeled by a five-layer thick slab, as it has the lowest surface energy non-polar surface,125 and exposes the Li intercalation channels. The slabs were separated by at least 13 Å of vacuum and a dipole correction was applied to eliminate spurious interactions across the periodic boundary in the direction perpendicular to the surface. The coordinates of the adsorbate and the two upmost surface layers were allowed to fully relax, while the three bottom layers were fixed at the bulk positions.
Author contribution
Conceptualization: Y. Z., Y. K., L. G., and Y. S.-H. Methodology: Y. Z., Y. K., R. T., L. G., Y. Y., J. G. S., and Y. S.-H. Investigation: Y. Z., R. T., L. G., Y. Y. Writing: Y. Z., R. T., L. G. Review & editing: Y. Z., Y. K., R. T., L. G., Y. Y., D. F., J. G. S., F. M., R. J., M. Z. B., and Y. S.-H. Supervision: Y. S.-H. Funding acquisition: Y. S.-H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by BMW group. This work made use of the MRSEC Shared Experimental Facilities at MIT (SEM), supported by the National Science Foundation under award number DMR-1419807. This research used resources of the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. This work also used resources of the Extreme Science and Engineering Discovery Environment (XSEDE),126 which is supported by National Science Foundation grant number ACI-1548562.References
- S. Menkin, D. Golodnitsky and E. Peled, Electrochem. Commun., 2009, 11, 1789–1791 CrossRef CAS.
- A. Smith, J. Burns, S. Trussler and J. Dahn, J. Electrochem. Soc., 2010, 157, A196–A202 CrossRef CAS.
- P. M. Attia, S. Das, S. J. Harris, M. Z. Bazant and W. C. Chueh, J. Electrochem. Soc., 2019, 166, E97–E106 CrossRef CAS.
- S. Das, P. M. Attia, W. C. Chueh and M. Z. Bazant, J. Electrochem. Soc., 2019, 166, E107–E118 CrossRef CAS.
- H.-J. Noh, S. Youn, C. S. Yoon and Y.-K. Sun, J. Power Sources, 2013, 233, 121–130 CrossRef CAS.
- S. Hwang, S. Y. Kim, K. Y. Chung, E. A. Stach, S. M. Kim and W. Chang, APL Mater., 2016, 4, 096105 CrossRef.
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2016, 2, 196–223 CrossRef.
- S. Hwang, S. M. Kim, S.-M. Bak, S. Y. Kim, B.-W. Cho, K. Y. Chung, J. Y. Lee, E. A. Stach and W. Chang, Chem. Mater., 2015, 27, 3927–3935 CrossRef CAS.
- Y.-K. Sun, S.-T. Myung, B.-C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320 CrossRef CAS.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef CAS.
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2016, 2, 196–223 CrossRef.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Phys. Chem. Lett., 2017, 8, 4820–4825 CrossRef CAS.
- L. Giordano, P. Karayaylali, Y. Yu, Y. Katayama, F. Maglia, S. Lux and Y. Shao-Horn, J. Phys. Chem. Lett., 2017, 8, 3881–3887 CrossRef CAS.
- Y. Yu, P. Karayaylali, Y. Katayama, L. Giordano, M. Gauthier, F. Maglia, R. Jung, I. Lund and Y. Shao-Horn, J. Phys. Chem. C, 2018, 122, 27368–27382 CrossRef CAS.
- M. Gauthier, P. Karayaylali, L. Giordano, S. Feng, S. F. Lux, F. Maglia, P. Lamp and Y. Shao-Horn, J. Electrochem. Soc., 2018, 165, A1377–A1387 CrossRef CAS.
- B. Han, T. Paulauskas, B. Key, C. Peebles, J. S. Park, R. F. Klie, J. T. Vaughey and F. Dogan, ACS Appl. Mater. Interfaces, 2017, 9, 14769–14778 CrossRef CAS.
- A. M. Wise, C. Ban, J. N. Weker, S. Misra, A. S. Cavanagh, Z. Wu, Z. Li, M. S. Whittingham, K. Xu and S. M. George, Chem. Mater., 2015, 27, 6146–6154 CrossRef CAS.
- J. Cho, T.-G. Kim, C. Kim, J.-G. Lee, Y.-W. Kim and B. Park, J. Power Sources, 2005, 146, 58–64 CrossRef CAS.
- X. Zhang, I. Belharouak, L. Li, Y. Lei, J. W. Elam, A. Nie, X. Chen, R. S. Yassar and R. L. Axelbaum, Adv. Energy Mater., 2013, 3, 1299–1307 CrossRef CAS.
- H.-M. Cheng, F.-M. Wang, J. P. Chu, R. Santhanam, J. Rick and S.-C. Lo, J. Phys. Chem. C, 2012, 116, 7629–7637 CrossRef CAS.
- Y.-K. Sun, B.-R. Lee, H.-J. Noh, H. Wu, S.-T. Myung and K. Amine, J. Mater. Chem., 2011, 21, 10108–10112 RSC.
- C. Hua, K. Du, C. Tan, Z. Peng, Y. Cao and G. Hu, J. Alloys Compd., 2014, 614, 264–270 CrossRef CAS.
- C. S. Yoon, S. J. Kim, U.-H. Kim, K.-J. Park, H.-H. Ryu, H.-S. Kim and Y.-K. Sun, Adv. Funct. Mater., 2018, 1802090 CrossRef.
- Y.-K. Sun, D.-H. Kim, H.-G. Jung, S.-T. Myung and K. Amine, Electrochim. Acta, 2010, 55, 8621–8627 CrossRef CAS.
- A. M. Haregewoin, A. S. Wotango and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 1955–1988 RSC.
- G.-Y. Kim and J. Dahn, J. Electrochem. Soc., 2015, 162, A437–A447 CrossRef CAS.
- R. Petibon, L. Rotermund and J. Dahn, J. Power Sources, 2015, 287, 184–195 CrossRef CAS.
- L. Ma, L. Ellis, S. L. Glazier, X. Ma and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A1718–A1724 CrossRef CAS.
- J. Li, H. Liu, J. Xia, A. R. Cameron, M. Nie, G. A. Botton and J. Dahn, J. Electrochem. Soc., 2017, 164, A655–A665 CrossRef CAS.
- C. Xu, S. Renault, M. Ebadi, Z. Wang, E. Björklund, D. Guyomard, D. Brandell, K. Edström and T. Gustafsson, Chem. Mater., 2017, 29, 2254–2263 CrossRef CAS.
- D. Aurbach, B. Markovsky, A. Rodkin, E. Levi, Y. S. Cohen, H.-J. Kim and M. Schmidt, Electrochim. Acta, 2002, 47, 4291–4306 CrossRef CAS.
- D. Aurbach, K. Gamolsky, B. Markovsky, G. Salitra, Y. Gofer, U. Heider, R. Oesten and M. Schmidt, J. Electrochem. Soc., 2000, 147, 1322–1331 CrossRef CAS.
- S. Feng, M. Huang, J. R. Lamb, W. Zhang, R. Tatara, Y. Zhang, Y. G. Zhu, C. F. Perkinson, J. A. Johnson and Y. Shao-Horn, Chem, 2019, 5(10), 2630–2641 CAS.
- K. Leung, J. Phys. Chem. C, 2012, 116, 9852–9861 CrossRef CAS.
- S. Xu, G. Luo, R. Jacobs, S. Fang, M. K. Mahanthappa, R. J. Hamers and D. Morgan, ACS Appl. Mater. Interfaces, 2017, 9, 20545–20553 CrossRef CAS PubMed.
- O. Borodin, M. Olguin, C. E. Spear, K. W. Leiter and J. Knap, Nanotechnology, 2015, 26, 354003 CrossRef.
- N. Kumar, K. Leung and D. J. Siegel, J. Electrochem. Soc., 2014, 161, E3059–E3065 CrossRef CAS.
- F. La Mantia, F. Rosciano, N. Tran and P. Novák, J. Appl. Electrochem., 2008, 38, 893–896 CrossRef CAS.
- D. J. Xiong, L. D. Ellis, J. Li, H. Li, T. Hynes, J. P. Allen, J. Xia, D. S. Hall, I. G. Hill and J. R. Dahn, J. Electrochem. Soc., 2017, 164, A3025–A3037 CrossRef CAS.
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H.-H. Chang, D. P. Fenning, S. F. Lux, O. Paschos and C. Bauer, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef.
- K. Leung, J. Phys. Chem. C, 2012, 116, 9852–9861 CrossRef CAS.
- A. Guéguen, D. Streich, M. He, M. Mendez, F. F. Chesneau, P. Novák and E. J. Berg, J. Electrochem. Soc., 2016, 163, A1095–A1100 CrossRef.
- R. Dedryvère, H. Martinez, S. Leroy, D. Lemordant, F. Bonhomme, P. Biensan and D. Gonbeau, J. Power Sources, 2007, 174, 462–468 CrossRef.
- K. Edström, T. Gustafsson and J. O. Thomas, Electrochim. Acta, 2004, 50, 397–403 CrossRef.
- P. Harks, F. M. Mulder and P. H. L. Notten, J. Power Sources, 2015, 288, 92–105 CrossRef CAS.
- B. Stuart, Kirk-Othmer Encycl. Chem. Technol., 2000, 1–18 Search PubMed.
- E. Goren, O. Chusid and D. Aurbach, J. Electrochem. Soc., 1991, 138, L6–L9 CrossRef CAS.
- G. V. Zhuang, H. Yang, B. Blizanac and P. N. Ross, Electrochem. Solid-State Lett., 2005, 8, A441–A445 CrossRef CAS.
- K. Hongyou, T. Hattori, Y. Nagai, T. Tanaka, H. Nii and K. Shoda, J. Power Sources, 2013, 243, 72–77 CrossRef CAS.
- M. Moshkovich, M. Cojocaru, H. E. Gottlieb and D. Aurbach, J. Electroanal. Chem., 2001, 497, 84–96 CrossRef CAS.
- K. Kanamura, S. Toriyama, S. Shiraishi, M. Ohashi and Z. Takehara, J. Electroanal. Chem., 1996, 419, 77–84 CrossRef CAS.
- Y. Akita, M. Segawa, H. Munakata and K. Kanamura, J. Power Sources, 2013, 239, 175–180 CrossRef CAS.
- K. Kanamura, T. Umegaki, M. Ohashi, S. Toriyama, S. Shiraishi and Z. Takehara, Electrochim. Acta, 2001, 47, 433–439 CrossRef CAS.
- M. Matsui, K. Dokko, Y. Akita, H. Munakata and K. Kanamura, J. Power Sources, 2012, 210, 60–66 CrossRef CAS.
- N. D. Phillip, R. E. Ruther, X. Sang, Y. Wang, R. R. Unocic, A. S. Westover, C. Daniel and G. M. Veith, ACS Appl. Energy Mater., 2019, 2, 1405–1412 CrossRef CAS.
- R. Sharabi, E. Markevich, V. Borgel, G. Salitra, D. Aurbach, G. Semrau and M. A. Schmidt, Electrochem. Solid-State Lett., 2010, 13, A32–A35 CrossRef CAS.
- M. A. Teshager, S. D. Lin, B.-J. Hwang, F.-M. Wang, S. Hy and A. M. Haregewoin, ChemElectroChem, 2016, 3, 337–345 CrossRef CAS.
- S. Hy, F. Felix, J. Rick, W.-N. Su and B. J. Hwang, J. Am. Chem. Soc., 2014, 136, 999–1007 CrossRef CAS.
- X. Zhang, J. K. Pugh and P. N. Ross, J. Electrochem. Soc., 2001, 148, E183–E188 CrossRef CAS.
- L. Xing, O. Borodin, G. D. Smith and W. Li, J. Phys. Chem. A, 2011, 115, 13896–13905 CrossRef CAS PubMed.
- O. Borodin and T. R. Jow, ECS Trans., 2011, 33, 77–84 CAS.
- M. Ue, M. Takeda, M. Takehara and S. Mori, J. Electrochem. Soc., 1997, 144, 2684–2688 CrossRef CAS.
- M. Egashira, H. Takahashi, S. Okada and J. Yamaki, J. Power Sources, 2001, 92, 267–271 CrossRef CAS.
- K. Xu, S. P. Ding and T. R. Jow, J. Electrochem. Soc., 1999, 146, 4172–4178 CrossRef CAS.
- J.-C. Lee and M. H. Litt, Macromolecules, 2000, 33, 1618–1627 CrossRef CAS.
- S. E. Sloop, J. B. Kerr and K. Kinoshita, J. Power Sources, 2003, 119, 330–337 CrossRef.
- L. Xing, W. Li, C. Wang, F. Gu, M. Xu, C. Tan and J. Yi, J. Phys. Chem. B, 2009, 113, 16596–16602 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- L. Yu, H. Liu, Y. Wang, N. Kuwata, M. Osawa, J. Kawamura and S. Ye, Angew. Chem., 2013, 125, 5865–5868 CrossRef.
- H. Yoshida, T. Fukunaga, T. Hazama, M. Terasaki, M. Mizutani and M. Yamachi, J. Power Sources, 1997, 68, 311–315 CrossRef CAS.
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2190–2200 RSC.
- L. Giordano, T. M. Østergaard, S. Muy, Y. Yu, N. Charles, S. Kim, Y. Zhang, F. Maglia, R. Jung, I. Lund, J. Rossmeisl and Y. Shao-Horn, Chem. Mater, 2019, 31(15), 5464–5474 CrossRef CAS.
- L. Giordano, P. Karayaylali, Y. Yu, Y. Katayama, F. Maglia, S. Lux and Y. Shao-Horn, J. Phys. Chem. Lett., 2017, 8, 3881–3887 CrossRef CAS PubMed.
- T. M. Østergaard, L. Giordano, I. E. Castelli, F. Maglia, B. K. Antonopoulos, Y. Shao-Horn and J. Rossmeisl, J. Phys. Chem. C, 2018, 122, 10442–10449 CrossRef.
- K. Leung, Y. Qi, K. R. Zavadil, Y. S. Jung, A. C. Dillon, A. S. Cavanagh, S.-H. Lee and S. M. George, J. Am. Chem. Soc., 2011, 133, 14741–14754 CrossRef CAS PubMed.
- Y.-C. Lu, A. N. Mansour, N. Yabuuchi and Y. Shao-Horn, Chem. Mater., 2009, 21, 4408–4424 CrossRef CAS.
- S. Verdier, L. El Ouatani, R. Dedryvere, F. Bonhomme, P. Biensan and D. Gonbeau, J. Electrochem. Soc., 2007, 154, A1088–A1099 CrossRef CAS.
- J. Cho, Y. J. Kim and B. Park, Chem. Mater., 2000, 12, 3788–3791 CrossRef CAS.
- S.-K. Hu, G.-H. Cheng, M.-Y. Cheng, B.-J. Hwang and R. Santhanam, J. Power Sources, 2009, 188, 564–569 CrossRef CAS.
- N. Yesibolati, M. Shahid, W. Chen, M. N. Hedhili, M. C. Reuter, F. M. Ross and H. N. Alshareef, Small, 2014, 10, 2849–2858 CrossRef CAS.
- H. Nohira, W. Tsai, W. Besling, E. Young, J. Pétry, T. Conard, W. Vandervorst, S. De Gendt, M. Heyns and J. Maes, J. Non-Cryst. Solids, 2002, 303, 83–87 CrossRef CAS.
- P. Karayaylali, R. Tatara, Y. Zhang, K.-L. Chan, Y. Yu, L. Giordano, F. Maglia, R. Jung, I. Lund and Y. Shao-Horn, J. Electrochem. Soc., 2019, 166, A1022–A1030 CrossRef CAS.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406–A2423 CrossRef CAS.
- Y. Yamada and A. Yamada, Chem. Lett., 2017, 46, 1056–1064 CrossRef.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- R. Tatara, Y. Yu, P. Karayaylali, A. K. Chan, Y. Zhang, R. Jung, F. Maglia, L. Giordano and Y. Shao-Horn, ACS Appl. Mater. Interfaces, 2019, 11, 34973–34988 CrossRef CAS PubMed.
- S.-D. Han, S.-H. Yun, O. Borodin, D. M. Seo, R. D. Sommer, V. G. Young Jr and W. A. Henderson, J. Phys. Chem. C, 2015, 119, 8492–8500 CrossRef CAS.
- A. K. Chan, R. Tatara, S. Feng, P. Karayaylali, J. Lopez, I. E. Stephens and Y. Shao-Horn, J. Electrochem. Soc., 2019, 166, A1867–A1874 CrossRef CAS.
- R. Tatara, D. G. Kwabi, T. P. Batcho, M. Tulodziecki, K. Watanabe, H.-M. Kwon, M. L. Thomas, K. Ueno, C. V. Thompson and K. Dokko, J. Phys. Chem. C, 2017, 121, 9162–9172 CrossRef CAS.
- S. Terada, K. Ikeda, K. Ueno, K. Dokko and M. Watanabe, Aust. J. Chem., 2019, 72, 70–80 CrossRef CAS.
- K. Ueno, R. Tatara, S. Tsuzuki, S. Saito, H. Doi, K. Yoshida, T. Mandai, M. Matsugami, Y. Umebayashi and K. Dokko, Phys. Chem. Chem. Phys., 2015, 17, 8248–8257 RSC.
- Z. Cao, M. Hashinokuchi, T. Doi and M. Inaba, J. Electrochem. Soc., 2019, 166, A82–A88 CrossRef CAS.
- T. Doi, Y. Shimizu, M. Hashinokuchi and M. Inaba, J. Electrochem. Soc., 2016, 163, A2211–A2215 CrossRef CAS.
- T. Doi, M. Hashinokuchi and M. Inaba, Curr. Opin. Electrochem., 2018, 9, 49–55 CrossRef CAS.
- C. P. Aiken, J. Harlow, L. Thompson, M. Bauer, T. Hynes, X. Ma and J. R. Dahn, Meeting Abstracts, The Electrochemical Society, 2019, pp. 223–223 Search PubMed.
- R. Petibon, L. Madec, D. W. Abarbanel and J. R. Dahn, J. Power Sources, 2015, 300, 419–429 CrossRef CAS.
- X. Ma, J. Harlow, A. S. Keefe, J. Li, S. Glazier, L. Ma, D. S. Hall, C. P. Aiken, M. Genovese and M. Cormier, Meeting Abstracts, The Electrochemical Society, 2019, pp. 210–210 Search PubMed.
- D. M. Seo, O. Borodin, S.-D. Han, P. D. Boyle and W. A. Henderson, J. Electrochem. Soc., 2012, 159, A1489–A1500 CrossRef CAS.
- A. von Wald Cresce, M. Gobet, O. Borodin, J. Peng, S. M. Russell, E. Wikner, A. Fu, L. Hu, H.-S. Lee and Z. Zhang, J. Phys. Chem. C, 2015, 119, 27255–27264 CrossRef CAS.
- S.-A. Hyodo and K. Okabayashi, Electrochim. Acta, 1989, 34, 1551–1556 CrossRef CAS.
- R. Chen, F. Wu, L. Li, X. Qiu, L. Chen and S. Chen, Vib. Spectrosc., 2007, 44, 297–307 CrossRef CAS.
- M. Shakourian-Fard, G. Kamath and S. K. Sankaranarayanan, ChemPhysChem, 2016, 17, 2916–2930 CrossRef CAS.
- R. Tatara, P. Karayaylali, Y. Yu, Y. Zhang, L. Giordano, F. Maglia, R. Jung, J. P. Schmidt, I. Lund and Y. Shao-Horn, J. Electrochem. Soc., 2019, 166, A5090–A5098 CrossRef CAS.
- E. R. Logan, E. M. Tonita, K. L. Gering, J. Li, X. Ma, L. Y. Beaulieu and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A21–A30 CrossRef CAS.
- R. P. Day, J. Xia, R. Petibon, J. Rucska, H. Wang, A. T. B. Wright and J. R. Dahn, J. Electrochem. Soc., 2015, 162, A2577–A2581 CrossRef CAS.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361–A1377 CrossRef CAS.
- A. T. Freiberg, M. K. Roos, J. Wandt, R. de Vivie-Riedle and H. A. Gasteiger, J. Phys. Chem. A, 2018, 122, 8828–8839 CrossRef CAS PubMed.
- S. Wiemers-Meyer, S. Jeremias, M. Winter and S. Nowak, Electrochim. Acta, 2016, 222, 1267–1271 CrossRef CAS.
- D. Aurbach, B. Markovsky, G. Salitra, E. Markevich, Y. Talyossef, M. Koltypin, L. Nazar, B. Ellis and D. Kovacheva, J. Power Sources, 2007, 165, 491–499 CrossRef CAS.
- R. Petibon, L. M. Rotermund and J. R. Dahn, J. Power Sources, 2015, 287, 184–195 CrossRef CAS.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS.
- M. Ender, J. Illig and E. Ivers-Tiffée, J. Electrochem. Soc., 2017, 164, A71–A79 CrossRef CAS.
- J. Costard, M. Ender, M. Weiss and E. Ivers-Tiffée, J. Electrochem. Soc., 2017, 164, A80–A87 CrossRef CAS.
- T. Osaka, D. Mukoyama and H. Nara, J. Electrochem. Soc., 2015, 162, A2529–A2537 CrossRef CAS.
- Y. Hoshi, Y. Narita, K. Honda, T. Ohtaki, I. Shitanda and M. Itagaki, J. Power Sources, 2015, 288, 168–175 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson and H. Nakatsuji, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- V. I. Anisimov, F. Aryasetiawan and A. Lichtenstein, J. Phys.: Condens. Matter, 1997, 9, 767 CrossRef CAS.
- S. Dudarev, G. Botton, S. Savrasov, C. Humphreys and A. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- Y.-L. Lee, J. Kleis, J. Rossmeisl and D. Morgan, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 224101 CrossRef.
- D. Kramer and G. Ceder, Chem. Mater., 2009, 21, 3799–3809 CrossRef CAS.
- J. Towns, T. Cockerill, M. Dahan, I. Foster, K. Gaither, A. Grimshaw, V. Hazlewood, S. Lathrop, D. Lifka and G. D. Peterson, Comput. Sci. Eng., 2014, 16, 62–74 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee02543j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |