
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterobimetallic propargyl gold complexes with π-bound copper or silver with enhanced anticancer activity†
Alice
Johnson
 a,
Isabel
Marzo
b and
M. Concepción
Gimeno
*a
a,
Isabel
Marzo
b and
M. Concepción
Gimeno
*a
aDepartamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, 50009 Zaragoza, Spain. E-mail: gimeno@unizar.es
bDepartamento de Bioquímica y Biología Celular, Universidad de Zaragoza-CSIC, 50009 Zaragoza, Spain
First published on 12th August 2020
Abstract
Several propargyl functionalised substrates with different heteroatoms (N, O or S) have been used for the preparation of propargyl gold(I) phosphine complexes. The complexes were prepared in high yields either by reaction of the substrate with [Au(acac)PPh3] or by reaction of [AuCl(PPh3)] with potassium hydroxide and the substrate in methanol. Several of the complexes have been characterised by X-ray diffraction showing the presence of secondary bonds such as π-stacking and aurophilic interactions. The reaction of the propargyl gold(I) phosphine complexes with [Cu(NO3)(PPh3)2] or [Ag(OTf)(PPh3)2] afforded heterobimetallic complexes with π-coordination of {Cu(PPh3)2} or {Ag(PPh3)2} to the alkyne bond. When the substituent of the propargyl unit contained more strongly coordinating pyridine moieties, [(PyCH2)2NCH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CAuPPh3], coordination of the heterometal to the pyridine units occurred, displacing the phosphine groups and giving rise to a dimeric structure. The antiproliferative activity of the complexes against cisplatin resistant lung cancer cell line A549 was determined by MTT assay. The mononuclear gold complexes showed excellent activities with IC50 values < 14 μM. Coordination of copper of silver to the alkynyl fragment resulted in a significant increase in activity suggesting a synergistic effect between the two metal centres.
CAuPPh3], coordination of the heterometal to the pyridine units occurred, displacing the phosphine groups and giving rise to a dimeric structure. The antiproliferative activity of the complexes against cisplatin resistant lung cancer cell line A549 was determined by MTT assay. The mononuclear gold complexes showed excellent activities with IC50 values < 14 μM. Coordination of copper of silver to the alkynyl fragment resulted in a significant increase in activity suggesting a synergistic effect between the two metal centres.
Introduction
Following the serendipitous discovery of the anticancer activity of cisplatin over 50 years ago1 and its subsequent approval by the FDA for the treatment of cancers in 1978, there has been increasing interest in the use of transition metal complexes in cancer treatments. Despite the success of the platinum complexes, there are several known side-effects and increasing reports of cisplatin resistant cancers and hence considerable research is now focussed on other metals. Interest in gold(I) complexes initially arose as a result of the anti-rheumatic drug Auranofin (Fig. 1(a)) which has shown potent anticancer activity.2 Gold(I) has not only been shown to be better tolerated in vivo than platinum, but the mechanism of action is significantly different since gold(I) targets cellular enzymes rather than DNA.3,4 Of the gold(I) complexes reported to have anticancer activity, alkynyl–gold(I)–phosphine complexes are among the most potent.5–14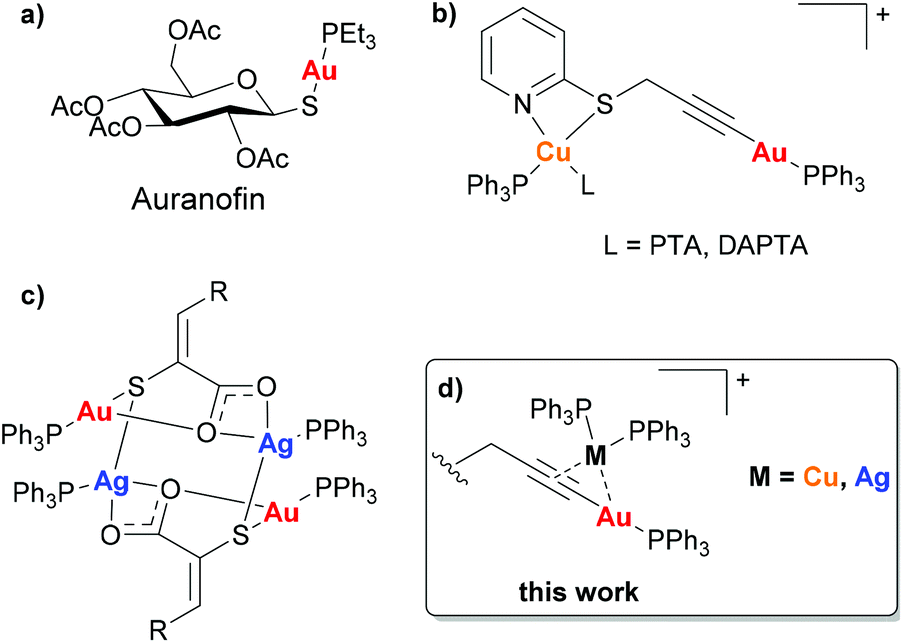 | ||
| Fig. 1 (a) Auranofin, (b) Au(I)–Cu(I) complexes reported by Laguna [PTA = 1,3,5-Triaza-7-phosphaadamantane, DAPTA = 3,7-diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane],16 (c) Au(I)–Ag(I) complexes reported by Couce [R = phenyl, 2-chlorophenyl, 3-methoxyphenyl, 3-hydroxyphenyl, 2-furyl, 2-thienyl],17 (d) this work. | ||
The incorporation of a second different metal centre into a complex can alter the physiochemical properties significantly. This can be advantageous in the development of chemotherapeutic agents as a means of enhancing cytotoxicity. In these bimetallic compounds, where both metals could be cytotoxic via different mechanisms of action, there is the possibility for a synergistic effect due to the interaction with multiple biological targets, modulation in redox properties and/or changes in compound stability that could result in the improvement of the antitumor activity in comparison with the mononuclear precursors.15
There have been several reports of heterometallic complexes exhibiting such a synergistic effect between the metal centres, and the presence of two metal centres which display cytotoxic activity through different mechanisms of action can also prevent resistance occurring. However, there have been very few reported studies of the anticancer activity of heterometallic complexes of the coinage metals. Laguna and co-workers reported the anticancer activity of four Au(I)–Cu(I) complexes (Fig. 1(b)) which all had IC50 values in the nanomolar range, significantly lower than cisplatin or Auranofin when tested under the same conditions. Although the mechanism of action of the heterometallic complexes was not fully understood the authors suggested that the complexes achieved the correct balance of hydrophilicity and lipophilicity to maximise the cytotoxicity.16 The only reported cytotoxic Au(I)–Ag(I) heterometallic complexes to date are a series of 3-(aryl)-2-sulfanylpropenoic acid derivatives reported by Couce and co-workers (Fig. 1(c)). The complexes exhibited greater cytotoxicity in the cervical cancer cell line A2780cisR, however no mechanistic studies were carried out.17 In general, the studies carried out with bimetallic complexes highlight that anti-proliferative properties are mainly driven by the most cytotoxic metal. However, the cooperative effect and synergism may be observed in metal-specific interactions with distinct biological targets or by improvement of the physicochemical properties of the final compounds.18
The π-coordination of a Cu(PPh3)2 or Ag(PPh3)2 group to an alkynyl–gold complex has previously been explored as a means of enhancing the luminescence.19,20 Such complexes can readily be synthesised and are known to be highly stable, however despite this their potential biological application has not been explored. Here were report the synthesis of a series of propargyl–Au(I)–PPh3 complexes and study their anticancer activity in a cisplatin resistant cancer cell line, A549. We then show how the alkyne bond can provide a coordination site for a second metal centre (Cu or Ag) giving complexes with improved cytotoxicity.
Results and discussion
Synthesis and characterisation
Propargyl functionalised substrates with different heteroatoms were chosen for the preparation of propargyl gold(I) phosphine complexes.Propargyl gold(I) triphenylphosphine complexes 1–8 could be prepared by two different methods (Scheme 1). Addition of [Au(acac)(PPh3)] to a solution of the propargyl substrate in dichloromethane led to the formation of the complexes which could be isolated by concentration of the reaction solution and precipitation with diethyl ether, and were obtained in good yields (48–99%).† Alternatively, the products could be prepared by the addition of [AuCl(PPh3)] and potassium hydroxide to a solution of the substrate in methanol. This method avoids the need to prepare the acac gold(I) precursor and the products are obtained with high purity since they precipitate from the reaction solution, however, in most cases the yields were lower (12–91%). All of the complexes were isolated as air-stable white solids. Successful reaction could be confirmed by the disappearance of the propargyl CH signal in the 1H NMR spectra, and the 31P NMR spectra all showed a sharp signal at around 42 ppm, a typical value for a triphenylphosphine group bound to gold(I) trans to a carbon-based ligand.
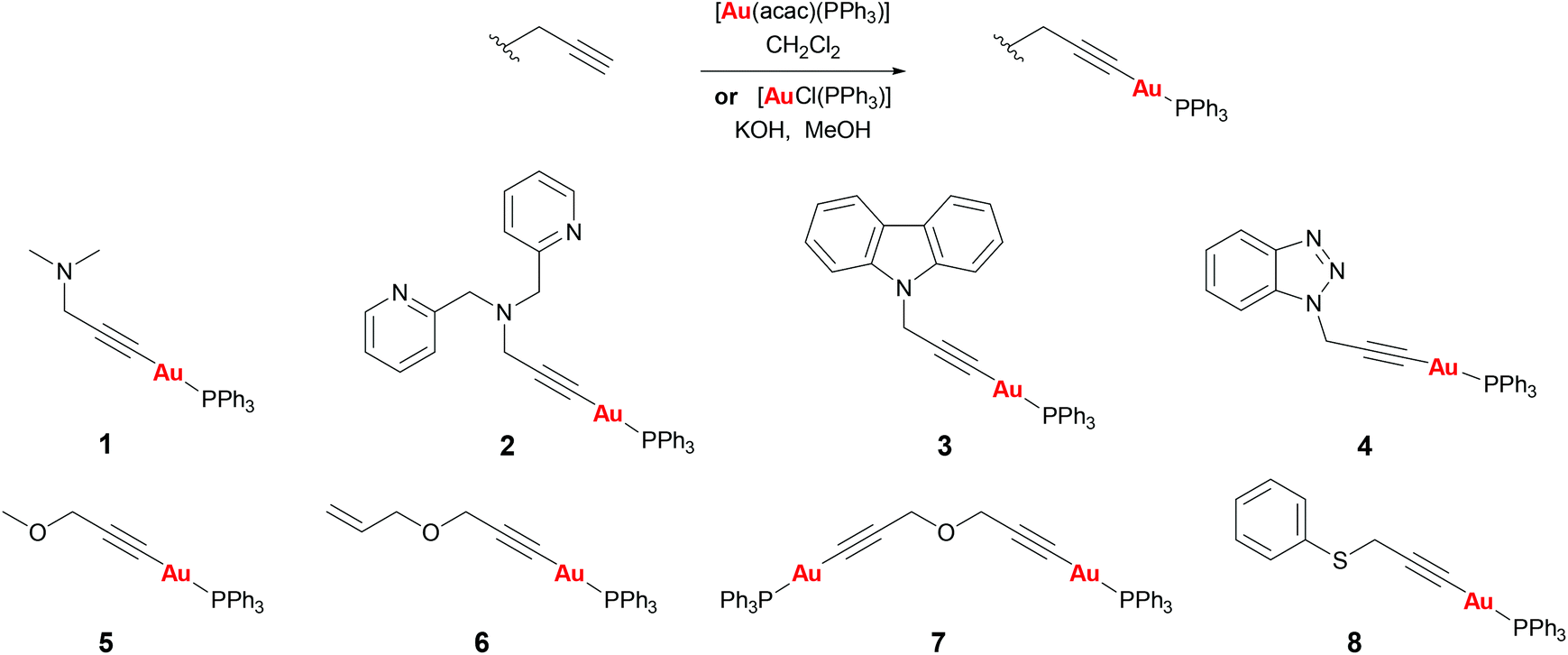 | ||
| Scheme 1 Synthesis of complexes 1–8. | ||
Complex 1 was found to be unstable in solution in dichloromethane, slowly converting into another product as observed by NMR studies (see ESI†). This is likely to be a dimeric or oligomeric species as a result of coordination of the sterically unhindered amine group to the gold centre. Complexes 2–8 were all stable in solution for at least 24 h.
The nitrogen based propargyl derivatives 2–4 were characterised by single crystal X-ray diffraction (Fig. 2). In all cases the propargyl bond distances are typical of those expected for triple and single C–C bonds. An almost linear geometry is observed about the gold centres and the trans influence of the phosphine ligand results in relatively long Au–C bonds. The Au–C distances are Au(1)–C(1) 2.024(5) Å for 2, Au(1)–C(1) 2.0004(18) Å for 3, and Au(1)–C(1) 1.997(3) Å for 4.
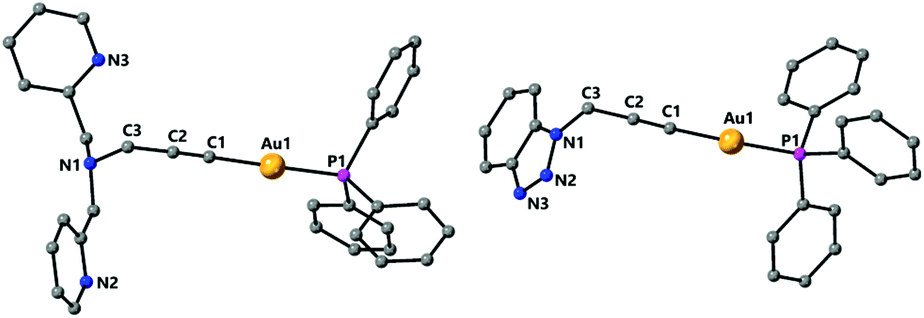 | ||
| Fig. 2 Molecular structures of complexes 2 and 4 determined by single crystal X-ray diffraction. Selected bond lengths [Å] and angles [°] 2: Au(1)–C(1) 2.024(5), Au(1)–P(1) 2.2711(13), C(1)–C(2) 1.173(7), C(2)–C(3) 1.487(8), C(1)–Au(1)–P(1) 178.95(15), C(1)–C(2)–C(3) 176.6(6); 4: Au(1)–C(1) 1.997(3), Au(1)–P(1) 2.2772(8), C(1)–C(2) 1.198(4), C(2)–C(3) 1.468(4), C(1)–Au(1)–P(1) 175.31(8), C(1)–C(2)–C(3) 178.7(3). | ||
Complexes 3 and 4 have a structure in which the molecules are arranged in dimers as a result of π-stacking interactions. For complex 3 the six-membered ring of the carbazole unit of one molecule lies over the five-membered ring of the carbazole unit of another molecule with intermolecular distances centroid–centroid 3.551 Å and C(7)–N(1)′ 3.546 Å observed (Fig. 3). For complex 4 a slipped π-stacking interaction between the benzotriazole rings with a distance of 3.654 Å is also observed (see ESI†).
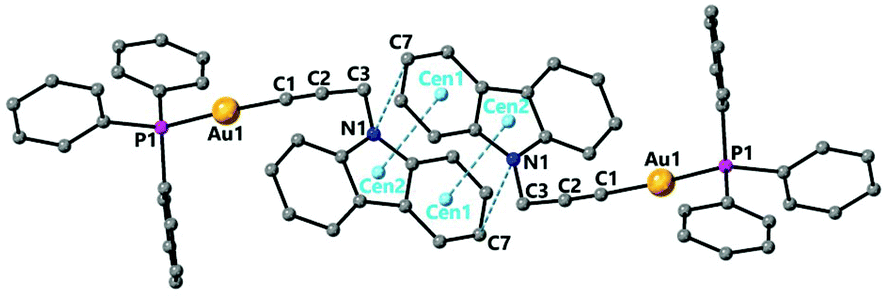 | ||
| Fig. 3 Molecular structures of complex 3 determined by single crystal X-ray diffraction showing π-stacking interactions. Selected bond lengths [Å] and angles [°]: Au(1)–C(1) 2.0004(18), Au(1)–P(1) 2.2686(8), C(1)–C(2) 1.200(2), C(2)–C(3) 1.472(2), C(1)–Au(1)–P(1) 175.56(5), C(1)–C(2)–C(3) 173.60(17), Centroid(1)–Centroid(2) 3.551, C(7)–N(1)′ 3.546. | ||
The structure of the propargyl ether and thioether gold complexes 5 and 8 is shown in Fig. 4. The general bond lengths and angles are similar to those found in the complexes above. The Au–C distances are 2.005(5) for 5, 2.060(9) for 7 (Fig. 4) or 1.994(3) Å for 8.
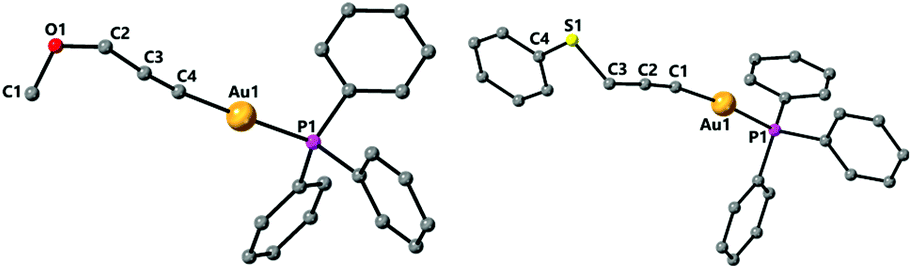 | ||
| Fig. 4 Molecular structures of complexes 5 and 8 determined by single crystal X-ray diffraction. Selected bond lengths [Å] and angles [°] 5: Au(1)–C(4) 2.005(5), Au(1)–P(1) 2.2870(12), O(1)–C(1) 1.393(7), O(1)–C(2) 1.416(6), C(2)–C(3) 1.489(7), C(3)–C(4) 1.205(7), C(4)–Au(1)–P(1) 178.38(15), C(4)–C(3)–C(2) 174.4(5); 8: Au(1)–C(1) 1.994(3), Au(1)–P(1) 2.2766(7), S(1)–C(4) 1.763(3), S(1)–C(3) 1.818(4), C(1)–C(2) 1.206(4), C(2)–C(3) 1.467(4), C(1)–Au(1)–P(1) 173.51(9), C(1)–C(2)–C(3) 175.1(3). | ||
The dinuclear complex 7 is the only compound to display intermolecular aurophilic interactions in the solid state. This results in the molecules being associated in chains with an intermolecular Au–Au distance of 3.2907(4) Å (Fig. 5).
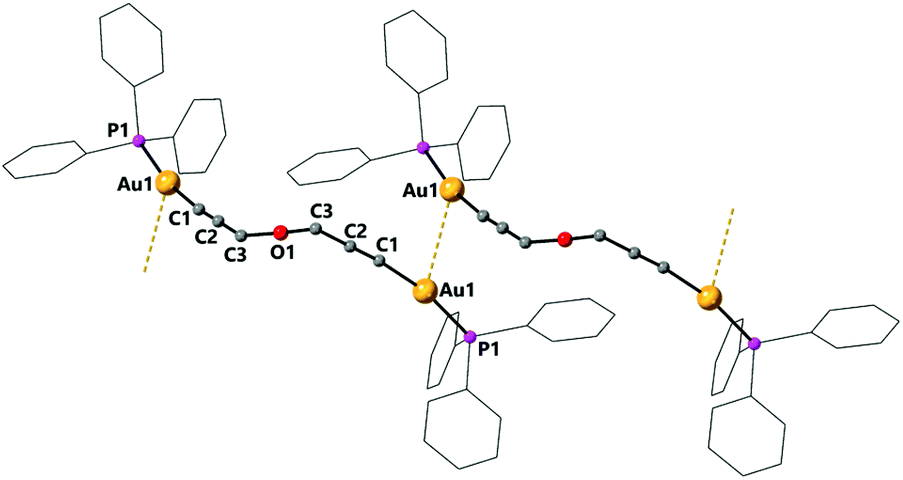 | ||
| Fig. 5 Molecular structure of complex 7 determined by single crystal X-ray diffraction showing intermolecular aurophilic interactions. Selected bond lengths [Å] and angles [°]: Au(1)–C(1) 2.060(9), Au1–C1B 1.951(9), Au(1)–P(1) 2.2733(12), Au(1)–Au(1)#1 3.2907(4), C(1)–C(2) 1.205(2), C(2)–C(3) 1.439(2), C(1)–Au(1)–P(1) 168.0(7), C(1)–C(2)–C(3) 177.6(18). In complex 7 the oxygen atom is disordered, only distances to the main component are included. | ||
Reaction of complex 2 with one equivalent of [Cu(NO3)(PPh3)2] in dichloromethane gave the dimeric complex 9 (Scheme 2). The copper successfully binds to the alkyne bond of the propargyl unit and the triphenylphosphine ligands at the copper centre are displaced by the nitrogen donor pyridine and amine groups. The analogous reaction with [Ag(OTf)(PPh3)] led to a mixture of products due to the stronger affinity of the silver centre for phosphine over nitrogen ligands.
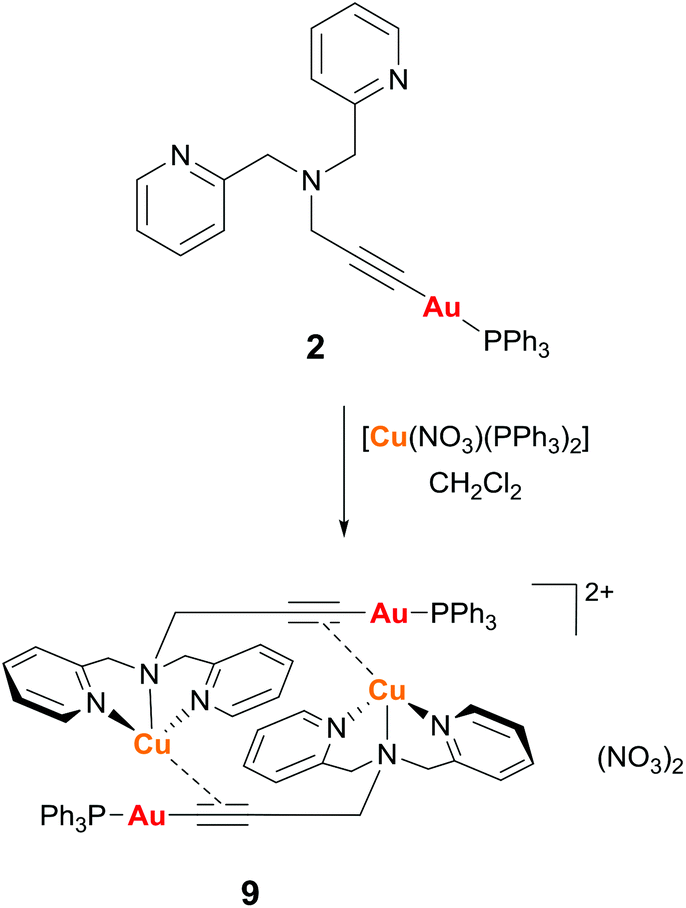 | ||
| Scheme 2 Synthesis of complex 9. | ||
Complex 9 was characterised by single crystal X-ray diffraction. The molecule lies in a symmetry centre and only half of the molecule corresponds to the asymmetric unit (Fig. 6). Coordination of the copper to the alkyne triple bond results in a lengthening of the bond to 1.233(4) Å compared to 1.173(7) Å for complex 2. The copper coordinates to the centre of the alkyne triple bond with distances Cu(1)–C(1) 2.019(2) Å and Cu(1)–C(2) 2.004(2) Å. In addition, the Au(1)–C(1) bond is 1.999(3) Å, which is very similar to that in the starting complex 2 suggesting that coordination of the copper fragment does not significantly affect the strength of the gold–carbon bond. There are two short Cu–N bonds, with lengths Cu(1)–N(1) 2.033(2) Å and Cu(1)–N(3) 2.017(2) Å to the nitrogens of the pyridyl groups, and one longer Cu(1)–N(2) 2.264(2) Å to the amine nitrogen. The coordination about the gold centre is slightly distorted from linearity due to steric hindrance between the bulky triphenylphosphine ligand and the pyridyl groups with an angle P(1)–Au(1)–C(1) 171.15(7)°. The propargyl angle is considerably distorted from linearity as a result of the coordination of the copper, with an angle C(1)–C(2)–C(3) 160.1(3)°. The Cu(1)–Au(1) distance of 3.535 Å is too long to indicate the presence of any metallophilic interaction between the two metal centres.
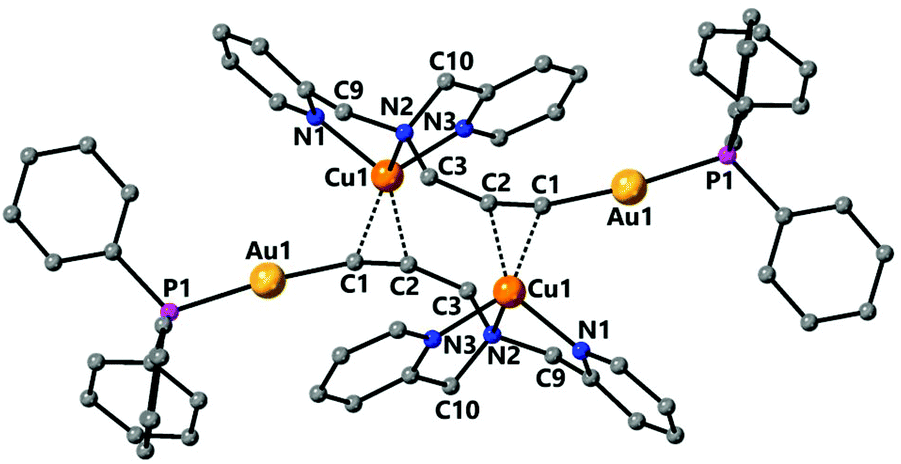 | ||
| Fig. 6 Molecular structure of complex 9 determined by single crystal X-ray diffraction. Selected bond lengths [Å] and angles [°]: Au(1)–C(1) 1.999(3), Au(1)–P(1) 2.2694(6), Cu(1)–C(2) 2.004(2), Cu(1)–N(3) 2.017(2), Cu(1)–C(1) 2.019(2), Cu(1)–N(1) 2.033(2), Cu(1)–N(2) 2.264(2), C(1)–C(2) 1.233(4), C(2)–C(3) 1.479(3), C(1)–Au(1)–P(1) 171.15(7), C(2)–Cu(1)–N(3) 106.54(9), C(2)–Cu(1)–C(1) 35.70(10), N(3)–Cu(1)–C(1) 142.18(9), C(2)–Cu(1)–N(1) 142.73(9), N(3)–Cu(1)–N(1) 109.17(8), C(1)–Cu(1)–N(1) 108.27(9), C(2)–Cu(1)–N(2) 116.78(9), N(3)–Cu(1)–N(2) 80.69(8), C(1)–Cu(1)–N(2) 111.27(9), N(1)–Cu(1)–N(2) 79.63(8), C(1)–C(2)–C(3) 160.1(3). | ||
The 31P NMR spectrum of 9 displays a broad signal at room temperature, indicative of a fluxional process, however at 193 K this signal becomes a sharp singlet and the protons of the CH2Py groups become inequivalent, in agreement with the solid-state structure.
Complexes 10–19 were prepared by reaction of mononuclear gold complexes 3–6 and 8 with [Cu(NO3)(PPh3)2] or [Ag(OTf)(PPh3)2] (Scheme 3). In these complexes the copper or silver binds to the alkyne triple bond, maintaining the two triphenylphosphine ligands. The identity of 10–19 can be confirmed by NMR studies. The 1H NMR spectra show additional aromatic protons for the triphenylphosphine ligands and two broad peaks are observed in the 31P{1H} NMR spectra.
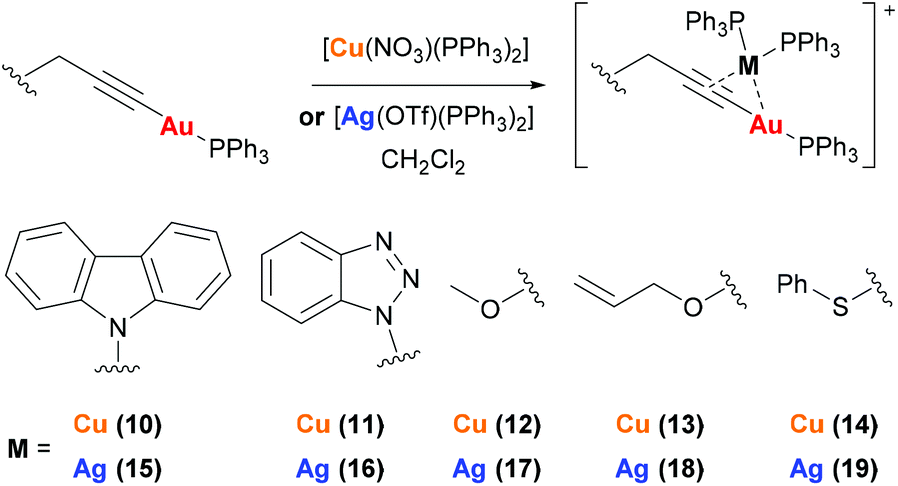 | ||
| Scheme 3 Synthesis of heterometallic complexes 10–19. | ||
Biological activity
Complexes 2–19 were not soluble in water but were soluble in DMSO and mixtures of DMSO/water used to carry out the tests. Stability studies have been done for the mononuclear complexes in DMSO by 1H NMR (Fig. S1–S9†) and the bimetallic complexes were tested in DMSO/PBS by UV-vis spectroscopy (Fig. S10–S16†) for a period of 24 h. No changes were observed between the initial 1H NMR or UV-vis spectra and the corresponding ones after 24 h, confirming the retention of the complexes in solution. The biological activity of the complexes was studied by MTT assay for lung carcinoma cell line A549. The calculated IC50 values for the complexes after 24 h incubation are shown in Table 1.| Au complex | IC50 (μM) | Au–Cu complex | IC50 (μM) | Au–Ag complex | IC50 (μM) |
|---|---|---|---|---|---|
| a Value for the mononuclear complex. | |||||
| 2 | 13.32 ± 0.51 | 9 | 1.37 ± 0.21 | ||
| 3 | >100 | 10 | 6.16 ± 0.18 | 15 | 4.92 ± 0.04 |
| 4 | 11.91 ± 1.54 | 11 | 2.50 ± 0.19 | 16 | 1.19 ± 0.03 |
| 5 | 10.27 ± 0.76 | 12 | 1.78 ± 0.18 | 17 | 1.44 ± 0.13 |
| 6 | 10.61 ± 0.93 | 13 | 1.60 ± 0.18 | 18 | 1.08 ± 0.12 |
| 7 | 12.28 ± 2.66 | ||||
| 8 | 9.11 ± 1.93 | 14 | 1.64 ± 0.10 | 19 | 1.44 ± 0.13 |
| [Cu(NO 3 )(PPh 3 ) 2 ] | 4.17 ± 0.19 | [Ag(OTf)(PPh 3 ) 2 ] | 2.54 ± 0.26 | ||
The carbazole derivative, complex 3, was the only complex not to show any antiproliferative activity against A549 cells at the concentrations tested with IC50 value of >100 μM. The other monometallic gold complexes (2, 4–6 and 8) showed excellent IC50 values for 24 h incubation with the cancer cells. No significant differences are observed upon changing the heteroatom of the propargyl substrate as all of the IC50 values are within a similar range 9–13 μM, the sulfur derivative, 8, having the lowest IC50 value of the gold triphenylphosphine complexes at 9.11 ± 1.93 μM. Dinuclear gold triphenylphosphine complex 7, derivative of dipropargylether did not show a greater activity than the mononuclear propargylether derivatives 5 and 6. There therefore appears to be no correlation between the number of gold-triphenylphosphine units in the complex and the overall activity. A comparison with the cytotoxicity of the reference cisplatin complex in the same conditions, 114.2 ± 9.1 μM,21 although measured in water, revealed a much higher activity for the gold complexes.
The introduction of the π-bound copper(I) or silver(I) ion had a huge effect on the antiproliferative activity of the complexes. In all cases the IC50 values were considerably lower, most notable for the carbazole derivatives where the gold complex 3 showed no activity but the gold–copper and gold–silver heterobimetallic complexes 10 and 15 did have antiproliferative activity. In all cases the derivatives with silver had slightly lower IC50 values than their copper analogues. It is also worth noting that other than with the carbazole derivatives, the heterometallic complexes all had IC50 values lower than the control compounds [Cu(NO)3(PPh3)2] or [Ag(OTf)(PPh3)2], therefore the combination of two metals, Au and Cu/Ag, is more potent than either complex separately.
Experimental section
Experimental details
Mass spectra were recorded on a BRUKER ESQUIRE 3000 PLUS, with the electrospray (ESI) technique. 1H, 13C{1H} and 31P NMR, including 2D experiments, were recorded at room temperature on a BRUKER AVANCE 400 spectrometer (1H, 400 MHz, 13C, 100.6 MHz, 31P, 162 MHz) or on a Bruker Avance II 300 spectrometer (1H 300.0 MHz, 13C 75.5 MHz, 31P 121.5 MHz, 19F 282.3 MHz), with chemical shifts (δ, ppm) reported relative to the solvent peaks of the deuterated solvent. Infrared spectra were recorded in the range 4000–250 cm−1 on a PerkinElmer Spectrum 100 FTIR spectrometer.Crystal structure determinations
Crystals were mounted in inert oil on glass fibres and transferred to the cold gas stream of an Xcalibur Oxford Diffraction (2) or a Smart APEX CCD diffractometers (3–5, 7, 8) equipped with a low-temperature attachment. Data were collected using monochromated MoKα radiation (λ = 0.71073 Å). Scan type ϖ. Absorption corrections based on multiple scans were applied using SADABS22 or spherical harmonics implemented in SCALE3 ABSPACK scaling algorithm.23 The structures were solved by direct methods and refined on F2 using the program SHELXT-2016,24 or and by using Olex2 as the graphical interface.25 All non-hydrogen atoms were refined anisotropically. CCDC deposition numbers 2009440 (2), 2009441 (3), 2009442 (4), 2009443 (5), 2009444 (7), 2009445 (8) and 2009446 (9) contain the supplementary crystallographic data.†Materials and procedures
The starting materials [AuCl(tht)],26 [AuCl(PPh3)],27 [Au(acac)(PPh3)],28 [Ag(OTf)(PPh3)2],29 [Cu(NO3)(PPh3)2],30N-propargyl-di(2-picolyl)amine31 and N-propargylcarbazole32 were prepared according to published procedures. All other reagents were commercially available and were used without further purification. Solvents were dried with a SPS solvent purification system.Cell culture
A549 (human lung carcinoma) cells were maintained in high glucose DMEM (Dulbecco's Modified Eagle's Medium) supplemented with 5% fetal bovine serum (FBS), 200 μg mL−1 penicillin, 100 μg mL−1 streptomycin and 2 mM L-glutamine.Antiproliferative studies: MTT assay
Exponentially growing cells (A549) were seeded at a density of approximately 104 cells per well in 96 well flat-bottomed microplates and allowed to attach for 24 h prior to addition of compounds. Various concentrations of the compounds (0.1–100 μM) were added and incubated for 24 h at 37 °C (total volume 200 μl). Stock solutions of the compounds were prepared as 10 mM DMSO solutions and diluted using DMEM media. The final concentration of DMSO in each well was ≤0.25%. After 24 h, 10 μl of MTT (5 mg ml−1 in PBS) was added to each well and the plates incubated for an additional 2 h at 37 °C. The media/MTT mixture was eliminated and DMSO (100 μl per well) was added to dissolve the formazan precipitates. The optical density was measured at 550 nm using a 96-well multiscanner autoreader (ELISA). Absorbance values were normalised to (DMSO-containing) control wells and plotted as concentration of compound versus % cell viability. IC50 values were calculated by nonlinear regression analysis. The reported IC50 values are the average of three independent experiments, each consisting of four replicates per concentration level (overall n = 12).General synthesis of [Au(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CCH2R)(PPh3)] via [Au(acac)(PPh3)]
CCH2R)(PPh3)] via [Au(acac)(PPh3)]
To a solution of the corresponding HCCCH2R (0.1 mmol) in CH2Cl2 (5 ml) was added [Au(acac)(PPh3)] (0.0559 g, 0.1 mmol) and the solution stirred for 2 h. The solution was filtered through Celite, the filtrate concentrated under reduced pressure to approximately 1 ml and Et2O added to precipitate a solid of the corresponding compound.
General synthesis of [Au(CCCH2R)(PPh3)] via [AuCl(PPh3)] and KOH
To a solution of the corresponding HCCCH2R (0.2 mmol) in MeOH (15 ml) was added [AuCl(PPh3)] (0.0989 g, 0.2 mmol) and KOH (0.0168 g, 0.3 mmol) and the mixture stirred for 12 h. A white precipitate formed which was collected, washed with Et2O and vacuum dried to give the corresponding product.
General synthesis of heterometallic complexes
To a solution of the corresponding gold complex (0.2 mmol) in dichloromethane (5 ml) was added [Cu(NO3)(PPh3)2] (0.1300 g, 0.2 mmol) or [Ag(OTf)(PPh3)2] (0.1563 g, 0.2 mmol) and the solution stirred for 1 h. The solution was concentrated under reduced pressure to approximately 1 ml and hexane (10 ml) added to precipitate a solid of the corresponding compound.Conclusions
We have prepared and characterised new propargyl gold triphenylphosphine complexes bearing different functional groups and tested the anticancer activity of these in A549 cells. The propargyl gold compounds could be prepared by two different methods: addition of [Au(acac)(PPh3)] to a solution of the substrate in dichloromethane, or reaction of the substrate with potassium hydroxide and [AuCl(PPh3)] in methanol, however the “acac” method generally resulted in higher yields. Several derivatives were structurally characterised by single crystal X-ray diffraction with intermolecular contacts observed.Heterometallic dinuclear gold(I) complexes with copper(I) and silver(I) were also prepared in which the copper or silver is bound to the triple bond of the propargyl unit.
We have for the first time used the alkyne bond to incorporate an additional metal centre into the complex in order to directly compare the antiproliferative activity of gold–copper and gold–silver heterobimetallic complexes. The use of the propargyl gold complexes as a tether for the cytotoxic copper or silver centres gave improved activities compared to the copper or silver starting complexes or the mononuclear gold complexes, suggesting a possible synergy between the two metal centres within the cell.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors also thank Ministerio de Investigación e Invovación (CTQ2016-75816-C2-1-P, PID2019-104379RB-C21 and RED2018-102471-T) and Gobierno de Aragón-Fondo Social Europeo (Research Group E07_20R) for financial support of our research.Notes and references
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- N. Liu, X. Li, H. Huang, C. Zhao, S. Liao, C. Yang, S. Liu, W. Song, X. Lu, X. Lan, X. Chen, S. Yi, L. Xu, L. Jiang, C. Zhao, X. Dong, P. Zhou, S. Li, S. Wang, X. Shi, P. Q. Dou, X. Wang and J. Liu, Oncotarget, 2014, 5, 5453–5471 CrossRef PubMed.
- C. Marzano, V. Gandin, A. Folda, G. Scutari, A. Bindoli and M. P. Rigobello, Free Radical Biol. Med., 2007, 42, 872–881 CrossRef CAS PubMed.
- K. Kahlos, Y. Soini, M. Säily, P. Koistinen, S. Kakko, P. Pääkkö, A. Holmgren and V. L. Kinnula, Int. J. Cancer, 2001, 95, 198–204 CrossRef CAS.
- R. A. Stockland, M. C. Kohler, I. A. Guzei, M. E. Kastner, J. A. Bawiec, D. C. Labaree and R. B. Hochberg, Organometallics, 2006, 25, 2475–2485 CrossRef CAS .4.
- C.-H. Chui, R. S.-M. Wong, R. Gambari, G. Y.-M. Cheng, M. C.-W. Yuen, K.-W. Chan, S.-W. Tong, F.-Y. Lau, P. B.-S. Lai, K.-H. Lam, C.-L. Ho, C.-W. Kan, K. S.-Y. Leung and W.-Y. Wong, Bioorg. Med. Chem., 2009, 17, 7872–7877 CrossRef CAS PubMed.
- E. Schuh, S. M. Valiahdi, M. A. Jakupec, B. K. Keppler, P. Chiba and F. Mohr, Dalton Trans., 2009, 10841–10845 RSC.
- E. Vergara, E. Cerrada, A. Casini, O. Zava, M. Laguna and P. J. Dyson, Organometallics, 2010, 29, 2596–2603 CrossRef CAS.
- R. G. Balasingham, C. F. Williams, H. J. Mottram, M. P. Coogan and S. J. A. Pope, Organometallics, 2012, 31, 5835–5843 CrossRef CAS.
- A. Meyer, C. P. Bagowski, M. Kokoschka, M. Stefanopoulou, H. Alborzinia, S. Can, D. H. Vlecken, W. S. Sheldrick, S. Wölfl and I. Ott, Angew. Chem., Int. Ed., 2012, 51, 8895–8899 CrossRef CAS PubMed.
- R. Gavara, E. Aguiló, J. Schur, J. Llorca, I. Ott and L. Rodríguez, Inorg. Chim. Acta, 2016, 446, 189–197 CrossRef CAS.
- N. Mirzadeh, S. H. Privér, A. Abraham, R. Shukla, V. Bansal and S. K. Bhargava, Eur. J. Inorg. Chem., 2015, 2015, 4275–4279 CrossRef CAS.
- I. Mármol, P. Castellnou, R. Alvarez, M. C. Gimeno, M. J. Rodriguez-Yoldi and E. Cerrada, Eur. J. Med. Chem., 2019, 183, 111661 CrossRef PubMed.
- E. Cerrada, V. Fernández-Moreira and M. C. Gimeno, Adv. Organomet. Chem., 2019, 71, 227–257 CrossRef CAS.
- J. Fernandez-Gallardo, B. T. Elie, M. Sanau and M. Contel, Chem. Commun., 2016, 52, 3155 RSC.
- E. García-Moreno, S. Gascón, M. J. Rodriguez-Yoldi, E. Cerrada and M. Laguna, Organometallics, 2013, 32, 3710–3720 CrossRef.
- E. Barreiro, J. S. Casas, M. D. Couce, A. Sánchez, J. Sordo and E. M. Vázquez-López, J. Inorg. Biochem., 2014, 131, 68–75 CrossRef CAS PubMed.
- N. Curado and M. Contel, Heterometallic Compounds as Anticancer Agents, in Metal-based Anticancer Agents, ed. A. Casini, A. Vessières and S. M. Meier-Menches, Royal Society of Chemistry, London, 2019, ch. 6 Search PubMed.
- M. C. Blanco, J. Cámara, M. C. Gimeno, P. G. Jones, A. Laguna, J. M. López-de-Luzuriaga, M. E. Olmos and M. D. Villacampa, Organometallics, 2012, 31, 2597–2605 CrossRef CAS.
- M. Hailmann, B. Hupp, A. Himmelspach, F. Keppner, P. T. Hennig, R. Bertermann, A. Steffen and M. Finze, Chem. Commun., 2019, 55, 9351–9354 RSC.
- M. Frik, J. Fernández-Gallardo, O. Gonzalo, V. Mangas-Sanjuan, M. González-Alvarez, A. Serrano del Valle, C. Hu, I. González-Alvarez, M. Bermejo, I. Marzo and M. Contel, J. Med. Chem., 2015, 58, 5825–5841 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, Program for adsorption correction, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- CrysAlisPro, Agilent Technologies, Version 1.171.35.11. Multi-scans absorption correction with SCALE3 ABSPACK scaling algorithm.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- R. Uson, A. Laguna, M. Laguna, D. A. Briggs, H. H. Murray and J. P. Fackler, in Inorg. Synth, John Wiley & Sons, Inc., 1989, pp. 85–91 Search PubMed.
- C. A. McAuliffe, R. V. Parish and P. D. Randall, J. Chem. Soc., Dalton Trans., 1979, 1730–1735 RSC.
- D. Gibson, B. F. G. Johnson and J. Lewis, J. Chem. Soc. A, 1970, 367–369 RSC.
- M. Bardají, O. Crespo, A. Laguna and A. K. Fisher, Inorg. Chim. Acta, 2000, 304, 7–16 CrossRef.
- F. A. Cotton and D. M. L. Goodgame, J. Chem. Soc., 1960, 5267–5269 CAS.
- S. Huang, R. J. Clark and L. Zhu, Org. Lett., 2007, 9, 4999–5002 CrossRef CAS PubMed.
- A. Balci, M. Arslan, A. R. Nixha, C. Bilen, A. Ergun and N. Gençer, J. Enzyme Inhib. Med. Chem., 2015, 30, 377–382 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2009440–2009446. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02113j |
| This journal is © The Royal Society of Chemistry 2020 |