
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAppraisal of calcium ferrites as cathodes for calcium rechargeable batteries: DFT, synthesis, characterization and electrochemistry of Ca4Fe9O17†
Ashley P.
Black
 ab,
Arturo
Torres
c,
Carlos
Frontera
ab,
M. Rosa
Palacín
ab and
M. Elena
Arroyo-de Dompablo
*c
ab,
Arturo
Torres
c,
Carlos
Frontera
ab,
M. Rosa
Palacín
ab and
M. Elena
Arroyo-de Dompablo
*c
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC) Campus UAB, E-08193 Bellaterra, Catalonia, Spain
bALISTORE-ERI European Research Institute, CNRS FR 3104, Hub de l’Energie, 15 Rue Baudelocque, 80039 Amiens, France
cDepartamento de Química Inorgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: e.arroyo@quim.ucm.es
First published on 12th February 2020
Abstract
Sustainability combined with high energy density prospects makes Fe-based oxides attractive as cathodes for calcium rechargeable batteries. This work presents a DFT evaluation of the CaFe2+nO4+n (0 < n < 3) family, for which both the average intercalation voltage and the theoretical specific capacity decrease with the increasing n value. The term n = 1/4, Ca4Fe9O17, meets the most appealing characteristics: a calculated average voltage of 4.16 V, a theoretical specific capacity of 230 mA h g−1 and the lowest energy barrier for Ca migration so far predicted for an existing oxide (0.72 eV). To overcome the previously reported synthesis difficulties, we employed a novel synthesis procedure in sealed quartz tubes followed by quenching in water. The XRD and SAED patterns of the prepared Ca4Fe9O17 powder reveal a certain degree of stacking defects along the c axis. Attempts to deinsert Ca ions from Ca4Fe9O17 by chemical means (NO2BF4 in ACN) and in electrochemical Ca cells were unsuccessful, although some hints of oxidation are observed in Li cells with the LP30 electrolyte. The suitability of Ca4Fe9O17 as a Ca cathode is pending further studies utilizing Ca-electrolytes with high anodic stability.
Introduction
The Li-ion technology currently dominates the rechargeable battery market for portable electronics and appears to be a possible near term solution for applications such as electric vehicles and grid storage, in parallel with the storage of energy from renewable and intermittent sources – solar, wind, waves, etc. However, Li-ion batteries are currently approaching their fundamental limits in terms of energy density, which in addition to the limited supply of Li instigate the race for the development of alternative technologies exhibiting advantages in terms of energy density, safety and cost. Rechargeable batteries based on a Ca-metal anode are attractive given the abundance of calcium in the Earth's crust, the high capacity of calcium anodes (1340 mA h g−1) and its negative reduction potential (only 170 mV above that of lithium metal).1,2 Advances in the development of such batteries are intricate due to the lack of competitive cathode materials and electrolytes.3,4 While recent findings demonstrate the feasibility of Ca anode operation using different electrolytes,5–8 the identification of suitable cathode materials has been facing several roadblocks.4,9,10 Limited reversible electrochemical Ca intercalation has been reported for a handful of compounds4 (V2O5,11 TiS2,12 MoO3,13 Ca0.5CoO2,14 and manganese hexacyanoferrate1), all of them showing poor cycling, if any. Parallel experimental and computational investigations establish a link between the lack of reversible electrochemical activity (experimentally observed) and hampered Ca diffusion (computationally estimated).15–20 In this regard, the poor diffusion of Ca2+ in inorganic structures is the major challenge for cathode discovery.21 Compared to Li+ diffusion, the hindrance to Ca mobility resides in the larger size of Ca2+ ions on one hand and its divalent charge on the other that increases the electrostatic repulsion/attraction with the neighbouring cations/anions in the structure.Sustainable cathode materials should include abundant and non-toxic elements, and at the same time, comply with the high energy density requirements. These criteria prompted the investigation of light and abundant oxidizing 3d-transition metal oxides as cathodes for Ca batteries. So far several Mn oxides and polyoxoanionic compounds have been reported, going from the virtual spinel CaMn2O422 to the CaMnO3 perovskite,16 its reduced derivative -Ca2Mn2O5,23 the known CaMn2O4 polymorphs (marokite and its high pressure form),15 a related oxidized phase, CaMn4O8,23 and the minerals K0.31MnO2 0.25H2O (birnessite),24 Mg0.19Na0.07MnO2 0.37H2O (todorokite),25 CaMn(CO3)2 (kutnahorite)26 and CaMnSi2O6 (johannsenite).26 In contrast, research on Fe oxides has been scarce; the perovskite CaFeO3 and the related brownmillerite Ca2Fe2O5 compounds have been only evaluated by computational means.16,23 It was found that Ca-perovskites are not competitive materials due to hindered Ca mobility (a calculated energy barrier of 2 V).16 In the brownmillerite structure the oxygen vacancies respective to the perovskite create a new pathway with much lower energy barrier (1 eV).23 The present investigation turns to other known oxides in the Ca–Fe–O system with general formulae CaFe2+nO4+n that might be worthy candidates (Fig. 1).
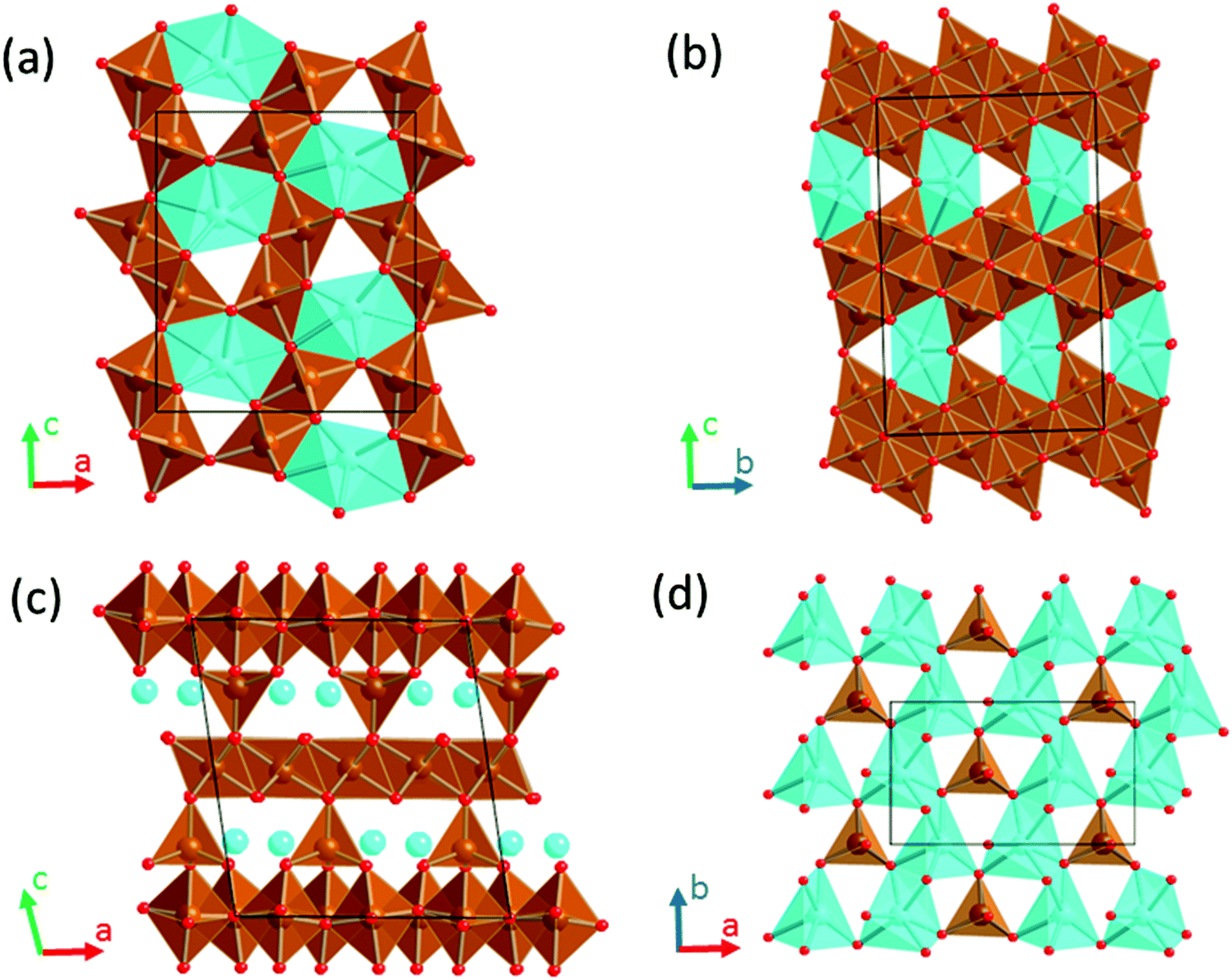 | ||
| Fig. 1 Crystal structures of CaFe2+nO4+n oxides. (a) n = 0, CaFe2O4 (b) n = 2, CaFe4O6 (c) n = 1/4, CaFe9O17 showing the [CaO6]–[FeO4] plane in (d). Color code: Ca in blue, Fe in brown and O in red. | ||
The crystal structure of CaFe2O4 (S.G. Pmna, no. 53)27,28 consists of edge-sharing chains of MnO6 octahedra, forming double rutile chains (see Fig. 1a). Each double-rutile chain is connected to two adjacent chains through edge-sharing oxygen. As a result, tunnels appear along the b axis where Ca ions reside in eight-fold coordination. The calcium ferrite CaFe2+nO4+n family is formed by stacking along the c axis (S.G. Cmcm, no. 63) of blocks with the CaFe2O4 composition and blocks with the FeO composition (see Fig. 1b). Within this family, the compounds with n = 0, 1, 3/2, 2, 5/2, and 3 have been reported.29,30 Interestingly, for n = 1/4, the compound Ca4Fe9O17 crystallizes in a different structure (S.G. C2, no. 5), built up by layers of Fe atoms in edge-sharing FeO6 octahedra and vertex-sharing Fe in trigonal-based bipyramid coordination (see Fig. 1c and d).31 These hexagonal layers stack along the c-axis direction, being cross-linked by iron atoms in tetrahedral coordination. The [FeO4]-tetrahedra are connected in the ab plane of the structure by hexa-coordinated Ca–O polyhedra (Fig. 1d). The CaO6 distorted octahedra share edges forming rings around the FeO4-tetrahedra. Unsurprisingly, this structure with Fe in three different coordination environments, and in an average oxidation state of 2.888, is thermodynamically quite unstable.32–34 The oxygen partial pressure is critical in the stability of Ca4Fe9O17, and therefore the complex synthesis of the material usually results in a mixture of a low crystalline Ca4Fe9O17 and more stable oxides (CaFe2O4, FeO, and Ca2Fe2O5). Indeed, the crystal structure determination was accomplished using single crystals grown under very specific conditions.31
To evaluate the performance of CaFe2+nO4+n compounds as cathode materials for Ca batteries, we have selected the compounds with n = 0, 1/4, 1 and 2 for computational investigations. Density Functional Theory (DFT) calculations are used to anticipate the average intercalation voltage so as to estimate their theoretical energy density, assuming the full Ca extraction from the structures. Ca diffusion is a key parameter, for which the migration energy barriers provide an approximate indication.3,35 Following vast literature in the field,18,21,22,35 in this work the energy barriers for Ca migration are extracted from DFT calculations. The results found for Ca4Fe9O17 motivated an experimental investigation comprising the synthesis, characterization and electrochemical testing of the material. We present a novel synthesis method to produce highly crystalline powder.
Materials and methods
Computational
The calculations have been carried out using the ab initio total-energy and molecular dynamics program VASP (Vienna ab initio simulation program) developed at the Universität Wien.36 Total energy calculations based on Density Functional Theory (DFT) were performed within the General Gradient Approximation (GGA), with the exchange and correlation functional form developed by Perdew, Burke and Ernzerhof (PBE).37 The interaction of core electrons with the nuclei is described by the Projector Augmented Wave (PAW) method.38 The energy cut off for the plane wave basis set is fixed at a constant value of 600 eV throughout the calculations. The integration in the Brillouin zone is done on an appropriate set of k-points determined using the Monkhors–Pack scheme. A convergence of the total energy close to 10 meV per formula unit is achieved with such parameters. Spin polarization calculations were performed in all cases. All crystal structures were fully relaxed (atomic positions, cell parameters and volume). The final energies of the optimized geometries were recalculated to correct the changes in the basis set of wave functions during relaxation. Average voltages for Ca deinsertion from CaFe2+nO4+n were calculated within the GGA+U method, following the methodology described by Aydinol et al.39. For consistency with the literature on this topic,16,21,22,40 the effective U value for the Fe-d orbitals was set to 5 eV (J = 1 eV).Calcium mobility is investigated using the Nudged Elastic Band method (NEB) as implemented in VASP. To study diffusion, a vacancy is created in the structural model of the stoichiometric oxides and a simple jumping mechanism from an occupied position to the vacant site is considered. Supercells 1 × 2 × 1 (Ca8Fe18O34) and 1 × 4 × 1 (Ca4Fe8O16) for the n = 1/4 and n = 0 compounds, respectively, were used for the calculations. Constant volume calculations were performed within the GGA approximation for three or five intermediate images initialized by linear interpolation between the two fully relaxed end-points. To calculate the energy at the saddle point, cubic splines were fit through the relaxed images along each hop.
Experimental
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 were thoroughly mixed in an agate mortar, pressed into a pellet and vacuum sealed in a quartz tube. First the samples were thermally treated at 1000° C for 24 h and quenched in water. Then, the purest sample was obtained after four consecutive regrinding and annealing processes of 24 h each at 1150 °C in evacuated quartz tubes. Ca2Fe2O5 was previously synthesized following the Pechini method. Ca(NO3)2·4H2O (Aldrich 99%), Fe(NO3)3·9H2O (Aldrich 99.95%), C6H8O7 (citric acid, Aldrich 99.5%) and C2H6O2 (ethylene glycol) were subsequently dissolved in water in a molar ratio of Ca:Fe:C6H8O7:C2H6O2 = 1:1:3:1. The solution was kept under continuous stirring at 80 °C until complete evaporation. The obtained citrate complexes were decomposed in air at 500 °C for 8 h. The decomposition process was carried out at a rate of 100 °C h−1. Finally, polycrystalline Ca2Fe2O5 was obtained by calcination of the amorphous carbonates at 1000° C for 24 h.
:1:1 were thoroughly mixed in an agate mortar, pressed into a pellet and vacuum sealed in a quartz tube. First the samples were thermally treated at 1000° C for 24 h and quenched in water. Then, the purest sample was obtained after four consecutive regrinding and annealing processes of 24 h each at 1150 °C in evacuated quartz tubes. Ca2Fe2O5 was previously synthesized following the Pechini method. Ca(NO3)2·4H2O (Aldrich 99%), Fe(NO3)3·9H2O (Aldrich 99.95%), C6H8O7 (citric acid, Aldrich 99.5%) and C2H6O2 (ethylene glycol) were subsequently dissolved in water in a molar ratio of Ca:Fe:C6H8O7:C2H6O2 = 1:1:3:1. The solution was kept under continuous stirring at 80 °C until complete evaporation. The obtained citrate complexes were decomposed in air at 500 °C for 8 h. The decomposition process was carried out at a rate of 100 °C h−1. Finally, polycrystalline Ca2Fe2O5 was obtained by calcination of the amorphous carbonates at 1000° C for 24 h.
Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray Spectroscopy (EDS) analyses were performed by using a FEI Quanta 200 FEG microscope equipped with an EDAX detector (resolution: 132 eV). Selected Area Electron Diffraction (SAED) studies were performed using a JEOL 1210 transmission electron microscope operating at 120 kV and equipped with a Gathan sample holder. The specimens for electron microscopy were prepared by dispersing the powders in dimethyl carbonate and depositing a droplet of this suspension on a carbon coated film supported on a copper grid.
The electrochemical tests were run in two and three electrode Swagelok cells assembled in an Ar-filled dry box, employing Al and stainless steel current collectors for the working and reference (RE)/counter (CE) electrodes, respectively, and a Whatman separator soaked in 0.6 ml of electrolyte solution. Mechanically polished Ca metal discs (obtained by pressing Ca pieces under an inert atmosphere) or Li metal were used as the counter/reference or counter electrodes, respectively.
Lithium hexafluorophosphate solutions in ethylene carbonate (EC) and dimethyl carbonate (DMC) (LP30, 1.0 M LiPF6 in EC/DMC = 50/50 (v/v), Solvionic) and the lithium bis(oxalato)borate salt (LiBoB, Chemetal, Battery grade) in a 1:1 v/v mixture of ethylene carbonate (EC) and propylene carbonate (PC) solvents (Solvionic, 99.9%) were used as electrolytes for the electrochemical tests in Li cells. The electrolyte used in Ca cells was a dry (<40 ppm H2O, Karl-Fischer titration) 0.45 M calcium tetrafluoroborate hydrate (Alfa Aesar) solution in EC/PC = 50/50 (v/v).
The electrochemical tests were performed on a Bio-Logic VMP3 potentiostat. Electrochemical deintercalation/intercalation tests were carried out in galvanostatic mode (i.e. Galvanostatic Cycling with Potential Limitation–GCPL). In order to avoid kinetic limitations the rate was set to C/100. A stabilization time of 5 hours at the OCV was applied before each measurement was carried out at 100 °C. Once tested, the cells were disassembled in an Ar-filled dry box and the electrodes were washed with dimethyl carbonate solvent (DMC, Sigma-Aldrich, ≥99%) and further collected in borosilicate glass capillaries of 0.5 mm diameter for ex situ XRD measurements.
:8. The dispersion was kept under reflux at 80 °C for 6 hours. Uninterrupted Ar bubbling served to maintain the reaction atmosphere inert and to eliminate the NO2 gas formed as the byproduct. Once the reactions were completed, the samples were washed with dry ACN, vacuum filtered, dried and collected for XRD analysis.
Results and discussion
DFT evaluation of basic electrode characteristics
The GGA+U method correctly reproduces the cell lattice parameters of CaFe2+nO4+n compounds (see Table S1†). The deinsertion of Ca can be expressed by the reaction:| CaFe2+nO4+n → Fe2+nO4+n + Ca | (1) |
The higher the n term, the lower the theoretical specific capacity is. These values are listed in Table 1, together with the average voltage predicted for n = 0, 1/4, 1 and 2. As n increases the Fe ions are in a lower oxidation state, and accordingly Ca deinsertion is predicted at lower voltages. Note that the average voltage depends primarily on the redox couple, but it is also influenced by the crystal structure.39 For instance, the voltages predicted for the Fe3+/Fe4+ couple are 4.16 V in brownmillerite-Ca2Fe2O5 (ref. 23) and 3.87 V in CaFe2O4. Taking the calculated voltage and the theoretical specific capacities into account, theoretical energy densities exceeding 600 mA h g−1 (the state of the art in the Li-ion technology) could be viable for n = 0, 1/4 and 1.
| CaFe2+nO4+n composition | n | Fe redox couple | Theoretical specific capacity (mA h g−1) | Calculated average voltage (V) | Theoretical specific energy (mW h g−1) |
|---|---|---|---|---|---|
| CaFe2O4 | 0 | +3/+4 | 249 | 3.87 | 964 |
| Ca4Fe9O17 | 1/4 | +2.88/3.77 | 230 | 4.16 | 957 |
| CaFe3O5 | 1 | +2.66/+3.33 | 186 | 3.66 | 680 |
| CaFe4O6 | 2 | +2.5/+3 | 149 | 2.37 | 353 |
One of the major concerns in cathode material design for the Ca battery technology is the mobility of Ca2+ ions in inorganic structures. Although other factors can certainly limit the success of Ca battery cathodes, a good cation diffusion, that is to say a low migration energy barrier, is a necessary prerequisite. It has been estimated that for a reasonable cell power rate (discharging time 2 hours) the energy barriers for cation diffusion should be below 0.525 eV in micrometer particles and 0.65 eV in nanosized-particles.3 The calculated energy barrier for CaFe2O4 (1.2 eV, see Fig. 1 in the ESI†) greatly surpasses this tolerable value, and therefore, the ferrites CaFe2+nO4+n built up from CaFe2O4 blocks will also suffer from poor Ca mobility. However, the n = 1/4 term might be a viable option. Fig. 2 shows the pathway for Ca diffusion in Ca4Fe9O17 and the calculated energy barrier. In the two-dimensional diffusion pathway in the [CaO6]–[FeO4] plane (shown in Fig. 1d) the diffusing Ca ions exit from their polyhedra across a triangular face to occupy the intermediate tetrahedral site (see Fig. 2a). The associated energy barrier for in-plane Ca diffusion is 0.72 eV (Fig. 2b), the lower energy ever reported for an existing transition metal oxide. The tetrahedral site represents a local energy minimum in the energy landscape. The similarity in Ca–Fe distances at this intermediate site and at the equilibrium sites (3.22 Å and 3.29 Å, respectively) suggests that the diffusing Ca ion does not experience large electrostatic repulsions with the neighbouring Fe+2.88 cations. Compared to other oxides, the lower repulsive forces between the diffusing Ca2+ and other cations in the structure reduce the migration energy.4 Although the barrier is relatively low for Ca diffusion, it is still too high for fast rate performance. Note that the barriers for Li diffusion in layered, olivine and spinel cathodes are below 0.3 eV.3,35,42,43
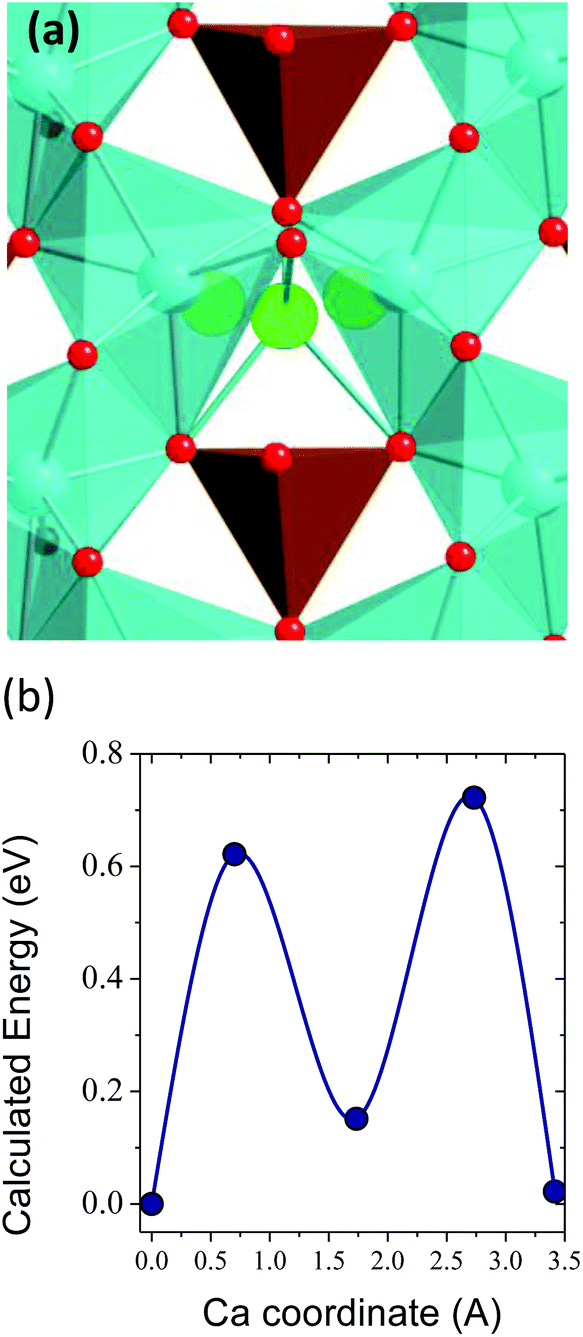 | ||
| Fig. 2 (a) View of the Ca diffusion pathway in Ca4Fe9O17, showing the tetrahedral site at the transition state. (b) Calculated energy barrier. Color code: Ca sites in blue, diffusing Ca in green, Fe in brown, O in red. | ||
Interestingly, a related compound, K2Fe4O7, has been recently reported and suggested as a K-superionic conductor.44 The framework of K2Fe4O7 consists of planes of edge-sharing FeO6 octahedra sandwiched between planes of FeO4 tetrahedra and octahedrally coordinated K ions. The K octahedra form rings around FeO4 tetrahedra, thus providing a 2D conduction plane similar to that in Ca4Fe9O17 (Fig. 1d and 2a). When tested in the K/K2Fe4O7/Pt cell no significant current due to the solid electrolyte decomposition was observed up to 5.0 V. Despite the presence of Fe3+, the authors found that the material is also stable upon reduction down to 0 V.
Synthesis
Ca4Fe9O17 was first reported by B. Malaman et al. in the 80's31 and its crystal structure was elucidated from a single crystal. Despite a few articles mentioning its existence (a summary of those is provided in Table S2†), no reliable bulk powder synthesis method is available in the literature. In all cases, this phase is reported to form, mixed with CaFe2O4 and Ca2Fe2O5 phases that are stable in the same region of the FeO–Fe3O4–CaO phase diagram.In view of the discrepancies present in the literature, in this work different synthesis methods were attempted at temperatures ranging from 800 to 1200 °C, including conventional solid state synthesis using CaCO3 or CaO as Ca sources and Fe2O3, Fe3O4 or FeO as iron precursors. The Pechini method was also attempted.45 Most reaction products consisted of a mixture of Fe2O3 and CaFe2O4 in all cases, whatever the reaction times (from 6 to 64 hours), except for the samples treated at 1200 °C for which melting and thermal reduction of iron oxide was observed. Further synthesis attempts were carried out in a tube sealed under vacuum, and the phase Ca4Fe9O17 was observed in small amounts after synthesis at 1000° C for 48 h followed by quenching. Optimization of this reaction protocol, modifying precursors, temperature and time, enabled the synthesis of pure Ca4Fe9O17 using a stoichiometric mixture of Ca2Fe2O5, Fe2O3 and Fe3O4 as precursors, targeting to control the iron oxidation state by tuning the amount of oxygen available in the precursors. However, a pure phase was achieved only after several treatments at 1150 °C for 24 h each in a sealed quartz tube followed by quenching in water (a summary of synthesis results is provided in Fig. S2†).
Structure and morphology
Fig. 3 shows a full pattern matching of X-ray diffraction patterns of the as prepared Ca4Fe9O17 sample in the monoclinic system, space group C2 using the Fullprof program. The cell parameters obtained by the Le Bail method were a = 10.44177(24) Å, b = 6.02422(21) Å, c = 11.36840(36) Å and β = 98.7853(16) which are in good agreement with those previously reported from the single crystal structural determination.31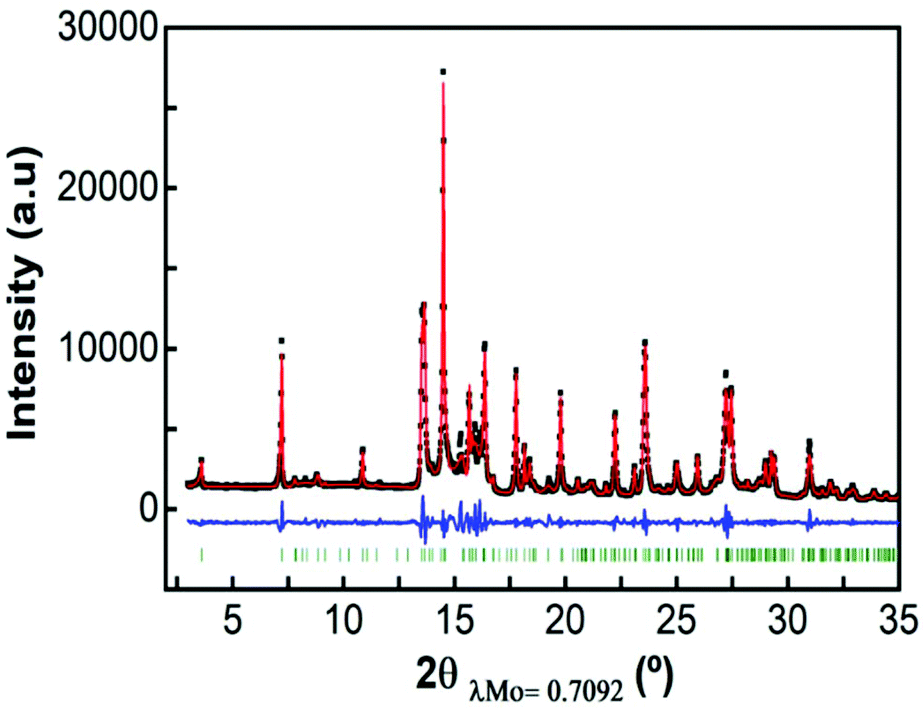 | ||
| Fig. 3 Pattern matching of Ca4Fe9O17 in the monoclinic system, space group C2. | ||
Attempts to perform a Rietveld refinement of this X-ray diffraction pattern using the structural model reported by B. Malaman et al.31 resulted in unsatisfactory fitting of the model to the experimental pattern, despite all efforts to modify cell parameters, occupation factors, etc. The differences in the observed and calculated integrated intensities of several reflections suggest that the prepared material might present some structural differences with respect to the single crystal studied by B. Malaman (see Fig. S3†).
In order to further characterize the structure of the as prepared Ca4Fe9O17 phase, SAED was performed and micrometric particles were observed, as shown in Fig. 4. The cell parameters obtained by electron diffraction were a = 10.4 Å, b = 6.02 Å and c = 11.4 Å. The ED image along the c zone axis presents the characteristic scattered lines indicative of the presence of stacking defects along the c axis. The occurrence of stacking faults could explain the differences between observed and calculated intensities in Rietveld refinement. The stacking faults are not expected to perturb the in-plane Ca conductivity, unless they imply inter-layer cation mixing or phase transformations.42,46
 | ||
| Fig. 4 SAED images of the Ca4Fe9O17 sample. | ||
Chemical and electrochemical tests on Ca4Fe9O17 and Ca2Fe2O5
Attempts to extract Ca2+ from Ca4Fe9O17 were carried out both by chemical oxidation with NO2BF4 (ref. 47) and by electrochemical methods at room temperature and 100 °C. Tape electrodes were prepared and cycled according to the electrochemical protocol described in the experimental section. The characteristic potential vs. capacity profiles of these electrodes upon galvanostatic cycling at RT and 100 °C in Li or Ca cells and in several Li or Ca electrolytes at C/100 are shown in Fig. 5(a and b) for Ca4Fe9O17 and in Fig. S4† (left panel) for Ca2Fe2O5. The utilization of a low rate (C/100) is in line with the calculated energy barrier of 0.72 eV, which as commented before is too high for fast-rate performance. Indeed, Ca intercalation in TiS2 (calculated energy barrier 0.75 eV) requires similarly slow rates.12 Upon oxidation, a short plateau-like region centered between 2.9 V and 4.8 V vs. Mn+/M (M = Li or Ca) is observed, with the potential at which the plateau appears depending on the nature of the electrolyte, the temperature and the counter electrode used. The ex situ X-ray powder diffraction patterns of the Ca4Fe9O17 samples collected after the electrochemical and chemical oxidation tests are shown in Fig. 5(c) and Fig. S4† (right panel), respectively. Compared with the pristine phase, neither peak shifts nor formation of extra phases is observed in any spectra except for the one corresponding to electrochemical measurement performed on Ca4Fe9O17 in the lithium cell with the LP30 electrolyte. Thus, except for that case, the electrochemical capacity observed is ascribed to side reactions most likely involving electrolyte decomposition, which are enhanced at high temperature. In contrast, the synchrotron X-ray diffraction patterns of the pristine sample and the sample electrochemically oxidized in a lithium/LP30 cell exhibit minor differences (see Fig. 6). Indeed, the pattern full width at half maximum for peaks situated at Q values of 2.09 Å−1, 2.24 Å−1 and 2.53 Å−1 is different.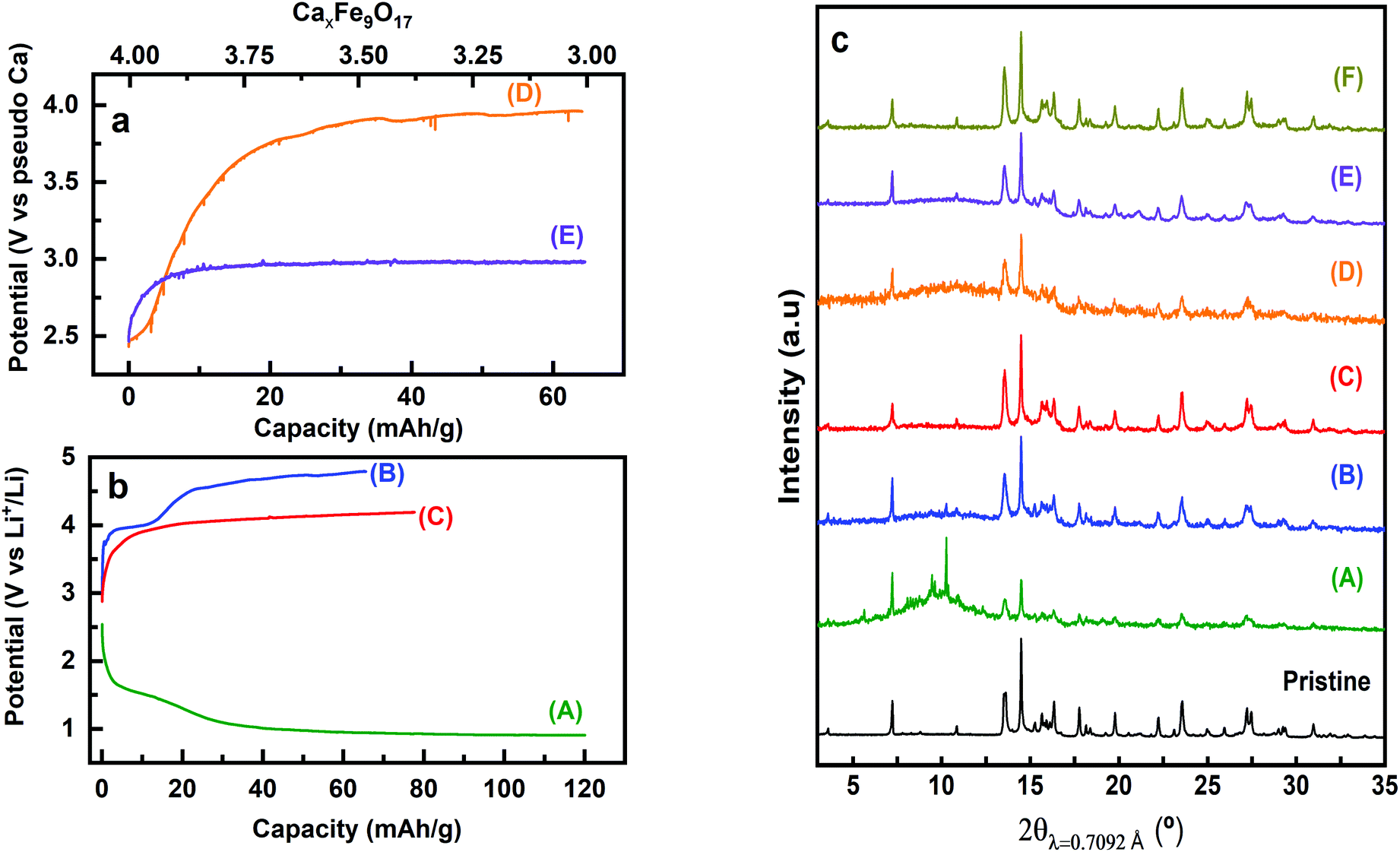 | ||
| Fig. 5 Characteristic voltage profiles versus specific capacity (bottom) and versus moles of virtually de-inserted Ca2+ (Δx, estimated that calcium deintercalation is the only reaction taking place) (top) for Ca4Fe9O17 in Ca cells (a), with 0.45 M Ca(BF4)2, EC/PC (1:1 vol) electrolyte at RT (D) and 100 °C (E) and in Li cells (b), with LP30 electrolyte at RT (A and B), 1 m LiBOB, EC/PC (1:1 vol) electrolyte at 100 °C (C). Corresponding ex situ XRD patterns in (c). XRD patterns of the pristine and chemically oxidized samples (F) are also displayed in (c). | ||
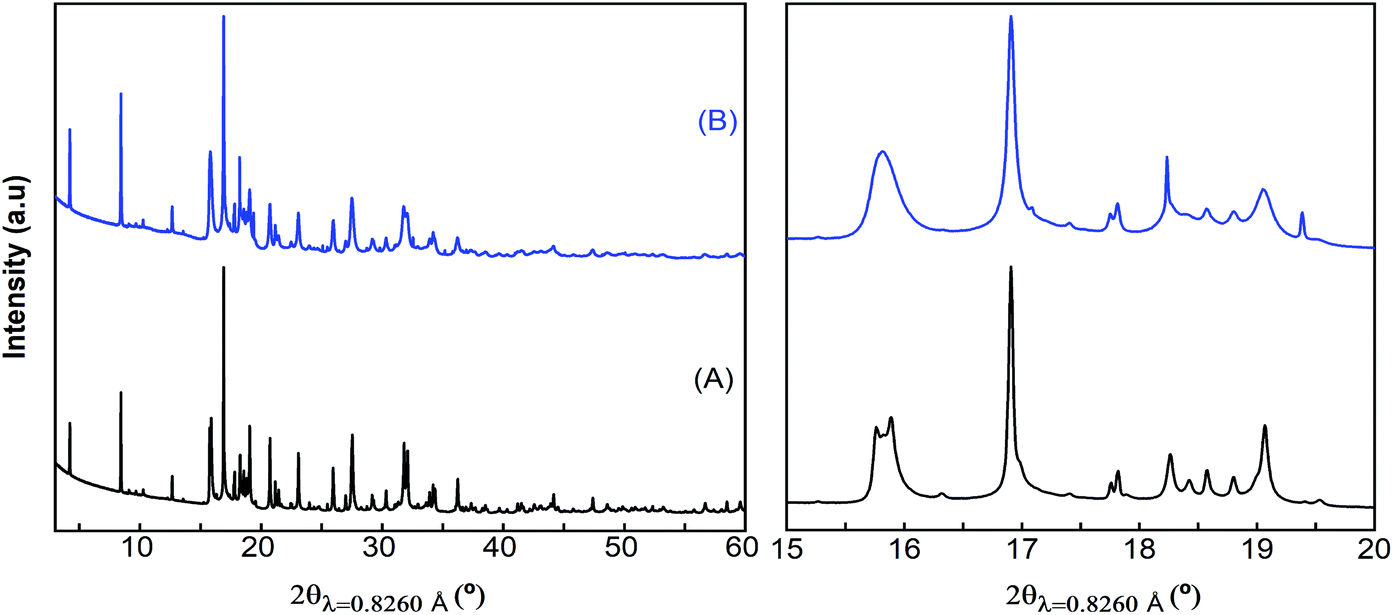 | ||
| Fig. 6 Synchrotron X-ray powder diffraction patterns of pristine (A) and electrochemically oxidized (in the LP30 electrolyte) (B) Ca4Fe9O17 samples. | ||
While the origin of these subtle changes has been unclear to date, one may speculate that they could be related to de-insertion of a small amount of calcium from Ca4Fe9O17 at the 4.0 V plateau (capacity 12 mA h g−1, curve B in Fig. 5b) concomitant with the oxidation of iron. In fact, hypothetical oxidation to yield Ca3.5Fe9O17 could be related to the Fe2+/Fe3+ redox pair, with an expected voltage considerably smaller than 4.16 V, and thus within the voltage window of the LP30 electrolyte stability. Further oxidation should involve the Fe3+/Fe4+ couple, with a higher voltage (ca. 4.2 V), which may be out of the voltage window of the electrolyte stability, especially if polarization is significant. Yet, in light of the low value of the capacity and the disorder present in the crystals, this phenomenon has not been further studied.
Regardless of the partial oxidation of Fe detected in Li cells (likely the Fe2+/Fe3+ couple), further oxidation has not been achieved either electrochemically or chemically. Noteworthily, the calculated average voltage for the full Ca deinsertion, the Fe+2.88/Fe+3.77 couple (4.16 V, see Table 1), is probably beyond the anodic stability of available Ca electrolytes5 and in the limit of Li electrolytes if cell polarization is significant. In this regard, we attempted the electrochemical and chemical oxidation of Ca2Fe2O5, for which DFT investigations found an average voltage of 4.16 V for the Fe3+/Fe4+ couple, although with a sluggish kinetics (calculated energy migration barrier 1 eV).23 The combination of electrochemical and XRD characterization (see Fig. 3 in the ESI†) reflects the inactivity of Ca2Fe2O5 in both Ca and Li electrolytes. A plausible explanation is that Ca deintercalation from these materials requires electrolytes operating in the 5 V range, so as to allow the oxidation of Fe to the +4 oxidation state.
To test the electrochemical stability window of Ca4Fe9O17, Li/LP30/Ca4Fe9O17 cells were discharged down to 1 V. Upon reduction, the potential vs. capacity profiles for Ca4Fe9O17 displayed in Fig. 5b(A) present a long plateau-like region centered at 1 V vs. Li+/Li and additional diffraction peaks are observed in the 2θ = 10° region of the corresponding ex situ X-ray powder diffraction pattern shown in Fig. 5c(A). This large capacity can be regarded as the combination of two competing processes: the low-voltage decomposition of the electrolyte and the reduction of Fe3+ to Fe2+ upon Li uptake. Noteworthily, a similar potential vs. capacity profile is observed for the reduction of Fe2O3 electrodes in Li cells;48 the electrochemical conversion reactions of Fe3+ compounds with lithium are known to evolve until the eventual formation of small Li2O and metallic iron domains.48,49 In the present case, the identification of the phases formed during the reduction remains undetermined due to the complex nature of the starting material. However, these results highlight the instability of Ca4Fe9O17 upon reduction, contrary to the reported stability of the related K-phase down to 0 V vs. K+/K.44
Conclusions
Sustainability combined with high energy density prospects makes Fe-based oxides appealing cathodes for calcium rechargeable batteries. The present DFT investigation reveals that among the CaFe2+nO4+n materials, Ca4Fe9O17 combines the most appealing characteristics. The favorable local topology for Ca diffusion in Ca4Fe9O17 bestows the lowest energy barrier so far reported for an existing oxide (0.72 eV). However, the predicted average deintercalation voltage (4.16 V for full Ca deinsertion) is beyond the stability range of current Ca electrolytes and, accordingly, electrochemical attempts to deintercalate Ca ions from Ca4Fe9O17 in Ca cells were unsuccessful. However, a small charge capacity probably associated with the oxidation of Fe2+ to Fe3+ is observed in Li cells using the LP30 electrolyte. The suitability of Ca4Fe9O17 as a Ca cathode is pending further studies utilizing electrolytes with higher anodic stability. Electrochemical and any other property investigations will no doubt benefit from the novel synthesis route reported here which, contrary to previous strategies, allows synthesizing pure and crystalline Ca4Fe9O17 powders.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the European Union's Horizon 2020 research and innovation programme H2020 FETOPEN-1-2016-2017 (CARBAT, grant agreement no. 766617). The authors are grateful for access to the computational facilities from Universidad de Oviedo (MALTA-Consolider cluster), to ALBA synchrotron for the provision of beam time and to Dr F. Fauth for his help during data collection and also acknowledge Dr. J. Oró-Solé for assistance with electron diffraction and Dr. Clemens Ritter for helpful discussions. ICMAB authors are also grateful to the Spanish Ministry for Economy, Industry and Competitiveness Severo Ochoa Programme for Centres of Excellence in R&D (SEV-2015-0496).Notes and references
- A. L. Lipson, B. Pan, S. H. Lapidus, C. Liao, J. Y. Vaughey and B. J. Ingram, Chem. Mater., 2015, 27, 8442–8447 CrossRef CAS.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- P. Canepa, G. S. Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS PubMed.
- M. E. Arroyo-de Dompablo, A. Ponrouch, P. Johansson and M. R. Palacin, Chem. Rev., 2019 DOI:10.1021/acs.chemrev.9b00339.
- A. Ponrouch, C. Frontera, F. Barde and M. R. Palacin, Nat. Mater., 2015, 15, 169–172 CrossRef PubMed.
- D. Wang, X. Gao, Y. Chen, L. Jin, C. Kuss and P. G. Bruce, Nat. Mater., 2018, 17, 16–20 CrossRef CAS PubMed.
- A. Shyamsunder, L. E. Blanc, A. Assoud and L. F. Nazar, ACS Energy Lett., 2019, 49, 2271–2276 CrossRef.
- Z. Li, O. Fuhr, M. Fichtner and Z. Zhao-Karger, Energy Environ. Sci., 2019, 12, 3496–3501 RSC.
- A. Ponrouch and M. R. Palacin, Curr. Opin. Electrochem., 2018, 9, 1–7 CrossRef CAS.
- R. J. Gummow, G. Vamvounis, M. B. Kannan and Y. He, Adv. Mater., 2018, 30, e1801702 CrossRef PubMed.
- G. G. Amatucci, F. Badway, A. Singhal, B. Beaudoin, G. Skandan, T. Bowmer, I. Plitza, N. Pereira, T. Chapman and R. Jaworski, J. Electrochem. Soc., 2001, 148, A940–A950 CrossRef CAS.
- D. S. Tchitchekova, A. Ponrouch, R. Verrelli, T. Broux, C. Frontera, A. Sorrentino, F. Barde, N. Biskup, M. E. Arroyo-de Dompablo and M. R. Palacin, Chem. Mater., 2018, 30, 847–856 CrossRef CAS.
- M. Cabello, F. Nacimiento, R. Alcantara, P. Lavela, C. Pérez-Vicente and J. L. Tirado, Chem. Mater., 2018, 30, 5853–5861 CrossRef CAS.
- M. Cabello, F. Nacimiento, J. R. González, G. Ortiz, R. Alcántara, P. Lavela, C. Pérez-Vicente and J. L. Tirado, Electrochem. Commun., 2016, 67, 59–64 CrossRef CAS.
- M. E. Arroyo-de Dompablo, C. Krich, J. Nava-Avendano, N. Biskup, M. R. Palacin and F. Barde, Chem. Mater., 2016, 28, 6886–6893 CrossRef.
- M. E. Arroyo-de Dompablo, C. Krich, J. Nava-Avendaño, M. R. Palacín and F. Bardé, Phys. Chem. Chem. Phys., 2016, 18, 19966–19972 RSC.
- R. Verrelli, A. P. Black, C. Pattanathummasid, D. S. Tchitchekova, A. Ponrouch, J. Oro-Sole, C. Frontera, F. Barde, P. Rozier and M. R. Palacin, J. Power Sources, 2018, 407, 162–172 CrossRef CAS.
- G. S. Gautam, P. Canepa, R. Malik, M. Liu, K. Perssonb and G. Ceder, Chem. Commun., 2015, 51, 13619–13622 RSC.
- D. S. Tchitchekova, C. Frontera, A. Ponrouch, C. Krich, F. Barde and M. R. Palacin, Dalton Trans., 2018, 47, 11298–11302 RSC.
- A. Torres, F. Bardé and M. E. Arroyo-de Dompablo, Solid State Ionics, 2019, 340, 115004 CrossRef.
- Z. Rong, R. Malik, P. Canepa, G. S. Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 6016–6021 CrossRef CAS.
- M. Liu, Z. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder and K. A. Persson, Energy Environ. Sci., 2015, 8, 964–974 RSC.
- A. Torres, F. J. Luque, J. Tortajada and M. E. Arroyo-de Dompablo, Energy Storage Mater., 2019, 21, 354–360 CrossRef.
- J. Hyoung, J. W. Heo and S.-T. Hong, J. Power Sources, 2018, 390, 127–133 CrossRef CAS.
- S. Suzuki, T. Kato, H. Kawabata and M. Miyayama, J. New Mater. Electrochem. Syst., 2016, 19, 51–55 CrossRef CAS.
- A. Torres, F. J. Luque, J. Tortajada and M. E. Arroyo-de Dompablo, Sci. Rep., 2019, 9, 9644 CrossRef CAS PubMed.
- E. F. Bertaut, P. Blum and G. Magnano, Acta Crystallogr., 1957, 10, 238–238 CrossRef.
- B. F. Decker and J. S. Kasper, Acta Crystallogr., 1957, 10, 332–337 CrossRef CAS.
- B. Malaman, H. Alebouyeh, F. Jeannot, A. Courtois, R. Gerardin and O. Evrard, Mater. Res. Bull., 1981, 16, 1139–1148 CrossRef CAS.
- O. Evrard, B. Malaman, F. Jeannot, A. Courtois, H. Alebouyeh and R. Gerardin, J. Solid State Chem., 1980, 35, 112–119 CrossRef CAS.
- B. Malaman, H. Alebouyeh, A. Courtois, R. Gerardin and O. Evrard, Mater. Res. Bull., 1982, 17, 795–800 CrossRef CAS.
- M. Aspiala, T. Hidayat, P. Taskinen and E. Jak, J. Alloys Compd., 2016, 658, 939–945 CrossRef CAS.
- H.-Y. Li and X.-M. Guo, Metall. Mater. Trans. B, 2015, 46, 278–285 CrossRef CAS.
- E. Rosen and B. Saitton, Scand. J. Metall., 1995, 24, 176–179 CAS.
- D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A30–A32 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Bloch, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1354–1365 CrossRef CAS.
- A. Torres and M. E. Arroyo-de Dompablo, J. Phys. Chem. C, 2018, 122, 9356–9362 CrossRef CAS.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, Powder Diffr., 2013, 28, 360–370 CrossRef.
- C. R. Fell, D. H. Lee, Y. S. Meng, J. M. Gallardo-Amores, E. Moran and M. E. Arroyo-de Dompablo, Energy Environ. Sci., 2012, 5, 6214–6224 RSC.
- Y. S. Meng and M. E. Arroyo-de Dompablo, Energy Environ. Sci., 2009, 2, 589–609 RSC.
- H. Yuan, H. Li, T. Zhang, G. Li, T. He, F. Du and S. Feng, J. Mater. Chem. A, 2018, 6, 8413–8418 RSC.
- M. P. Pechini, US Pat, 3330697, 1967 Search PubMed.
- B. Mortemard de Boisse, M. Reynaud, J. Ma, J. Kikkawa, S.-i. Nishimura, M. Casas-Cabanas, C. Delmas, M. Okubo and A. Yamada, Nat. Commun., 2019, 10, 2185 CrossRef PubMed.
- A. R. Wizansky, P. E. Rauch and F. J. Disalvo, J. Solid State Chem., 1989, 81, 203–207 CrossRef CAS.
- D. Larcher, D. Bonnin, R. Cortes, I. Rivals, L. Personnaz and J.-M. Tarascon, J. Electrochem. Soc., 2003, 150, A1643–A1650 CrossRef CAS.
- J. M. Tarascon, S. Grugeon, S. Laruelle, D. Larcher and P. Poizot, in Lithium Batteries: Science and Technology, ed. G. Nazri and G. Pistoia, Kluwer Academic Publishers, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: DFT and lattice parameters, Fig. S1: calculated energy migration barrier for CaFe2O4, Table S2 and Fig. S2: summary of synthesis methods, Fig. S3: Rietveld refinement, and Fig. S4: electrochemistry and XRD of Ca2Fe2O5. See DOI: 10.1039/C9DT04688G |
| This journal is © The Royal Society of Chemistry 2020 |