
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dimolybdenum paddlewheel as a building block for heteromultimetallic structures†
Nicolai D.
Knöfel
 ,
Christoph
Schoo
,
Tim P.
Seifert
and
Peter W.
Roesky
*
,
Christoph
Schoo
,
Tim P.
Seifert
and
Peter W.
Roesky
*
Institute for Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 10th January 2020
Abstract
Diphenylphosphine functionalized propionic acid was applied for the synthesis of heteromultimetallic dimolybdenum(II) complexes. The ligand features both carboxylic acid and phosphine functionalities, allowing the selective synthesis of a tetracarboxylate bridged Mo2(II)-paddlewheel structure in a first step. Due to the symmetrically arranged phosphine functionalities, the dimolybdenum(II) complex was utilized as a metalloligand. Subsequent coordination of late transition metal ions, such as gold(I), rhodium(I), iridium(I) or ruthenium(II) to the phosphine moieties allowed the formation of heteromultimetallic structures. The flexibility of the diphenylphosphino propionate ligand system enabled intermolecular aurophilic interactions in the Au(I) functionalized dimolybdenum(II) complexes. Depending on the Au(I) species applied, either a dimeric structure or a 1D coordination polymer was formed in the solid state. These structures represent the first examples of heterometallic dimolybdenum(II) complexes, forming supramolecular structures via aurophilic interactions.
Introduction
In the last decades molybdenum has been extensively studied regarding the formation of multiply-bonded M2n+ compounds, due to the complexes’ unique scaffold, redox behavior, spectroscopic properties1 as well as catalytic applications.2–4 The formation of dimolybdenum(II) paddlewheel structures requires ligands, which support a bidentate bridging coordination mode, e.g. carboxylates (O2CR−) or amidinates ((RN)2C(R′)−).1 Both ligand systems are easily adjustable regarding their solubility behavior, steric demand as well as the introduction of additional functionalities. However, for the formation of heteromultimetallic structures containing a dimolybdenum(II) core unit, the application of bifunctional ligands, exhibiting orthogonal functionalities is mandatory. This was e.g. demonstrated by Mashima et al., who utilized the bifunctional ligand 6-diphenylphosphino-2-pyridonate (pyphos) for the formation of Mo24+ centered heterometallic structures via the phosphine coordination of Pd(II) and Pt(II) (Scheme 1A).5 Additionally, our group investigated the application of bifunctional ligand 4-(diphenylphosphino) benzoic acid for the formation of heterometallic dimolybdenum(II) complexes. The synthetic strategy resulted in heteromultimetallic structures of the general formula [Mo2(O2C-Ph-PPh2)4(MLx)4] (M = Au, Rh, Ir), which have been studied in detail regarding their tunable photophysical properties.6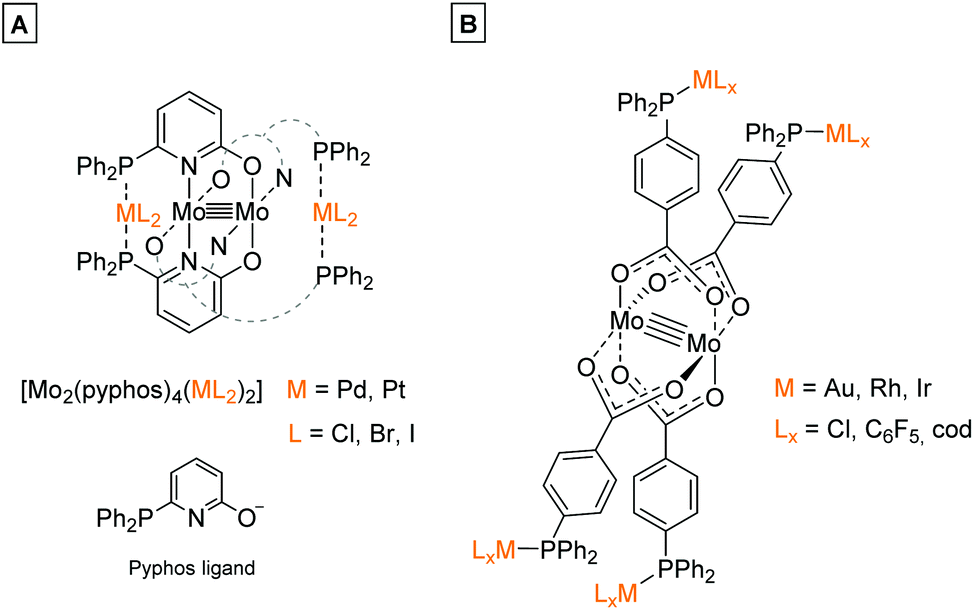 | ||
| Scheme 1 (A) Dinuclear metalloligand [Mo2(pyphos)4] (pyphos = 6-diphenylphosphino-2-pyridonat) allowed the formation of heterometallic structures via coordination of Pd(II) or Pt(II) to the phosphine moieties.5 (B) Phosphine-functionalized dimolybdenum(II) metalloligand [Mo2(O2C-Ph-PPh2)4] was suitable for the coordination of late transition metal ions (e.g. Au(I), Rh(I), Ir(I)) by the symmetrically arranged phosphine groups, resulting in heteromultimetallic structures containing a Mo24+ core unit.6 | ||
In general, phosphine functionalized carboxylic acids have been proven advantageous for the synthesis of heterometallic complexes,7,8 as they are able to selectively coordinate via different bonding modes to various metal ions by their hard carboxylic and their soft phosphine donor centers.9–12 Such ligand systems allow the combination of early and late transition metals within an organometallic structure, a research field, which has been comprehensively studied in the last years. Hereby, cooperative effects, caused by an electronic communication between metal ions, can amongst others lead to enhanced catalytic systems or interesting photophysical properties.9,13
The bifunctional ligand 3-(diphenylphosphino)propionic acid is sterically less demanding in comparison to the previously discussed 4-(diphenylphosphino) benzoic acid (Scheme 1B), thus exhibiting a higher flexibility between the two functional moieties. Hence, 3-(diphenylphosphino)propionic acid is able to function as a flexible hemilabile chelating system.14–17 Here, both the carboxylic acid and phosphine functionality are able to coordinate with different bonding strength to a metal center (Scheme 2B), which is of high interest regarding the construction of organometallic catalysts.7,17–19 3-(Diphenylphosphino)propionic acid has already been successfully applied as a bifunctional ligand for the construction of supramolecular structures as well as heterometallic complexes, applying different metal combinations.8,20–22
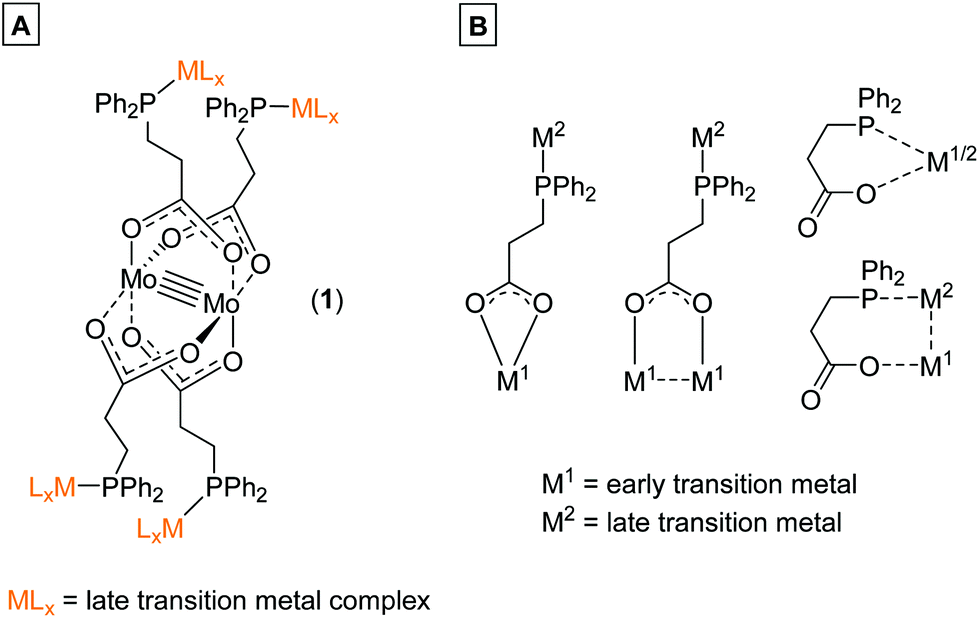 | ||
| Scheme 2 (A) Dimolybdenum(II) metalloligand (1) allowed the subsequent build-up of heteromultimetallic structures via metal coordination of the phosphine donor sites. (B) Variable coordination modes of the bifunctional ligand 3-(diphenylphosphino)propionic acid towards different metal centers. Due to the ethylene-spacer moiety between phosphine and carboxylic acid functionality the ligand can act as a flexible hemilabile ligand system. | ||
Herein, we report the synthesis of tetracarboxylate dimolybdenum(II) complex [Mo2(O2C-C2H4-PPh2)4] (1) and its application as a fourfold metalloligand, applying 3-(diphenylphosphino)propionic acid. The four additional phosphine donor sites allowed for subsequent treatment with late transition metals, obtaining heteromultimetallic complexes (Scheme 2A). Due to the ethylene-spacer between the phosphine and the carboxylate unit, a significantly enhanced flexibility of the ligand, compared to those shown in Scheme 1, was anticipated. This was proven by the coordination of different Au(I) species, which enabled the formation of dimeric as well as 1D polymeric structures via aurophilic interactions.
Results and discussion23
For the synthesis of dimolybdenum(II) carboxylate paddlewheel complexes of the general formula [Mo2(O2CR)4] starting materials such as [Mo(CO)6]1,24,25 and K4[Mo2Cl8]26 are commonly applied. Additionally, the easily accessible tetraacetate [Mo2(OAc)4] has proven to be an ideal starting material,1,27–31e.g. in case of phosphine functionalized carboxylic acids.6 Here, four acetate groups act as bidentate bridging ligands, forming a homoleptic paddlewheel structure around a Mo24+ unit,32 a structural motif prototypical for most dimolybdenum(II) tetra-carboxylate complexes.The phosphine functionalized homoleptic metalloligand 1 was obtained by a carboxylate exchange reaction, applying [Mo2(OAc)4] and 3-(diphenylphosphino)propionic acid (Scheme 3). Interestingly, compound 1 was not accessible via a reaction with K4[Mo2Cl8]. This is most likely due to the formation of side products such as [Mo2Cl4(PR3)4], which has been sufficiently investigated for sterically less demanding phosphine ligands (R = Me, n-Pr, n-Bu).33,34 Yellow single crystals of 1 were obtained from slow diffusion of n-pentane into a solution of 1 in THF, allowing characterization by X-ray diffraction. Compound 1 crystallizes in the monoclinic space group P21/n with half of a molecule in the asymmetric unit and no additional solvent molecules coordinating to the axial sites of the dimolybdenum unit. The molecular structure in the solid state, depicted in Fig. 1, reveals a homoleptic tetracarboxylate-bridged Mo24+ paddlewheel scaffold. The molybdenum–molybdenum bond length is 2.1134(4) Å, which is in the expected range for a quadruple Mo–Mo bond in a bidentate bridged coordination geometry (2.06–2.13 Å).35 All molybdenum–oxygen bond lengths to the carboxylic units are almost identical (Mo–O = 2.109–2.128 Å), indicating a highly symmetric arrangement in the solid state. The individual carboxylate ligands are oriented in a 90° angle to each other, as it is expected in a paddlewheel structure.
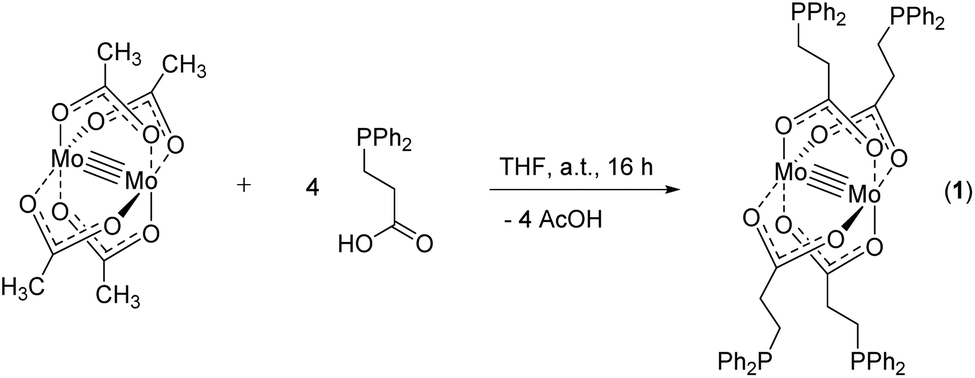 | ||
| Scheme 3 Synthesis of metalloligand 1via carboxylate exchange from molybdenum(II) acetate with 3-(diphenylphosphino)propionic acid in THF. | ||
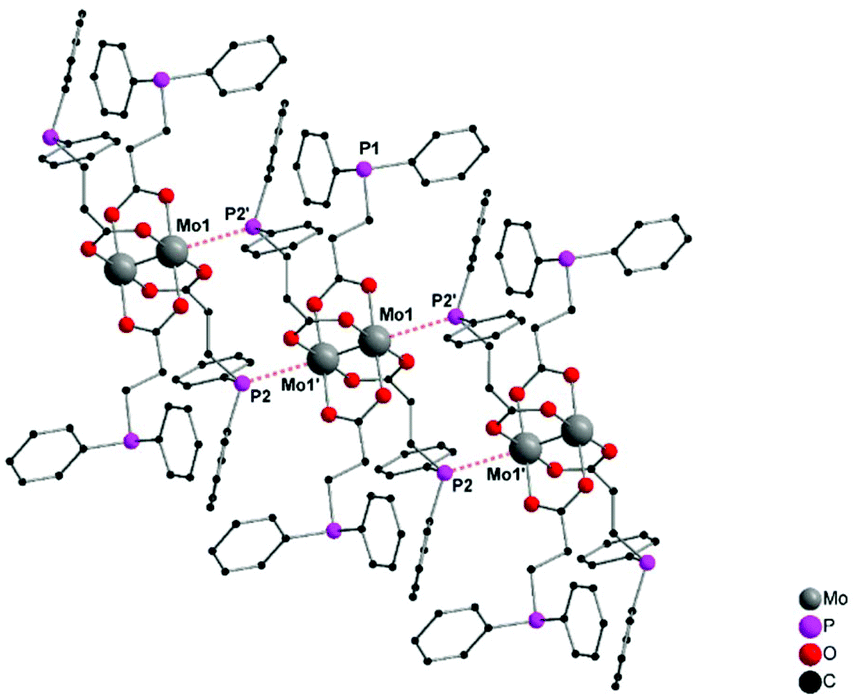 | ||
| Fig. 1 Molecular structure of 1 in the solid state. Formation of a 1D coordination polymeric structure, due to an axial coordination of phosphine moieties to neighboring dimolybdenum units. An excerpt of three units is displayed and all hydrogen atoms are omitted for clarity. Distance P2–Mo1 (red, dotted): 3.0162(6) Å. | ||
Interestingly, a 1D coordination polymeric structure is formed in the solid state, which is due to an axial coordination of phosphine ligands (P2, P2′) to neighboring Mo24+ units (Fig. 1). The distance between the phosphine ligands and dimolybdenum moieties (Mo1–P2) is 3.0162(6) Å. Hence, the flexible diphenylphosphino moiety allows an intermolecular axial coordination and formation of a supramolecular structure. However, the axial phosphine coordination does not seem to have a significant influence upon the Mo–Mo quadruple bond. The intermolecular phosphine coordination to the axial coordination sites of a bridged Mo24+ moiety is a known structural motif,36 leading to 1D polymeric structures in specific cases. The application of the sterically more demanding diphenylphosphino benzoate ligand resulted, in earlier studies, in a similar 1D coordination structure in the solid state.6 However, in the latter case a Mo–P distance of ca. 3.5 Å was observed, indicating only weak axial interactions compared to 1. Additionally, an axial attachment of two PPh3 ligands to [Mo2(O2CCF3)4] was reported by Cotton, exhibiting similar, slightly longer Mo–P distances of 3.07(5) Å.36
Additionally, 31P{1H} NMR measurements of 1 (CDCl3) indicate no direct phosphor molybdenum interaction in solution, as merely a single resonance at δ = −14.7 ppm is detected, which displays the typical range for non-coordinating diphenylphosphino moieties and is in agreement with the results obtained for the structurally related [Mo2(p-O2C-C6H4-PPh2)4].6 In the corresponding 1H and 13C{1H} NMR spectra of 1, one set of resonances is observed for the ligand system, indicating the symmetric behavior of compound 1 in solution. Further information regarding the structural arrangement of 1 was obtained by vibrational spectroscopy. In a bridged coordination of a carboxylate unit (RCO2−), the two C–O bonds are equivalent and the corresponding frequencies of the symmetric and anti-symmetric stretching modes can normally be detected as strong bands in the region of 1400–1600 cm−1, mainly depending on the ligand and coordinated metal ions, respectively.25,36,37 However, the evaluation of the corresponding IR data of 1 is complicated by the aromatic skeletal vibrations of the diphenylphosphino moieties.25 For compound 1, the characteristic antisymmetric C–O stretching mode can be detected at ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) a = 1517 cm−1 and the symmetric mode at s = 1427/1410 cm−1, respectively. This is in agreement with data obtained for structurally related dimolybdenum complexes25 and refers to a bridged carboxylate coordination mode.
a = 1517 cm−1 and the symmetric mode at s = 1427/1410 cm−1, respectively. This is in agreement with data obtained for structurally related dimolybdenum complexes25 and refers to a bridged carboxylate coordination mode.
The symmetrically placed phosphine functionalities in metalloligand 1 enable the subsequent coordination of additional metal ions, e.g. late transition metals such as Au(I), Rh(I), Ir(I) and Ru(II). Thus, the formation of heteromultimetallic complexes with a dimolybdenum core unit can be realized. Furthermore, the choice of 3-(diphenylphosphino)propionic acid as a bifunctional ligand allows the investigation of potential metallophilic interactions of respective Au(I) functionalized structures. In a first approach, metalloligand 1 was reacted with four equivalents of [AuC6F5(tht)] (tht = tetrahydrothiophene), obtaining the heterometallic complex [Mo2(O2C-C2H4-PPh2)4(AuC6F5)4]2 (2) by ligand exchange reactions (Scheme 4).
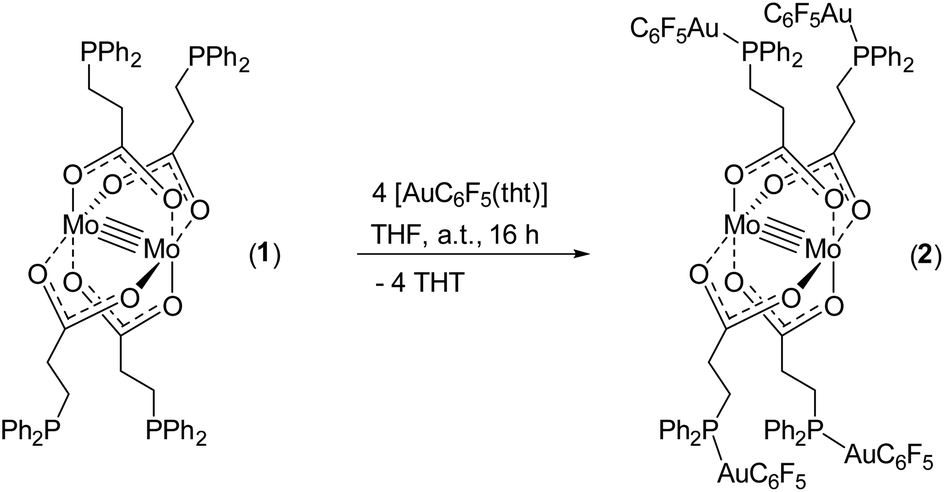 | ||
| Scheme 4 Synthesis of the heteromultimetallic complex 2, reacting the phosphine-functionalized metalloligand 1 with [AuC6F5(tht)]. Compound 2 forms a dimeric complex [Mo2(O2C-C2H4-PPh2)4(AuC6F5)4]2·2 thf in the solid state (scheme merely exhibits the respective monomer). | ||
Yellow single crystals were obtained by slow diffusion of n-pentane into a solution of 2 in THF. The heterometallic complex 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one monomer, which is part of a dimeric structure, in the asymmetric unit (Fig. 2). The formation of a dimeric structure in the solid state is due to aurophilic interactions between Au1 and Au2′ (respectively Au1′ und Au2). The distance between Au1–Au2 is 3.2198(4) Å and hence below the corresponding van der Waals distance of two gold atoms (3.32 Å), suggesting the presence of intermolecular aurophilic interactions.38,39 The aurophilic contacts between the Au(I) centers in 2 are most likely facilitated by the high flexibility of the applied ligand system, since no aurophilic contacts have been observed in the similar Au(I) functionalized complexes shown in Scheme 1B.
with one monomer, which is part of a dimeric structure, in the asymmetric unit (Fig. 2). The formation of a dimeric structure in the solid state is due to aurophilic interactions between Au1 and Au2′ (respectively Au1′ und Au2). The distance between Au1–Au2 is 3.2198(4) Å and hence below the corresponding van der Waals distance of two gold atoms (3.32 Å), suggesting the presence of intermolecular aurophilic interactions.38,39 The aurophilic contacts between the Au(I) centers in 2 are most likely facilitated by the high flexibility of the applied ligand system, since no aurophilic contacts have been observed in the similar Au(I) functionalized complexes shown in Scheme 1B.
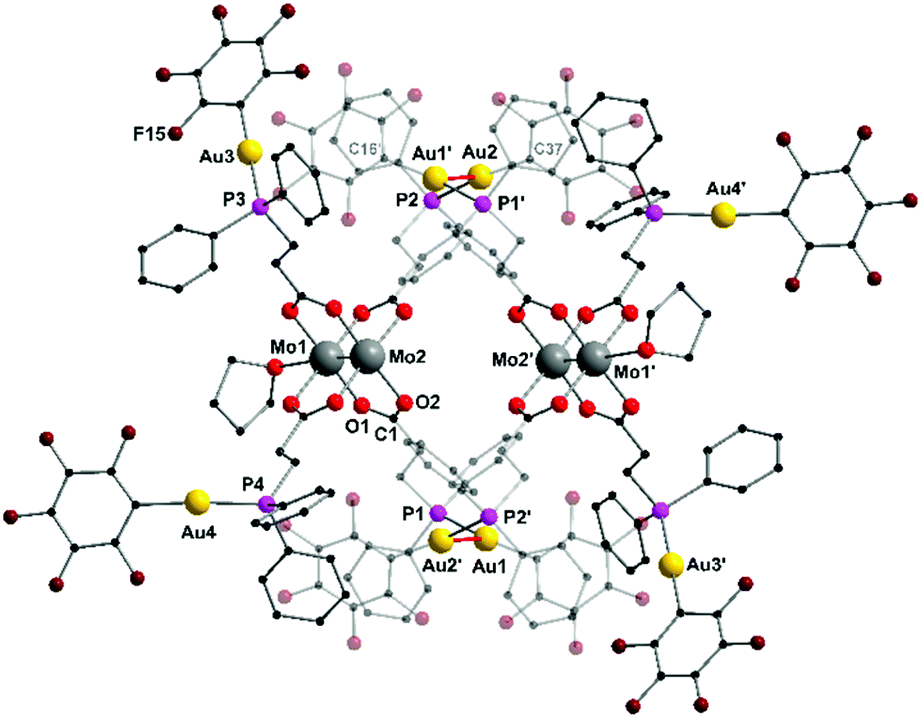 | ||
| Fig. 2 Molecular structure of 2 (·2 thf) in the solid state. Formation of a dimeric structure, due to intermolecular aurophilic interactions (marked red) between Au1 and Au2. Metal–metal distance [Å]: Au1–Au2′ 3.2198(4). Hydrogen atoms and non-coordinating solvent molecules (THF) are omitted for clarity. | ||
The main paddlewheel scaffold remains unchanged upon the AuC6F5 complexation with a molybdenum–molybdenum quadruple bond length of 2.0904(9) Å, which is slightly shorter compared to 1. Regarding the Mo–Mo bond length, the influence of axially coordinating solvent molecules has to be considered.35 Interestingly, in the case of 2, merely one THF molecule coordinates axially to each Mo24+ moiety due to the dimeric structure, which shields the axial coordination site of Mo2 (Mo2′). Additionally, the remaining two gold atoms (Au3, Au4), which do not participate in aurophilic interactions, are ligated in an almost linear fashion as indicated by the P–Au–C6F5 bond angles (P3–Au3–C58 177.8(3)° and P4–Au4–C79 175.8(3)°). Accordingly, the coordination geometry of the gold atoms, which are connected via aurophilic interactions deviate significantly from a linear arrangement (P1–Au1–C16 and P2–Au2–C37 169.8(2)°).
The composition of compound 2 was further confirmed by multi-nuclei NMR measurements (1H, 13C{1H}, 31P{1H} 19F{1H}) as well as IR spectroscopy. In the 1H NMR spectrum a downfield shift of the aromatic resonances, compared to metalloligand 1, is observed upon coordination of the AuC6F5 units. In the 31P{1H} NMR spectrum a single resonance, significantly downfield shifted compared to metalloligand 1 (δ = −14.7 ppm), is detected at δ = 37.7 ppm in the splitting of a pseudo-quintet, indicating a sole phosphine–gold coordination (Fig. 4). The resonance splitting is caused by a long-range phosphor coupling to the fluorine atoms of the C6F5 moieties, an effect, which has already been observed in comparable complexes.40–42 Additionally, in the 19F{1H} NMR spectrum, a characteristic set of three resonances for the C6F5 units, with an integration ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2, is observed at δ = −116.1 (o-F), −157.8 (t, 3JF,F = 20.0 Hz, p-F) and −161.9 ppm (m-F), respectively. In the IR spectrum of 2 no significant shift of the symmetric and antisymmetric C–O stretching modes is detected (a = 1501 cm−1, s = 1435/1409 cm−1), hence remaining nearly unaffected upon gold(I) coordination.
:1:2, is observed at δ = −116.1 (o-F), −157.8 (t, 3JF,F = 20.0 Hz, p-F) and −161.9 ppm (m-F), respectively. In the IR spectrum of 2 no significant shift of the symmetric and antisymmetric C–O stretching modes is detected (a = 1501 cm−1, s = 1435/1409 cm−1), hence remaining nearly unaffected upon gold(I) coordination.
To obtain further heteromultimetallic complexes, metalloligand 1 was reacted with two equivalents of the metal precursors [RhCl(cod)]2, [IrCl(cod)]2 and [RuCl2(p-cymene)]2, respectively. Upon phosphine coordination the dimeric Rh(I)-, Ir(I)- and Ru(II)-precursors dissociate to obtain the heterometallic complexes [Mo2(O2C-C2H4-PPh2)4(RhCl(cod))4] (3), [Mo2(O2C-C2H4-PPh2)4(IrCl(cod))4] (4) and [Mo2(O2C-C2H4-PPh2)4(RuCl2(p-cymene))4] (5) (Scheme 5).
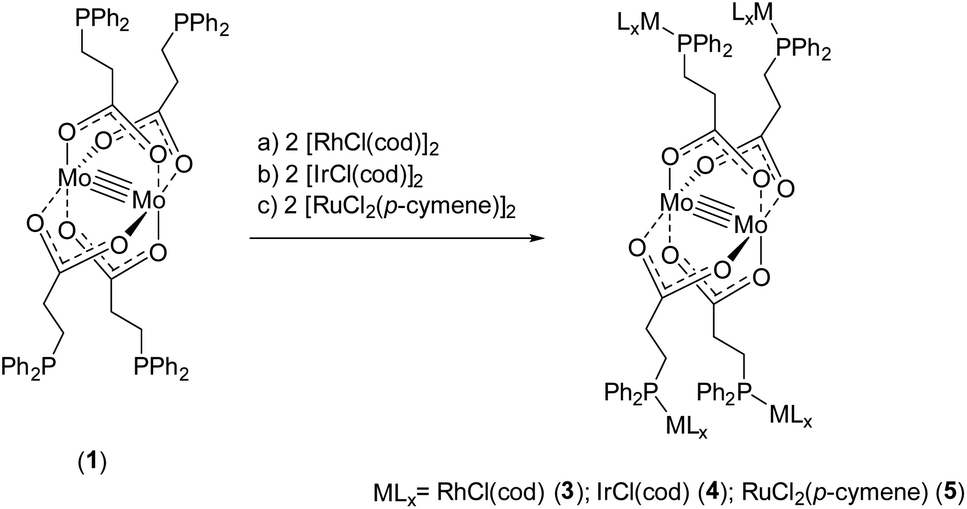 | ||
| Scheme 5 Synthesis of heteromultimetallic complexes 3–5, applying metalloligand 1 and the corresponding metal precursors [RhCl(cod)]2, [IrCl(cod)]2 and [RuCl2(p-cymene)]2. | ||
Single crystals of compounds 3 and 4 were obtained after cooling of respective hot THF solutions. Both complexes crystallize in the triclinic space group P, in each case with half a molecule in the asymmetric unit. The corresponding molecular structures of 3 and 4 in the solid state are isostructural (3 is exhibited in Fig. 3, left; 4 see ESI, Fig. S5†). The tetracarboxylate-bridged dimolybdenum structure remains unchanged upon Rh(I) or Ir(I) coordination, yet two THF molecules coordinate axially to the Mo24+ units. The bond length of the Mo–Mo quadruple bond is 2.1045(8) Å (3) and 2.1005(8) Å (4), respectively. Every Rh(I)- or Ir(I) center is formally in a square planar coordination sphere, composed of a phosphine ligand, chloride and bidentate cyclooctadiene (cod).
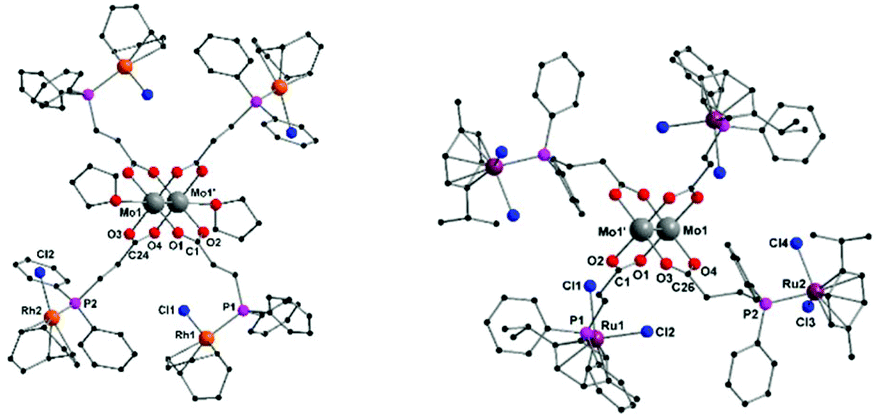 | ||
| Fig. 3 Molecular structures of compounds 3 and 5 in the solid state. Hydrogen atoms and non-coordinating solvent molecules (THF, DCM) are omitted for clarity. Molecular structure of Ir(I) functionalized compound 4 is isostructural to 3 in the solid state (ESI, Fig. S5†). | ||
Furthermore, the isostructural compounds 3 and 4 were investigated by NMR and IR spectroscopy. Due to a coupling to the NMR active 103Rh nuclei (s = ½), a doublet resonance at δ = 25.8 ppm is detected (1JP,Rh = 149.2 Hz) in the 31P{1H} NMR spectrum of Rh(I) compound 3 (Fig. 4). Analogously, a singlet resonance at δ = 15.6 ppm is observed for the Ir(I) compound 4 in the corresponding 31P{1H} NMR spectrum, confirming a complete phosphine–metal coordination and symmetric behavior in solution (Fig. 4). The IR spectra of both compounds 3 and 4 are almost identical and exhibit the characteristic C–O stretching modes, confirming the isostructural composition of the two complexes.
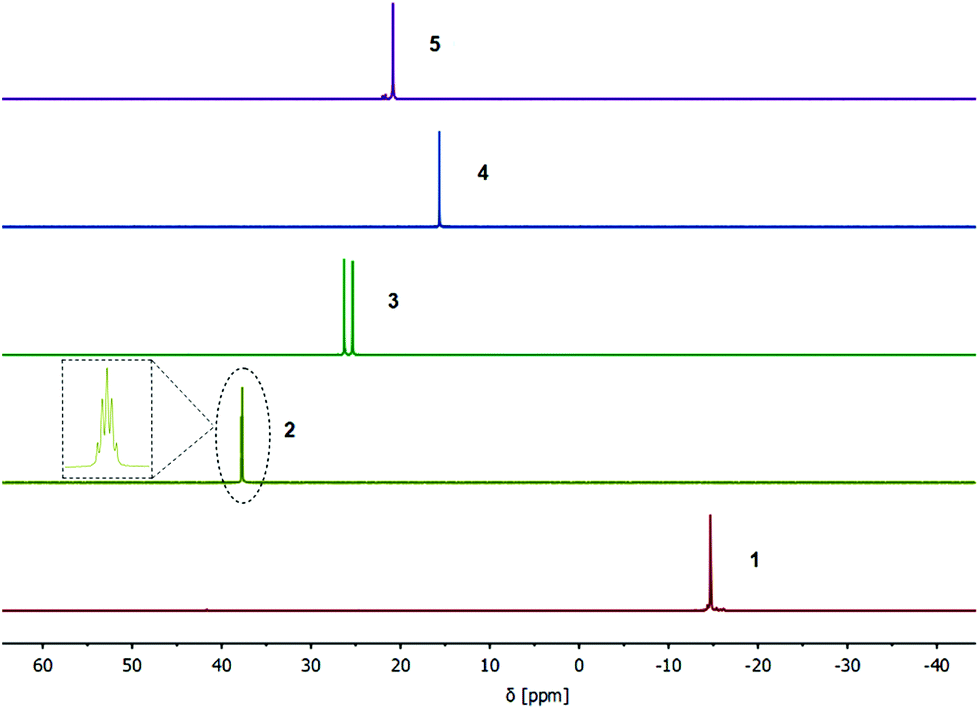 | ||
| Fig. 4 31P{1H} NMR spectra of the compounds 1–5 in CDCl3 (the intensity of the corresponding resonances is adjusted with regards to compound 1). Complex 2 exhibits a resonance at δ = 37.7 ppm in the splitting of a pseudo-quintet, which is due to a long-range coupling of the phosphorous atoms with the fluorine atoms of the C6F5 moieties. The 31P resonance of 3 is split to a doublet due to a coupling with the 103Rh nuclei (d, 1JP,Rh = 149.2 Hz). | ||
Red single crystals of the Ru(II) functionalized complex 5, suitable for X-ray diffraction, were obtained by slow evaporation of a DCM solution of 5. The compound crystallizes in the triclinic space group P with half of a molecule of 5 in the asymmetric unit (Fig. 3, right). The molecular structure in the solid state exhibits the expected heteromultimetallic Mo2Ru4 paddlewheel structure. Every Ru(II) center is hereby coordinated by a phosphine ligand, two chlorides and η6-coordinated p-cymene. Interestingly, the Mo–Mo quadruple bond length of 2.079(2) Å in 5 is the shortest of the dimolybdenum complexes presented here (1–6), yet still in the lower region of the expected range for a bridged coordination geometry (2.06–2.13 Å).35 Due to crystallization in the non-coordinating solvent DCM, no axial donor coordination is observed in the solid state, which may amongst others explain the rather short Mo–Mo bond distance. Additionally, compound 5 was investigated by NMR and IR spectroscopy. In the 1H and 13C{1H} NMR spectra (CDCl3) additional resonances for the p-cymene ligand, in comparison to metalloligand 1, are observed. In the corresponding 31P{1H} NMR spectrum of 5 a singlet resonance at δ = 20.8 ppm is detected, referring to a complete complexation of the phosphine moieties with Ru(II) (Fig. 4). Additionally, the symmetric and antisymmetric C–O stretching modes (a = 1508 cm−1, s = 1424/1413 cm−1) are detected in the IR spectrum, confirming the proposed carboxylate coordination mode.
To introduce yet another Au(I) species besides AuC6F5 and further investigate the potential formation of aurophilic interactions, as observed in 2, metalloligand 1 was reacted with [AuCl(tht)]. However, following this synthesis route no product formation was observed, most likely due to redox induced side reactions. Hence, in a different approach, utilizing the orthogonal properties of the bifunctional ligand system, the literature known complex [3-(diphenylphosphino)propionic acid-gold(I)chloride]43 was synthesized via reaction of 3-(diphenylphosphino)propionic acid with [AuCl(tht)]. A subsequent conversion with [Mo2(OAc)4] led to the formation of heterometallic compound [Mo2(O2C-C2H4-PPh2)4(AuCl)4] (6) (Scheme 6).
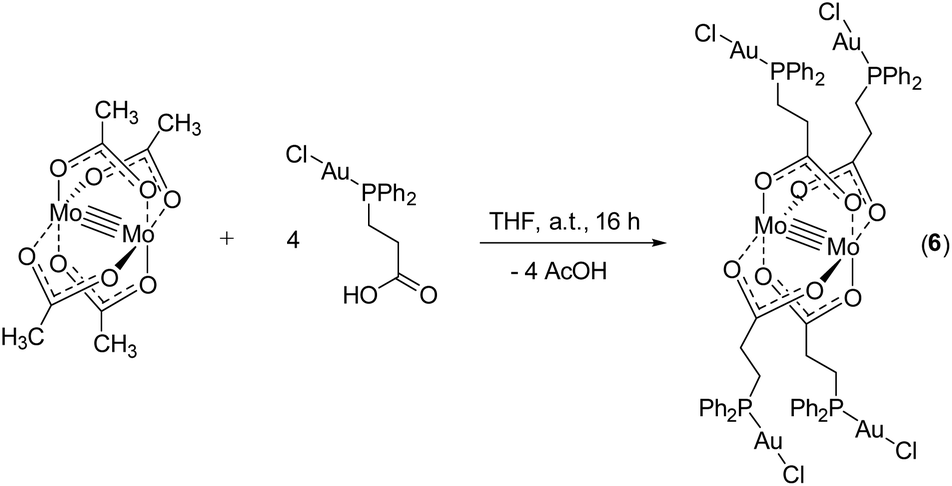 | ||
| Scheme 6 Synthesis of heteromultimetallic Mo2Au4 compound 6via carboxylate substitution from molybdenum(II) acetate. Compound 6 forms a 1D coordination polymeric structure in the solid state (scheme merely exhibits the respective monomer). | ||
Yellow single crystals of compound 6, suitable for X-ray diffraction, were obtained from a hot THF solution. Compound 6 crystallizes in the orthorhombic space group Pbcn with half of a molecule in the asymmetric unit and two THF solvent molecules axially coordinated to the dimolybdenum scaffold (ESI; Fig. S7†). Interestingly, a 1D coordination polymeric structure is formed in the solid state due to intermolecular aurophilic interactions between Au2–Au2′ (Fig. 5), explaining the low solubility of 6 in common organic solvents. The distance of the respective gold atoms is 3.1427(12) Å and therefore below the corresponding van der Waals distance (3.32 Å), suggesting the formation of aurophilic interactions.38,39
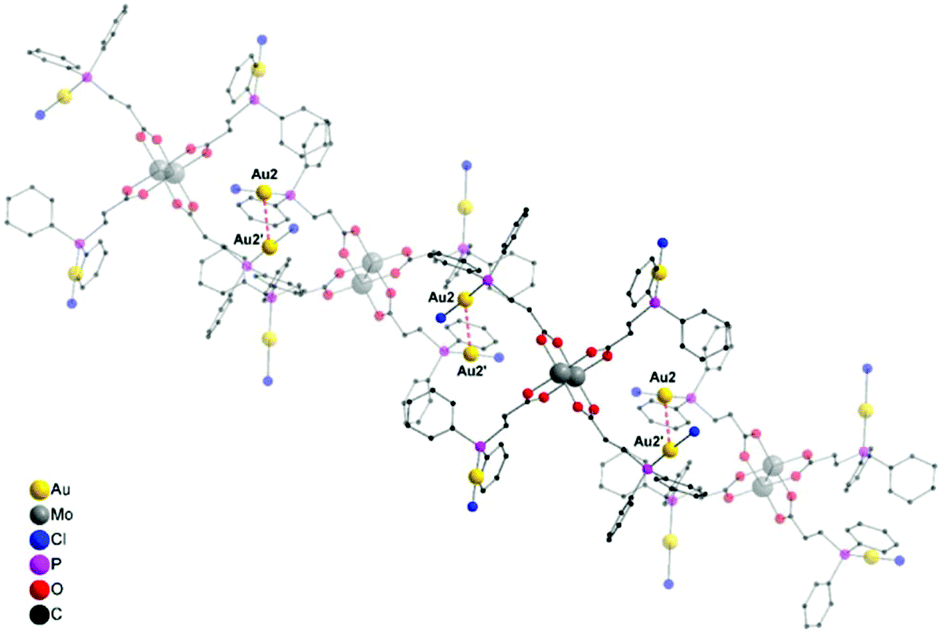 | ||
| Fig. 5 Excerpt of the extended molecular structure of 6 (·2 thf) in the solid state. Hydrogen atoms and solvent molecules (THF) are omitted for clarity. Compound 6 forms a 1D coordination polymer structure in the solid state due to intermolecular aurophilic interactions between Au2 and Au2′. Metal–metal distance Au2–Au2′ (red, dotted): 3.1427(12) Å. | ||
Because of the sterically less demanding AuCl fragments, compound 6, in comparison to 2 (dimeric structure), forms a supramolecular 1D polymer structure via aurophilic contacts. Additionally, the observed gold–gold distances in 6 are significantly shorter than in 2 (Au–Au distance: 3.2198(4) Å), which is attributed to the different steric demand of the gold substituents. In compound 6, all the gold atoms are coordinated in a near linear fashion, as indicated by the P–Au–Cl bond angles (P1–Au1–Cl1 178.0(2)°, P2–Au2–Cl2 177.8(2)°). The corresponding Mo–Mo quadruple bond length is 2.100(3) Å and lies in the expected region (see ESI; Fig. S7†).
Compound 6 was further characterized by NMR and IR spectroscopy. In the 31P{1H} NMR spectrum (CDCl3) of compound 6, a main resonance at δ = 30.0 ppm is detected, which is downfield shifted compared to metalloligand 1 (δ = −14.7 ppm), indicating a complete coordination of the gold(I) chloride units to the phosphine moieties. This is in agreement with the chemical shift of the gold(I) precursor complex [3-(diphenylphosphino)propionic acid-gold(I)chloride] (δ = 30.9 ppm; DMSO-d6).43 Interestingly, additional resonances in the region δ = 30.0–29.0 ppm are observed, which can most likely be attributed to partial aurophilic interactions in solution, causing a slightly different chemical environment (see ESI; Fig. S31†). This effect is accordingly observed, performing NMR measurements at elevated temperatures (up to 333 K; see Fig. S32†). The antisymmetric and symmetric C–O stretching modes of the bridged carboxylate units are detected in the IR spectrum (a = 1522 cm−1, s = 1432/1405 cm−1) and are in the expected range.
Conclusion
In summary, we present the synthesis of heteromultimetallic dimolybdenum(II) complexes, utilizing the bifunctional ligand 3-(diphenylphosphino)propionic acid. The orthogonal carboxylic acid and phosphine functionalities allowed the selective synthesis of a tetracarboxylate bridged Mo2(II)-paddlewheel structure (1) in a first step, which was subsequently applied as a dinuclear metalloligand. Coordination of late transition metal precursors (gold(I), rhodium(I), iridium(I) or ruthenium(II)) to the phosphine moieties of the metalloligand led to the formation of heteromultimetallic complexes of the general composition [Mo2(O2C-C2H4-PPh2)4(MLx)4]. Interestingly, the flexibility of the propionate ligand system helped, enabling the formation of intermolecular aurophilic interactions in the Au(I) functionalized dimolybdenum(II) complexes, whereupon either a dimeric (2) or a 1D coordination polymer structure (6) was formed in the solid state. These structures represent the first examples of heterometallic quadruply bonded dimolybdenum complexes, forming supramolecular structures via aurophilic interactions.Experimentals
General procedures
All manipulations were performed under exclusion of moisture and oxygen in flame-dried Schlenk-type glassware or in an argon-filled MBraun glovebox. Prior to use, DCM was distilled under nitrogen from CaH2, and MeOH from magnesium. Hydrocarbon solvents (THF, n-pentane) were dried using an MBraun solvent purification system (SPS-800). THF was additionally distilled under nitrogen from potassium and benzophenone before storage over 4 Å molecular sieves. Deuterated solvents were obtained from Carl Roth GmbH (99.5 atom% D). Prior to use, CDCl3 was stored over molecular sieves (4 Å). NMR spectra were recorded on a Bruker Ascend 400 MHz spectrometer. 1H and 13C{1H} NMR chemical shifts were referenced to the residual 1H and 13C resonances of the deuterated solvents and are reported relative to tetramethyl silane (TMS). 31P{1H} and 19F{1H} NMR resonances were referenced to external 85% phosphoric acid and CFCl3, respectively. IR spectra were obtained on a Bruker Tensor 37 FTIR spectrometer equipped with a room temperature DLaTGS detector and a diamond ATR (attenuated total reflection) unit. Elemental analyses were carried out with a Micro Cube from Elementar Analysensysteme GmbH.[Mo2(OAc)4],25 [AuCl(tht)]44 and [AuC6F5(tht)]45 were prepared according to literature procedures. Bis(1,5-cyclooctadiene)diiridium(I) dichloride (97%), molybdenum hexacarbonyl (98%) and 3-(diphenylphosphino)propionic acid (97%) were purchased from Sigma-Aldrich. Bis(1,5-cyclooctadiene)dirhodium(I) dichloride and dichloro(p-cymene)ruthenium(II) dimer (98%) were purchased from Alfa Aesar. They were all used as received.
Synthesis23
[Mo2(O2C-C2H4-PPh2)4] (1). Molybdenum(II) acetate (200 mg, 0.47 mmol, 1.00 equiv.) and 3-(diphenylphosphino)propionic acid (507 mg, 1.96 mmol, 4.20 equiv.) were dissolved in THF (10 mL) and stirred at room temperature for 16 h. The solvent was removed under reduced pressure, and the yellow solid subsequently washed two times with dry MeOH (5 mL). The solid was recrystallized two times from THF (3 mL), storing the solution at −30 °C for three days. Finally, the mother liquor was decanted off and the solid dried under vacuum. Crystalline yield: 310 mg (49%), relating to 1 (·2 thf). Single crystals suitable for X-ray analysis were obtained from slow diffusion of n-pentane into a solution of 1 in THF. The formation of a 1D coordination polymer is observed in the corresponding solid state structure.
1H NMR (400 MHz, CDCl3): δ [ppm] = 7.41–7.32 (m, 16H, Ph), 7.28–7.18 (m, 24H, Ph), 3.09–2.95 (m, 8H, CH2CO2), 2.64–2.51 (m, 8H, CH2PPh2). – Additional resonances (m) for residually coordinated THF are observed at δ = 3.76–3.65 and 1.90–1.78 ppm. 13C{1H} NMR (101 MHz, CDCl3): δ [ppm] = 184.4 (d, 3JC,P = 14.1 Hz, CO2R), 137.6 (d, 1JC,P = 10.8 Hz, i-CP), 133.0 (d, 2JC,P = 18.3 Hz, o-CPh), 128.8 (p-CPh), 128.6 (d, 3JC,P = 6.8 Hz, m-CPh), 33.4 (d, 2JC,P = 18.2 Hz, CH2CO2), 24.7 (d, 1JC,P = 10.7 Hz, CH2PPh2). – Additional resonances (s) for residually coordinated THF are observed at δ = 68.1 and 25.7 ppm. 31P{1H} NMR (162 MHz, CDCl3): δ [ppm] = −14.7 (s, PPh2). IR (ATR): [cm−1] = 3067 (w), 3048 (w), 2981 (m), 2949 (w), 2893 (vw), 2874 (w), 1584 (w), 1517 (s), 1480 (m), 1427 (vs), 1410 (s), 1333 (vw), 1305 (s), 1263 (w), 1180 (w), 1160 (vw), 1136 (vw), 1093 (w), 1068 (m), 1047 (m), 1027 (m), 998 (w), 935 (w), 923 (w), 879 (m), 828 (m), 774 (vw), 746 (s), 698 (s), 677 (m), 658 (m), 610 (m), 586 (w), 509 (s), 481 (w), 459 (w), 430 (w). Elemental analysis calcd (%) for [C60H56Mo2O8P4] (1220.92 g mol−1): C 59.03, H 4.62; found C 58.93, H 4.90.
[Mo2(O2C-C2H4-PPh2)4(AuC6F5)4]2 (2). The phosphine functionalized metalloligand 1 (·2 thf) (75.0 mg, 0.06 mmol, 1.00 equiv.) and [AuC6F5(tht)] (111 mg, 0.25 mmol, 4.00 equiv.) were dissolved in THF (10 mL) and stirred at room temperature for 16 h. The solvent was removed under reduced pressure, the obtained yellow solid washed with n-pentane (10 mL) and subsequently dried under vacuum. Single crystals suitable for X-ray analysis were obtained from slow diffusion of n-pentane into a solution of 2 in THF. Crystalline yield: 87.0 mg (58%), relating to 2 (·2 thf).
1H NMR (400 MHz, CDCl3): δ [ppm] = 7.80–7.69 (m, 16H, Ph), 7.53–7.42 (m, 24H, Ph), 3.38–3.23 (m, 8H, CH2), 3.11–2.97 (m, 8H, CH2). – Additional resonances (m) for residually coordinated THF are observed at δ = 3.60–3.51 and 1.82–1.72 ppm. 13C{1H} NMR (101 MHz, CDCl3): δ [ppm] = 182.4 (d, 3JC,P = 20.8 Hz, CO2R), 133.5 (d, JC,P = 13.3 Hz, CHPh), 132.1 (d, 4JC,P = 2.3 Hz, p-CPh), 129.9 (d, 1JC,P = 53.2 Hz, i-CP), 129.6 (d, JC,P = 11.1 Hz, CHPh), 32.8 (d, 2JC,P = 7.8 Hz, CH2CO2), 24.8 (d, 1JC,P = 34.4 Hz, CH2PPh2). – Carbon atoms of the C6F5 moieties cannot be observed. – Additional resonances (s) for residually coordinated THF are observed at δ = 68.1 and 25.6 ppm. 31P{1H} NMR (162 MHz, CDCl3): δ [ppm] = 37.7 (pseudo-quintet, P-AuC6F5). 19F{1H} NMR (377 MHz, CDCl3): δ [ppm] = −116.1 (m, 8F, o-F), −157.8 (t, 3JF,F = 20.0 Hz, 4F, p-F), −161.9 (m, 8F, m-F). IR (ATR): [cm−1] = 3077 (vw), 3056 (vw), 1635 (w), 1607 (vw), 1526 (m), 1501 (s), 1454 (vs), 1435 (s), 1409 (s), 1367 (w), 1354 (m), 1312 (m), 1277 (w), 1259 (w), 1204 (vw), 1187 (w), 1161 (vw), 1141 (vw), 1104 (m), 1059 (s), 1052 (s), 1000 (w), 952 (s), 891 (vw), 821 (vw), 791 (m), 738 (m), 691 (s), 516 (m), 484 (m), 436 (vw), 416 (w). Elemental analysis calcd (%) for [C84H56Au4F20Mo2O8P4·C4H8O] (2749.12 g mol−1): C 38.45, H 2.35; found C 37.88, H 2.49.
[Mo2(O2C-C2H4-PPh2)4(RhCl(cod))4] (3). The phosphine functionalized metalloligand 1 (·2 thf) (50.0 mg, 0.04 mmol, 1.00 equiv.) and bis(1,5-cyclooctadiene)-dirhodium(I) dichloride (40.4 mg, 0.08 mmol, 2.00 equiv.) were dissolved in THF (10 mL) and stirred at room temperature for 16 h. A yellow crystalline solid precipitated; the mother liquor was decanted off and the product was recrystallized from hot THF, yielding single crystals suitable for X-Ray analysis. Crystalline yield: 56.0 mg (65%), relating to 3 (·2 thf).
1H NMR (400 MHz, CDCl3): δ [ppm] = 7.67–7.53 (m, 16H, Ph), 7.34–7.24 (m, 24H, Ph), 5.51–5.42 (m, 8H, COD-CH), 3.71–3.61 (m, 8H, CH2), 3.15–3.06 (m, 8H, CH2) 3.06–2.99 (m, 8H, COD-CH), 2.40–2.23 (m, 16H, COD-CH2), 2.04–1.92 (m, 8H, COD-CH2), 1.84–1.77 (m, 8H, COD-CH2). – Additional resonances (m) for residually coordinated THF are observed at δ = 3.75–3.68 and 1.87–1.81 ppm and are partially overlaying with resonances from –CH2CO2 and COD-CH2. 13C{1H} NMR (101 MHz, CDCl3): δ [ppm] = 183.4 (d, 3JC,P = 17.5 Hz, CO2R), 133.7 (d, JC,P = 10.3 Hz, CHPh), 132.6 (d, 1JC,P = 40.3 Hz, i-CP), 130.1 (d, 4JC,P = 1.6 Hz, p-CPh), 128.4 (d, JC,P = 9.4 Hz, CHPh), 105.3 (dd, 1JC,Rh = 12.0 Hz, 2JC,P = 7.1 Hz, dept(+), COD-CH), 70.3 (d, 1JC,Rh = 13.6 Hz, dept(+), COD-CH), 33.8 (d, 2JC,P = 1.9 Hz, dept(−), CH2CO2), 33.2 (d, 2JC,Rh = 2.2 Hz, dept(−), COD-CH2), 28.9 (dept(−), COD-CH2), 24.0 (d, 1JC,P = 25.6 Hz, dept(−), CH2PPh2). – A hyperfine splitting of the CH2 resonances due to a coupling with 103Rh is not adequately resolved. Additional resonances (s) for residually coordinated THF are observed at δ = 68.1 and 25.7 ppm. 31P{1H} NMR (162 MHz, CDCl3): δ [ppm] = 25.8 (d, 1JP,Rh = 149.2 Hz, P-RhCl(cod)). IR (ATR): [cm−1] = 3051 (w), 2936 (w), 2915 (w), 2873 (m), 2827 (m), 1520 (s), 1481 (m), 1429 (s), 1407 (vs), 1332 (w), 1308 (m), 1271 (vw), 1238 (vw), 1182 (w), 1155 (vw), 1098 (m), 1066 (w), 1044 (w), 1029 (w), 996 (w), 957 (w), 889 (vw), 862 (w), 816 (w), 785 (vw), 739 (s), 694 (s), 518 (s), 488 (m), 456 (vw), 420 (w). Elemental analysis calcd (%) for [C92H104Rh4Cl4Mo2O8P4·C4H8O] (2279.18 g mol−1): C 50.59, H 4.95; found C 50.76, H 5.05.
[Mo2(O2C-C2H4-PPh2)4(IrCl(cod))4] (4). The phosphine functionalized metalloligand 1 (·2 thf) (50.0 mg, 0.04 mmol, 1.00 equiv.) and bis(1,5-cyclooctadiene)diiridium(I) dichloride (55.0 mg, 0.08 mmol, 2.00 equiv.) were dissolved in THF (5 mL) and stirred at room temperature for 3 h until an orange crystalline solid precipitated. Subsequently, the solid was recrystallized from hot THF, yielding single crystals suitable for X-Ray analysis. Crystalline yield: 77.0 mg (78%), relating to 4 (·2 thf).
1H NMR (400 MHz, CDCl3): δ [ppm] = 7.67–7.56 (m, 16H, Ph), 7.41–7.31 (m, 24H, Ph), 5.12–5.03 (m, 8H, COD-CH), 3.60–3.44 (m, 8H, CH2), 3.27–3.13 (m, 8H, CH2) 2.71–2.60 (m, 8H, COD-CH), 2.26–2.06 (m, 16H, COD-CH2), 1.82–1.75 (m, 8H, COD-CH2), 1.57–1.48 (m, 8H, COD-CH2). – Additional resonances (m) for residually coordinated THF are observed at δ = 3.73–3.67 and 1.87–1.81 ppm and are partially overlaying with resonances from COD-CH2. 13C{1H} NMR (101 MHz, CDCl3): δ [ppm] = 183.7 (d, 3JC,P = 16.2 Hz, CO2R), 134.0 (d, JC,P = 10.1 Hz, CHPh), 131.4 (d, 1JC,P = 48.4 Hz, i-CP), 130.4 (d, 4JC,P = 2.0 Hz, p-CPh), 128.5 (d, JC,P = 9.8 Hz, CHPh), 94.0 (d, 2JC,P = 14.1 Hz, dept(+), COD-CH), 53.5 (dept(+), COD-CH), 33.6 (d, 3JC,P = 3.0 Hz, dept(−), COD-CH2), 33.4 (dept(−), CH2CO2), 29.6 (d, 3JC,P = 1.4 Hz, dept(−), COD-CH2), 23.7 (d, 1JC,P = 32.1 Hz, dept(−), CH2PPh2). – Additional resonances (s) for residually coordinated THF are observed at δ = 68.1 and 25.7 ppm. 31P{1H} NMR (162 MHz, CDCl3): δ [ppm] = 15.6 (s, P-IrCl(cod)). IR (ATR): [cm−1] = 3073 (vw), 3052 (w), 2933 (m), 2914 (m), 2874 (m), 2829 (m), 1521 (s), 1481 (m), 1429 (s), 1406 (vs), 1328 (w), 1308 (m), 1272 (vw), 1212 (vw), 1182 (w), 1155 (vw), 1099 (m), 1066 (w), 1044 (w), 1029 (vw), 998 (w), 969 (vw), 948 (vw), 895 (w), 886 (w), 816 (w), 779 (vw), 738 (s), 693 (s), 530 (m), 509 (m), 491 (m), 465 (vw), 433 (vw), 416 (vw). Elemental analysis calcd (%) for [C92H104Ir4Cl4Mo2O8P4·2 C4H8O] (2708.53 g mol−1): C 44.35, H 4.47; found C 44.35, H 4.58.
[Mo2(O2C-C2H4-PPh2)4(RuCl2(p-cymene))4] (5). The phosphine functionalized metalloligand 1 (·2 thf) (80.0 mg, 0.07 mmol, 1.00 equiv.) and dichloro(p-cymene)ruthenium(II) dimer (80.0 mg, 0.13 mmol, 2.00 equiv.) were dissolved in THF (10 mL) and stirred at room temperature for 16 h. An orange solid precipitated; the mother liquor was decanted off and the solid dried under vacuum. The solid was dissolved in DCM (10 mL) and the red solution subsequently filtered. The product was crystallized by slow evaporation of the DCM solution of 5, yielding single crystals suitable for X-Ray analysis. Crystalline yield: 82.0 mg (57%).
1H NMR (400 MHz, CDCl3): δ [ppm] = 7.92–7.82 (m, 16H, Ph), 7.47–7.40 (m, 24H, Ph), 5.25 (d, 3JH,H = 5.3 Hz, 8H, CHcym), 5.08 (d, 3JH,H = 6.1 Hz, 8H, CHcym), 3.20–3.01 (m, 8H, CH2), 2.81–2.67 (m, 8H, CH2) 2.54 (hept, 3JH,H = 6.9 Hz, 4H, CH(CH3)2), 1.83 (s, 12H, p-CH3), 0.88 (d, 3JH,H = 6.9 Hz, 24H, CH(CH3)2). 13C{1H} NMR (101 MHz, CDCl3): δ [ppm] = 183.5 (d, 3JC,P = 14.2 Hz, CO2R), 133.5 (d, JC,P = 8.6 Hz, CHPh), 132.8 (d, 1JC,P = 43.1 Hz, i-CP), 130.8 (d, 4JC,P = 2.1 Hz, p-CPh), 128.6 (d, JC,P = 9.5 Hz, CHPh), 109.0 (Cquart-cym), 94.4 (Cquart-cym), 90.8 (d, 2JC,P = 2.7 Hz, CHcym), 85.6 (d, 2JC,P = 5.9 Hz, CHcym), 31.3 (d, 2JC,P = 3.1 Hz, CH2CO2), 30.2 (CH(CH3)2), 22.9 (d, 1JC,P = 28.7 Hz, CH2PPh2), 21.7 (CH(CH3)2), 17.6 (p-CH3). 31P{1H} NMR (162 MHz, CDCl3): δ [ppm] = 20.8 (s, P-RuCl2(cym)). IR (ATR): [cm−1] = 3075 (vw), 3038 (m), 2961 (m), 2917 (vw), 2872 (vw), 1508 (s), 1483 (m), 1472 (w), 1424 (s), 1413 (vs), 1388 (m), 1375 (m), 1325 (w), 1302 (w), 1280 (w), 1239 (vw), 1201 (w), 1188 (vw), 1160 (w), 1097 (m), 1057 (w), 1030 (w), 999 (w), 955 (w), 869 (w), 812 (m), 775 (w), 748 (s), 730 (m), 697 (s), 677 (w), 639 (w), 565 (vw), 522 (s), 492 (s), 449 (w), 432 (w). Elemental analysis calcd (%) for [C100H112Mo2O8P4Ru4Cl8] (2445.68 g mol−1): C 49.11, H 4.62; found C 48.66, H 4.77.
[Mo2(O2C-C2H4-PPh2)4(AuCl)4] (6). In a first step, 3-(diphenylphosphino)propionic acid (75.0 mg, 0.29 mmol, 1.00 equiv.) and [AuCl(tht)] (93.1 mg, 0.29 mmol, 1.00 equiv.) were dissolved in DCM (10 mL) and stirred at room temperature for 3 h. The solvent of the colorless solution was removed under reduced pressure and the remaining solid subsequently washed with n-pentane (10 mL). The synthesized complex [HO2C-C2H4-PPh2-AuCl]43 was subsequently reacted with molybdenum(II) acetate (24.9 mg, 0.06 mmol, 0.20 equiv.) in THF (10 mL). After stirring for 16 h, the solvent was removed under reduced pressure and the yellow solid washed with MeOH (10 mL). Thereafter, the solid was recrystallized from hot THF (5 mL), yielding single crystals of 6 suitable for X-Ray analysis. Crystalline yield: 107 mg (80%), relating to 6 (·2 thf).
1H NMR (400 MHz, CDCl3): δ [ppm] = 7.74–7.56 (m, 16H, Ph), 7.53–7.37 (m, 24H, Ph), 3.38–3.21 (m, 8H, CH2), 3.13–2.95 (m, 8H, CH2). – Additional resonances (m) for residually coordinated THF are observed at δ = 3.77–3.70 and 1.88–1.82 ppm. 13C{1H} NMR (101 MHz, CDCl3): δ [ppm] = 181.8 (d, 3JC,P = 22.4 Hz, CO2R), 133.5 (d, JC,P = 13.3 Hz, CHPh), 132.5 (d, 4JC,P = 2.2 Hz, p-CPh), 129.6 (d, JC,P = 11.8 Hz, CHPh), 128.5 (d, 1JC,P = 61.1 Hz, i-CP), 32.8 (d, 2JC,P = 6.3 Hz, CH2CO2), 25.1 (d, 1JC,P = 39.1 Hz, CH2PPh2). – Additional resonances (s) for residually coordinated THF are observed at δ = 68.1 and 25.8 ppm. 31P{1H} NMR (162 MHz, CDCl3): δ [ppm] = 30.0 (s, P-AuCl). IR (ATR): [cm−1] = 3051 (w), 2921 (vw), 1522 (s), 1482 (m), 1432 (s), 1405 (vs), 1311 (m), 1268 (w), 1239 (w), 1204 (vw), 1185 (w), 1158 (vw), 1140 (w), 1103 (s), 1041 (w), 1027 (w), 997 (m), 949 (w), 883 (vw), 823 (w), 795 (w), 740 (s), 690 (s), 617 (vw), 541 (vw), 519 (s), 485 (m), 417 (w). Elemental analysis calcd (%) for [C60H56Au4Cl4Mo2O8P4·C4H8O] (2222.69 g mol−1): C 34.58, H 2.90; found C 34.15, H 2.89.
X-ray crystallographic studies
Detailed XRD measurement description as well as crystal and structure refinement data are provided as ESI.†46–48 Additionally, selected bond lengths and angles of all compounds (1–6) are given in the ESI (Fig. S1–S8†).
104 reflections measured, 5444 unique (Rint = 0.0383) which were used in all calculations. The final wR2 was 0.0709 (all data) and R1 was 0.0294 (I > 2(I)).
(no. 2), a = 16.7915(4) Å, b = 18.7734(5) Å, c = 19.3826(5) Å, α = 115.497(2)°, β = 92.100(2)°, γ = 110.887(2)°, V = 5019.9(2) Å3, T = 100 K, Z = 2, Z′ = 1, μ(MoKα) = 6.224 mm−1, 42389 reflections measured, 19696 unique (Rint = 0.0469) which were used in all calculations. The final wR2 was 0.1579 (all data) and R1 was 0.0560 (I > 2(![[I with combining low line]](https://www.rsc.org/images/entities/i_char_0049_0332.gif) )).
(no. 2), a = 10.8868(5) Å, b = 16.6804(9) Å, c = 20.7303(11) Å, α = 105.130(4)°, β = 102.169(4)°, γ = 104.645(4)°, V = 3358.0(3) Å3, T = 210 K, Z = 1, Z′ = 0.5, μ(MoKα) = 0.842 mm−1, 23197 reflections measured, 13020 unique (Rint = 0.0590) which were used in all calculations. The final wR2 was 0.1868 (all data) and R1 was 0.0595 (I > 2(I)).
(no. 2), a = 10.8365(3) Å, b = 16.7623(5) Å, c = 20.7559(6) Å, α = 105.261(2)°, β = 102.501(2)°, γ = 104.496(2)°, V = 3356.71(17) Å3, T = 210 K, Z = 1, Z′ = 0.5, μ(MoKα) = 4.311 mm−1, 23004 reflections measured, 13040 unique (Rint = 0.0341) which were used in all calculations. The final wR2 was 0.1125 (all data) and R1 was 0.0365 (I > 2(I)).
(no. 2), a = 11.4314(10) Å, b = 17.3797(13) Å, c = 17.3723(16) Å, α = 91.633(7)°, β = 100.343(7)°, γ = 102.881(6)°, V = 3301.4(5) Å3, T = 150 K, Z = 1, Z′ = 0.5, μ(MoKα) = 1.255 mm−1, 23611 reflections measured, 12919 unique (Rint = 0.0811) which were used in all calculations. The final wR2 was 0.2812 (all data) and R1 was 0.0932 (I > 2(I)).
023.7(6) Å3, T = 150 K, Z = 4, Z′ = 0.5, μ(MoKα) = 6.299 mm−1, 31447 reflections measured, 9867 unique (Rint = 0.1089) which were used in all calculations. The final wR2 was 0.2283 (all data) and R1 was 0.0737 (I > 2(I)).
)).
(no. 2), a = 10.8868(5) Å, b = 16.6804(9) Å, c = 20.7303(11) Å, α = 105.130(4)°, β = 102.169(4)°, γ = 104.645(4)°, V = 3358.0(3) Å3, T = 210 K, Z = 1, Z′ = 0.5, μ(MoKα) = 0.842 mm−1, 23197 reflections measured, 13020 unique (Rint = 0.0590) which were used in all calculations. The final wR2 was 0.1868 (all data) and R1 was 0.0595 (I > 2(I)).
(no. 2), a = 10.8365(3) Å, b = 16.7623(5) Å, c = 20.7559(6) Å, α = 105.261(2)°, β = 102.501(2)°, γ = 104.496(2)°, V = 3356.71(17) Å3, T = 210 K, Z = 1, Z′ = 0.5, μ(MoKα) = 4.311 mm−1, 23004 reflections measured, 13040 unique (Rint = 0.0341) which were used in all calculations. The final wR2 was 0.1125 (all data) and R1 was 0.0365 (I > 2(I)).
(no. 2), a = 11.4314(10) Å, b = 17.3797(13) Å, c = 17.3723(16) Å, α = 91.633(7)°, β = 100.343(7)°, γ = 102.881(6)°, V = 3301.4(5) Å3, T = 150 K, Z = 1, Z′ = 0.5, μ(MoKα) = 1.255 mm−1, 23611 reflections measured, 12919 unique (Rint = 0.0811) which were used in all calculations. The final wR2 was 0.2812 (all data) and R1 was 0.0932 (I > 2(I)).
023.7(6) Å3, T = 150 K, Z = 4, Z′ = 0.5, μ(MoKα) = 6.299 mm−1, 31447 reflections measured, 9867 unique (Rint = 0.1089) which were used in all calculations. The final wR2 was 0.2283 (all data) and R1 was 0.0737 (I > 2(I)).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. K.'s PhD study was funded by the Fonds der Chemischen Industrie (198271). Financial support by the DFG-funded transregional collaborative research center SFB/TRR 88 “Cooperative Effects in Homo and Heterometallic Complexes (3MET)” project C3 is gratefully acknowledged.Notes and references
- F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds Between Metal Atoms, Springer Science and Business Media, Inc., New York, 3rd edn, 2005 Search PubMed.
- S. Rej, M. Majumdar, S. Kando, Y. Sugino, H. Tsurugi and K. Mashima, Inorg. Chem., 2017, 56, 634 CrossRef CAS PubMed.
- H. Toshikazu, F. Yoshimi, T. Shingo, O. Yoshiki and A. Toshio, Chem. Lett., 1984, 13, 367 CrossRef.
- Y. Yamashita, M. M. Salter, K. Aoyama and S. Kobayashi, Angew. Chem., Int. Ed., 2006, 45, 3816 CrossRef CAS PubMed.
- K. Mashima, H. Nakano and A. Nakamura, J. Am. Chem. Soc., 1996, 118, 9083 CrossRef CAS.
- N. D. Knöfel, C. Schweigert, T. J. Feuerstein, C. Schoo, N. Reinfandt, A.-N. Unterreiner and P. W. Roesky, Inorg. Chem., 2018, 57, 9364 CrossRef PubMed.
- P. Štěpnička and I. Císařová, Organometallics, 2003, 22, 1728 CrossRef.
- G. E. Lewis and C. S. Kraihanzel, Inorg. Chem., 1983, 22, 2895 CrossRef CAS.
- D. W. Stephan, Coord. Chem. Rev., 1989, 95, 41 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 643 CrossRef CAS.
- R. G. Pearson, Inorg. Chim. Acta, 1995, 240, 93 CrossRef CAS.
- R. Peters, Cooperative Catalysis – Designing Efficient Catalysts for Synthesis, Wiley-VCH, Weinheim, 2015 Search PubMed.
- S. B. Gajera, J. V. Mehta and M. N. Patel, Med. Chem. Res., 2016, 25, 526 CrossRef CAS.
- F. Refosco, F. Tisato, G. Bandoli and E. Deutsch, J. Chem. Soc., Dalton Trans., 1993, 2901 RSC.
- M. Ravera, E. Gabano, M. Sardi, E. Monti, M. B. Gariboldi and D. Osella, Eur. J. Inorg. Chem., 2012, 3441 CrossRef CAS.
- G. J. P. Britovsek, W. Keim, S. Mecking, D. Sainz and T. Wagner, J. Chem. Soc., Chem. Commun., 1993, 1632 RSC.
- S. Mecking, Angew. Chem., Int. Ed., 2001, 40, 534 CrossRef CAS PubMed.
- M. Peuckert and W. Keim, Organometallics, 1983, 2, 594 CrossRef CAS.
- G. Amenuvor, J. Darkwa and B. C. E. Makhubela, Catal. Sci. Technol., 2018, 8, 2370 RSC.
- X.-C. Shan, F.-L. Jiang, H.-b. Zhang, X.-Y. Qian, L. Chen, M.-Y. Wu, S. A. Al-Thabaiti and M.-C. Hong, Chem. Commun., 2013, 49, 10227 RSC.
- M.-C. Tse, K.-K. Cheung, M. C.-W. Chan and C.-M. Che, Chem. Commun., 1998, 2295 RSC.
- N. D. Knöfel, Dissertation (Karlsruher Institut für Technologie), Cuvillier Verlag, Göttingen, 2019.
- E. Hochberg, P. Walks and E. H. Abbott, Inorg. Chem., 1974, 13, 1824 CrossRef CAS.
- T. A. Stephenson, E. Bannister and G. Wilkinson, J. Chem. Soc., 1964, 2538 RSC.
- S.-M. Kuang, P. E. Fanwick and R. A. Walton, Inorg. Chem. Commun., 2002, 5, 134 CrossRef CAS.
- F. A. Cotton, J. G. Norman, B. R. Stults and T. R. Webb, J. Coord. Chem., 1976, 5, 217 CrossRef CAS.
- R. H. Cayton, M. H. Chisholm, J. C. Huffman and E. B. Lobkovsky, Angew. Chem., Int. Ed. Engl., 1991, 30, 862 CrossRef.
- J. Hicks, S. P. Ring and N. J. Patmore, Dalton Trans., 2012, 41, 6641 RSC.
- F. A. Cotton, L. R. Falvello, A. H. Reid and J. H. Tocher, J. Organomet. Chem., 1987, 319, 87 CrossRef CAS.
- N. D. Knöfel, H. Rothfuss, C. Barner-Kowollik and P. W. Roesky, Polym. Chem., 2019, 10, 86 RSC.
- F. A. Cotton, Z. C. Mester and T. R. Webb, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2768 CrossRef.
- F. A. Cotton, M. W. Extine, T. R. Felthouse, B. W. S. Kolthammer and D. G. Lay, J. Am. Chem. Soc., 1981, 103, 4040 CrossRef CAS.
- H. D. Glicksman, A. D. Hamer, T. J. Smith and R. A. Walton, Inorg. Chem., 1976, 15, 2205 CrossRef CAS.
- F. A. Cotton, L. M. Daniels, E. A. Hillard and C. A. Murillo, Inorg. Chem., 2002, 41, 2466 CrossRef CAS PubMed.
- F. A. Cotton and D. G. Lay, Inorg. Chem., 1981, 20, 935 CrossRef CAS.
- J. Catterick and P. Thornton, in Advances in Inorganic Chemistry and Radiochemistry, Vol. 20, Academic Press, 1977 Search PubMed.
- F. Scherbaum, A. Grohmann, G. Müller and H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1989, 28, 463 CrossRef.
- H. Schmidbaur, Gold Bull., 1990, 23, 11 CrossRef CAS.
- X. C. Cambeiro, T. C. Boorman, P. Lu and I. Larrosa, Angew. Chem., Int. Ed., 2013, 52, 1781 CrossRef CAS.
- S. Bestgen, C. Schoo, C. Zovko, R. Köppe, R. P. Kelly, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2016, 22, 7115 CrossRef CAS PubMed.
- S. Bestgen, M. T. Gamer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2015, 21, 601 CrossRef CAS PubMed.
- B. Kemper, L. Zengerling, D. Spitzer, R. Otter, T. Bauer and P. Besenius, J. Am. Chem. Soc., 2018, 140, 534 CrossRef CAS PubMed.
- S. Ahrland, K. Dreisch, B. Norén and Å. Oskarsson, Mater. Chem. Phys., 1993, 35, 281 CrossRef CAS.
- R. Uson, A. Laguna, M. Laguna, D. A. Briggs, H. H. Murray and J. P. Fackler, (Tetrahydrothiophene)Gold(I) or Gold(III) Complexes, John Wiley & Sons, Inc., 2007 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional XRD and structure refinement data; selected bond lengths and angles of all compounds; NMR- and IR-spectra. CCDC 1961131–1961136. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt04167b |
| This journal is © The Royal Society of Chemistry 2020 |