
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the mechanochemical synthesis of the perovskite LaMnO3 and its catalytic behaviour†‡
Rachel H.
Blackmore
 ab,
Maria Elena
Rivas
c,
Tugce
Eralp Erden
c,
Trung
Dung Tran
c,
Huw R.
Marchbank
c,
Dogan
Ozkaya
cd,
Martha
Briceno de Gutierrez
c,
Alison
Wagland
c,
Paul
Collier
c and
Peter P.
Wells
*abe
ab,
Maria Elena
Rivas
c,
Tugce
Eralp Erden
c,
Trung
Dung Tran
c,
Huw R.
Marchbank
c,
Dogan
Ozkaya
cd,
Martha
Briceno de Gutierrez
c,
Alison
Wagland
c,
Paul
Collier
c and
Peter P.
Wells
*abe
aUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratories, Harwell Science & Innovation Campus, Didcot, Oxfordshire OX11 0FA, UK. E-mail: P.P.Wells@soton.ac.uk
bSchool of Chemistry, University of Southampton, Southampton SO17 1BJ, UK
cJohnson Matthey Technology Centre, Blounts Court Road, Sonning Common, Reading RG4 9NH, UK
dElectron Physical Sciences Imaging Centre (ePSIC), Diamond Light source Ltd, Didcot, Oxfordshire OX11 0DE, UK
eDiamond Light Source Ltd, Harwell Science & Innovation Campus, Didcot, Oxfordshire OX11 0FA, UK
First published on 9th December 2019
Abstract
Mechanochemistry offers a solventless, ‘waste free’ route to preparing metal oxide catalysts, however, there is limited information on the chemical steps involved. In this work, the perovskite LaMnO3 has been successfully synthesized via mechanochemistry from metal oxide powders, La2O3 and Mn2O3, at room temperature, using a planetary ball mill. Separate ex situ ‘time slices’ were taken during the milling procedure to provide insights into the underlying chemistry. The crystalline material was assessed using XRD, which identified 100% perovskite phase after 3 h of milling. Conversely, characterization by X-ray absorption spectroscopy (XAS) at both the Mn K-edge and La L3-edge provides a very different picture. The XAS data shows that there are significant structural alterations as early as 30 min of milling, with the La precursor dispersed over Mn2O3. Increasing milling time then allows for mechanical activation of both precursors and the formation of powdered LaMnO3, with no calcination step required. The XAS highlights that there is a significant amount of amorphous, oxygen deficient, content even when XRD has identified 100% perovskite phase. The samples were tested for the decomposition of the environmental pollutant N2O; at a milling time of 3 h, the LaMnO3 catalyst displays a much early onset production of N2 compared to a traditional sol–gel synthesized LaMnO3, resulting from increased oxygen deficiency at the surface, confirmed by XPS and STEM-EELS. This is an encouraging sign that mechanochemical routes can be harnessed to provide a sustainable route to preparing mixed metal oxide catalysts with enhanced catalytic performance.
1. Introduction
Mechanochemistry is an area rapidly growing in interest as an alternative ‘one-step’ synthesis process as it has remarkable advantages in terms of sustainability, both environmental and economical, which can also produce materials with enhanced properties for applications in catalysis.1,2 The benefits within industrial applications arise as a consequence of the removal of process steps, e.g. filtration, pH control and washing.3,4 It offers a rapid and solvent-less route for the efficient mixing, particle size reduction and formation of new powered materials.5 Recent research has focused on the preparation of nanomaterials, whereby mechanical action – in the form of compression, shear or friction between the wall of the jars and milling media – induce chemical transformations.6Mixed metal oxides with perovskite structures, ABO3, are known to readily form via mechanochemical grinding from their single metal oxide precursors.7–9 These perovskite structures have long been recognized for their catalytic capabilities, with the potential to become suitable substitutes for noble metals in electrocatalysis and automotive exhaust applications.10,11 Their structural versatility provides a wide range of possibilities in tailoring specific properties or designing new perovskites.12 Within catalytic applications of perovskites their performance is attributed to the redox capabilities of the B cation and therefore in turn its oxygen mobility.13 The properties of catalysts are highly dependent on the method of preparation; changing the synthetic route results in different phase composition, surface area and particle size, all of which affects crystallinity, texture and morphology.14 Traditional synthetic routes, e.g. co-precipitation and sol–gel, require a final high temperature calcination step to achieve the crystalline perovskite phase, resulting in sintering, low surface area and reduced activity as a catalyst.7,15 However, the mechanochemical synthesis allows for the absence of a high temperature calcination step, therefore, limiting the sintering of particles. This provides a high surface concentration of –OH, high levels of distortion, along with a significant volume of intergranular amorphous phase with high oxygen mobility and therefore produce improved catalytically active materials.1
Perovskite materials are promising catalysts for the decomposition of nitrous oxide, deN2O, due to their low cost, thermal stability and good activity.16–19 N2O is an environmental pollutant contributing to the depletion of the stratospheric ozone layer and to the greenhouse effect.20 It itself has a global warming potential (GWP) that is roughly 310 times higher than that of CO2 and an atmospheric half-life of over 115 years.21,22 N2O is unfortunately a common by-product during nitric acid production, used for fertilisers, and is also produced in the synthesis of adipic acid, a precursor for nylon.23,24 Through direct catalytic decomposition it will allow for a simple method for industries to reduce N2O emissions.16
Developing sustainable synthetic routes to produce ‘earth-abundant’ catalysts can help with finding alternatives to the commercial platinum group metals currently used.25 Furthermore, despite the extensive possible uses, commercial success for perovskite-type materials is yet to be achieved.26 This presents a clear opportunity to progress the mechanochemical synthesis of perovskites and to optimize the performance in renewable energy conversion technologies and emission control strategies.21 However, there are significant challenges in mechanochemical reactions; controlling uniform stoichiometry and structure of the resultant materials has proved difficult.15 To further the technology an improved understanding of the underlying chemical steps is required. This is further complicated by the nature of using grinding media and the formation of amorphous structures during milling.27
The mechanochemical synthesis of LaMnO3 has been well reported within literature, with the work focussing on the resultant functional properties of the material.8,28,29 These studies are helpful in showing the general applicability of mechanochemical synthesis but do not adequately describe the intermediates produced through milling or understanding of the perovskite formation process via mechanochemistry. Furthermore, the characterisation in these studies is typically limited to lab-based techniques, e.g. XRD, which can only assess the crystalline material, excluding any proportion of amorphous material, which is commonly known to be produced by milling.
Our approach is to use advanced characterization techniques, i.e. X-ray Absorption Spectroscopy (XAS), to understand the chemical transformations occurring within ball-milling. XAS is highly sensitive to the local structure of materials without the need for periodic ordering; it is ideally suited to studying samples prepared through ball-milling as it provides information on both amorphous and crystalline phases.30
Herein, we detail the mechanochemical synthesis of LaMnO3, from La2O3 and Mn2O3 precursors, and provide a more in-depth insight into the steps for perovskite formation by XAS, which has not previously been reported. Moreover, we demonstrate the performance of these materials towards deN2O and highlight how mechanochemical routes introduce active sites for catalysis.
2. Experimental
2.1 LaMnO3 sample preparation
High energy planetary ball milling was performed at room temperature in a 4-station Fritsch Pulverisette 5 Planetary Ball Mill. ZrO2 vessels were prepared with precursors Mn2O3 and La2O3 at the correct proportions to synthesise stoichiometric LaMnO3, along with 5 mm ZrO2 spheres at a sphere![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :powder ratio of 10:1. Milling was then conducted up to 4 hours at 400 rpm in sessions of 20 min. For the sol–gel synthesis stoichiometric amounts of Mn(NO3)2·4H2O, and La(NO3)3·6H2O were dissolved in deionised water and added to a gel prepared by mixing equimolecular amounts of citric acid (99.5%) and ethylene glycol (99.5%) as a polydentate ligand. The excess water was slowly removed on a hot plate until a viscous liquid was obtained. Subsequently, the final slurry was slowly heated in air (1 °C min−1) from room temperature up to 700 °C and kept at this temperature for 4 h. These conditions are essential to obtain a crystalline material.
:powder ratio of 10:1. Milling was then conducted up to 4 hours at 400 rpm in sessions of 20 min. For the sol–gel synthesis stoichiometric amounts of Mn(NO3)2·4H2O, and La(NO3)3·6H2O were dissolved in deionised water and added to a gel prepared by mixing equimolecular amounts of citric acid (99.5%) and ethylene glycol (99.5%) as a polydentate ligand. The excess water was slowly removed on a hot plate until a viscous liquid was obtained. Subsequently, the final slurry was slowly heated in air (1 °C min−1) from room temperature up to 700 °C and kept at this temperature for 4 h. These conditions are essential to obtain a crystalline material.
2.2 Catalyst characterisation
X-ray diffraction (XRD) patterns were collected a Bruker AXS D8 diffractometer (Johnson Matthey, Sonning Common, UK) with Cu Kα radiation over a range of 2θ = 10–130° with 0.044° step size. Phase identification was conducted using Bruker-AXS Diffrac Eva V4.2 with Rietveld refinement performed using Bruker-AXS Topas 4.2. Crystallite sizes were calculated using the volume weighted column height LVol-IB method. The specific surface area of the samples was determined by krypton adsorption (BET method) using a Quantachrome Autosorb iQ “E”. Samples were heated at 368 K under vacuum for 12 h before adsorption-desporption experiments. Krypton adsorption measurements were performed at 77 K up to a relative pressure P/P0 = 0.175. Inductively coupled plasma mass spectrometry (ICP-MS) was performed using 100 mg of sample in concentrated HCl. It was then ramped to 240 °C and held for 40 min. X-ray absorption spectroscopy (XAS) measurements were performed at the B18 Beamline, Diamond Light Source. XAS measurements were performed at the Mn K-edge (6539 eV) and La L3-edge (5483 eV) in transmission mode using QEXAFS setup with fast scanning Si(111) double crystal monochromator. All XAS spectra were acquired concurrently with the appropriate foil placed between It and Iref. Mn K-edge XAS spectra were acquired with a time resolution of 20 min per spectrum (kmax = 14) averaged over 3 scans. La L3-edge XAS spectra were acquired with a time resolution of 5 min per spectrum (kmax = 10) averaged over 3 scans. Data processing was performed using IFEFFIT31 with Horae package32 (Athena and Artemis). The amplitude reduction factor, S02 was derived using EXAFS analysis of appropriate reference spectra foil of known coordination numbers. X-ray photoelectron spectroscopy (XPS) was carried out with a Thermo Escalab 250. The radiation used was monochromatised aluminium Kα radiation with a 650 μm spot size. Charge compensation was provided by the in-lens electron flood gun at a 2 eV setting and the “401” unit for “zero energy” argon ions. Sensitivity factors after Scofield used in quantification performed by Johnson Matthey, Sonning Common, UK.33Electron Energy Loss Spectroscopy (EELS) was performed using GIF Quantum 965ER spectrometer equipped on the probe-corrected JEOL ARM200CF Transmission Electron Microscope (TEM) operating at 200 kV (Johnson Matthey's at I14 ePSIC, Diamond Light Source). EELS elemental mapping was performed in Scanning TEM (STEM) mode at the atomic resolution. For the oxidation states, Mn L3,2-edge was acquired at high energy-resolution (<0.5 eV FWHM) using 0.025 eV energy dispersion. EELS spectrum images were acquired with average beam doses less than 25 × 106 e− per nm2 to avoid beam damage which can induce a change in oxidation states. For imaging, the High-Angle-Annular-Dark-Field (HAADF) detector was used.2.3 Catalytic testing
N 2 O decomposition (deN 2 O) was carried out in a Hiden CATLAB fixed-bed quartz reactor in the temperature range of 100–800 °C with a ramp of 10 °C min−1. The reaction was performed at a flow of 30 mL min−1 with GHSV = 18000 h−1 composed of 0.5% N2O in He. The composition of exhaust gases was measured using a Hiden QGA mass spectrometer for He (m/z = 4), N2 (m/z = 28), O2 (m/z = 32), N2O (m/z = 46).
3. Results and discussion
3.1 Understanding the mechanochemical synthesis of LaMnO3
To assess the composition of the milled materials, inductively coupled plasma mass spectrometry (ICP-MS) was performed and confirmed that the correct molar quantities of La (0.393 mol) and Mn (0.397 mol) to form a stoichiometric LaMnO3 phase. Low contamination from the milling jar and media was also observed by ICP-MS analysis (Table S1‡).To assess the structural changes as a function of time during milling, X-ray diffraction (XRD) studies were performed on separate time-slices throughout the reaction (Fig. 1). Separate grinding jars were milled for each time-slice up to 4 h. The first clear insight was the nature of La precursor, which at time-zero was already present as La(OH)3 as opposed to La2O3; this is clearly attributed to the well-established moisture/atmospheric sensitivity of La.34 Care was taken to use fresh/dried La precursor to achieve La2O3 starting material. On subsequent extended exposure to air after milling La(OH)3 formed. ICP-MS, as stated above, yields the correct stoichiometry of final materials to correspond with La2O3 phase as the starting precursor. A minor proportion of stabilised La2O3 is observed from 0.5–1.5 h of milling, before further transformation at increased milling times.
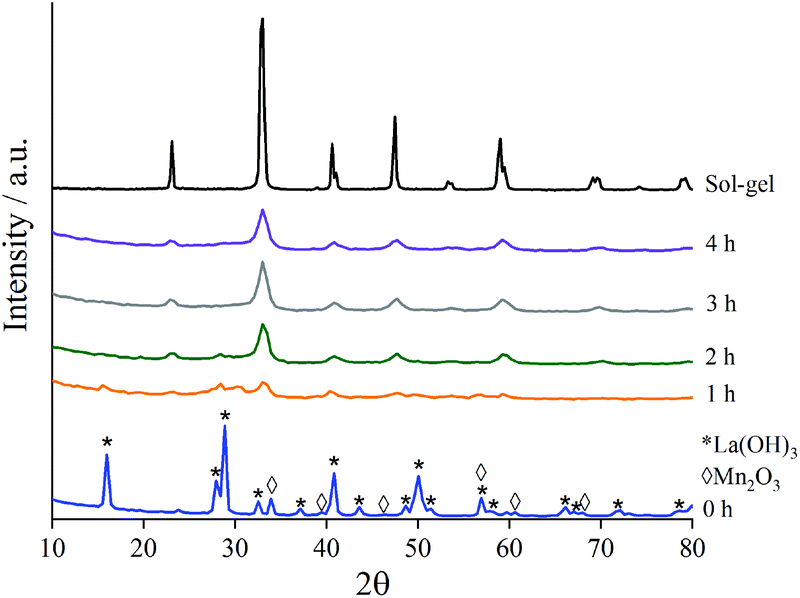 | ||
| Fig. 1 XRD patterns to show the formation of crystalline LaMnO3 at time-slices during the mechanochemical synthesis compared to sol–gel prepared LaMnO3. | ||
As milling time is increased the peaks in the diffraction patterns corresponding to La(OH)3 and Mn2O3 decrease in intensity, and are completely absent after 3 h milling (Fig. S1‡). Peaks attributed to LaMnO3 start to appear at 1 h with their intensity increasing with further milling time. At time 3 h 100% conversion to crystalline LaMnO3, by Rietveld analysis (Fig. S1‡), is reached; the mechanochemical action of ball-milling has achieved the transformation of single oxide precursors to an ordered perovskite phase without the need of high temperature thermal annealing.
However, there are clear indications in the XRD data that these samples are more complex than a 100% crystalline perovskite phase. The final material (i.e. 4 h milling) has low intensity XRD peaks and broad features in the base line, compared to the sol–gel synthesised analogue, suggesting smaller crystallite size and/or low crystallinity.7 Calculating crystallite size by the LVol-IB method confirmed the size to be 6.2 nm after 4 h of milling. However, the broad features, often attributed to amorphous content, are largely undetectable by XRD. Amorphous content is a commonly known consequence of ball-milling and it often influences the catalytic properties of the material.3
Using X-ray absorption spectroscopy (XAS), which does not rely on periodic ordering, ex situ measurements at the Mn K-edge and La L3-edge were performed on time-slices to analyse the local environment surrounding the absorbing atom. The reaction series has been compared to Mn2O3, La(OH)3 and to sol–gel synthesised LaMnO3.
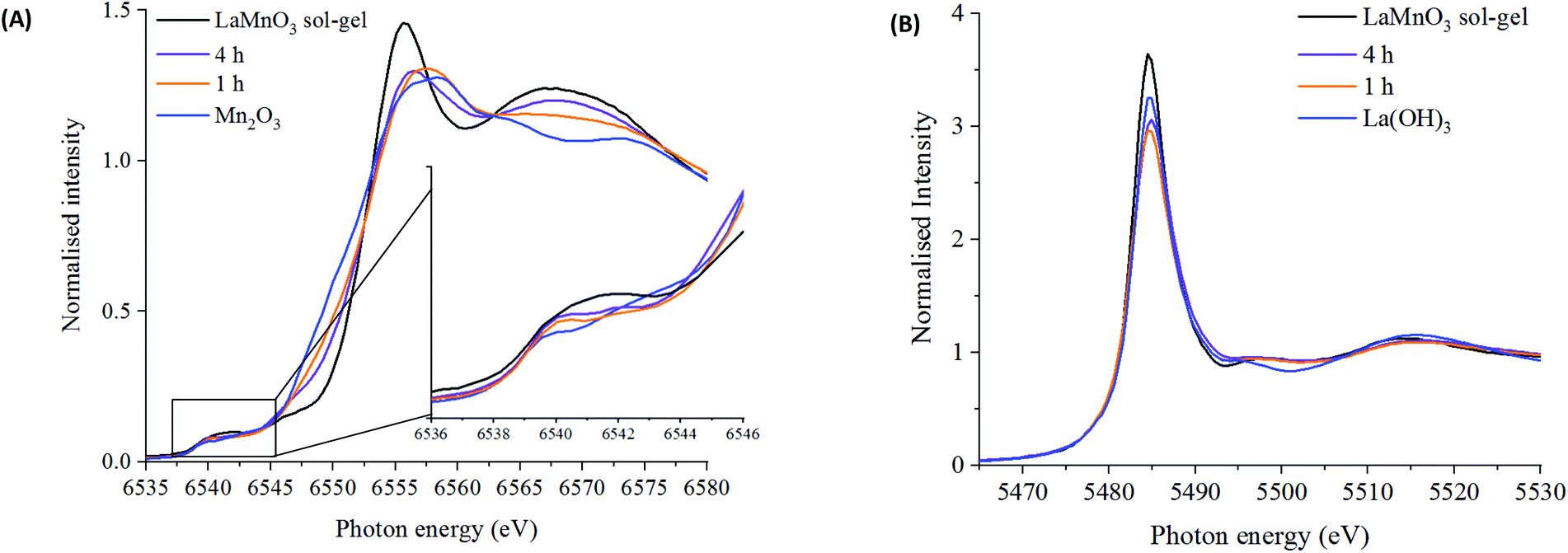 | ||
| Fig. 2 XANES spectra at the (A) Mn K-edge for LaMnO3 prepared by sol–gel synthesis and ball milling after 1 and 4 hours compared to Mn2O3 precursor with highlighted pre-edge region and (B) La L3-edge for LaMnO3 prepared by sol–gel synthesis and ball milling after 1 and 4 hours compared to La(OH)3 precursor. | ||
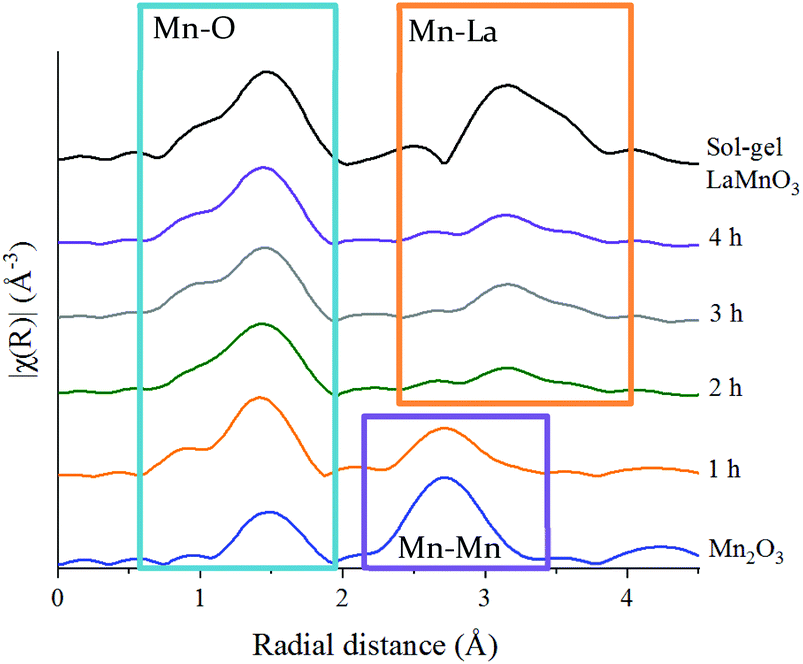 | ||
| Fig. 3 Non phase-corrected Fourier transform at the Mn K-edge for LaMnO3 synthesised by sol–gel and by ball milling from 1 h and 4 h compared to precursor Mn2O3. | ||
These features have been further investigated by fitting the EXAFS data at the Mn K-edge (Table S2‡). A reliable fitting model for Mn2O3 and time 0 h was achieved using 4 single scattering paths, Mn–O1, Mn–O2, Mn–Mn1 and Mn–Mn2. The pure crystalline perovskite phase, i.e. the sol–gel synthesised LaMnO3, was modelled with a good degree of fit using three single scattering paths, Mn–O1, Mn–La1 and Mn–La2.
The Fourier transform EXAFS data for the 1 h milled sample is similar in appearance to Mn2O3 (time 0 h) (Fig. 3) and therefore a similar fitting model has been applied. By XRD, crystalline Mn2O3 is still detected after 1 h of milling, however, observations within the Mn K-edge XANES region (Fig. 2A) suggest there are changes occurring to the Mn local environment. This is primarily reflected in the EXAFS fit by the absence of the longer Mn–O2 scattering path. The absence of this scattering path arises as a consequence of the increased structural disorder introduced via the milling process. Oxygen has a limited back-scattering amplitude at these longer distances, which is compounded by an increase in the mean square disorder parameter, σ2, that makes any inclusion on this path unreliable. Therefore, the absence of the path in the fitting model does not preclude the existence of longer Mn–O bond distances but implies a significant degree of structural disorder.
For the samples milled at 2, 3 and 4 h there is a clear shift to a longer distance for the second coordination shell in the Fourier transform EXAFS data. This increase in distance can now be ascribed to the presence of Mn–La scattering paths. Reliable fitting models have been achieved using a single Mn–O, and two Mn–La single scattering paths (Fig. S3‡ and Fig. 4C, D). Observations within the XANES region suggests clear electronic and geometric differences within the first coordination sphere surrounding the central Mn atom between ball milled and sol–gel samples, but it is unclear what causes these changes due to the broad, unresolved features. Also, there are differences in the XRD between the final ball milled material and the sol–gel synthesised perovskite, but both are considered as 100% crystalline LaMnO3. These differences can be rationalised by the change in oxygen coordination found in the simulated models for the 2, 3 and 4 h milled materials (Fig. 4C and D) (Table S2‡). As the structure moves towards a perovskite with a more regular octahedral Mn–O environment, a reduced CN of 5 for the 2, 3 and 4 h milled materials can be linked to a perovskite phase with oxygen deficiency.
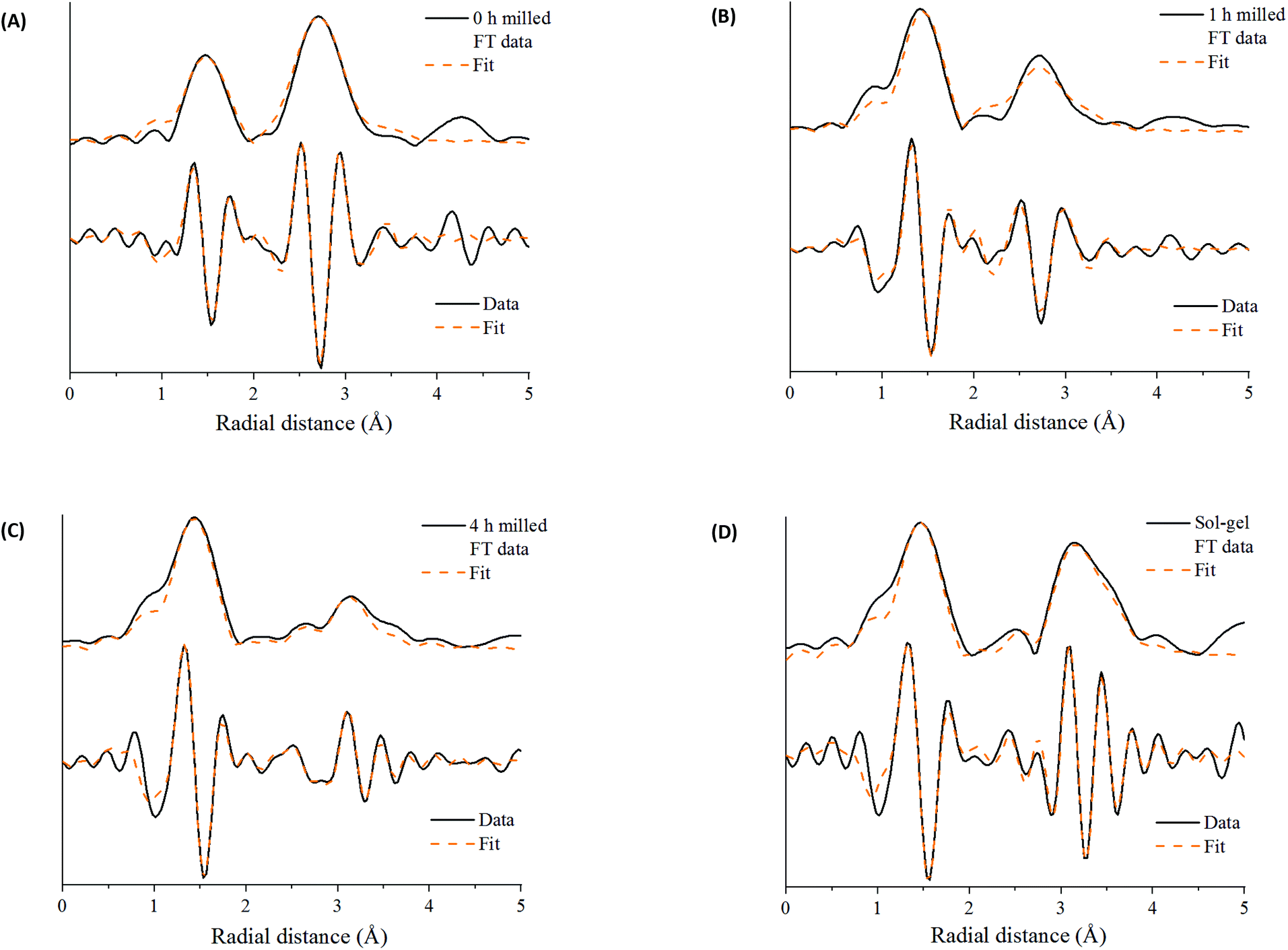 | ||
| Fig. 4 Mn K-edge EXAFS data after (A) time 0 h of milling (Mn2O3) and after time (B) 1 h of milling (C) 4 h of milling compared to (D) sol–gel synthesized LaMnO3 showing the magnitude and imaginary components of the k2-weighted Fourier transform data and simulated fits. | ||
Further discrepancies between the 4 h milled and the sol–gel prepared LaMnO3 have been modelled in the second coordination shell with respect to the Mn–La scattering paths. Distances for the sol–gel sample are consistent with crystallographic data (3.34 and 3.70 Å). However, for the 4 h milled material the Mn–La distances are observed at 3.24 Å (Mn–La3) and 3.37 Å (Mn–La1), which can be ascribed as amorphous and crystalline material, respectively. The shorter scattering path has a corresponding high disorder, σ2, of 0.014 Å−2 indicative of a disordered species. This amorphous content is known to be produced via mechanochemical grinding, as well as a proportion of crystalline material, which has also been suggested by broad diffraction peaks in the XRD.
The La L3-edge EXAFS Fourier transform data also allows insights into the underlying steps in perovskite formation during the mechanochemical synthesis of LaMnO3. However, the short k range imparted by the overlap of L3 and L2 edges does not allow for reliable simulation of the data. The La L3-edge EXAFS data does show significant structural alterations after 1 h of milling indicative of La–Mn scattering (Fig. 5), with no further changes observed from 1 to 4 h (Fig. S2‡ and Fig. 5). At the Mn K-edge (Fig. 3) the structural model after 1 h of milling is, however, largely consistent with Mn2O3. The combined data supports a model where La has initially been dispersed over the Mn2O3 surface. Considering the EXAFS provides a per atom average and with the size domains of Mn2O3 present it would not be expected to see any Mn–La interactions at the Mn K-edge after 1 h of milling. However, where La is present at the surface many La–Mn interactions would be expected. As milling time increases there is further disruption of Mn2O3 and an increased association between Mn and La. At the Mn K-edge after 2 h of milling a shift is observed within the second coordination shell corresponding to a Mn–La scattering path, indicating the formation of LaMnO3.
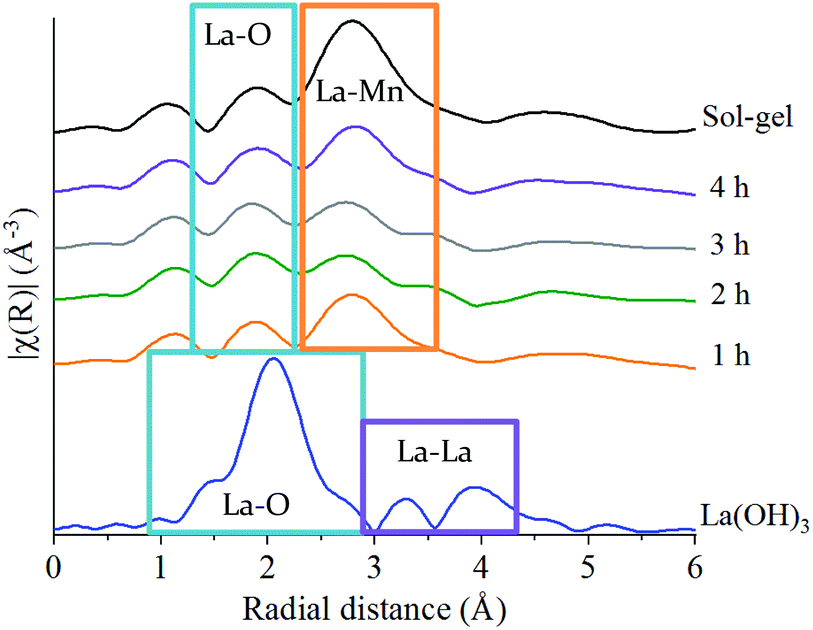 | ||
| Fig. 5 Non phase-corrected Fourier transform at the La L3-edge for LaMnO3 synthesised by sol–gel and by ball milling from 1 h to 4 h compared to the precursor La(OH)3. | ||
These XAS observations of the intermediates during milling further highlight the differences in information obtained and shows the importance of advanced characterisation for the understanding of amorphous content. They provide further insights into the mechanochemical synthesis of LaMnO3, not previously reported.
3.2 Understanding catalytic activity
Fig. 6 shows the oxygen 1s XPS signals at the surface of ball milled and sol–gel synthesised LaMnO3. On curve fitting, the spectra show three features at 529.3 eV–529.6 eV, 531.1 eV–531.7 eV and 533.4–534.0 eV. The lower energy binding peak is due to lattice-type oxygens in the perovskite and the higher energy features arises due to adsorbed species such as hydroxyls, chemisorbed oxygen and organic oxygen.37–39 The small feature at 533.6 eV results from carbon–oxygen interactions from adventitious carbon, which is further confirmed by corresponding carbon peaks at 286.2 eV and 288.6 eV.40 On the subtraction of this adventitious carbon, a change in the peak intensity at 531.1 eV–531.7 eV has previously been correlated to oxygen vacancies on the perovskite surface.39,41 These studies used a ratio of the peak areas of these oxygen features (adsorbed oxygen: lattice oxygen) to give a value for the percentage of oxygen vacancies.42 In our study, the relative intensity of the feature associated with adsorbed oxygen increases in the order: 3 h milled >4 h milled ≈ sol–gel preparation. The surface areas of the materials were calculated using Brunauer–Emmett–Teller (BET) theory and were determined to be 4.6 ± 1.1 m2 g−1 for ball milled samples, compared to 7.6 ± 1.8 m2 g−1 for the sample prepared through the sol–gel route. Considering that the sol–gel prepared sample has the greatest surface area of the three samples, it is clear that there are profound changes to the surface chemistry that provides the active centres for adsorption; others have eventually concluded that the perturbation of the surface, through oxygen vacancies, are a significant cause to increasing the number of adsorbed oxygen species. Elsewhere, we also performed O2-TPD studies that also quantified the extent of adsorbed oxygen species, (Fig. S4‡) which agreed with the trend observed for the XPS data. Further XPS analysis in the La 3d region (Fig. S5‡) indicate a 3+ oxidation state for all samples, which is further confirmed by no shift in the XANES La L3-edge position (Fig. 2B). The magnitude of the multiplet splitting for the ball milled and sol–gel LaMnO3 is 4.6 eV, indicating the same splitting for the perovskite as for the La2O3 species. Spectra at the Mn 3s region (Fig. S6‡) indicate the presence of some Mn4+ species, along with Mn3+, at the surface for all samples due to a peak splitting of ∼4.9 eV. This suggests significant structural variance at the surface in both the ball milled and sol–gel LaMnO3, compared to the bulk structure seen by XAS. It is these redox proprieties of Mn based perovskites that are said to be responsible for its catalytic activity.43
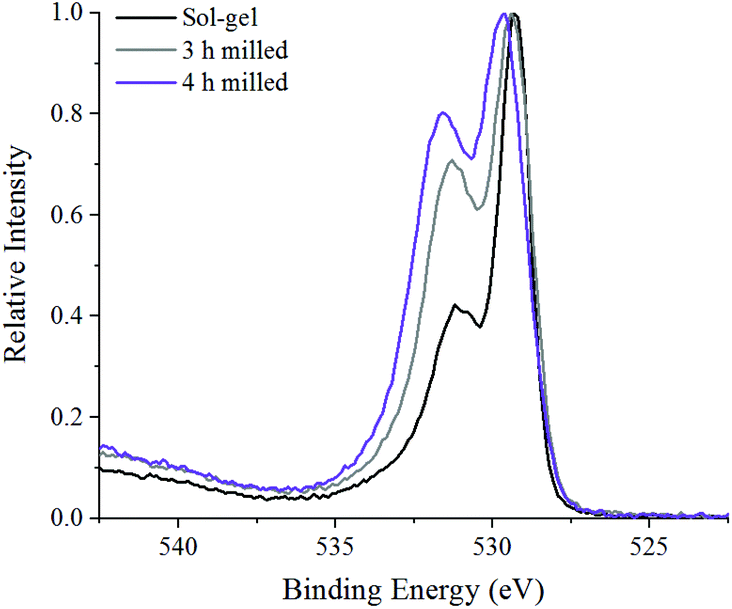 | ||
| Fig. 6 XPS spectra in the O 1s region of 3, 4 h ball milled samples compared to sol–gel synthesised LaMnO3. | ||
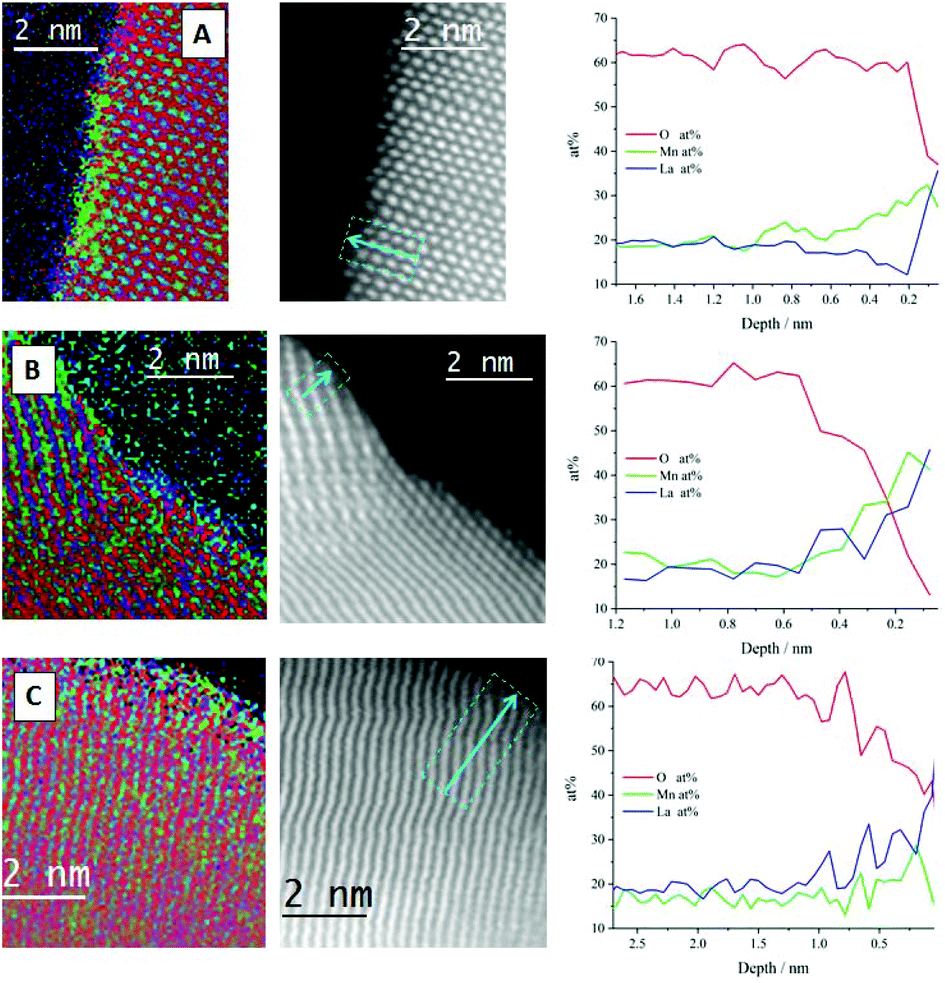 | ||
| Fig. 7 STEM-EELS atomic percentage elemental mapping (left column), HAADF-STEM images with integrated profiling lines (central column) and line profiling along the arrow, for the atomic percentage of La (blue), Mn (green) and O (red) for (A) sol–gel synthesised, (B) 3 h milled and (C) 4 h milled LaMnO3. | ||
The line profile of the sol–gel synthesised LaMnO3 confirms that the interior of the particle is consistent with the stoichiometric composition, i.e. La (20 at%), Mn (20 at%) and O (60 at%). As the line profile approaches the edge of the particle (depth = 0.8 nm) it goes through a Mn-enriched area (∼28 at%), with a corresponding decrease in La content (∼12 at%). Further towards the surface, an oxygen deficiency is observed within the last few atomic layers of ∼2 Å. The STEM-EELS imaging (Fig. 7a) has confirmed a level of structural heterogeneity, with the enriched Mn areas not present across the entirety of the catalyst sub-surface. This structural heterogeneity makes any assignment of the origin of the Mn4+content difficult to ascribe.
The nature of the oxygen deficiency in the ball-milled samples extends slightly further into the sample (∼5 Å) compared to the sol–gel analogue. This deficiency is more pronounced for the 3 h milled sample (Fig. 7b) compared to the sample milled at 4 h (Fig. 7c). The La and Mn concentration within the ball-milled samples tend to have a consistent fluctuation (20 ± 3 at%) within the crystal bulk and both increase at the same rate as they enter the O vacancy layer. However, at the surface of the 4 h milled sample (Fig. 7c, depth 0.1 nm) there is an apparent change in the composition. Here, La dominates the content at the end of the line profile, suggesting the presence of La oxide residue and a reduction in Mn species at the surface of the perovskite crystal. These different phases detected by STEM-EELS of all samples suggest a significant structural variance at the surface that results in a Mn4+/Mn3+, stated by XPS within the Mn 3s region.
The STEM-EELS data needs to be taken in the context of the sample population presented, however, the observation of Mn surface enrichment is consistent throughout the samples presented, which is an encouraging sign regarding the reliability of this observation.
3.3 Catalytic testing
N2O decomposition (deN2O) was performed on ball milled samples after 3 and 4 h of milling, due the maximum conversion of precursor materials, and compared to the sol–gel synthesised LaMnO3 (Fig. 8). The LaMnO3 catalysts start to show activity for deN2O above 300 °C, with 100% conversion to N2 and O2 reached by 550 °C. Similar activity is observed between the 4 h milled sample and the sol–gel synthesised LaMnO3. However, the 3 h milled sample shows a much earlier onset decomposition of N2O at lower temperatures (<500 °C). Furthermore, two distinct regions within the light-off curve are observed with ball milled LaMnO3 during the deN2O. Good reproducibility of deN2O on another batch of the catalysts are shown in Fig. S7.‡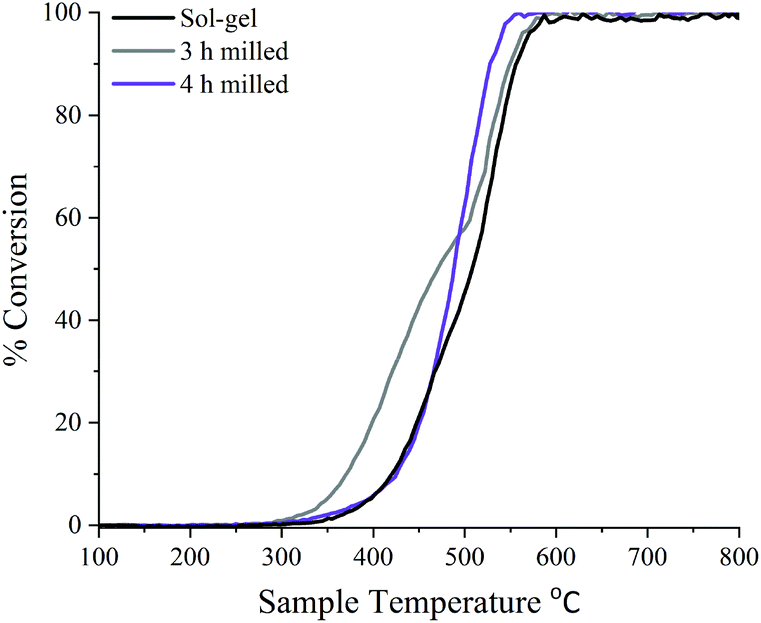 | ||
| Fig. 8 Light-off curve showing the percentage of deN2O to N2 over 3 h, 4 h ball milled and sol–gel synthesised LaMnO3 using 0.5% N2O/He at 30 mL min−1 with a pre-treatment of He at 30 mL min−1. | ||
With the active sites for this reaction ascribed to Mn and oxygen vacancies, during the reaction Mn is expected to partially reduce, providing reaction oxygen. This reduced Mn will then re-oxidise by gas phase oxygen or from mobile oxygen from the perovskite lattice.43 The ease at which the Mn can fluctuate between two oxidation states relates to oxygen mobility of the sample. With all samples initially having the same Mn4+/Mn3+ at the surface, it demonstrates here that the activity can be linked to oxygen vacancies. This is reflected in the percentage conversion of N2O to N2 (Fig. 8) where the 3 h milled LaMnO3 catalyst, which has the higher proportion of oxygen vacancies to total oxygen atoms, has the higher conversion at lower temperatures. Here the increase in oxygen vacancies provides additional active sites for deN2O. With oxygen desorption said to be the rate determining step for deN2O, the reaction can be an indication of the effectiveness of Mn redox ability.18 It suggests at lower temperatures the 3 h milled catalyst also has increased oxygen mobility.44 Furthermore, surface area studies by Brunauer–Emmett–Teller (BET) theory were calculated to be 4.6 ± 1.1 m2 g−1 for ball milled samples, compared to 7.6 ± 1.8 m2 g−1 for sol–gel synthesised LaMnO3. Performing scanning electron microscopy (SEM) imaging showed the 3 and 4 h milled samples have predominately similar agglomerated morphology on the same size scale (Fig. S8‡). The sol–gel synthesised LaMnO3, however, has a much larger size scale but with a cage like structure. This suggests the morphology does not have a contributing effect to the catalytic activity. With the 3 h milled LaMnO3 having a higher activity with a lower surface area to the sol–gel sample it further emphasises the effect of surface vacancies on the catalytic activity of LaMnO3 to deN2O.
As the temperature increases above 500 °C a feature in the light-off curve suggests a phase transformation is occurring, and thus altering the catalytic activity. XRD performed on ball milled samples post deN2O demonstrates a higher level of crystallinity for the LaMnO3 phase, i.e. increased peak intensity and reduced line broadening (Fig. S9‡). This suggests that at higher temperatures oxygen ions, generated at the surface during deN2O, diffuse into the bulk to fill the internal oxygen vacancies, reported in the XAS, to produce the highly crystalline perovskite, LaMnO3, orthorhombic phase. Due to the high temperatures towards the end of the catalytic reaction a phase transformation had occurred, and the light-off profiles are not reversible. However, these materials show ample scope to be effective at lower temperatures where the observed phase transformation would not occur.
4. Conclusions
This work demonstrates new insights into the formation of mixed-metal oxides through mechanochemistry and how this method of preparation affords catalytically active centres. By developing an understanding of the chemical steps and active sites produced through mechanochemistry it will further advance the use of this preparation method for industrial catalytic applications.LaMnO3 has been successful synthesised via mechanical grinding from La2O3 and Mn2O3 in the absence of high temperature thermal annealing. Performing XRD on time-slices suggested that 100% crystalline perovskite phase is present from 3 h of milling. However, observations within the EXAFS show La–Mn scattering at the La L3-edge from 1 h and Mn–La scattering at the Mn K-edge from 2 hours. This indicates that early in the milling process La species disperse over the Mn2O3 bulk, with increasing milling time resulting in further disruption and increased interactions between Mn and La. From 2 h of milling the Mn K-edge EXAFS indicates the formation of perovskite-like structures. EXAFS analysis further revealed a degree of oxygen deficiency in ball milled LaMnO3. Through STEM-EELS significant structural variation is observed at the surface which can be reflected in a mixed valence state of Mn by XPS along with oxygen vacancies. During the deN2O by the different LaMnO3 catalysts, ball milled samples showed promising activity compared to their sol–gel analogue. The 3 h milled shows a much earlier onset conversion of N2O; this difference in performance is attributed to an increased level of surface oxygen vacancies found by both STEM-EELS and XPS.
Data access statement
All data supporting this study are openly available from the University of Southampton repository at https://doi.org/10.5258/SOTON/D1128.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Diamond Light Source and The UK Catalysis Hub for provision of beam time (Experiment sp15151-1 and sp15151-4). The staff on B18 at Diamond Light Source, particularly Dr Diego Gianolio are thanked for their assistance in collecting data. The RCaH are acknowledged for use of facilities and staff support. Johnson Matthey is acknowledged for their provision of precursor materials and milling equipment. The Johnson Matthey advanced analytical department are thanked for their help and support throughout the project. The UK Catalysis Hub is thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium (portfolio grants EP//K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/I019693/1). The University of Southampton and EPSRC are thanked for the iCASE studentship of RHB.References
- Perovskites and Related Mixed Oxides Concepts and Applications, ed. P. Granger, V. Parvulescu, S. Kaliaguine and W. Prellier, Wiley-VCH, 2015 Search PubMed.
- Q. Zhang and F. Saito, Adv. Powder Technol., 2012, 23, 523–531 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- X. Guo, D. Xiang, G. Duan and P. Mou, Waste Manag., 2010, 30, 4–10 CrossRef PubMed.
- C. Xu, S. De, A. M. Balu, M. Ojeda and R. Luque, Chem. Commun., 2015, 51, 6698–6713 RSC.
- L. Takacs, Chem. Soc. Rev., 2013, 42, 7649–7659 RSC.
- S. Kaliaguine, A. Van Neste, V. Szabo, J. E. Gallot, M. Bassir and R. Muzychuk, Appl. Catal., A, 2001, 209, 345–358 CrossRef CAS.
- Q. Zhang and F. Saito, J. Alloys Compd., 2000, 297, 99–103 CrossRef CAS.
- B. D. Stojanovic, J. Mater. Process. Technol., 2003, 143–144, 78–81 CrossRef CAS.
- W. F. Libby, Science, 1971, 171, 499–500 CrossRef CAS PubMed.
- R. Voorhoeve, D. Johnson, J. Remeika and P. Gallagher, Science, 1977, 195, 827–833 CrossRef CAS PubMed.
- Y. Teraoka, H. Nii, S. Kagawa, K. Jansson and M. Nygrenb, J. Mater. Chem., 1996, 6, 97–102 RSC.
- V. Szabo, M. Bassir, J. E. Gallot, A. Van Neste and S. Kaliaguine, Appl. Catal., B, 2003, 42, 265–277 CrossRef CAS.
- S. Keav, S. Matam, D. Ferri and A. Weidenkaff, Catalysts, 2014, 4, 226–255 CrossRef.
- P. Baláž, M. Achimovičová and M. Baláž, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC.
- N. Labhasetwar, G. Saravanan, S. K. Megarajan, N. Manwar, R. Khobragade, P. Doggali and F. Grasset, Sci. Technol. Adv. Mater., 2015, 16 Search PubMed.
- S. Kumar, A. Vinu, J. Subrt, S. Bakardjieva, S. Rayalu, Y. Teraoka and N. Labhsetwar, Catal. Today, 2012, 198, 125–132 CrossRef CAS.
- C. S. Swamy and J. Christopher, Catal. Rev., 1992, 34, 409–425 CrossRef CAS.
- M. A. Pena and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2017 CrossRef CAS PubMed.
- S. Kumar, Y. Teraoka, A. G. Joshi, S. Rayalu and N. Labhsetwar, J. Mol. Catal. A: Chem., 2011, 348, 42–54 CrossRef CAS.
- M. Konsolakis, ACS Catal., 2015, 5, 6397–6421 CrossRef CAS.
- T. Yamashita and A. Vannice, J. Catal., 1996, 161, 254–262 CrossRef CAS.
- R. A. Reimer, C. S. Slaten, M. Seapan, M. W. Lower and P. E. Tomlinson, Environ. Prog., 1994, 13, 134–137 CrossRef CAS.
- S. Van de Vyver and Y. Román-Leshkov, Catal. Sci. Technol., 2013, 3, 1465–1479 RSC.
- S. Royer, D. Duprez, F. Can, X. Courtois, C. Batiot-Dupeyrat, S. Laassiri and H. Alamdari, Chem. Rev., 2014, 114, 10292–10368 CrossRef CAS PubMed.
- K. Simmance, D. Thompsett, W. Wang and B. Thiebaut, Catal. Today, 2019, 320, 40–50 CrossRef CAS.
- V. Šepelák, S. Bégin-Colin and G. Le Caër, Dalton Trans., 2012, 41, 11927 RSC.
- A. M. Bolarín, F. Sánchez, A. Ponce and E. E. Martínez, Mater. Sci. Eng. A, 2007, 454–455, 69–74 CrossRef.
- C. A. C. Escobedo, F. S. De Jesús, A. M. Bolarín Miró and J. Muñoz-Saldaña, Phys. Status Solidi C, 2007, 4, 4054–4063 CrossRef CAS.
- F. Ali, A. V. Chadwick and M. E. Smith, J. Mater. Chem., 1997, 7, 285–291 RSC.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- Y. Zhao, Materials, 2012, 5, 1413–1438 CrossRef CAS.
- T. Yamamoto, X-Ray Spectrom., 2008, 37, 572–584 CrossRef CAS.
- H. Aritani, H. Yamada, T. Yamamoto, T. Tanaka and S. Imamura, J. Synchrotron Radiat., 2001, 8, 593–595 CrossRef CAS PubMed.
- J. Niu, J. Deng, W. Liu, L. Zhang, G. Wang, H. Dai, H. He and X. Zi, Catal. Today, 2007, 126, 420–429 CrossRef CAS.
- P. R. N. Silva and A. B. Soares, Ecletica Quim. J., 2009, 34, 31–38 CrossRef CAS.
- S. K. Gupta, M. Sahu, P. S. Ghosh, D. Tyagi, M. K. Saxena and R. M. Kadam, Dalton Trans., 2015, 44, 18957–18969 RSC.
- P. A. W. Van Der Heide, Surf. Interface Anal., 2002, 33, 414–425 CrossRef CAS.
- Y. Yang, S. Zhang, S. Wang, K. Zhang, H. Wang, J. Huang, S. Deng, B. Wang, Y. Wang and G. Yu, Environ. Sci. Technol., 2015, 49, 4473–4480 CrossRef CAS PubMed.
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS.
- J. Chen, M. Shen, X. Wang, G. Qi, J. Wang and W. Li, Appl. Catal., B, 2013, 134–135, 251–257 CrossRef CAS.
- N. Gunasekaran, S. Rajadurai and J. J. Carberry, Catal. Lett., 1995, 35, 373–382 CrossRef CAS.
Footnotes |
| † All data presented in the manuscript is freely available at https://doi.org/10.5258/SOTON/D1128. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt03590g |
| This journal is © The Royal Society of Chemistry 2020 |