
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterodinuclear complexes featuring Zn(II) and M = Al(III), Ga(III) or In(III) for cyclohexene oxide and CO2 copolymerisation†
Arron C.
Deacy
,
Christopher B.
Durr
and
Charlotte K.
Williams
 *
*
Department of Chemistry, Chemistry Research Laboratory, 12 Mansfield Rd, Oxford OX1 3TA, UK. E-mail: charlotte.williams@chem.ox.ac.uk
First published on 9th December 2019
Abstract
The ring opening copolymerisation of CO2 and epoxides is a useful means to valorise waste emissions and to reduce pollution in polymer manufacturing. Heterodinuclear catalysts, particularly those of Zn(II)/Mg(II), have shown better performances than homodinuclear analogues in this reaction. As part of on-going efforts to better understand the catalytic synergy, this work describes a series of heterodinuclear complexes, combining Zn(II) with a metal from Group 13 (M = Al(III), Ga(III) or In(III)). The complexes are synthesised from a symmetrical macrocyclic ligand in high yields via sequential metalation steps and are the thermodynamic reaction products. The Zn(II)/Group 13 complexes are effective homogeneous catalysts for the ring opening copolymerisation (ROCOP) of cyclohexene oxide at 1 bar pressure of carbon dioxide, but all show inferior performances compared to the di-zinc analogue. The CO2 uptake into the polymer increases in the order Al(III) < Ga(III) < In(III) which is attributed to lower Lewis acidity heavier Group 13 homologues showing a reduced tendency to form ether linkages. Concurrently, polycarbonate activity increases down the Group 13 series consistent with weaker metal–oxygen bonds which show enhanced lability to insertion reactions.
Introduction
The transformation of CO2 into valuable products is a cornerstone of sustainable chemistry,1 driven by the necessity to reduce green-house gas emissions and fossil fuel consumption in chemical manufacturing.2 In contrast to the plethora of esoteric stoichiometric CO2 transformations, CO2/epoxide ring-opening copolymerisation (ROCOP) is a promising utilisation as it is truly catalytic and allows for CO2 uptake of 30–50% by weight into the polymer.3,4 The major immediate term opportunity for these materials is as low molecular mass, hydroxyl terminated polycarbonate or polyether-carbonate polyols.5 These polyols are used to manufacture a range of coatings, urethanes and thermosets which show equivalent or better properties than incumbent materials that derive only from petrochemicals.6–9 Importantly, the process of CO2/epoxide copolymerisation to yield polycarbonate polyols is amenable to large-scale manufacturing.5,10 Furthermore, life cycle analysis has shown a ‘triple win’ in terms of CO2 emissions, for every CO2 molecule incorporated into the polymer backbone, two more are saved by reducing epoxide consumption.2 Catalysis is central to the success of CO2/epoxide ROCOP but overall the field suffers from a lack of diversity in catalyst structures with most studies focussing on either di-Zn(II) or bicomponent Co(III)/Cr(III)/Al(III) complex/co-catalyst systems.11,12 These bicomponent systems can show high activity but are known to enchain by complex mechanisms and require the use of expensive and corrosive co-catalysts. Our research group have focused on single-component dinuclear catalysts which obviate co-catalyst requirements; di-Zn(II), Mg(II), Co(II/III) and Fe(III) macrocyclic complexes are all active and selective in the low pressure regime (1 bar CO2 pressure).13–16 In 2015, we reported the first Zn(II)/Mg(II) heterodinuclear catalyst which showed superior performances to either di-Zn(II) or di-Mg(II) counterparts, or any combinations of them.17 Recently we investigated a series of these Zn(II)/Mg(II) heterocatalysts and proposed a chain shuttling mechanism whereby magnesium coordinates and activates the epoxide whilst zinc provides the nucleophilic carbonate group.18 In 2018, Okuda and Mashima reported a series of hetero-tetranuclear catalysts of the form Zn3Ln which were also highly active.19 Given the promise of these mixed metal catalysts it is important to understand which metal combinations are most effective and how synergy functions. Naturally, not all metal combinations are synergic as demonstrated by recent investigation of heterocomplexes featuring Zn(II) with Group 1 elements [Li(I), Na(I) and K(I)] or Zn(II) with Group 2 elements [Mg(II) and Ca(II)]. Within the series of complexes, only the Zn(II)/Mg(II) combination showed synergy and all others were less active than the Zn(II)/Zn(II) homodinuclear complex.20 In this work, we investigate a series of complexes featuring Zn(II) and Group 13 elements. The metal selection is motivated by some precedent for Al(III) complexes in this catalysis, for example Al(III) salen,21–23 porphyrin24–28 or trisphenolate29 complexes are all active as part of bicomponent systems (i.e. with co-catalysts). Very recently, the first In(III) catalysts were reported and operate without co-catalyst, attributed to an unusual mononuclear mechanism.30 Al-catalyst systems, with co-catalysts, are also widely investigated in the ring-opening copolymerisation of epoxides and cyclic anhydrides.31–36 Group 13 complexes also show good activity for mechanistically related reactions such as in the ring-opening polymerisation of lactide, lactones or cyclic carbonates.37–42 For the target macrocyclic catalysts, it was envisaged that combining the labile Zn(II) site with Group 13 elements could allow modification of metal Lewis acidity and alkoxide lability. When targeting new catalysts, it is proposed that both metals should have sufficiently small ionic radii to enable in-plane coordination in the macrocycle cavity.20Results and discussion
Firstly, the di-zinc complex (1) was prepared to allow for comparison with the target heterodinuclear complexes (2–4); all complexes were synthesised using a sequential metalation procedure (Fig. 1). The synthesis involved ligand deprotonation with diethyl zinc, in THF at 25 °C, to yield in situ a mono-zinc complex (not isolated). Next, ZnCl2 (for (1)) or the appropriate MCl3 {for (2–4) where M = Al(III), Ga(III) or In(III)} precursor was added and the reaction heated to 100 °C for 16 h. The complexes were isolated, without further purification, as white powders in reasonable isolated yields (>70%). To understand the importance of providing a free coordination site for monomers, cationic complex 5 was synthesised by chloride abstraction from complex 4, by reaction with K[B(C6F5)4] in THF at 25 °C, and was isolated as an off-white powder in 90% yield. To understand the influence of the co-ligand on initiation rates complex 6, featuring a bridging carboxylate co-ligand, was synthesised via a metathesis reaction between complex 4 and KOBzpCF3. The reaction was conducted in MeOH, at 25 °C, and enabled isolation of the product as a white powder in 95% yield. All complexes were characterised by solution state NMR spectroscopy (Fig. S1–S25†), mass spectrometry (Fig. S26–S29†), elemental analysis and complexes 1–4 were also characterised by single crystal X-ray diffraction experiments (Fig. S30–S33†).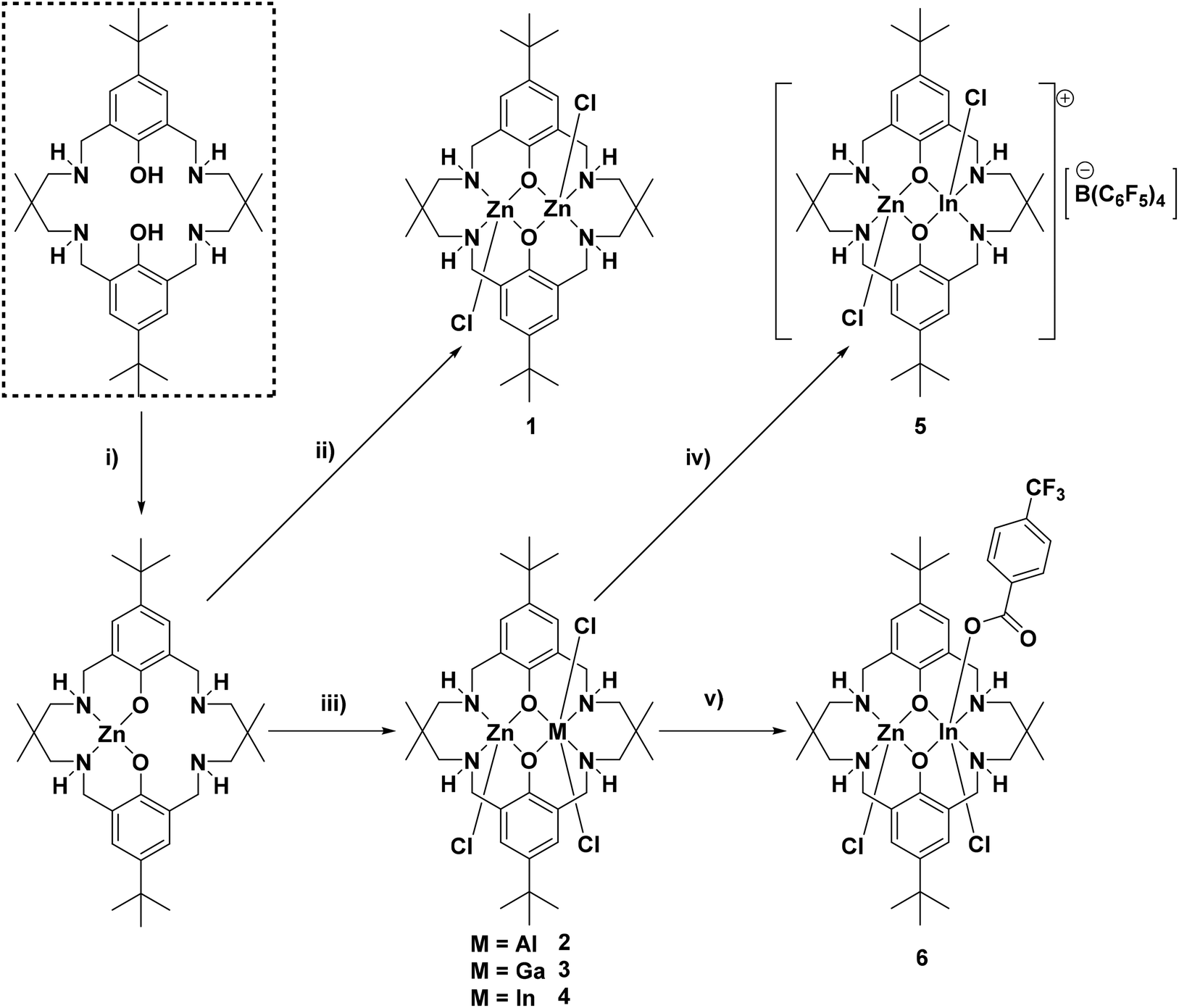 | ||
| Fig. 1 Synthesis of complexes 1–6. (i) ZnEt2, THF, 25 °C, 16 h. (ii) ZnCl2, THF, 100 °C, 16 h, 81%. (iii) MCl3 (M = Al, Ga or In), THF, 100 °C, 16 h, >70%. (iv) K[B(C6F5)4], THF, 25 °C, 16 h, 90%. (v) KOBzpCF3, MeOH, 25 °C, 16 h, 95%. | ||
The 1H NMR spectrum of 1 shows multiple, complex signals at room temperature in all solvents tested (THF, methanol, TCE) but was successfully determined in tetrachloroethane (TCE) at 403 K, under these conditions the signals coalesce to give an assignable spectrum. The spectrum shows five resonances between 2.2 ppm and 5.0 ppm, representative of the benzylic, methylene and amine proton environments. There is a single resonance at 7.00 ppm corresponding to the phenyl protons and this peak is indicative of a homodinuclear complex coordinated by a symmetrical ligand. Its 13C NMR spectrum displays two signals, at 58.7 and 64.5 ppm, corresponding to the benzylic and methylene carbon atoms, along with four phenyl carbon signals, at 160.5 (ipso, phenolic carbon), 140.3 (para), 125.5 (ortho) and 128.4 ppm (meta), due to the complex's symmetry. The 1H and 13C NMR spectra are in-line with related macrocycle di-zinc complexes with co-ligands such as acetate, phenyl, benzoate and iodide.13,20,43,44 The MALDI-ToF mass spectrum displays a peak at 717 m/z, corresponding to the molecular cation, [LZn2(Cl)]+.
NMR spectroscopy is a useful means to determine the success of the heterodinuclear complex synthesis and comparisons between the signals for the heterodinuclear complexes and homodinuclear (1) are useful to determine purity. The homodinuclear complex is C2 symmetric but heterodinuclear complexes 2–4 lack this symmetry and as a result show different environments for all the protons in the 1H NMR spectra (Fig. 2). Another indicator of heterodinuclear complex formation is the presence of coupling between the two phenyl proton resonances, at ∼7.00 ppm, as confirmed by 2D COSY NMR spectroscopy (Fig. S5, S9 and S14†). If a mixture of homodinuclear complexes were formed, it would be expected to display two phenyl resonances but these would not couple with one another. The presence of coupled aromatic signals is a strong indicator of heterodinuclear complex formation; that said, in some circumstances this coupling is more difficult to unambiguously assign as it depends upon the magnitude of the 4JH–H coupling constant. The 1H NMR spectra for the heterodinuclear complexes can be used to infer the approximate purity, by comparing the integrals of signals at 6.79 (if any complex 1 is present) and ∼6.97 ppm (for heterocomplexes 2–4). This analysis indicates that the heterodinuclear complexes are all formed as the major product, with purity values, within the detection limits of NMR spectroscopy, of 90% (2), 95% (3) and >99% (4), respectively.
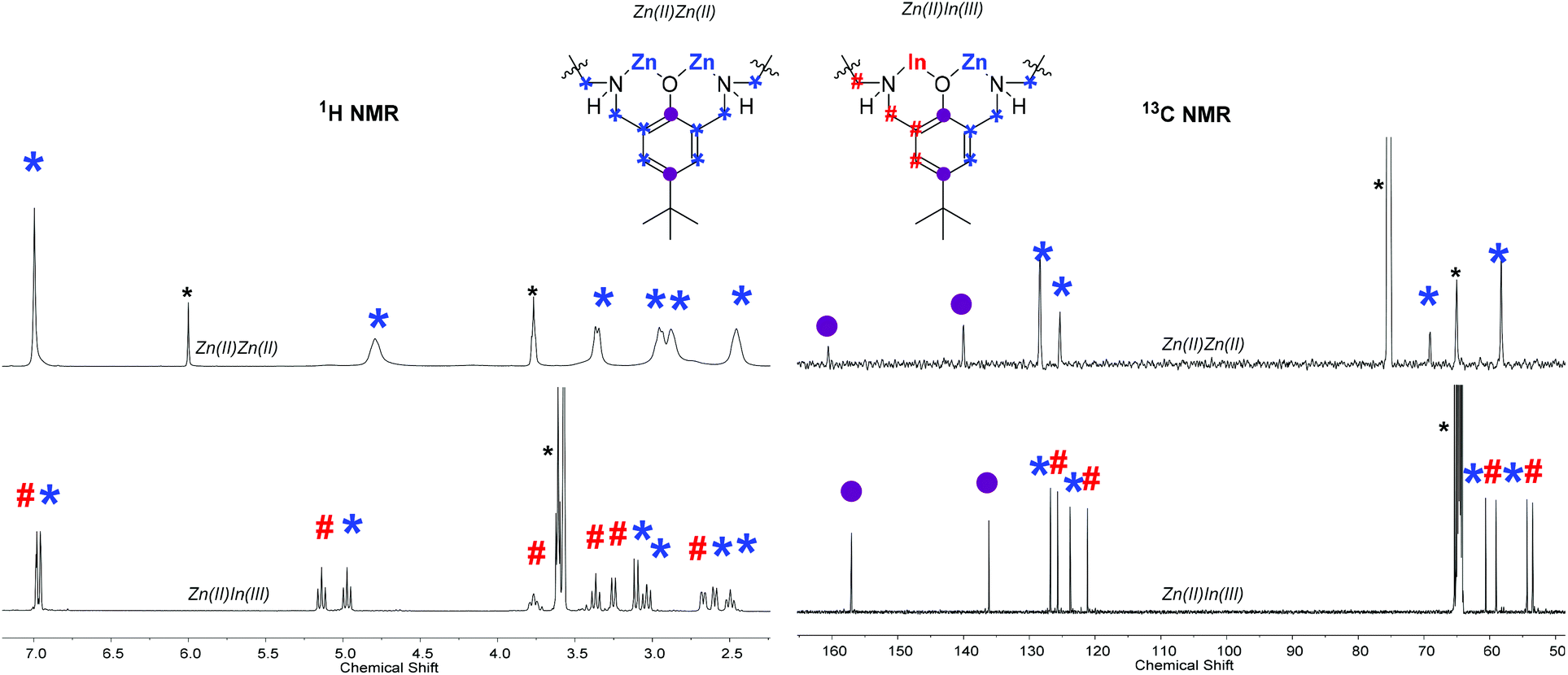 | ||
| Fig. 2 Overlaid selected regions of the 1H and 13C NMR spectra of di-zinc complex (1) and heterodinuclear Zn(II)In(III) complex (4). Note the full spectra and assignment is available in the ESI (Fig. S1, S2, S12 and S15†). *Residual TCE and THF. | ||
The 13C NMR spectra also show the loss of C2 symmetry compared to 1, with the heterodinuclear complexes displaying double the number of signals for nearly all the carbon atoms (Fig. 2). Another diagnostic feature of heterodinuclear complex formation is the retention of a single resonance for the ipso- and para-phenyl carbons, at approx. 160 and 140 ppm, respectively (Fig. 2). A mixture of homodinuclear complexes would be expected to display two signals each for these carbons. The 13C NMR can be used in conjunction with 1H and COSY NMR to confirm heterodinuclear complex formation.
2D DOSY NMR spectra for complexes 2–4 all show a single diffusion coefficient, consistent with a discrete monomeric heterodinuclear complex in THF solution (as a representative solvent considering that polymerizations are conducted in neat epoxide) (Fig. S11, S16 and S25†). MALDI-ToF mass spectra showed molecular ions corresponding to the cation [LZnM(Cl2)]+ at 714 m/z, 755 m/z and 800 m/z, where M = Al, Ga and In, for complexes 2, 3 and 4, respectively (Fig. S27–S29†).
The 1H NMR spectrum of cationic complex 5 displays significant shifts to higher chemical shift values for all NH resonances compared to complex 4 (Fig. S17†), consistent with the increased positive charge on the complex. The NMR data clearly confirm that the heterodinuclear complex is maintained and that there are not any metal re-distribution reactions.
The 11B, 13C and 19F NMR spectra also all indicate the formation of a single complex (Fig. S19–S21†). The 1H NMR spectrum of complex 6 indicates the success of the metathesis reaction and is consistent with the solid state structure where the pCF3benzoate is located at the bridging position (Fig. S22 and S34;† single crystal analysis by X-ray diffraction gave insufficient data to fully resolve the structure, though its connectivity could still be obtained). The 1H NMR data confirms that the heterodinuclear complex is retained without any detectable metal redistribution. The NH resonances are not observed due to H–D exchange processes occurring between the complex and the solvent (d4-MeOD). A major and minor set of resonances are observed for the pCF3benzoate protons which is attributed to both κ1vs. κ2 coordination modes, as was previously observed for an analogous Mg(II)/Zn(II) complex.18
Single crystals, suitable for X-ray crystallography, were isolated by diffusion of pentane into a saturated solution of the complexes in acetonitrile (1, 2 and 3) or THF (4 and 6), at −40 °C in a glovebox (Fig. S30–S34 and Tables S1–S5†). The structures of 2–4 confirm the formation of heterodinuclear complexes, which are monomeric in the solid state, and which feature three chloride co-ligands. The structures show the macrocycle adopts a ‘bowl’ conformation in each case. For complexes 1–4, the zinc atom(s) adopts a pentacoordinate, distorted square based pyramidal geometry (τ5 = 0.35 Zn1 (1), τ5 = 0.15 Zn2 (1), τ5 = 0.07 Zn1 (2), τ5 = 0.02 Zn1 (3), τ5 = 0.01 Zn1 (4)) and is coordinated by a chloride co-ligand. The Group 13 metals show octahedral coordination geometries and are coordinated by two chloride co-ligands in line with the higher oxidation state of these metals. For 1, the Zn–Zn separation is 3.04(1) Å, consistent with previous di-zinc complexes coordinated by this ligand and with the proposed distance for effective catalysis.20,44–46 For 2–4, Zn–M(III) separations increase with the increasing ionic radius of the Group 13 element and are 3.02(3) Å, 3.12(3) Å and 3.15(8) Å for complexes 2, 3 and 4, respectively (Fig. 3).
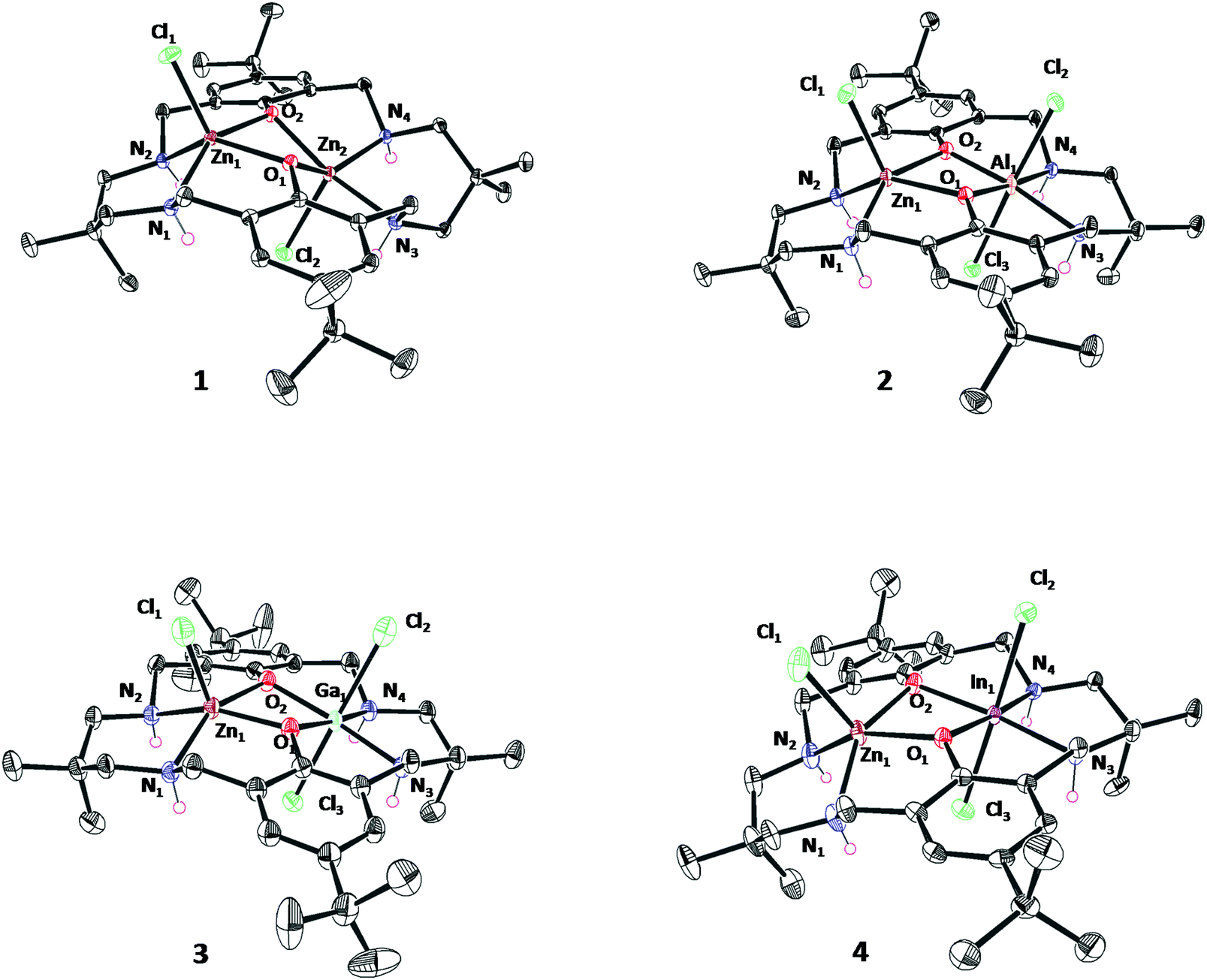 | ||
| Fig. 3 ORTEP representation of the crystallographic structure of complex 1–4 and 6 obtained by single crystal X-ray diffraction. Disorder and H-atoms (excluding NH) have been omitted for clarity, with thermal ellipsoids represented at 40% probability. Selected bond lengths (Å) and angles (°) see Tables S1–S5.† | ||
The heterodinuclear complexes, 2–6, were tested for CO2/CHO ROCOP and compared against the di-zinc catalyst (1). The polymerisations were all conducted at 80 °C, 0.1 mol% catalyst loading and using 1 bar CO2 pressure. The conditions were selected as they have previously been shown to result in reasonable activity for related catalysts and to enable comparison against the existing literature.13,20 Within the series of compounds, the di-zinc catalyst (1) shows the highest activity (9 h−1) and yields the highest carbonate linkage content (CO2 uptake) and polymer selectivity (minor quantities of trans-cyclohexene carbonate are observed by 1H NMR spectroscopy). Indeed, its performance is equivalent to the previously reported di-zinc acetate analogue.13 Heterodinuclear tri-chloride complexes (2–4) all show low activity and yield polymers with significant ether linkage contents and/or polyether contamination (up to 32%). These polyethers form as a result of the metal-alkoxide catalytic intermediate under-going sequential epoxide ring opening of an epoxide molecule (see Fig. S36,† for illustrations of the reactions occurring in the catalytic cycle and side-reactions). All heterodinuclear complexes show high polymer selectivity and there is very little cyclic carbonate by-product formation.
The polymers’ molecular masses were analysed using GPC and, in most cases, bimodal molecular mass distributions were apparent. Multi-modal molecular mass distributions are very common in ROCOP reactions and are attributed to two series of chains resulting from different initiating groups.49 It is proposed that the lower molecular mass series are initiated from the catalyst M–Cl, whereas the higher molecular mass series are from 1,2-cyclohexanediol. The 1,2-cyclohexanediol is proposed to form by reaction between cyclohexene oxide and residual water (e.g. in the CO2). Darensbourg and co-workers have established the rates of such hydrolyses typically exceed polymerization initiation and propagation rates.50–52 In the case of catalyst 4 at 1 bar CO2 pressure a trimodal molecular mass distribution was observed, with peaks being tentatively attributed to polyether and two polycarbonate series, respectively (Table 1, entry 4).30 At 20 bar pressure, catalyst 4 does not produce any polyether and the expected bimodal molecular mass distribution is observed (Table 1, entry 5).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| Entry | Catalyst | Time (d) | TONb | TOFc (h−1) | CO2d (%) |
Polym.e (%) | M n [Đ]f (g mol−1) |
|---|---|---|---|---|---|---|---|
|
a Catalysis conditions: catalyst:CHO 1:1000, 80 °C, 1 bar pressure CO2 and in neat epoxide.
b Turnover number (TON) = number of moles of cyclohexene oxide consumed/number of moles of catalyst.
c Turnover frequency (TOF) = TON/time (h).
d Expressed as a percentage of CO2 uptake vs. the theoretical maximum (100%), determined by comparison of the relative integrals of the 1H NMR proton resonances due to carbonate (δ 4.65 ppm) and ether (δ 3.45 ppm) linkages in the polymer backbone.
e Expressed as a percentage of polymer formation vs. the theoretical maximum (100%), determined by comparison of the relative integrals of the 1H NMR proton resonances due to polymer (4.65 ppm) and trans-cyclic carbonate (4.00 ppm).
f Determined by SEC, in THF, calibrated using narrow-Mn polystyrene standards. Values for Zn/Al and Zn/Ga were not determined as the conversion was too low.
g 20 bar CO2.
h 60 °C.
i 35 bar CO2.
j 50 bar CO2.
k 15 bar CO2, 40 °C, 0.5 mol% cat loading (Fig. S35† for molecular structures).
|
|||||||
| 1 | 1 | 2 | 417 | 9 | >99 | 98 | 20500[1.04] |
| 8800 [1.13] | |||||||
| 2 | 2 | 3 | 24 | 0.3 | 68 | 88 | n.d |
| 3 | 3 | 3 | 69 | 1 | 85 | 95 | n.d |
| 4 | 4 | 3 | 399 | 6 | 85 | 91 | 16800 [1.07] |
| 6600 [1.07] | |||||||
| 2700 [1.12] | |||||||
| 5 | 4 | 0.33 | 221 | 28 | >99 | >99 | 12400 [1.03] |
| 5100 [1.18] | |||||||
| 6 | 5 | 0.25 | 696 | 116 | <1 | >99 | n.d |
| 7 | 6 | 3 | 227 | 3 | >99 | 90 | 11200 [1.04] |
| 4900 [1.12] | |||||||
| 8 | ZnMgBr218 |
0.25 | 247 | 78 | >99 | >99 | 3000 [1.18] |
| 9 | PSalan-InClh30 |
2 | 350 | 15 | >99 | 95 | 19700 [2.00] |
| 3400 [1.32] | |||||||
| 10 | Salan-AlCl + Bu4NCli21 |
0.33 | 404 | 35 | 98 | 3 | n.d |
| 11 | β-Keto-Al2Me + Bu4NOAcj47 |
0.25 | 36 | 15 | >99 | 97 | 4200 [1.21] |
| 12 | Amino tris(phenolate)AlCl + PPNClk48 |
2 | 154 | 3 | >99 | >99 | 11900 [1.49] |
In terms of polymerization selectivity, the catalysts show decreasing amounts of ether linkages on descending the series and the complexes of Ga(III) and In(III) show reasonable/good selectivity. This change in selectivity is proposed to result from the lower Lewis acidity of heavier Group 13 congeners compared to Al(III) and highlights the importance of appropriate balance of Lewis acidity in controlling side-reactions. The high ether content may also signal that CO2 insertion is implicated in the rate limiting step and thus complex 4 was tested with 20 bar CO2 pressure. Under these conditions a perfectly alternating copolymer formed, with no ether linkages. In the higher pressure regime, there is also a significant increase in activity (28 h−1) which may be a preliminary indication of a rate law dependent on CO2 pressure or from the improved reaction stirring in the high pressure autoclaves.
All polymerisation catalysts show induction periods prior to the start of the catalysis. To rule out that metal redistribution reactions occur during these induction periods, catalysts 2–4 were heated at 100 °C for 3 days in THF (Fig. S37–39†). In all cases, the 1H NMR spectra were identical before and after prolonged heating, i.e. there was no evidence for the formation of dizinc 1. The low polymerisation activity and significant induction periods of these ZnM(III) complexes could be due to coordinative saturation retarding epoxide coordination and activation. To investigate whether this could be overcome, cationic complex 5 showing a ‘free’ coordination site was prepared. Unfortunately, it rapidly forms a polymer which is almost entirely polyether, probably due to the increased Lewis acidity of the In(III) site.53
A recent investigation into analogous ZnMg catalysts showed that by applying carboxylate ligands, in place of halides, it was possible to significantly enhance turn-over-frequency values.18 Nonetheless, in this series of catalysts complex 6, featuring a p-CF3benzoate ligand, showed no discernible difference in rate or selectivity, but a slight decrease in ether linkages, compared with 4. Attempts to further substitute the two remaining chloride ligands in 6 with p-CF3benzoate groups were unsuccessful and resulted in broad, complex NMR spectra.
In terms of the catalytic activity of the series of ZnM(III) catalysts, there is a marginal increase on descending the series of Group 13 elements from ∼1 h−1 (Al(III)) to 9 h−1 (In(III)). This increasing activity may correlate with the decreasing M–O bond strengths and the increasing lability of the metal alkoxide intermediates. Overall, it is quite clear that the activity values for these complexes are very low and particularly compared to leading catalysts in this field.18,19,54,55 The Zn(II)Mg(II) catalyst shows a TOF of ∼78 h−1 and quantitative selectivity for carbonate linkages, under the same conditions (Table 1). Nonetheless, it is worth noting that the activities are in-line with other (previously reported) Group 13 catalysts (Table 1).56 For example, the high pressure CO2 activity of 4 (ZnIn(III) is approximately equivalent to that of a salanAl/Bu4NCl system and significantly greater than a di-Al(III) catalyst system (β-ketoAl2Et/Bu4NOAc) or the amino tris(phenolate)Al/PPNCl systems (Table 1).21,47,48 Overall, these heterodinuclear ZnM(III) catalysts show some promise in terms of activity and selectivity, particularly compared to other Group 13 catalysts. Future investigations should target ancillary ligands capable of reducing the Group 13 element Lewis acidity, and associated M–O bond strength, so as to increase activity and selectivity for carbonate linkage formation.
Conclusions
The syntheses and characterisations of a series of heterodinuclear Zn(II)/Al(III), Ga(III) or In(III) complexes, coordinated by a symmetrical macrocycle are reported. The compounds are synthesised using a sequential metalation route, operating under thermodynamic control, which yields the heterodinuclear complexes as the major reaction products. The complexes are applied as homogeneous catalysts for cyclohexene oxide/CO2 ring opening copolymerisation but all show worse performance than the di-zinc analogue. It seems that the enhanced Lewis acidity and significant M–O bond strengths of these Group 13 elements, compared to Zn(II), are less desirable in this catalysis. The ability to use inexpensive, colourless and abundant elements, such as Al(III) and Mg(II), remains an important goal for the field. Future work needs to address optimising the ligand design so as to improve catalytic activity and selectivity.Experimental section
All experimental manipulations were performed using a dual-manifold nitrogen-vacuum Schlenk line or in a nitrogen filled glovebox. All solvents and reagents were obtained from commercial sources and used as received unless stated otherwise. THF, acetonitrile and pentane were obtained from an SPS system, degassed by several freeze–pump–thaw cycles, further dried with 3 Å molecular sieves and stored under nitrogen. Cyclohexene oxide was dried over calcium hydride (2 days) and purified via fractional distillation prior to use and stored under a nitrogen atomsphere.Low pressure copolymerisation studies were performed using a triple manifold youngs tap CO2/N2/vacuum Schlenk line where research-grade carbon dioxide was dried through a drierite column, and two addition drying columns (Micro Torr, Model number: MC1-804FV) in series, before use in copolymerisation studies. High pressure copolymerisation studies were performed in a 25 mL Parr 5500 HP Compact Reactor using research-grade carbon dioxide that was dried by passing through two drying columns in series (VICI, Thames Restek).
1H, 11B, 13C, 19F and 2D NMR spectra were obtained using a Bruker AV 400 MHz spectrometer at 298 K, unless stated otherwise. MALDI-ToF analysis was performed on a Micromass MALDI micro MX spectrometer. The matrix used was trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malonitrile.
Gel permeation chromatrography analysis was carried out on a two mixed bed PSS SDV linear S column in series, with THF as the eluent, at a flow rate of 1 mL min−1, on a Shimadzu LC-20AD instrument, at 40 °C. Polymer molecular mass values (Mn) were obtained by calibration of the instrument using a series of narrow molecular mass polystyrene standards and are reported without correction.
Elemental Analysis was determined by Mr Stephen Boyer at London Metropolitan University.
General procedure of complexes 1–4 synthesis
ZnEt2 (0.11 g, 0.91 mmol) was added dropwise to a solution of H2L (0.50 g, 0.91 mmol) in THF (10 mL) and stirred for 16 h at 25 °C. ZnCl2 or MCl3 (M = Al, Ga or In) (0.91 mmol) in THF (5 mL) was added to the stirring solution (LZn) and the reaction heated to 100 °C for 16 h. The solvent was removed in vacuo to afford the products as white powders.General procedure for the synthesis of polycarbonates from CO2/epoxide, at 1 bar CO2 pressure
The catalyst (15 μmol) was dissolved in CHO (15 mmol) and placed under 1 bar CO2 in a Schlenk tube and was heated to 80 °C, at a stirring rate of 350 RPM. Aliquots were taken at regular intervals and quenched by addition, in air, into chloroform.General procedure for the synthesis of polycarbonates from CO2/epoxide, at 20 bar CO2 pressure
The catalyst (60 μmol) was dissolved in CHO (60 mmol) and placed into a 25 mL Parr Reactor inside a nitrogen filled glovebox. The reactor was removed from the glove box and charged with CO2, to a pressure of 20 bar, and heated to 80 °C.X-ray crystallography
Crystalline samples were isolated in a glovebox, under a pool of fluorinated oil, and mounted on MiTeGen MicroMounts. The crystals were cooled to 150 K, using an Oxford Cryostream nitrogen cooling device. Data collection was carried out with an Oxford Diffraction supernova diffractometer using Cu Kα (λ = 1.5417 Å) radiation. The resulting raw data was processed using CrysAlis Pro.58 Structures were solved using SHELXT and Full-matrix least-squares refinements based on F2 were performed in SHELXL-14,59 as incorporated in the WinGX package.60 For each methyl group, the hydrogen atoms were added at calculated positions, using a riding model with U(H) = 1.5Ueq (bonded carbon atom). The rest of the hydrogen atoms were included in the model at calculated positions, using a riding model with U(H) = 1.2Ueq (bonded atom). Neutral atom scattering factors were used and included terms for anomalous dispersion.61 All crystal structures have been registered with the Cambridge structural database: CCDC 1939693–1939697.†Conflicts of interest
CKW is a director of Econic Technologies.Acknowledgements
The EPSRC (EP/L017393/1, EP/K014668/1) and econic technologies (CASE award to AD) are acknowledged for research funding.References
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- N. von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- J. Langanke, A. Wolf, J. Hofmann, K. Böhm, M. A. Subhani, T. E. Müller, W. Leitner and C. Gürtler, Green Chem., 2014, 16, 1865–1870 RSC.
- W. Kuran, M. Sobczak, T. Listos, C. Debek and Z. Florjanczyk, Polymer, 2000, 41, 8531–8541 CrossRef CAS.
- P. Alagi, R. Ghorpade, Y. J. Choi, U. Patil, I. Kim, J. H. Baik and S. C. Hong, ACS Sustainable Chem. Eng., 2017, 5, 3871–3881 CrossRef CAS.
- M. Scharfenberg, J. Seiwert, M. Scherger, J. Preis, M. Susewind and H. Frey, Macromolecules, 2017, 50, 6577–6585 CrossRef CAS.
- M. DeBolt, A. Kiziltas, D. Mielewski, S. Waddington and M. J. Nagridge, J. Appl. Polym. Sci., 2016, 133, 9 CrossRef.
- M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, ACS Catal., 2019, 9, 7937–7956 CrossRef CAS.
- Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS.
- C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed.
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 50, 4164–4167 RSC.
- G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618–4627 RSC.
- H. Nagae, R. Aoki, S. Akutagawa, J. Kleemann, R. Tagawa, T. Schindler, G. Choi, T. P. Spaniol, H. Tsurugi, J. Okuda and K. Mashima, Angew. Chem., Int. Ed., 2018, 57, 2492–2496 CrossRef CAS PubMed.
- A. C. Deacy, C. B. Durr, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2018, 57, 15575–15583 CrossRef CAS PubMed.
- D. J. Darensbourg and D. R. Billodeaux, Inorg. Chem., 2005, 44, 1433–1442 CrossRef CAS PubMed.
- N. Ikpo, S. M. Barbon, M. W. Drover, L. N. Dawe and F. M. Kerton, Organometallics, 2012, 31, 8145–8158 CrossRef CAS.
- T. Ohkawara, K. Suzuki, K. Nakano, S. Mori and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 10728–10735 CrossRef CAS PubMed.
- T. Aida and S. Inoue, J. Am. Chem. Soc., 1983, 105, 1304–1309 CrossRef CAS.
- T. Aida, M. Ishikawa and S. Inoue, Macromolecules, 1986, 19, 8–13 CrossRef CAS.
- H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172–4186 CrossRef CAS.
- C. Chatterjee and M. H. Chisholm, Inorg. Chem., 2012, 51, 12041–12052 CrossRef CAS PubMed.
- H. Sugimoto, H. Goto, S. Honda, R. Yamada, Y. Manabe and S. Handa, Polym. Chem., 2016, 7, 3906–3912 RSC.
- L. Peña Carrodeguas, J. González-Fabra, F. Castro-Gómez, C. Bo and A. W. Kleij, Chem. – Eur. J., 2015, 21, 6115–6122 CrossRef PubMed.
- A. Thevenon, A. Cyriac, D. Myers, A. J. P. White, C. B. Durr and C. K. Williams, J. Am. Chem. Soc., 2018, 140, 6893–6903 CrossRef CAS PubMed.
- E. H. Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef CAS.
- N. J. Van Zee and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 2665–2668 CrossRef CAS PubMed.
- J. Li, Y. Liu, W. M. Ren and X. B. Lu, J. Am. Chem. Soc., 2016, 138, 11493–11496 CrossRef CAS PubMed.
- M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed.
- F. Isnard, F. Santulli, M. Cozzolino, M. Lamberti, C. Pellecchia and M. Mazzeo, Catal. Sci. Technol., 2019, 9, 3090–3098 RSC.
- J. Li, B. H. Ren, S. Y. Chen, G. H. He, Y. Liu, W. M. Ren, H. Zhou and X. B. Lu, ACS Catal., 2019, 9, 1915–1922 CrossRef CAS.
- C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Inorg. Chem., 2013, 52, 12561–12567 CrossRef CAS PubMed.
- K. Press, I. Goldberg and M. Kol, Angew. Chem., 2015, 127, 15071–15074 CrossRef.
- K. M. Osten and P. Mehrkhodavandi, Acc. Chem. Res., 2017, 50, 2861–2869 CrossRef CAS PubMed.
- M. Chwatko and N. A. Lynd, Macromolecules, 2017, 50, 2714–2723 CrossRef CAS.
- D. Myers, A. J. P. White, C. M. Forsyth, M. Bown and C. K. Williams, Angew. Chem., 2017, 129, 5361–5366 CrossRef.
- T. Stößer, D. Mulryan and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 16893–16897 CrossRef PubMed.
- Y. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 CrossRef CAS PubMed.
- C. Romain, J. A. Garden, G. Trott, A. Buchard, A. J. P. White and C. K. Williams, Chem. – Eur. J., 2017, 23, 7367–7376 CrossRef CAS PubMed.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- K. Nishioka, H. Goto and H. Sugimoto, Macromolecules, 2012, 45, 8172–8192 CrossRef CAS.
- C. Martín and A. W. Kleij, Macromolecules, 2016, 49, 6285–6295 CrossRef.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2861–2871 CrossRef CAS.
- G.-P. Wu and D. J. Darensbourg, Macromolecules, 2016, 49, 807–814 CrossRef CAS.
- D. J. Darensbourg, Green Chem., 2019, 21, 2214–2223 RSC.
- D. Franz and S. Inoue, Chem. – Eur. J., 2019, 25, 2898–2926 CrossRef CAS PubMed.
- S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579–4582 RSC.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS PubMed.
- N. Ikpo, J. C. Flogeras and F. M. Kerton, Dalton Trans., 2013, 42, 8998–9006 RSC.
- P. C. Andrikopoulos, D. R. Armstrong, H. R. L. Barley, W. Clegg, S. H. Dale, E. Hevia, G. W. Honeyman, A. R. Kennedy and R. E. Mulvey, J. Am. Chem. Soc., 2005, 127, 6184–6185 CrossRef CAS PubMed.
- CrysAlis CCD, CrysAlis RED and associated programs: Oxford Diffraction, Oxford Diffraction Ltd, Abingdon, England, 2006 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. J. C. Wilson, International Tables for Crystallography, Kluwer Academic Publishers, Dordrecht, 1st edn, 1992, vol. C Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1939693–1939697. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt02918d |
| This journal is © The Royal Society of Chemistry 2020 |