
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shining light on the solid–liquid interface: in situ/operando monitoring of surface catalysis
Leila
Negahdar
*ab,
Christopher M. A.
Parlett
 cde,
Mark A.
Isaacs
ab,
Andrew M.
Beale
ab,
Karen
Wilson
f and
Adam F.
Lee
*f
cde,
Mark A.
Isaacs
ab,
Andrew M.
Beale
ab,
Karen
Wilson
f and
Adam F.
Lee
*f
aDepartment of Chemistry, UCL, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: l.negahdar@ucl.ac.uk
bUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, Harwell, Oxfordshire OX11 0FA, UK
cDepartment of Chemical Engineering & Analytical Science, The University of Manchester, Manchester, M13 9PL, UK
dDiamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
eDiamond Light Source, University of Manchester at Harwell, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
fCentre for Advanced Materials and Industrial Chemistry (CAMIC), School of Science, RMIT University, Melbourne, VIC3000, Australia. E-mail: adam.lee2@rmit.edu.au
First published on 11th June 2020
Abstract
Many industrially important chemical transformations occur at the interface between a solid catalyst and liquid reactants, despite which relatively little attention has been paid to spectroscopic methods for interrogating the working solid–liquid interface. This partly reflects a limited number of analytical techniques that give access to interface-specific information. Direct observation of surface species at catalytic solid–liquid interfaces is a daunting challenge for many in situ techniques due to the low concentration and/or short lifetime of chemical species in dynamic reactions. This review discusses the application of in situ and operando spectroscopies to probe solid–liquid interfaces, with a focus on the resulting mechanistic insight in the context of catalysis for sustainable chemistry.
1. Introduction
The rational design of heterogeneous catalysts with optimal performance heavily relies on a detailed understanding of atomic and molecular interactions and associated reactions at solid surfaces, for which in situ (in place) and operando (under working conditions) studies can offer unparalleled insight.1–3 Heterogeneous catalysis, wherein the reactants and catalyst are in different phases, occurs at solid–gas, solid–liquid, or solid–liquid–gas interfaces4 (and exceptionally at the liquid–gas interface5) and involves a complex interdependency between adsorption/desorption, diffusion, and speciation processes, which dictate reaction kinetics and mechanisms.6 Heterogeneous catalysis underpins chemicals, pharmaceuticals, and fuels production from fossil and biomass resources.7,8Numerous industrial catalytic processes are conducted in the liquid phase, however the number of in situ and/or operando studies of catalytic transformations at the solid–liquid interface are limited, in part due to the lack of techniques able to provide quantitative and chemically specific information on such interfaces. Over the past decade, infrared, Raman, UV-vis and X-ray spectroscopies have been extended to elucidate the nature of active sites in reacting catalyst surfaces (e.g. their phase, oxidation state and density) and molecular processes such as adsorption of reactants and products (strength and geometry) and the identification of reactive surface intermediates. This review highlights the emerging mechanistic insight available through applying in situ/operando spectroscopies to catalytic reactions at the solid–liquid interfaces, and identifies creative solutions to current challenges in liquid phase heterogeneous catalysis. Popular spectroscopic techniques and state-of-the-art methodologies, and their advantages and limitations are critically discussed for exemplar systems, particularly in the context of sustainable chemistry. Requisite conditions for reaction analysis, and the types of information that can be determined, notably reaction mechanisms and catalytically active species, are outlined. Concluding remarks address the current status and future perspectives for emerging spectroscopic techniques as applied to liquid phase heterogeneous catalysis.
2. Spectroscopic techniques
In situ catalytic studies at the solid–liquid interface are extremely challenging.9 First, the reaction medium plays a significantly greater role than occurs in solid–gas catalysis. The high density of liquids results in increased absorption of incident and transmitted/radiated photons, and greater scattering/attenuation of photoexcited electrons in electronic spectroscopies, and hence poorer signal:noise in resulting spectra. Where water is the solvent of choice, such as for water-splitting to H2, such issues are particularly problematic when using IR spectroscopy. Second, a multitude of species may co-exist at solid–liquid interfaces, including reactants, intermediates, spectators, products and solvents, some of which may occur at concentrations high enough to obscure signals from other species. Third, the dynamic nature of solid–liquid interfaces arising from exchange between the bulk media and local environment can induce broadening of spectral features. For example, at 0 °C, the vapour pressure of water is associated with the exchange of around three million monolayers per second of molecules between a liquid surface and vapour phase.10 Finally, under most experimental conditions a solvent is contacted by other phases (such as vapour/gas) or insoluble/immiscible reactants and products which can alter the density of liquid at the solid surface.11–13 Common in situ/operando spectroscopies applied to catalysis at the solid–liquid interface are summarised below.2.1 Attenuated total reflection infrared spectroscopy (ATR-IR)
The vibrational spectrum of a molecule adsorbed on a solid surface includes information about the surface–adsorbate bonds, the intermolecular interactions and the local orientation of the molecule. Such detailed information makes vibrational spectroscopy, especially in the infrared (IR), a valuable technique in the field of heterogeneous catalysis. IR spectroscopy has been extensively applied to catalytic solid–gas interfaces. However, studies of liquid phase catalysis by IR spectroscopy are more limited due to large spectral interference from the liquid phase. Much of the experimental IR spectroscopic data available on solid–liquid catalyst interfaces has been obtained using attenuated total reflection infrared spectroscopy (ATR-IR).14 ATR-IR spectroscopy is suitable for studying molecular vibrations at the solid–liquid interface because the IR light is restricted to the surface vicinity which minimises spectral contributions from the bulk liquid. Using ATR-IR, spectra are obtained from the absorption of light penetrating only a few microns orthogonal to the direction of travel through an optically transparent crystal, the internal reflection element (IRE) (Fig. 1), into a catalyst film. A multiple-bounce IRE increases the intensity of such evanescent IR light reaching the sample, and hence improves the signal to noise ratio, important for samples exhibiting low absorbances, such as heterogeneous catalysts containing a small number of surface sites.12 ATR-IR spectra of a heterogeneous catalyst in the liquid phase can provide information on both soluble and adsorbed reactants, products and reactive intermediates. The detailed background and theory of ATR-IR spectroscopy has been recently reviewed.4,6,14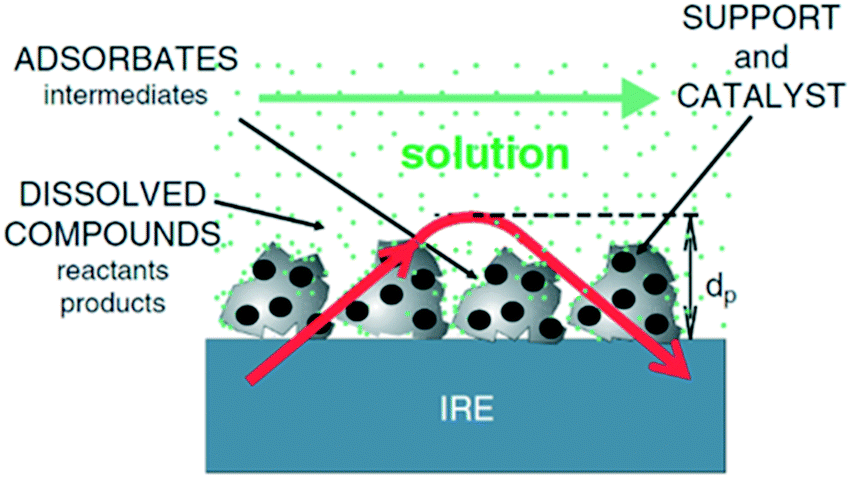 | ||
| Fig. 1 Schematic of ATR-IR spectroscopy at the solid–liquid interface; the red arrow is the evanescent light propagating through the crystal and catalyst film, reproduced from ref. 14 with permission from Elsevier, copyright 2006. | ||
Moreover, ATR-IR spectroscopy can probe the volume of sample very close to the IRE, and hence is valuable for investigating powder catalysts coatings. However, the catalyst particle size, packing and the coating thickness influences the relative signals from the catalyst surface and bulk media.16 Despite these complexities, ATR-IR spectroscopy can offer unique insight into competing catalytic reaction pathways.17
ATR-IR spectroscopy at the solid–liquid interface can however be challenging due to differences in the sensitivity of IR absorbing species and complexity of some reaction networks. Spectra collected at the solid–liquid interface comprise signals from both soluble and adsorbed species, including reactants, reaction intermediates, products and spectators. This array of species and resulting spectral complexity may necessitate additional experimental and/or computational investigations to discriminate between different species.4 Low conversions, arising from limitations in the thickness of catalyst films that can be uniformly deposited on IRE crystals, may also necessitate additional analytic strategies, such as modulation excitation spectroscopy in conjunction with phase-sensitive detection, to decouple weak periodically varying reaction signals from large static ones associated with the bulk media or catalytic spectators.18 Where multi-bounce IRE crystals are employed, varying concentration gradients can arise across the catalyst coatings, resulting in inconsistent surface chemistry and complicating analysis of the (spatially averaged) spectra. Changes in the reflectivity of liquids due to dissolved gas (e.g. H2, N2 or O2) and/or catalysts due to surface oxidation or reduction present additional challenges to spectral interpretation.4,14 The limit of detection of ATR-IR extends from a single monolayer (typically 10–20 Å of a surfactant or polymer, or a few tens of nanograms).19,20 Surface sensitivity is relatively poor since penetration depths into the coated catalyst and/or bulk media are on the order of the wavelength (up to a few microns). However, depth profiling is possible if the sample is suitably prepared and spectral parameters carefully chosen.21 Selected examples of successful applications of operando ATR-IR spectroscopy, and resulting mechanistic insight, are described in the following section.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibrations at 1582 cm−1, concomitant with the formation of formic acid, acetic acid and levulinic acid with C–O vibrations at around 1213, 1280, and 1172 cm−1 respectively. A vibration assigned to an aldehyde CO stretch was also observed at 1664 cm−1.
C vibrations at 1582 cm−1, concomitant with the formation of formic acid, acetic acid and levulinic acid with C–O vibrations at around 1213, 1280, and 1172 cm−1 respectively. A vibration assigned to an aldehyde CO stretch was also observed at 1664 cm−1.
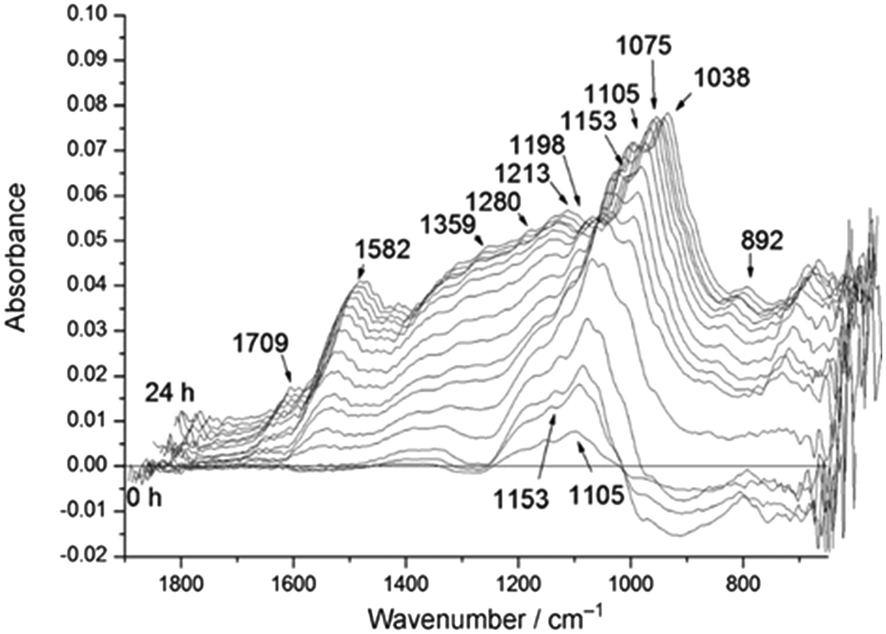 | ||
| Fig. 2 Time-resolved differential ATR-IR spectra during solid acid catalysed hydrolysis of cellulose in water, reproduced from ref. 22 with permission from Wiley-VCH, copyright 2012. | ||
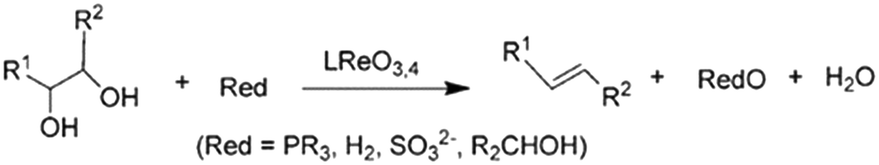 | ||
| Scheme 1 Reduction of glycols to olefins, reproduced from ref. 23 with permission from Wiley-VCH, copyright 2013. | ||
 | ||
| Fig. 3 Stirred batch reactor incorporating ATR-IR probe for in situ spectroscopy of liquid phase catalytic reductions, reproduced from ref. 23 with permission from Wiley-VCH, copyright 2013. | ||
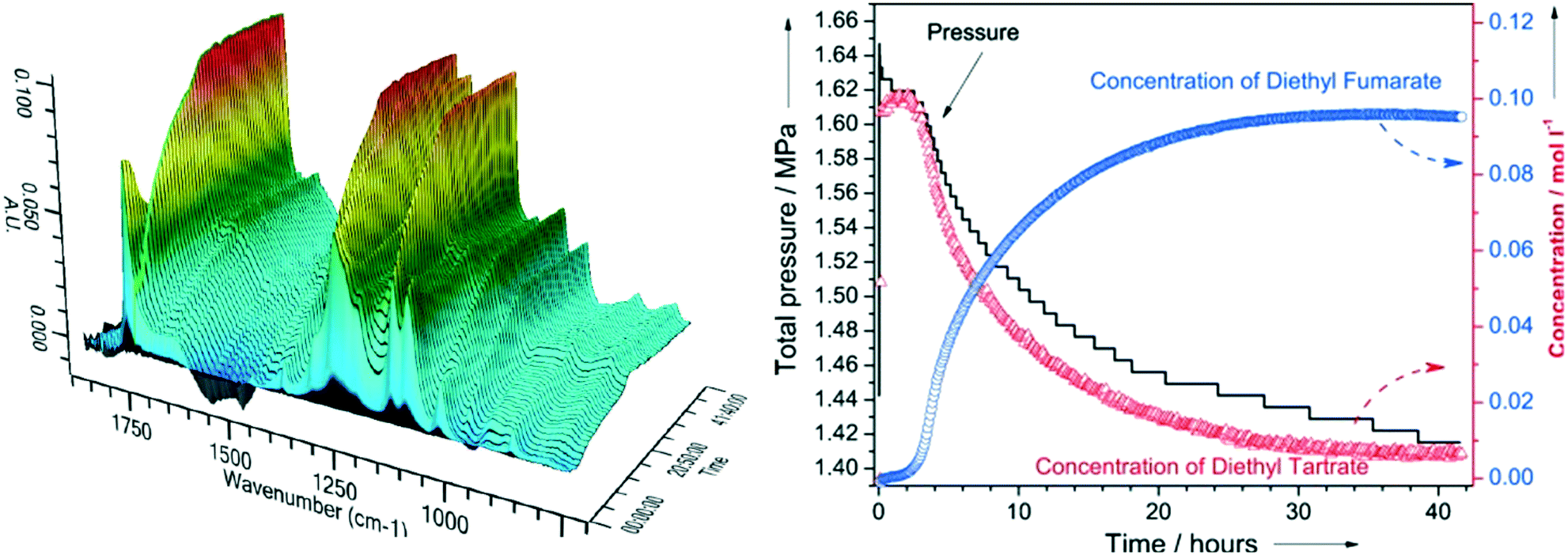 | ||
| Fig. 4 (left) In situ ATR-IR spectra during DODH of diethyl tartrate over ReOx–C at 150 °C; (right) corresponding reactant and product reaction profiles and H2 pressure, reproduced from ref. 23 with permission from Wiley-VCH, copyright 2013. | ||
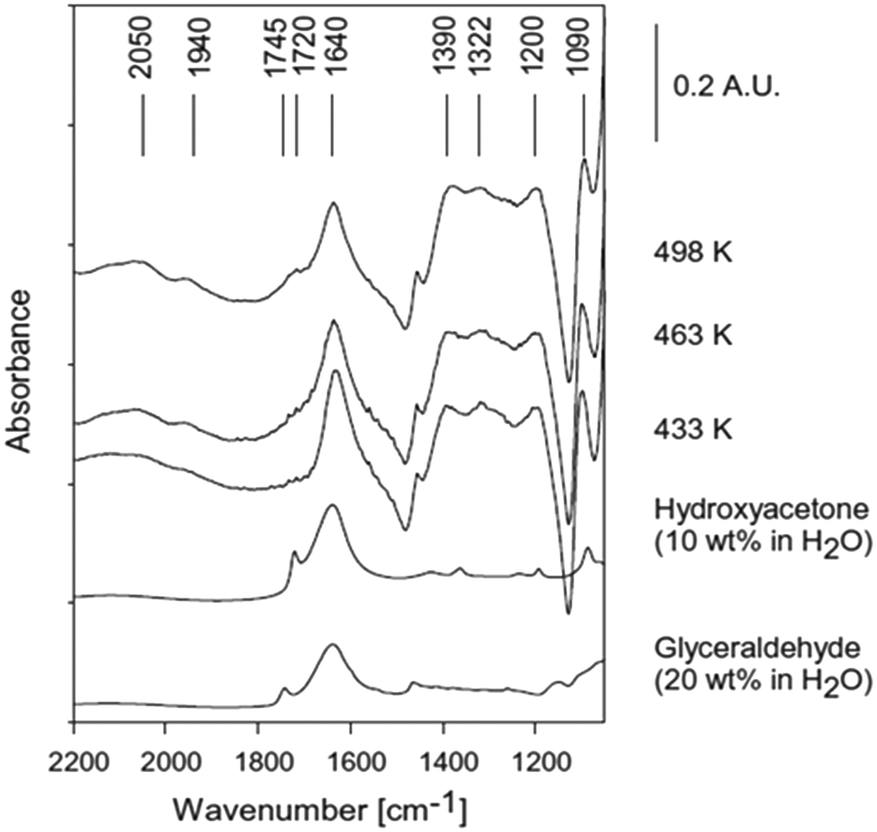 | ||
| Fig. 5 ATR-IR spectra during reforming of 20 wt% glycerol on 3 wt% Pt/Al2O3, reproduced from ref. 24 with permission from Elsevier, copyright 2013. | ||
Operando ATR-IR spectroscopy was also employed to study methanol reforming over Pt/Al2O3 at 423 K in the vapour (1.36 or 3.08 bar) or liquid (5.84 bar) phases.25 Spectra were recorded using a horizontal flow cell, wherein the catalyst-coated ATR crystal was sealed by a Viton O-ring to a Teflon-coated aluminum frame. Different methanol partial pressures resulted in significant differences in adsorbed CO. The surface coverage of CO increased by 35% when switching from liquid to vapour reaction conditions, suggesting that methanol decomposition is more kinetically limiting in the liquid than vapour phase reaction. ATR-IR also implicated hydrogen co-adsorbed with CO on the Pt surface, evidenced by a shift in the linearly bound CO band. The same reactor configuration and catalyst was investigated for water–gas shift (WGS) to understand the effects of CO, H2 and H2O partial pressures in the vapour or liquid phase on the reaction mechanism.25 In the vapour, Pt surfaces are covered by adsorbed CO, and hence reactions were zero order in CO pressure. A negative order was observed with respect to H2 due to surface saturation by H adatoms. A fractional positive order with respect to water pressure confirmed the non-equilibrated dissociation of H2O over Pt. Liquid phase studies revealed a slight decrease in the CO coverage for the same CO partial pressure, however the catalytic performance for the WGS reaction was comparable under liquid and vapour phase conditions. Pt/Al2O3 catalysed APR of ethylene glycol was similarly studied in a continuous flow, fixed bed reactor (Fig. 6) over a ZnSe IRE spray-coated with catalyst and placed inside a tunnel cell connected to a gas manifold.26 The alumina support is unstable under most APR conditions, phase transitioning to boehmite. This transition occurred under more mild conditions than expected, resulting in irreversible catalyst deactivation. The resulting alumina restructuring escalated the rate of formation of hydrogen per surface Pt atom, with the enhancement correlating with the increase in concentration of surface OH groups and enhanced oxidation of Pt.
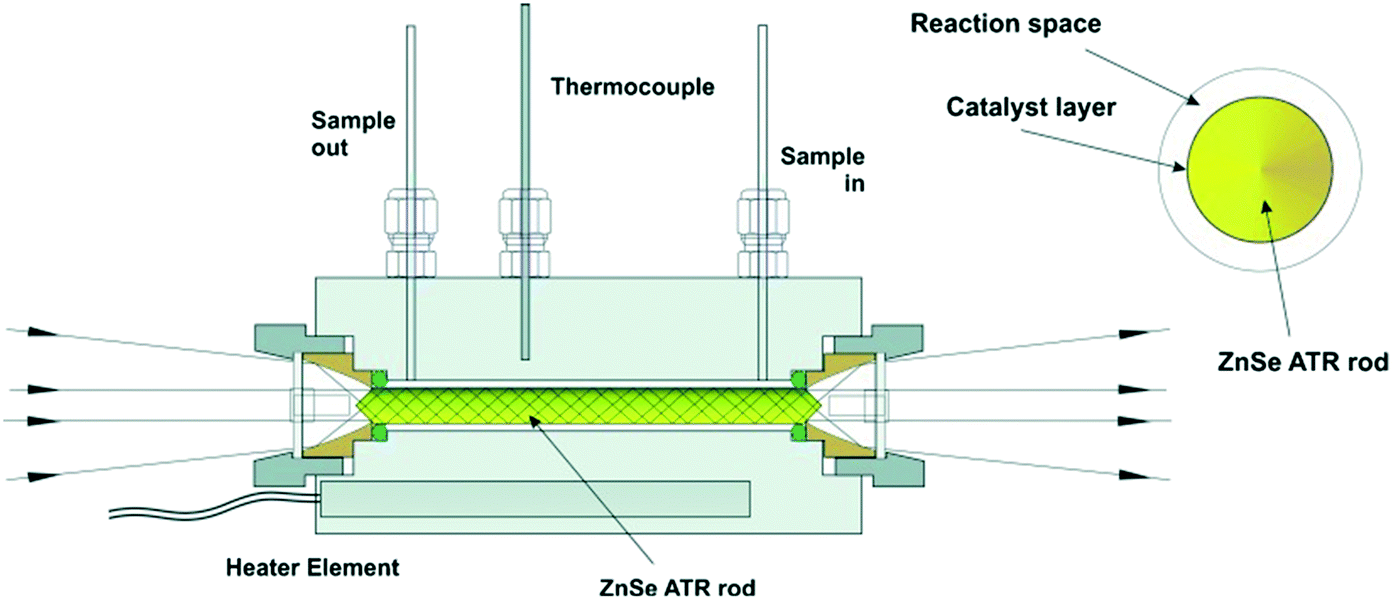 | ||
| Fig. 6 ATR-IR tunnel cell and cross-section of the catalyst coated ZnSe IRE rod, reproduced from ref. 26 with permission from Wiley-VCH, copyright 2013. | ||
Pretreatment of catalyst films deposited on IRE crystals is critical to the success of liquid phase in situ/operando ATR-IR studies.27 Such pretreatments typically comprise two steps: (i) flowing only the solvent (i.e. without dissolved reactants) for up to several hours to ensure mechanical stability of the catalyst film and the removal of any weakly bound adsorbates; and if required (ii) addition of dissolved H2 or O2 to the solvent stream to respectively remove oxidised or carbonaceous surface residues.13,28,29 Copeland et al. studied the effect of pretreatment on Pt/γ-Al2O3 catalysts for glycerol APR,27 identifying alternating flows of H2 and O2 saturated water as the most effective cleaning protocol. Subsequent ATR-IR spectroscopy showed that Pt/γ-Al2O3 activates glycerol at room temperature in water to form adsorbed CO. Optimal rates of CO formation correlated with the concentration of available surface Pt sites, some of which exhibited unique high reactivity. The highly reactive Pt sites preferentially adsorbed hydrogen and bridge-bound CO from glycerol reforming and directed the removal of bridging CO species via the water–gas shift reaction.
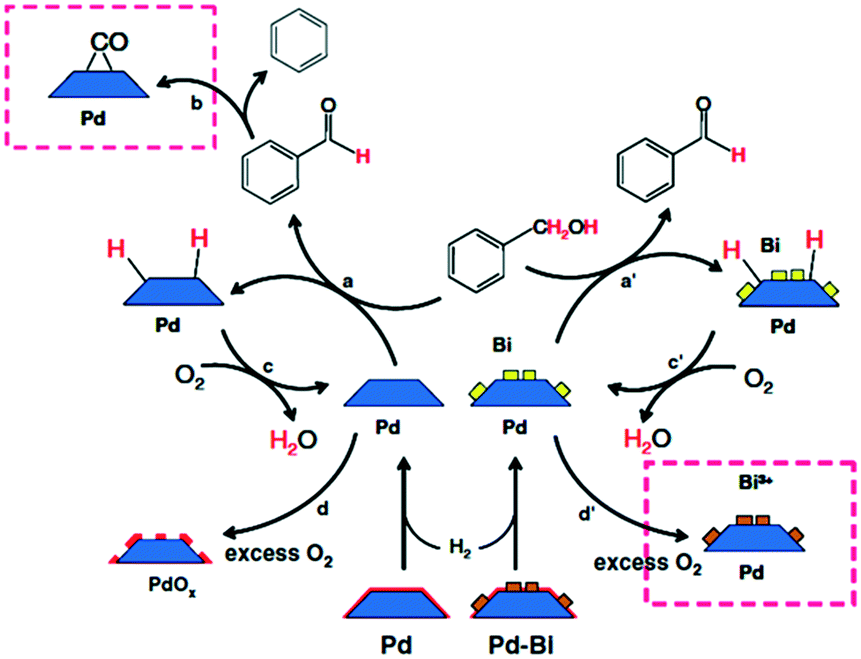 | ||
| Scheme 2 Proposed mechanism of benzyl alcohol oxidation evinced by ATR-IR spectroscopy, reproduced from ref. 15 with permission from Springer Nature, copyright 2009. | ||
 | ||
| Fig. 7 (a)–(h) In situ ATR-IR spectra of Pt/Al2O3 in cyclohexane during CO2/H2 adsorption at 40 °C and (i) following exposure to N2-saturated cyclohexane, reproduced from ref. 31 with permission from the Royal Society of Chemistry, copyright 2002. | ||
CO2 was proposed to adsorb on Al2O3, to form carbonate-like species, which subsequently react with either dissociatively adsorbed H2 over Pt (Fig. 8a) or an oxygen defect (Fig. 8b) to form CO at the metal–oxide interface; reactively-formed CO was thus inferred as a selective probe of such interfacial sites.
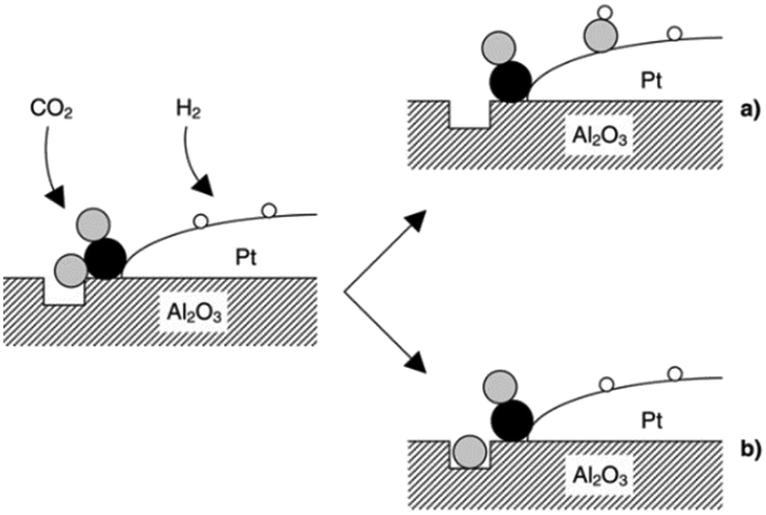 | ||
| Fig. 8 Schematic of CO2 reduction over a Pt/Al2O3 model catalyst by the reaction of an oxygen atom with either (a) adsorbed hydrogen to form a surface hydroxyl, or (b) a defect in the oxide surface. Black, grey and white balls depict carbon, oxygen and hydrogen atoms, respectively, reproduced from ref. 31 with permission from the Royal Society of Chemistry, copyright 2002. | ||
A valuable approach to detailed insight into supported noble metal catalysts is achieved via investigations of small and simple molecule conversion, such as CO. Lefferts et al. investigated the CO adsorption and oxidation on noble metal catalysts such as Pt/Al2O3 and Pd/Al2O3 with ATR-IR in both the gas and liquid phase.28 Parameters such as the effect of water on adsorbed CO and the effect of pH on CO adsorption and oxidation were studied. Water and pH impart significant effects on the adsorption and oxidation of CO over Pt/Al2O3 and Pd/Al2O3. Furthermore, water also affected the metal particle potential and the adsorbed CO molecule, resulting in higher oxidation rates when conducted in liquid water compared to gas-phase conditions. pH also influenced the metal particle potential which in turn effected the observed oxidation rates.
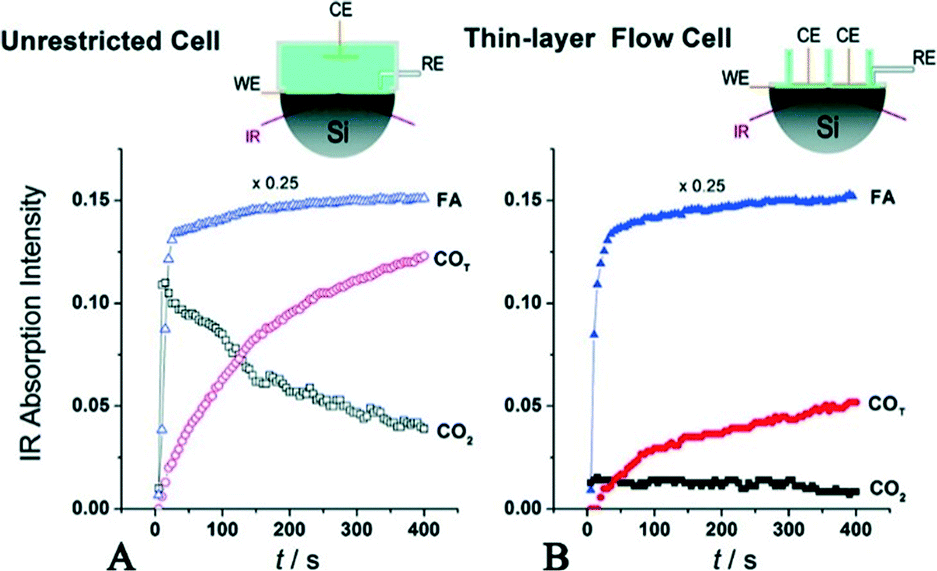 | ||
| Fig. 9 Evolution of IR bands associated with near-surface CO2 product, interfacial FA molecules in the solution phase and reactively-formed adsorbed COT species on the Pd surface. In (A), FA was added to the regular unrestricted ATR cell a fixed amount of HClO4 electrolyte. In (B), FA-containing solution was pumped into the flow cell to replace the supporting base electrolyte, reproduced from ref. 32 with permission from the American Chemical Society, copyright 2011. | ||
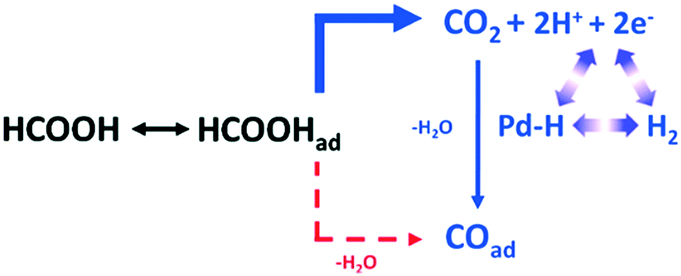 | ||
| Scheme 3 Proposed reaction pathway from HCOOH to COad over a Pd electrode via CO2 as a reactive intermediate, reproduced from ref. 32 with permission from the American Chemical Society, copyright 2011. | ||
This in situ mechanistic study may inform the development of new Pd based catalysts for hydrogen energy utilisation.
Collins et al. constructed an ATR-IR microreactor cell using CO chemisorption over a thin platinum film to validate the design.33 The cell comprised a jacketed liquid top plate for temperature control, and inlet and outlet ports to feed reagents and collect the resulting product stream. The IRE was fitted into the cell body, incorporating mirrors to focus the IR beam to/from the IRE crystal; these components were sealed by a Viton O-ring, and the resulting cavity between the body and top determined by the O-ring dimensions (Fig. 10 left).
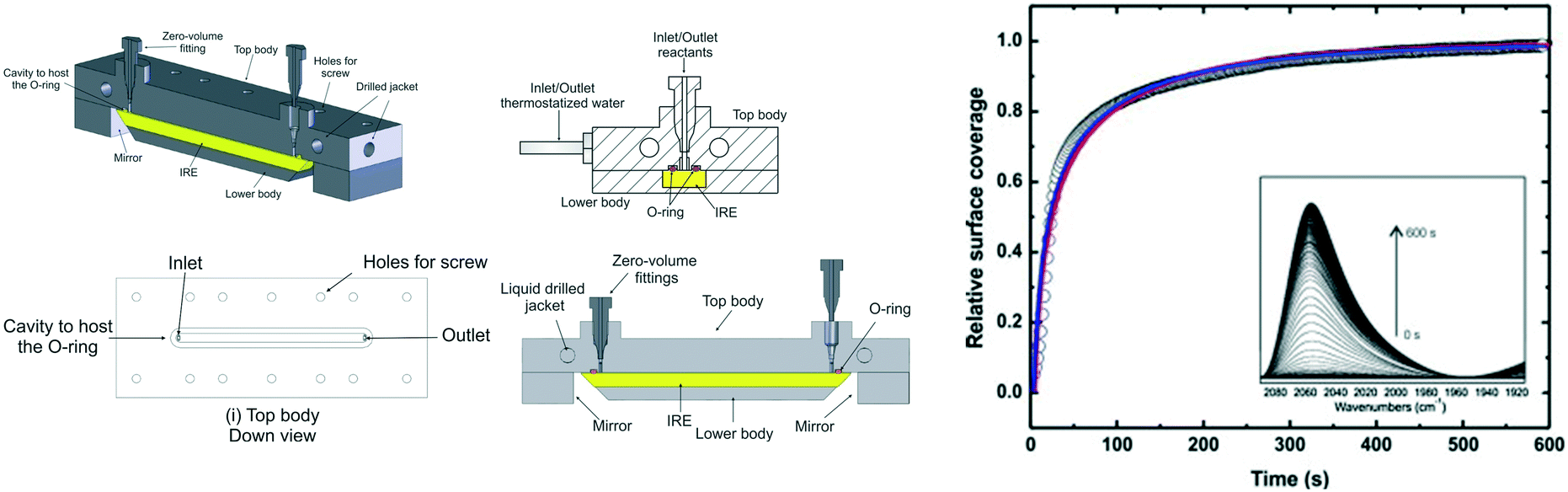 | ||
| Fig. 10 (left) Schematic of ATR flow cell, and (right) time-dependent normalised IR signal of Pt–CO band at 2048 cm−1. Lines are best fits to the data assuming a convection-diffusion model, reproduced from ref. 33 with permission from Elsevier, copyright 2014. | ||
2.2 Raman spectroscopy
The high sensitivity of IR spectroscopy to changes in dipole moment result in a strong signal from water, in contrast to Raman spectroscopy. This difference favours the use of Raman spectroscopy for in situ/operando studies of aqueous phase catalysis.34,35 Conventional Raman, UV Raman and surface/tip-enhanced Raman spectroscopies (SERS and TERS) have been widely used to study heterogeneously catalysed reactions as illustrated in the following case studies.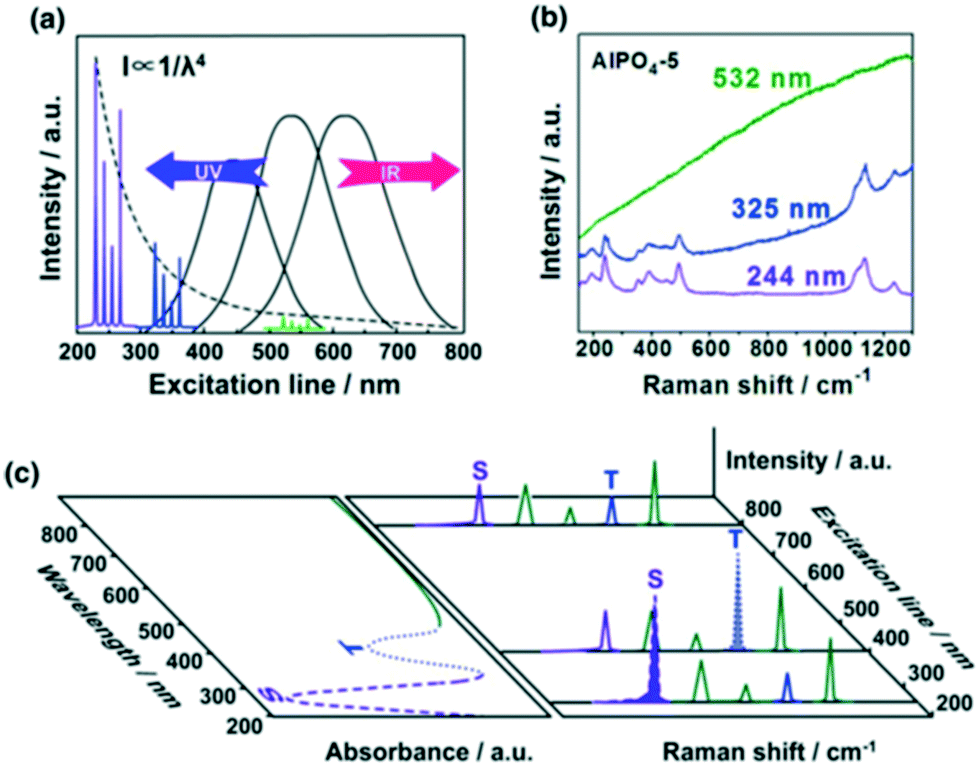 | ||
| Fig. 11 a) UV Raman spectroscopy avoiding fluorescence interference while increasing Raman scattering intensity; b) Raman spectra of AlPO4-5 excited at 532, 325, and 244 nm; c) a schematic description of UV resonance Raman spectroscopy, the Raman bands attributed to S and T components can be selectively enhanced by shifting the excitation laser lines towards their UV-visible absorption bands, reproduced from ref. 37 with permission from Springer Nature, copyright 2014. | ||
Since electronic transitions of chemical compounds are also usually excited by UV irradiation, resonance Raman effects can enhance spectral quality by several orders of magnitude (Fig. 11c).
2.2.2.1 Zeolite synthesis. Elucidating mechanisms of catalytic synthesis is challenging. Fan et al. studied the synthesis of transition metal containing microporous and mesoporous materials using in situ UV Raman spectroscopy.40 The evolution of precursors and intermediates was followed during the synthesis of zeolite X using a hydrothermal reactor specially designed for coupling to a UV Raman spectrometer (Fig. 12a1). A silicon rubber-sealed lens built into the reactor lid enabled laser focusing on to samples, enhancing the Raman signal by 3–4 times that achievable with a plane window. Two sample holders, of different depths, allowed the focal point to be shifted between the liquid phase (Fig. 12a2) and solid–liquid interface (Figure 12a3). Initial spectra (Fig. 12b) showed an amorphous solid phase in equilibrium with monomeric silicate species in the liquid phase (774 and 920 cm−1 bands). Four-membered rings and a small amount of branched derivatives (575 cm−1 band) were observed during in the early stage of nucleation, whose subsequent disappearance coincided with the emergence of polycyclic structures and zeolite seed crystals (Fig. 12c); these branched derivatives are believed to be a key intermediate in zeolite X formation (Fig. 12d). Such insight could guide the rational design and solution-phase synthesis of porous catalysts.
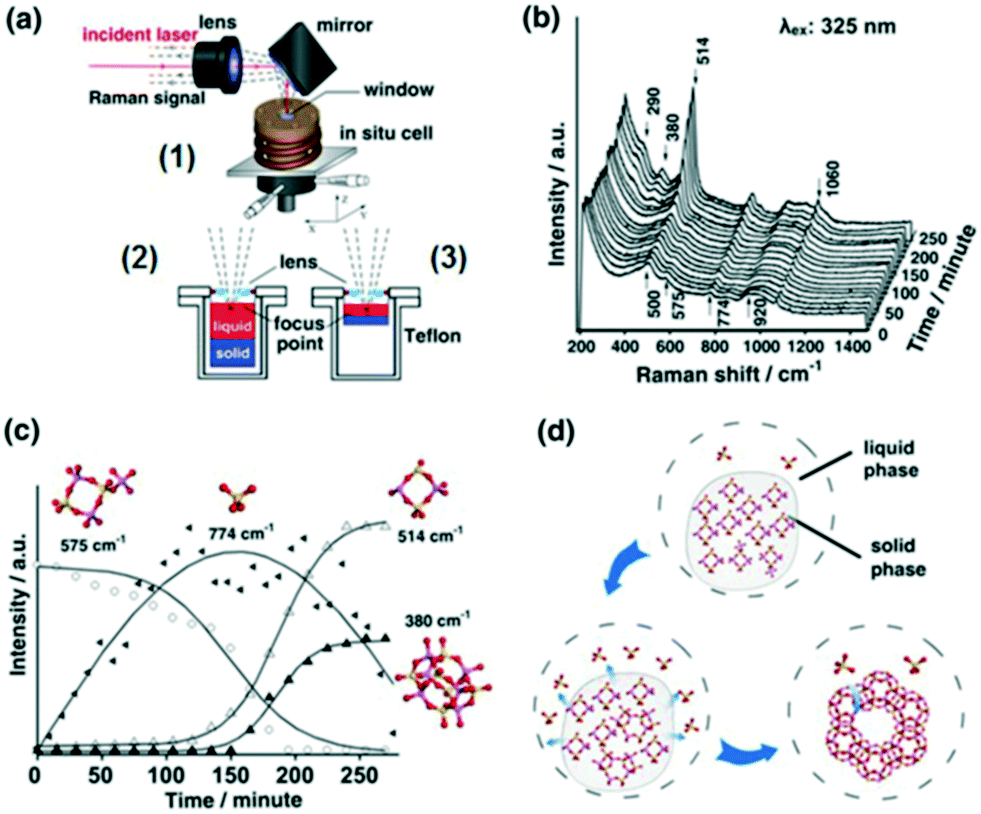 | ||
| Fig. 12 (a) In situ Raman reactor (1) for zeolite crystallisation offering varying focal points (2 and 3); (b) UV Raman spectra (325 nm excitation) during zeolite X synthesis at 373 K; (c) time-dependent evolution of molecular species; (d) proposed mechanism for zeolite X formation, adapted from ref. 37 with permission from Spring Nature, copyright 2015. | ||
2.2.2.2 Furfuryl alcohol polymerization. The effect of ethanol on the acid-catalysed polymerisation of furfuryl alcohol (FA) has been studied by time-resolved UV Raman spectroscopy41 wherein UV excitation circumvented a strong fluorescence background (Fig. 13). For pure furfural (FS1 and FS4), the 1504 cm−1 band was characteristic of the symmetric C
C stretch in the furan ring of the monomer, whereas that at 1655 cm−1 was indicative of a CC stretch in the FA polymer. The intensity ratio of these bands, I1654/1504, was subsequently employed to obtain quantitative information on the extent of polymerisation in different solvents (Fig. 13). Increasing the ethanol concentration (FES7-FES10), or switching to butanol, inhibited room temperature polymerisation. The time-dependence of 1/I1504 suggested that polymerisation followed second-order kinetics (Fig. 14).
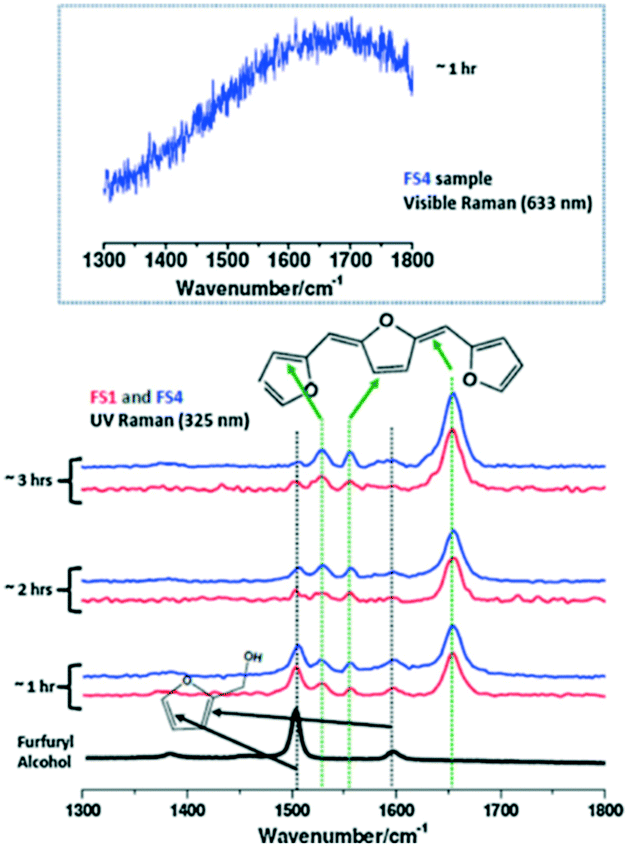 | ||
| Fig. 13 (left) Raman spectra of furfuryl alcohol (FS1 and FS4) during polymerisation under UV excitation (325 nm) at room temperature and ambient pressure, reproduced from ref. 41 with permission from Elsevier, copyright 2013. | ||
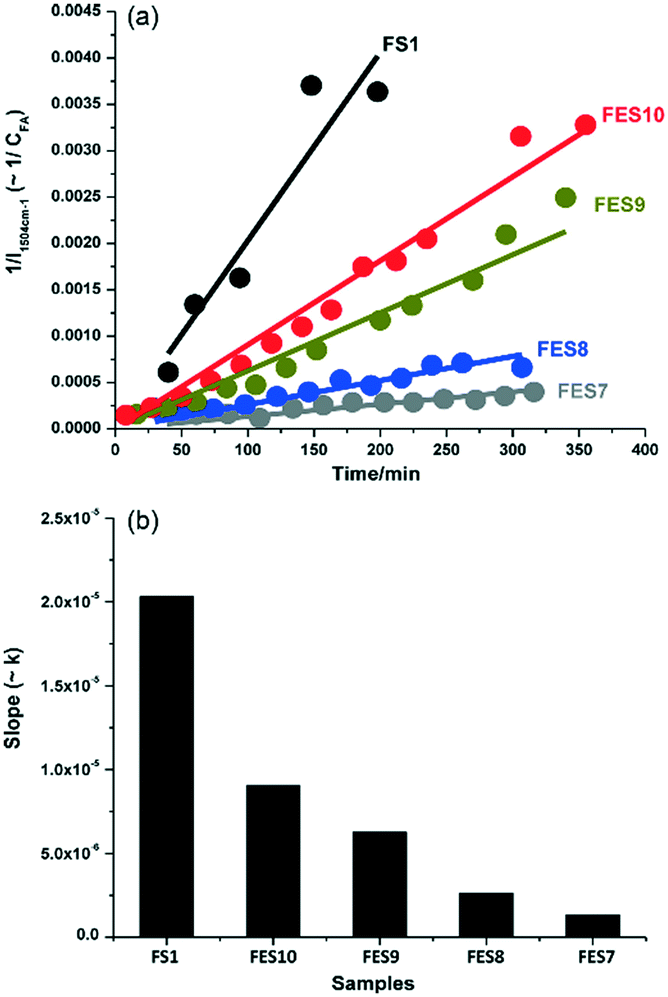 | ||
| Fig. 14 Effect of ethanol concentration on rate of furfuryl alcohol polymerisation versus FS1: (a) 1/I1504cm−1versus reaction time; and (b) corresponding second-order rate constant, reproduced from ref. 41 with permission from Elsevier, copyright 2013. | ||
2.2.3.1 Glycerol oxidation over gold nanoshell catalysts. SERS was applied to study the room-temperature oxidation of glycerol in water and associated role of pH.45 Au nanoshells were used as both an active oxidation catalyst and SERS enabling substrate, and comprised 100–200 nm silica particles encapsulated by a shell of Au metal several nm thick, which provided SERS enhancements exceeding those of a roughened metal surface (108–109vs. 104). Glycerol oxidation in deoxygenated solutions at pH 11 resulted in SERS features between 800 and 1600 cm−1, with sharp bands ∼1000 and 1200 cm−1 assigned to oxygen-derived species and that at 1500 cm−1 to physisorbed O2 (Fig. 15a); vibrational bands for the C
O bond of carboxylic acids were not observed. Introducing high concentrations of dissolved O2 led to the appearance of a weak peak between 1900–2100 cm−1, ascribed to the C–O stretch of chemisorbed CO (Fig. 15c).
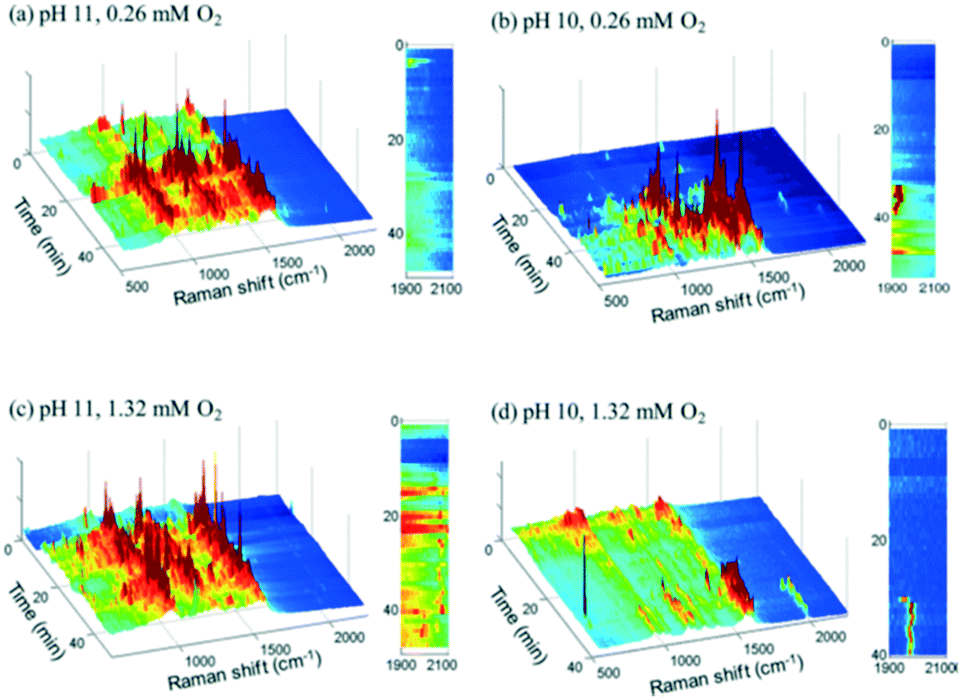 | ||
| Fig. 15 Time-resolved Au nanoshell SER spectra of 1.3 mM glycerol solutions (a) at pH 11 with 0.26 mM O2, (b) at pH 10 with 0.26 mM O2, (c) at pH 11 with 1.32 mM O2, and (d) at pH 10 with 1.32 mM O2, reproduced from ref. 45 with permission from the American Chemical Society, copyright 2013. | ||
SERS indicates that glycerol and glycerolate species do not bind to the catalyst surface in the absence of oxygen, but rather participates in an Eley–Rideal mechanism in which near surface glycerolate reacts directly with adsorbed O2 or surface bound hydroxyls to form carboxylate species in solution. In addition to deprotonating glycerol, surface hydroxyls increased the surface negative charge on the Au shell, which may enhance O2 adsorption/activation explaining the high activity of Au-derived catalysts their dependence on basic reaction conditions.
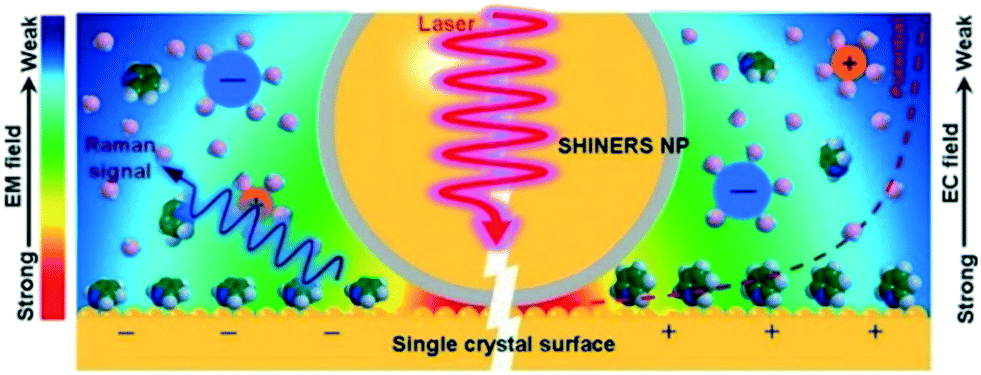 | ||
| Fig. 16 Operating principle of SHINERS on a single-crystal-electrode surface, reproduced from ref. 48 with permission from the American Chemical Society, copyright 2019. | ||
Formation of surface oxides often hinders electrocatalyst performance, and hence molecular insight into electrochemical oxidation can aid the design of improved catalyst systems. The electrochemical oxygen reduction reaction (ORR) over Pt-based catalyst surfaces is an important challenge for fuel cells research, and the SHINERS technique has been utilised to study this over low-index Pt(hkl) surfaces.49Fig. 17a shows SHINERS results during the ORR on a Pt(111) surface as a function of applied potential under acidic conditions. Only one Raman peak was observed at 933 cm−1 at high potentials, assigned to the symmetric-stretching mode of the perchlorate ion electrolyte, however a second band appeared at 732 cm−1 attributed to adsorbed 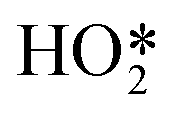 on decreasing the potential to 0.8 V. Under identical conditions, Pt(110) and Pt(100) surfaces exhibited the perchlorate band and Raman features at 1030 and 1081 cm−1, but not the
on decreasing the potential to 0.8 V. Under identical conditions, Pt(110) and Pt(100) surfaces exhibited the perchlorate band and Raman features at 1030 and 1081 cm−1, but not the 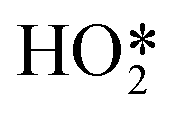 feature (Fig. 17b), and under alkaline conditions only a single Raman peak at 1150 cm−1 attributed to superoxide O2− anions (Fig. 17c). In conjunction with quantum chemical DFT calculations, the SHINER technique demonstrated that Pt(111) electrodes stabilise
feature (Fig. 17b), and under alkaline conditions only a single Raman peak at 1150 cm−1 attributed to superoxide O2− anions (Fig. 17c). In conjunction with quantum chemical DFT calculations, the SHINER technique demonstrated that Pt(111) electrodes stabilise 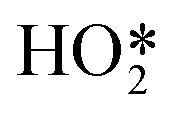 whereas Pt(110) and Pt(100) electrodes favour OH*, however under alkaline conditions the ORR reaction occurs over all three surfaces via surface O2−.
whereas Pt(110) and Pt(100) electrodes favour OH*, however under alkaline conditions the ORR reaction occurs over all three surfaces via surface O2−.
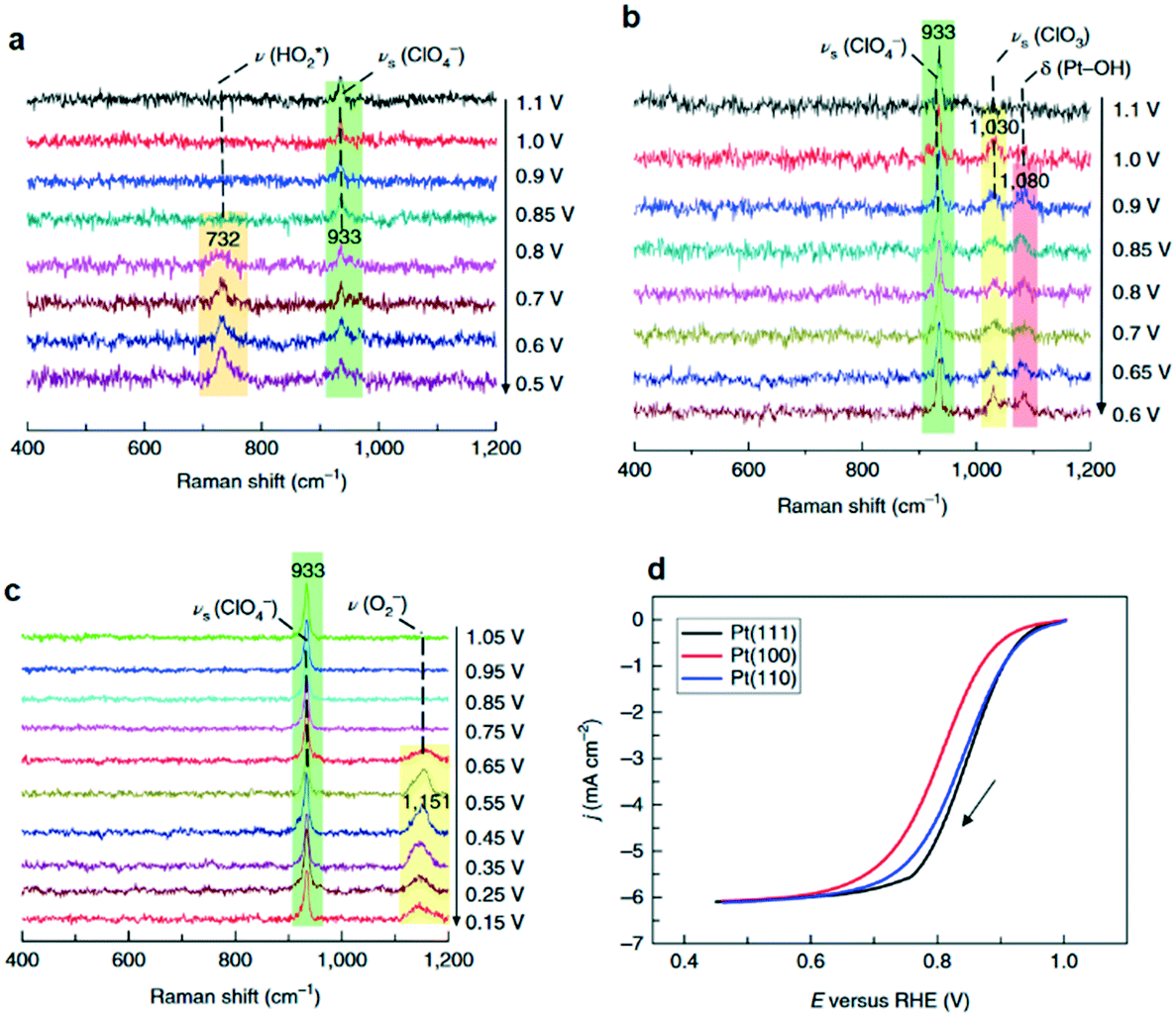 | ||
| Fig. 17 Electrochemical SHINERS spectra of ORR process over (a) Pt(111) and (b) Pt(110) electrodes in 0.1 M HClO4 solution saturated with O2, and (c) a Pt(110) electrode in 0.1 M NaClO4 solution (pH ∼ 10.3) saturated with O2. (d) Polarization curves of the ORR reaction on Pt(hkl) rotating-disk electrodes in saturated 0.1 M HClO4 solution, adapted from ref. 49 with permission from Springer Nature, copyright 2018. | ||
SHINERS was recently adapted to enable operando SERS monitoring of electrochemical reactions, extending its application to surface reactions at heterogeneous interfaces. Kim et al. placed gold microshells, electrically isolated from an underlying flat Pt electrode by a layers of 11-mercaptoundecanol (MCU) grafted on the microshell surface to perform SERS.38 The MCU buffer layer facilitates numerous hot spots, without itself participating in the electrochemical reaction (Fig. 18). Such MCU modified gold microshells were used to obtain operando Raman spectra during the electrochemical reduction of 4-nitrobenzenethiol (4-NBT) over a flat polycrystalline Pt electrode (Fig. 19). Unreduced 4-NBT was indicated by bands at 1080, 1331 (–NO2 symmetric stretch) and 1570 cm−1 (CC stretching mode). At negative potentials the NO2 band diminished and the CC stretch shifted to 1587 cm−1 consistent with electrochemical reduction to 4-aminobenzenethiol. Modified gold microshells offer the opportunity to observe interfacial reactions by SERS without restrictions on the electrode structure or material.
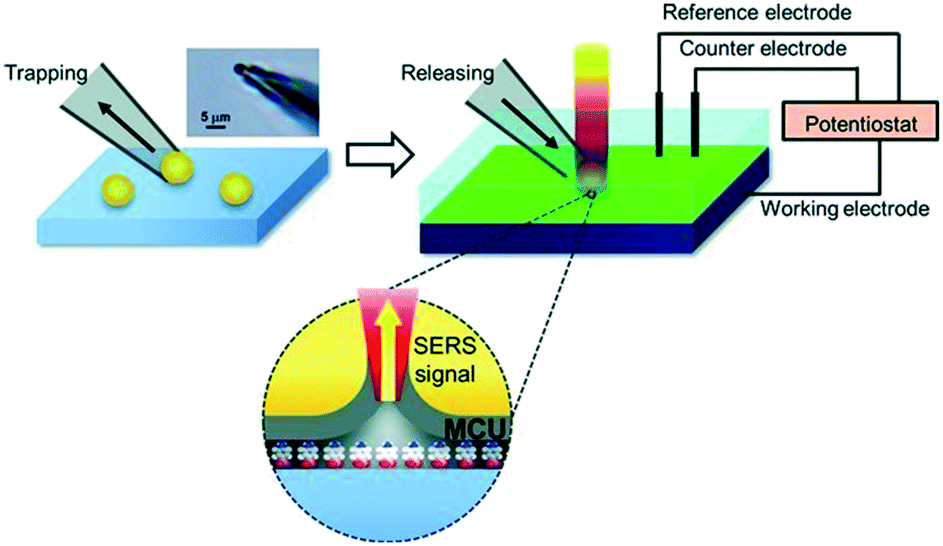 | ||
| Fig. 18 Schematic showing trapping and placement of individual 11-mercaptoundecanol (MCU) modified gold microshells on a Pt electrode. Upper left: Optical microscopic image of gold micro-shell trapped at the end of a micropipette. Upper right: Electrochemical system with SERS integration; grey layer in the lower part represents MCU self-assembled monolayer on the gold microshell in contact with nitrobenzenethiol molecules, reproduced from ref. 38 with permission from Wiley-VCH, copyright 2013. | ||
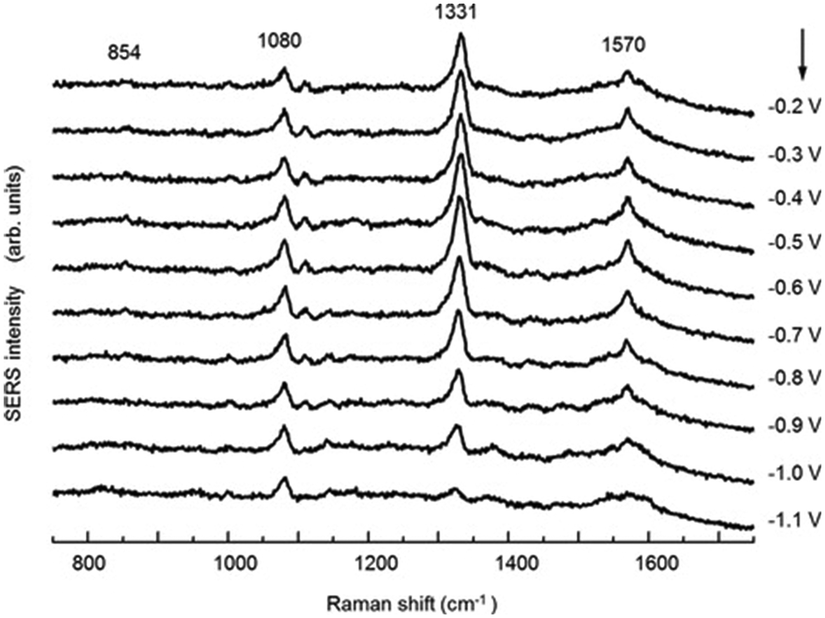 | ||
| Fig. 19 Potential-dependent SERS spectra of a 4-NBT of adsorbed on a flat Pt electrode in 0.1 M NaOH, reproduced from ref. 38 with permission from Wiley-VCH, copyright 2013. | ||
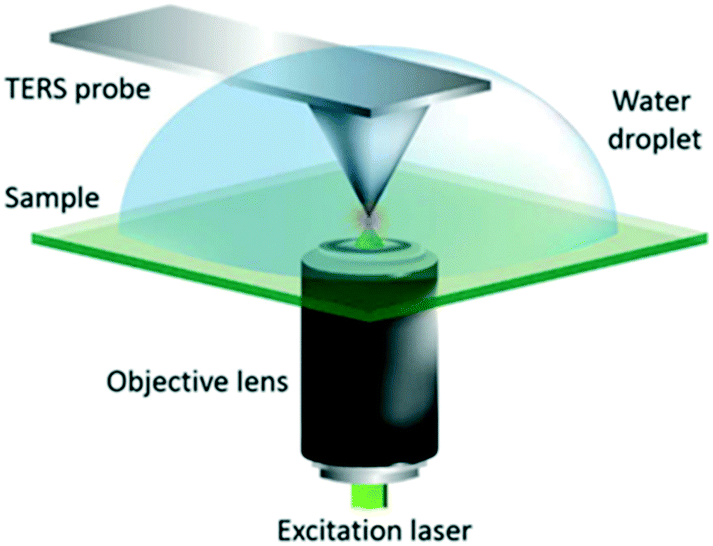 | ||
| Fig. 20 Schematic diagram of the AFM-TERS system employed. For TERS measurements in liquid, a water droplet was placed on the sample and measurements were performed with the TERS probe fully immersed, reproduced from ref. 53 with permission from the Royal Society of Chemistry, copyright 2018. | ||
2.2.5.1 In situ photocatalytic oxidation of p-aminothiophenol. Weckhuysen and co-workers recently developed a novel atomic force microscopy (AFM)-TERS probe wherein the multilayer metal tip coating was protected by an ultrathin layer of zirconia.54 This design overcomes TERS limitations such as a short tip lifetime, chemical inertness, and instability in a liquid environment. The zirconia coated (AFM)-TERS probe was used to map the photocatalytic oxidation of a p-aminothiophenol (pATP) self-assembled monolayer to p,p′-dimercaptoazobenzene (DMAB) across a heterogeneous metal surface in water. TERS measurements were performed inside a water droplet on the catalyst surface, which interestingly resulted in a 210 times stronger signal compared to air measurement (Fig. 21). Additionally, their design extended the lifetime of the probes over 850 times while rendering them chemically inert for the investigation of catalytic reactions in the liquid phase. Such improvements in probe lifetime and structural and chemical stability expand the potential application of AFM-TERS for nanoscale chemical characterization of catalysts under reactive environments.
 | ||
| Fig. 21 Maps of pATP → DMAB at the TERS probe apex obtained using the intensity of the 1437 cm−1 (νNN) DMAB Raman band measured from the pATP self-assembled monolayer on a Ag substrate in (a) air and (b) water. Corresponding TERS (red) and SERS (blue) spectra measured at the position of maximum DMAB signal at the TERS probe apex in (c) panel (a) and (d) panel (b), reproduced from ref. 54 with permission from the American Chemical Society, copyright 2019. | ||
2.2.5.2 Electrochemical tip-enhanced Raman spectroscopy. Cobalt-derived molecular catalysts are promising candidates for electrochemical reactions such as the ORR. Electrochemical (EC)-TERS was employed to investigate the catalytic activity of cobalt phthalocyanine (CoPc) over Au(111) as a function of substrate potential and excitation wavelength (Fig. 22).55 The TERS signal was sensitive to both potential and excitation wavelength: under 632.8 nm excitation the Raman intensity was proportional to potential; under 700 nm excitation an inverse relationship was observed; and under 720 nm excitation the intensity was independent of potential. For all excitation wavelengths, no TERS peaks were apparent at 0.1 V. This behaviour was interpreted in terms of CoPc adsorption at the solid–liquid interface. The surface charge density increases with negative potential, weakening the molecule–substrate interaction and inducing an ordered-to-diffuse phase transition (Fig. 22b). During the ORR, the electronic transition of CoPc redshifted from 657 to 700 nm which changes the resonance conditions at different excitation wavelengths. These observations supported a molecular understanding of CoPc catalytic behaviour during the ORR.55
 | ||
| Fig. 22 (a) EC-TERS study of CoPc on Au(111) acquired at different potentials with three excitation wavelengths in 0.1 M HClO4. (b) Sequential STM images of the CoPc adlayer adsorbed on Au(111) at different substrate potentials in 0.1 M HClO4, reproduced from ref. 55 with permission from the American Chemical Society, copyright 2019. | ||
2.3 X-ray absorption spectroscopy
X-ray absorption spectroscopy (XAS) can provide invaluable insight into both the electronic and local structural properties of catalytically active species through the excitation and subsequent scattering of core-level electrons.56 The measurement proceeds through the photoexcitation of electrons most commonly from 1s (K-edge) or 2s/2p (L-edges) into either unoccupied atomic orbitals below or above the Fermi level, or into the continuum at energies significantly above the adsorption edge. This results in two distinct analysis regions associated with X-ray absorption near edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS). XANES, spectra typically extend from ∼50 eV before the adsorption edge to ∼100 eV above it and provide information on molecular geometry (pre-edge features) and oxidation state (edge position and intensity) and is often used for chemical fingerprinting against reference materials. EXAFS extends from ∼100 eV above the adsorption edge to ∼1000 eV (depending on the element and core-level), and exhibits features arising from scattering interactions between ejected photoelectrons and neighbouring atoms which provide local structural information (coordination number, neighbouring elements, bond geometry and distances, and disorder). Photon-in/photon-out techniques facilitate in situ analysis of catalysts (particularly those containing transition metals interrogable by hard X-rays i.e. 5–20 keV) under real-world environments, in contrast to e.g. X-ray photoelectron spectroscopy (XPS), wherein vacuum or low pressures are necessary,57 due to the relatively weak attenuation of X-rays versus low energy electrons. However, although the penetrating nature of (particularly hard) X-rays is beneficial for investigating catalysts within fluids, and active sites at buried interfaces or within porous architectures, a caveat is that transmission and fluorescence XAS detection are not surface sensitive, indeed spectral contributions may be a summation over several thousand nanometers of material. Analysis of highly dispersed nanoparticles (a few nms diameter), clusters and single sites offers an indirect means to impart surface sensitivity, through increasing the proportion of surface atoms in the catalytically phase. Alternatively, X-ray excited Auger and secondary scattered electrons can provide direct surface sensitive information through total electron yield (TEY) detection.58 TEY requires either the detection of such electrons either near to the surface (prior to their absorption by surrounding media, necessitating vacuo/near ambient pressure configurations59), or measurement of the compensating current by a picoammeter wherein the catalyst is part of an electrical circuit. In both configurations TEY gives spectral information from the first few nanometres (≤3 nm) of a catalyst surface due to the short electron escape depth.60 Soft X-rays (∼0.1 to 3 keV) have also been used to study catalyst surfaces, allowing K-edge measurements of light elements e.g. C, O and N (common reactants/products) and S, P, Cl (common active site poisons), and L and M edges of 1st and 2nd row transition metals respectively.61 As with hard X-rays, the sampling depth for X-ray detection depends on the photon mean-free path, which for soft X-rays is ca. 100 nm62 necessitating thin samples, on the order of tens of nanometers, and gas environments.59 Historically, such measurements have been the domain of synchrotron radiation sources, in part due to their provision of tunable and high brilliance X-ray photon energies,63 however, laboratory XAS instruments have enjoyed a recent resurgence driven by the development of commercial systems exploiting bremsstrahlung radiation from modern X-ray tubes.64,65In contrast, operando XAS studies of liquid phase catalysts are rarer, in part reflecting stronger X-ray absorption by the reaction media, especially for halogenated solvents, but also due to added complexity in the reactor design. Nevertheless, valuable insight has been obtained for example, the nature of catalytically active sites and deactivation mechanisms in the liquid phase selective aerobic oxidation of alcohols over Pd catalysts.79,80 Although such measurements can be conducted at ambient pressure, thereby simplifying the experimental set-up and use of standard commercial or in-house reaction cells. In contrast, selective hydrogenations typically require elevated hydrogen pressure, and hence bespoke autoclaves in which high-pressure reactions can be conducted in concert with the collection of X-ray spectra are required.81 Additional engineering challenges for liquid phase XAS measurements include interfacing multiple techniques to mirror the gas phase experiments described above, and rapidly changing the reaction environment to permit modulation excitation experiments. Strong attenuation by the reaction media and reactor windows is also particularly problematic for catalysts requiring soft(er) X-rays. Microfluidics82 and/or the development of novel window materials with high X-ray transparency such as graphene and beryllium83 are possible solutions, and will benefit from increases in brilliance, through the roll-out of 4th generation synchrotron sources,84 and detector sensitivity, and further improvements in X-ray optics.85 Together these will afford greater sensitivity for active species and promotors at realistic concentrations (ppm to ppb levels), and improved temporal and spatial resolutions moving to sub-milli/microseconds and sub-μm respectively; such progress is necessary to elucidate quantitative structure–reactivity relations, map active site/promoter/poison distributions, and evaluate active site leaching/migration.
Examples of liquid phase operando XAS will be discussed to highlight the current state-of-the-art and inspire future investigations particularly in renewable energy and sustainable chemistry.
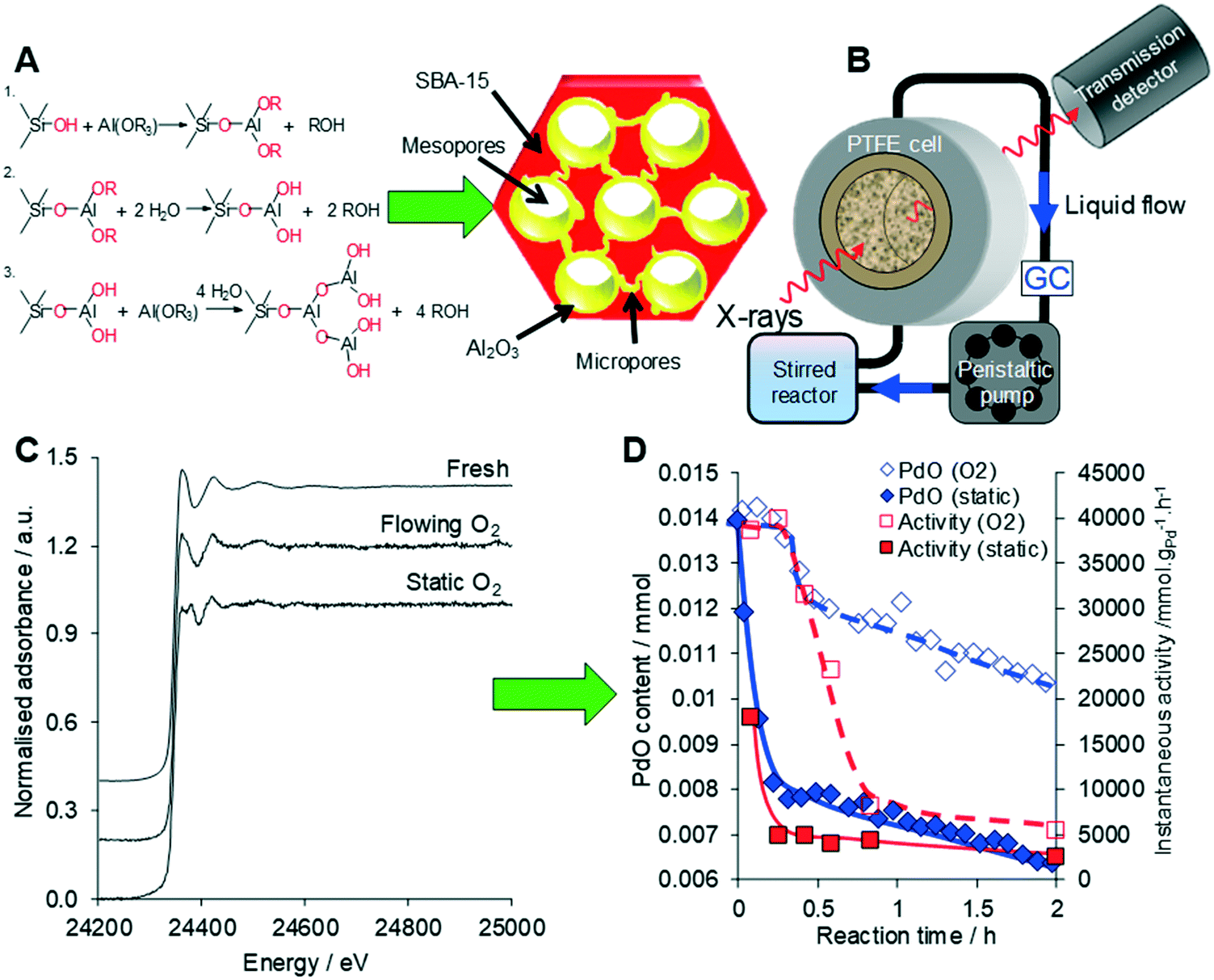 | ||
| Fig. 23 (A) Synthesis of a conformal adlayer of alumina over a mesoporous SBA-15 silica framework. (B) Operando liquid phase XAS setup. (C) Liquid phase operando Pd K-edge XAS of Pd/Al2O3-SBA-15 during cinnamyl alcohol selox. (D) Correlation between PdO concentration from XAS and selox activity under static versus flowing oxygen. Modified from ref. 80 with permission from Elsevier, copyright 2014. | ||
Allylic alcohol selox has also been investigated by Scott and co-workers through Pd LIII-, P K- and Au LIII-edge XAS to probe the nature of PVP-stabilised AuPd colloidal nanoparticles. In situ XAS measurements necessitated the use of a bespoke PEEK cell with Kapton windows alongside commercial XRF liquid cells, with X-ray attenuation minimised through judicious choice of cell windows. XAS studies revealed the critical role of sequential reduction of the bimetallic nanoparticles during catalyst synthesis, which resulted in an electron-deficient Pd-enriched surface over a Au core.97In situ XAS was also used to study the synthesis of a AuPd bimetallic catalyst from a Pd(II) salt in the presence of crotyl alcohol. Two possible mechanisms were hypothesised: (i) redox cycling of Pd(2+) and Pd(0), in which Pd(0) deposition from K2PdCl4 was driven by the alcohol reductant, evidenced by concomitant crotonaldehyde formation, and subsequent dissolution of Pd(2+) from the Au surface driven by reaction with O2; or (ii) an entirely heterogeneous pathway in which the adsorbed alcohol reacts with electron-deficient Pd surface atoms to form the allylic aldehyde via β-hydrogen elimination. The stability of the Pd oxidation state from Pd LIII-edge XAS studies upon introducing oxygen to the reaction solution favours the heterogeneous mechanism.98
O versus CC hydrogenation in Pt/SiO2 catalysed cinnamaldehyde and crotonaldehyde reduction,99,100 charge transfer in active sites for biomass-derived substrate hydrogenation in ambient pressure,101 and the presence of metallic Au in structure-sensitive hydrogenation of and the presence of metallic Au in structure-sensitive hydrogenation of nitroaromatics.102In situ XAS gas phase measurements before and after 773 K reduction in flowing H2 provided some insight the influence of Co on the electronic properties of Pt in bimetallic alloy nanoparticles for citral hydrogenation.103 Although the latter represents a significant improvement on ex situ characterisation, the low H2 pressures (ambient versus 10 bar during kinetic studies) and absence of solvent/reagent(s) during XAS analysis limits the utility of such measurements.
To address this, van Bokhoven and co-workers constructed a bespoke autoclave reactor capable of simultaneous XAS and ATR-IR (via a fibre optic dip probe) measurement (Fig. 24) for high pressure hydrogenation.81 This was applied to study the evolution of a Au/CeO2 catalyst during nitrobenzene hydrogenation. A metallic Au active phase was identified, and ATR-IR revealed a stepwise reaction via an azoxybenzene intermediate. The same operando XAS reactor has been used to evaluate: the effect of Au particle size on nitrobenzene hydrogenation;104 shifts in the Fermi level of Pt nanoparticles due to allylic carbonyl adsorption (during aldehyde reduction) as a descriptor of catalyst selectivity;105 and the mechanism of Pd(2+) reduction and aggregation within a [Pd(2-pymo)2]n, (2-pymo = 2-pyrimidinolate) metal–organic framework during 1-octene hydrogenation at 2 bar H2.106 Continuous flow high pressure (22 bar) furfural hydrogenation within a glassy carbon reactor tube107 enabled observation of on-stream sintering of Cu nanoparticles and resulting catalyst deactivation.108
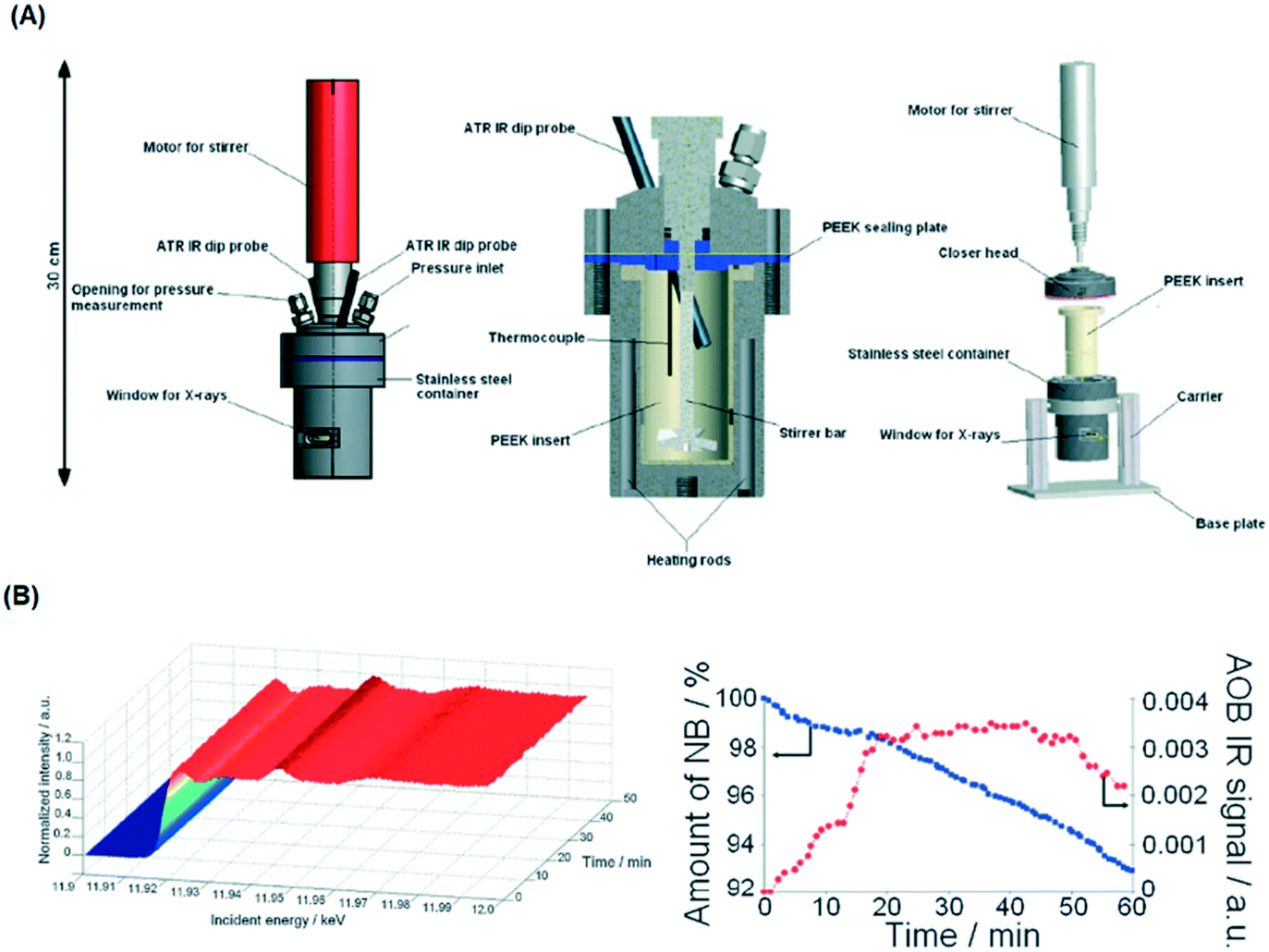 | ||
| Fig. 24 (A) HERFD XAS/ATR-FTIR autoclave schematic. (B) (left) Time-dependent Au LIII-edge HERFD XAS and (right) nitrobenzene (NB) and azoxybenzene (AOB) concentrations during aqueous phase hydrogenation over Au/CeO2 under 10 bar H2. Reproduced from ref. 81 with permission from the Royal Society of Chemistry, copyright 2012. | ||
This reaction cell was evaluated for H2O2 production from H2 and O2 (diluted in CO2) in methanol over a commercial Pd catalyst, however a lack of quantitation of the catalytic reaction prohibited determination of structure–reactivity relationships. Despite this, Pd K-edge fluorescence spectra evidenced the ability of Br− to promote reduction of PdO to Pd metal and concomitant leaching, which improves peroxide selectivity. Ex situ XAS measurements on Pd/TiO2 systems also postulated roles for both Pd metal and oxide, speculating that the interface between these phases was responsible for optimal H2O2 productivity.112 Grunwaldt and co-workers commissioned a similar continuous flow reactor, but in which spectra could be collected at different positions along the catalyst bed and capable of pressures up to 10 bar. Liquid phase H2O2 synthesis in water saturated with H2 and O2 (from separate streams) was evaluated over Pd/TiO2 in the presence of Br− promotor, and similarly identified the presence of Pd metal and oxide phases, attributed to a metal core@oxide shell structure. Under reaction conditions, the core was transformed to Pd hydride, with the hydride phase (α or β) dependent on the H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2; however H2O2 production was only associated with α-PdH.113 Bimetallic AuPd nanoparticles were also screened for H2O2 production continuous flow within a microfluidic reactor, wherein PdHx was also observed. In the latter case, it was postulated that hydride was inactive (although which phase is unclear), with Pd metal proposed as the active species with Au prohibiting hydride formation.82
:O2; however H2O2 production was only associated with α-PdH.113 Bimetallic AuPd nanoparticles were also screened for H2O2 production continuous flow within a microfluidic reactor, wherein PdHx was also observed. In the latter case, it was postulated that hydride was inactive (although which phase is unclear), with Pd metal proposed as the active species with Au prohibiting hydride formation.82
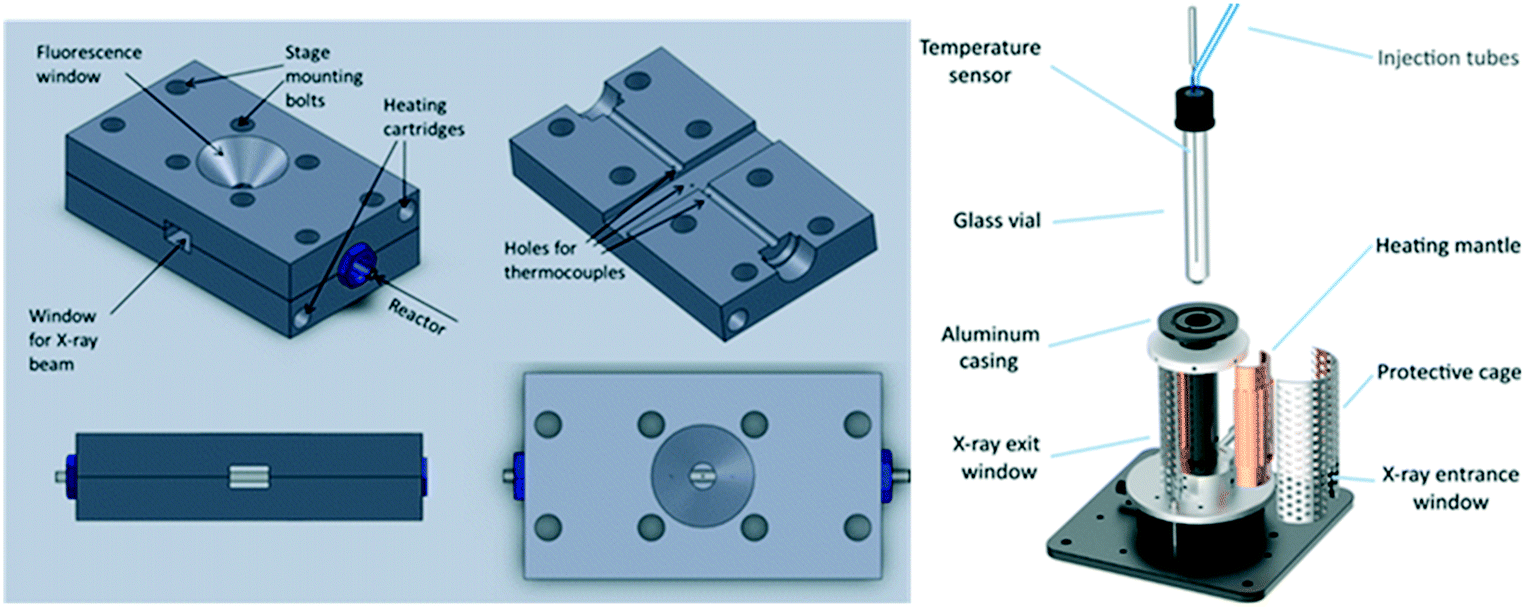
The Suzuki–Miyaura cross-coupling of iodonanisole and phenylboronic acid by PVP-stabilised Pd nanoparticles was studied by operando XAS in a bespoke PTFE cell to evaluate whether Pd leaching (and associated homogeneous catalysis) occurred during reaction.118 Under mild reaction conditions, the Pd–Pd coordination number remained constant, indicating a common size and negligible dissolution from low coordination edge and corner sites of nanoparticles, which in conjunction with kinetic and spectroscopic studies,119 provided strong evidence for a purely heterogeneous active site. In situ liquid phase XAS measurements also identified Pd(0) clusters as the active phase for ligandless Suzuki–Miyaura cross-coupling of 4-chlorobromobenzene and 4-fluorophenylboronic acid.120
Hii and co-workers also studied the stability of supported Pd nanoparticles during Suzuki–Miyaura cross-coupling by operando K-edge measurements along a packed catalyst bed under continuous flow. Herein, hot flowing ethanol resulted in Pd dissolution and subsequent downstream redeposition as metallic Pd nanoparticles,121 an important observation given the widespread use of ethanol as a solvent/reactant,122 and medium for catalyst preparation and characterisation.123 Pd/Al2O3 catalysts have also been evaluated for enantioselective allylic alkylation of (E)-1,3-diphenylallyl acetate, and Heck coupling of bromobenzene with styrene.124,125 Catalyst stability in the former reaction was dependent on the Pd oxidation state, with Pd(2+) leaching exacerbated under oxidising (air) environments or halogenated solvents thereby opening the possibility of a homogeneous pathway, while deactivation in the latter reaction coincided with formation of a bromopalladate complex. Heck coupling has also been evaluated over Pd@MOF catalysts by operando XAS and PXRD within a bespoke batch reactor (Fig. 25).126 Migration and agglomeration of Pd within the MOF collapsed the porous framework, however this was not responsible for catalyst deactivation, which rather occurred through halide poisoning and site blocking of Pd clusters.
2.4 X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) permits elemental quantification, and information regarding the oxidation/chemical state and electronic band structure of materials, and in some cases their structural arrangement.127 The technique involves photoexcitation of a core (or sometimes valence) electron, typically using soft X-rays (0.1 to 3 keV) and measurement of the kinetic energy of resulting photoelectrons ejected from a material. XPS is an inherently surface sensitive technique due to the relatively short inelastic mean free path (λ) of emitted photoelectrons, which usually span 0.1 to 2 nm, and hence has found routine application in heterogeneous catalysis where surface phenomena dominate.57,128A major drawback of the aforementioned techniques is the necessity of ‘tender’ or ‘hard’ X-rays (approximately 2 keV to 7 keV) to produce photoelectrons able to escape the liquid layer. Such X-rays have reduced photoionisation cross-sections for lighter elements and can result in spectra dominated by the underlying catalyst surface.139 Liquid jets, which have a rich history of application to study pure liquids,144 have also been used to study solid–liquid interfaces. Addition of SiO2 nanoparticles to a liquid stream enabled analysis of the local charge density at the water/silica interface.145 Such methods may also utilise soft X-rays, but with greatly diminished signals, and tend to be more effective for photon energies >2 keV. They also require highly specialised equipment and a dilute suspension, which may be problematic for low loading catalyst systems. In situ cells can exploit pressure differentials between UHV and liquids across a Nafion® proton exchange membrane to transport vapour and target molecules to catalyst surfaces for analysis.146 Such liquid–solid layers permit operando measurements of electrocatalysts, revealing surface species present in the oxygen evolution reaction (OER) over Pt146 and Ir147 electrodes. Potentially the most promising technological advance for liquid–solid XPS studies is the use of graphene-based cells (Fig. 26),148 wherein the high mechanical strength of ultra-thin graphene sheets/nanobubbles offer good signal quality using soft X-rays at pressures reaching 1 bar, even under continuous liquid flow.149,150 Such systems have exhibited excellent sensitivity even at photon energies as low as 275 eV.149 The use of graphene in the cell architecture renders such cells ideal for in situ studies of metal–graphene catalysts for OER.150,151 For example, the observation of carbonyl-like hybrid interfaces forming in situ between Co and graphene has helped identify the electrocatalytic active site.151 Deposited nanoparticle catalysts can also be interrogated in the liquid phase by this approach, unlocking diverse applications across heterogeneous catalysis.152 Graphene nanobubbles have also been utilised to study the operando thermal reduction of FeCl3 to FeCl2 in aqueous solution, although improved flow capabilities are desirable.153 Production of graphene windows is time- and labour-intensive and remains a significant hurdle to wider adoption for real-time solid–liquid catalysis.
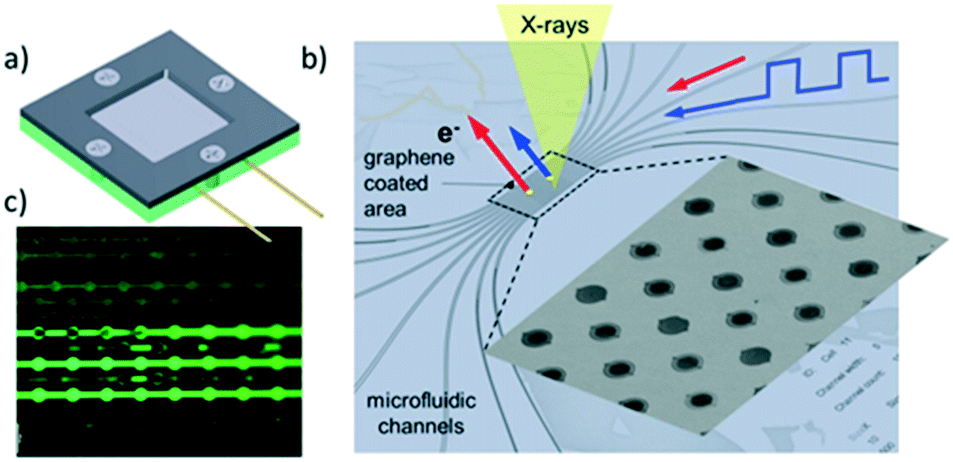 | ||
| Fig. 26 Fluidic multichannel graphene cell for photoelectron spectroscopy under UHV conditions. a) Overall design; b) SEM image of water filled micro-channels with graphene capped orifices; c) fluidic tests with fluorescein dye, reproduced from ref. 148 with permission from Cambridge University Press, copyright 2018. | ||
2.5 UV-vis spectroscopy
The use of UV-vis spectroscopic in catalytic reactions is challenging: it usually does not provide unequivocal information on molecular structure, bands are often broad and suffer from interference by minority species hindering spectral interpretation, and quantitation may be impossible due to a lack of molar absorption coefficients. There are few studies on the application of UV-vis spectroscopy in solid–liquid catalysis, and these are typically in conjunction with complementary techniques to help differentiate possible structures. Some examples of time-resolved UV-vis spectroscopic studies of heterogeneously catalysed liquid phase reactions are described below.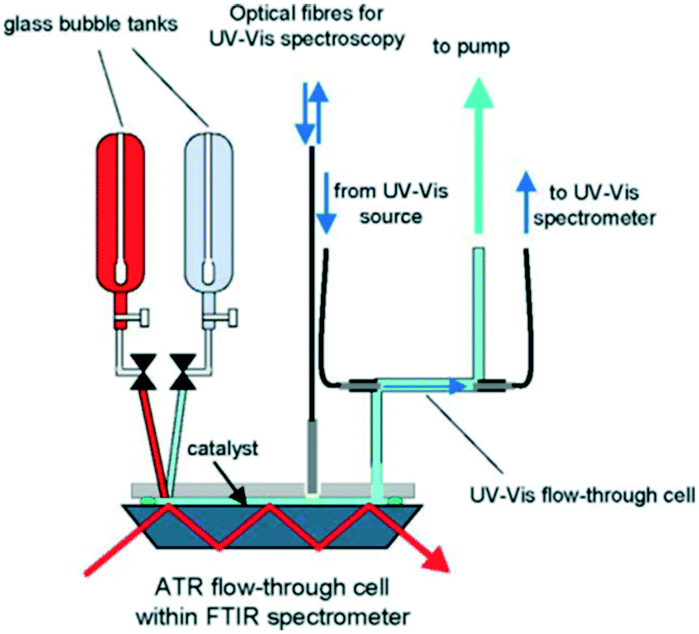 | ||
| Fig. 27 Schematic of continuous flow reactor for simultaneous in situ ATR-IR and UV-vis analysis during ethanol oxidation over Pd/Al2O3, reproduced from ref. 154 with permission from Elsevier, copyright 2005. | ||
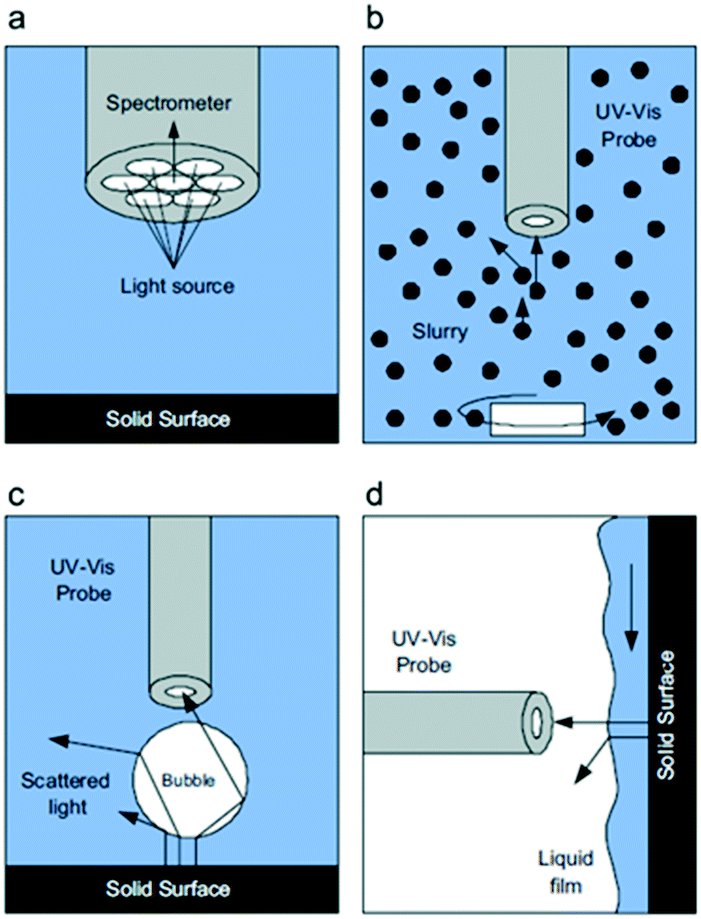 | ||
| Fig. 28 a) Details of the optical probe tip and schematic representations during in situ UV-vis analysis, (b) in a slurry reactor, (c) a packed bubble column, and (d) a trickle bed reactor, highlighting interferences at the gas–liquid and solid–liquid interfaces that scatter light away from the optical probe, reproduced from ref. 155 with permission from Elsevier, copyright 2010. | ||
2.6 Second-order nonlinear optical spectroscopy
Sum frequency generation (SFG) vibrational spectroscopy (VS) is one of few techniques able to provide molecular information at the solid–liquid interface that is directly relatable to catalytic activity.156,157 This is a consequence of selection rules which allow molecules in a centrosymmetric environment in the bulk (liquid) media to be differentiated from those at a surface. In SFG-VS, visible and IR lasers are simultaneously focused on the interface of interest. An optical (SFG) signal is generated by combining the visible and IR light in a predetermined direction to the interface. This SFG signal is resonantly enhanced when the IR frequency is tuned across the frequency of a molecular vibration, thereby isolating the vibrational spectra of molecules at an interface156,157 which can be combined with computer modelling to determine absolute molecular orientations. However, SFG-VS is challenging since large, non-resonant signals from metallic and semiconductor surfaces can obscure the weaker molecular fingerprint,157 and for thick (opaque) substrates, a thin solvent layer is required to obtain data on adsorbates.158 Improved modelling of the interfacial refractive index is also required to derive more accurate experimental spectra. Some examples of SFG-VS applied to liquid phase heterogenous catalysis are discussed below.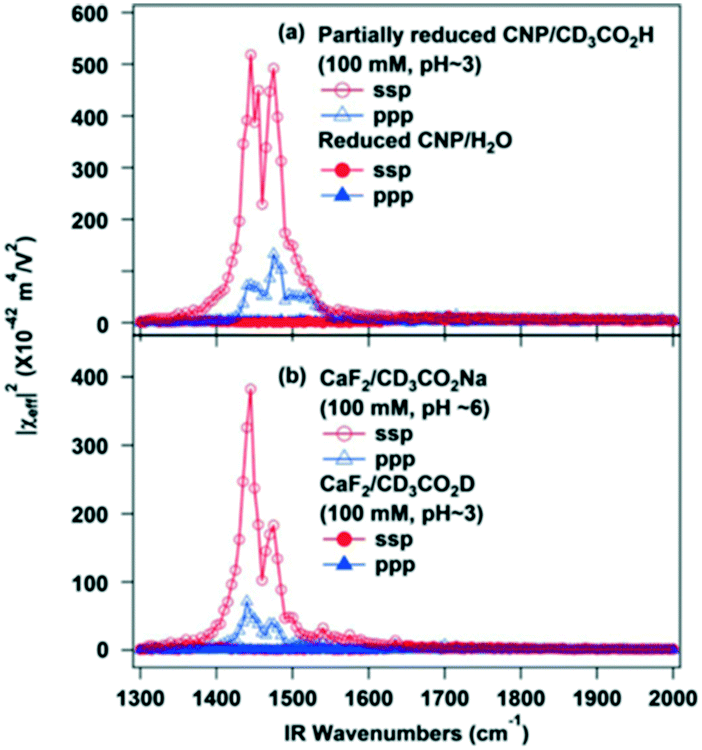 | ||
| Fig. 29 SFG-VS spectra of (a) partially reduced ceria nanoparticles in contact with water or a CD3CO2H aqueous solution (pH ∼ 3), and (b) bare CaF2 substrate in contact with either CD3CO2H (pH ∼ 3) or CD3CO2Na (pH ∼ 6) aqueous solutions, reproduced from ref. 159 with permission from the American Chemical Society, copyright 2013. | ||
3. Conclusions
In situ and operando techniques can provide unique and often invaluable insight into catalytic reaction mechanisms, the nature of active and spectator species, and deactivation pathways in liquid phase heterogeneous catalysis. Such studies have been facilitated by the design and development of advanced catalytic reactors and their integration with time- and spatially resolved spectroscopies which have been enabled by modern synchrotron radiation facilities.A molecular level understanding of reaction mechanisms at the solid–liquid interface requires careful kinetic analysis and correlation between evolving physicochemical properties and adsorbed species. For kinetic studies, quantitative analysis of spectra is required, however this is not trivial, and in practice studies often result in empirical/semi-empirical correlations between signal intensities and concentrations of species for individual techniques. To improve the accuracy of kinetic information, and hence reliability of associated models, calibration of the detected signal intensities is essential. The combination of multiple in situ/operando techniques such as ATR-IR or Raman with UV-vis and/or scanning probe techniques in a single experimental configuration improves the reliability of kinetic information and affords greater insight into chemical processes. There are many authoritative articles and texts that compare such complementary spectroscopies.9,161
Studies of structure–activity relationships in solid catalysed liquid-phase reactions require careful consideration of multiple interactions at the solid–liquid interface including solvation, adsorption, and intermolecular bonding. In porous catalysts such as zeolites, the solvent composition can significantly affect the concentration of reactants throughout the pore network and compete for acid sites during reactions. In electrocatalysis, the concentrations of species at the electrochemical interface may differ substantially from those in the bulk media. Quantitative understanding of mass transport and solvation effects near the electrode surface are therefore important to develop meaningful models of electrode-mediated reactions wherein intrinsic kinetics are measured in the absence of diffusion limitations. Spectroscopic techniques able to selectivity analyse interfaces, such as surface enhanced infrared absorption reaction spectroscopy will become increasingly valuable tools for understanding surface–adsorbate interaction,162 and are best complemented by techniques such as XAS and XPS that interrogate the catalyst electronic and geometric structure.
Improved temporal resolution, particularly through the wider exploitation of pump–probe techniques,163 into the pico- and femtosecond domains will expand our knowledge of elementary reaction steps in surface catalysis. Conversely, better approaches to undertaking stability testing to mimic industrial processes over days, weeks and even years, will better elucidate on-stream deactivation (and reactivation) mechanisms, but require new access routes to synchrotrons and related national facilities to enable acquisition of extended data-sets.164 Closer alignment of operando conditions with those in used in industry is also necessary to enable better translation of laboratory findings to the ‘real-world’; this is contingent on the design and commercial availability of reaction cells able to operate at elevated pressures >10 MPa (e.g. esterification and Diels–Alder165) and temperature >400 °C and with improved chemical tolerance to corrosive environments. Spatial resolution is another critical consideration, with spectral acquisition at the sub-micron scale by e.g. photoemission electron microscopy166 enabling mapping of active site distributions, catalyst restructuring and concentration gradients at the solid–liquid interface, and at the cm to metre scale allowing chemical mapping of e.g. active site leaching and chemical speciation by XAS analysis across a reactor bed.122
The application of modulation excitation methods, in which one or more reaction parameters (such as temperature, reactant composition, pressure or voltage) are switched between different states, will provide unique insight into the nature of reactive intermediates in solid–liquid catalysis. Such studies have proven valuable for gas-phase systems,167 and furthermore recent modelling168,169 and experiments170 have demonstrated that careful tuning of the modulation frequency can result in catalytic resonance offering the tantalising possibility of overcoming the Sabatier principle for static reactions, thereby unlocking rate enhancements of 10–10000× for reactions such as formic acid electrooxidation. Inducing rapid switching of temperature, pressure and concentration will prove difficult for conventional liquid phase reactors, due to slow heat/mass transfer compared to gas phase catalysis, however the use of microfluidic reactors, and light or electrical170 excitation for catalytic state-switching are promising solutions. Kinetic studies of rapid chemical transformations employing multidimensional spectroscopies will also generate vast datasets, requiring high capacity data compression/transfer/storage, and automated data processing and mining algorithms in conjunction with modelling/simulations (Machine Learning approaches171,172) to extract key descriptors for the development of next-generation catalysts.173Operando methodologies facilitate online analysis of catalytic processes and rapid feedback to optimise productivity, but require ultrafast data transfer and real-time data analysis. Advances in reactor design will aid the integration of in situ and operando spectroscopies, notably through advanced manufacturing techniques such as additive (3D) printing to fabricate novel reactor configurations with improved heat/mass transport and optical interfacing.174 High throughput 3D printing directed by computational fluid dynamic modelling will permit the rapid evaluation of diverse reactor configurations that ensure spectroscopic measurements are acquired under real-world conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. N. has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 837794. The UK Catalysis Hub is kindly thanked for resources and support provided via membership of the UK Catalysis Hub Consortium and funded by EPSRC (portfolio grants EP/K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/I019693/1).Notes and references
- B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4557–4559 RSC.
- U. Bentrup, Chem. Soc. Rev., 2010, 39, 4718–4730 RSC.
- A. M. Beale, S. D. Jacques and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4656–4672 RSC.
- J. M. Andanson and A. Baiker, Chem. Soc. Rev., 2010, 39, 4571–4584 CAS.
- S.-M. Xia, K.-H. Chen, H.-C. Fu and L.-N. He, Front. Chem., 2018, 6, 462 CrossRef CAS PubMed.
- A. R. Hind, S. K. Bhargava and A. McKinnon, Adv. Colloid Interface Sci., 2001, 93, 91–114 CAS.
- R. A. Sheldon and R. S. Downing, Appl. Catal., A, 1999, 189, 163–183 CrossRef CAS.
- K. J. Carpenter, Chem. Eng. Sci., 2001, 56, 305–322 CAS.
- J. D. Grunwaldt and A. Baiker, Phys. Chem. Chem. Phys., 2005, 7, 3526–3539 RSC.
- S. Gopalakrishnan, D. F. Liu, H. C. Allen, M. Kuo and M. J. Shultz, Chem. Rev., 2006, 106, 1155–1175 CAS.
- T. Burgi, Chimia, 2003, 57, 623–627 Search PubMed.
- B. L. Mojet, S. D. Ebbesen and L. Lefferts, Chem. Soc. Rev., 2010, 39, 4643–4655 RSC.
- I. Ortiz-Hernandez, D. J. Owens, M. R. Strunk and C. T. Williams, Langmuir, 2006, 22, 2629–2639 CrossRef CAS PubMed.
- T. Burgi and A. Baiker, Adv. Catal., 2006, 50, 227–283 CAS.
- D. Ferri and A. Baiker, Top. Catal., 2009, 52, 1323–1333 CrossRef CAS.
- I. Ortiz-Hernandez and C. T. Williams, Langmuir, 2003, 19, 2956–2962 CrossRef CAS.
- T. Burgi and A. Baiker, J. Phys. Chem. B, 2002, 106, 10649–10658 CrossRef.
- A. Urakawa, T. Bürgi and A. Baiker, Chem. Eng. Sci., 2008, 63, 4902–4909 CrossRef CAS.
- M. W. Urban, J. Adhes. Sci. Technol., 1993, 7, 1–47 CrossRef CAS.
- J. Yarwood, Anal. Proc., 1993, 30, 13–18 RSC.
- L. J. Fina and G. Chen, Vib. Spectrosc., 1991, 1, 353–361 CrossRef CAS.
- J. Zakzeski, R. J. H. Grisel, A. T. Smit and B. M. Weckhuysen, ChemSusChem, 2012, 5, 430–437 CrossRef CAS PubMed.
- A. L. Denning, H. Dang, Z. M. Liu, K. M. Nicholas and F. C. Jentoft, ChemCatChem, 2013, 5, 3567–3570 CrossRef CAS.
- A. Wawrzetz, B. Peng, A. Hrabar, A. Jentys, A. A. Lemonidou and J. A. Lercher, J. Catal., 2010, 269, 411–420 CAS.
- R. He, R. R. Davda and J. A. Dumesic, J. Phys. Chem. B, 2005, 109, 2810–2820 CrossRef CAS PubMed.
- K. Koichumanova, A. K. K. Vikla, D. J. M. de Vlieger, K. Seshan, B. L. Mojet and L. Lefferts, ChemSusChem, 2013, 6, 1717–1723 CrossRef CAS.
- J. R. Copeland, G. S. Foo, L. A. Harrison and C. Sievers, Catal. Today, 2013, 205, 49–59 CrossRef CAS.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, Langmuir, 2006, 22, 1079–1085 CrossRef CAS PubMed.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, J. Catal., 2007, 246, 66–73 CrossRef CAS.
- C. Keresszegi, D. Ferri, T. Mallat and A. Baiker, J. Phys. Chem. B, 2005, 109, 958–967 CrossRef CAS PubMed.
- D. Ferri, T. Burgi and A. Baiker, Phys. Chem. Chem. Phys., 2002, 4, 2667–2672 RSC.
- J. Y. Wang, H. X. Zhang, K. Jiang and W. B. Cai, J. Am. Chem. Soc., 2011, 133, 14876–14879 CrossRef CAS PubMed.
- A. Aguirre, P. A. Kler, C. L. A. Berli and S. E. Collins, Chem. Eng. J., 2014, 243, 197–206 CrossRef CAS.
- H. Knozinger and G. Mestl, Top. Catal., 1999, 8, 45–55 CrossRef CAS.
- I. E. Wachs and C. A. Roberts, Chem. Soc. Rev., 2010, 39, 5002–5017 RSC.
- P. C. Stair, Advances in Catalysis, 2007, vol. 51, pp. 75–98 Search PubMed.
- S. Q. Jin, Z. C. Feng, F. T. Fan and C. Li, Catal. Lett., 2015, 145, 468–481 CrossRef CAS.
- S. Kim, L. Piao, D. Han, B. J. Kim and T. D. Chung, Adv. Mater., 2013, 25, 2056–2061 CrossRef CAS PubMed.
- L. Opilik, T. Schmid and R. Zenobi, Annu. Rev. Anal. Chem., 2013, 6, 379–398 CrossRef CAS PubMed.
- F. T. Fan, Z. C. Feng and C. Li, Acc. Chem. Res., 2010, 43, 378–387 CrossRef CAS PubMed.
- T. Kim, R. S. Assary, H. Kim, C. L. Marshall, D. J. Gosztola, L. A. Curtiss and P. C. Stair, Catal. Today, 2013, 205, 60–66 CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- H. Y. H. Chan, C. T. Williams, M. J. Weaver and C. G. Takoudis, J. Catal., 1998, 174, 191–200 CrossRef CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- K. N. Heck, B. G. Janesko, G. E. Scuseria, N. J. Halas and M. S. Wong, ACS Catal., 2013, 3, 2430–2435 CrossRef CAS.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- H. Zhang, S. Duan, P. M. Radjenovic, Z. Q. Tian and J. F. Li, Acc. Chem. Res., 2020, 53, 729–739 CrossRef CAS PubMed.
- Y. H. Wang, J. Wei, P. Radjenovic, Z. Q. Tian and J. F. Li, Anal. Chem., 2019, 91, 1675–1685 CrossRef CAS PubMed.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, Nat. Energy, 2019, 4, 60–67 CrossRef CAS.
- T. Deckert-Gaudig, A. Taguchi, S. Kawata and V. Deckert, Chem. Soc. Rev., 2017, 46, 4077–4110 RSC.
- P. Verma, Chem. Rev., 2017, 117, 6447–6466 CrossRef CAS PubMed.
- A. Hartschuh, Angew. Chem., Int. Ed., 2008, 47, 8178–8191 CrossRef CAS PubMed.
- N. Kumar, W. T. Su, M. Vesely, B. M. Weckhuysen, A. J. Pollard and A. J. Wain, Nanoscale, 2018, 10, 1815–1824 RSC.
- N. Kumar, C. S. Wondergem, A. J. Wain and B. M. Weckhuysen, J. Phys. Chem. Lett., 2019, 10, 1669–1675 CrossRef CAS PubMed.
- S. Jiang, Z. Chen, X. Chen, D. Nguyen, M. Mattei, G. Goubert and R. P. Van Duyne, J. Phys. Chem. C, 2019, 123, 9852–9859 CrossRef CAS.
- D. C. Koningsberger, X-ray absorption: principles, applications, techniques of EXAFS, SEXAFS, and XANES, John Wiley and Sons, United States, 1988 Search PubMed.
- A. F. Lee, V. Prabhakaran and K. Wilson, Chem. Commun., 2010, 46, 3827–3842 RSC.
- G. A. Somorjai, S. K. Beaumont and S. Alayoglu, Angew. Chem., Int. Ed., 2011, 50, 10116–10129 CrossRef CAS PubMed.
- R. Toyoshima and H. Kondoh, J. Phys. Condens. Matter, 2015, 27, 15 CrossRef PubMed.
- H. L. Han, G. Melaet, S. Alayoglu and G. A. Somorjai, ChemCatChem, 2015, 7, 3625–3638 CrossRef CAS.
- D. Bazin and L. Guczi, Appl. Catal., A, 2001, 213, 147–162 CrossRef CAS.
- J. W. Smith and R. J. Saykally, Chem. Rev., 2017, 117, 13909–13934 CrossRef CAS PubMed.
- D. H. Bilderback, P. Elleaume and E. Weckert, J. Phys. B: At., Mol. Opt. Phys., 2005, 38, S773–S797 CrossRef CAS.
- G. T. Seidler, D. R. Mortensen, A. J. Remesnik, J. I. Pacold, N. A. Ball, N. Barry, M. Styczinski and O. R. Hoidn, Rev. Sci. Instrum., 2014, 85, 12 CrossRef PubMed.
- E. P. Jahrman, W. M. Holden, A. S. Ditter, D. R. Mortensen, G. T. Seidler, T. T. Fister, S. A. Kozimor, L. F. J. Piper, J. Rana, N. C. Hyatt and M. C. Stennett, Rev. Sci. Instrum., 2019, 90, 15 Search PubMed.
- M. W. Tew, M. Nachtegaal, M. Janousch, T. Huthwelker and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2012, 14, 5761–5768 RSC.
- A. L. Bugaev, A. A. Guda, A. Lazzarini, K. A. Lomachenko, E. Groppo, R. Pellegrini, A. Piovano, H. Emerich, A. V. Soldatov, L. A. Bugaev, V. P. Dmitriev, J. A. van Bokhoven and C. Lamberti, Catal. Today, 2017, 283, 119–126 CrossRef CAS.
- V. M. Gonzalez-delaCruz, R. Pereniguez, F. Ternero, J. P. Holgado and A. Caballero, J. Phys. Chem. C, 2012, 116, 2919–2926 CrossRef CAS.
- M. M. Gunter, T. Ressler, R. E. Jentoft and B. Bems, J. Catal., 2001, 203, 133–149 CrossRef.
- N. E. Tsakoumis, A. Voronov, M. Ronning, W. van Beek, O. Borg, E. Rytter and A. Holmen, J. Catal., 2012, 291, 138–148 CrossRef CAS.
- M. Bron, D. Teschner, A. Knop-Gericke, A. Scheybal, B. Steinhauer, M. Havecker, R. Fodisch, D. Honicke, R. Schlogl and P. Claus, Catal. Commun., 2005, 6, 371–374 CrossRef CAS.
- H. J. Wei, C. Gomez, J. J. Liu, N. Guo, T. P. Wu, R. Lobo-Lapidus, C. L. Marshall, J. T. Miller and R. J. Meyer, J. Catal., 2013, 298, 18–26 CrossRef CAS.
- M. A. Newton, D. G. Burnaby, A. J. Dent, S. Diaz-Moreno, J. Evans, S. G. Fiddy, T. Neisius and S. Turin, J. Phys. Chem. B, 2002, 106, 4214–4222 CrossRef CAS.
- M. A. Newton, C. Belver-Coldeira, A. Martinez-Arias and M. Fernandez-Garcia, Nat. Mater., 2007, 6, 528–532 CrossRef CAS PubMed.
- D. Ferri, M. S. Kumar, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff and M. A. Newton, Phys. Chem. Chem. Phys., 2010, 12, 5634–5646 RSC.
- M. A. Newton, M. Di Michiel, A. Kubacka and M. Fernández-García, J. Am. Chem. Soc., 2010, 132, 4540–4541 CrossRef CAS PubMed.
- S. T. Korhonen, A. M. Beale, M. A. Newton and B. M. Weckhuysen, J. Phys. Chem. C, 2011, 115, 885–896 CrossRef CAS.
- A. F. Lee, C. V. Ellis, J. N. Naughton, M. A. Newton, C. M. A. Parlett and K. Wilson, J. Am. Chem. Soc., 2011, 133, 5724–5727 CrossRef CAS PubMed.
- A. F. Lee and K. Wilson, Green Chem., 2004, 6, 37–42 RSC.
- C. M. A. Parlett, L. J. Durndell, A. Machado, G. Cibin, D. W. Bruce, N. S. Hondow, K. Wilson and A. F. Lee, Catal. Today, 2014, 229, 46–55 CrossRef CAS.
- M. Makosch, C. Kartusch, J. Sa, R. B. Duarte, J. A. van Bokhoven, K. Kvashnina, P. Glatzel, D. L. A. Fernandes, M. Nachtegaal, E. Kleymenov, J. Szlachetko, B. Neuhold and K. Hungerbuhler, Phys. Chem. Chem. Phys., 2012, 14, 2164–2170 RSC.
- S. Kanungo, L. van Haandel, E. J. M. Hensen, J. C. Schouten and M. F. N. d'Angelo, J. Catal., 2019, 370, 200–209 CrossRef CAS.
- R. S. Weatherup, Top. Catal., 2018, 61, 2085–2102 CrossRef CAS.
- G. Pacchioni, Nat. Rev. Phys., 2019, 1, 100–101 CrossRef.
- E. de Smit, I. Swart, J. F. Creemer, G. H. Hoveling, M. K. Gilles, T. Tyliszczak, P. J. Kooyman, H. W. Zandbergen, C. Morin, B. M. Weckhuysen and F. M. F. de Groot, Nature, 2008, 456, 222–225 CrossRef CAS PubMed.
- J. D. Grunwaldt, C. Keresszegi, T. Mallat and A. Baiker, J. Catal., 2003, 213, 291–295 CrossRef CAS.
- J. D. Grunwaldt, M. Caravati and A. Baiker, J. Phys. Chem. B, 2006, 110, 25586–25589 CrossRef CAS PubMed.
- J.-D. Grunwaldt, M. Caravati and A. Baiker, J. Phys. Chem. B, 2006, 110, 9916–9922 CrossRef CAS PubMed.
- A. F. Lee, Abstr. Pap. Am. Chem. Soc., 2001, vol. 221, p. 187-COLL Search PubMed.
- A. F. Lee, J. J. Gee and H. J. Theyers, Green Chem., 2000, 2, 279–282 RSC.
- A. F. Lee, Z. Chang, P. Ellis, S. F. J. Hackett and K. Wilson, J. Phys. Chem. C, 2007, 111, 18844–18847 CrossRef CAS.
- A. F. Lee, J. N. Naughton, Z. Liu and K. Wilson, ACS Catal., 2012, 2, 2235–2241 CrossRef CAS.
- C. V. Gaskell, C. M. A. Parlett, M. A. Newton, K. Wilson and A. F. Lee, ACS Catal., 2012, 2, 2242–2246 CrossRef CAS.
- C. M. A. Parlett, C. V. Gaskell, J. N. Naughton, M. A. Newton, K. Wilson and A. F. Lee, Catal. Today, 2013, 205, 76–85 CrossRef CAS.
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS PubMed.
- H. Chen, S. He, M. Xu, M. Wei, D. G. Eyans and X. Duan, ACS Catal., 2017, 7, 2735–2743 CrossRef CAS.
- T. Balcha, J. R. Strobl, C. Fowler, P. Dash and R. W. J. Scott, ACS Catal., 2011, 1, 425–436 CrossRef CAS.
- A. Maclennan, A. Banerjee, Y. F. Hu, J. T. Miller and R. W. J. Scott, ACS Catal., 2013, 3, 1411–1419 CrossRef CAS.
- R. Y. Zheng, M. D. Porosoff, J. L. Weiner, S. L. Lu, Y. X. Zhu and J. G. G. Chen, Appl. Catal., A, 2012, 419, 126–132 CrossRef.
- K. Taniya, H. Jinno, M. Kishida, Y. Ichihashi and S. Nishiyama, J. Catal., 2012, 288, 84–91 CrossRef CAS.
- S. Nishimura, N. Ikeda and K. Ebitani, Catal. Today, 2014, 232, 89–98 CrossRef CAS.
- K. Shimizu, Y. Miyamoto, T. Kawasaki, T. Tanji, Y. Tai and A. Satsuma, J. Phys. Chem. C, 2009, 113, 17803–17810 CrossRef CAS.
- N. M. Bertero, A. F. Trasarti, B. Moraweck, A. Borgna and A. J. Marchi, Appl. Catal., A, 2009, 358, 32–41 CrossRef CAS.
- U. Hartfelder, C. Kartusch, M. Makosch, M. Rovezzi, J. Sa and J. A. van Bokhoven, Catal. Sci. Technol., 2013, 3, 454–461 RSC.
- H. G. Manyar, R. Morgan, K. Morgan, B. Yang, P. Hu, J. Szlachetko, J. Sa and C. Hardacre, Catal. Sci. Technol., 2013, 3, 1497–1500 RSC.
- S. Schuster, E. Klemm and M. Bauer, Chem. – Eur. J., 2012, 18, 15831–15837 CrossRef CAS PubMed.
- V. F. Kispersky, A. J. Kropf, F. H. Ribeiro and J. T. Miller, Phys. Chem. Chem. Phys., 2012, 14, 2229–2238 RSC.
- B. J. O'Neill, J. T. Miller, P. J. Dietrich, F. G. Sollberger, F. H. Ribeiro and J. A. Dumesic, ChemCatChem, 2014, 6, 2493–2496 CrossRef.
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef CAS.
- P. Centomo, C. Meneghini and M. Zecca, Rev. Sci. Instrum., 2013, 84, 5 CrossRef PubMed.
- P. Centomo, C. Meneghini, S. Sterchele, A. Trapananti, G. Aquilanti and M. Zecca, Catal. Today, 2015, 248, 138–141 CrossRef CAS.
- L. Ouyang, P. F. Tian, G. J. Da, X. C. Xu, C. Ao, T. Y. Chen, R. Si, J. Xu and Y. F. Han, J. Catal., 2015, 321, 70–80 CrossRef CAS.
- M. Selinsek, B. J. Deschner, D. E. Doronkin, T. L. Sheppard, J. D. Grunwaldt and R. Dittmeyer, ACS Catal., 2018, 8, 2546–2557 CrossRef CAS.
- A. M. Karim, C. Howard, B. Roberts, L. Kovarik, L. Zhang, D. L. King and Y. Wang, ACS Catal., 2012, 2, 2387–2394 CrossRef CAS.
- Z. H. Wei, A. Karim, Y. Li and Y. Wang, ACS Catal., 2015, 5, 7312–7320 CrossRef CAS.
- Z. H. Wei, A. M. Karim, Y. Li, D. L. King and Y. Wang, J. Catal., 2015, 322, 49–59 CrossRef CAS.
- P. J. Dietrich, R. J. Lobo-Lapidus, T. P. Wu, A. Sumer, M. C. Akatay, B. R. Fingland, N. Guo, J. A. Dumesic, C. L. Marshall, E. Stach, J. Jellinek, W. N. Delgass, F. H. Ribeiro and J. T. Miller, Top. Catal., 2012, 55, 53–69 CrossRef CAS.
- P. J. Ellis, I. J. S. Fairlamb, S. F. J. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820–1824 CrossRef CAS PubMed.
- A. F. Lee, P. J. Ellis, I. J. S. Fairlamb and K. Wilson, Dalton Trans., 2010, 39, 10473–10482 RSC.
- L. A. Adrio, B. N. Nguyen, G. Guilera, A. G. Livingston and K. K. Hii, Catal. Sci. Technol., 2012, 2, 316–323 RSC.
- J. B. Brazier, B. N. Nguyen, L. A. Adrio, E. M. Barreiro, W. P. Leong, M. A. Newton, S. J. A. Figueroa, K. Hellgardt and K. K. M. Hii, Catal. Today, 2014, 229, 95–103 CrossRef CAS.
- M. A. Newton, J. B. Brazier, E. M. Barreiro, S. Parry, H. Emmerich, L. A. Adrio, C. J. Mulligan, K. Hellgardt and K. K. Hii, Green Chem., 2016, 18, 406–411 RSC.
- M. A. Newton, J. B. Brazier, E. M. Barreiro, H. Emerich, L. A. Adrio, C. J. Mulligan, K. Hellgardt and K. K. Hii, Catal. Sci. Technol., 2016, 6, 8525–8531 RSC.
- S. Reimann, J. D. Grunwaldt, T. Mallat and A. Baiker, Chem. – Eur. J., 2010, 16, 9658–9668 CrossRef CAS PubMed.
- S. Reimann, J. Stotzel, R. Frahm, W. Kleist, J. D. Grunwaldt and A. Baiker, J. Am. Chem. Soc., 2011, 133, 3921–3930 CrossRef CAS PubMed.
- N. Yuan, V. Pascanu, Z. H. Huang, A. Valiente, N. Heidenreich, S. Leubner, A. K. Inge, J. Gaar, N. Stock, I. Persson, B. Martin-Matute and X. D. Zou, J. Am. Chem. Soc., 2018, 140, 8206–8217 CrossRef CAS PubMed.
- M. P. Seah, Vacuum, 1984, 34, 463–478 CrossRef CAS.
- A. M. Venezia, Catal. Today, 2003, 77, 359–370 CrossRef CAS.
- K. Siegbahn, C. Nordling, G. Johansson, J. Hedman, P. F. Heden, K. Hamrin, U. Gelius, T. Bergmark, L. O. Werme, R. Manne and Y. Baer, ESCA Applied to Free Molecules, North-Holland, 1969 Search PubMed.
- D. F. Ogletree, H. Bluhm, G. Lebedev, C. S. Fadley, Z. Hussain and M. Salmeron, Rev. Sci. Instrum., 2002, 73, 3872–3877 CrossRef CAS.
- M. Salmeron and R. Schlögl, Surf. Sci. Rep., 2008, 63, 169–199 CrossRef CAS.
- H. Bluhm, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 71–84 CrossRef CAS.
- D. Starr, Z. Liu, M. Hävecker, A. Knop-Gericke and H. Bluhm, Chem. Soc. Rev., 2013, 42, 5833–5857 RSC.
- A. Knop-Gericke, V. Pfeifer, J.-J. Velasco-Velez, T. Jones, R. Arrigo, M. Hävecker and R. Schlögl, J. Electron Spectrosc. Relat. Phenom., 2017, 221, 10–17 CrossRef CAS.
- O. Karslıoğlu, S. Nemšák, I. Zegkinoglou, A. Shavorskiy, M. Hartl, F. Salmassi, E. Gullikson, M. Ng, C. Rameshan and B. Rude, Faraday Discuss., 2015, 180, 35–53 RSC.
- P. Kolb, D. Rath, R. Wille and W. Hansen, Berichte der Bunsengesellschaft für physikalische Chemie, 1983, 87, 1108–1113 CrossRef.
- K. Roy, L. Artiglia and J. A. van Bokhoven, ChemCatChem, 2018, 10, 666–682 CrossRef CAS.
- M. Favaro, F. F. Abdi, E. J. Crumlin, Z. Liu, R. van de Krol and D. E. Starr, Surfaces, 2019, 2, 78–99 CrossRef.
- S. Axnanda, E. J. Crumlin, B. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. Edwards, M. Lundqvist, R. Moberg and P. Ross, Sci. Rep., 2015, 5, 9788 CrossRef CAS PubMed.
- K. Arima, P. Jiang, X. Deng, H. Bluhm and M. Salmeron, J. Phys. Chem. C, 2010, 114, 14900–14906 CrossRef CAS.
- S. Nemšák, A. Shavorskiy, O. Karslioglu, I. Zegkinoglou, A. Rattanachata, C. S. Conlon, A. Keqi, P. K. Greene, E. C. Burks and F. Salmassi, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- Y. Takagi, H. Wang, Y. Uemura, T. Nakamura, L. Yu, O. Sekizawa, T. Uruga, M. Tada, G. Samjeské and Y. Iwasawa, Phys. Chem. Chem. Phys., 2017, 19, 6013–6021 RSC.
- H. Ali-Löytty, M. Hannula, M. Valden, A. Eilert, H. Ogasawara and A. Nilsson, J. Phys. Chem. C, 2019, 123, 25128–25134 CrossRef.
- M. Faubel, B. Steiner and J. P. Toennies, J. Chem. Phys., 1997, 106, 9013–9031 CrossRef CAS.
- M. A. Brown, N. Duyckaerts, A. B. Redondo, I. Jordan, F. Nolting, A. Kleibert, M. Ammann, H. J. Wörner, J. A. Van Bokhoven and Z. Abbas, Langmuir, 2013, 29, 5023–5029 CrossRef CAS PubMed.
- R. Arrigo, M. Hävecker, M. E. Schuster, C. Ranjan, E. Stotz, A. Knop-Gericke and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 11660–11664 CrossRef CAS PubMed.
- V. Pfeifer, T. E. Jones, J. J. V. Vélez, R. Arrigo, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2017, 8, 2143–2149 RSC.
- A. Kolmakov, E. Strelcov, H. Guo, A. Yulaev, B. Hoskins, G. Holland, S. Nemsak, C. M. Schneider, J. Wang, N. Appathurai, S. Urquhart, S. Gunther, L. Gregoratti, M. Amati and M. Kiskinova, Microsc. Microanal., 2018, 24, 64–67 Search PubMed.
- R. S. Weatherup, B. Eren, Y. Hao, H. Bluhm and M. B. Salmeron, J. Phys. Chem. Lett., 2016, 7, 1622–1627 CrossRef CAS PubMed.
- R. Mom, L. Frevel, J.-J. S. Velasco-Vélez, M. Plodinec, A. Knop-Gericke and R. Schlögl, J. Am. Chem. Soc., 2019, 141, 6537–6544 CrossRef CAS PubMed.
- J. J. Velasco-Velez, V. Pfeifer, M. Hävecker, R. S. Weatherup, R. Arrigo, C. H. Chuang, E. Stotz, G. Weinberg, M. Salmeron and R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 14554–14558 CrossRef CAS PubMed.
- M. Al-Hada, M. Amati, H. Sezen, L. Cozzarini and L. Gregoratti, Catal. Lett., 2018, 148, 2247–2255 CrossRef CAS.
- S. Nappini, A. Matruglio, D. Naumenko, S. Dal Zilio, F. Bondino, M. Lazzarino and E. Magnano, Nanoscale, 2017, 9, 4456–4466 RSC.
- T. Burgi, J. Catal., 2005, 229, 55–63 CrossRef.
- C. P. Stemmet, J. C. Schouten and T. A. Nijhuis, Chem. Eng. Sci., 2010, 65, 267–272 CrossRef CAS.
- C. D. Bain, J. Chem. Soc., Faraday Trans., 1995, 91, 1281–1296 RSC.
- A. J. Hopkins, C. L. McFearin and G. L. Richmond, Curr. Opin. Solid State Mater. Sci., 2005, 9, 19–27 CrossRef CAS.
- S. B. Waldrup and C. T. Williams, Catal. Commun., 2007, 8, 1373–1376 CrossRef CAS.
- Z. Lu, A. Karakoti, L. Velarde, W. N. Wang, P. Yang, S. Thevuthasan and H. F. Wang, J. Phys. Chem. C, 2013, 117, 24329–24338 CrossRef CAS.
- J. F. Gomes, K. Bergamaski, M. F. S. Pinto and P. B. Miranda, J. Catal., 2013, 302, 67–82 CrossRef CAS.
- R. Schlögl, in Basic Principles in Applied Catalysis, ed. M. Baerns, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 321–360, DOI:10.1007/978-3-662-05981-4_9.
- N. S. Gould and B. J. Xu, Chem. Sci., 2018, 9, 281–287 RSC.
- J. Bredenbeck, A. Ghosh, H. K. Nienhuys and M. Bonn, Acc. Chem. Res., 2009, 42, 1332–1342 CrossRef CAS PubMed.
- C. Hammond, Green Chem., 2017, 19, 2711–2728 RSC.
- A. Adeyemi, J. Bergman, J. Brånalt, J. Sävmarker and M. Larhed, Org. Process Res. Dev., 2017, 21, 947–955 CrossRef CAS.
- H. Guo, E. Strelcov, A. Yulaev, J. Wang, N. Appathurai, S. Urquhart, J. Vinson, S. Sahu, M. Zwolak and A. Kolmakov, Nano Lett., 2017, 17, 1034–1041 CrossRef CAS PubMed.
- M. A. Newton, Catalysts, 2017, 7, 58 CrossRef.
- M. A. Ardagh, T. Birol, Q. Zhang, O. A. Abdelrahman and P. J. Dauenhauer, Catal. Sci. Technol., 2019, 9, 5058–5076 RSC.
- M. A. Ardagh, O. A. Abdelrahman and P. J. Dauenhauer, ACS Catal., 2019, 9, 6929–6937 CrossRef CAS.
- G. Joshua, A. Matthew, S. Manish, B. Sean, D. Paul and A. Omar, Resonance-Promoted Formic Acid Oxidation via Dynamic Electrocatalytic Modulation, 2020 Search PubMed.
- J. R. Kitchin, Nat. Catal., 2018, 1, 230–232 CrossRef.
- B. R. Goldsmith, J. Esterhuizen, J.-X. Liu, C. J. Bartel and C. Sutton, AIChE J., 2018, 64, 2311–2323 CrossRef CAS.
- M. Basham, J. Filik, M. T. Wharmby, P. C. Y. Chang, B. El Kassaby, M. Gerring, J. Aishima, K. Levik, B. C. A. Pulford, I. Sikharulidze, D. Sneddon, M. Webber, S. S. Dhesi, F. Maccherozzi, O. Svensson, S. Brockhauser, G. Naray and A. W. Ashton, J. Synchrotron Radiat., 2015, 22, 853–858 CrossRef PubMed.
- E. Achilli, A. Minguzzi, A. Visibile, C. Locatelli, A. Vertova, A. Naldoni, S. Rondinini, F. Auricchio, S. Marconi, M. Fracchia and P. Ghigna, J. Synchrotron Radiat., 2016, 23, 622–628 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |