
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inhibition in multicopper oxidases: a critical review†
Morgane
Valles‡§
 *ab,
Amirah F.
Kamaruddin‡
acd,
Lu Shin
Wong
ab and
Christopher F.
Blanford
*ac
*ab,
Amirah F.
Kamaruddin‡
acd,
Lu Shin
Wong
ab and
Christopher F.
Blanford
*ac
aManchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: mvalles@ibecbarcelona.eu; christopher.blanford@manchester.ac.uk
bDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cDepartment of Materials, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
dDepartment of Chemistry, Faculty of Science, Universiti Teknologi Malaysia, Skudai, 81310, Johor, Malaysia
First published on 17th July 2020
Abstract
Multicopper oxidases (MCO), such as laccases and bilirubin oxidases, are catalysts of wide interest in a range of biotechnological applications, especially bioremediation and energy transduction. Many of these processes take place in the presence of dissolved species, particularly halides, at concentrations high enough to inhibit the catalytic activity of MCOs. Despite this, MCO inhibition is rarely considered the focus of reviews on these enzymes. This review critically analyses the scientific literature on modes of MCO inhibition and current hypotheses on the structural origins of inhibition. The article provides a comprehensive overview of what is known from the different techniques applied to the study of MCO inhibition. These techniques include solution-based enzymatic assays, electrochemical methodologies, various spectroscopic approaches, X-ray crystallography and computational analyses. This review highlights gaps in the literature, identifies nine challenges to accurately benchmarking MCO inhibition and gives recommendations to address these challenges and to interpret published data on the subject.
 Morgane Valles | Morgane Valles holds a BSc in Biology (2013) and an MSc in Applied Biosciences and Biotechnology (2014) from Imperial College London. She then moved to the University of Manchester for a BBSRC-sponsored doctoral training program under the direction of Drs Wong and Blanford, completing her thesis on engineering charge transport in multicopper oxidases in early 2019. That same year, after a brief placement in the lab of Dr Miguel Alcalde in Madrid, she took up a postdoctoral position in the group of Dr Samuel Sánchez at IBEC Barcelona, working on developing enzyme-powered micro- and nanomotors for biomedical applications. |
 Amirah F. Kamaruddin | Amirah F. Kamaruddin is currently a PhD candidate under the supervision of Drs Wong and Blanford. She received her bachelor's and master's degrees in chemistry from Universiti Teknologi Malaysia, in 2014 and 2017, respectively. She worked at the pharmaceutical company Ain Medicare after she finished her undergraduate studies. Her work focused on separations using nanomaterials such as mesoporous carbons. She is currently continuing research begun by Dr Valles on applying electrochemical methods to study inhibition in multicopper oxidases. She will move into a lectureship at Universiti Teknologi Malaysia, Johor, Malaysia upon successful completion of her PhD. |
 Lu Shin Wong | Lu Shin Wong graduated with his BPharm (Nottingham), Postgraduate Diploma in Clinical Pharmacy (Bradford) and PhD (Southampton, with Prof. Mark Bradley) in 1997, 2001 and 2005, respectively. Subsequently, he undertook postdoctoral research at the University of Manchester with Prof. Jason Micklefield on biomolecular array technologies. In 2008, Lu Shin was awarded an EPSRC Life Science Interface research fellowship to work with Prof. Chad A. Mirkin at Northwestern University. In 2011, he returned to the UK and was appointed to the Department of Chemistry at the University of Manchester, where he is now a senior lecturer. |
 Christopher F. Blanford | Chris Blanford studies and engineers the interface between biomacromolecules and conductors. He received his B.S. in chemical engineering from the University of Notre Dame in 1995 and his Ph.D. in 2000 from the University of Minnesota – Twin Cities working under Andreas Stein and C. Barry Carter. He then held post-doctoral appointments at the University of Oxford's Department of Chemistry, first with Robert G. Denning then with Fraser A. Armstrong. In 2008, he was awarded a Career Acceleration Fellowship from the UK's Engineering and Physical Sciences Research Council. He joined the University of Manchester's Department of Materials in 2011. |
1. Introduction
Multicopper oxidases (MCOs) are a broad family of oxidoreductases that effectively harness the strong oxidizing power of O2 for the catalytic oxidation of a broad range of substrates including monomeric phenolic compounds and polyphenols (including lignin), diamines and metal ions.1,2 This reaction is made possible by the arrangement of the four Cu co-factors, characteristic of all MCOs, into two spatially separated but electronically connected active sites.1–3 A single-copper site near the protein surface extracts electrons from the soluble organic substrate or metal ion and supplies them to a buried three-copper cluster where O2 binds and is reduced to water.The MCO family includes ascorbate oxidases (AO), laccases, bilirubin oxidases (BOD), bacterial endospore coat proteins such as CotA, and various metal oxidases, such as ceruloplasmin, Fet3p ferroxidases, the copper efflux protein CueO and the manganese oxidase CumA.1,4–7 MCOs are widely distributed in nature and can be found in all kingdoms of life.4,5,8–12
The many desirable properties of multicopper oxidases, such as their broad substrate range, their strong oxidizing power (i.e., high reduction potential) and, in the case of BODs and bacterial MCOs, their near-neutral pH optimum and halide tolerance, make them suitable candidates for a variety of industrial applications. These primarily include bioremediation (29% of publications); bioelectrocatalytic devices, such as biofuel cells and biosensors (22%); the processing of textiles (20%), and pulp and paper processing (11%).13
Applied research into MCOs has been driven mainly by their applications in bioremediation of industrial waste streams14–18 including polychlorinated biphenyls (PCBs),19 phenolic compounds,19–21 and industrial dyes.22,23 However, these complex mixtures are rich in chemical species, particularly halides, that can inhibit MCO biocatalysis. Enzyme inhibition is known to be challenge in bioremediation, textile processing and in bioelectrocatalytic systems, which together represent 71% of the industrial applications of MCOs.
In this review, we distinguish between compounds that bind specifically to MCOs and result in a reversible or irreversible loss of catalytic activity (herein labelled as “inhibitors”, in line with international standards)24,25 and compounds that bind non-specifically to many classes of protein (if not all) and lead to enzyme inactivation (e.g., denaturants, chelating agents, reducing agents, chaotropic agents). We use “irreversible inhibitors” for those that act specifically on a particular protein class. Enzyme inactivation can be both reversible and irreversible. For example, a reducing/oxidizing agent may block a redox-active protein in an oxidation state such that it is not able to turn over the substrate readily, but catalytic activity may be regained upon electrochemical re-oxidation of the enzyme.
The most commonly studied MCO inhibitors are halides and pseudohalides (e.g., azide and cyanide),26–31 but the activity of MCOs is also affected by metal ions (e.g., Ca2+, Mn2+, Fe2+/3+, Co2+, Cu+/2+, Ag+, Zn2+, Hg2+, Al3+ and As3+/5+),32–40 reactive oxygen species (ROS) such as O2− and H2O2,41 reducing agents (e.g., L-cysteine, dithiothreitol, 2-mercaptoethanol),36,42–44 chelating agents (e.g., EDTA),36,41,44,45 chaotropic agents (e.g., SDS and urea)32,36,41,45,46 and others (e.g., short alcohols, ionic liquids and polymer end groups).41,47–50 Misleadingly, these enzyme inactivators are routinely surveyed alongside the more specific inhibitors of MCO activity.32,36,37,41,44–46,51 This review focuses on reversible MCO inhibitors, and does not examine loss of catalytic activity resulting from protein unfolding or loss of Cu co-factors.
Inhibitors may bind near either active site (i.e. either the electron-donor or oxygen binding sites), or may allosterically regulate enzyme function. The possibility of multiple inhibitor-binding sites makes it challenging to derive mechanistic information about the site and mode of inhibition using a single technique. The variety of methods and conditions used to study inhibition in MCOs has led to a wealth of data, but some of the results contradict each other, and values for the strength of binding differ by orders of magnitude in some cases (section 3.4).
Since Solomon's influential review of multicopper oxidases and oxygenases in 1996,1 there have been numerous review articles that cover MCOs.9,52–58 The dozens of reviews that were published from 2017 to the present include both general summaries13,59,60 and more focused reviews that cover wastewater and soil remediation (including dye and pharmaceutical degradation);38,39,41,61–75 pulp, paper, lignin and wood processing (including lignin valorization);67,68,76–80 chemical synthesis;80–84 polymer synthesis (including lignin formation);79,85–89 enzyme-catalyzed fuel cells, (a.k.a., “biofuel cells”);90–95 aspects of invertebrate and mammalian metabolism;96–99 biosensors;100–102 slate degradation;103 scaling up enzyme production;73,104,105 and catalytic mechanism.106 Some of these cover inhibition only as a brief discussion,60,61,66,69,70,73,79,92,105,107,108 but three make special mention of inhibition in MCOs in a subsection.41,80,94 Baldrian's review published in 2006 included a then-comprehensive list substrates and inhibitors,56 but this list has yet to be updated. Apart from these examples, there is not a single published review that focuses solely on MCO inhibition, despite the ca. 1400 research articles describing some form of MCO inhibition.
This review aims to address this gap in the study of MCO inhibition by presenting a critical, holistic examination of the techniques used to study inhibition, quantitatively and qualitatively, to determine the sites and strengths of inhibitor binding; and to provide some best-practice recommendations. In the following section, this review will describe the classification and structure of MCOs, along with a list of MCO-specific considerations, providing the reader with the tools to understand the challenges of studying MCO inhibition. Sections 3–7 then cover and critique the methods used to investigate inhibition in MCOs, before section 8 presents our proposals for benchmarking in this field.
2. Background
2.1. Multicopper oxidases
Laccases are the largest MCO group, and have been identified in a range of organisms, including plants, fungi, insects, lichen and sponges; and can oxidize a structurally diverse set of aromatic substrates.1,4,5,8–10,112,113 The biological roles, and hence expression patterns, of laccase isoforms are often overlapping in their native organisms, making it difficult to isolate single isoforms from the native organism for detailed study.114 Laccases are also classified according to their color. Typically, laccases are blue (due to the ligand-to-metal charge transfer (LMTC) at the T1 site, see section 2.1.2), but “white” and “yellow” laccases have also been described.9,51,115–120 These atypical laccases have the same function and are structurally very similar to blue laccases (they are often isoenzymes of blue laccases), but differ in their spectroscopic properties (section 2.1.2).115,116,121–124
BODs are a sub-group of MCOs characterized by their ability to catalyze the oxidation of bilirubin to biliverdin, and have been found in bacteria and fungi.125 MCOs are not systematically tested for BOD activity, so it is likely that many laccases can also act as BODs.94 Although they share much of the same structural and spectroscopic characteristics with laccases, BODs differ from laccases in their biophysical properties. BODs typically have higher thermostability, pH optima nearer to neutral, and higher chloride tolerance than laccases.46,94,125–133
Bacterial MCOs can be categorized into two groups based on their functionality: metallo-oxidases, which oxidize cytotoxic metals ions, such as Cu(I), Fe(II) and Mn(II), and laccase-like bacterial MCOs.134–137 Laccase-like bacterial MCOs can be mostly found in two bacterial genera: Bacillus (CotA-like) and Streptomyces (two-domain or “small” laccases). CotA-like bacterial MCOs have a wider substrate range than metallo-oxidases and also display characteristics of BODs, including high chloride tolerance.138,139
2.1.2.1. Co-factor organization. Although there is little sequence homology between different classes of MCO,4,140 the arrangement and coordination geometry of their four copper co-factors is highly conserved,1 and underpins their mechanism of catalysis.
The Cu atoms are described as various “types” by the UV-vis and EPR properties.1 One type 1 (T1) Cu is responsible for the enzymes' blue color and oxidizing the electron-donor substrates. A ligand-to-metal charge transfer (LMCT) between the Cys–S and the T1 copper results in an intense absorption (5000 M−1 cm−1) band at ∼600 nm.1,9 In addition to the cysteine, the T1 copper is coordinated by two or three additional residues: two highly conserved histidine residues, and in some cases, an axial methionine (Fig. 1). MCOs with axial methionine residues typically have a low redox potential (ErevT1 < 0.46 V vs. NHE),141 while in MCOs with medium (ErevT1 = 0.46–0.71 V vs. NHE) or high redox potential (ErevT1 = 0.73–0.79 V vs. NHE), such as “blue” laccases, the methionine is replaced by a non-coordinating leucine or phenylalanine.13 The three other Cu ions, two type 3 (T3) and one type 2 (T2), are arranged to form a trinuclear cluster (TNC) (Fig. 1) and catalyze the reduction of one O2 to two H2O (Section S2†). The TNC receives electrons from the T1 Cu (through the highly conserved His–Cys–His tripeptide, cyan residues in Fig. 1B) and has an open coordination site allowing it to make bridged intermediates necessary for the oxygen reduction reaction (ORR).142
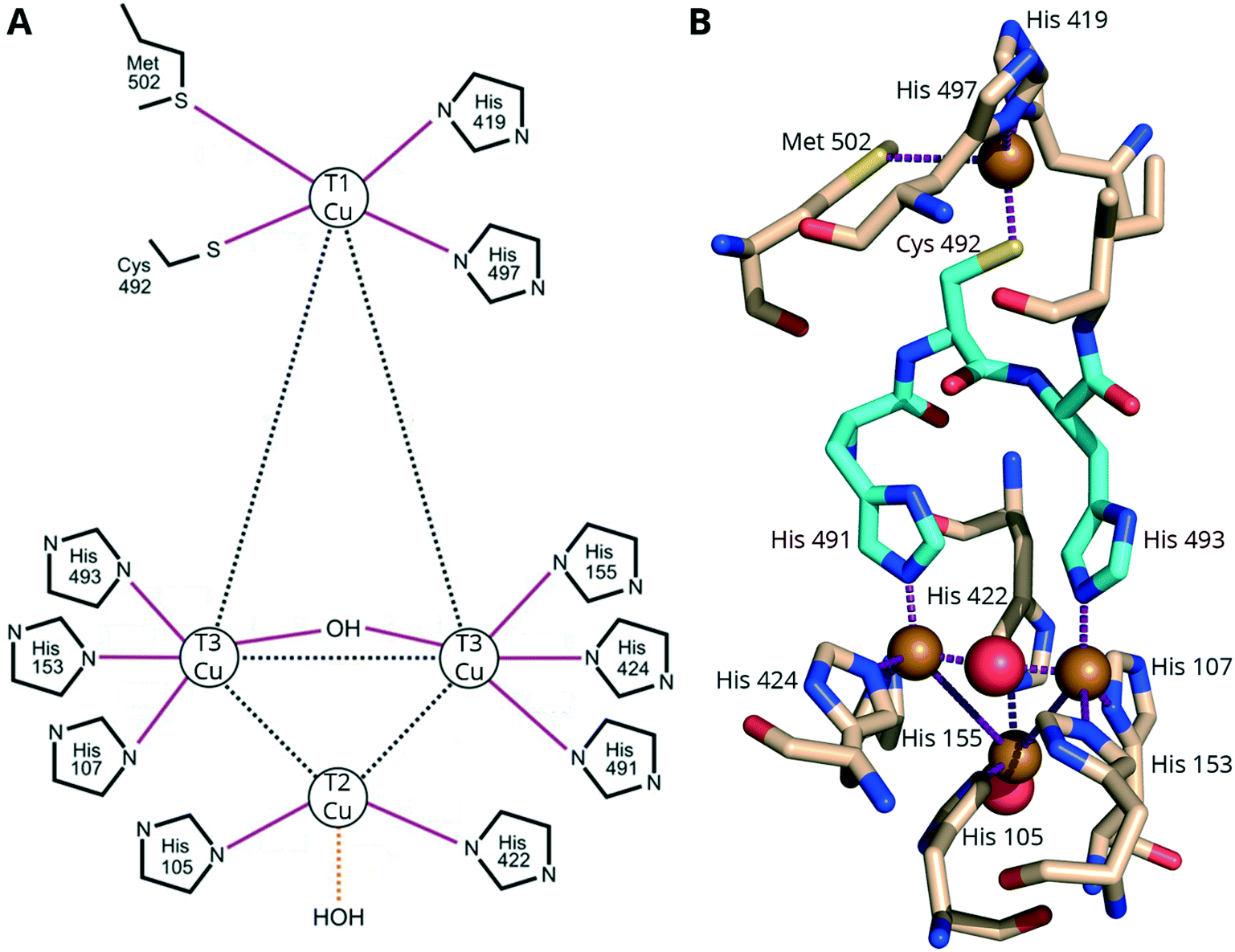 | ||
| Fig. 1 Copper coordination in CotA from B. subtilis. A Schematic adapted with permission from ref. 143. Copyright 2003, American Society for Biochemistry and Molecular Biology. B A representation from the associated X-ray crystal structure (PDB: 1gsk, see section 4) rotated 180° along the vertical axis to make the TNC more visible. Yellow and red spheres represent copper and oxygen atoms, respectively. Red, blue and yellow atoms in the amino acid stick models represent oxygen, nitrogen and sulfur, respectively. The His–Cys–His linkage between the T1 Cu and TNC is shown in cyan. | ||
Despite having the functional characteristics of “blue” laccases (i.e., they are able to oxidize characteristic laccase substrates), “white” and “yellow” laccases lack the absorption band 600 nm.9 “Yellow” laccases have the same copper content as “blue” laccase, but form an adduct with an electron-donor radical substrate, which distorts the structure of the T1 site such that the LMCT is perturbed or even eliminated.124 “White” laccases, on the other hand, do not have the same copper content as “blue” laccases. Instead, they are generally characterized by their metal heterogeneity.116,123 For example, manganese replaces the T1 Cu in “white” laccase from Trametes hirsuta.123
2.1.2.2. MCO surface and channels. Compared to the copper coordination sites in MCOs, there has been relatively little focus on how the surface chemistry or quaternary structure influences steady-state kinetics and inhibition. The surface residues near the T1 Cu can vary considerably depending on the MCO due to differences in substrate specificity. The binding pocket for electron-donor substrates in a BOD from Myrothecium verrucaria, for example, is particularly hydrophilic and has a medium reduction potential (ErevT1 = 0.66 V vs. SHE), which is unusual for an MCO containing an axial methionine ligand (section 2.1.2.1).144,145 Akter et al. identified a covalent bond between a surface tryptophan and one of the histidine residues coordinating the T1 Cu, and proposed that it raises the redox potential of the T1 site to match that of the bilirubin substrate.146 In a “yellow” laccase from Sclerotinia sclerotiorum, Mot et al. identified a surface tyrosine residue at the T1 site, which forms a reversible adduct with the electron-donor substrate.124
The surface residues near the T1 site also influence the dipole moment of MCOs. Lojou et al. found that BODs from M. verrucaria and Bacillus pumilus with opposing dipole moments were attracted to oppositely charged electrode surfaces, such that the MCO with greater negative charge around the T1 site was attracted to positively charged functional groups on the electrode surface, and vice versa.147 This discovery suggests that control over orientation can block inhibition sites, and not only help unravel the structural origins and modes of inhibition (Section S1†) but also convey tolerance to inhibitors in biotechnological applications such as enzyme-catalyzed fuel cells.148
A methionine-rich helix found near the T1 site is a characteristic feature of MCOs with cuprous oxidase activity (such as CueO from E. coli) and is the binding site of a fifth copper ion.134,149–154 This additional Cu is labile and essential for cuprous oxidation. Deletion of the methionine-rich region in CueO reduces cuprous oxidase activity by 90% compared to the wild-type, but also significantly enhances these enzymes' activity towards organic electron-donor substrates.155 Thus, the hypothesized role of the methionine-rich region is to obstruct the T1 site and restrict substrate specificity to cuprous ions.
Channels allow the O2 substrate access to the TNC and the H2O product to exit the protein.143,156,157 In most MCOs for which the structure has been determined, there are two channels, one that is narrow and runs from the surface of the protein to the T2 Cu and another broader channel that starts at one of the T3 Cu ions and ends at the surface (Fig. 2). Some research groups assign the former of the two channels as the water exit channel and the latter the O2 entrance route.158 However, evidence for this proposal is sparse because it is experimentally difficult to determine O2 diffusion routes in protein matrices.159 Nevertheless, molecular dynamics simulations and molecular probes such as xenon have shown that hydrophobic residues in the channel leading up to the T3 Cu ions make O2 diffusion by this route thermodynamically favorable.160,161
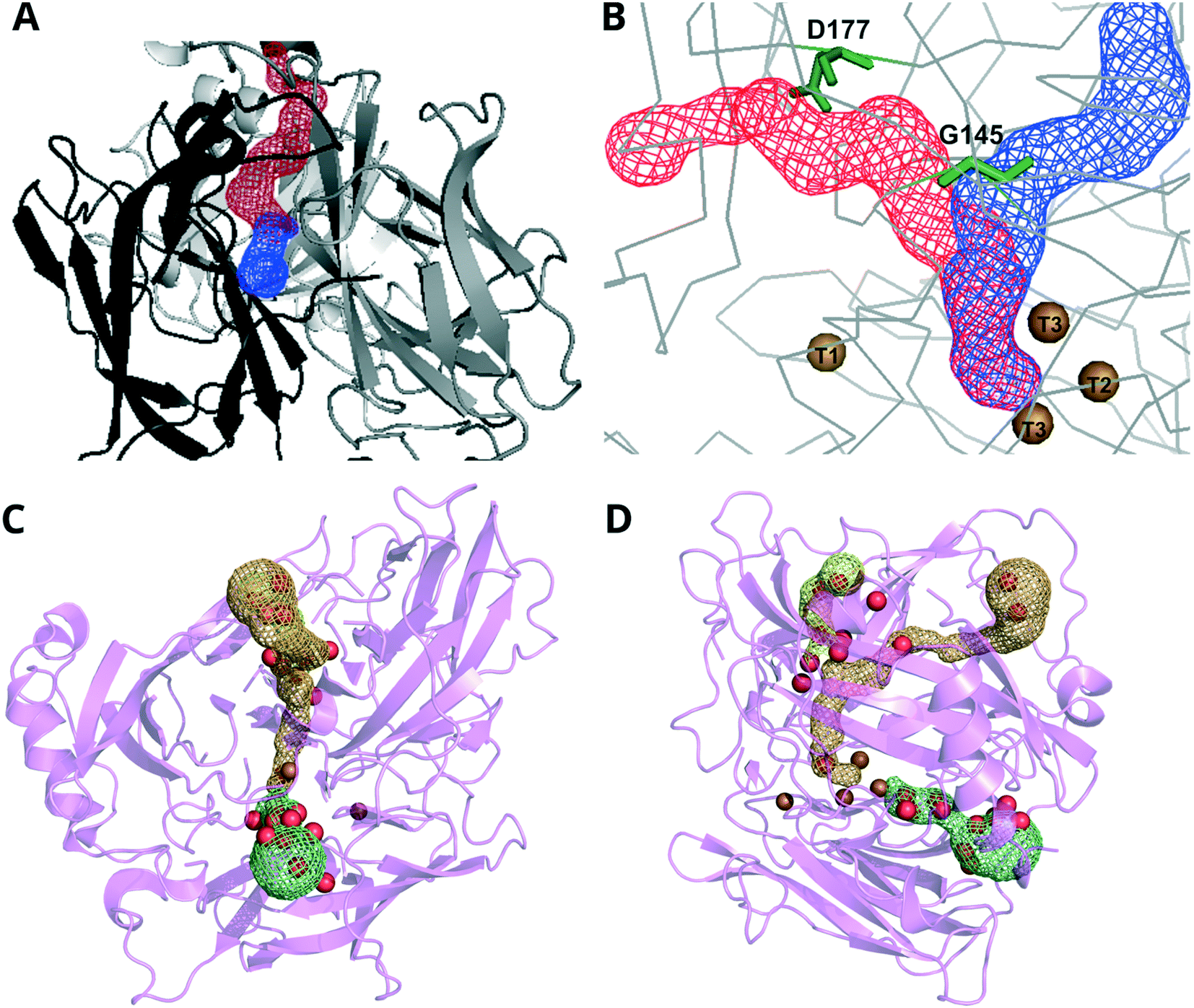 | ||
| Fig. 2 Representations of the channels between the surface and the TNC (colored meshes) in A and B CueO from Ochrobactrum sp. (PDB: 6evg) and C and D BOD from M. verrucaria (PDB: 2xll).145 Images in A and C show a view looking down to the TNC. Images in B and D are rotated 90° and positioned with the T1 Cu to the left of the representation. Key: Cu = brown, O (water) = red. Panels A and B adapted with permission from ref. 162. Copyright 2018, John Wiley & Sons. | ||
Inhibitors that compete with O2 are thought to gain access to the TNC by permeation through these channels.163–165 Thus, enzymes with narrower, more negatively charged channels are likely to have greater tolerance to anion inhibition.163,166 Xu suggested that plant laccases have increased sensitivity to (pseudo)halide inhibition because their solvent channels relatively wide, but there is no crystal structure of a plant laccase to validate this hypothesis.163,166 In contrast, bacterial MCOs have increased tolerance against small anion inhibitors, a characteristic attributed to the negative residues at the channel opening that may electrostatically repel anionic inhibitors.165 Mutations made in the second coordination sphere of the T1 site in a high redox potential laccase were found to confer higher tolerance to Cl− inhibition, indicating that residues that are distant from the TNC (but possibly found within the solvent channels) can have a role in tolerance against anion inhibition.167
2.2. A priori considerations for MCO inhibition
Linear data representations such as Lineweaver–Burk168 and Dixon169 plots are frequently used for to calculate an enzyme's kinetic parameters, mode of inhibition and inhibitor dissociation constants.170 Non-linear fits, algorithms that minimize residuals or χ2, allow uncertainty estimates to be assigned to kinetic and inhibition values and allow the contribution of data points in a fit to be weighted based on measurement precision. Several approaches have been used for model discrimination, such as an F-test and the Akaike information criterion.171,172
However, MCO catalysis is more complex than the simplified Michaelis–Menten foundation described Section S1.†
First, for MCOs in solution, there are three substrates: an electron donor, protons, and the terminal electron acceptor, O2. (For electrochemically driven MCO O2 reduction, discussed in section 4.1, the electrode on which the MCO is immobilized supplies the electrons.) Consequently, interpreting steady-state solution-based analyses requires deconvoluting the effects of substrate and product diffusion, differing binding strengths of multiple reactants and products, and the presence of at least two sites for competitive inhibitor binding. For O2 reduction, the values of kcat and KM for MCOs also depend on the electrochemical driving force for reduction.144 Electron-donor substrates typically used for solution-based spectrophotometric assays of MCO kinetics differ in their reductive driving force, and this driving force may change with pH. So, electron-donor substrates will produce different kinetic parameters for the same enzymes not only because of their binding affinity but also because of their effective reduction potentials.173
Second, MCOs can enter at least one “resting” state (the RO intermediate in Fig. S1†).174–176 This state is a form of an MCO that requires more reductive driving force for it to carry out O2 reduction compared to enzyme in the main catalytic cycle. Steady-state catalysis can take tens of seconds to reach depending on the source of and driving force provided by reducing equivalents, the type of MCO, the solution pH and the presence of inhibitors.174 The time for the MCO catalysis to reach steady state is comparable to the time for a typical solution-based spectrophotometric assay, which can lead to apparent reaction rates that are lower than their steady-state values.
Third, O2 is poorly soluble in water. The Henry's law constant for O2 in pure water is 13 μmol m−3 Pa−1 and it has a temperature coefficient (ΔsolnH/R) of −1500 K.177 So, water in equilibrium with air at atmospheric pressure contains 0.28 mM O2 at 25 °C and 0.40 mM at 4 °C. Many MCOs of industrial relevance (that is, kcat ≫ 1 s−1) have KM values for O2 close to these concentrations,144 so the rate of O2 reduction is limited to about half its maximum. As a result, O2 is almost never in large excess for MCOs with high KM,O2 values. The rate of O2 reduction cannot therefore be factored out in the determination of the parameters related to donor oxidation unless the limiting rate for O2 reduction is extrapolated from the rate of reaction at several [O2] values and in an excess of electron-donor substrate.
Fourth, inhibition strength determined by spectroscopic (section 3.1) or electrochemical (section 4.3) techniques is frequently reported as an IC50 value, that is, the concentration of inhibitor required to achieve 50% inhibition. Eqn (1) presents a common way in which relative inhibition is calculated.
 | (1) |
The IC50 value for an inhibitor–enzyme combination can be a useful and easily determined metric for a single enzyme's performance under a fixed set of conditions, but it cannot be used to compare the strength of inhibitor binding between MCOs or even different measurement conditions for the same MCO. The value of IC50 depends not only on the strength of inhibitor binding (Ki), but also on the substrate concentration relative to KM (which varies with pH and temperature) and the mechanism of enzyme inhibition. For example, the apparent IC50 values – even with the same combination of MCO, pH, inhibitor and electron donor – will differ if the O2 comes from air or pure O2 because the enzyme's uninhibited catalytic rate will be below vmax.
An apparent IC50 value is always larger than the true value because [O2] is never infinite (Section S1†). The mathematical relationships between Ki and either the true or apparent IC50 are given in Table S2 (ESI†). Values of KM,O2 varies with MCO, pH and driving force for reduction,144 so IC50 values are biased estimates of variations in inhibitor binding strength compared to Ki.178
There are two steps in the O2 reduction reaction of MCOs that require protons to be donated by acidic amino acids near the TNC (e.g., D116 and E498 in Fig. S1, ESI†).182 These steps become rate-limiting at higher pH values.144 Cracknell and Blanford developed a mathematical model that captures these differences in MCO pH optima.174,179 The O2/H2O reduction potential ( ) decreases by about 59 mV pH−1 unit, while ErevT1 varies by only ≤0.1 V over 8 pH units.144,179,183 For high-potential MCOs, the
) decreases by about 59 mV pH−1 unit, while ErevT1 varies by only ≤0.1 V over 8 pH units.144,179,183 For high-potential MCOs, the  potential equals ErevT1 at pH 7–8, introducing an energetically “uphill” electron-transfer step between the donor and O2 and/or the TNC. Low-potential MCOs, in contrast, would still have some driving force for the ORR even under alkaline conditions.
potential equals ErevT1 at pH 7–8, introducing an energetically “uphill” electron-transfer step between the donor and O2 and/or the TNC. Low-potential MCOs, in contrast, would still have some driving force for the ORR even under alkaline conditions.
Halide inhibition in MCOs is pH dependent. As exemplified by the work of Mano et al., the presence of Cl− in concentrations as low as 1.5 mM causes a significant drop in the electrocatalytic current of BOD-mediated O2 reduction at pH 4, while at neutral pH the electrocatalytic current is largely unaffected by Cl− (discussed further in section 4.4).29,184 This behavior has mostly been studied in BODs and low potential MCOs due to their higher pH optimum than fungal laccases.29,125,176,184
3. Solution-based techniques
3.1. Steady-state kinetics
Solution-based assays are routinely used to screen the oxidation activity of an MCO against a range of electron-donor substrates.163,179,185,186 The prevalence of the technique stems from its ease of implementation and the very low enzyme concentrations required to get reliable data. Measurements are typically carried out spectrophotometrically by monitoring changes in UV-vis absorption of an electron-donor substrate such as 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) or a redox-coupled indicator dye. Alternatively, oxygen consumption can be measured using a dissolved O2 sensor such as Clark electrode.Most of the data describing the sensitivity of an MCO to an inhibitor in quantitative terms (i.e., IC50 or Ki) has been acquired by means of solution-based assays. The results of 18 studies reporting quantitative values for MCO inhibition are given in tables in the ESI.†
To date, the most extensive work to study the inhibition kinetics of MCOs using solution-based assays has been reported by Xu.163 In this seminal study, Xu evaluated the inhibitory effect of fluoride, chloride and bromide on ABTS oxidation in six different MCOs: two high-potential laccases, two low-potential laccases, and a BOD, all of fungal origin, and a plant laccase (low redox potential). Xu suggested that the extent of inhibition depended on the size of the anion, with the smallest anions gaining better access to the TNC (order of potency was F− > Cl− > Br−). Xu also observed different potencies (as quantified by a Ki) for the same inhibitor between the different MCOs and attributed these discrepancies to variations in the size of the channels leading up to the TNC (section 2.1.2.2).
Consistent with Xu's hypothesis, the IC50 or Ki values reported for the smaller narrower anionic (pseudo)halide inhibitors (N3−, CN−, F−) are manifold more potent than the larger ions (Cl−, Br−, I−). For example, Holmberg et al. reported Ki for F− inhibition of two isoforms of a Pycnoporus sanguineus MCO (72.4 μM and 18.4 μM for the LAC1 and LAC2 isoforms, respectively, Table S3†) were an order of magnitude lower than the Ki for Cl− inhibition (64.7 mM and 15.2 mM for LAC1 and LAC2, respectively, see Table S4†).187 However, there is a large amount of variations between MCOs for the same inhibitor (as illustrated in each of the tables).
Two factors that Xu did not take into account are the electron-donor used and the pH of the assay solution,163 though both of these variables affect IC50 or Ki values.29,184,188,189 For example, the IC50 for Cl− binding to the enzyme–substrate complex of a T. versicolor laccase was calculated to be 18 times higher at pH 3 than pH 6 (Table S4†).188 Similarly, Garzillo et al. showed a clear electron-donor effect for Cl− inhibition of a T. trogii laccase;190 they reported an IC50 of 5 mM for 2,6-dimethylphenol (2,6-DMP) and 70 mM for ABTS. They observed no difference in IC50 for F− under analogous conditions. The authors considered size effects when comparing F− and Cl−, but their data suggest that the Cl− may bind near the T1 Cu rather than the TNC; the larger dianion ABTS may displace Cl− and produce an weaker enzyme–substrate–inhibitor complex (Ki (ABTS) = 52 mM; Ki (2,6-DMP) = 17 mM).
Comparisons between reports are further complicated by the use of different terms to quantify inhibition, because IC50 and Ki values are not directly comparable (section 2.2.1). Furthermore, variations in the conditions used to quantify MCO inhibition have therefore resulted in widely varying IC50 or Ki values, making cross-species studies challenging.
3.2. Semi-quantitative assays
A full steady-state kinetics characterization of enzyme inhibition is time and labor intensive. Thus, relative comparisons of inhibition are often made by describing the “relative activity” of an enzyme against inhibitor concentration where all other experimental parameters are fixed (section 2.2.1). This relative activity value is generally expressed as a percentage of the enzyme's initial rate of electron-donor oxidation (its “activity”) at a given inhibitor concentration, against its activity in the absence of inhibitor. This semi-quantitative assay is usually employed with a limited range of electron-donor substrates, pH and temperature conditions.31,36,38,39,44,46,139,165,167,189,191–202 Examples of how this data is presented are shown in Fig. 3. | ||
| Fig. 3 Solution-based assays presenting relative enzyme activity for MCO systems that show either enhanced tolerance against or activation by halides. A ABTS oxidation activity of a Candidatus Solibacter usitatus laccase at various concentrations of a Cl− at pH 4 and 25 °C. B and C 2,6-DMP oxidation rate by polyphenol oxidase PPO1 from Marinomonas mediterranea in the presence of NaF (black bars), NaCl (hashed bars) or KBr (white bars). Activity is relative to activity in halide-free buffer. Panel A reproduced with permission from ref. 196. Copyright 2017, Springer Nature. Panels B and C reproduced with permission from ref. 189. Copyright 2005, Elsevier. | ||
Some researchers studying inhibition in terms of relative activity have described an activation phenomenon whereby an inhibitor enhances MCO activity,34,45,46,165,192–194,198,199 exemplified with KBr in Fig. 3B and NaCl in Fig. 3C. Chloride activation is a characteristic that has been increasingly observed in newly discovered MCOs isolated from extremophiles and marine organisms, but its mechanistic origin remains poorly understood.107 For example, Fang et al. found a laccase in a marine metagenomic library and expressed it heterologously in E. coli. They determined the KM and vmax for laccase-catalyzed syringaldazine (SGZ) oxidation with up to 500 mM Cl− (i.e., concentrations that would inhibit most MCOs) and found that the reaction rate increased with concentrations of Cl− up to 80 mM.198 Additionally, this chloride-induced enhancement in the catalytic activity of MCOs appears to depend on pH, reaching optimal efficiency at neutral to slightly alkaline pH.165,194 Solomon et al. recently reported that Cl− binds preferentially to the partially reduced TNC of a human ceruloplasmin under physiological NaCl concentrations and pH.203 They also noted a 10-fold increase in the reaction rate for o-dianisidine oxidation in the presence of 100 mM NaCl (at pH 7), confirming that Cl− can enhance the catalytic activity of ceruloplasmin under certain conditions.
Similarly to Cl− activation, Durand et al. have also observed urea-induced activation of bilirubin oxidation from Magnaporthe oryzae.46 Urea is a chaotropic agent that can irreversibly inactivate enzymes, but it was used at sub-denaturing concentration (<2 M).46,204 The specificity and mode of action of urea against MCOs requires further study.
In addition to these examples, there have been reports of substrate activation for both “yellow” laccases and methionine-rich bacterial MCOs (the latter described in section 4.4). Mot et al. identified a biphasic behavior in the Michaelis–Menten saturation curve of the S. sclerotium “yellow” laccase and associated a “high-affinity phase” (characterized by a lower KM) with the formation of an adduct with the electron-donor substrate, 2,3-dimethoxy-5-methyl-p-benzoquinone (Q0H2). This biphasic behavior was also observed for the enzyme adducts with ABTS, TMB and guaiacol. All the adducts had the same KM in the high-affinity phase for the Q0H2 substrate as the bare yellow laccase.124 The authors therefore suggested that adduct formation with an electron-donor substrate enhances the catalytic properties of the “yellow” laccase.
3.3. Oxygen consumption assays
An alternative to measuring rate by spectrophotometric quantification of changes to electron-donor substrates or their oxidized products is to monitor O2 depletion.205 The technique is most advantageous when studying inhibitors that may bind to the TNC and compete with O2 binding. Despite this use, examples of this type of assay are rare.For laccase from Toxicodendron vernicifluum (formerly Rhus vernicifera) at pH 6, the reductant-dependent Ki of F− (3.1 mM) was similar to that of the reductant independent Ki (5.1 mM), implying that F− is likely to be affecting both substrate oxidation and O2 reduction.206 This agrees with Xu's conclusion that the mode of F− inhibition is mixed (as found using an ABTS substrate at pH 5, Table S3†). At higher pH values, however, the Ki of the O2 reduction step is about an order of magnitude smaller than the reductant-dependent step, which suggests that at neutral to slightly basic pH, F− is predominantly inhibiting O2 reduction. This effect of pH is consistent with later quantum mechanical models of halide binding in the TNC (section 7).27
3.4. Critique
Solution-based methods are the preferred technique used to study MCO inhibition because they are quantitative, do not require complicated apparatus and can be easily executed in a high-throughput format.Most studies describing MCO inhibition in solution report a mixed mode of inhibition This mode was generally assigned by analyzing the graphical characteristics of linear inhibition plots (Section S3†). The type of line plots used to analyze the data could account for some of the inconsistencies found in the inhibition type assigned for a particular inhibitor (fourth column in Tables S3–S5†). For example, analysis using the Lineweaver–Burk method suggested different modes of F− and Cl− inhibition in high-potential laccases187 compared to Hanes–Woolf plots.190 Linear transformations based on reciprocal rates or concentrations can introduce large errors because extrapolation is frequently necessary in order to determine the kinetic and inhibition parameters.207 These limitations have a disproportionate effect on efforts to determine the type of inhibition, because non-competitive inhibition is only discernable from the mixed (unbalanced) case by having its lines intercept on the x-axis.
Some analyses employed the deprecated term “non-competitive”.49,187,200,208,209 (Details on terminology are given in Section S1.†) Thus, cases in which a non-competitive model was assigned need to be critically reanalyzed to determine the relative strengths of the competitive (specific) and uncompetitive (catalytic) components.28,188,190,200
Interpreting inhibition results based on solution-based assays requires considerations beyond those listed in section 2.2.
The reaction products of certain typical MCO substrates, such as the ABTS and 2,6-DMP, have been found to react with sulfhydryl organic compounds (e.g., dithiothreitol (DTT), thioglycolic acid and cysteine), and can form complexes with copper, and therefore are believed to be MCO inhibitors. Indeed, Johannes and Majcherczyk demonstrated that the ABTS cation radical product and the diquinone oxidation product of 2,6-DMP can be reduced by these sulfhydryl compounds43 resulting in an underestimation of the electron-donor oxidation rate of an MCO in the presence of these compounds.196,210,211 Moreover, Zhang and Hess have reported impurities found in commercially available ABTS, specifically precursors and analogs of ABTS that can inhibit the enzymatic activity of peroxidases.212 Therefore, the commercial preparations of the electron-donor substrates themselves might be an underexplored source of MCO inhibition. These confounding factors may be an underlying reason numerous studies report widely varying IC50 and Ki values for (pseudo)halide inhibition in solution, and particularly for chloride inhibition (Table S4†).
The reaction rates of MCOs depend on the electrochemical driving force, which in the case of solution-based assays, is set by the electron-donor substrate (e.g., ABTS).173 Hypothetically, the inhibition mode and sensitivity to an inhibitor could change as a function of the driving force of the dominant oxidation state of the metal co-factors.213,214 For example, the electrostatic force between a monovalent anionic inhibitor (such as F− or Cl−) and an ionic copper co-factor of an MCOs in either +1 or +2 oxidation state will differ by a factor of 2. Small molecules and ionic electron donors also provide different driving forces, so different probes may give different values for Ki and even different modes of inhibition (Table 1). Moreover, for electron-donor substrates that involve a proton-coupled electron transfer, the measured steady-state kinetics of the enzymatic reaction (and hence the Ki) will be influenced by the pH.173 For example, the redox potential of a phenolic substrate decreases with increasing pH (as is the case with SGZ, catechol and caffeic acid in Table 1). The potential of the T1 Cu also has a pH dependence to its reversible potential,179 so the relative driving force for electron transfer from the substrate to the T1 Cu varies with pH. Thus, the Ki obtained for a specific enzyme/inhibitor combination using solution-based assays is only valid for that particular electron-donor substrate and within the conditions of the experiment.
| Electron donor | E°′ (mV vs. SHE) | pH | Buffer | Ref. |
|---|---|---|---|---|
| ABTS | 669 | 4.0 | 0.05 M sodium citrate | 216 |
| 690 | 4.7 | 0.01 M sodium acetate, 0.1 M LiClO4 in 4% (v/v) CH3CN/H2O | 217 | |
| 681 | 5.0 | Britton–Robinson buffer | 218 | |
| 700 | 5.3 | 8 mM sodium 2-(N-morpholino)ethanesulfonate (MES) | 186 | |
| 682 | 9.0 | 0.1 M potassium carbonate | 219 | |
| Syringaldazine | 600 | 4.0 | 0.1 M sodium phosphate | 220 |
| 511 | 5.0 | Britton–Robinson buffer | 218 | |
| 630 | 5.3 | 8 mM sodium 2-(N-morpholino)ethanesulfonate (MES) | 186 | |
| 410 | 7.0 | 0.1 M sodium phosphate | 220 | |
| 396 | 7.1 | NEt4BF4/NMe4OH in 50% (v/v) methanol/water | 221 | |
| 320 | 9.0 | 0.1 M sodium phosphate | 220 | |
| Catechol | 670 | 2.0 | 0.1 M hydrochloric acid (unbuffered) | 222 |
| 570 | 4.0 | 0.1 M sodium citrate | 222 | |
| 608 | 5.0 | Britton–Robinson buffer | 218 | |
| 395 | 7.0 | 0.1 M sodium phosphate | 222 | |
| Caffeic acid | 701 | 2.0 | 50 mM citric acid, 50 mM potassium phosphate | 223 |
| 475 | 5.5 | 50 mM citric acid, 50 mM potassium phosphate | 223 | |
| 380 | 7.4 | 50 mM citric acid, 50 mM potassium phosphate | 223 | |
| Fe(CN6)4−/3– | 414 | 4.75 | 0.1 M sodium acetate | 224 |
| 419 | 5.0 | Britton–Robinson buffer | 218 | |
| 426 | 7.8 | 0.1 M sodium/potassium phosphate | 224 | |
| 415 | 10.0 | 0.1 M sodium borate | 224 | |
| 2,6-DMP | 688 | 5.6 | 0.5 M sodium acetate in 50% (v/v) 2-propanol/H2O | 225 |
4. Electrochemical techniques
4.1. General remarks
Electrochemistry is a powerful tool to probe the kinetics of redox-active proteins.226 In contrast to chemical redox reactions, where the driving force for electron transfer is determined by the electron donor–acceptor pair, the driving force for electron transfer between an electrode and a redox center can be controlled by the electrode potential (i.e., the applied voltage at the working electrode). In the case of studies using immobilized enzymes, known as protein film electrochemistry (PFE), mass-transport effects can be factored out by rotating the electrode.227Detailed introductions to electrochemistry are available in recent primers,228,229 but the points most relevant to understanding electrochemically driven catalysis of enzymes (i.e., bioelectrocatalysis) can be succinctly summarized: (i) electronic current is directly proportional to enzyme turnover; (ii) the thermodynamic driving force for electron transfer is directly proportional to the difference in the reduction potential between an electron donor and acceptor; (iii) to a first approximation, the rate of electron transfer from an electrode increases exponentially with potential difference; and (iv) the IUPAC convention for electrochemical reactions is that more negative potentials equate to more driving force for reductions, and electronic current that drives reduction reactions (cathodic current) has a negative value.
Two electrochemical techniques are principally used to study MCOs: chronoamperometry and cyclic voltammetry (CV). Chronoamperometry is used to visualize the time-dependent response of the catalytic current to changes in the experimental conditions, such as variations in substrate concentration, inhibitor concentration, temperature and pH.29,42,230,231 CV can be used to measure the current response of immobilized enzymes to a scanning potential applied to the electrode.226,232 The shape of the CV (sigmoidal in the simplest case) can be indicative of electron transfer events taking place at the enzyme–electrode interface.226 The potential at which the rate of change in current with potential is highest has been designated Ecat.226 Aspects of the inhibition mechanism can be deduced from scan hysteresis between the reductive and oxidative traces and shifts in Ecat in the presence of an enzyme inhibitor (section 4.4).
4.2. Non-turnover electrochemical measurements
Non-turnover electrochemical measurements are those that measure spectroscopic signals as a function of electrochemical potential in the absence of catalysis. These are run anaerobically in the presence of electron-transfer mediators. An optically transparent thin film electrode can be used to control the system's reduction potential. Alternatively, soluble electron donors or acceptors titrated in and the resultant reduction potential is measured. At the reversible potential (Erev) the copper ion has an equal probability of being in the +1 or +2 state. Values for the reversible potential of the T1 and T2 Cu ions (ErevT1 and ErevT2) have been determined using UV-vis233 or EPR234 spectroscopy (section 5). Electrochemistry allows MCOs to be classified by ErevT1 (section 2.1.1). The values of ErevT1 and Ecat often coincide, consistent with the hypothesis that the reduction T1 Cu controls what happens before TNC reduction, so Ecat is frequently used as a proxy for ErevT1.Examples of spectroelectrochemical measurements of MCOs in the presence of inhibitors are rare. The electrochemical non-turnover signals from three fungal and one tree laccase that were immobilized in an anion-exchange polymer.235 The authors hypothesized that N3− bound to the TNC based on the disappearance of a signal they associated with the T2 Cu in two Trametes laccases.
FTIR spectroelectrochemistry was used to study F− and Cl− binding sites in laccase from T. hirsuta and a BOD from Magnaporthe oryzae, using N3− as an infrared probe.26 Using this technique, the interaction between the inhibitors and copper co-factor can be measured at a specified potential and fixed inhibitor concentrations. The authors concluded that N3− bound only to the oxidized TNC and F− competed with N3− for binding at the TNC (Fig. 4A). The shape of the N3− absorption did not change with Cl− binding, suggesting that Cl− was not a competitive inhibitor of O2 binding under the reported measurement conditions.
 | ||
| Fig. 4 Position of N3− in the MCO TNC based on spectroscopic data. A Model assigned to N3− binding and subsequent F− displacement in T. hirsuta laccase and a BOD from Magnaporthe oryzae based on spectroelectrochemical FT-IR spectroscopy. Adapted with permission from ref. 26. Copyright 2017, Springer Nature. B Spectroscopically effective models for N3− binding in tree laccase based on EPR and (magnetic) circular dichroism measurements. Adapted with permission from ref. 236. Copyright 1990, American Chemical Society. | ||
4.3. Chronoamperometric measurements
Chronoamperometry can be used to determine inhibitor sensitivity of an MCO by quantifying how an increase in inhibitor concentration affects electrocatalytic current. The electrocatalytic current of MCOs is not only sensitive to variations in experimental conditions such as pH and substrate concentration, but also responds rapidly to these changes, so-called “instant dialysis”.226,237 Chronoamperometry is commonly used to evaluate the resistance of an MCO-modified cathode to common halide inhibitors.176,231,238–240 For example, by injecting increasing concentrations of NaCl or NaF into the electrochemical cell over time, both Beneyton et al. and Vaz-Dominguez et al. were able to demonstrate that their MCO-modified electrodes were highly resistant to Cl− inhibition, but not to F− inhibition (Fig. 5A and B).238,239 The authors of both studies suggested that Cl− was competitively binding to the T1 site in the two MCOs studied, because it appears to be only affecting mediated electron transfer (when ABTS is present, Fig. 5C). Conversely, the authors hypothesized that F− is likely to be binding to the TNC because the presence of this inhibitor seems to completely impede the biocatalytic activity of the enzymes. | ||
| Fig. 5 Chronoamperometry traces showing the effect of MCO inhibitors on O2 reduction current. A Effect of halides on B. subtilis CotA in two types of redox polymers, represented by the two lines (conditions: O2-equilibtrated 100 mM phosphate–citrate pH 4.0, 0.1 V Ag|AgCl|3 M KCl, 40 °C). B Effect of incremental Cl− addition the biocatalytic current of T. hirsuta laccase covalently attached to the electrode by an aminophenyl redox mediator (conditions: 50 mM acetate pH 4.2 + 100 mM NaClO4, equilibrated with O2 by stirring, 0.2 V vs. Ag|AgCl|3 M KCl, 27 °C). C Current response of immobilized T. hirsuta laccase to the addition of 0.2 mM ABTS (redox mediator), followed by 150 mM Cl− and 13 mM F− (conditions same as B). Panel A reproduced with permission from ref. 239. Copyright 2011, John Wiley & Sons. Panels B and C reproduced with permission from ref. 238. Copyright 2008, Elsevier. | ||
Milton et al. found similar results for Cl− and F− inhibition of a laccase and a BOD by chronoamperometry, as well as demonstrating inhibition by H2O2.241 In this case, H2O2 was found to reversibly inhibit laccase based on the recovery of activity on addition of the peroxide-degrading enzyme catalase. Catalase can be used to generate supersaturated O2 in solution,242 so some of the apparent recovery could be a result of temporarily elevated O2 concentration. Also, they concluded that H2O2 irreversibly inhibited the BOD, but their results could also be explained by the lower stability of the enzyme on their electrode. Based on the increase in catalytic current after mediator addition, the authors hypothesized that the mode of inhibition was likely to be non-competitive with respect to the T1 site, but acknowledged that further evidence is required.241,243,244
MCOs impregnated within a polymer hydrogel incorporating redox mediators (and thus electronically “wired”) generally display better resistance to halide inhibition than systems in which the enzyme is immobilized on an electrode.128,245,246 These hydrogels have therefore been investigated as a means to confer Cl− resistance for environments which have high Cl– concentrations, such as fuel cells operating in blood.128,246–248 The authors of these studies often attribute this observation to the complexation between the moieties of the redox polymer and the T1 site, shielding this site from halide binding and subsequent inhibition of electron transfer.30,249 This conclusion is consistent with the hypothesis that Cl− not acting on the TNC, but the observed current could also be explained by the mediators allowing direct transfer of electrons to the TNC, bypassing the T1 Cu.
4.4. Cyclic voltammetry measurements
MCO inhibitors can affect the shape of CV traces in a number of ways, which can indicate the binding location of the inhibitor. Fig. 6 illustrates how the presence of Cl− shifts Ecat to more negative potentials (i.e., a higher driving force is required for O2 reduction when inhibitor is present) for a BOD from B. pumilus and a laccase from T. versicolor. This effect diminishes when the MCO is driven by lower, more reducing potentials where the enzyme is more active, so that the oxidative CV trace appears close to the position of the uninhibited enzyme. de Poulpiquet et al. concluded that reductive potentials drove the enzyme to overcome Cl− inhibition, which is why the biocatalytic current recovers on the oxidative sweep of the CV. They also associated Cl− inhibition with the formation of a catalytically inactive “alternative resting” (AR) state (Section S2†), which when exposed to reducing potentials (or an equally reducing chemical agent) regain their activity.175,250 The CV trace of the AR state is characterized by cross-over features as well as a shift in the Ecat from the first to the second scan, and has been observed by others reporting Cl− inhibition of BODs.176,184,251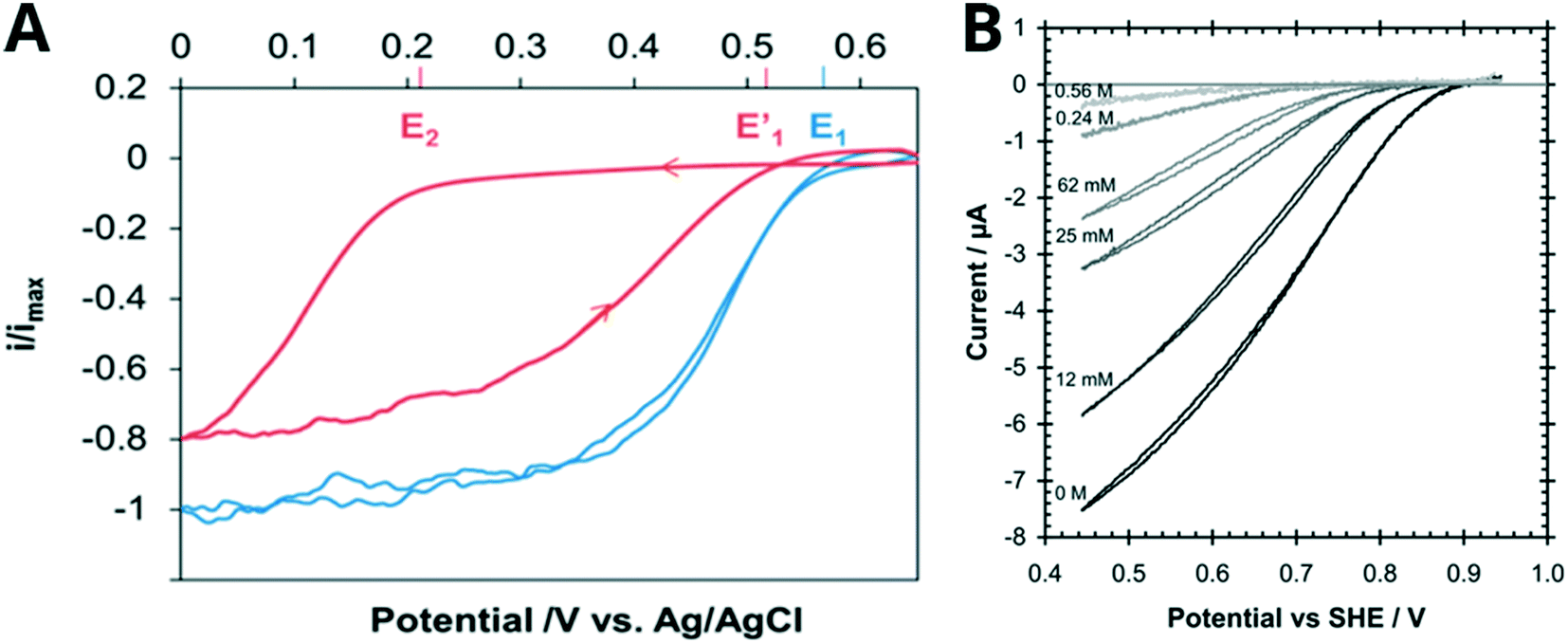 | ||
| Fig. 6 Cyclic voltammograms demonstrating the response in the biocatalytic current of MCOs to increasing inhibitor concentrations. A Bioelectrocatalytic O2 reduction current of a BOD from B. pumilus before (blue line) and after (red) the addition of 15 mM NaCl (100 mM citrate 200 mM phosphate pH 4, scan speed 5 mV s−1). Reproduced with permission from ref. 29. Copyright 2017, American Chemical Society. B Effect of Cl− on the electrocatalytic reduction of O2 by a laccase from T. versicolor at various chloride concentrations (conditions: 0.20 M citrate pH 4.0, 25 °C, scan speed 5 mV s−1). Reproduced with permission from ref. 230. Copyright 2009, The Royal Society of Chemistry. | ||
The forward and reverse traces can loop or cross over each other, as they appear on the 12 mM Cl− trace in Fig. 6B at around 0.5 V. This hysteresis between the reductive and oxidative scans has been proposed to be the result of an inhibitor binding near the T1 site, which creates an electrostatic barrier to interfacial electron transfer to the T1 Cu. Thus, more reducing potentials are required in order to surmount this barrier and initiate catalysis.30,148,184,230
Mano et al. have also demonstrated that the effect of chloride inhibition is less pronounced at high pH (Fig. 7) and argue that this effect could be due to the deprotonation of residues in the vicinity of the TNC and the T1 sites (section 2.2). It was therefore hypothesized that these same residues in their anionic (deprotonated) form block anionic inhibitors from accessing the TNC at high pH.184,252 Cosnier et al. have hypothesized that a histidine found in the proximity of the T1 site is involved in proton-coupled electron transfer, and thus, in its deprotonated form, might also have a protective effect against Cl− inhibition of the T1 site above pH 6.184,253,254
 | ||
| Fig. 7 Cyclic voltammograms showing how the O2 reduction current of a BOD from Magnaporthe oryzae responds to increasing Cl− concentrations in solutions of different pH. A Oxygen-saturated phosphate–citrate buffer pH 4, B oxygen-saturated phosphate–citrate buffer pH 6 and C oxygen-saturated phosphate–citrate buffer pH 7. Additional experimental conditions: GC/MWNT electrode, scan rate: 10 mV s−1, second scan shown. Reproduced with permission from ref. 184. Copyright 2019, American Chemical Society. | ||
Hitaishi et al. used CV measurements to deconvolute the electron relay events of a Thermus thermophilus MCO with Cu+-activated cuprous oxidase activity. Contrary to what was found with CueO, cuprous oxidase activity of the MCO was unaffected by the deletion of the methionine-rich domain, so the Cu+ binding site remains unknown.40
4.5. Critique
PFE measurements solve the problem of having two active sites, both of which may interact with an inhibitor (section 2.2.1). Thus, PFE is very well suited at determining the effects of inhibition on the O2-reduction (rather than electron-donor–oxidation) part of the catalytic reaction. However, quantitative measurement of kcat (i.e., the specific turnover frequency) is challenging because it is difficult to quantify the electroactive enzyme concentration on the surface of the electrode.174,255,256Further complications arise from protein desorption from the electrode surface or by irreversible inactivation of the enzyme.257,258 This behavior was often called “film loss,” but this term implies a purely desorptive process and should be avoided. The resulting decrease in current magnitude must be accounted for with control experiments and replicates. In chronoamperometry experiments, current loss can be modelled as an exponential decay that is independent from catalysis.257,259
The measurements described in section 4.3 appear to provide quantitative information about the capacity of a biocathode to tolerate halide inhibition, but these interpretations are relatively simplistic given the complex catalytic cycle of MCOs. For example, the fact that “wired” biocathodes have better resistance to Cl− inhibition does not necessarily signify that this particular inhibitor is binding at the T1 site.30,249 The evidence from these experiments does indeed suggest that Cl− does not inhibit electron transfer to the “wired” MCO, but this does not imply that this is the only binding site for chloride, nor would it be the case for soluble enzyme. Another possibility is that “wired” MCOs have greater tolerance to Cl− inhibition because the redox polymer blocks chloride from diffusing into the redox-enzyme layer, and thus the local concentration of Cl− is lower than in the bulk solution.
The shift in the Ecat on a cyclic voltammogram in the presence of weaker inhibitors like Cl− has been attributed to inhibitor binding near the T1 Cu, affecting the rate of electron transfer from the electrode to the T1 Cu (section 4.3).29,30,148,184,230,238,239 Stronger inhibitors such as F− greatly decrease the current amplitude of a CV without causing a shift in Ecat, so it often assumed that this inhibitor is competitively binding to the TNC.30,238,239 However, if the former hypothesis is correct, then the lack of shift in Ecat can only be attributed to the inhibitor binding somewhere besides the T1 site rather than necessarily specifically binding to the TNC.
As outlined in section 2.2.1, MCO catalysis is more complex than the basic Michaelis–Menten equation because of the role of oxidation state of the MCO, that is, each enzyme molecule needs to be reduced by four electrons before it can reduce O2. The rate of electron transfer depends on applied potential, so a more complete kinetic scheme would include both a potential-dependent rate constant for copper reduction144 and reversible inhibition equilibria.
5. Spectroscopy
5.1. Spectroscopic evidence for inhibitor binding locations in MCOs
A number of spectroscopic methods have been used to study the inhibition in MCOs, including nuclear magnetic resonance (NMR) spectroscopy; electron paramagnetic resonance (EPR) spectroscopy including multifrequency, pulsed EPR (e.g., electron-nuclear double resonance (ENDOR)); UV-vis absorption spectroscopy; circular dichroism (CD) and magnetic circular dichroism (MCD) spectroscopy; and vibrational spectroscopies, particularly Raman spectroscopy and Fourier-transform infrared (FTIR) spectroscopy.26,236,260–263Research to understand the mechanism of O2 reduction by MCOs has most often used forms of the enzyme that are catalytically inactive, either by removing/substituting of one or more of the copper co-factors, using inhibitor concentrations much higher than stated or apparent Ki, or using frozen enzyme solutions.182 MCO specimens are prepared in this way either to stop the enzyme at a point in its catalytic cycle so it can be probed spectroscopically or because of the physical limitations of the technique.
The archetypical study using catalytically inactive laccase comes from Cole et al.236 In this study, CD/MCD, continuous-wave EPR, UV-vis absorption and magnetic susceptibility measurements were used to investigate F−and N3− binding to a plant laccase from Toxicodendron vernicifluum in which the T1 Cu was replaced by a mercury(II) ion (termed T1Hg). They concluded from EPR analysis that a single F− ion binds equatorially to the T2 Cu in a 50% glycerol solution. This contrasts with earlier work in aqueous solution showing two F− ions sequentially bind to the T2 Cu.264 UV-vis absorption spectra showed that both the T2 and T3 sites are perturbed by F− binding.236 Later work assigned the F− to a μ3 coordination, connected to all three coppers in the TNC, based on pulsed EPR measurements and DFT models.263 For N3−, they deduced that two N3− groups were bound based on EPR and (magnetic) circular dichroism. They assigned a first N3−binding between the T2 Cu and one of the T3 Cu ions. Higher concentrations of N3− led to a second N3− ion binding between the T2 Cu and the other T3 Cu (Fig. 4B). Their conclusions were consistent with earlier work from Morpurgo et al. and Winkler et al., both of whom studied the UV-vis and EPR spectra of F− inhibition of a tree laccase with a full complement of copper ions.265,266 Their model was also consistent with the spectroelectrochemical N3− displacement study described in section 4.2, depicted in Fig. 4A.
Santagostini et al. looked at inhibition involving the T1 Cu site using zucchini AO and fluorophenols that act as competitive inhibitors for ascorbate binding.262 Their enzyme was inactivated by replacing the T2 Cu with Zn leaving the T1 Cu available for EPR and visible absorption analyses. They used paramagnetic relaxation of the 19F nucleus to get information about the Cu–F distances and, combined with computational docking studies, EPR and CD, concluded that the site of inhibition was near the T1 Cu rather than the TNC.
Other inhibitors have been less well characterized spectroscopically. The addition of 20 eq. of 13CN− to tree laccase produced a hyperfine spectrum consistent with two pseudohalides and two histidine residues binding at the T2 Cu.267 Morpurgo and co-workers assigned binding constants for CNS− and CNO− to tree laccase, but the spectroscopic method they used for this determination was not clear.268
5.2. Critique
The spectroscopic techniques covered in this section show where inhibitors can bind, but the results may not be applicable to MCOs actively turning over substrate rather than in a “resting state”. MCOs are known to form “resting states” (Section S2†) that are outside the normal catalytic cycle and have a different arrangement of copper ions in the TNC,175,176,269 so conclusions based on MCOs that were not frozen or studied quickly after active catalysis may not reflect inhibitor locations during catalysis. Thus, spectroscopic measurements of inhibition of MCOs under electrochemical control need to be more prevalent. The Vincent group have used infrared spectrometry in unmediated systems to study the catalytic mechanisms in hydrogen-cycling metalloenzymes270,271 and other metalloproteins.272,273 Their methods could be readily applied to the study of MCO inhibition because MCOs generally remain active toward O2 reduction on immobilization (section 4).6. X-ray crystallography
6.1. Crystallographic structures of MCO–inhibitor complexes
Protein crystallography gives a three-dimensional, near-atomic-resolution snapshot of an enzyme, on its own or bound to substrates or inhibitors.274 X-ray crystallography of MCOs has driven structure–function studies since the first MCO structure was solved in the early 1990s, that of zucchini AO275 and the first laccase structures a decade later.276,277 Not only can the crystal structures directly show a binding site for an inhibitor (and thus suggest a mechanism for inhibition), the structures are used as the basis for computational models of inhibition (section 7). Associated temperature factors (a.k.a. B factors) indicate more positional uncertainty in the atomic coordinates and thus regions where the structure is more flexible, an important consideration for molecular dynamics (MD) simulations (section 7.2).Table 2 lists the MCO structures that include inhibiting species in the Protein Data Bank (PDB) to date.278 The location assigned to charged inhibitors (N3−, F−, Cl−) in crystal structures depends on both the MCO type and the particular inhibitor. For example, Cl− ions have been resolved near the T1 Cu in CotA from B. subtilis (Fig. 8A), between the T3 Cu pair in CueO from E. coli (Fig. 8B), replacing the hydroxide at the T2 Cu apex of the TNC in a laccase from M. albomyces (Fig. 8C). In the case of Aspergillus niger McoG and Lentinus tigrinus laccase, the Cl− ion was found on the surface of the protein (Fig. 8D and E).153,279–281 The Cl− appears most consistently at the TNC apex in fungal laccases, with the exception of the L. tigrinus laccase. In the limited number of MCO structures incorporating F−, the halide also occupied this apical site.282
| PDB code | MCO (source organism) | Inhibitor | Location |
|---|---|---|---|
| 5o4q | 2DL (S. griseoflavus) | N3− | Surface near T1 Cu |
| 4naj, 4nb7 | 2DL (S. lividans) | N3− | Surface near T1 Cu (1 per subunit) |
| 1w6w | CotA (B. subtilis) | N3− | Between T3 Cu pair |
| 1asq | AO (Cucurbita pepo) | N3− × 2 | 1 × between T3 Cu pair, 1 × next to one T3 Cu |
| —283 | Ceruloplasmin (Homo sapiens) | N3− | Next to one T3 Cu |
| See text | Laccase (Steccherinum murashkinskyi) | F− | T2 Cu (TNC apex) (2 sites) |
| See text | Laccase (Steccherinum murashkinskyi) | Cl− | T2 Cu (TNC apex) |
| 1n68 | CueO (E. coli) | Cl− | Between T3 Cu pair |
| 5lww | McoG (A. niger) | Cl− | [ZnCl]+ complex near T1 Cu surface |
| 4q89 | CotA (B. subtilis) | Cl− × 2 | Surface × 2 |
| 2zwn | 2DL (metagenome) | Cl− | Surface |
| 2lhl, 5lhl | 2DL (S. griseoflavus) | Cl− × 2 | Surface × 2 |
| 4n8u | 2DL (S. viridochromogenes) | Cl− × 6 | Surface × 6 |
| 1gw0 & others | Laccase (M. albomyces) | Cl− | T2 Cu apex |
| 4gyb | 2DL (S. lividans) | Cl− × 3 | Surface × 3 |
| 4q8b | CotA (B. subtilis) | Cl− × 2 | Surface × 2 |
| 2qt6 | Laccase (Lentinus tigrinus) | Cl− × 7 | Surface × 7 |
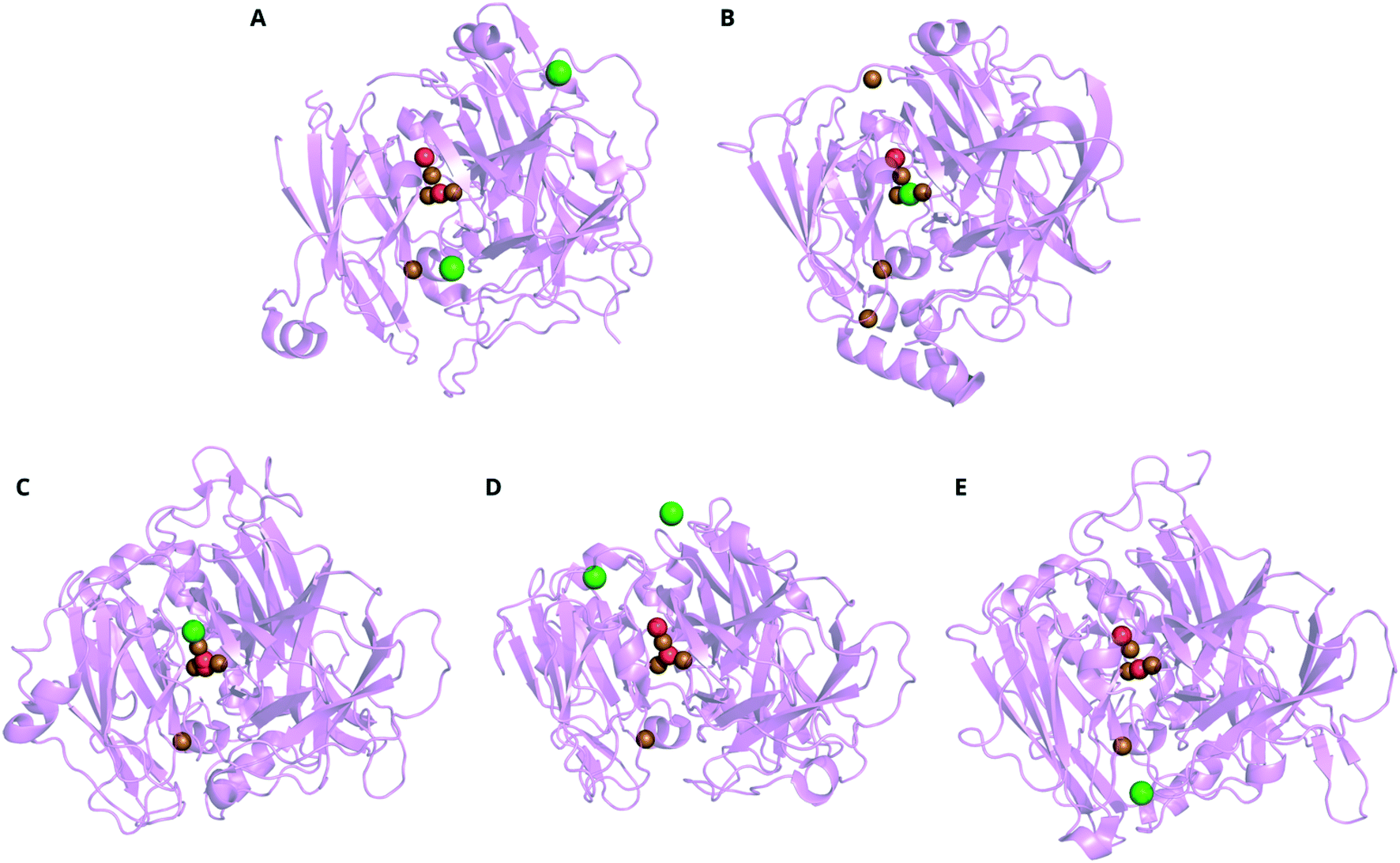 | ||
| Fig. 8 Ribbon representations of representative X-ray crystal structures of MCOs incorporating Cl−, aligned with the TNC toward the top center of each image. A CotA from B. subtilis (PDB: 4q89), B CueO from E. coli (PDB: 1n68),153C laccase from M. albomyces (PDB: 2q9o),279D laccase from L. tigrinus (PDB: 2 qt6)280 and E McoG from A. niger (PDB: 5Llww).281 Key: O = red, Cl = green, Cu = brown. | ||
Like Cl−, the position of N3− is MCO-dependent. In CotA from B. subtilis and zucchini AO, one N3− species is resolved between the T3 Cu pair (Fig. 9A and B). A second N3− appears near the TNC in the latter case. In SLACs for Streptomyces spp., N3− ions are assigned to many locations including one ordered near to the surface by the T1 Cu (Fig. 9C). Zaitsev et al. assigned the position of N3− in human ceruloplasmin to one of the T3 Cu ions,283 similar to the binding in zucchini AO.275 Unlike AO, ceruloplasmin does not appear to bind a second N3− based on soaking studies. Structures in which Cl− and N3− are observed on the protein surface may still contribute to inhibition, because they may influence substrate transport through channels to the TNC or have a coulombic effect on electron transfer to the T1 Cu.
 | ||
| Fig. 9 Ribbon representations of X-ray crystal structures of MCOs incorporating fluoride ions, aligned with the TNC toward the top center of each image. A CotA from B. subtilis soaked in 50 mM buffered NaN3 for 7 d (PDB: 1w6w),164B AO from Cucurbita pepo soaked in 10 mM NaN3 for 24 h (PDB: 1asq)275 and C a two-domain laccase from S. lividans after 180 min soaking in NaN3 at an unspecified concentration (PDB: 4nb7). The representation of the two-domain laccase only shows one monomer of the homotrimer. Key: N = blue, O = red, Cu = brown. | ||
Hakulinen et al. used Xe to infer the position of transient O2-transport sites in a laccase from Melanocarpus albomyces.161 Similarly, there are numerous MCO structures incorporating electron-donor substrates (or mimics of them), including 2,6-DMP in M. albomyces laccase;284 sinapic acid285 and ABTS286,287 in CotA; and phenol,288 xylidine277 and methyl benzoate289 in laccases from Trametes species. These enzyme–substrate complexes can be used to understand the interactions that might lead to competitive binding interactions in the same way that they previously have been suggested as a route to engineering substrate scope in laccases.277
There have been studies on X-ray induced O2 reduction in MCOs from M. albomyces,290Steccherinum murashkinskyi282 and Thermus thermophilus.291 Radiolysis of water provides both reducing equivalents and protons for O2 reduction.291 X-ray crystallography was used to create atomic-resolution snapshots of how ordered Cl− or F− ions at the T2 apex of the TNC (Fig. 10) affected X-ray-induced O2 reduction in S. murashkinskyi laccase.292
 | ||
| Fig. 10 Comparative X-ray structures of a laccase from Steccherinum murashkinskyi presented as (top) cartoon representations of the whole macromolecule and (bottom) a closeup of the TNC with the coordinating histidine residues. A Laccase with no inhibitor after 4.065 MGy X-ray dose (PDB: 6rh0), B laccase complexed with fluoride (83% occupancy) after 5.200 MGy X-ray dose (PDB: 6rik), C laccase complexed with chloride (45% occupancy) after 4.065 MGy X-ray dose (PDB: 6ri2).292 Key: H = white, N = blue, O = red, F = cyan, Cl = green, Cu = brown. | ||
6.2. Critique
The main limitation of X-ray crystallographic studies is that the structure is a static snapshot that is not necessarily representative of the active form in solution. This is especially relevant to structures based on crystals soaked in inhibitors, such as for azide soaked into an MCO crystal for hours275 or days.164 X-radiation also induces water splitting and reduces metals. Therefore, the structure of an MCO changes as its being probed by X-rays.293 Ash et al. presented a route to studying metalloprotein crystals in a defined oxidation state by coupling them to an electrode through redox mediators.294 This technique, demonstrated with a [NiFe] hydrogenase and coupled to infrared spectroscopic measurements could address the limitations of oxidation state and catalytic form, and could be applied to the study of MCOs.X-ray structures present ensemble-average data that only reflect the positions of atoms that are in the same place in (nearly) every unit cell. For example, a non-competitive inhibitor that does not bind to a specific point on the protein with high regularity will not appear in the diffraction pattern. The position of light atoms like hydrogen must be inferred from the rest of the structure and ordered water appears only as oxygen atoms. Atoms with similar atomic number (e.g., –OH versus F− near the TNC or aspartic acid versus asparagine in a protein backbone) must be differentiated by bond length (if the crystallographic resolution is sufficient), knowledge of the protein sequence or information from independent analyses,274 therefore there is some subjectivity in devising models.
7. Computational modelling and simulation
7.1. Application to MCO inhibition
Computational studies of MCOs have mainly been applied to analyzing the oxygen-reduction reaction in the absence of inhibitors.295–297 Fewer papers consider inhibition in MCOs. Kepp reported how the TNC's reorganization energy (RE, the summed energies required to distort the reduced state to the oxidized geometry and vice versa) obtained from DFT could explain the mechanism of halide and OH− binding and inhibition in two MCOs, a BOD from M. verrucaria and CotA from Bacillus sp.27 His results suggested that the inhibitors bound to the T2 Cu increase the RE of the second reduction of the resting state (Section S2†) and reduce the reduction potentials, providing a possible mechanism for inhibition. The RE obtained is larger with F− (203 kJ mol−1/48.5 kcal mol−1) or Cl− (154 kJ mol−1/36.8 kcal mol−1) bound to the T2 site than for an uninhibited catalytic intermediate with OH− bound (112 kJ mol−1/26.8 kcal mol−1). This is consistent with studies on other MCOs.179,298 Reduction leads to the release of Cl− from the T2 Cu (Fig. 11), suggesting a mechanism for thermal or reductive activation of laccases by dissociation of inhibiting halides or OH− from the T2 site. | ||
| Fig. 11 Geometry-optimized structures showing how two-electron reduction of the fully oxidized state causes the T2 Cu's ligand to dissociate (Cl−, top) or remain bound (OH−, bottom). The behavior for Cl− was also observed for F− and H2O. Reproduced with permission from ref. 27. Copyright 2015, American Chemical Society. | ||
DFT has been applied to the understanding the mechanism of Cl− activation of O2 reduction in ceruloplasmin (section 3.2).203 The calculations showed Cl− bridging between the T2 Cu and one T3 Cu (Fig. 12) and changing the ligand geometry around the T2 Cu, consistent with the authors' EPR measurements. The position of the Cl− in this human protein contrasts with the position of halides in fungal MCOs determined by X-ray crystallography (section 6.1). Despite the Cl− seemingly blocking O2 access to the TNC in ceruloplasmin, the authors attributed the Cl− activation to an increased intramolecular electron transfer rate between the T1 Cu and the TNC.203
 | ||
| Fig. 12 Geometry optimized structure of chloride bound to the partially reduced TNC. Copper ions are shown as orange spheres, Cl− as a central green sphere. Reproduced with permission from ref. 203. Copyright 2019, American Chemical Society. | ||
Martins et al. used a combined docking and DFT approach to model inhibition of laccase from T. versicolor with a pesticide FMT known to inhibit its activity by binding to a weak acid near the T1 Cu.299 Santagostini et al., whose 19F NMR measurements was described in section 5.1, used docking and QM/MM to conclude that F− competitively binds at the surface near the T1 Cu of ascorbate oxidase262 instead of TNC as reported in other papers.300,301 Molecular docking calculations were also used to explain the inhibitory effect of the tree-derived antifungal compound medicarpin on T. versicolor laccase.302 Medicarpin was predicted to block both the solvent channel in the laccase preventing the rapid access of O2 to the TNC and bind in near the T1 Cu which then blocks the access of oligolignol molecules.
7.2. Critique
For molecular docking, the flexibility of the target binding sites (i.e., “induced fit”) is frequently overlooked and treats both protein and inhibitor molecules as rigid structures. MD simulations, which include this necessary flexibility, are still relatively rare in the literature, though they are able to provide alternative conformational states that correspond to induced-binding structures303 and to explore in silico mutations that confer tolerance against inhibitors.3048. Conclusions and recommendations for benchmarking
MCO inhibition has been studied for decades and reported in hundreds of papers. For F− and N3−, there is general consensus (based mostly spectroscopic and crystallographic evidence, sections 5 and 6) that both inhibitors bind at the TNC and compete with the O2 substrate. This consensus has been so long-standing that there has been relatively little investigation on the possibility of these inhibitors binding at multiple locations. The handful of solution-based assays showing a mixed mode of inhibition for F− and N3− (Tables S3 and S5†) serve as an indication that further investigation is required. Moreover, as explained in section 4.5, the electrochemical evidence for F− inhibition only suggests that F− does not bind to the T1 site (because it does not affect electron transfer at the enzyme–electrode interface). Therefore, the possibility of these strong inhibitors binding at a location other than the T1 or TNC sites (allosteric inhibition) cannot be ruled out.How MCOs are inhibited by Cl−, on the other hand, has always been contentious. Only now are some general patterns emerging, most notably the close link between pH and Cl− inhibition: in bacterial and fungal MCOs Cl− inhibition is more potent at lower pH values. The lack of a high-resolution crystallographic structure of any tree laccase hinders efforts to understand the structural origins of its anomalously high sensitivity to Cl−. The surprising observation of Cl−activation (and hints of nitrate activation) in some MCOs requires further systematic investigation to robustly quantify and identify the origin as well as to determine if this behavior is also exhibited by higher-potential MCOs.
Inhibition of O2 reduction in MCOs can arise not only from direct interaction with the TNC but also through the blocking of the substrate relays and channels (i.e., binding-site accessibility), and alterations of the relative driving forces (e.g., Erev for the copper cofactors and electron donors), electrostatics and surface interactions. These additional effects mean that the mechanisms and models for inhibition from one MCO cannot be uncritically applied to others. Table 3 summarizes pervasive problems in current studies of MCO inhibition, and some suggestions to mitigate against these issues.
| Challenge | Recommendations |
|---|---|
| 1. MCOs have two actives sites that can be inhibited by different mechanisms | a. Use techniques that study one active site only such as PFE, or spectroscopic studies on enzymes where one of the active sites has been disabled |
| b. Use more than one concentration of O2 (e.g., inert atmosphere, air and pure O2 at atmospheric pressure) as well as more than one electron-donor substrates | |
| c. Specify the substrate being analyzed when reporting KM and Ki data (e.g., KM,O2, Ki,Cl−) | |
| d. Specify/report the reduction potential of any electron-donor substrate and ErevT1 | |
| 2. pH and temperature affect the strength of inhibition | a. Study inhibition effects at two or more pH values |
| b. Control and report measurement temperatures | |
| c. Study inhibition effects at two or more temperatures | |
| 3. Native organisms may express many different MCO isoforms that have different tolerance to inhibition | a. Purify crude enzyme samples |
| b. Recombinantly express single MCO isoforms | |
| c. Consider post-translational modification (e.g. glycosylation) when selecting heterologous host | |
| 4. MCOs have at least one “resting” state | a. Activate the MCO before running catalytic assays in which the MCO is in solution or consider activation when interpreting the substrate conversion data from the first ca. 30 s of turnover |
| b. Activate the MCO before running PFE-based catalytic assays using chronoamperometry or repeated CV scans until a consistent current response is observed | |
| c. Use potential-controlled spectroscopic analyses | |
| 5. Impure electron-donor reagents can affect apparent mode of inhibition | a. Analyze reagents, particularly ABTS,124 for impurities |
| b. Compare solution-based assays using multiple batches/lots of electron-donor substrate | |
| 6. Inhibitors can irreversibly bind and/or alter an MCO's structure | a. Re-determine catalytic parameters on samples that have be dialyzed into inhibitor-free buffers |
| b. Determine effect of inhibitor on MCO structure (e.g., using circular dichroism) | |
| c. Measure the metal content of MCO before and after exposure to an inhibitor (e.g., using EPR, EDXRF305 and/or chemical titration) | |
| 7. More than one kinetic model may fit the data | a. Apply and report statistical tests for goodness of fit and overfitting |
| b. Employ non-linear fits to the data that include error weighting and that generate uncertainty estimates (i.e., error bars or plus/minus values) on the fitting parameters | |
| 8. Multiple inhibitors may bind to a single MCO macromolecule | a. Include hyperbolic and parabolic inhibition models in comparisons |
| 9. MCOs structures are not rigid | a. Use dynamic and flexible for computational models of inhibition |
| b. Verify results from PFE using assays in which the MCO is not immobilized on an electrode |
The emerging “critical mass” of data suggest some techniques to engineer greater tolerance against inhibitors. First, engineering higher pH optima in the enzymes themselves may mitigate some of the halide-inhibition effects, but may come at a cost of reduced oxidizing power from the lower reduction potential of the T1 copper or reduced activity from impaired proton transfer to the TNC. Second, controlled binding-site accessibility can be engineered through mutagenesis around the channels leading to the TNC (section 2.1.2.2) or the surfaces closest to the lone, T1 copper. Finally, engineering the electrode or the MCO surface, encapsulation (especially in polymers incorporating redox mediators), and control of orientation in immobilized enzymes all contribute to increased tolerance against anionic inhibitors.
PFE offers advantages over solution assays when studying inhibition of O2 reduction in MCOs (section 4), but implicitly assumes that the immobilized enzyme behaves comparably to enzyme in solution. Electroactive coverage of the enzyme layer changes between measurements and may change with inhibitor addition, so specific enzyme activity values need to be treated with caution. Analytical PFE inhibition studies should include both CV and chronoamperometry measurements, use controlled-atmosphere electrochemical cells and rotating electrodes. Related potential-controlled spectroscopic measurements need to be more widely employed (section 5.2).
A change in experimental practice is essential to resolve the inconsistent and contradictory results, contribute results that can be used as benchmarks by the wider MCO research community, allow this community to deduce the overarching trends, and engineer these biocatalysts for industrial use.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors thank the U. K.'s Biotechnology and Biological Sciences Research Council for M. V.'s studentship (BB/J014478/1) and the Ministry of Higher Education Malaysia and Universiti Teknologi Malaysia for A. K.'s studentship. The authors thank Rachel Heath for her comments on the manuscript.References
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed.
- E. I. Solomon, R. K. Szilagyi, S. DeBeer George and L. Basumallick, Chem. Rev., 2004, 104, 419–458 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- P. J. Hoegger, S. Kilaru, T. Y. James, J. R. Thacker and U. Kues, FEBS J., 2006, 273, 2308–2326 CrossRef CAS PubMed.
- D. Sirim, F. Wagner, L. Wang, R. D. Schmid and J. Pleiss, Database, 2011, 2011, bar006 CrossRef PubMed.
- T. Sakurai and K. Kataoka, Chem. Rec., 2007, 7, 220–229 CrossRef CAS PubMed.
- J. A. T. Ramos, S. Barends, R. M. Verhaert and L. H. de Graaff, Microb. Cell Fact., 2011, 10, 78 CrossRef PubMed.
- Z. Laufer, R. P. Beckett, F. V. Minibayeva, S. Lüthje and M. Böttger, Bryologist, 2009, 112, 418–426 CrossRef.
- P. Giardina, V. Faraco, C. Pezzella, A. Piscitelli, S. Vanhulle and G. Sannia, Cell. Mol. Life Sci., 2010, 67, 369–385 CrossRef CAS PubMed.
- M. Lang, M. R. Kanost and M. J. Gorman, PLoS One, 2012, 7, e33985 CrossRef CAS PubMed.
- Q. Li, X. Wang, M. Korzhev, H. C. Schroder, T. Link, M. N. Tahir, B. Diehl-Seifert and W. E. Muller, Biochim. Biophys. Acta, 2015, 1850, 118–128 CrossRef CAS PubMed.
- S. Rodríguez-Couto, in Current Developments in Biotechnology and Bioengineering, Elsevier, Amsterdam, 2018, pp. 211–234, DOI:10.1016/B978-0-444-63990-5.00011-6.
- D. M. Maté and M. Alcalde, Microb. Biotechnol., 2016, 10, 1457–1467 CrossRef PubMed.
- R. Chandra and P. Chowdhary, Environ. Sci.: Processes Impacts, 2015, 17, 326–342 RSC.
- N. Chandra, S. Srivastava, A. Srivastava and S. Kumar, in Environmental Pollution of Paddy Soils, ed. M. Z. Hashmi and A. Varma, Springer International Publishing, Basel, 2018, pp. 229–243, DOI:10.1007/978-3-319-93671-0_15.
- V. Gupta, N. Capalash and P. Sharma, in Advances in Biodegradation and Bioremediation of Industrial Waste, CRC Press, Boca Raton, 2015, pp. 111–140 Search PubMed.
- A. Sekretaryova, S. M. Jones and E. I. Solomon, J. Am. Chem. Soc., 2019, 141, 11304–11314 CrossRef CAS PubMed.
- C. H. Okino-Delgado, M. R. Zanutto-Elgui, D. Z. do Prado, M. S. Pereira and L. F. Fleuri, in Microbial Metabolism of Xenobiotic Compounds, ed. P. K. Arora, Springer Singapore, Singapore, 2019, pp. 79–101, DOI:10.1007/978-981-13-7462-3_4.
- Q. Husain, in Emerging and Eco-Friendly Approaches for Waste Management, Springer, 2019, pp. 329–358, DOI:10.1007/978-981-10-8669-4_15.
- M. Kumari, P. Ghosh and I. S. Thakur, in Bioremediation: Applications for Environmental Protection and Management, ed. S. J. Varjani, A. K. Agarwal, E. Gnansounou and B. Gurunathan, Springer Singapore, Singapore, 2018, pp. 223–241, DOI:10.1007/978-981-10-7485-1_11.
- K. Pandey, B. Singh, A. K. Pandey, I. J. Badruddin, S. Pandey, V. K. Mishra and P. A. Jain, Int. J. Curr. Microbiol. Appl. Sci., 2017, 6, 1243–1254 CrossRef.
- R. L. Singh, P. K. Singh and R. P. Singh, Int. Biodeterior. Biodegrad., 2015, 104, 21–31 CrossRef CAS.
- M. Trovaslet, E. Enaud, Y. Guiavarc'h, A.-M. Corbisier and S. Vanhulle, Enzyme Microb. Technol., 2007, 41, 368–376 CrossRef CAS.
- A. Cornish-Bowden, H. B. F. Dixon, K. J. Laidler, I. H. Segel, J. Ricard, S. F. Velick and E. C. Webb, Biochem. J., 1983, 213, 561 CrossRef PubMed.
- A. Cornish-Bowden, Perspect. Sci., 2014, 1, 74–87 CrossRef.
- C. Di Bari, N. Mano, S. Shleev, M. Pita and A. L. De Lacey, JBIC, J. Biol. Inorg. Chem., 2017, 22, 1179–1186 CrossRef CAS PubMed.
- K. P. Kepp, Inorg. Chem., 2015, 54, 476–483 CrossRef CAS PubMed.
- E. Enaud, M. Trovaslet, F. Naveau, A. Decristoforo, S. Bizet, S. Vanhulle and C. Jolivalt, Enzyme Microb. Technol., 2011, 49, 517–525 CrossRef CAS PubMed.
- A. de Poulpiquet, C. H. Kjaergaard, J. Rouhana, I. Mazurenko, P. Infossi, S. Gounel, R. Gadiou, M. T. Giudici-Orticoni, E. I. Solomon, N. Mano and E. Lojou, ACS Catal., 2017, 7, 3916–3923 CrossRef CAS PubMed.
- M. Tominaga, A. Sasaki and M. Togami, Electrochemistry, 2016, 84, 315–318 CrossRef CAS.
- R. Kittl, K. Mueangtoom, C. Gonaus, S. T. Khazaneh, C. Sygmund, D. Haltrich and R. Ludwig, J. Biotechnol., 2012, 157, 304–314 CrossRef CAS PubMed.
- G. N. Kumar and K. Srikumar, Appl. Biochem. Biotechnol., 2012, 167, 662–676 CrossRef CAS PubMed.
- J. Juárez-Gómez, E. Rosas-Tate, G. Roa-Morales, P. Balderas-Hernández, M. Romero-Romo and M. Ramírez-Silva, J. Chem., 2018, 2018, 7462697 Search PubMed.
- K. Murugesan, Y.-M. Kim, J.-R. Jeon and Y.-S. Chang, J. Hazard. Mater., 2009, 168, 523–529 CrossRef CAS PubMed.
- T. Wang, R. D. Milton, S. Abdellaoui, D. P. Hickey and S. D. Minteer, Anal. Chem., 2016, 88, 3243–3248 CrossRef CAS PubMed.
- A. Robles, R. Lucas, M. Martínez-Cañamero, N. Ben Omar, R. Pérez and A. Gálvez, Enzyme Microb. Technol., 2002, 31, 516–522 CrossRef CAS.
- Q. Zhang, R. Miao, T. Liu, Z. Huang, W. Peng, B. Gan, X. Zhang and H. Tan, 3 Biotech, 2019, 9, 171 CrossRef PubMed.
- Y.-J. Kim and J. A. Nicell, J. Chem. Technol. Biotechnol., 2006, 81, 1344–1352 CrossRef CAS.
- A. Chapple, L. N. Nguyen, F. I. Hai, A. Dosseto, M. H.-O. Rashid, S. Oh, W. E. Price and L. D. Nghiem, Biocatal. Biotransform., 2019, 37, 10–17 CrossRef CAS.
- V. P. Hitaishi, R. Clément, L. Quattrocchi, P. Parent, D. Duché, L. Zuily, M. Ilbert, E. Lojou and I. Mazurenko, J. Am. Chem. Soc., 2020, 142, 1394–1405 CrossRef CAS PubMed.
- M. B. Asif, F. I. Hai, J. Hou, W. E. Price and L. D. Nghiem, J. Environ. Manage., 2017, 201, 89–109 CrossRef CAS PubMed.
- D. Leech and K. O. Feerick, Electroanalysis, 2000, 12, 1339–1342 CrossRef CAS.
- C. Johannes and A. Majcherczyk, J. Biotechnol., 2000, 78, 193–199 CrossRef CAS.
- S. Sondhi, P. Sharma, S. Saini, N. Puri and N. Gupta, PLoS One, 2014, 9, e96951 CrossRef PubMed.
- G. Yasar, U. G. Guven, E. Guduk and F. Aktas, Biocatal. Biotransform., 2019, 37, 268–277 CrossRef CAS.
- F. Durand, S. Gounel, C. H. Kjaergaard, E. I. Solomon and N. Mano, Appl. Microbiol. Biotechnol., 2012, 96, 1489–1498 CrossRef CAS PubMed.
- Y.-X. Si, S. Ji, W. Wang, N.-Y. Fang, Q.-X. Jin, Y.-D. Park, G.-Y. Qian, J. Lee, H.-Y. Han and S.-J. Yin, Process Biochem., 2013, 48, 152–161 CrossRef CAS.
- N. Mano and F. Durand, Bacillus pumilus Bilirubin Oxidase and Applications Thereof, US9617577B2, 2017.
- J. C. Stevens, L. Das, J. K. Mobley, S. O. Asare, B. C. Lynn, D. W. Rodgers and J. Shi, ACS Sustainable Chem. Eng., 2019, 7, 15928–15938 CrossRef CAS.
- M. Hijazi, E. Türkmen and J. C. Tiller, ChemBioChem, 2019, 21, 874–882 CrossRef PubMed.
- A. Zerva, E. Koutroufini, I. Kostopoulou, A. Detsi and E. Topakas, New Biotechnol., 2019, 49, 10–18 CrossRef CAS PubMed.
- E. I. Solomon, P. Chen, M. Metz, S. K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570–4590 CrossRef CAS PubMed.
- A. Leonowicz, N. Cho, J. Luterek, A. Wilkolazka, M. Wojtas-Wasilewska, A. Matuszewska, M. Hofrichter, D. Wesenberg and J. Rogalski, J. Basic Microbiol., 2001, 41, 185–227 CrossRef CAS PubMed.
- A. M. Mayer and R. C. Staples, Phytochemistry, 2002, 60, 551–565 CrossRef CAS PubMed.
- N. Durán, M. A. Rosa, A. D'Annibale and L. Gianfreda, Enzyme Microb. Technol., 2002, 31, 907–931 CrossRef.
- P. Baldrian, FEMS Microbiol. Rev., 2006, 30, 215–242 CrossRef CAS PubMed.
- S. Riva, Trends Biotechnol., 2006, 24, 219–226 CrossRef CAS PubMed.
- S. R. Couto and J. L. T. Herrera, Biotechnol. Adv., 2006, 24, 500–513 CrossRef PubMed.
- P. S. Chauhan, B. Goradia and A. Saxena, 3 Biotech, 2017, 7, 323 CrossRef PubMed.
- K. Agrawal, V. Chaturvedi and P. Verma, Bioresour. Bioprocess., 2018, 5, 4 CrossRef.
- J. O. Unuofin, A. I. Okoh and U. U. Nwodo, Molecules, 2019, 24, 2064 CrossRef CAS PubMed.
- M. Bilal and H. M. N. Iqbal, Sci. Total Environ., 2019, 690, 447–459 CrossRef CAS PubMed.
- M. Bilal, H. M. N. Iqbal and D. Barcelo, Sci. Total Environ., 2019, 689, 160–177 CrossRef CAS PubMed.
- M. Bilal, T. Rasheed, F. Nabeel, H. M. Iqbal and Y. Zhao, J. Environ. Manage., 2019, 234, 253–264 CrossRef CAS PubMed.
- B. Varga, V. Somogyi, M. Meiczinger, N. Kováts and E. Domokos, J. Cleaner Prod., 2019, 221, 306–322 CrossRef CAS.
- M. Alshabib and S. A. Onaizi, Curr. Pollut. Rep., 2019, 5, 52–65 CrossRef CAS.
- X. Wang, B. Yao and X. Su, Int. J. Mol. Sci., 2018, 19, 3373 CrossRef PubMed.
- Z.-B. Guan, Q. Luo, H.-R. Wang, Y. Chen and X.-R. Liao, Cell. Mol. Life Sci., 2018, 75, 3569–3592 CrossRef CAS PubMed.
- S. Voběrková, V. Solčány, M. Vršanská and V. Adam, Chemosphere, 2018, 202, 694–707 CrossRef PubMed.
- M. Naghdi, M. Taheran, S. K. Brar, A. Kermanshahi-Pour, M. Verma and R. Y. Surampalli, Environ. Pollut., 2018, 234, 190–213 CrossRef CAS PubMed.
- C. Barrios-Estrada, M. de Jesús Rostro-Alanis, B. D. Muñoz-Gutiérrez, H. M. Iqbal, S. Kannan and R. Parra-Saldívar, Sci. Total Environ., 2018, 612, 1516–1531 CrossRef CAS PubMed.
- J. Singh, V. Saharan, S. Kumar, P. Gulati and R. K. Kapoor, Crit. Rev. Biotechnol., 2018, 38, 883–901 CrossRef CAS PubMed.
- J. Yang, W. Li, T. B. Ng, X. Deng, J. Lin and X. Ye, Front. Microbiol., 2017, 8, 832 CrossRef PubMed.
- T. Kadri, T. Rouissi, S. K. Brar, M. Cledon, S. Sarma and M. Verma, J. Environ. Sci., 2017, 51, 52–74 CrossRef PubMed.
- S. Ba and V. Vinoth Kumar, Crit. Rev. Biotechnol., 2017, 37, 819–832 CrossRef PubMed.
- G. Singh and S. K. Arya, Int. J. Biol. Macromol., 2019, 134, 1070–1084 CrossRef CAS PubMed.
- J. Becker and C. Wittmann, Biotechnol. Adv., 2019, 37, 107360 CrossRef PubMed.
- S. K. Chaurasia and N. K. Bhardwaj, J. Carbohydr. Chem., 2019, 38, 87–108 CrossRef CAS.
- A. Sharma, K. K. Jain, A. Jain, M. Kidwai and R. C. Kuhad, Appl. Microbiol. Biotechnol., 2018, 102, 10327–10343 CrossRef CAS PubMed.
- M. D. Cannatelli and A. J. Ragauskas, Chem. Rec., 2017, 17, 122–140 CrossRef CAS PubMed.
- R. Javaid, A. Sabir, N. Sheikh and M. Ferhan, Molecules, 2019, 24, 786 CrossRef PubMed.
- T. Kudanga, B. Nemadziva and M. Le Roes-Hill, Appl. Microbiol. Biotechnol., 2017, 101, 13–33 CrossRef CAS PubMed.
- C. Romero-Guido, A. Baez and E. Torres, Catalysts, 2018, 8, 223 CrossRef.
- K. A. Nguyen, S. Wikee and S. Lumyong, Mycosphere, 2018, 9, 1073–1088 CrossRef.
- P. Walde, K. Kashima and G. Ćirić-Marjanović, Front. Bioeng. Biotechnol., 2019, 7, 165 CrossRef PubMed.
- J. Su, J. Fu, C. Silva and A. Cavaco-Paulo, Trends Biotechnol., 2019, 37, 683–686 CrossRef CAS PubMed.
- Y. Tobimatsu and M. Schuetz, Curr. Opin. Biotechnol., 2019, 56, 75–81 CrossRef CAS PubMed.
- S. Slagman, H. Zuilhof and M. C. Franssen, ChemBioChem, 2018, 19, 288–311 CrossRef CAS PubMed.
- J. Su, J. Fu, Q. Wang, C. Silva and A. Cavaco-Paulo, Crit. Rev. Biotechnol., 2018, 38, 294–307 CrossRef CAS PubMed.
- A. Nasar and R. Perveen, Int. J. Hydrogen Energy, 2019, 44, 15287–15312 CrossRef CAS.
- S. Tsujimura, Biosci., Biotechnol., Biochem., 2019, 83, 39–48 CrossRef PubMed.
- X. Huang, L. Zhang, Z. Zhang, S. Guo, H. Shang, Y. Li and J. Liu, Biosens. Bioelectron., 2018, 124–125, 40–52 Search PubMed.
- Y. Zhang, Z. Lv, J. Zhou, F. Xin, J. Ma, H. Wu, Y. Fang, M. Jiang and W. Dong, Appl. Microbiol. Biotechnol., 2018, 102, 10409–10423 CrossRef CAS PubMed.
- N. Mano and A. de Poulpiquet, Chem. Rev., 2018, 118, 2392–2468 CrossRef CAS PubMed.
- X. Xiao, H.-q. Xia, R. Wu, L. Bai, L. Yan, E. Magner, S. Cosnier, E. Lojou, Z. Zhu and A. Liu, Chem. Rev., 2019, 119, 9509–9558 CrossRef CAS PubMed.
- T. Asano, Y. Seto, K. Hashimoto and H. Kurushima, Insect Biochem. Mol. Biol., 2019, 108, 61–70 CrossRef CAS PubMed.
- V. B. Vasilyev, BioMetals, 2019, 32, 195–210 CrossRef CAS PubMed.
- K. Kaur, A. Sharma, N. Capalash and P. Sharma, Microbiol. Res., 2019, 222, 1–13 CrossRef CAS PubMed.
- A. Luna-Acosta, M. Breitwieser, T. Renault and H. Thomas-Guyon, Mar. Pollut. Bull., 2017, 122, 5–16 CrossRef CAS PubMed.
- A. L. Campaña, S. L. Florez, M. J. Noguera, O. P. Fuentes, P. Ruiz Puentes, J. C. Cruz and J. F. Osma, Biosensors, 2019, 9, 41 CrossRef PubMed.
- S. Yashas, B. Shivakumara, T. Udayashankara and B. Krishna, Orient. J. Chem., 2018, 34, 631–637 CrossRef CAS.
- V. Hooda, A. Gahlaut, A. Gothwal and V. Hooda, Biotechnol. Lett., 2017, 39, 1453–1462 CrossRef CAS PubMed.
- J. Kirtzel, D. Siegel, K. Krause and E. Kothe, Adv. Appl. Microbiol., 2017, 99, 83–101 CAS.
- S. Zhou, P. Rousselot-Pailley, L. Ren, Y. Charmasson, E. C. Dezord, V. Robert, T. Tron and Y. Mekmouche, Methods Enzymol., 2018, 613, 17–61 Search PubMed.
- B. Bertrand, F. Martinez-Morales and M. R. Trejo-Hernández, Biotechnol. Prog., 2017, 33, 1015–1034 CrossRef CAS PubMed.
- C. Stines-Chaumeil, E. Roussarie and N. Mano, Biochim. Open, 2017, 4, 36–40 CrossRef PubMed.
- M. Theerachat, D. Guieysse, S. Morel, M. Remaud-Siméon and W. Chulalaksananukul, Appl. Biochem. Biotechnol., 2019, 187, 583–611 CrossRef CAS PubMed.
- D. M. Maté and M. Alcalde, Biotechnol. Adv., 2015, 33, 25–40 CrossRef PubMed.
- H. B. Gray, B. G. Malmström and R. J. P. Williams, J. Biol. Inorg. Chem., 2000, 5, 551–559 CrossRef CAS PubMed.
- N. M. Marshall, D. K. Garner, T. D. Wilson, Y.-G. Gao, H. Robinson, M. J. Nilges and Y. Lu, Nature, 2009, 462, 113–116 CrossRef CAS PubMed.
- P. Hosseinzadeh, N. M. Marshall, K. N. Chacón, Y. Yu, M. J. Nilges, S. Y. New, S. A. Tashkov, N. J. Blackburn and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 262 CrossRef CAS PubMed.
- V. Madhavi and S. S. Lele, BioResources, 2009, 4, 1694–1717 Search PubMed.
- U. Kües and M. Rühl, Curr. Genomics, 2011, 12, 72–94 CrossRef PubMed.
- C. M. Rivera-Hoyos, E. D. Morales-Álvarez, R. A. Poutou-Piñales, A. M. Pedroza-Rodríguez, R. RodrÍguez-Vázquez and J. M. Delgado-Boada, Fungal Biol. Rev., 2013, 27, 67–82 CrossRef.
- A. Leontievsky, N. Myasoedova, N. Pozdnyakova and L. Golovleva, FEBS Lett., 1997, 413, 446–448 CrossRef CAS.
- G. Palmieri, P. Giardina, C. Bianco, A. Scaloni, A. Capasso and G. Sannia, J. Biol. Chem., 1997, 272, 31301–31307 CrossRef CAS PubMed.
- N. N. Pozdnyakova, J. Rodakiewicz-Nowak and O. V. Turkovskaya, J. Mol. Catal. B: Enzym., 2004, 30, 19–24 CrossRef CAS.
- D. Zhao, X. Zhang, D. Cui and M. Zhao, PLoS One, 2012, 7, e38817 CrossRef CAS PubMed.
- S.-N. Wang, Q.-J. Chen, M.-J. Zhu, F.-Y. Xue, W.-C. Li, T.-J. Zhao, G.-D. Li and G.-Q. Zhang, Biochimie, 2018, 148, 46–54 CrossRef CAS PubMed.
- K. Agrawal, N. Bhardwaj, B. Kumar, V. Chaturvedi and P. Verma, Int. J. Biol. Macromol., 2019, 125, 1042–1055 CrossRef CAS PubMed.
- A. A. Leontievsky, T. Vares, P. Lankinen, J. K. Shergill, N. N. Pozdnyakova, N. M. Myasoedova, N. Kalkkinen, L. A. Golovleva, R. Cammack, C. F. Thurston and A. Hatakka, FEMS Microbiol. Lett., 1997, 156, 9–14 CrossRef CAS PubMed.
- A. A. Leontievsky, N. M. Myasoedova, B. P. Baskunov, N. N. Pozdnyakova, T. Vares, N. Kalkkinen, A. I. Hatakka and L. A. Golovleva, Biochemistry, 1999, 64, 1150–1156 CAS.
- Z. Haibo, Z. Yinglong, H. Feng, G. Peiji and C. Jiachuan, Biotechnol. Lett., 2009, 31, 837–843 CrossRef PubMed.
- A. C. Mot, C. Coman, N. Hadade, G. Damian, R. Silaghi-Dumitrescu and H. Heering, PLoS One, 2020, 15, e0225530 CrossRef CAS PubMed.
- N. Mano, Appl. Microbiol. Biotechnol., 2012, 96, 301–307 CrossRef CAS PubMed.
- A. Roucher, E. Roussarie, R. M. Gauvin, J. Rouhana, S. Gounel, C. Stines-Chaumeil, N. Mano and R. Backov, Enzyme Microb. Technol., 2019, 120, 77–83 CrossRef CAS PubMed.
- Y. Z. Xiao, Q. Chen, J. Hang, Y. Y. Shi, Y. Z. Xiao, J. Wu, Y. Z. Hong and Y. P. Wang, Mycologia, 2004, 96, 26–35 CrossRef CAS PubMed.
- N. Mano, H.-H. Kim, Y. Zhang and A. Heller, J. Am. Chem. Soc., 2002, 124, 6480–6486 CrossRef CAS PubMed.
- V. Coman, R. Ludwig, W. Harreither, D. Haltrich, L. Gorton, T. Ruzgas and S. Shleev, Fuel Cells, 2010, 10, 9–16 CAS.
- C. Santoro, S. Babanova, B. Erable, A. Schuler and P. Atanassov, Bioelectrochemistry, 2016, 108, 1–7 CrossRef CAS PubMed.
- C. Kang, H. Shin and A. Heller, Bioelectrochemistry, 2006, 68, 22–26 CrossRef CAS PubMed.
- G. Gupta, C. Lau, V. Rajendran, F. Colon, B. Branch, D. Ivnitski and P. Atanassov, Electrochem. Commun., 2011, 13, 247–249 CrossRef CAS.
- M. Rasmussen, S. Abdellaoui and S. D. Minteer, Biosens. Bioelectron., 2016, 76, 91–102 CrossRef CAS PubMed.
- L. O. Martins, P. Durao, V. Brissos and P. F. Lindley, Cell. Mol. Life Sci., 2015, 72, 911–922 CrossRef CAS PubMed.
- C. N. Butterfield, A. V. Soldatova, S.-W. Lee, T. G. Spiro and B. M. Tebo, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11731 CrossRef CAS PubMed.
- W. M. Huston, M. P. Jennings and A. G. McEwan, Mol. Microbiol., 2002, 45, 1741–1750 CrossRef CAS PubMed.
- G. Grass and C. Rensing, J. Bacteriol., 2001, 183, 2145–2147 CrossRef CAS PubMed.
- S.-i. Sakasegawa, H. Ishikawa, S. Imamura, H. Sakuraba, S. Goda and T. Ohshima, Appl. Environ. Microbiol., 2006, 72, 972–975 CrossRef CAS PubMed.
- F. Durand, C. H. Kjaergaard, E. Suraniti, S. Gounel, R. G. Hadt, E. I. Solomon and N. Mano, Biosens. Bioelectron., 2012, 35, 140–146 CrossRef CAS PubMed.
- K. Nitta, K. Kataoka and T. Sakurai, J. Inorg. Biochem., 2002, 91, 125–131 CrossRef CAS PubMed.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed.
- E. I. Solomon, A. J. Augustine and J. Yoon, Dalton Trans., 2008, 3921–3932, 10.1039/b800799c.
- F. J. Enguita, L. O. Martins, A. O. Henriques and M. A. Carrondo, J. Biol. Chem., 2003, 278, 19416–19425 CrossRef CAS PubMed.
- L. dos Santos, V. Climent, C. F. Blanford and F. A. Armstrong, Phys. Chem. Chem. Phys., 2010, 12, 13962–13974 RSC.
- J. A. Cracknell, T. P. McNamara, E. D. Lowe and C. F. Blanford, Dalton Trans., 2011, 40, 6668–6675 RSC.
- M. Akter, T. Tokiwa, M. Shoji, K. Nishikawa, Y. Shigeta, T. Sakurai, Y. Higuchi, K. Kataoka and N. Shibata, Chem. – Eur. J., 2018, 24, 18052–18058 CrossRef CAS PubMed.
- I. Mazurenko, K. Monsalve, J. Rouhana, P. Parent, C. Laffon, A. L. Goff, S. Szunerits, R. Boukherroub, M.-T. Giudici-Orticoni, N. Mano and E. Lojou, ACS Appl. Mater. Interfaces, 2016, 8, 23074–23085 CrossRef CAS PubMed.
- M. Dagys, A. Laurynėnas, D. Ratautas, J. Kulys, R. Vidžiūnaitė, M. Talaikis, G. Niaura, L. Marcinkevičienė, R. Meškys and S. Shleev, Energy Environ. Sci., 2017, 10, 498–502 RSC.
- S. A. Roberts, A. Weichsel, G. Grass, K. Thakali, J. T. Hazzard, G. Tollin, C. Rensing and W. R. Montfort, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2766 CrossRef CAS PubMed.
- K. Miyazaki, Extremophiles, 2005, 9, 415–425 CrossRef CAS PubMed.
- A. T. Fernandes, L. O. Martins and E. P. Melo, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 75–83 CrossRef CAS PubMed.
- S. K. Singh, S. A. Roberts, S. F. McDevitt, A. Weichsel, G. F. Wildner, G. B. Grass, C. Rensing and W. R. Montfort, J. Biol. Chem., 2011, 286, 37849–37857 CrossRef CAS PubMed.
- S. A. Roberts, G. F. Wildner, G. Grass, A. Weichsel, A. Ambrus, C. Rensing and W. R. Montfort, J. Biol. Chem., 2003, 278, 31958–31963 CrossRef CAS PubMed.
- T. Classen, J. Pietruszka and S. M. Schuback, Protein Expression Purif., 2013, 89, 97–108 CrossRef CAS PubMed.
- K. Kataoka, H. Komori, Y. Ueki, Y. Konno, Y. Kamitaka, S. Kurose, S. Tsujimura, Y. Higuchi, K. Kano, D. Seo and T. Sakurai, J. Mol. Biol., 2007, 373, 141–152 CrossRef CAS PubMed.
- A. Messerschmidt, R. Ladenstein, R. Huber, M. Bolognesi, L. Avigliano, R. Petruzzelli, A. Rossi and A. Finazzi-Agró, J. Mol. Biol., 1992, 224, 179–205 CrossRef CAS PubMed.
- N. Hakulinen, L.-L. Kiiskinen, K. Kruus, M. Saloheimo, A. Paananen, A. Koivula and J. Rouvinen, Nat. Struct. Biol., 2002, 9, 601–605 CAS.
- N. E. Zhukhlistova, Y. N. Zhukova, A. V. Lyashenko, V. N. Zaĭtsev and A. M. Mikhaĭlov, Crystallogr. Rep., 2008, 53, 92–109 CrossRef CAS.
- J. Cohen, A. Arkhipov, R. Braun and K. Schulten, Biophys. J., 2006, 91, 1844–1857 CrossRef CAS PubMed.
- J. M. Damas, A. M. Baptista and C. M. Soares, J. Chem. Theory Comput., 2014, 10, 3525–3531 CrossRef CAS PubMed.
- J. P. Kallio, J. Rouvinen, K. Kruus and N. Hakulinen, Biochemistry, 2011, 50, 4396–4398 CrossRef CAS PubMed.
- R. S. Granja-Travez, R. C. Wilkinson, G. F. Persinoti, F. M. Squina, V. Fülöp and T. D. H. Bugg, FEBS J., 2018, 285, 1684–1700 CrossRef CAS PubMed.
- F. Xu, Biochemistry, 1996, 35, 7608–7614 CrossRef CAS PubMed.
- I. Bento, L. O. Martins, G. Gato Lopes, M. Arménia Carrondo and P. F. Lindley, Dalton Trans., 2005, 3507–3513, 10.1039/B504806K.
- S. Brander, J. D. Mikkelsen and K. P. Kepp, PLoS One, 2014, 9, e99402 CrossRef PubMed.
- F. Xu, Appl. Biochem. Biotechnol., 1996, 59, 221–230 CrossRef CAS.
- D. M. Maté, D. Gonzalez-Perez, M. Falk, R. Kittl, M. Pita, A. L. De Lacey, R. Ludwig, S. Shleev and M. Alcalde, Chem. Biol., 2013, 20, 223–231 CrossRef PubMed.
- H. Lineweaver and D. Burk, J. Am. Chem. Soc., 1934, 56, 658–666 CrossRef CAS.
- M. Dixon, Biochem. J., 1953, 55, 170 CrossRef CAS PubMed.
- M. Dixon and E. C. Webb, Enzymes, Spottiswoode, Ballantyne and Co., London, 2nd revised edn, 1964 Search PubMed.
- A. Dias, P. Pinto, I. Fraga and R. Bezerra, J. Chem. Educ., 2014, 91, 1017–1021 CrossRef CAS.
- P. A. Pinto, R. M. Bezerra and A. A. Dias, Biocatal. Biotransform., 2018, 36, 401–407 CrossRef CAS.
- C. J. Rodgers, C. F. Blanford, S. R. Giddens, P. Skamnioti, F. A. Armstrong and S. J. Gurr, Trends Biotechnol., 2010, 28, 63–72 CrossRef CAS PubMed.
- J. A. Cracknell and C. F. Blanford, Chem. Sci., 2012, 3, 1567–1581 RSC.
- C. H. Kjaergaard, F. Durand, F. Tasca, M. F. Qayyum, B. Kauffmann, S. Gounel, E. Suraniti, K. O. Hodgson, B. Hedman, N. Mano and E. I. Solomon, J. Am. Chem. Soc., 2012, 134, 5548–5551 CrossRef CAS PubMed.
- F. Tasca, D. Farias, C. Castro, C. Acuna-Rougier and R. Antiochia, PLoS One, 2015, 10, e0132181 CrossRef PubMed.
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS.
- P.-P. Champagne, M. E. Nesheim and J. A. Ramsay, Appl. Microbiol. Biotechnol., 2013, 97, 6263–6269 CrossRef CAS PubMed.
- F. Xu, J. Biol. Chem., 1997, 272, 924–928 CrossRef CAS PubMed.
- F. Xu, Appl. Biochem. Biotechnol., 2001, 95, 125–133 CrossRef CAS PubMed.
- F. Xu, R. M. Berka, J. A. Wahleithner, B. A. Nelson, J. R. Shuster, S. H. Brown, A. E. Palmer and E. I. Solomon, Biochem. J., 1998, 334, 63–70 CrossRef CAS PubMed.
- S. M. Jones and E. I. Solomon, Cell. Mol. Life Sci., 2015, 72, 869–883 CrossRef CAS PubMed.
- J. A. Fee, B. G. Malmström and T. Vänngård, Biochim. Biophys. Acta, Bioenerg., 1970, 197, 136–142 CrossRef CAS.
- F. A. Al-Lolage, P. N. Bartlett, S. Gounel, P. Staigre and N. Mano, ACS Catal., 2019, 9, 2068–2078 CrossRef CAS.
- F. Xu, A. E. Palmer, D. S. Yaver, R. M. Berka, G. A. Gambetta, S. H. Brown and E. I. Solomon, J. Biol. Chem., 1999, 274, 12372–12375 CrossRef CAS PubMed.
- F. Xu, W. Shin, S. H. Brown, J. A. Wahleithner, U. M. Sundaram and E. I. Solomon, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1996, 1292, 303–311 CrossRef.
- S. Holmberg, M. Rodriguez-Delgado, R. D. Milton, N. Ornelas-Soto, S. D. Minteer, R. Parra and M. J. Madou, ACS Catal., 2015, 5, 7507–7518 CrossRef CAS.
- N. Raseda, S. Hong, O. Y. Kwon and K. Ryu, J. Microbiol. Biotechnol., 2014, 24, 1673–1678 CrossRef CAS PubMed.
- N. Jimenez-Juarez, R. Roman-Miranda, A. Baeza, A. Sánchez-Amat, R. Vazquez-Duhalt and B. Valderrama, J. Biotechnol., 2005, 117, 73–82 CrossRef CAS PubMed.
- A. M. Garzillo, M. C. Colao, C. Caruso, C. Caporale, D. Celletti and V. Buonocore, Appl. Microbiol. Biotechnol., 1998, 49, 545–551 CrossRef CAS PubMed.
- S. Brander, J. D. Mikkelsen and K. P. Kepp, J. Mol. Catal. B: Enzym., 2015, 112, 59–65 CrossRef CAS.
- S. Basheer, N. Rashid, R. Ashraf, M. S. Akram, M. A. Siddiqui, T. Imanaka and M. Akhtar, Extremophiles, 2017, 21, 563–571 CrossRef CAS PubMed.
- V. Gupta, S. Balda, N. Gupta, N. Capalash and P. Sharma, Int. J. Biol. Macromol., 2019, 123, 1052–1061 CrossRef CAS PubMed.
- J. Li, Y. Xie, R. Wang, Z. Fang, W. Fang, X. Zhang and Y. Xiao, Eur. Biophys. J., 2018, 47, 225–236 CrossRef CAS PubMed.
- Y. Zhang, W. Dong, Z. Lv, J. Liu, W. Zhang, J. Zhou, F. Xin, J. Ma and M. Jiang, Mol. Biotechnol., 2018, 60, 681–689 CrossRef CAS PubMed.
- L. Ausec, F. Berini, C. Casciello, M. S. Cretoiu, J. D. van Elsas, F. Marinelli and I. Mandic-Mulec, Appl. Microbiol. Biotechnol., 2017, 101, 6261–6276 CrossRef CAS PubMed.
- F. Rosconi, L. Franco Fraguas, G. Martínez-Drets and S. Castro-Sowinski, Enzyme Microb. Technol., 2005, 36, 800–807 CrossRef CAS.
- Z.-M. Fang, T.-L. Li, F. Chang, P. Zhou, W. Fang, Y.-Z. Hong, X.-C. Zhang, H. Peng and Y.-Z. Xiao, Bioresour. Technol., 2012, 111, 36–41 CrossRef CAS PubMed.
- J. O. Unuofin, H. A. Moubasher, A. I. Okoh and U. U. Nwodo, Biotechnol. Rep., 2019, 22, e00337 CrossRef CAS PubMed.
- T. Ters, T. Kuncinger and E. Srebotnik, J. Mol. Catal. B: Enzym., 2009, 61, 261–267 CrossRef CAS.
- I. Eichlerová, J. Šnajdr and P. Baldrian, Chemosphere, 2012, 88, 1154–1160 CrossRef PubMed.
- S. Sadhasivam, S. Savitha, K. Swaminathan and F.-H. Lin, Process Biochem., 2008, 43, 736–742 CrossRef CAS.
- S. Tian, S. M. Jones, A. Jose and E. I. Solomon, J. Am. Chem. Soc., 2019, 141, 10736–10743 CrossRef CAS PubMed.
- B. J. Bennion and V. Daggett, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5142 CrossRef CAS PubMed.
- L. C. Clark, R. Wolf, D. Granger and Z. Taylor, J. Appl. Physiol., 1953, 6, 189–193 CrossRef CAS PubMed.
- G. B. Koudelka and M. J. Ettinger, J. Biol. Chem., 1988, 263, 3698–3705 CAS.
- J. E. Dowd and D. S. Riggs, J. Biol. Chem., 1965, 240, 863–869 CAS.
- J. Hirose, K. Inoue, H. Sakuragi, M. Kikkawa, M. Minakami, T. Morikawa, H. Iwamoto and K. Hiromi, Inorg. Chim. Acta, 1998, 273, 204–212 CrossRef CAS.
- K. Uematsu and H. Katano, Anal. Sci., 2013, 29, 25–29 CrossRef CAS PubMed.
- D. D'Souza-Ticlo, D. Sharma and C. Raghukumar, Mar. Biotechnol., 2009, 11, 725–737 CrossRef PubMed.
- M. Kumar, A. Mishra, S. S. Singh, S. Srivastava and I. S. Thakur, Int. J. Biol. Macromol., 2018, 115, 308–316 CrossRef CAS PubMed.
- Y. Zhang and H. Hess, Anal. Chem., 2020, 92, 1502–1510 CrossRef CAS PubMed.
- L. Quintanar, C. Stoj, A. B. Taylor, P. J. Hart, D. J. Kosman and E. I. Solomon, Acc. Chem. Res., 2007, 40, 445–452 CrossRef CAS PubMed.
- S. Shleev, V. Andoralov, M. Falk, C. T. Reimann, T. Ruzgas, M. Srnec, U. Ryde and L. Rulíšek, Electroanalysis, 2012, 24, 1524–1540 CrossRef CAS.
- H. T. S. Britton and R. A. Robinson, J. Chem. Soc., 1931, 1456–1462, 10.1039/JR9310001456.
- R. Bourbonnais, D. Leech and M. G. Paice, Biochim. Biophys. Acta, Gen. Subj., 1998, 1379, 381–390 CrossRef CAS.
- M. Fabbrini, C. Galli and P. Gentili, J. Mol. Catal. B: Enzym., 2002, 16, 231–240 CrossRef CAS.
- M. Frasconi, G. Favero, H. Boer, A. Koivula and F. Mazzei, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 899–908 CrossRef CAS PubMed.
- I. Schröder, E. Steckhan and A. Liese, J. Electroanal. Chem., 2003, 541, 109–115 CrossRef.
- W. Nogala, E. Rozniecka, J. Rogalski and M. Opallo, J. Electroanal. Chem., 2007, 608, 31–36 CrossRef CAS.
- P. Hapiot, J. Pinson, P. Neta and C. Rolando, J. Electroanal. Chem., 1993, 353, 225–235 CrossRef CAS.
- Q. Lin, Q. Li, C. Batchelor-McAuley and R. G. Compton, J. Phys. Chem. C, 2015, 119, 1489–1495 CrossRef CAS.
- C. Giacomelli, K. Ckless, D. Galato, F. S. Miranda and A. Spinelli, J. Braz. Chem. Soc., 2002, 13, 332–338 CrossRef CAS.
- J. E. O'Reilly, Biochim. Biophys. Acta, Bioenerg., 1973, 292, 509–515 CrossRef.
- A. S. Pavitt, E. J. Bylaska and P. G. Tratnyek, Environ. Sci.: Processes Impacts, 2017, 19, 339–349 RSC.
- C. Léger and P. Bertrand, Chem. Rev., 2008, 108, 2379–2438 CrossRef PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Fundamental and Applications, John Wiley & Sons, Hoboken, NJ, 2001 Search PubMed.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- R. G. Compton and C. E. Banks, Understanding Voltammetry, World Scientific, London, U.K., 3rd edn, 2018 Search PubMed.
- C. F. Blanford, C. E. Foster, R. S. Heath and F. A. Armstrong, Faraday Discuss., 2009, 140, 319–335 RSC.
- S. Chumillas, B. Maestro, J. M. Feliu and V. Climent, Front. Chem., 2018, 6, 358 CrossRef PubMed.
- C. Léger, S. J. Elliott, K. R. Hoke, L. J. C. Jeuken, A. K. Jones and F. A. Armstrong, Biochemistry, 2003, 42, 8653–8662 CrossRef PubMed.
- N. Makino and Y. Ogura, J. Biochem., 1971, 69, 91–100 CrossRef CAS PubMed.
- J. A. Fee, R. Malkin, B. G. Malmström and T. Vänngård, J. Biol. Chem., 1969, 244, 4200–4207 CAS.
- M. Frasconi, H. Boer, A. Koivula and F. Mazzei, Electrochim. Acta, 2010, 56, 817–827 CrossRef CAS.
- J. L. Cole, P. A. Clark and E. I. Solomon, J. Am. Chem. Soc., 1990, 112, 9534–9548 CrossRef CAS.
- J. Hirst, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 225–239 CrossRef CAS PubMed.
- C. Vaz-Dominguez, S. Campuzano, O. Rudiger, M. Pita, M. Gorbacheva, S. Shleev, V. M. Fernandez and A. L. De Lacey, Biosens. Bioelectron., 2008, 24, 531–537 CrossRef CAS PubMed.
- T. Beneyton, Y. Beyl, D. A. Guschin, A. D. Griffiths, V. Taly and W. Schuhmann, Electroanalysis, 2011, 23, 1781–1789 CrossRef CAS.
- U. Salaj-Kosla, S. Pöller, W. Schuhmann, S. Shleev and E. Magner, Bioelectrochemistry, 2013, 91, 15–20 CrossRef CAS PubMed.
- R. D. Milton, F. Giroud, A. E. Thumser, S. D. Minteer and R. C. T. Slade, Chem. Commun., 2014, 50, 94–96 RSC.
- M. R. Chapman, S. C. Cosgrove, N. J. Turner, N. Kapur and A. J. Blacker, Angew. Chem., Int. Ed., 2018, 57, 10535–10539 CrossRef CAS PubMed.
- P. Scodeller, R. Carballo, R. Szamocki, L. Levin, F. Forchiassin and E. J. Calvo, J. Am. Chem. Soc., 2010, 132, 11132–11140 CrossRef CAS PubMed.
- R. D. Milton, F. Giroud, A. E. Thumser, S. D. Minteer and R. C. T. Slade, Phys. Chem. Chem. Phys., 2013, 15, 19371–19379 RSC.
- S. Calabrese Barton, H.-H. Kim, G. Binyamin, Y. Zhang and A. Heller, J. Phys. Chem. B, 2001, 105, 11917–11921 CrossRef.
- N. Mano, H.-H. Kim and A. Heller, J. Phys. Chem. B, 2002, 106, 8842–8848 CrossRef CAS.
- R. Szamocki, V. Flexer, L. Levin, F. Forchiasin and E. J. Calvo, Electrochim. Acta, 2009, 54, 1970–1977 CrossRef CAS.
- H. Shin, S. Cho, A. Heller and C. Kang, J. Electrochem. Soc., 2009, 156, F87–F92 CrossRef CAS.
- Y. Beyl, D. A. Guschin, S. Shleev and W. Schuhmann, Electrochem. Commun., 2011, 13, 474–476 CrossRef CAS.
- C. H. Kjaergaard, S. M. Jones, S. Gounel, N. Mano and E. I. Solomon, J. Am. Chem. Soc., 2015, 137, 8783–8794 CrossRef CAS PubMed.
- R. Antiochia, D. Oyarzun, J. Sánchez and F. Tasca, Catalysts, 2019, 9, 1056 CrossRef CAS.
- J. Filip and J. Tkac, Bioelectrochemistry, 2014, 96, 14–20 CrossRef CAS PubMed.
- S. Cosnier, A. Le Goff and M. Holzinger, Electrochem. Commun., 2014, 38, 19–23 CrossRef CAS.
- N. Lalaoui, A. Le Goff, M. Holzinger and S. Cosnier, Chem. – Eur. J., 2015, 21, 16868–16873 CrossRef CAS PubMed.
- T. McArdle, T. P. McNamara, F. Fei, K. Singh and C. F. Blanford, ACS Appl. Mater. Interfaces, 2015, 7, 25270–25280 CrossRef CAS PubMed.
- I. Mazurenko, K. Monsalve, P. Infossi, M.-T. Giudici-Orticoni, F. Topin, N. Mano and E. Lojou, Energy Environ. Sci., 2017, 10, 1966–1982 RSC.
- V. Fourmond and C. Léger, in Biophotoelectrochemistry: From Bioelectrochemistry to Biophotovoltaics. Advances in Biochemical Engineering/Biotechnology, ed. L. J. C. Jeuken, Springer International Publishing, Cham, Switzerland, 2016, vol. 158 Search PubMed.
- K. Singh, T. McArdle, P. R. Sullivan and C. F. Blanford, Energy Environ. Sci., 2013, 6, 2460–2464 RSC.
- V. Fourmond, T. Lautier, C. Baffert, F. Leroux, P.-P. Liebgott, S. Dementin, M. Rousset, P. Arnoux, D. Pignol, I. Meynial-Salles, P. Soucaille, P. Bertrand and C. Léger, Anal. Chem., 2009, 81, 2962–2968 CrossRef CAS PubMed.
- T. Sakurai, S. Sawada, S. Suzuki and A. Nakahara, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1987, 915, 238–245 CrossRef CAS.
- S. Hirota, H. Matsumoto, H.-W. Huang, T. Sakurai, T. Kitagawa and O. Yamauchi, Biochem. Biophys. Res. Commun., 1998, 243, 435–437 CrossRef CAS PubMed.
- L. Santagostini, M. Gullotti, L. De Gioia, P. Fantucci, E. Franzini, A. Marchesini, E. Monzani and L. Casella, Int. J. Biochem. Cell Biol., 2004, 36, 881–892 CrossRef CAS PubMed.
- L. Quintanar, J. Yoon, C. P. Aznar, A. E. Palmer, K. K. Andersson, R. D. Britt and E. I. Solomon, J. Am. Chem. Soc., 2005, 127, 13832–13845 CrossRef CAS PubMed.
- D. J. Spira-Solomon, M. D. Allendorf and E. I. Solomon, J. Am. Chem. Soc., 1986, 108, 5318–5328 CrossRef CAS.
- L. Morpurgo, E. Agostinelli, M. Senepa and A. Desideri, J. Inorg. Biochem., 1985, 24, 1–8 CrossRef CAS.
- M. E. Winkler, D. J. Spira, C. D. LuBien, T. J. Thamann and E. I. Solomon, Biochem. Biophys. Res. Commun., 1982, 107, 727–734 CrossRef CAS PubMed.
- C. S. Peyratout, J. C. Severns, S. R. Holm and D. R. McMillan, Arch. Biochem. Biophys., 1994, 314, 405–411 CrossRef CAS PubMed.
- L. Morpurgo, G. Rotilio, A. F. Agrò and B. Mondoví, Biochim. Biophys. Acta, Protein Struct., 1974, 336, 324–328 CrossRef CAS.
- L.-E. Andréasson, R. Brändén and B. Reinhammar, Biochim. Biophys. Acta, Enzymol., 1976, 438, 370–379 CrossRef.
- A. J. Healy, P. A. Ash, O. Lenz and K. A. Vincent, Phys. Chem. Chem. Phys., 2013, 15, 7055–7059 RSC.
- R. Hidalgo, P. A. Ash, A. J. Healy and K. A. Vincent, Angew. Chem., Int. Ed., 2015, 54, 7110–7113 CrossRef CAS PubMed.
- D. B. Grabarczyk, P. A. Ash and K. A. Vincent, J. Am. Chem. Soc., 2014, 136, 11236–11239 CrossRef CAS PubMed.
- D. B. Grabarczyk, P. A. Ash, W. K. Myers, E. L. Dodd and K. A. Vincent, Dalton Trans., 2019, 48, 13960–13970 RSC.
- A. Wlodawer, W. Minor, Z. Dauter and M. Jaskolski, FEBS J., 2013, 280, 5705–5736 CrossRef CAS PubMed.
- A. Messerschmidt, H. Luecke and R. Huber, J. Mol. Biol., 1993, 230, 997–1014 CrossRef CAS PubMed.
- K. Piontek, M. Antorini and T. Choinowski, J. Biol. Chem., 2002, 277, 37663–37669 CrossRef CAS PubMed.
- T. Bertrand, C. Jolivalt, P. Briozzo, E. Caminade, N. Joly, C. Madzak and C. Mougin, Biochemistry, 2002, 41, 7325–7333 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- N. Hakulinen, M. Andberg, J. Kallio, A. Koivula, K. Kruus and J. Rouvinen, J. Struct. Biol., 2008, 162, 29–39 CrossRef CAS PubMed.
- M. Ferraroni, N. M. Myasoedova, V. Schmatchenko, A. A. Leontievsky, L. A. Golovleva, A. Scozzafava and F. Briganti, BMC Struct. Biol., 2007, 7, 60 CrossRef PubMed.
- M. Ferraroni, H. Westphal Adrie, M. Borsari, A. Tamayo-Ramos Juan, F. Briganti, H. D. Graaff Leo and J. H. V. Berkel Willem, Biocatal. Biotransform., 2017, 3, 1 Search PubMed.
- K. M. Polyakov, S. Gavryushov, S. Ivanova, T. V. Fedorova, O. A. Glazunova, A. N. Popov and O. V. Koroleva, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 388–401 CrossRef CAS PubMed.
- V. Zaitsev, I. Zaitseva, M. Papiz and P. Lindley, JBIC, J. Biol. Inorg. Chem., 1999, 4, 579–587 CrossRef CAS PubMed.
- J. P. Kallio, S. Auer, J. Jänis, M. Andberg, K. Kruus, J. Rouvinen, A. Koivula and N. Hakulinen, J. Mol. Biol., 2009, 392, 895–909 CrossRef CAS PubMed.
- T. Xie, Z. C. Liu and G. G. Wang, Protein Data Bank, 2015, DOI:10.2210/pdb4Q8B/pdb.
- F. J. Enguita, D. Marçal, L. O. Martins, R. Grenha, A. O. Henriques, P. F. Lindley and M. A. Carrondo, J. Biol. Chem., 2004, 279, 23472–23476 CrossRef CAS PubMed.
- F. J. Enguita, D. Marcal, R. Grenha, L. O. Martins, A. O. Henriques and M. A. Carrondo, Protein Data Bank, 2004, DOI:10.2210/pdb1OF0/pdb.
- M. Orlikowska, M. de J. Rostro-Alanis, A. Bujacz, C. Hernández-Luna, R. Rubio, R. Parra and G. Bujacz, Int. J. Biol. Macromol., 2018, 107, 1629–1640 CrossRef CAS PubMed.
- I. Matera, A. Gullotto, S. Tilli, M. Ferraroni, A. Scozzafava and F. Briganti, Inorg. Chim. Acta, 2008, 361, 4129–4137 CrossRef CAS.
- N. Hakulinen, K. Kruus, A. Koivula and J. Rouvinen, Biochem. Biophys. Res. Commun., 2006, 350, 929–934 CrossRef CAS PubMed.
- H. Serrano-Posada, S. Centeno-Leija, S. P. Rojas-Trejo, C. Rodriguez-Almazan, V. Stojanoff and E. Rudino-Pinera, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 2396–2411 CrossRef CAS PubMed.
- K. M. Polyakov, S. Gavryushov, T. V. Fedorova, O. A. Glazunova and A. N. Popov, Acta Crystallogr., Sect. D: Struct. Biol., 2019, 75, 804–816 CrossRef CAS PubMed.
- E. De la Mora, J. E. Lovett, C. F. Blanford, E. F. Garman, B. Valderrama and E. Rudino-Pinera, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 564–577 CrossRef CAS PubMed.
- P. A. Ash, S. B. Carr, H. A. Reeve, A. Skorupskaitė, J. S. Rowbotham, R. Shutt, M. D. Frogley, R. M. Evans, G. Cinque, F. A. Armstrong and K. A. Vincent, Chem. Commun., 2017, 53, 5858–5861 RSC.
- M. Srnec, U. Ryde and L. Rulíšek, Faraday Discuss., 2011, 148, 41–53 RSC.
- L. Rulíšek and U. Ryde, Coord. Chem. Rev., 2013, 257, 445–458 CrossRef.
- J. Li, M. Farrokhnia, L. Rulíšek and U. Ryde, J. Phys. Chem. B, 2015, 119, 8268–8284 CrossRef CAS PubMed.
- D. M. Maté, D. Gonzalez-Perez, R. Kittl, R. Ludwig and M. Alcalde, BMC Biotechnol., 2013, 13, 38 CrossRef PubMed.
- A. C. V. Martins, F. W. P. Ribeiro, G. Zanatta, V. N. Freire, S. Morais, P. de Lima-Neto and A. N. Correia, Bioelectrochemistry, 2016, 108, 46–53 CrossRef CAS PubMed.
- L. Casella, E. Monzani, L. Santagostini, L. de Gioia, M. Gullotti, P. Fantucci, T. Beringhelli and A. Marchesini, Coord. Chem. Rev., 1999, 185, 619–628 CrossRef.
- J. L. Cole, D. P. Ballou and E. I. Solomon, J. Am. Chem. Soc., 1991, 113, 8544–8546 CrossRef CAS.
- C. Martínez-Sotres, J. G. Rutiaga-Quiñones, R. Herrera-Bucio, M. Gallo and P. López-Albarrán, Wood Sci. Technol., 2015, 49, 857–868 CrossRef.
- F. R. Salsbury, Jr., Curr. Opin. Pharmacol., 2010, 10, 738–744 CrossRef PubMed.
- V. G. Giacobelli, E. Monza, M. Fatima Lucas, C. Pezzella, A. Piscitelli, V. Guallar and G. Sannia, Catal. Sci. Technol., 2017, 7, 515–523 RSC.
- S. Fruncillo, M. Trande, C. F. Blanford, A. Astegno and L. S. Wong, Anal. Chem., 2019, 91, 11502–11506 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Introduction to enzyme inhibition kinetics including formulas relating IC50 values to Ki for linear and hyperbolic inhibition models; introduction to O2 reduction in MCOs; tables of inhibition constants determined for MCOs by solution-based assays. See DOI: 10.1039/d0cy00724b |
| ‡ These authors contributed equally to this article. |
| § Current address: Institute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology (BIST), Baldiri Reixac 10-12, 08028 Barcelona, Spain. |
| This journal is © The Royal Society of Chemistry 2020 |