
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhere silylene–silicon centres matter in the activation of small molecules
Changkai
Shan
,
Shenglai
Yao
 and
Matthias
Driess
*
and
Matthias
Driess
*
Department of Chemistry, Metalorganics and Inorganic Materials, Technische Universität Berlin, Strasse des 17. Juni 135, Sekr. C2, 10623 Berlin, Germany. E-mail: matthias.driess@tu-berlin.de
First published on 19th August 2020
Abstract
Small molecules such as H2, N2, CO, NH3, O2 are ubiquitous stable species and their activation and role in the formation of value-added products are of fundamental importance in nature and industry. The last few decades have witnessed significant advances in the chemistry of heavy low-coordinate main-group elements, with a plethora of newly synthesised functional compounds, behaving like transition-metal complexes with respect to facile activation of such small molecules. Among them, silylenes have received particular attention in this vivid area of research showing even metal-free bond activation and catalysis. Recent striking discoveries in the chemistry of silylenes take advantage of narrow HOMO–LUMO energy gap and Lewis acid–base bifunctionality of divalent Si centres. The review is devoted to recent advances of using isolable silylenes and corresponding silylene–metal complexes for the activation of fundamental but inert molecules such as H2, COx, N2O, O2, H2O, NH3, C2H4 and E4 (E = P, As).
 Changkai Shan | Changkai Shan is currently a PhD candidate under the supervision of Prof. Matthias Driess at the Department of Metalorganics and Inorganic Materials, Technical University of Berlin. He received his MSc in organic chemistry from Xiamen University, China in 2018. His current research focuses on the synthesis of novel silylene-ligands and their reactivity towards catalysis and small molecule activation. |
 Shenglai Yao | Shenglai Yao attained his PhD degree in 2005 at the Johannes Gutenberg University of Mainz (Germany) under supervision of Professor Karl W. Klinkhammer. Afterwards he joined the Driess group at the Department of Metalorganics and Inorganic Materials, Technical University of Berlin and works in the group as a senior researcher. His research interests lie in the coordination chemistry of low-valent main-group elements and transition metals. |
 Matthias Driess | Matthias Driess is a full professor of metalorganics and inorganic materials at the Department of Chemistry of Technische Universität Berlin. He obtained his PhD degree and completed his habilitation at the University of Heidelberg in Germany. He serves as a deputy of the Cluster of Excellence UniSysCat and is a Director of the UniSysCat-BASF SE joint lab BasCat, and of the Chemical Invention Factory (CIF) for Start-ups in Green Chemistry. He is a member of the German National Academy of Sciences (Leopoldina), the Berlin-Brandenburg Academy of Sciences and Humanities, and the European Academy of Sciences. |
1. Introduction
Nowadays, small molecules such as H2, COx, N2O, O2, H2O, etc. are ubiquitous as well as economically available building blocks for fundamental chemical processes in the production of value-added fine chemicals and play a pivotal role in maintaining the prospects of industrial society.1 They are very stable, that is, the activation of their relatively inert bonds for selective chemical transformations requires a suitable catalyst. Learning from biocatalysts in nature and taking advantage of new synthetic methods in organometallic and coordination chemistry have paved the way to numerous artificial molecular catalysts for selective activation and transformation of small molecules.1.1 Activation of small molecules in nature
Nature has developed a myriad of cellular enzymes, the majority of which harbour a transition-metal cofactor and are capable of small-molecule conversion with high efficiency under mild conditions.2 For example, nitrogenase, one of the most important enzymes, allows for dinitrogen fixation and further synthesis of fundamental building blocks of cellular molecules. The molybdenum-dependent nitrogenase consists of electron-delivery protein components and MoFe cofactor, where N2 is reduced to NH3. At the same time, vanadium-dependent or heterometal-independent forms show less reactivity.3 Hydrogenases are remarkable catalysts for reversible H2 oxidation and proton reduction along with an energy-yielding process. They can be divided into two major classes, namely [NiFe]- and [FeFe]-hydrogenases.3b,4 H2O and O2 are essential components in both photosystem II (PSII) and cytochromes P450 (P450). PSII, a metal-bound protein complex, catalyses the light-driven oxidation of water by taking advantage of the four-electron redox-active Mn4CaO5 cluster,5 whereas P450 catalyses the introduction of O2 into non-activated C–H bonds with the assistance of a protein partner to deliver one or more electrons to the Fe reactive site.6 CO dehydrogenase (CODH) with a [Ni4Fe–5S] or [4Fe–4S] cluster can convert CO2 to the more valuable synthetic feedstock CO.7 Taking the aforementioned few examples as an inspiration for synthetic chemists, the design of artificial catalysts that are approximate to or even better than their natural counterparts is highly desirable.1.2 Artificial transition-metal-mediated activation of small molecules
Coordination chemists were much inspired by biocatalysis and developed an enormous number of artificial metalloenzymes for small molecule activation (e.g., oxygenases, hydrogenases, carboxodehydrogenases, nitrogenases) mimicking their counterparts in nature.8 To mention only a few selected ones, artificial metalloenzymes consisting of Fe or Mn centres have been synthesised for O2 activation;9 for example, the well-known O2-dependent phenol oxidase A (Fig. 1), bearing a “Due Ferri” (two-iron; DF) diiron cofactor, is capable to catalyse the oxidative transformation of 4-aminophenol to the corresponding quinone monoimine.10 Mimicking photosystem II (PSII), among many other examples, a molecular dinuclear Mn2H5L cluster allows for photocatalytic oxygen evolution reaction (OER) from water oxidation.11 Oxidation of H2 seems more complex than its back reaction (hydrogen evolution reaction (HER) from water reduction).12 In this regard, the designed diiron complex B (Fig. 1), modelling [FeFe] hydrogenase, participates in the oxidation of H2 only with a slow conversion rate.13 CO2 is the primary C1 source in nature and the electrochemical reduction of CO2 with, for example, [NiII(cyclam)]2+ (cyclam = 1,4,8,11-tetraazacyclotetradecane) C (Fig. 1), affords CO in aqueous and dimethylformamide (DMF) solutions.11 Notably, many synthetic Fe–S clusters mimicking nitrogenases have been reported, and these artificial clusters may one day approximate or surpass the ability of natural nitrogenases on the fixation and conversion of N2.14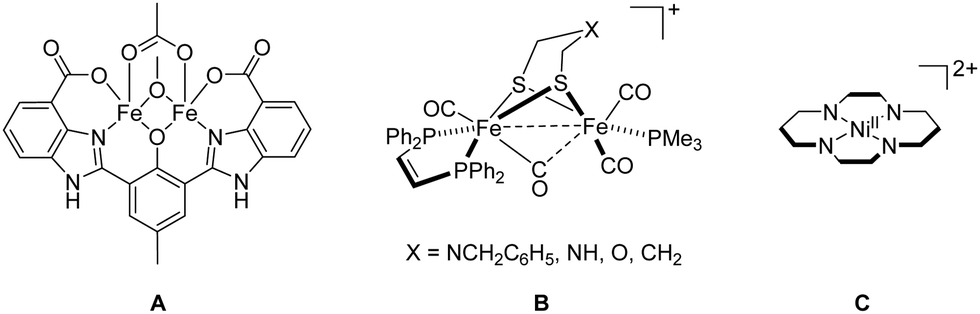 | ||
| Fig. 1 Examples of artificial metalloenzyme cofactors A, B and C (four protons on the N atoms are omitted for clarity). | ||
1.3 Main-group elements mimicking transition-metals in small molecule activation
The seminal review entitled “Main-Group Elements as Transition Metals” published by Power15 reflected a renaissance of main-group chemistry aiming at the development of even metal-free, benign catalysts as alternative mediators for the synthesis of fine chemicals compared to those relying on transition-metals. At first, the valence s or p orbitals of main-group compounds are thought to be far apart energetically. However, dozens of isolable low-coordinate main-group species exist, possessing frontier orbitals with small-energy separations, which show transition-metal-like properties towards small molecules.16 A striking example for the activation of H2 was reported by Power in 2005, where a heavy alkyne analogue with Ge [Ge≡Ge; Ar = 2,6-Trip2-C6H3 (Trip = 2,4,6-iPr3C6H2)] splits the H–H bond at room temperature to give Ar(H)Ge![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ge(H)Ar, Ar(H)2Ge–Ge(H)2Ar, and ArGeH3, respectively.17 Electron donation from the σ-orbital of H2 into the LUMO of the Ge species as well as a synergistic electron donation from the π-HOMO of the Ge species into the σ*-orbital of H2 are involved in the disruption of H–H bond, reminiscent of interactions between H2 and a transition-metal site in complexes (Fig. 2). Bertrand also showed that acyclic alkyl amino carbenes (aCAAC), such as [:C(tBu)NiPr2], can readily break the H–H bond to give the corresponding [H2C(tBu)NiPr2] addition product.18 Related stable carbenes also take part in the activation of unactivated bonds, acting also as ligands towards transition metals for the stabilisation of reactive intermediates.19 Stephan et al. reported the activation of H2 by the use of frustrated Lewis pairs (FLPs). FLPs bearing both an available acceptor and donor orbitals mimic a similar function of frontier orbitals in transition-metal complexes.20 Singlet tetrylenes, the heavy congeners of carbenes, possessing a lone pair of electrons and a vacant p-orbital also show similar reactivity to transition-metal centres.16d The past three decades have witnessed tremendous advances in the development of isolable divalent silicon species (named silylenes) which show a fascinating reactivity towards small molecules and are even suitable for metal-free catalysis.
Ge(H)Ar, Ar(H)2Ge–Ge(H)2Ar, and ArGeH3, respectively.17 Electron donation from the σ-orbital of H2 into the LUMO of the Ge species as well as a synergistic electron donation from the π-HOMO of the Ge species into the σ*-orbital of H2 are involved in the disruption of H–H bond, reminiscent of interactions between H2 and a transition-metal site in complexes (Fig. 2). Bertrand also showed that acyclic alkyl amino carbenes (aCAAC), such as [:C(tBu)NiPr2], can readily break the H–H bond to give the corresponding [H2C(tBu)NiPr2] addition product.18 Related stable carbenes also take part in the activation of unactivated bonds, acting also as ligands towards transition metals for the stabilisation of reactive intermediates.19 Stephan et al. reported the activation of H2 by the use of frustrated Lewis pairs (FLPs). FLPs bearing both an available acceptor and donor orbitals mimic a similar function of frontier orbitals in transition-metal complexes.20 Singlet tetrylenes, the heavy congeners of carbenes, possessing a lone pair of electrons and a vacant p-orbital also show similar reactivity to transition-metal centres.16d The past three decades have witnessed tremendous advances in the development of isolable divalent silicon species (named silylenes) which show a fascinating reactivity towards small molecules and are even suitable for metal-free catalysis.
 | ||
| Fig. 2 Frontier orbital interactions of H2 with (a) transition-metals, (b) multiply bonded main-group compounds, (c) frustrated Lewis pairs, and (d) carbenes and its heavy congeners, tetrylenes. Reprinted from ref. 16e with permission from AAAS, Copyright 2019. | ||
1.4 Divalent silicon employed in bond activation
Why using silicon for small-molecule activation? Silicon is the second most abundant element of the Earth's crust, and subvalent states (e.g., SiII) are well accessible. For a long time, silylenes represented laboratory curiosities and could only be studied in argon or hydrocarbon matrices at cryogenic temperatures.21 Since the isolation of the first N-heterocyclic silylene (NHSi) reported by West and Denk,21 a plethora of stable silylenes have been synthesised. They proved to be suitable and selectively adoptable for the activation of small molecules and metal-free catalysis. It was shown that SiII species can approximate characteristics of and even become alternatives of transition metals due to its facile availability of donor/acceptor orbitals.16 Acyclic silylenes with larger bite angles and smaller HUMO–LUMO gaps are considered as more reactive species than cyclic ones,22 at the same time, bis-silylenes show splendid reaction scope with two SiII centres synergistically interacting with substrates.23 Examples of SiII-mediated industrial transformations also show their potential for catalysis.24 Perhaps most challenging is the regeneration of a SiII site if a silylene acts as a catalyst.16d As several excellent reviews are available ranging from the synthesis to the reactivity of SiII species,16a–d,25 this review discusses most recent advances in the activation of H2, CO, CO2, N2O, O2, NH3, H2O, ethylene and P4 by isolable silylenes and particular silylene–transition-metal complexes which show silicon–transition-metal cooperativity in the activation of bonds.2. Isolable silylenes and their reactivity towards small molecules
2.1 Frontier orbitals for the activation of small molecules
In contrast to carbenes which can be in a triplet or singlet ground states, the Si atom in silylenes prefers the (3s)2(3p)2 valence electron configurations due to its inherent reluctance to undergo s,p-orbital hybridisation.26 Thus, silylenes have a singlet ground state with an in-plane non-bonding lone pair of electrons featuring a high 3s-character. Accordingly, the mostly 3p out-of-plane orbital is prone to accept electrons to obey the “octet rule”. Some strategies on thermodynamic and kinetic stabilisation of the reactive SiII atom enables access to isolable silylenes, through introduction of heteroatoms such as N, Si and P, etc. and/or sterically encumbered substituents (Fig. 3). Beside the electronic and steric effects, the singlet–triplet energy differences ΔEST also account for the reactivity of silylenes. The obtuse bond angle at the SiII atom leads to an increase of the p-character of the lone-pair orbital, which lowers the energy upon promoting a non-bonding lone-pair electron to a 3p-orbital.27 | ||
| Fig. 3 Features of stabilisation in substituted silylenes. | ||
Indeed, the modest to narrow HOMO–LUMO energy gap can be modified by combining different steric and electronic effects by the choice of suitable substituents and adjusting the angle at the SiII atom.
2.2 Activation of H2
Since the germanium alkyne analogue ArGe![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) GeAr (Ar = 2,6-(2,6-iPr2C6H3)2C6H3) was shown to undergo oxidative addition with dihydrogen in 2005,17 the activation of H2 by other multiply-bonded main-group compounds,15 or specific cyclic and acyclic carbenes,18 and FLP systems28 has also been accomplished successfully. Since the SiII centre in N-heterocyclic silylenes is stabilised by the adjacent N atoms of the substituents and has a relatively small endocyclic angle, it cannot add H2. In 2012, Jones and Aldridge reported the first room temperature-stable two-coordinate acyclic silylene 1, bearing the strong σ-donating and high sterically demanding B(NDippCH)2 substituent (Dipp = 2,6-iPr2C6H3) (Scheme 1).22a This acyclic silylene undergoes facile oxidative addition of H2, being the first example of experimentally observed dihydrogen activation by a silylene. Irreversibility of this reaction suggests it to be thermodynamically strongly exergonic (ΔG = −122.2 kJ mol−1), and the parallel reaction with HD gives 1-HD as the sole product in accordance with DFT calculations which indicate a concerted bimolecular process. The computed value of ΔG‡ (+97.2 kJ mol−1) suggests a substantially lower activation energy than for other related cyclic bis(amido)silylenes. Significant widening of the bond angle at the SiII atom (109.7(1)°) compared to the values for N-heterocyclic silylenes revealed a narrower singlet–triplet gap (103.9 kJ mol−1) and higher reactivity, which is reminiscent of transition-metals.29
GeAr (Ar = 2,6-(2,6-iPr2C6H3)2C6H3) was shown to undergo oxidative addition with dihydrogen in 2005,17 the activation of H2 by other multiply-bonded main-group compounds,15 or specific cyclic and acyclic carbenes,18 and FLP systems28 has also been accomplished successfully. Since the SiII centre in N-heterocyclic silylenes is stabilised by the adjacent N atoms of the substituents and has a relatively small endocyclic angle, it cannot add H2. In 2012, Jones and Aldridge reported the first room temperature-stable two-coordinate acyclic silylene 1, bearing the strong σ-donating and high sterically demanding B(NDippCH)2 substituent (Dipp = 2,6-iPr2C6H3) (Scheme 1).22a This acyclic silylene undergoes facile oxidative addition of H2, being the first example of experimentally observed dihydrogen activation by a silylene. Irreversibility of this reaction suggests it to be thermodynamically strongly exergonic (ΔG = −122.2 kJ mol−1), and the parallel reaction with HD gives 1-HD as the sole product in accordance with DFT calculations which indicate a concerted bimolecular process. The computed value of ΔG‡ (+97.2 kJ mol−1) suggests a substantially lower activation energy than for other related cyclic bis(amido)silylenes. Significant widening of the bond angle at the SiII atom (109.7(1)°) compared to the values for N-heterocyclic silylenes revealed a narrower singlet–triplet gap (103.9 kJ mol−1) and higher reactivity, which is reminiscent of transition-metals.29
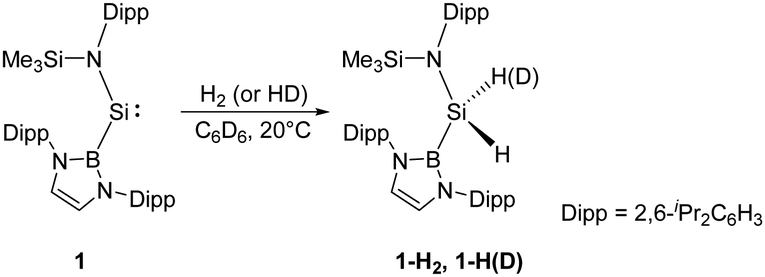 | ||
| Scheme 1 H–H bond activation by the acyclic silylene 1. | ||
In the same year, Power reported another isolable acyclic silylene, Si(SArMe6)2 [ArMe6 = C6H3-2,6(C6H2-2,4,6-Me3)2]. However, it does not react with H2 at ambient reaction condition as the relatively high electronegativity of the thiolato substituents increases the energy gap between the lone pair and the vacant 3p orbitals of the Si atom, hampering this reaction to occur.22b
By taking advantage of the robust reducing agent (thf)2K[Si(SiMe3)3], the acyclic Si{Si(SiMe3)3}{N(SiMe3)Dipp} silylene 2 was successfully isolated as purple-coloured crystals by Aldridge and co-workers (Scheme 2).22c Notably, the N–Si–Si bond angle of 116.91° in 2 is even larger than the corresponding N–Si–B angle of 1. The higher degree of steric crowding in 2 is evidenced from the observation of two sets of multinuclear NMR spectra in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which result from conformers related by restricted rotation along the Si–N bond. One set of the 1H NMR signals is derived from the isomer in which the Dipp group lies syn to the hypersilyl group while the other set is due to the corresponding anti one. The relatively narrow energetic gap between its singlet ground state and first excited triplet state (103.7 kJ mol−1) is consistent with its reactivity towards H2 at room temperature, affording the corresponding dihydrosilane 2-H2.
:1, which result from conformers related by restricted rotation along the Si–N bond. One set of the 1H NMR signals is derived from the isomer in which the Dipp group lies syn to the hypersilyl group while the other set is due to the corresponding anti one. The relatively narrow energetic gap between its singlet ground state and first excited triplet state (103.7 kJ mol−1) is consistent with its reactivity towards H2 at room temperature, affording the corresponding dihydrosilane 2-H2.
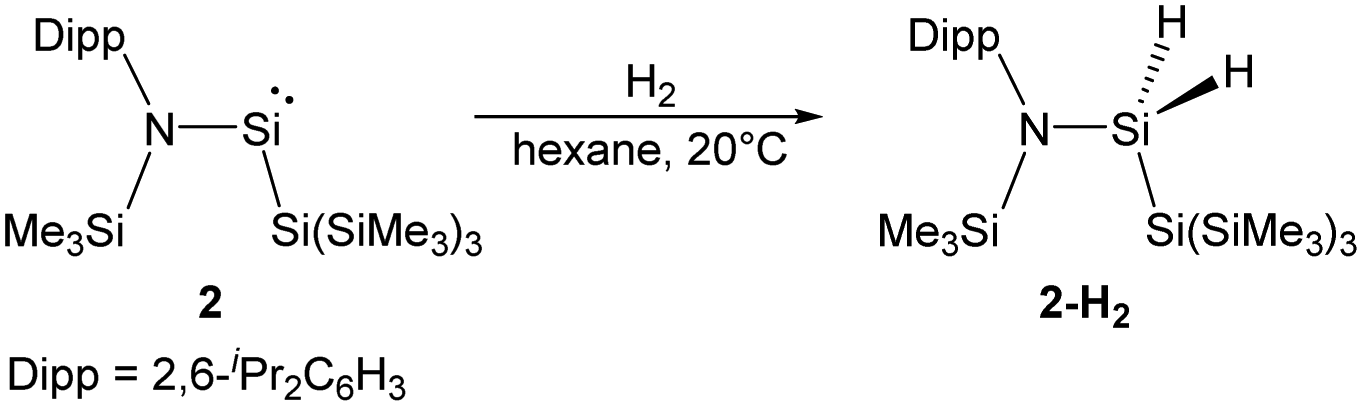 | ||
| Scheme 2 Addition of H2 to the amido(silyl)silylene 2. | ||
In 2017, the Driess group reported that the silylene–Ni0 complex 3 [(TMSL)ClSi: → Ni(NHC)2] (TMSL = N-(SiMe3)Dipp; Dipp = 2,6-iPr2C6H3, NHC = :C[(iPr)NC(Me)]2) is also effective in a H2 activation process (Scheme 3).30 The narrow singlet–triplet gap of 77.82 kJ mol−1 for 3 suggests that it may be suitable for facile dihydrogen activation. Indeed, exposure of a C6D6 solution of 3 to H2 atmosphere gives immediately and quantitatively 3-H2. Its 1H NMR spectrum reveals the presence of both Si–H (δ 5.69 ppm) and Ni–H (δ −10.23 ppm, J(1H,29Si) = 16 Hz) protons as doublets. The molecular structure of 3-H2 and DFT calculations suggest the initial H2 activation occurrence at the Ni centre and the following insertion of the silylene ligand into one Ni–H bond.
 | ||
| Scheme 3 Activation of H2 by the silylene–Ni complex 3 to form 3-H2. | ||
The Driess group also investigated the reactivity of the bis-silylene coordinated Ni complexes [SiII(Xant)SiII]Ni(η2-1,3-cod) 4 and [SiII(Xant)SiII]–Ni(PMe3)26 towards H2 (Scheme 4).23f Exposure of both Ni complexes to 1 bar H2 at room temperature affords different kinds of activation products. The reaction of 4 with H2 affords the unexpected diamagnetic dinuclear Ni complex 5 as yellow-brown crystals in 67% isolated yields featuring a four-membered planar Ni2Si2 core with two bridging hydride atoms lying almost in the same plane, whereas the reaction of 6 in a H2 atmosphere gives the SiII-stabilised Ni dihydride complex 7. Upon removal of H2 complex 7 converts to 6 in 29% NMR yields, suggesting a reversible H2-activation process. Notably, the SiII site plays an essential role in the activation of dihydrogen according to the DFT calculations. Indeed, the SiII and Ni0 sites show not only cooperativity in H2 activation but also in further chemoselective hydrogenation of olefins.23f
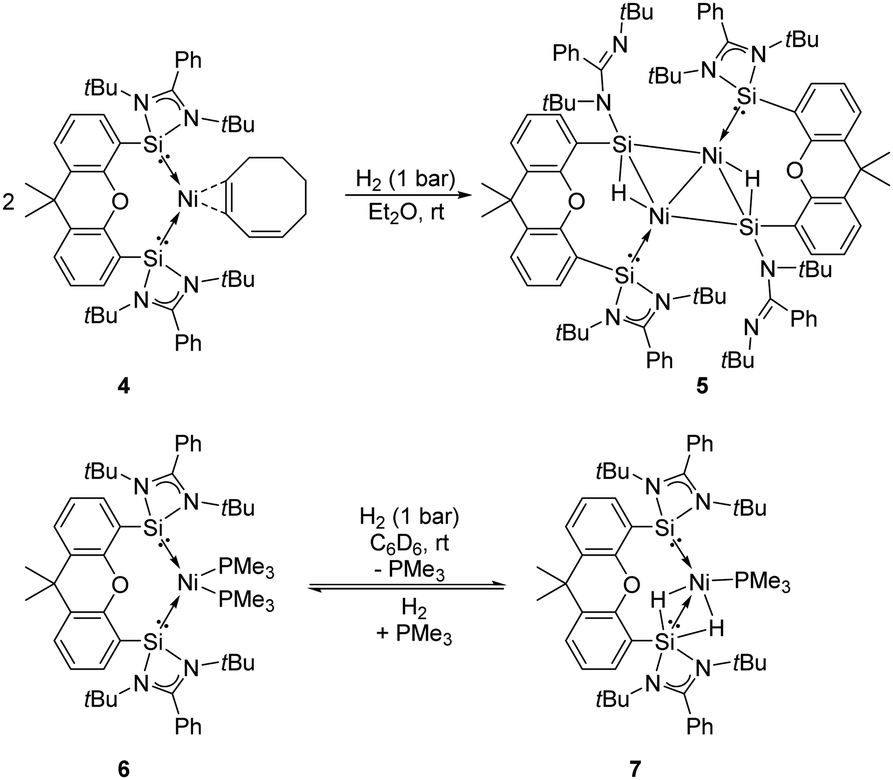 | ||
| Scheme 4 Activation of H2 by the bis-silylene–Ni complexes 4 and 6. | ||
The same group also reported cooperativity of SiII and BIII in the activation of H2: the N-heterocyclic silylene–borane complex 8 LSi-R-BMes2 (L = PhC(NtBu)2; R = 1,12-xanthendiyl spacer; Mes = 2,4,6-Me3C6H2), bearing a Lewis acidic boryl group, enables H2 splitting in the style of FLP chemistry, affording the silane borane 8-H2 (Scheme 5).31 To gain further insights into the necessity of the intramolecular presence of SiII and BIII atoms for H2 activation, the electronically similar N,N′-di-tert-butyl(phenylamidinato) phenylsilylene 9 as well as the intermolecular silylene borane system Mes2BPh/9 towards dihydrogen were both investigated. However, no hydrogenation product was observed, reflecting the importance of the indispensable role of an intramolecularly pre-organised Si–B separation in 8.
 | ||
| Scheme 5 Addition of H2 by the NHSi–borane complex 8. | ||
Inoue and co-workers reported the highly reactive acyclic silylene 10′ stabilised by an N-heterocyclic imino ligand (Scheme 6). It readily undergoes an intramolecular CC insertion into its aromatic ligand framework, affording the room temperature stable silacycloheptatriene (silepin) 10.22h Moreover, variable temperature UV-vis measurements and DFT calculations were conducted and suggested thermally accessible interconversion between silepin and silylene at higher temperatures (e.g. 100 °C). This equilibrium was also evidenced by isolation of a silylene–borane adduct upon addition of B(C6F5)3. Therefore, silepin 10 acts as a “masked” silylene and takes part in the H–H bond activation process. The reaction of 10 and H2 is slow at room temperature but leads to full conversion upon heating to 50 °C within two days. Isolation of the silane product through recrystallisation was problematic probably due to similar solubilities of all species.
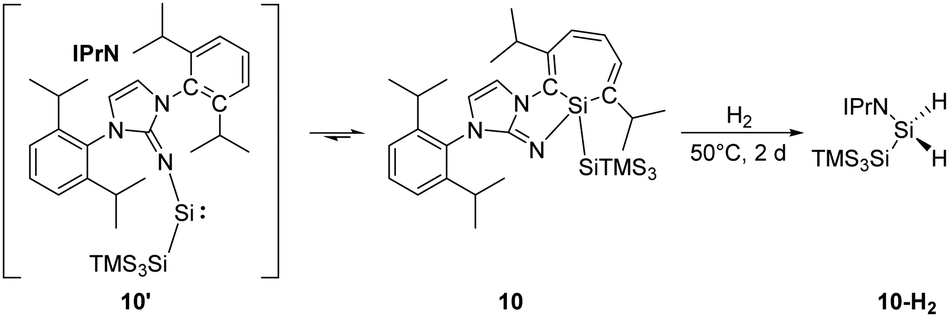 | ||
| Scheme 6 Activation of H2 by the “masked” silylene 10 to form 10-H2. | ||
More recently, the Inoue group reported the synthesis of a novel tetrasilyldisilene 11 displaying bis(silyl)silylene reactivity.22j Reductive debromination of [(TMS)3Si](tBu3Si)SiBr2 with two molar equivalents of potassium graphite at low temperatures results in the formation of tetrasilyldisilene 11, the isomer of (hypersilyl)(supersilyl)silylene 11′ (Scheme 7). Indeed, an equilibrium between 11′ and 11 in solutions is suggested, and the disilene/silylene equilibrium mixture is capable of H2 activation at low temperatures. Treatment of an n-hexane solution of 11/11′ at −40 °C with H2 results in the quantitative formation of the corresponding silane, and no formation of the hypothetical disilene addition product is detectable. Although silylene 11′ reacts smoothly with H2, it exhibits a sizeable HOMO–LUMO energy gap of 403.3 kJ mol−1, which contradicts the assumption that a low HOMO–LUMO gap facilitates the activation of H2 for an acyclic silylenes. Then, further computational calculations were performed and determined a quite low effective barrier of 17.99 kJ mol−1, which rationalise the reaction between 11′ and H2.
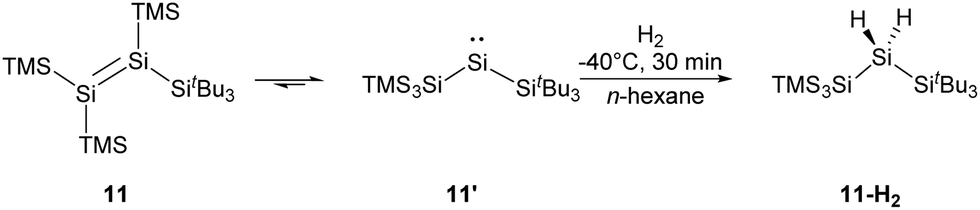 | ||
| Scheme 7 Activation of H2 by a mixture of tetrasilyldisilene 11 and bis(silyl)silylene 11′. | ||
In 2019, Kato et al. isolated a cyclic (amino)metal-substituted di-coordinate silylene 12, which is stabilised by an amino group and a RhI fragment (Scheme 8).32 Interestingly, the molecular structure exhibits a distorted tetrahedral geometry around the rhodium atom rather than a classical square planar reside. A considerably shortened Si–Rh distance (2.138 Å) was observed compared to classical Si–Rh single bonds (ca. 2.30–2.35 Å), indicating an increased Si–Rh multiple bond character. According to theoretical calculations, geometrical deviation around the Rh centre increases the π-donating and σ-accepting character of the RhI fragment, which efficiently stabilises the silylene and leads to a sizeable HOMO–LUMO energy gap. However, 12 remains highly reactive towards H–H bond insertion, affording the corresponding cyclic dihydrosilane 12-H2.
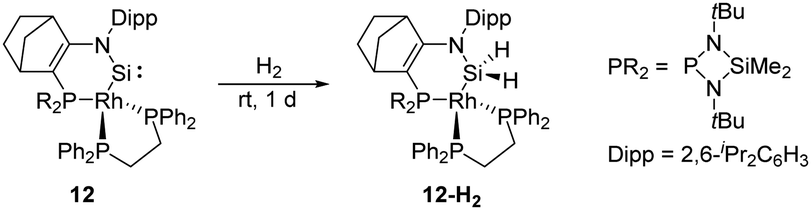 | ||
| Scheme 8 Activation of H2 by the cyclic (imino)metal substituted silylene 12. | ||
More recently, Schultz et al. reported H2 activation by an acyclic transient dimetallasilylene 14 [L(Br)Ga]2Si: (L = HC[C(Me)N(2,6-iPr2-C6H3)]2) (Scheme 9).22l14 exhibits the lowest HOMO–LUMO energy gap (260.5 kJ mol−1) and smallest ΔEST (−5.86 kJ mol−1) so far, due to the kinetic and thermodynamic stabilisation by the ‘metallic’ LGa substituents. Therefore, it undergoes hydrogenation smoothly in the presence of H2, otherwise giving C–C bond activation product under an inert atmosphere of argon. Notably, 14 reacts with CO to form the first isolable [L(Br)Ga]2Si–CO (silylene–CO) complex (see below, Scheme 13), which acts as a “masked” silylene and activates, under liberation of CO, H2 to give the same hydrogenation product 14-H2.
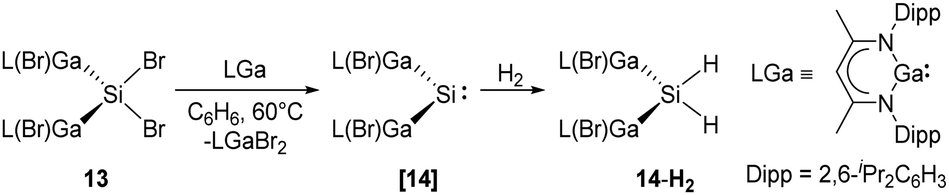 | ||
| Scheme 9 Synthesis of 14-H2 by the acyclic silylene 14. | ||
2.3 Activation of CO
Activation of CO has always been the domain of transition metals as the dissociation energy of CO (BDE = 1077 kJ mol−1) is so high that scission of the strongest neutral triple bond is particularly challenging.33 The well-known Fischer–Tropsch synthesis with the assistance of transition-metal catalysts involves CO bond scission in the presence of H2.34 On the contrary, main-group elements mediated CO activation remains rare, given its limited redox capability. Examples includes activation of CO by the highly reactive cyclic and acyclic alkyl amino carbenes to give ketenes, and germylenes to give germyloxy ketones.16a Reactions with CO by other main-group species are inaccessible, presumably due to their larger HOMO–LUMO energy gap. Surprisingly, splitting and reductive homo-coupling of CO by bis-silylene (LSi:)2Xant 15a [Xant = 9,9-dimethyl-xanthene-4,5-diyl; L = PhC(NtBu)2] and (LSi:)2Fc 15b (Fc = 1,1′-ferrocenyl) were reported by the Driess group (Scheme 10).23g Exposure of Xant(LSi:)215a to CO atmosphere at room temperature for 12 hours affords Xant(LSi)2(μ-O)(μ-CCO) 16a which is isolated as colourless crystals in 83% yields. A strong infrared absorption band at ν = 2069 cm−1 assignable to the CO stretching vibration can be observed. In the case of 15b, it also splits CO smoothly to afford Fc(LSi)2(μ-O)(μ-CCO) 16b as confirmed by multinuclear NMR and XRD analysis. DFT calculations reveal that the initial step of CO binding and scission involved CO as a Lewis acid (four-electron acceptor), contrary to the Lewis base (two-electron donor) role during activation processes mediated by transition metals, and both silylene units of 15a synergistically act as Lewis donors. Notably, the related bis(NHSi)dibenzofuran 15c shows no reactivity towards CO, mainly due to the long distance between two SiII atoms. In a related work, exposure of 16b to NH3 and benzylamine yields Fc-disiloxanediamines and respective acetamides.35
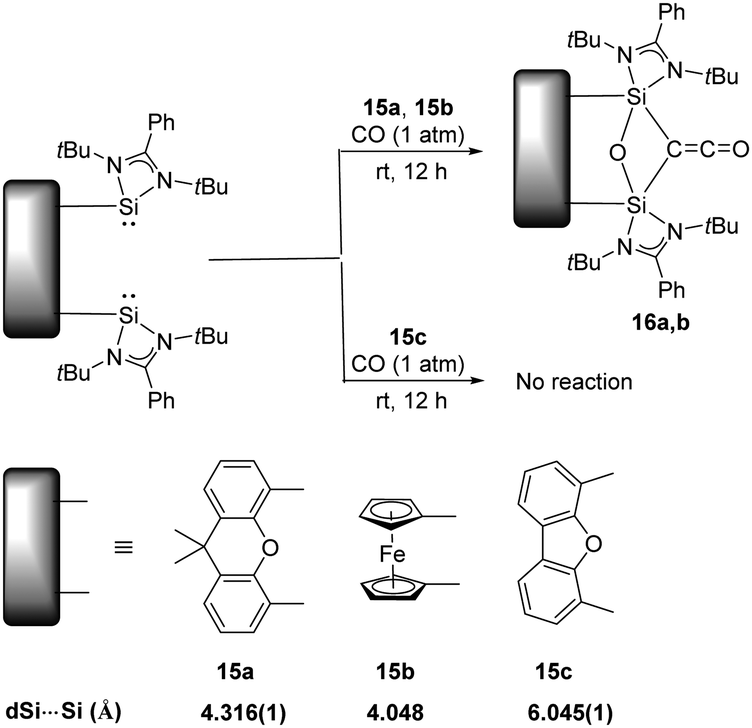 | ||
| Scheme 10 Activation of CO by the bis-silylenes 15a–c. | ||
More recently, the same group reported another example of homocoupling of CO mediated by the bis-silylene 17 [(LSi:)C]2B10H10 {L= PhC(tBuN)2} (Scheme 11).23h Treatment of toluene solutions of 17 to CO atmosphere at −20 °C leads to a colour change from yellow to brown. The unprecedented head-to-tail coupling and C–O cleavage product 18 is obtained in the form of colourless crystals in 62% yields. Although the proposed disilaketene intermediate 17′ has not been isolated or even observed, DFT calculations revealed the initial formation of 17′ which undergoes migration of one Ccage from Si to the central Cketene to afford another intermediate 17′′ featuring a SiC–CO moiety. Head-to-tail dimerisation of 17′′ furnishes the final CO homo-coupling product 18.
 | ||
| Scheme 11 Activation of CO by the bis-silylene 17. | ||
Compared to N-heterocyclic silylenes, acyclic silylenes are thought to be more reactive SiII species due to their more obtuse angle at silicon and narrower HOMO–LUMO gap.27 Aldridge et al. reported the reductive coupling of CO with the boryl-substituted acyclic silylene 1, generating four- and six-membered ethynediolate 19 through the formation of intra- and intermolecular Si–O interactions (Scheme 12).36 Notably, the reductive coupling of CO to give the ethynediolate dianion [OCCO]2− could only be achieved for alkali, d- and f-block metals before.37
 | ||
| Scheme 12 Activation of CO by the acyclic silylene 1. | ||
Recently, the Schultz group reported the aforementioned acyclic transient dimetallasilylene 14 which reacts with CO to give the first isolable silylene carbonyl complex 20 (Scheme 13).22l Reaction of [L(Br)Ga]2SiBr213 and one equivalent of LGa in benzene at 60 °C in CO atmosphere results in the formation of 20. CO serves as a mild Lewis acid in the reaction while [L(Br)Ga]2Si: shows strong electron-donating properties due to the electropositive L(Br)Ga substituents and the wide Ga–Si–Ga bond angle. DFT calculations were performed for the comparison of non-covalent H2C/Si:–CO adducts. While ketene is highly favoured for carbon (−347.69 versus −323.84 kJ mol−1), silaketene is considerably less stable than the [H2Si:–CO] adduct (−44.77 versus −113.0 kJ mol−1).
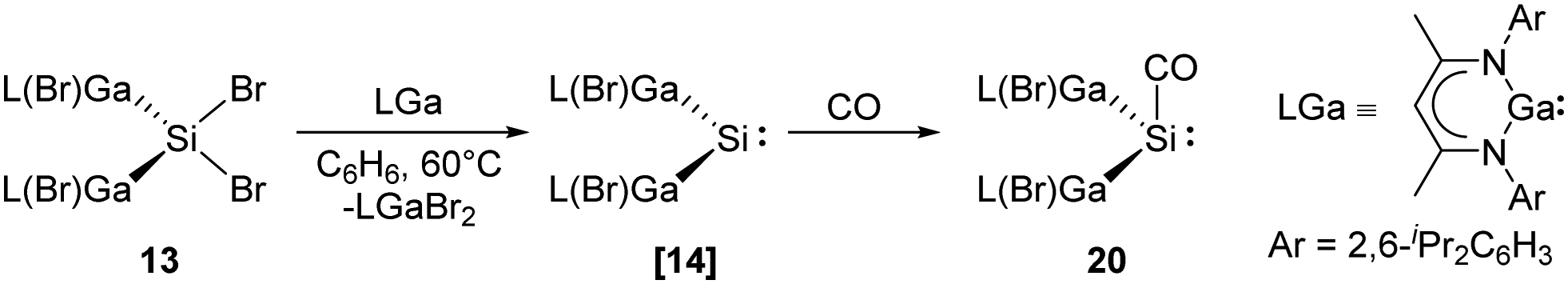 | ||
| Scheme 13 Coordination of CO to the acyclic silylene 14 to form the first isolable silylene–CO complex 20. | ||
2.4 Activation of CO2 and N2O
CO2 and N2O activation has become a hot topic and underwent intense research due to environmental considerations. Advances involving heterogeneous, homogeneous, transition metal-free and biological catalytic systems to use CO2 and N2O for the formation of value-added oxygenation products have been achieved.38,39 In main-group chemistry, multiply-bonded main-group compounds, such as disilene, and low-valent p-block compounds, such as gallium species and germylene, are also developed and found to show reactivity towards CO2 and N2O.16a,c Meanwhile, silylenes have a high oxophilicity and thus can react very efficiently with these thermodynamically very stable species.In 1996, Jutzi et al. reported the first reductive activation of CO2 with (Me5C5)2Si 21 under release of CO. Stoichiometric formation of the carbodisiloxane SiIV complex 22 and the bis-carbonato SiIV complex 23 are obtained depending on the solvent used, respectively (Scheme 14).40 The pathway is proposed to undergo a reactive [2+1] cycloaddition intermediate or its ring-opened isomer, which easily releases CO.
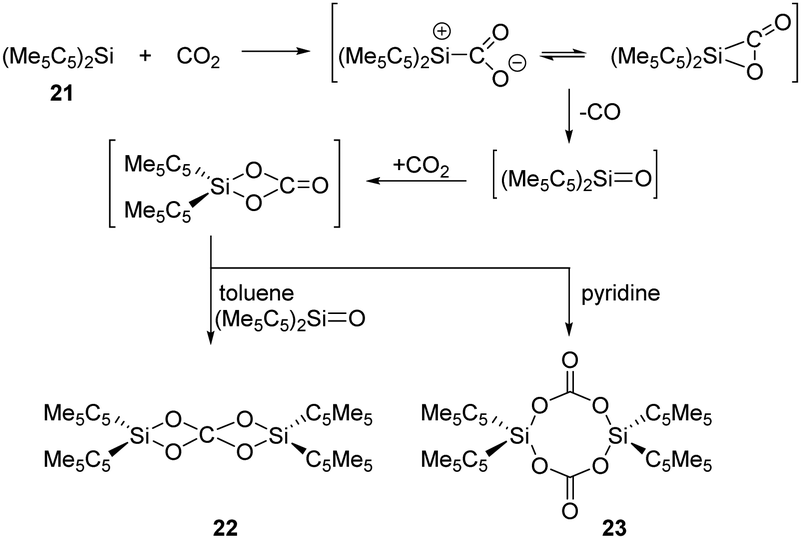 | ||
| Scheme 14 Activation of CO2 by the decamethylsilicocene 21. | ||
In 2007, the Driess group reported the selective monooxygenation of the siloxysilylene 24 by exposing a brown solution of the siloxysilylene 24 to N2O and CO2 at −78 °C, respectively, affording silanoic silylester 25 in 79% yields (Scheme 15).41 Besides, the same group reported a new NHC–silylene adduct 27 (NHC = 1,3,4,5-tetramethylimidazol-2-ylidene)42 which undergoes facile oxygenation with N2O to give the first isolable NHC-supported silanone 28 featuring an ylidic SiO bond.43 No reaction occurs between silylene 26 and N2O even after several days at room temperature.
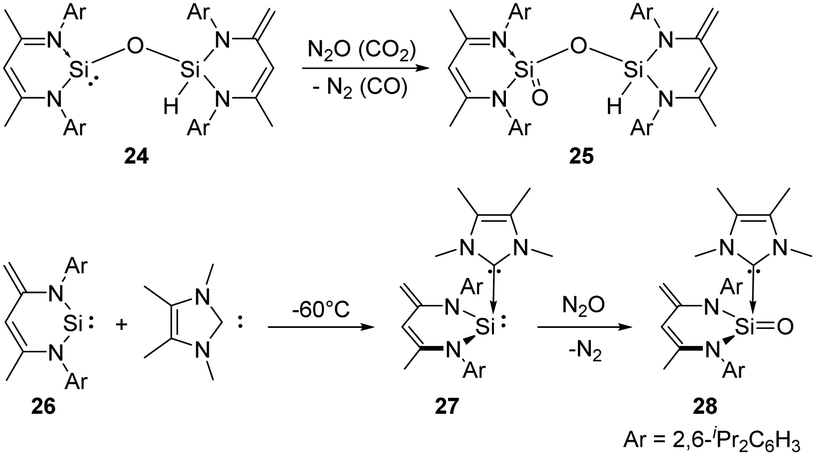 | ||
| Scheme 15 Activation of N2O/CO2 by the siloxysilylene 24 and the NHC–silylene adduct 27. | ||
The Driess group also synthesised a Lewis-base coordinated SiO complex 30 which can be obtained by oxygenation of the silylene p-dimethylaminopyridine (DMAP) adduct 29 with N2O.4430 was further used for the activation of ammonia, affording a unique pair of the tautomer products silahemiaminal 31 and silanoic amide 32 with DMAP being ultimately released, respectively (Scheme 16). The two products are in equilibrium in solutions, and undergo intermolecular stabilisation via SiOH⋯OSi hydrogen bond.
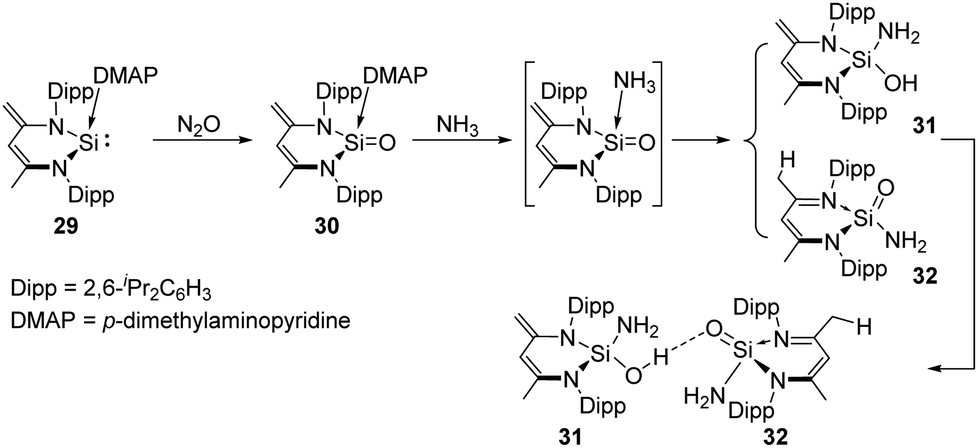 | ||
| Scheme 16 Activation of NH3 by the SiO complex 30. | ||
The ring compound 34 featuring two four-membered disiloxane rings bridged by two oxygen atoms were synthesised by exposure of red solutions of the base-stabilised bis-silylene 33 (LSi–SiL, L = PhC(NtBu)2) in toluene to N2O at room temperature (Scheme 17).45 A mechanism is suggested where the Si–Si bond is cleaved under insertion of an oxygen atom from N2O. Then the two Si atoms bearing the lone pairs of electrons react with N2O to give SiO moieties, followed by dimerisation to give the final ring compound 34. Meanwhile, the reaction of monosilylene 35 with N2O affords 36 comprising a Si3O3 six-membered ring.46 Unlike 34, compound 36 features a paddle-wheel arrangement.
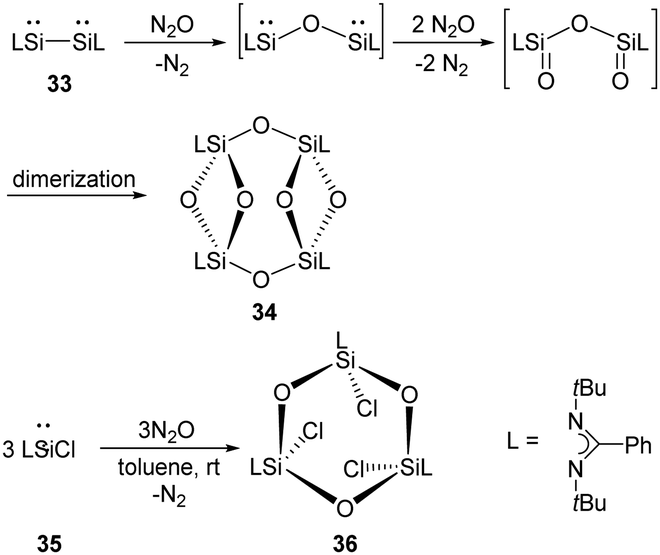 | ||
| Scheme 17 Oxygenation of the mono- and bis-silylenes 35 and 33 by N2O. | ||
Kato and Baceiredo reported the phosphine-stabilised silylene 37 in 2011, which acts as either a sila-Wittig reagent or a nucleophilic silylene complex. It reduces CO2 rapidly at room temperature, affording the original P-chiral tricyclic phosphine 38 with an oxygen atom bridging Si and P (Scheme 18).47 The plausible mechanism involves a phosphine centred sila-Wittig type reaction. Starting from a phosphine-stabilised silylene, the same group also synthesised the donor-stabilised silacycloprop-1-ylidene 39,48 and its reaction with N2O allows access to the silacyclopropanan-1-one 40, representing the smallest membered ring silaketone known to date.49
 | ||
| Scheme 18 Activation of CO2 by the phosphine-stabilised silylene 37 and silacycloprop-1-ylidene 39. | ||
The synthesis and characterisation of the first metallosilylene 41 containing a two-coordinate silicon centre was reported by Filippou and co-workers in 2014. It shows a high reactivity towards the cleavage of the σ-bonds of H2, H2O, etc. due to the small HOMO–LUMO energy gap (Scheme 19).50 Furthermore, exposure of 41 to N2O gives the metallosilanone product 42. Theoretical calculations of the σ and π NBO orbital of the SiO bond in 42 revealed 85% of the NBO density on the O atom, suggesting a substantial contribution of a zwitterionic resonance structure (Si+–O−).
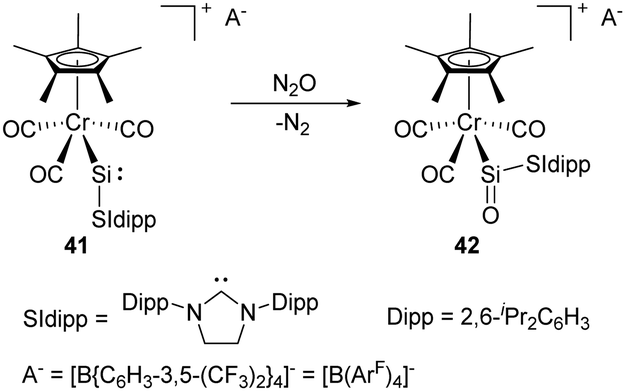 | ||
| Scheme 19 Oxygenation of the metallosilylene 41 with N2O. | ||
Kira demonstrated that the dicoordinate dialkylsilylene 43, 2,2,5,5-tetrakis(trimethylsilyl)-silacyclopentane-1,1-diyl, reacts with CO2 smoothly at room temperature, followed by hydrolysis by the water contamination in MeOH, to give the corresponding bis(silyl)carbonate 44 in high yields (Scheme 20).51 DFT calculations showed that the reaction involves the formation of a SiO bonded intermediate, which is similar to that of the decamethylsilicocene reported by Jutzi et al.40
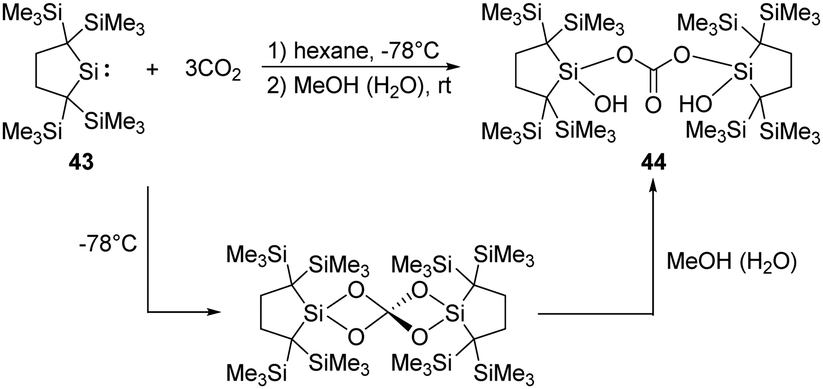 | ||
| Scheme 20 Synthesis of the bis(silyl)carbonate 44 by the reaction of dialkylsilylene 43 with CO2. | ||
As for the bis(amidinato)silylene 45 (Scheme 21), its reaction with an excess of N2O at low temperatures affords the dinuclear five-coordinate SiIV complex 46, while the mononuclear six-coordinate SiIV complex 47 is obtained by treatment of 45 with an excess of CO2 at room temperature.52 The mechanism can be rationalised through the formation of a five-coordinate silanone species, followed by its dimerisation to give 46, or reaction with an additional equivalent of CO2 to give 47. Similarly, treatment of the analogous bis(guanidinato)silylene 48 with N2O and CO2 in toluene at low temperatures affords the six-coordinate SiIV complex 49 and the dinuclear four-coordinate SiIV complex 50, respectively.53 Treatment of the guanidinatosilylene 51 with N2O and CO2 in toluene at low temperatures affords the unsaturated four-coordinate SiIV compound 52 featuring a SiN bond, and the five-coordinate SiIV compound 53.54 The silylene 54 bearing a bulky bidentate benzamidinato ligand also reacts with N2O to give the dinuclear five-coordinate SiIV compound 55,55 similar to those of 45 and 51.
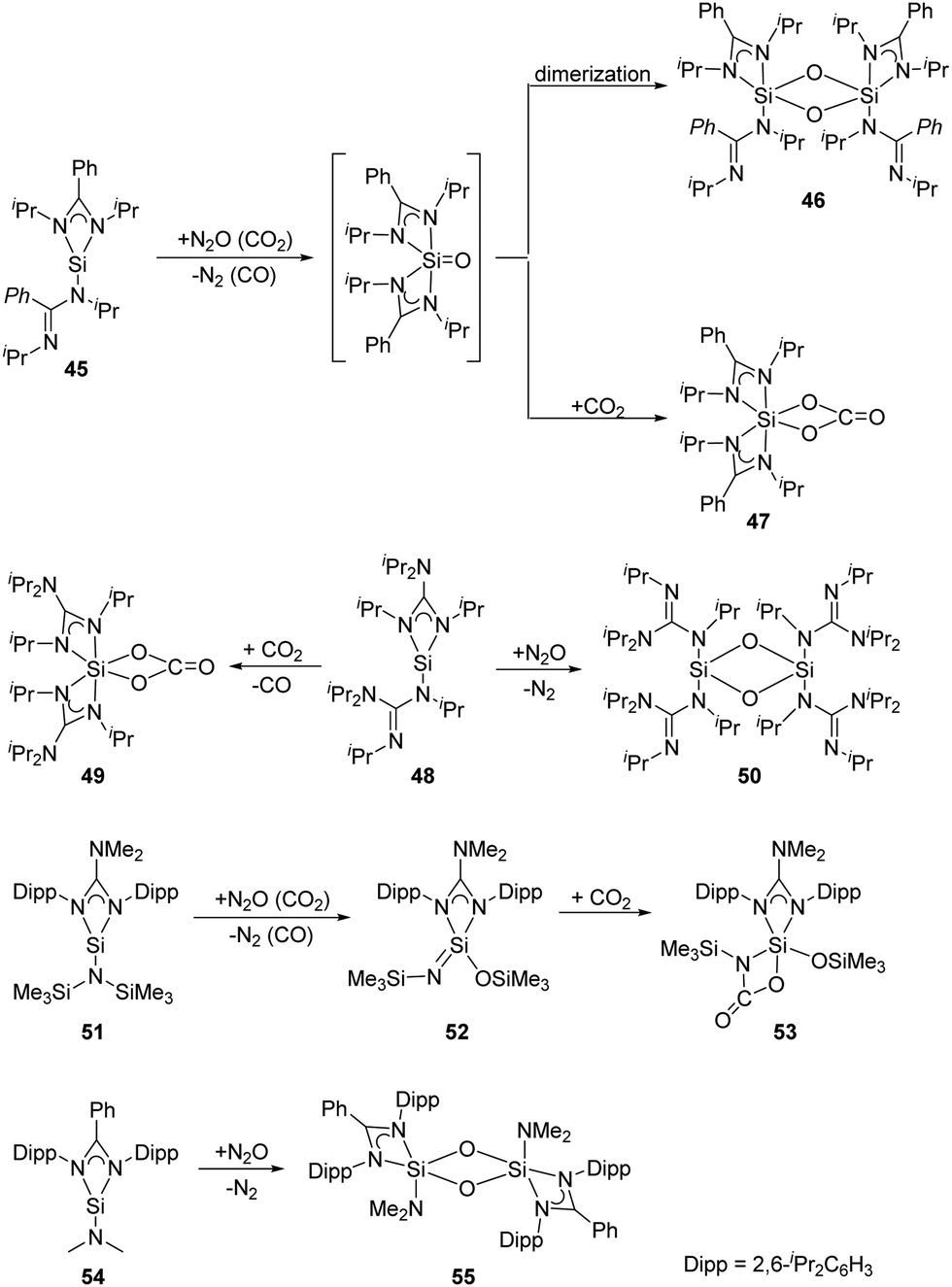 | ||
| Scheme 21 Activation of N2O and CO2 by the amidinato silylenes 45, 48, 51 and 54. | ||
Heterocyclic silylene 56a featuring a particularly electron-rich Si centre reacts smoothly with N2O in THF at −80 °C to give the silanone product 57a (Scheme 22), which is stable below −50 °C. However, it undergoes dimerisation at room temperature to afford the head-to-tail cyclodisiloxane 58a, with the estimated t1/2 being 30 min.56 The more sterically hindered 57b shows an enhanced persistence with t1/2 = 5 h at room temperature. Notably, the strong electron-donating ability of the phosphonium ylide moiety is inferred to account for the electron-rich Si centre and related reactivity. Therefore, by taking advantage of the better donating ability of phosphonium bora-ylide function, the (amino)(bora-ylide)silylene 59 reacts immediately with N2O at −20 °C to give the corresponding silanone 60.57 Even though the electronic effects of phosphonium bora-ylide and sterically bulky substituents play a great role for the stabilisation of silanone 60, its dimerisation remains exergonic (ΔG61–60 = −66.9 kJ mol−1) and furnishes slowly the cyclodisiloxane 61 at room temperature (t1/2 = 4 d).
 | ||
| Scheme 22 Oxygenation of the phosphonium ylide and phosphonium bora-ylide silylenes 56 and 59 by N2O. | ||
The “masked” form of acyclic silylenes, silepin 10 and its analogue 10a (Scheme 23), demonstrated by Inoue et al., also undergo oxygenation with N2O and CO2, and the processes follow the mechanism which is parallel to that of the aforementioned reactions.22h,i Exposure of n-hexane solutions of the silepin to N2O atmosphere at low temperatures gives the colourless silanone compounds 62 and 62a, and they can be further converted quantitatively to the silicon carbonate complexes 63 and 63a at room temperature, respectively. The synthesis of 63 can also be directly achieved by reaction of 10 with CO2.22h No dimerisation of carbonato silicon compound was observed due to the bulky protection.
 | ||
| Scheme 23 Activation of N2O and CO2 by the acyclic silylenes 10 and 10a. | ||
The silylene borane complex 8 reported by the Driess group acts as an intramolecular FLP and allows access to the silanone–borane 64 featuring a SiO → B interaction through reductive activation of N2O and CO2, respectively (Scheme 24).31 Notably, isolation of 64 is also possible via reaction of 8 with dioxygen.
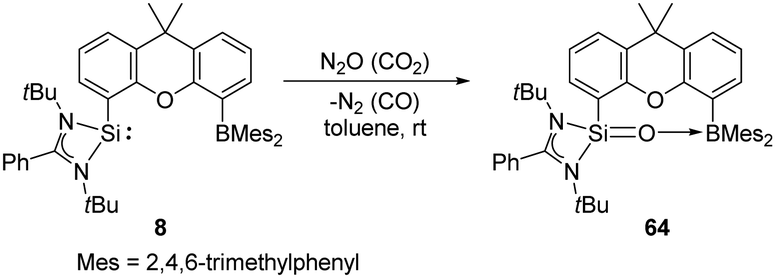 | ||
| Scheme 24 Activation of N2O and CO2 by the silylene borane complex 8. | ||
The novel β-diketiminato-supported silaacyl chloride 66 featuring a unique Lewis-acid-free Si(O)Cl unit was synthesised by oxidation of chlorosilylene 65 with N2O at room temperature (Scheme 25).58 Treatment of 66 with sources of either H− or tBuO− allows access to the nucleophilic substituted silaaldehyde 67 and silaester 68. Analogous to the acyl chloride chemistry, silaacyl chloride 66 offers a systematic methodology to sila-carbonyl derivatives by nucleophilic substitution at the Si centre.
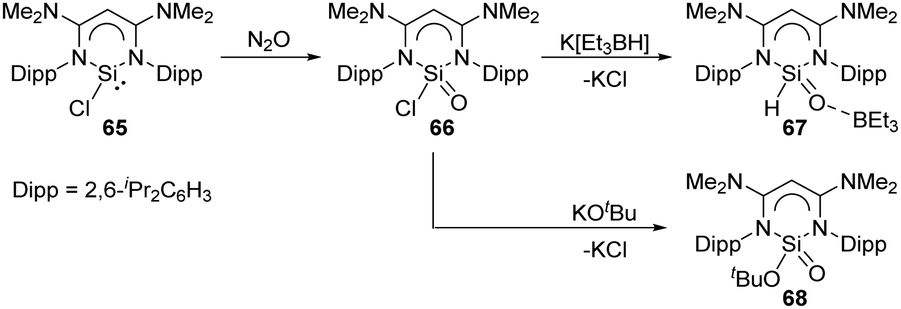 | ||
| Scheme 25 Activation of N2O by the chlorosilylene 65 and further metathesis reaction at the Si centre. | ||
Another example of reductive activation of N2O and CO2 by the acyclic silylene 1 shows a net oxygen atom abstraction process (Scheme 26).36 The authors inferred the formation of a short-lived silanone intermediate followed by a rapid N-to-O silyl group transfer to give the product 69. The transition state corresponding to intramolecular silyl group transfer was identified by DFT calculations with an activation energy of 71.9 kJ mol−1.
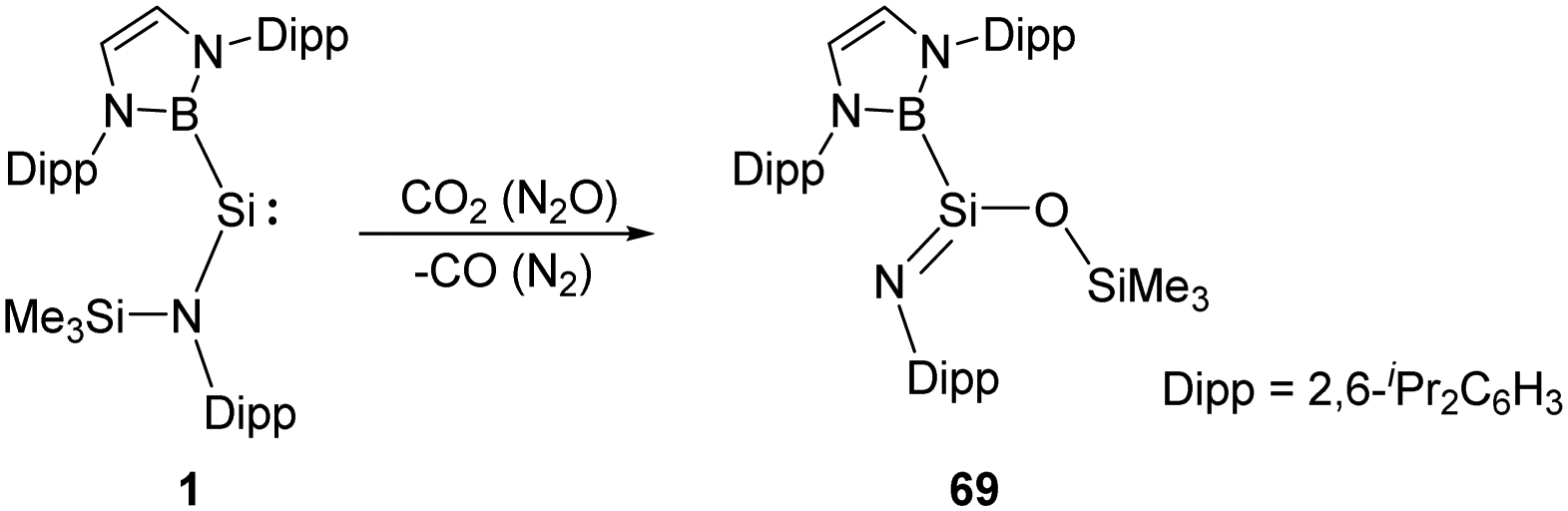 | ||
| Scheme 26 Activation of N2O and CO2 by the acyclic silylene 1. | ||
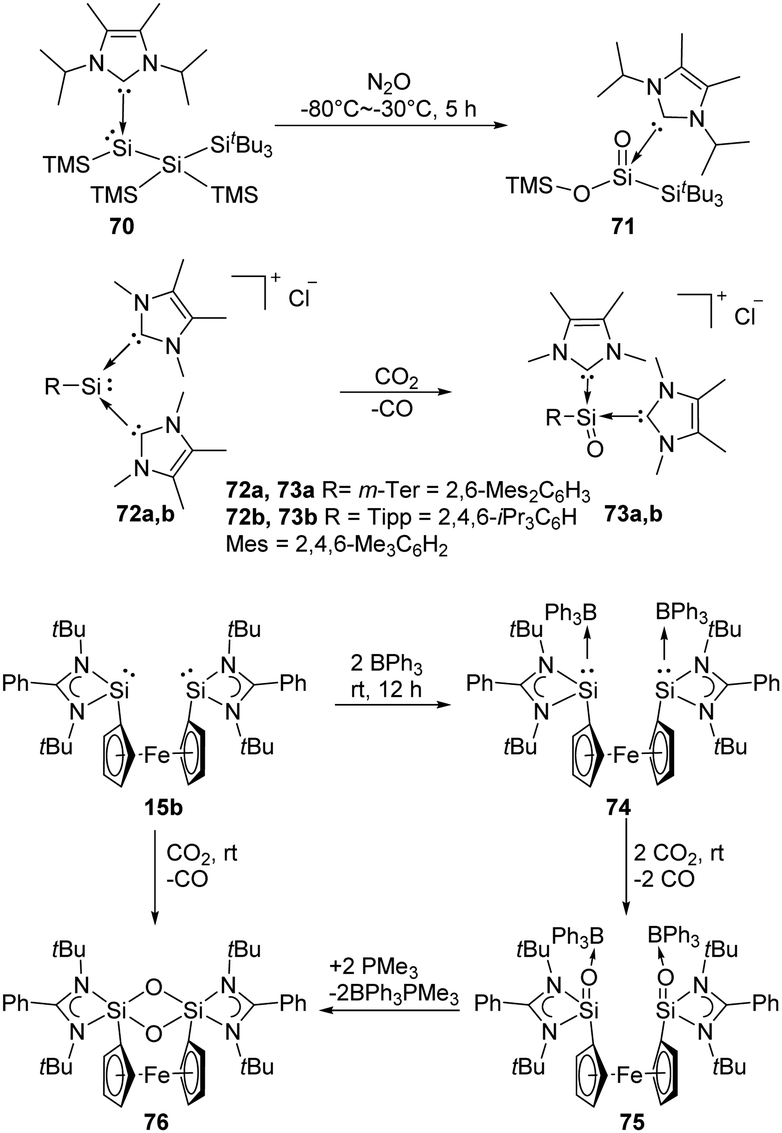
Lewis acids or bases are often introduced for the stabilisation of some reactive species. For example, Inoue et al. isolated the first acyclic NHC-stabilised silanoic ester 71 by exposure of the NHC-supported silylene 70 bearing two silyl groups to N2O atmosphere (Scheme 27).22j Even though the exact mechanism is so far not clear yet, the authors inferred that the formation of a silanone with subsequent silyl migration, oxidation and rearrangement eventually lead to the ester product. The same group also reported the reduction of CO2 using the corresponding silyliumylidene cations 72, resulting in the successful synthesis of silaacylium ions 73 stabilised by two NHCs (NHC = 1,3,4,5-tetramethylimidazol-2-ylidene).59 Computational calculations suggested strong SiO bond character in 73. Another Lewis acid-stabilised bis-silylene 15b reported by Driess and co-workers also undergoes reductive activation of CO2 to give silanone product 75,60 similar to that of intramolecular silylene borane complex 8. Without the presence of Lewis acids, with N2O and CO2 the 1,3,2,4-disiloxane product 76 is obtained instead.
 | ||
| Scheme 27 Activation of N2O and CO2 by Lewis acid/base-stabilised SiII species 70, 72a, b, 15b and 74. | ||
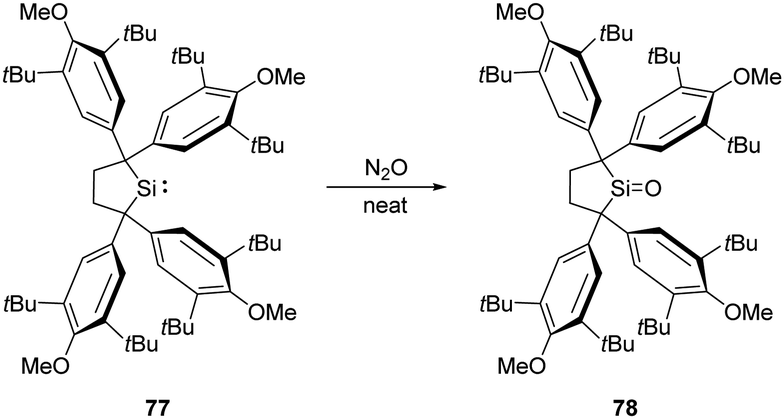
Recently, Iwamoto et al. reported the isolation of a genuine silicon analogue of a ketone, compound 78, featuring a three-coordinate Si centre and an unperturbed SiO bond (Scheme 28).61 Exposing solid silylene 77 to N2O atmosphere at room temperature affords the silanone product 78 quantitatively as a white solid. Remarkably, coordination by Lewis bases and acids or the introduction of electron-donating groups are not indispensable for this silanone case.
 | ||
| Scheme 28 Activation of N2O by the cyclic silylene 77 to give the isolable genuine silanone 78. | ||
In 2020, Kato and Baceiredo reported the synthesis of the N-hetero-RhI-metallacyclic silylene 79 featuring a tetrahedral Rh centre.62 It shows an improved stability compared with 12 and undergoes oxidation after being exposed to N2O atmosphere at low temperatures to furnish the corresponding silanone 80 (Scheme 29). Even though silanone 80 shows a large dimerisation energy with ΔG = +360.66 kJ mol−1, it displays considerable stability due to the presence of the σ- and π-donating and sterically protective Rh fragment. Cooperative involvement of both SiO and RhI reactive sites was observed on its further reactivity towards H2.
 | ||
| Scheme 29 Oxygenation of the cyclic (amino)(rhodium) silylene 79 by N2O. | ||
2.5 Activation of O2
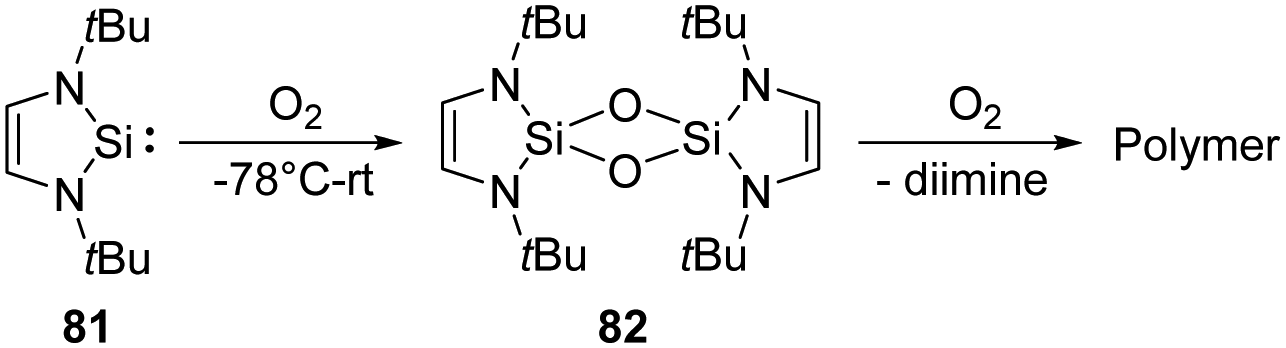
Activation of the O–O bond in dioxygen is non-trivial and crucial in both biology and industry. Nature has evolved several strategies to achieve this goal, such as cytochrome oxidase63 and metalloenzymes taking Cu-64 and Fe-sites65 as cofactors. A lot of homo- and hetero-multinuclear metal complexes were also developed to achieve a controllable activation of O2.66 As expected by the large oxophilicity of silicon, silylenes are very reactive towards O2.The first isolable N-heterocyclic silylene 81 reacts with dry O2 rapidly, giving rise to a colourless and insoluble polymer. At the same time, a small amount of minor product 82 could also be observed and verified by 1H NMR data and mass spectrometry (Scheme 30).67 However, any further characterisation was prevented due to insufficient amounts. The proposed pathway reveals the initial formation of a transient silanone, which then undergoes dimerisation to give 82.
 | ||
| Scheme 30 Activation of O2 by the NHSi 81. | ||
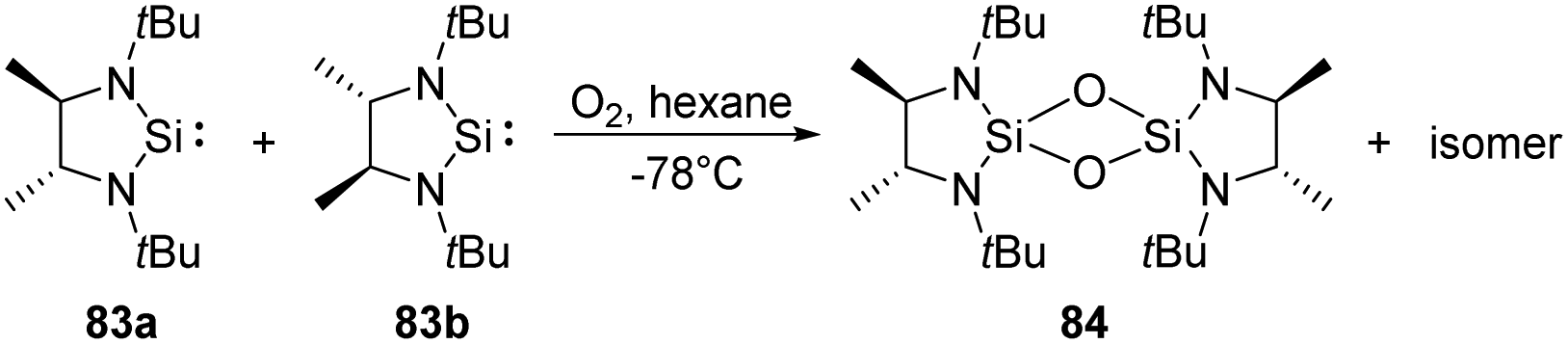
West et al. reported the new isolable silylenes 83a–b, rac-N,N′-di-tert-butylethylene-4,5-dimethyl-1,3-diaza-2-silacyclopentan-2-ylide (Scheme 31), which have been utilised for the reaction with a stoichiometric amount of O2 at low temperatures, affording the corresponding 1,3,2,4-disiladioxetane.68 Interestingly, the meso isomer 84 was isolated as solid, while the rac isomer being a colourless liquid.
 | ||
| Scheme 31 Oxygenation of the NHSis 83a and 83b by O2. | ||
On the exploration of isolable silanoic ester synthesis from the siloxysilylene 24, its reaction with dioxygen at −78 °C readily gives an unexpected new type of strained cyclodisildioxane 85 featuring a remarkably clean deoxygenation process (Scheme 32).41 Compounds 24′ and 24′′ are assumed to be reactive intermediates during the formation of this cyclodisildioxane compound.
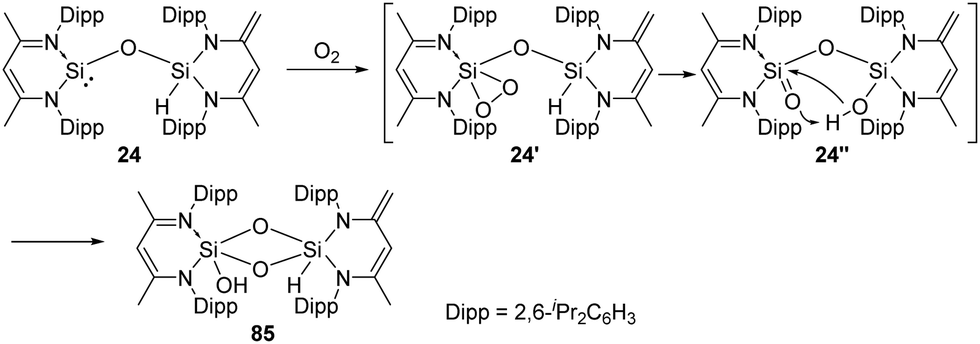 | ||
| Scheme 32 Activation of the siloxysilylene 24 by O2. | ||
2,3-Dioxasilirane with a SiO2-peroxo ring has always been elusive species for a long time and could only be generated and studied in cryogenic argon matrices. This situation has changed since Driess et al. reported the isolable adducts 86 and 86a of transient dioxasilirane featuring fascinating five-coordinate square-pyramidal silicon atoms and “side-on” coordinated peroxo ligands (Scheme 33).4486 and 86a can be obtained selectively through facile O2 activation with the NHC-stabilised silylenes 27 and 27a, respectively. Remarkably, 86 undergoes internal oxygen atom transfer at room temperature to afford a cyclourea → sila-urea adduct 87 featuring a CO → SiO dative interaction, which highlights the importance of donor–acceptor stabilization for the successful isolation of elusive silicon–oxygen species.
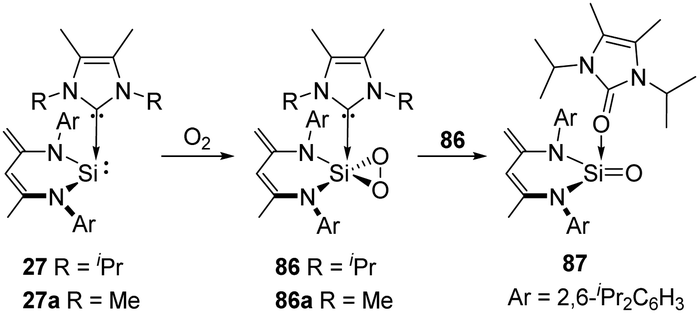 | ||
| Scheme 33 Activation of O2 by the NHC-activated silylenes 27 and 27a. | ||
The base-stabilised silacycloprop-1-ylidene 39 was reported by Kato and Baceiredo and demonstrated to react with O2, leading to the base-stabilised sila-β-lactone product 88 featuring a planar β-lactone four-membered ring (Scheme 34).69 Driven by the ring strain and the high polarity of silacarbonyl function, the reactive lactone 88 reacts with ethanol to afford donor/acceptor-stabilised silanoic acid.
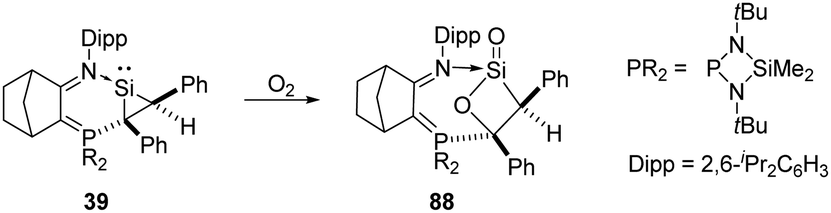 | ||
| Scheme 34 Activation of O2 by the silacycloprop-1-ylidene 39. | ||
As an alternative route to an acyclic silanone, Jones and Aldridge et al. treated the acyclic diaminosilylene 89 with an excess of dry O2, leading to an unexpected unsymmetrical silanol 90 (Scheme 35).22g This reaction is assumed to proceed via a transient dioxasilirane followed by O–O bond cleavage and a subsequent radical-induced alkyl C–H activation, though the attempts to observe the transient silirane were unsuccessful.
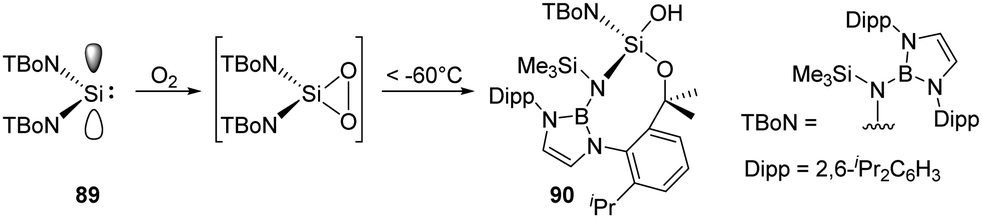 | ||
| Scheme 35 Synthesis of the silanol 90. | ||
2.6 Activation of H2O, H2O·B(C6F5)3 and H2S
Activation of water is most commonly observed in nature, existing in a series of hydrolase enzymes.70 H2O and H2S are also fundamental to our industrial society due to their inherent availability. Comparatively, introduction of hydroxy and sulfide can be easily achieved by relatively reactive isolable silylenes.West and co-workers demonstrated that the first isolable N-heterocyclic silylene 81 inserts into O–H bonds of H2O easily (Scheme 36).71 Isolation of the colourless disiloxane 91 can be achieved from the reaction of silylene 81 with H2O. The corresponding silanol compound is proposed to be an intermediate which eventually self-condenses to the product.
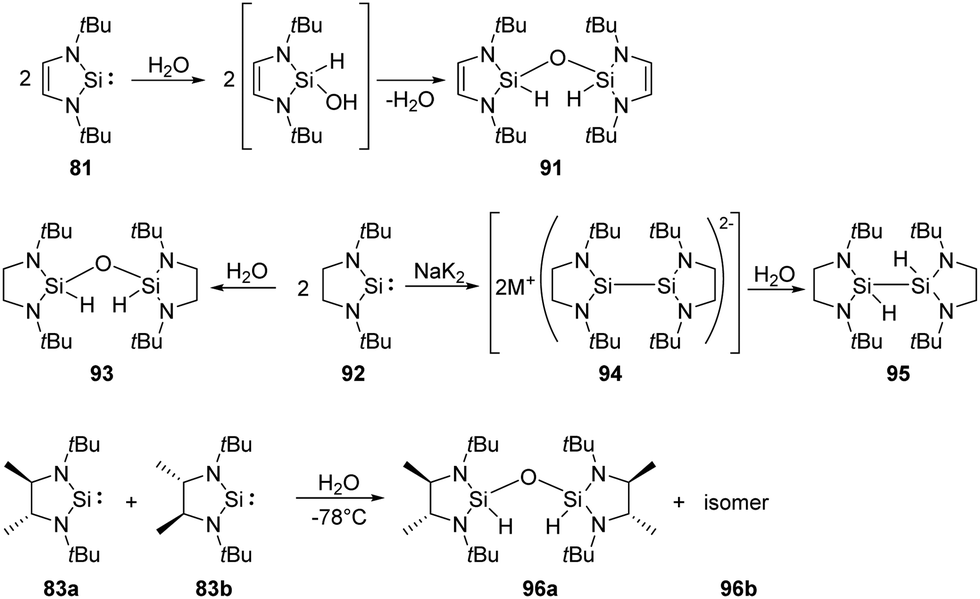 | ||
| Scheme 36 Activation of H2O by the NHSis 81, 92, 83a, and 83b. | ||
Similarly, the saturated analogue of 81, silylene 92, also undergoes the same insertion into O–H bond of water, affording disiloxane 93.72 Notably, the overreduction of silylene 92 competes with the reduction of its dichloro precursors, resulting in a lower yield of silylene. The overreduction product 94 can be trapped by proton abstraction from water, giving the dihydrido compound 95. As for the racemic saturated silylenes 83a and 83b, both rac and meso disiloxane isomers (96a and 96b) are formed, resulting from the reaction of the silylenes and H2O via the corresponding silanol intermediate.68
The zwitterionic silylene 26 also undergoes H2O activation in the molar ratio of 2:1, affording solely the siloxy silylene 98 which represents an unprecedented type of mixed-valent disiloxane compound containing di- and tetravalent silicon atoms bridged by an O atom (Scheme 37).73 Although the mechanism is still unclear, the formation of 98 suggests a faster proton migration from a terminal methyl group in the proposed reactive intermediate 97 than from the OH group in 97′ to the divalent silicon atom. Meanwhile, the presence of a strong Lewis acid bound to oxygen atom of water leads to the drastically increased acidity of the OH group and fast proton migration from the OH group to Si. This assumption is verified by addition of the water–borane adduct H2O·B(C6F5)3 to 26 in a molar ratio of 1:1, which affords exclusively the silaformamide–borane complex 99 featuring a unique short Si-O interatomic distance of 1.552(2) Å. IR measurements by means of isotope labelling experiments and DFT calculations were performed to verify the SiO bond character in 99. The observed bands for 18O-labeled (1112 cm−1) and 16O-99 (1165 cm−1) to a SiO stretching mode are far above frequencies typical for Si–O single bonds (800–900 cm−1). Silylene 26 also shows reactivity towards H2S at low temperatures in the molar ratio of 1:1, resulting in the surprisingly simple formation of the donor-stabilised silathioformamide 100.74
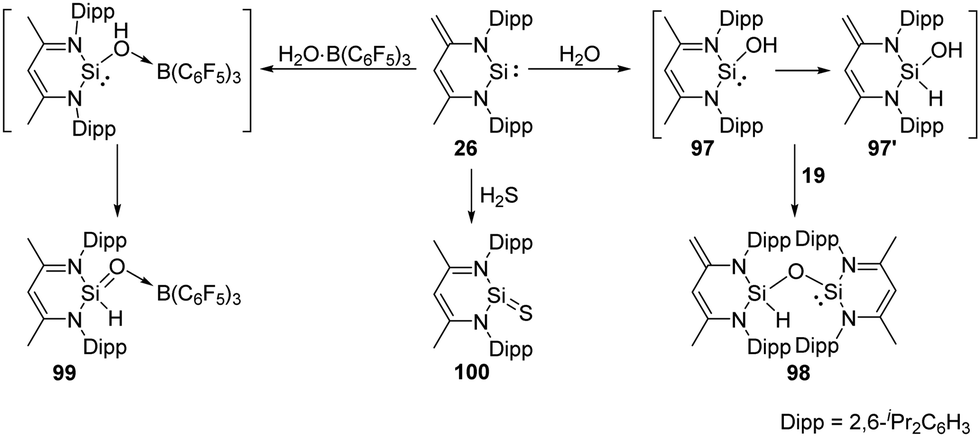 | ||
| Scheme 37 Activation of H2O, H2S and H2O·B(C6F5)3 by the zwitterion-like NHSi 26. | ||
Different from the well-studied reactions of silylene 26 with water, the addition of H2O to complex 101 enables the donor/acceptor abilities of the silicon ligand to be tuned while still coordinated to the Ni centre and without changing its ligand sphere, affording the corresponding SiII hydroxide–Ni complex 102 (Scheme 38).75 This process is in contrast to the well-studied reactions of other metal silylene complexes with H2O where the Si–metal bond is either broken by 1,1-addition to the SiII centre or remains intact by 1,2-addition across the Si–metal bond.76 Besides, complex 101 is also capable of activating H2S in the molar ratio of 1:1, solely affording the 1,4-addition product 103 despite the high thiophilicity of nickel.74
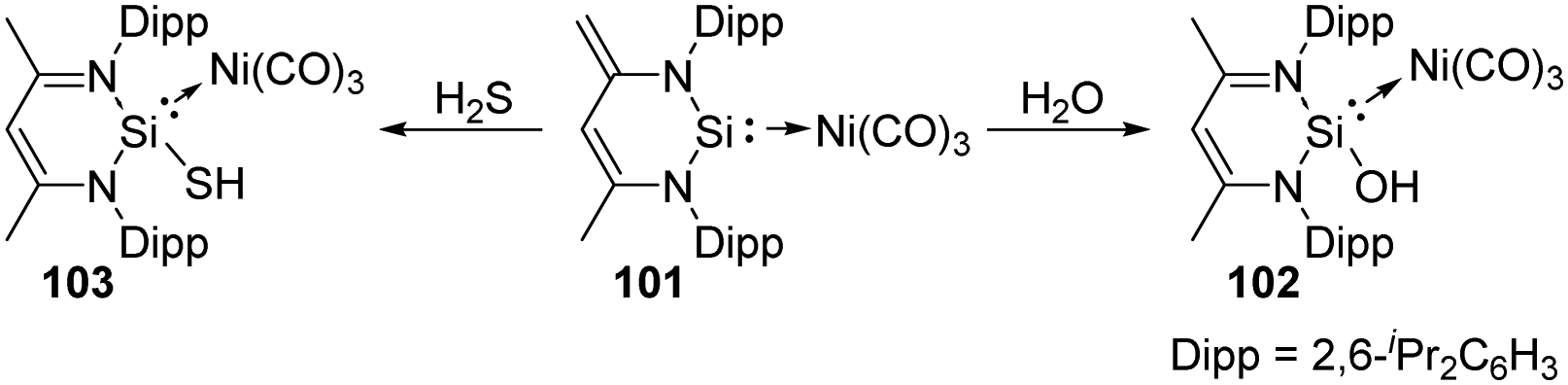 | ||
| Scheme 38 Additions of H2O and H2S by the silylene–Ni0 complex 101. | ||
The reaction of the monochlorosilylene 35 with H2O·B(C6F5)3 in the presence of IPr [1,3-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene] leads to the formation of a stable silicon analogue of an acid anhydride 106 featuring an OSi–O–SiO core (Scheme 39).77 The presumed mechanism reveals the involvement of a silaformaldehyde 104b and a silacarboxylic acid 105. The reaction of 35 and H2O·B(C6F5)3 affords 104a along with the elimination of HCl as IPr·HCl. 104b is then obtained by subsequent rearrangement of 104a and undergoes isomerisation to give 104c. Oxygen abstraction of cationic silicon of 104c affords 105, and finally protonation of the amidinate ligand of 104b by 105 affords finally the acid anhydride analogue 106.
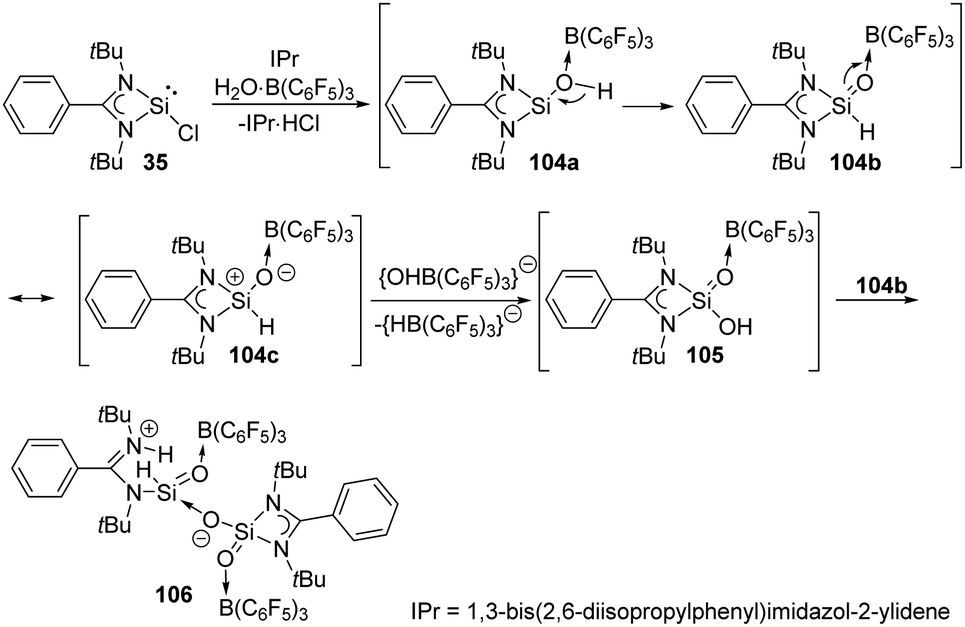 | ||
| Scheme 39 Reaction of H2O·B(C6F5)3 with the monochlorosilylene 35. | ||
The first acyclic silacarbonyl halide compound 108 stabilised by Lewis donor was reported by Roesky et al. (Scheme 40).78108 can be prepared by insertion of silylene 107 into the O–H bond of H2O·B(C6F5)3 adduct and subsequent elimination of HCl assisted by IPr (1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene). The stable silaformyl chloride 108 features a significant SiO bond character and a distorted tetrahedral Si centre.
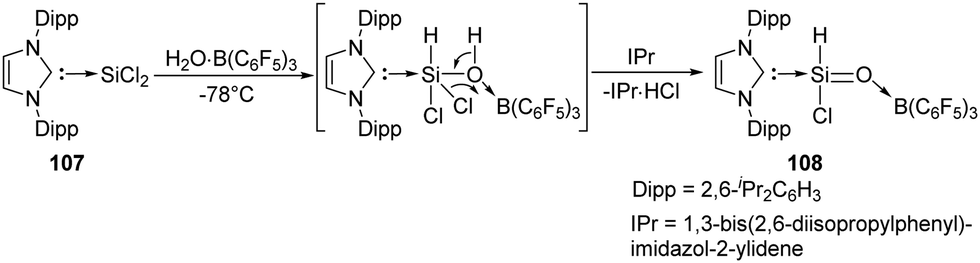 | ||
| Scheme 40 Reaction of H2O·B(C6F5)3 with the NHC–dichlorosilylene adduct 107. | ||
Müller et al. have developed a previously unknown synthetic pathway for the stabilisation of silylenes by taking advantage of the NHC-induced fragmentation of silanorbornadiene derivatives. The new way allows the isolation and characterisation of new hydridosilylenes.79 The hydridosilylene 109 synthesised in this way is highly reactive and shows significant reactivity after being exposed to H2O in THF solution at low temperatures, giving cleanly the tetrahydridodisiloxane 110 as the sole product (Scheme 41).
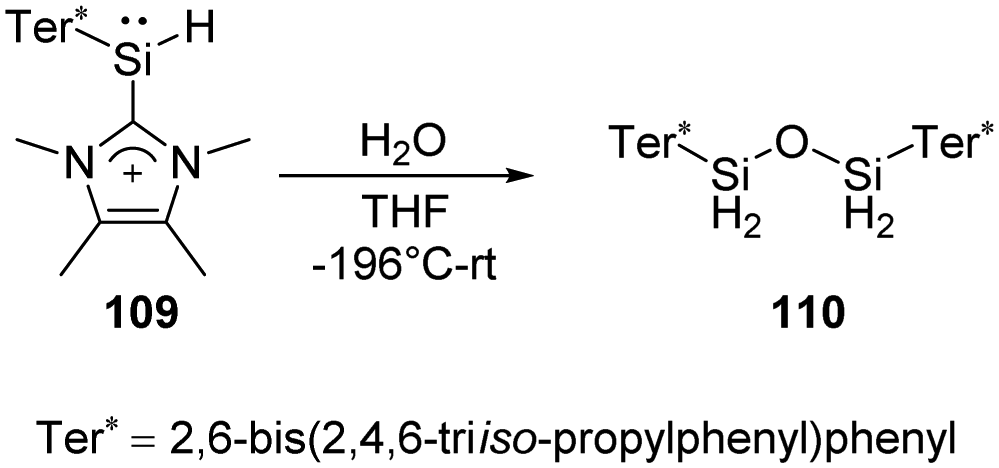 | ||
| Scheme 41 Activation of H2O by the hydridosilylene adduct 109. | ||
The pre-organised intramolecular silylene borane complex 8 isolated by the Driess group acts as a FLP and undergoes unexpected metal-free dehydrogenation of H2O to give silanone–borane 111 as an isolable product (Scheme 42).31 Release of H2 was observed by in situ1H NMR measurements with a proton resonance at δ 4.46 ppm. DFT calculations suggested that initial coordination of the H2O molecule to the B centre forms 8A, which further undergoes oxidation of O–H to the Si centre to yield 8B. Subsequently, migration of OH from B to Si gives 8C, followed by dehydrogenation to form product 111 along with release of H2.
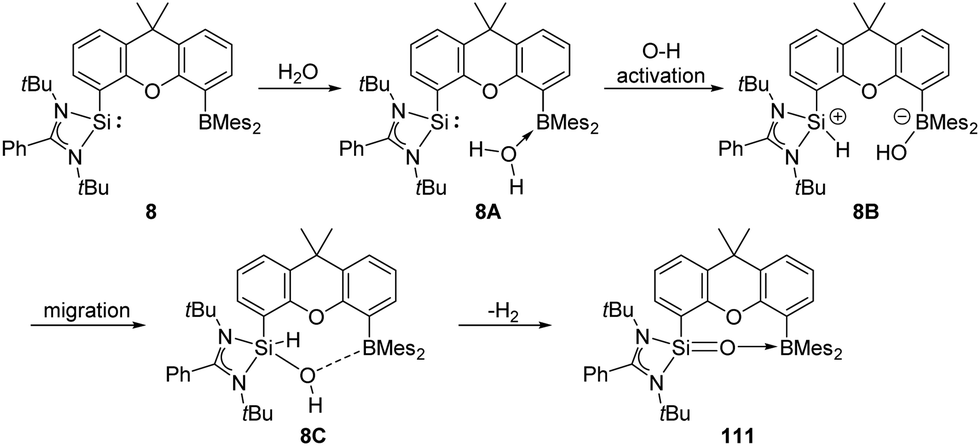 | ||
| Scheme 42 Activation of H2O by the silylene borane FLP 8. | ||
Synthesis and characterisation of the donor/acceptor complex of an aryl silaaldehyde 112 were demonstrated by Inoue (Scheme 43).80112 can be achieved by reaction of silyliumylidene complex 72a with an equimolar amount of H2O at low temperatures in the presence of the Lewis acid GaCl3. The proton NMR spectrum of 72a shows a signal of Si-bound H at δ 4.98 ppm with a coupling constant of 1JSi,H = 234 Hz. In the absence of GaCl3, however, hydrolysis of silyliumylidene 72a with the hope of isolating Lewis-acceptor free silaaldehyde resulted in the formation of a sterically hindered spirosiloxane 113 in low yields and other unknown products.
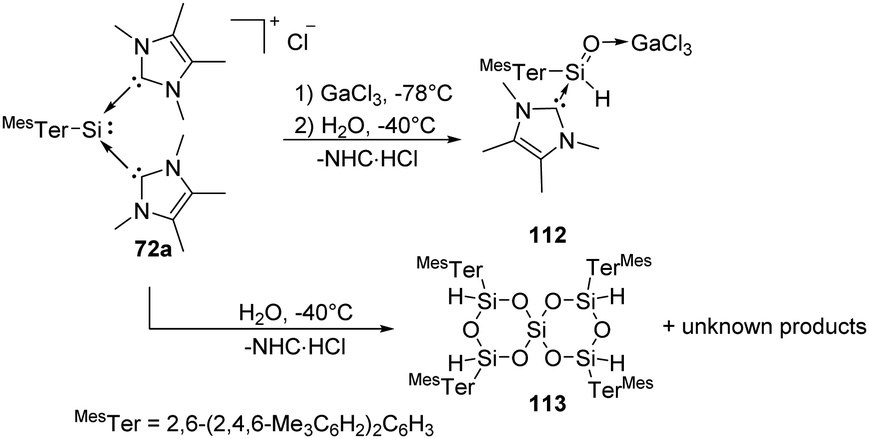 | ||
| Scheme 43 Activation of H2O by the silyliumylidene-NHC complex 72a. | ||
2.7 Activation of NH3
As coordination of the Lewis base ammonia to transition-metals is more favourable, its N–H bond activation is therefore more challenging. In 2005, the first example of an intermolecular ammonia activation by a transition metal has been reported by the Hartwig group.81 Since the activation of ammonia could also be achieved by a carbene in 2007,18 low-valent group 13 to 15 compounds, such as digallene, germylene and phosphorus species featuring a bent geometry were found to activate ammonia.16a,b,d A few cyclic and acyclic silylenes also proved to be suitable for the activation of the N–H bond of ammonia. The ylide-like N-heterocyclic silylene LSi: 26 (LCH[(CCH2)CMe(NDipp)2]; Dipp = 2,6-iPrC6H3), isolated by the Driess group in 2006,73a reacts with ammonia to afford the 1,1-addition product 114 as described by Roesky and co-workers (Scheme 44).82 Notably, the NH2 group in 114 is not involved in any kind of hydrogen bonding.
 | ||
| Scheme 44 Activation of NH3 by the ylide-like NHSi 26 and its Ni complex 101. | ||
The silylene–Ni(CO)3 complex 101 is also capable of activating ammonia and amine, affording the 1,4-addition β-diketiminato SiII–Ni(CO)3 complexes 115 without rupture of the Si–Ni bond or ligand exchange at the Ni atom (Scheme 44).74 The proposed mechanism could be explained by the initial formation of an acid–base pair, increasing acidity of the N–H protons and basicity of the exocyclic methylene group. Deprotonation of N–H can then be achieved by another molecule of 101. This process may also occur simultaneously.
Acyclic low-valent group 14 species show higher reactivity towards NH3, for example, coordination of NH3 to a bis(boryl) tin(II) compound followed by N–H cleavage leads to the amidotin(IV) hydride as described by Aldridge.83 Similarly, acyclic silylenes are also good candidates for NH3 activation. Reaction of acyclic diaminosilylene :Si(TBoN)2 (TBoN = [N(SiMe3){B(DAB)}]; DAB = (DippNCH)2; Dipp = 2,6-iPr2C6H3) 89 with NH3 gives a 1:1 mixture of the triaminosilane 116 and the secondary amine, TBoNH (Scheme 45).22g The plausible mechanism reveals a transient diaminosilylene from a σ-bond metathesis reaction between 89 and NH3, which adds NH3 oxidatively to yield the triaminosilane product 116. The acyclic two-coordinate N,O-silylene 117 also cleaves N–H bond of ammonia, affording aminosilane 118 as described by Inoue very recently (Scheme 45).84
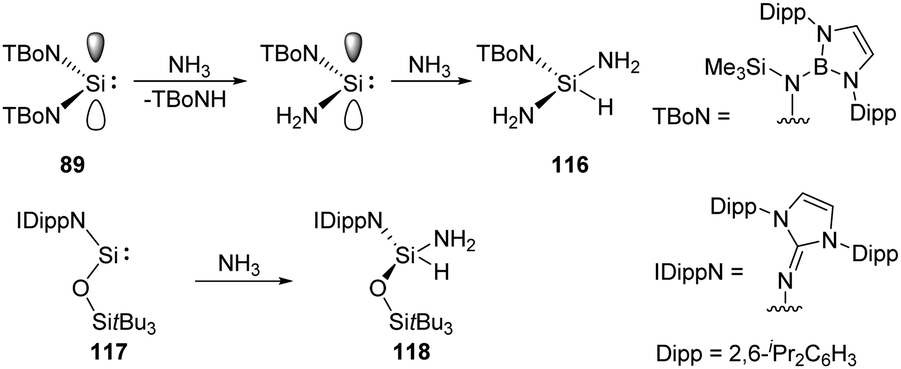 | ||
| Scheme 45 Activation of NH3 by the acyclic diaminosilylene 89 and N,O-silylene 117. | ||
The novel tetrasilyldisilene displaying bis(silyl)silylene reactivity also shows high reactivity towards NH3. The hydroamination is achieved by treating an n-hexane solution of 11/11′ with one molar equivalent of NH3 along with an apparent colour change from blood red to pale yellow and the formation of 119. However, the addition product was identified by NMR spectroscopy to be only derived from tetrasilyldisilene 11 (Scheme 46).22j
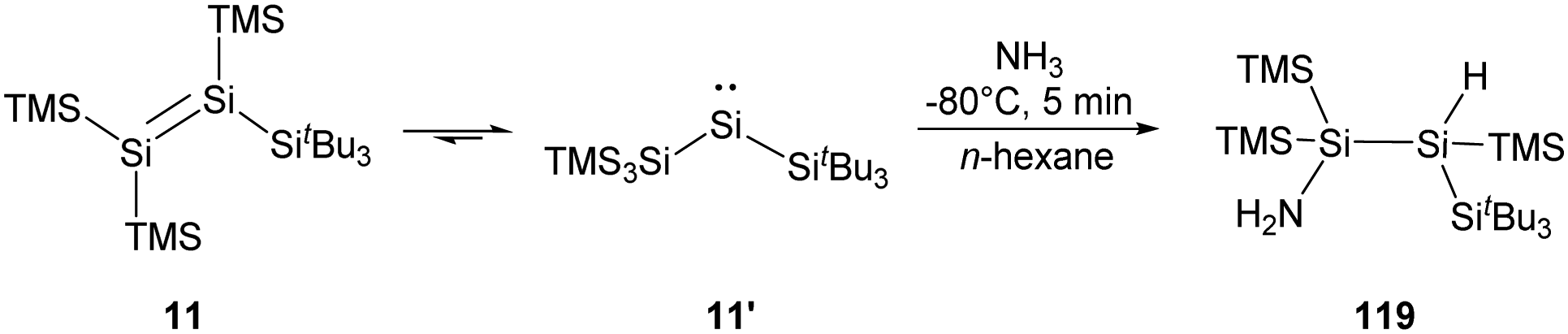 | ||
| Scheme 46 Activation of NH3 by tetrasilyldisilene 11 and bis(silyl)silylene 11′. | ||
In 2020, the Aldridge group investigated the hydroamination of [(N-nacnac)Si]+120 when being exposed to ammonia, representing the first example of oxidative addition at a two-coordinate silicon(II) cation (Scheme 47).85 In the cases of Ge(II) and Sn(II), only Werner coordination complexes are obtained after being treated with tBuNH2. Differences in EII/EIV redox chemistry on descending group 14 are inferred to account for the results.
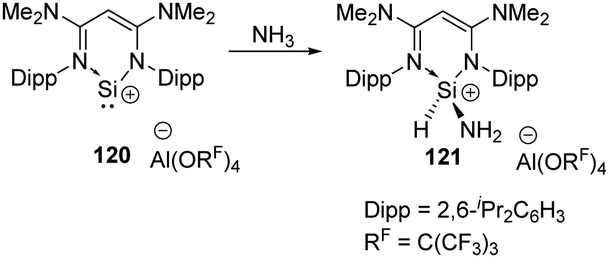 | ||
| Scheme 47 Activation of NH3 by the two-coordinate Si cation 120. | ||
2.8 Activation of C–H bonds
C–H bond activation is one of the most potent methodologies to achieve C–C and C–heteroatom bond formation, and recent major advancement in this area has always been the domain of transition metals. Since Arduengo reported the activation of C–H bond by carbenes, some N-heterocyclic and cyclic amino carbenes are used for this purpose.16b With the establishment of low-coordinate main-group chemistry, a lot of main-group species, especially silylenes, appear to be good candidates for the activation of C–H bonds. Their activation towards organic substrates might also inspire future catalytic application and narrow the catalytic gap between silylenes and transition metals.Kira revealed that irradiation of silylene 43 with light of wavelengths longer than 420 nm allows access to the 1,1-biradical excited state (Scheme 48).86 Thus, irradiation of 43 in the presence of mesitylene results in the insertion to a benzylic C–H bond rather than the expected silepin compound. The formation of the latter is inferred to be hampered by the steric hindrance at CC double bonds of mesitylene.
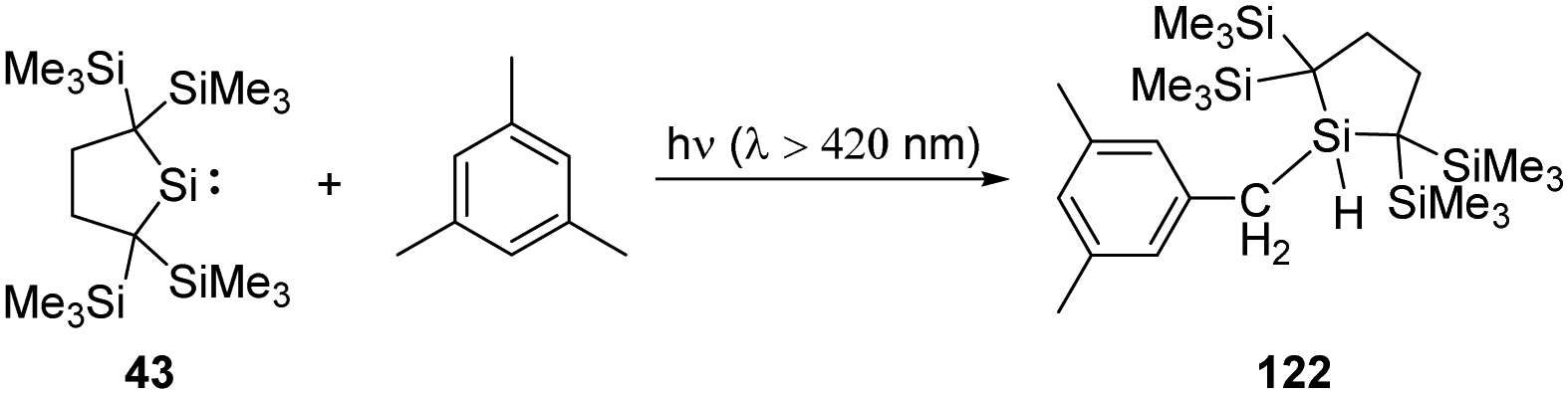 | ||
| Scheme 48 Activation of a benzylic C–H bond by photochemical insertion of silylene 43. | ||
Though aromatic C–F bond activation by N-heterocyclic silylene 26 in the absence of any additional catalyst allows access to the preparation of silicon fluorine compounds, it reacts preferentially with partially fluorinated aromatic substrates to give 123 and 124via C–H bond activation (Scheme 49),87 probably due to the weaker C–H bond.
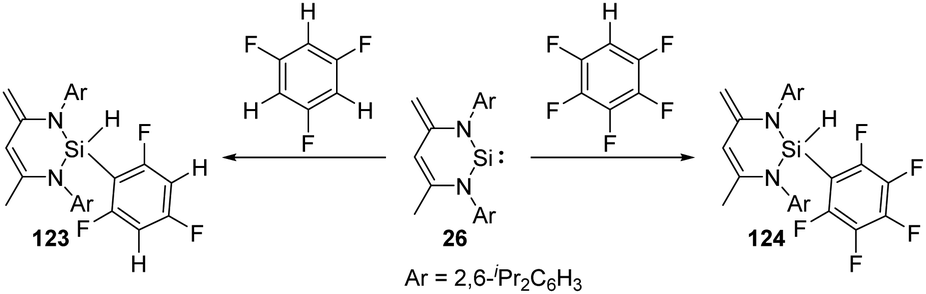 | ||
| Scheme 49 Activation of aromatic C–H bond by the NHSi 26. | ||
The NHC-coordinated silylene adduct 27 (NHC = 1,3,4,5-tetramethylimidazol-2-ylidene and 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) reported by the Driess group is found to be thermo-labile and undergoes rearrangement above −20 °C, leading to the asymmetric N-heterocyclic silylcarbene 125 (Scheme 50).88 One of the possible pathways reveals the insertion to an NMe group of NHC moiety by the reactive Si centre, followed by the cleavage of the C → Si dative bond to give the final silylcarbene product.
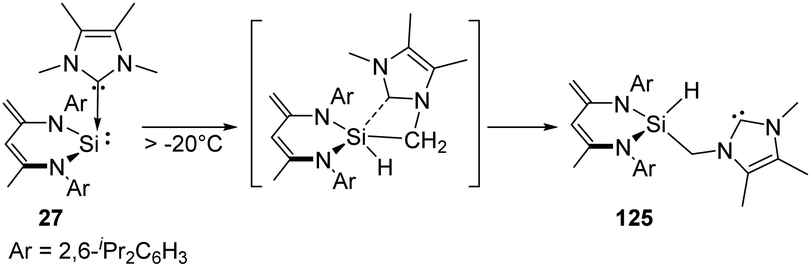 | ||
| Scheme 50 Preparation of N-heterocyclic silylcarbene via Si-mediated activation of C–H bond with the NHC–NHSi adduct 27. | ||
Two-coordinate acyclic silylenes with narrower HOMO–LUMO energy gap compared to cyclic ones are considered as more reactive species and more likely to undergo isomerisation or intramolecular C–H bond activation. As has been discussed that RSi(SiR3) type silylenes are prone to undergo rearrangement to give isomeric disilenes, the stable acyclic silylsilylene 2 isolated by Aldridge and co-workers undergoes C–H activation over five days at 80 °C to give silaindoline 126 (Scheme 51).22c The bis(silyl)silylene 11′ which is in equilibrium with tetrasilyldisilene 11via 1,2-silyl migrations mentioned above also undergoes intramolecular C–H activation over four days to give the full conversion to disiletane 127.22j Another acyclic silysilylene 128 reported by Rivard reacts with one molar equivalent of tert-butyl isocyanide via intermolecular C–H activation, leading to the quantitative transformation of the silyl-cyanide product 129, representing an intermolecular activation of a primary C–H bond by a silylene.22k
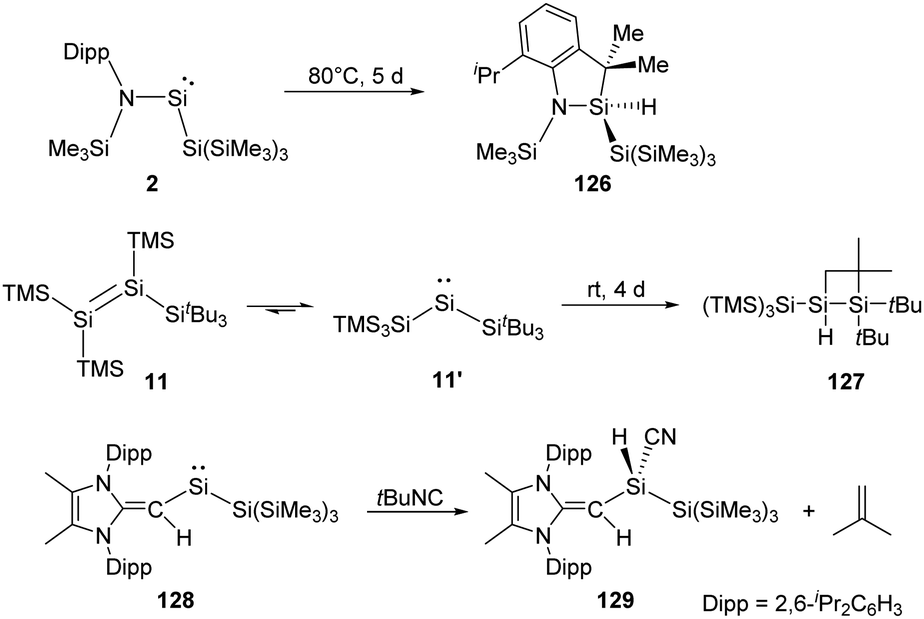 | ||
| Scheme 51 Activation of C–H bond by the silylsilylenes 2, 11′ and 128. | ||
Intermolecular activation of benzylic C–H bond by silylenes is rare. Iwamoto et al. successfully realised a benzylic C–H insertion reaction with the cyclic (alkyl)(amino)silylene 130 in toluene solutions at elevated temperatures, furnishing hydridobenzylsilane 131 (Scheme 52).89 Notably, the benzylic C–H insertion also competes with dehydrogenation of 9,10-dihydroanthracene by 130, affording silanes 132, 133 and anthracene as products.
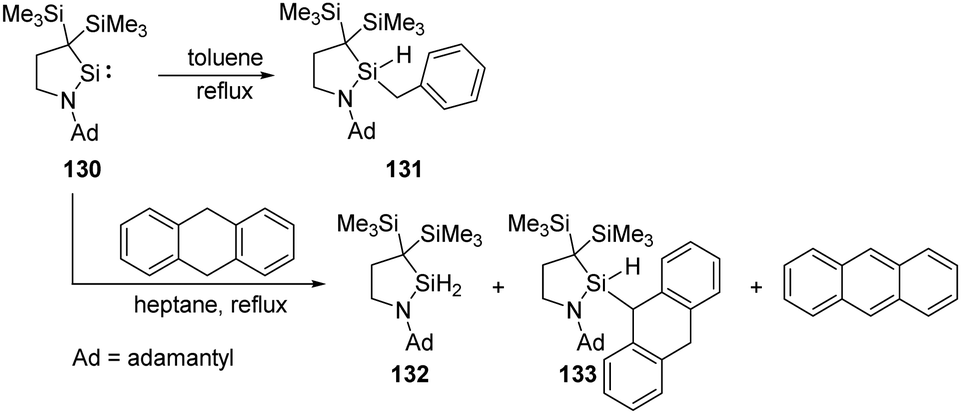 | ||
| Scheme 52 Activation of benzylic C–H bond by the cyclic (alkyl)(amino)silylene 130. | ||
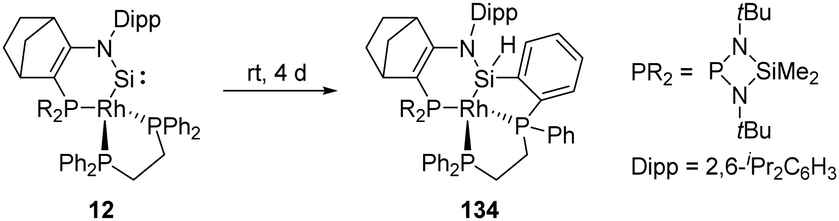
As aforementioned, the N-hetero-RhI-metallacyclic silylene 12 featuring a tetrahedral Rh centre has been described by Kato and Baceiredo (Scheme 53).32 The geometry of the Rh atom increases the π-donating and σ-accepting character of the metal fragment, which leads to a sizeable HOMO–LUMO gap and stabilises the Si moiety. Nevertheless, 12 undergoes insertion over four days to the ortho-C–H bond of phenyl group bound to phosphine, furnishing the silane derivative 134.
 | ||
| Scheme 53 Activation of intramolecular aromatic C–H bond by the metallacyclic silylene 12. | ||
2.9 Activation of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CH2
CH2
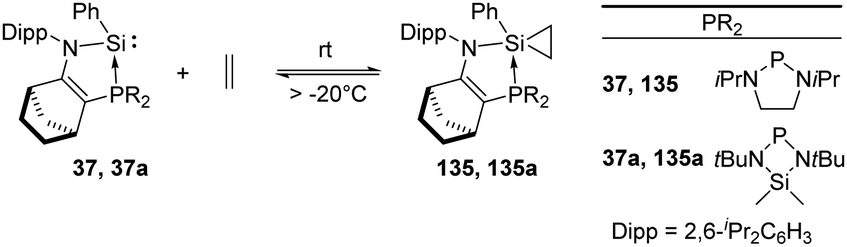
Ethylene is produced from methionine in nature and acts as an essential natural plant hormone to force the ripening of fruits.90 It is also widely used in the chemical industry as its worldwide production exceeds that of any other raw chemicals. Major industrial reactions of ethylene include polymerisation, oxidation, halogenation etc. The famous Ziegler–Natta catalyst based on titanium and aluminium as active sites is extensively employed in the polymerisation of ethylene.91 Up to now, similarly active catalysts for the polymerisation of ethylene based on silicon are still unknown. Nevertheless, a few examples for the activation of ethylene with silylenes have been reported. This includes the phosphine silylene complexes 37 and 37a which react with ethylene to give the corresponding silirane products 135 and 135a and the conversion is strongly dependent on the ethylene pressure (Scheme 54).92 Notably, the two resonance signals in the 13C NMR spectrum at δ −0.06 and −1.68 ppm are consistent with a silirane structure rather than a η2-coordination mode. Interestingly this reaction shows reversibility by regeneration of the phosphine silylene 37 at low ethylene pressure. A small value of Gibbs free energy for the addition reaction was calculated [ΔG20°C = (−0.717 ± 0.452) kcal mol−1], which shows the thermoneutrality of the reaction, in accordance with the observed reversibility of the reaction.
 | ||
| Scheme 54 Reversible addition of ethylene to the phosphine silylene complexes 37 and 37a. | ||
Reversible ethylene binding by stable, two-coordinate acyclic silylenes was also demonstrated by Power et al.22e Exposure of toluene solutions of Si(SArMe6)2 (ArMe6 = C6H3-2,6(C6H2-2,4,6-Me3)2) 136 and Si(SAriPr4)2 (AriPr4 = C6H3-2,6(C6H3-2,6-iPr2)2) 136a to ethylene atmosphere results in the formation of the corresponding silirane compounds 137 and 137a, respectively (Scheme 55). The 29Si NMR spectrum of 137a displays a signal at δ +270.7 ppm (free silylene 136a), indicating a dissociation equilibrium in solutions. A van’t Hoff analysis of the association of ethylene with 136 using variable-temperature 1H NMR spectroscopy afforded ΔHassn = −83.61(8.4) kJ mol−1 and ΔGassn = −24.9(2.5) kJ mol−1 at 300 K, which is more favourable than that of phosphine silylene complex 37. A van’t Hoff analysis of the association of ethylene with 136a was hampered because of the solubility characteristics of 136a and 137a.
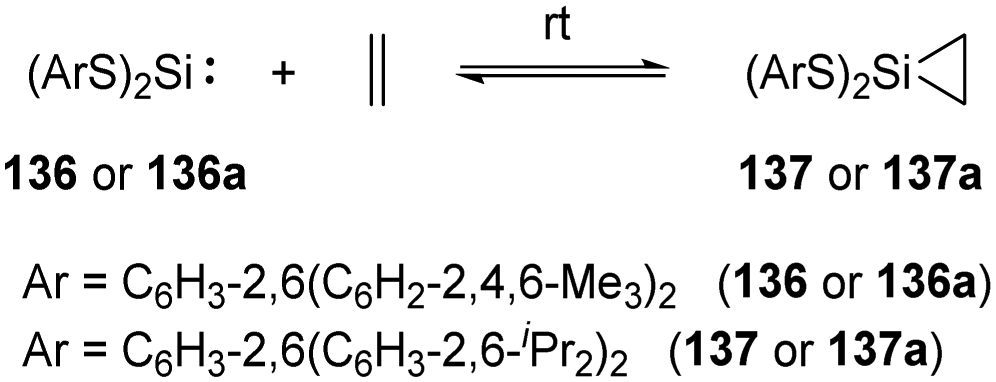 | ||
| Scheme 55 Reversible addition of ethylene to the acyclic silylenes 136 and 136a. | ||
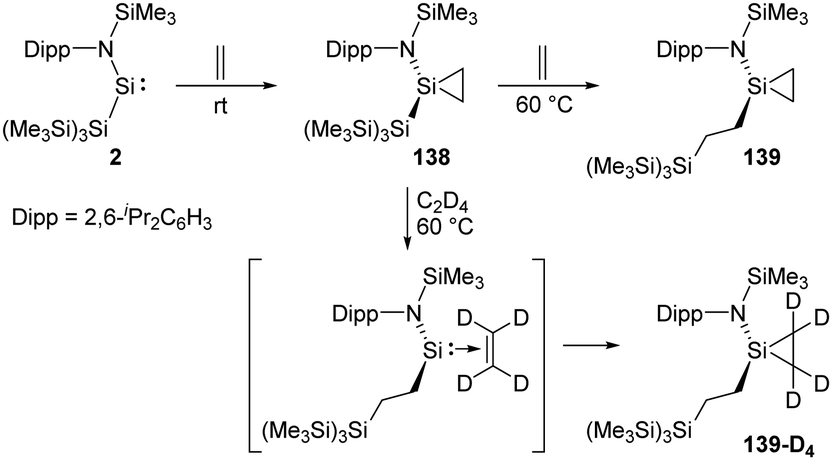
Rieger et al. demonstrated the first migratory insertion of ethylene into a Si–Si bond in the acyclic two-coordinate silylene 2 mimicking typical reactions of transition-metal complexes (Scheme 56).22f A quantitative conversion to the silirane product 138, Si{CH2–CH2}{NDipp(SiMe3)}{Si(SiMe3)3}, is achieved at room temperature from the reaction of stable, acyclic silylsilylene 2 in the presence of ethylene atmosphere. Upon being heated to 60 °C unexpected formation of the modified silirane Si{CH2–CH2}{NDipp-(SiMe3)}{CH2–CH2–Si(SiMe3)3} 139via a Si–Si bond insertion was observed. In order to investigate the mechanism, a 1H NMR experiment using C2D4 was performed and suggested a migratory insertion of the coordinated ethylene of 138 into the Si–Si bond of the ligand and subsequent addition of a second ethylene molecule.
 | ||
| Scheme 56 Activation of ethylene by the acyclic silylene 2via ethylene insertion into Si–Si bond. | ||
Inoue and Rieger et al. have demonstrated the reversibility between the highly reactive acyclic iminosilylsilylene 10' and silepin 10, which can act as a “masked” silylene.22h Treatment of a freshly prepared solution of silepin 10 with ethylene at room temperature affords the corresponding silirane compound 140 (Scheme 57), even though it could not be purified by crystallisation due to its similar solubility to side products. Moreover, the silirane structure can be confirmed by observation of the coupling of the central Si to the ring-bound protons in the 1H–29Si HMBC spectrum.
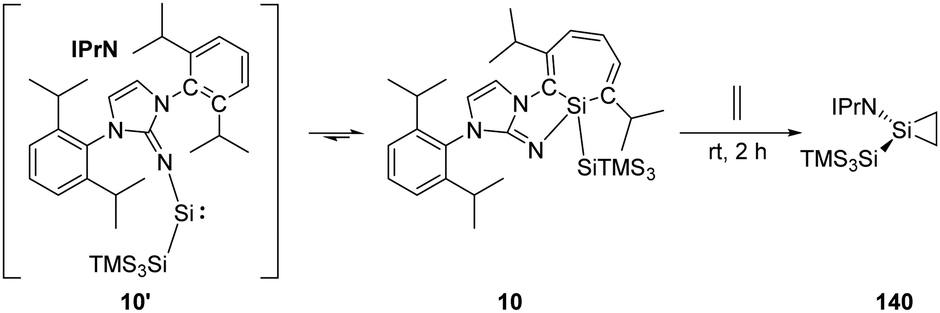 | ||
| Scheme 57 Activation of ethylene by the “masked” silylene 10. | ||
The long-term pursued acyclic and neutrally charged silanone 62a was isolated by the Inoue group and found to perform rearrangement to give the acyclic two-coordinate N,O-silylene 117 bearing a siloxy ligand (Scheme 58).22i Silylene 117 undergoes irreversible transformation to give the corresponding silirane 141 when being exposed to ethylene. Irreversibility of this reaction probably benefits from the reduced steric shielding at the Si centre.57
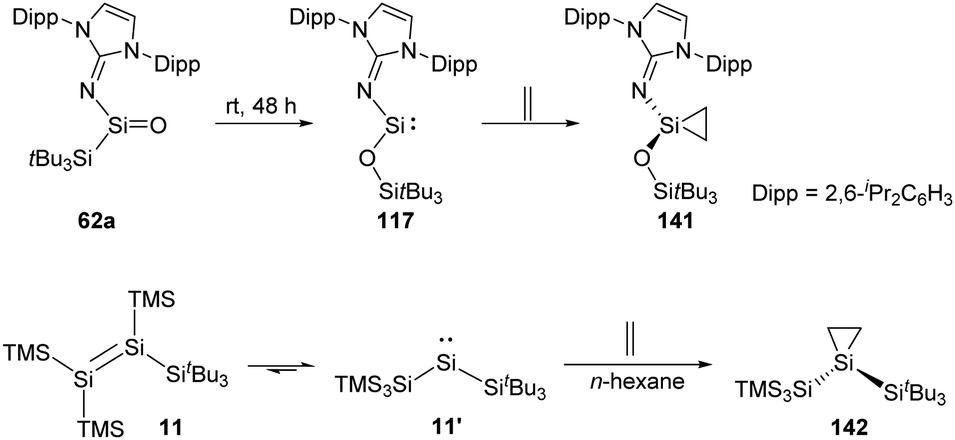 | ||
| Scheme 58 Activation of ethylene by the acyclic silylenes 117 and 11′. | ||
Insights into the disilene–silylene equilibrium were also gained by the same group. Logically, exposure of 11 to ethylene results in the clean formation of the corresponding silirane 142 featuring an expansive Si–Si–Si bond angle of 131.7(1)° because of the steric hindrance of the silyl groups.22j
More recently, the reactivity of the silylene–Ni0 complex 3 [(TMSL)ClSi: → Ni(NHC)2] (TMSL = N-(SiMe3)Dipp; Dipp = 2,6-iPr2C6H3, NHC =:C[(iPr)NC(Me)]2) towards ethylene was also investigated by the Driess group (Scheme 59).93 The addition of ethylene to 3 affords the four-membered nickelasilacyclobutane 143. Notably, the addition reaction is reversible, and a [2+2+2] cycloaddition product, the six-membered metallasilacycle 144, can also be achieved via the cleavage of the Si–Ni bond. 144 is quite unstable and undergoes a consecutive β-hydride elimination/reductive elimination to give compound 145.
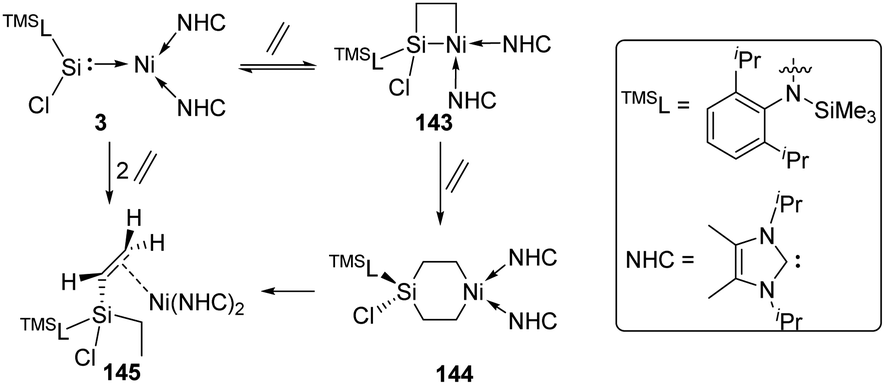 | ||
| Scheme 59 Reversible activation of ethylene by the silylene–Ni complex 3. | ||
2.10 Degradation of P4 and As4
Phosphorus is one of the essential biogenic elements required by every living organism,94 and phosphorus compounds play a significant role in the industry, including ligands design, organic synthesis, drugs, optoelectronic materials and fertilisers etc., due to their diverse array of useful chemical, physical and biological properties.95 However, these phosphorus-containing species are currently prepared via hazardous and wasteful multi-step procedures involving the initial transformation of P4 to PCl3 and PCl5.94 Though degradation of P4 has been established by transition-metal systems,96 highly reactive main-group compounds also show reactivity towards P4, for example, cyclic alkyl amino carbenes and N-heterocyclic carbenes undergo a nucleophilic attack at P4 to give degradation products, low-valent aluminium and gallium compounds were also found to cleave P–P bonds.16b Silylene-mediated degradation procedures show excellent prospects due to their inherent advantages including mild conditions and easier purification processes.In 2007, the Driess group reported the consecutive P4 degradation with the zwitterionic silylene 26, which results in the formation of the first SiP4146 and Si2P4147 cage compounds (Scheme 60).97146 can be synthesised by the reaction of 26 with P4 in toluene in the molar ratio of 1:1 at room temperature and isolated in the form of colourless crystals, featuring a tricyclic SiP4 core with pyramidally coordinated P and Si atoms in a tetrahedral environment. It bears three chemically different sorts of P nuclei according to the three temperature-invariant resonance signals (δX = 131.9, δA = −342.4, δB = −348.0 ppm) in the 31P NMR spectrum, and the low-field resonance signal at δX splits into a doublet of doublets (1J(PX,PA) = 146.8, 1J(PX,PB) = 144.7 Hz) while each of the two high-field signals shows a doublet of triplets (1J(PX,PA) = 144.7, 1J(PA,PB) = 188.0 Hz). An X-ray diffraction analysis confirmed the molecular structure. Further reaction of 146 with silylene 26 in toluene at room temperature affords the Si2P4147 in the form of colourless crystals. The second insertion step is kinetically unfavourable due to steric congestion, and 147 shows two multiplets (δA = 153, δB = 154 ppm) in the 31P NMR spectrum, representing an AA′BB′ splitting pattern of higher order with unusually small magnitudes of coupling constants.
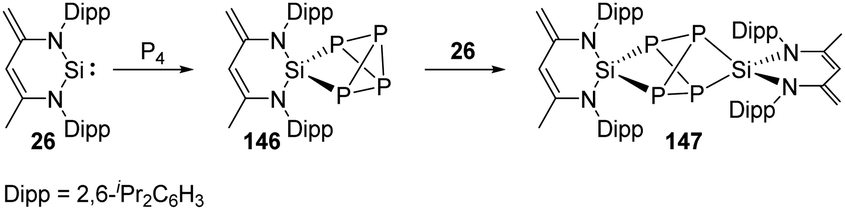 | ||
| Scheme 60 Consecutive activation of P4 by the zwitterionic NHSi 26. | ||
Synthesis and characterisation of 148 featuring the planar Si–P–Si–P four-membered ring through the activation of P4 by monochlorosilylene 35 were reported by Roesky and Stalke et al (Scheme 61).98 The formation of 148 is suggested by treatment of 35 and P4 in toluene overnight, and the structure of 148 shows a bis-ylide ring in which the two Si atoms are positively charged while the two P atoms carry partial negative charges. The natural charges obtained from the NBO analysis are +1.12 for the Si and −0.69 for the P atoms. The value of the four equivalent Si–P bonds (2.174 Å) is between a Si–P single (2.25 Å) and double bond (2.09 Å).99 The bis-silylene 33 bearing two lone pairs of electrons and a labile Si–Si bond also reacts with P4 at room temperature in the molar ratio of 1:2, affording quantitatively the same product 148.98 In contrast to the four-membered ring products, a neutral acyclic P4 chain species 150 could be achieved by the degradation of P4 with SiII bis(trimethylsilyl)amido silylene 149 in toluene solutions.100 Purple-coloured crystals can be obtained from concentrated toluene solutions at low-temperature featuring a neutral acyclic SiP–PP–PSi chain with 6π electrons contained in a diphosphene and two phosphasilene units.101 By contrast, the reaction of 149 with As4 in toluene leads to the formation of the unprecedented As10 cage compound 151 featuring a nortricyclane core.102 The Si–As bond of the exocyclic substituents in 151 can be regarded as an elongated double bond or an ion compound with the negative charge localised at the As atom and the positive charge over both N atoms in the heterocycle.
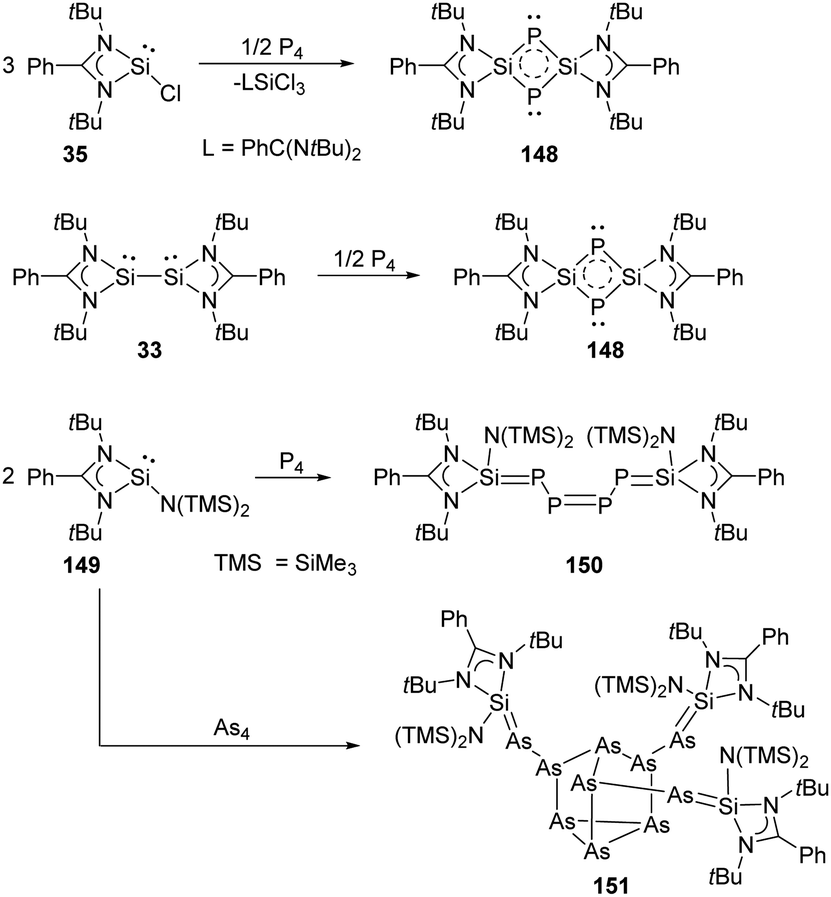 | ||
| Scheme 61 Activation of P4 and As4 by the NHSis 33, 35, and 149. | ||
Driess and West have already investigated the reactivity of tetraaryldisilenes 152a–c, e with white phosphorus and arsenic (Scheme 62).103 The disilenes 152a–c react with P4 to give bicycle[1.1.0] butane compounds 154a–c, while the more sterically encumbered disilene 152e reacts slowly with P4 to afford two isomers 154e and 154e′via intermediate 153. Besides, 152a reacts with arsenic to afford 155 and 156 in a ratio of about 3:1, 155 converts to 156 quantitatively when heated at 85 °C for 24 days. Comparatively, the N(SiMe3)2-substituted disilene 157 exists in equilibrium with the corresponding silylene in solutions,104 and its reactivity towards P4 was also investigated in a 1:1 molar ratio in toluene at ambient temperature, affording the Si–P cage compound 158 through the insertion of two molecules of silylenes into the P4 tetrahedron.101 Besides, disilene 157 also reacts with As4 in toluene at room temperature to give [Cp*{(SiMe3)2N}SiAs]2159 containing a butterfly-like diarsadisilabicyclo[1.1.0]butane unit.102
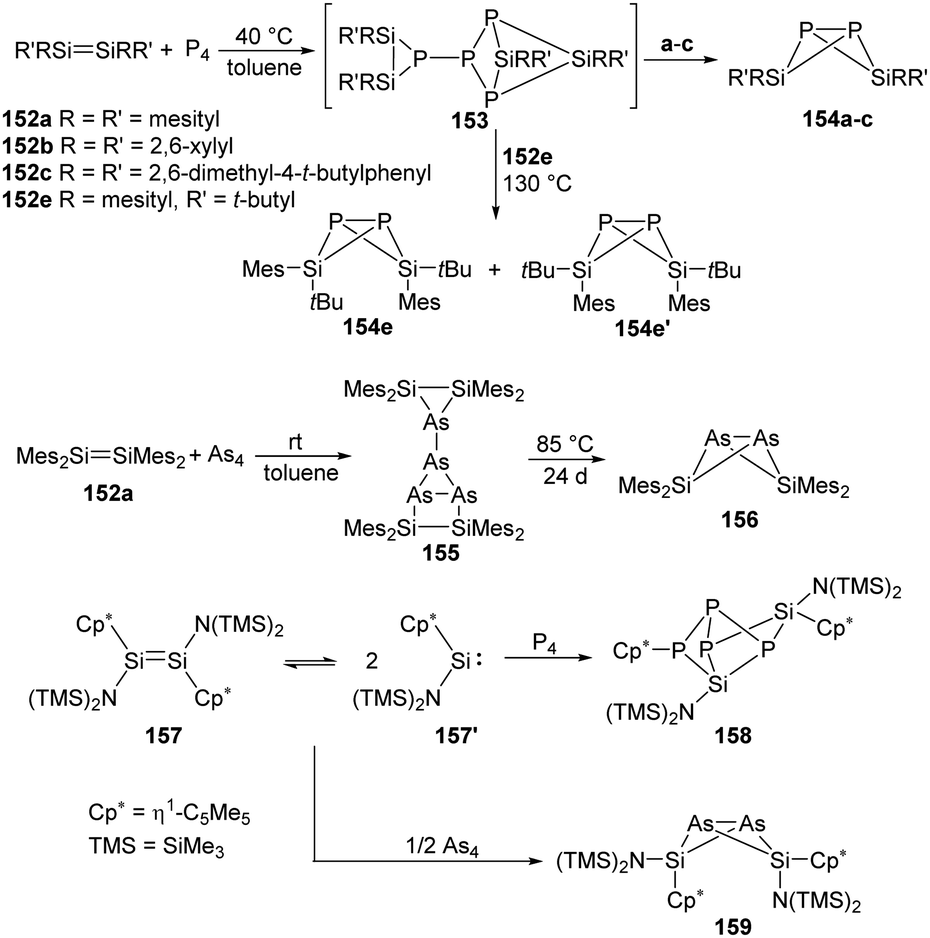 | ||
| Scheme 62 Activation of P4 and As4 by the disilenes 152 and 157. | ||
A new isolable heterocyclic silylene 56a stabilised by an amino group and a more π-donating and electropositive phosphonium ylide moiety was reported by Kato et al. (Scheme 63).105 The silylene 56a shows thermal stability and a robust nucleophilic character, and its reaction with P4 occurs at room temperature via insertion of Si into the σ P–P bond to give the SiP4 cage compound 160.
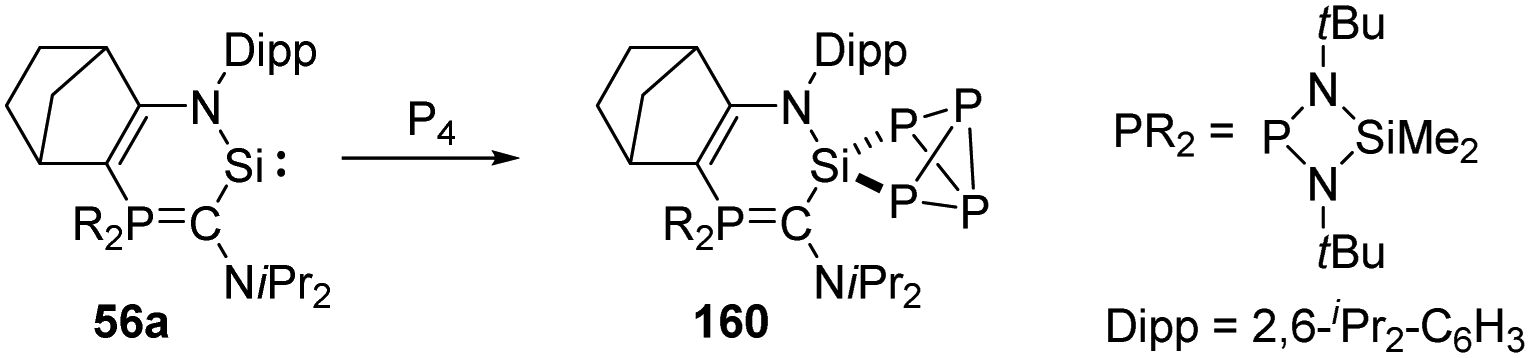 | ||
| Scheme 63 Activation of P4 by the new heterocyclic silylene 56a. | ||
While the cyclic silylene-mediated P4 degradation is limited to the oxidative addition of a single P–P bond across the corresponding Si centre, the acyclic two-coordinate silylene 128 stabilised by a bulky vinylic N-heterocyclic olefin ligand and the σ-donating hypersilyl group shows high reactivity towards P4 to give (MeIPrCH)Si(P4){Si(SiMe3)3} (MeIPr = [(MeCNDipp)2C]; Dipp = 2,6-iPr2C6H3) 161 as the final product (Scheme 64).22j The proposed mechanism probably involves the initial oxidative addition of a P–P bond followed by a subsequent 1,2-silyl migration regarding the polarised SiII–Si(SiMe3)3 bond.
 | ||
| Scheme 64 Activation of P4 by the acyclic silylene 128. | ||
More recently, the Driess group investigated the facile metal-free degradation of P4 to the zero-valent diphosphorus complex 162 stabilised by two cooperative divalent Si centres of the bis-silylene 15a (Scheme 65).106 The two lone-pairs of electrons on each P atom in 162 allow access to facile construction of P–C, P–H and P–B bonds upon its reactions with CO2, H2O and a borane. Notably, 162 also serves as a monophosphorus anion (P−) transfer reagent, leading to the phosphaketenide ligand (PCO−) and a phosphinidene germylene complex in the presence of M(CO)6 (M = Cr, W) and LGeCl (L = PhC(NtBu)2), respectively.
 | ||
| Scheme 65 Degradation of P4 by the bis(NHSi) 15a to give the bis-silylene-stabilised zero-valent P2 complex 162. | ||
2.11 A dream in Si(II) chemistry: how to achieve fixation of N2?
Activation of dinitrogen could so far be realised by natural nitrogenases with MoFeco3 or industrial Haber–Bosch technology107 under extremely high pressure and temperature. Alternative (artificial) transition-metal mediated activation of N2 has always been laboratory curiosities since the synthesis and isolation of the first isolable transition-metal–dinitrogen complex with a (η1-N2) end-on coordination mode.108 By far, numerous transition metal–N2 complexes taking advantage of Fe,109a–h,j,k Mo,109c–e,g–k W109d,g,h have been developed for consecutive conversion of N2 to ammonia, hydrazine and other fundamental nitrogen-containing chemicals.109 Main-group elements supported fixation and functionalisation of N2 is scarce, examples like borabenzene intermediate under flash-thermolysis condition,110 as well as phenyl cation111 and phenylborylene112 under cryogenic argon matrix condition were observed by IR spectroscopy and isotopic labelling experiments. Very recently, Braunschweig and co-workers have demonstrated that coordination of N2 to an empty 2p-orbital of a transient borylene complex gives rise to a borylene–N2 adduct, which either reacts with the second equivalent of borylene to afford the reductive B2N2 product,113 or is reduced by excess KC8 to give a nitrene-like radical followed by dimerisation to afford a [N4]2− coupling product caped with two boron centres.114 Strong π-backbonding from boron to N2 is revealed to be critical to end-on N2 binding to boron atoms. Besides, Schulz has reported the isolation of an isolable silylene–CO adduct (CO is isolobal with N2) more recently and demonstrated that the enlarged sp2 lone pair is more available for backbonding the CO moiety (nS1 → π*CO).22l All above examples reveal backbonding to be a critical factor for the successful fixation of N2 as well as the weakening and functionalisation of the dinitrogen bond. Therefore, it is also inferred that modified silylenes featuring occupied and vacant orbitals proximal in space and energy can show more backbonding effects as well as play a critical role for the binding and activation of the non-polar and strong N–N triple bond. It remains to be seen whether this summit can be reached by new tricks in silylene chemistry!3. Conclusions and outlook
We have discussed very recent progress in small molecule activation mediated by divalent silicon without and in the presence of a transition metal. Small molecules such as H2, COx, N2O, O2, H2O, C2H4 and NH3 are ideal and readily available resources in synthetic chemistry but require specific activation because they are constituted by relatively strong chemical bonds. Though nature has developed numerous enzymatic systems which are capable of activating most of the aforementioned small molecules but these processes are limited to physiological reaction conditions. Inspired by the latter, artificial transition-metal complexes mimicking the characteristics of metalloenzymes were developed. With the motivation of pursuing cheaper and more sustainable catalysts, heavy main-group multiply bonded compounds, FLPs and group 14 congeners of carbenes have been introduced recently, among which silylenes show an outstanding potential for metal-free activation of small molecules. The bifunctionality at the SiII centre in silylenes originating from a vacant 3p orbital and a lone pair of electrons in a non-bonding orbital with high 3s character, as well a relatively narrow and tunable HOMO–LUMO energy gap are superior for mimicking transition-metal active sites. Even though in combination with transition-metals, divalent silicon centres are capable to cooperatively assist and boost bond activation and atom transfer which cannot be achieved by a transition-metal alone (e.g. H2 activation and subsequent hydrogenation of olefins). Particular efforts are needed to develop suitable silylenes for the activation of extremely stable and non-polar chemical bonds, such as in N2. Moreover, the regeneration of SiII as active sites in many of the stoichiometric bond activation reactions, as herein discussed, remains challenging and key to achieve genuine low-valent silicon-based catalysis competitive to transition-metal systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1-390540038. CS gratefully acknowledges financial support by the China Scholarship Council.Notes and references
- A. Holleman, E. Wiberg, N. Wiberg and G. Fischer, Lehrbuch der anorganischen Chemie. 2007 Search PubMed.
- Z. Thompson and J. A. Cowan, Small, 2020, e2000392 CrossRef PubMed.
- (a) F. Mus, D. R. Colman, J. W. Peters and E. S. Boyd, Free Radical Biol. Med., 2019, 140, 250–259 CrossRef CAS PubMed; (b) J. W. Peters and J. B. Broderick, Annu. Rev. Biochem., 2012, 81, 429–450 CrossRef CAS PubMed; (c) P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2019, 21, 15747–15759 RSC; (d) R. D. Milton and S. D. Minteer, Acc. Chem. Res., 2019, 52, 3351–3360 CrossRef CAS PubMed.
- (a) P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed; (b) M. J. Lacasse, S. Sebastiampillai, J. P. Cote, N. Hodkinson, E. D. Brown and D. B. Zamble, J. Biol. Chem., 2019, 294, 15373–15385 CrossRef CAS PubMed; (c) O. Lampret, J. Esselborn, R. Haas, A. Rutz, R. L. Booth, L. Kertess, F. Wittkamp, C. F. Megarity, F. A. Armstrong and M. Winkler, et al. , Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15802–15810 CrossRef CAS PubMed.
- (a) I. D. Young, M. Ibrahim, R. Chatterjee, S. Gul, F. Fuller, S. Koroidov, A. S. Brewster, R. Tran, R. Alonso-Mori and T. Kroll, et al. , Nature, 2016, 540, 453–457 CrossRef CAS PubMed; (b) T. Cardona, P. Sanchez-Baracaldo, A. W. Rutherford and A. W. Larkum, Geobiology, 2019, 17, 127–150 CrossRef CAS PubMed; (c) N. Cox, D. A. Pantazis and W. Lubitz, Annu. Rev. Biochem., 2020, 89, 795–820 CrossRef CAS PubMed.
- (a) D. C. Lamb, A. H. Follmer, J. V. Goldstone, D. R. Nelson, A. G. Warrilow, C. L. Price, M. Y. True, S. L. Kelly, T. L. Poulos and J. J. Stegeman, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12343–12352 CrossRef CAS PubMed; (b) V. B. Urlacher and M. Girhard, Trends Biotechnol., 2019, 37, 882–897 CrossRef CAS PubMed.
- (a) J. G. Rebelein, M. T. Stiebritz, C. C. Lee and Y. Hu, Nat. Chem. Biol., 2017, 13, 147–149 CrossRef CAS PubMed; (b) M. Yuan, M. J. Kummer and S. D. Minteer, Chem. – Eur. J., 2019, 25, 14258–14266 CrossRef CAS PubMed.
- (a) F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Kohler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed; (b) J. M. Le and K. L. Bren, ACS Energy Lett., 2019, 4, 2168–2180 CrossRef CAS.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- (a) J. Kaplan and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11566–11570 CrossRef CAS PubMed; (b) F. Nastri, M. Chino, O. Maglio, A. Bhagi-Damodaran, Y. Lu and A. Lombardi, Chem. Soc. Rev., 2016, 45, 5020–5054 RSC; (c) B. Battistella and K. Ray, Coord. Chem. Rev., 2020, 408, 213176 CrossRef CAS.
- F. Moller, S. Piontek, R. G. Miller and U. P. Apfel, Chem. – Eur. J., 2018, 24, 1471–1493 CrossRef PubMed.
- (a) M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed; (b) M. P. Stewart, M. H. Ho, S. Wiese, M. L. Lindstrom, C. E. Thogerson, S. Raugei, R. M. Bullock and M. L. Helm, J. Am. Chem. Soc., 2013, 135, 6033–6046 CrossRef CAS PubMed.
- M. T. Olsen, B. E. Barton and T. B. Rauchfuss, Inorg. Chem., 2009, 48, 7507–7509 CrossRef CAS PubMed.
- (a) N. S. Sickerman, K. Tanifuji, Y. Hu and M. W. Ribbe, Chem. – Eur. J., 2017, 23, 12425–12432 CrossRef CAS PubMed; (b) F. Mus, A. B. Alleman, N. Pence, L. C. Seefeldt and J. W. Peters, Metallomics, 2018, 10, 523–538 RSC.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- (a) S. Yadav, S. Saha and S. S. Sen, ChemCatChem, 2016, 8, 486–501 CrossRef CAS; (b) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed; (c) T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176–4197 RSC; (d) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS; (e) R. L. Melen, Science, 2019, 363, 479–484 CrossRef CAS PubMed.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232–12233 CrossRef CAS PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
- (a) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC; (b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; (c) G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed; (d) S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233–1252 RSC; (e) M. Soleilhavoup and G. Bertrand, Chem, 2020, 6, 1275–1282 CrossRef CAS.
- (a) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed; (b) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed; (c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed; (d) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed; (e) A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef.
- M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS PubMed.
- (a) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed; (b) B. D. Rekken, T. M. Brown, J. C. Fettinger, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2012, 134, 6504–6507 CrossRef CAS PubMed; (c) A. V. Protchenko, A. D. Schwarz, M. P. Blake, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Angew. Chem., Int. Ed., 2013, 52, 568–571 CrossRef CAS PubMed; (d) B. D. Rekken, T. M. Brown, J. C. Fettinger, F. Lips, H. M. Tuononen, R. H. Herber and P. P. Power, J. Am. Chem. Soc., 2013, 135, 10134–10148 CrossRef CAS PubMed; (e) F. Lips, J. C. Fettinger, A. Mansikkamaki, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2014, 136, 634–637 CrossRef CAS PubMed; (f) D. Wendel, W. Eisenreich, C. Jandl, A. Pöthig and B. Rieger, Organometallics, 2015, 35, 1–4 CrossRef; (g) T. J. Hadlington, J. A. Abdalla, R. Tirfoin, S. Aldridge and C. Jones, Chem. Commun., 2016, 52, 1717–1720 RSC; (h) D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134–8137 CrossRef CAS PubMed; (i) D. Wendel, D. Reiter, A. Porzelt, P. J. Altmann, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 17193–17198 CrossRef CAS PubMed; (j) D. Reiter, R. Holzner, A. Porzelt, P. J. Altmann, P. Frisch and S. Inoue, J. Am. Chem. Soc., 2019, 141, 13536–13546 CrossRef CAS PubMed; (k) M. M. D. Roy, M. J. Ferguson, R. McDonald, Y. Zhou and E. Rivard, Chem. Sci., 2019, 10, 6476–6481 RSC; (l) C. Ganesamoorthy, J. Schoening, C. Wolper, L. Song, P. R. Schreiner and S. Schulz, Nat. Chem., 2020, 12, 608–614 CrossRef CAS PubMed.
- (a) W. Wang, S. Inoue, S. Yao and M. Driess, J. Am. Chem. Soc., 2010, 132, 15890–15892 CrossRef CAS PubMed; (b) W. Wang, S. Inoue, S. Enthaler and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 6167–6171 CrossRef CAS PubMed; (c) W. Wang, S. Inoue, E. Irran and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 3691–3694 CrossRef CAS PubMed; (d) D. Gallego, S. Inoue, B. Blom and M. Driess, Organometallics, 2014, 33, 6885–6897 CrossRef CAS; (e) Y. P. Zhou, S. Raoufmoghaddam, T. Szilvasi and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 12868–12872 CrossRef CAS PubMed; (f) Y. Wang, A. Kostenko, S. Yao and M. Driess, J. Am. Chem. Soc., 2017, 139, 13499–13506 CrossRef CAS PubMed; (g) Y. Wang, A. Kostenko, T. J. Hadlington, M. P. Luecke, S. Yao and M. Driess, J. Am. Chem. Soc., 2019, 141, 626–634 CrossRef CAS PubMed; (h) Y. Xiong, S. Yao, T. Szilvási, A. Ruzicka and M. Driess, Chem. Commun., 2020, 56, 747–750 RSC.
- E. Fritz-Langhals, Org. Process Res. Dev., 2019, 23, 2369–2377 CrossRef CAS.
- (a) B. Blom, M. Stoelzel and M. Driess, Chem. – Eur. J., 2013, 19, 40–62 CrossRef CAS PubMed; (b) R. Tacke and T. Ribbeck, Dalton Trans., 2017, 46, 13628–13659 RSC.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- P. P. Gaspar, M. Xiao, D. H. Pae, D. J. Berger, T. Haile, T. Chen, D. Lei, W. R. Winchester and P. Jiang, J. Organomet. Chem., 2002, 646, 68–79 CrossRef CAS.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- Y. Wang and J. Ma, J. Organomet. Chem., 2009, 694, 2567–2575 CrossRef CAS.
- T. J. Hadlington, T. Szilvasi and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 7470–7474 CrossRef CAS PubMed.
- Z. Mo, T. Szilvasi, Y. P. Zhou, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 3699–3702 CrossRef CAS PubMed.
- S. Takahashi, E. Bellan, A. Baceiredo, N. Saffon-Merceron, S. Massou, N. Nakata, D. Hashizume, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2019, 58, 10310–10314 CrossRef CAS PubMed.
- R. Kalescky, E. Kraka and D. Cremer, J. Phys. Chem. A, 2013, 117, 8981–8995 CrossRef CAS PubMed.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- M. P. Luecke, A. Kostenko, Y. Wang, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 12940–12944 CrossRef CAS PubMed.
- A. V. Protchenko, P. Vasko, D. C. H. Do, J. Hicks, M. A. Fuentes, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 1808–1812 CrossRef CAS PubMed.
- (a) W. Büchner, Helv. Chim. Acta, 1963, 46, 2111–2120 CrossRef; (b) S. Coluccia, E. Garrone, E. Guglielminotti and A. Zecchina, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 1063–1073 RSC; (c) P. W. Lednor and P. C. Versloot, J. Chem. Soc., Chem. Commun., 1983, 284–285 RSC; (d) P. A. Bianconi, I. D. Williams, M. P. Engeler and S. J. Lippard, J. Am. Chem. Soc., 1986, 108, 311–313 CrossRef CAS; (e) R. N. Vrtis, C. P. Rao, S. G. Bott and S. J. Lippard, J. Am. Chem. Soc., 1988, 110, 7564–7566 CrossRef CAS; (f) J. D. Protasiewicz and S. J. Lippard, J. Am. Chem. Soc., 1991, 113, 6564–6570 CrossRef CAS; (g) A. S. Frey, F. G. Cloke, P. B. Hitchcock, I. J. Day, J. C. Green and G. Aitken, J. Am. Chem. Soc., 2008, 130, 13816–13817 CrossRef CAS PubMed; (h) P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2011, 2, 77–79 RSC; (i) S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed; (j) B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265–9270 CrossRef CAS PubMed.
- (a) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (b) A. Cherubini-Celli, J. Mateos, M. Bonchio, L. Dell'Amico and X. Companyó, ChemSusChem, 2018, 11, 3056–3070 CrossRef CAS PubMed; (c) X. Su, X. F. Yang, Y. Huang, B. Liu and T. Zhang, Acc. Chem. Res., 2019, 52, 656–664 CrossRef CAS PubMed; (d) M. A. A. Aziz, A. A. Jalil, S. Wongsakulphasatch and D.-V. N. Vo, Catal. Sci. Technol., 2020, 10, 35–45 RSC.
- (a) W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018–1024 CrossRef CAS PubMed; (b) E. I. Solomon, R. Sarangi, J. S. Woertink, A. J. Augustine, J. Yoon and S. Ghosh, Acc. Chem. Res., 2007, 40, 581–591 CrossRef CAS PubMed.
- P. Jutzi, D. Eikenberg, A. Möhrke, B. Neumann and H.-G. Stammler, Organometallics, 1996, 15, 753–759 CrossRef CAS.
- S. Yao, Y. Xiong, M. Brym and M. Driess, J. Am. Chem. Soc., 2007, 129, 7268–7269 CrossRef CAS PubMed.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS.
- (a) Y. Xiong, S. Yao and M. Driess, J. Am. Chem. Soc., 2009, 131, 7562–7563 CrossRef CAS PubMed; (b) S. Yao, Y. Xiong and M. Driess, Chem. – Eur. J., 2010, 16, 1281–1288 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, R. Müller, M. Kaupp and M. Driess, Nat. Chem., 2010, 2, 577–580 CrossRef CAS PubMed.
- S. S. Sen, G. P. Tavčar, H. W. Roesky, D. Kratzert, J. Hey and D. Stalke, Organometallics, 2010, 29, 2343–2347 CrossRef CAS.
- A. Jana, R. Azhakar, S. P. Sarish, P. P. Samuel, H. W. Roesky, C. Schulzke and D. Koley, Eur. J. Inorg. Chem., 2011, 5006–5013 CrossRef CAS.
- D. Gau, R. Rodriguez, T. Kato, N. Saffon-Merceron, A. de Cozar, F. P. Cossio and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 1092–1096 CrossRef CAS PubMed.
- R. Rodriguez, T. Troadec, T. Kato, N. Saffon-Merceron, J. M. Sotiropoulos and A. Baceiredo, Angew. Chem., Int. Ed., 2012, 51, 7158–7161 CrossRef CAS PubMed.
- R. Rodriguez, T. Troadec, D. Gau, N. Saffon-Merceron, D. Hashizume, K. Miqueu, J. M. Sotiropoulos, A. Baceiredo and T. Kato, Angew. Chem., Int. Ed., 2013, 52, 4426–4430 CrossRef CAS PubMed.
- A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2014, 53, 565–570 CrossRef CAS PubMed.
- X. Liu, X.-Q. Xiao, Z. Xu, X. Yang, Z. Li, Z. Dong, C. Yan, G. Lai and M. Kira, Organometallics, 2014, 33, 5434–5439 CrossRef CAS.
- K. Junold, M. Nutz, J. A. Baus, C. Burschka, C. Fonseca Guerra, F. M. Bickelhaupt and R. Tacke, Chem. – Eur. J., 2014, 20, 9319–9329 CrossRef CAS PubMed.
- (a) F. M. Mück, J. A. Baus, M. Nutz, C. Burschka, J. Poater, F. M. Bickelhaupt and R. Tacke, Chem. – Eur. J., 2015, 21, 16665–16672 CrossRef PubMed; (b) F. M. Mück, D. Kloss, J. A. Baus, C. Burschka, R. Bertermann, J. Poater, C. Fonseca Guerra, F. M. Bickelhaupt and R. Tacke, Chem. – Eur. J., 2015, 21, 14011–14021 CrossRef PubMed.
- (a) F. M. Mück, A. Ulmer, J. A. Baus, C. Burschka and R. Tacke, Eur. J. Inorg. Chem., 2015, 1860–1864 CrossRef; (b) F. M. Mück, J. A. Baus, A. Ulmer, C. Burschka and R. Tacke, Eur. J. Inorg. Chem., 2016, 1660–1670 CrossRef.
- R. Tacke, C. Kobelt, J. A. Baus, R. Bertermann and C. Burschka, Dalton Trans., 2015, 44, 14959–14974 RSC.
- I. Alvarado-Beltran, A. Rosas-Sanchez, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 10481–10485 CrossRef CAS PubMed.
- A. Rosas-Sanchez, I. Alvarado-Beltran, A. Baceiredo, N. Saffon-Merceron, S. Massou, D. Hashizume, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 15916–15920 CrossRef CAS PubMed.
- D. C. H. Do, A. V. Protchenko, M. Angeles Fuentes, J. Hicks, E. L. Kolychev, P. Vasko and S. Aldridge, Angew. Chem., Int. Ed., 2018, 57, 13907–13911 CrossRef CAS PubMed.
- S. U. Ahmad, T. Szilvási, E. Irran and S. Inoue, J. Am. Chem. Soc., 2015, 137, 5828–5836 CrossRef CAS PubMed.
- M. P. Luecke, E. Pens, S. Yao and M. Driess, Chem. – Eur. J., 2020, 26, 4500–4504 CrossRef CAS PubMed.
- R. Kobayashi, S. Ishida and T. Iwamoto, Angew. Chem., Int. Ed., 2019, 58, 9425–9428 CrossRef CAS PubMed.
- T. Kato, S. Takahashi, K. Nakaya, A. Baceiredo, N. Saffon-Merceron, S. Massou, N. Nakata, D. Hashizume, V. Branchadell and M. F. Pastor, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006088.
- B. E. Schultz and S. I. Chan, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 23–65 CrossRef CAS PubMed.
- C. H. Kjaergaard, M. F. Qayyum, S. D. Wong, F. Xu, G. R. Hemsworth, D. J. Walton, N. A. Young, G. J. Davies, P. H. Walton and K. S. Johansen, et al. , Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8797–8802 CrossRef CAS PubMed.
- M. Y. Pau, J. D. Lipscomb and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18355–18362 CrossRef CAS PubMed.
- (a) H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2019, 141, 9516–9520 CrossRef CAS PubMed; (b) L. Wang, M. Gennari, F. G. Cantu Reinhard, S. K. Padamati, C. Philouze, D. Flot, S. Demeshko, W. R. Browne, F. Meyer and S. P. de Visser, et al. , Inorg. Chem., 2020, 59, 3249–3259 CrossRef CAS PubMed; (c) M. L. Wind, S. Hoof, B. Braun-Cula, C. Herwig and C. Limberg, Inorg. Chem., 2020, 59, 6866–6875 CrossRef CAS PubMed.
- M. Haaf, A. Schmiedl, T. A. Schmedake, D. R. Power, A. Millevolte, J. M. Denk and R. West, J. Am. Chem. Soc., 1998, 120, 12714–12719 CrossRef CAS.
- W. Li, N. J. Hill, A. C. Tomasik, G. Bikzhanova and R. West, Organometallics, 2006, 25, 3802–3805 CrossRef CAS.
- R. Rodriguez, D. Gau, T. Troadec, N. Saffon-Merceron, V. Branchadell, A. Baceiredo and T. Kato, Angew. Chem., Int. Ed., 2013, 52, 8980–8983 CrossRef CAS PubMed.
- (a) T. D. Bugg, Bioorg. Chem., 2004, 32, 367–375 CrossRef CAS PubMed; (b) T. Nagasawa and H. Yamada, Biochem. Biophys. Res. Commun., 1987, 147, 701–709 CrossRef CAS PubMed.
- M. Haaf, A. Schmiedl, T. A. Schmedake, D. R. Powell, A. J. Millevolte, M. Denk and R. West, J. Am. Chem. Soc., 1998, 120, 12714–12719 CrossRef CAS.
- M. Haaf, T. A. Schmedake, B. J. Paradise and R. West, Can. J. Chem., 2000, 78, 1526–1533 CrossRef CAS.
- (a) M. Driess, S. Yao, M. Brym, C. van Wullen and D. Lentz, J. Am. Chem. Soc., 2006, 128, 9628–9629 CrossRef CAS PubMed; (b) S. Yao, M. Brym, C. van Wullen and M. Driess, Angew. Chem., Int. Ed., 2007, 46, 4159–4162 CrossRef CAS PubMed.
- A. Meltzer, S. Inoue, C. Prasang and M. Driess, J. Am. Chem. Soc., 2010, 132, 3038–3046 CrossRef CAS PubMed.
- A. Meltzer, C. Präsang and M. Driess, J. Am. Chem. Soc., 2009, 131, 7232–7233 CrossRef CAS PubMed.
- M. Okazaki, H. Tobita and H. Ogino, Dalton Trans., 2003, 493–506 RSC.
- R. S. Ghadwal, R. Azhakar, H. W. Roesky, K. Propper, B. Dittrich, S. Klein and G. Frenking, J. Am. Chem. Soc., 2011, 133, 17552–17555 CrossRef CAS PubMed.
- R. S. Ghadwal, R. Azhakar, H. W. Roesky, K. Propper, B. Dittrich, C. Goedecke and G. Frenking, Chem. Commun., 2012, 48, 8186–8188 RSC.
- D. Lutters, C. Severin, M. Schmidtmann and T. Müller, J. Am. Chem. Soc., 2016, 138, 6061–6067 CrossRef CAS PubMed.
- D. Sarkar, V. Nesterov, T. Szilvasi, P. J. Altmann and S. Inoue, Chem. – Eur. J., 2019, 25, 1198–1202 CrossRef CAS PubMed.
- J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080–1082 CrossRef CAS PubMed.
- A. Jana, C. Schulzke and H. W. Roesky, J. Am. Chem. Soc., 2009, 131, 4600–4601 CrossRef CAS PubMed.
- A. V. Protchenko, J. I. Bates, L. M. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed.
- D. Reiter, P. Frisch, D. Wendel, F. M. Hormann and S. Inoue, Dalton Trans., 2020, 49, 7060–7068 RSC.
- D. C. H. Do, A. V. Protchenko, M. A. Fuentes, J. Hicks, P. Vasko and S. Aldridge, Chem. Commun., 2020, 56, 4684–4687 RSC.
- M. Kira, S. Ishida, T. Iwamoto and C. Kabuto, J. Am. Chem. Soc., 2002, 124, 3830–3831 CrossRef CAS PubMed.
- A. Jana, P. P. Samuel, G. Tavčar, H. W. Roesky and C. Schulzke, J. Am. Chem. Soc., 2010, 132, 10164–10170 CrossRef CAS PubMed.
- Y. Xiong, S. Yao and M. Driess, Chem. – Asian J., 2010, 5, 322–327 CrossRef CAS PubMed.
- T. Kosai, S. Ishida and T. Iwamoto, Angew. Chem., Int. Ed., 2016, 55, 15554–15558 CrossRef CAS PubMed.
- K. L. Wang, H. Li and J. R. Ecker, Plant Cell, 2002, 14, S131–151 CrossRef CAS PubMed.
- A. G. Fisch, Kirk-Othmer Encyclopedia of Chemical Technology, J. Wiley & Sons, New York, 2019, vol. 26, pp. 502–554 Search PubMed.
- R. Rodriguez, D. Gau, T. Kato, N. Saffon-Merceron, A. De Cozar, F. P. Cossio and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 10414–10416 CrossRef CAS PubMed.
- T. J. Hadlington, A. Kostenko and M. Driess, Chem. – Eur. J., 2020, 26, 1958–1962 CrossRef CAS PubMed.
- U. Lennert, P. B. Arockiam, V. Streitferdt, D. J. Scott, C. Rodl, R. M. Gschwind and R. Wolf, Nat. Catal., 2019, 2, 1101–1106 CrossRef CAS PubMed.
- (a) Y. Ren and T. Baumgartner, J. Am. Chem. Soc., 2011, 133, 1328–1340 CrossRef CAS PubMed; (b) C. Wang, M. Taki, Y. Sato, A. Fukazawa, T. Higashiyama and S. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 10374–10381 CrossRef CAS PubMed; (c) N. M. Wu, M. Ng and V. W. Yam, Angew. Chem., Int. Ed., 2019, 58, 3027–3031 CrossRef CAS PubMed.
- M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, M. Brym and M. Driess, Angew. Chem., Int. Ed., 2007, 46, 4511–4513 CrossRef CAS PubMed.
- S. S. Sen, S. Khan, H. W. Roesky, D. Kratzert, K. Meindl, J. Henn, D. Stalke, J. P. Demers and A. Lange, Angew. Chem., Int. Ed., 2011, 50, 2322–2325 CrossRef CAS PubMed.
- (a) H. R. G. Bender, E. Niecke and M. Nieger, J. Am. Chem. Soc., 1993, 115, 3314–3315 CrossRef CAS; (b) M. Driess, Adv. Organomet. Chem., 1996, 39, 193–229 CrossRef CAS; (c) M. Driess, H. Pritzkow, S. Rell and U. Winkler, Organometallics, 1996, 15, 1845–1855 CrossRef CAS; (d) P. Pyykko and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- S. S. Sen, J. Hey, R. Herbst-Irmer, H. W. Roesky and D. Stalke, J. Am. Chem. Soc., 2011, 133, 12311–12316 CrossRef CAS PubMed.
- S. Khan, R. Michel, S. S. Sen, H. W. Roesky and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 11786–11789 CrossRef CAS PubMed.
- A. E. Seitz, M. Eckhardt, S. S. Sen, A. Erlebach, E. V. Peresypkina, H. W. Roesky, M. Sierka and M. Scheer, Angew. Chem., Int. Ed., 2017, 56, 6655–6659 CrossRef CAS PubMed.
- (a) M. Driess, A. D. Fanta, D. R. Powell and R. West, Angew. Chem., Int. Ed. Engl., 1989, 28, 1038–1040 CrossRef; (b) A. D. Fanta, R. P. Tan, N. M. Comerlato, M. Driess, D. R. Powell and R. West, Inorg. Chim. Acta, 1992, 198–200, 733–739 CrossRef CAS.
- (a) P. Jutzi, A. Mix, B. Neumann, B. Rummel, W. W. Schoeller, H. G. Stammler and A. B. Rozhenko, J. Am. Chem. Soc., 2009, 131, 12137–12143 CrossRef CAS PubMed; (b) S. Khan, S. S. Sen, H. W. Roesky, D. Kratzert, R. Michel and D. Stalke, Inorg. Chem., 2010, 49, 9689–9693 CrossRef CAS PubMed.
- I. Alvarado-Beltran, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2016, 55, 16141–16144 CrossRef CAS PubMed.
- Y. Wang, T. Szilvási, S. Yao and M. Driess, Nat. Chem., 2020 DOI:10.1038/s41557-020-0518-0.
- V. Smil, Nature, 1999, 400, 415 CrossRef CAS.
- A. D. Allen and C. V. Senoff, Chem. Commun., 1965, 621–622 RSC.
- (a) J. L. Crossland and D. R. Tyler, Coord. Chem. Rev., 2010, 254, 1883–1894 CrossRef CAS; (b) N. Hazari, Chem. Soc. Rev., 2010, 39, 4044–4056 RSC; (c) K. C. Macleod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed; (d) H. P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC; (e) Y. Nishibayashi, Inorg. Chem., 2015, 54, 9234–9247 CrossRef CAS PubMed; (f) M. J. Bezdek and P. J. Chirik, Angew. Chem., Int. Ed., 2016, 55, 7892–7896 CrossRef CAS PubMed; (g) Y. Tanabe and Y. Nishibayashi, Chem. Rec., 2016, 16, 1549–1577 CrossRef CAS PubMed; (h) P. Bhattacharya, D. E. Prokopchuk and M. T. Mock, Coord. Chem. Rev., 2017, 334, 67–83 CrossRef CAS; (i) M. Holscher and W. Leitner, Chem. – Eur. J., 2017, 23, 11992–12003 CrossRef PubMed; (j) Y. Roux, C. Duboc and M. Gennari, ChemPhysChem, 2017, 18, 2606–2617 CrossRef CAS PubMed; (k) L. J. Taylor and D. L. Kays, Dalton Trans., 2019, 48, 12365–12381 RSC; (l) A. J. Kendall and M. T. Mock, Eur. J. Inorg. Chem., 2020, 1358–1375 CrossRef CAS.
- G. Maier, H. P. Reisenauer, J. Henkelmann and C. Kliche, Angew. Chem., Int. Ed. Engl., 1988, 27, 295–296 CrossRef.
- M. Winkler and W. Sander, J. Org. Chem., 2006, 71, 6357–6367 CrossRef CAS PubMed.
- K. Edel, M. Krieg, D. Grote and H. F. Bettinger, J. Am. Chem. Soc., 2017, 139, 15151–15159 CrossRef CAS PubMed.
- M. A. Legare, G. Belanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef CAS PubMed.
- M. A. Legare, M. Rang, G. Belanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |