
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopy, microscopy, diffraction and scattering of archetypal MOFs: formation, metal sites in catalysis and thin films†
Miguel
Rivera-Torrente
,
Laurens D. B.
Mandemaker
,
Matthias
Filez
,
Guusje
Delen
,
Beatriz
Seoane
,
Florian
Meirer
and
Bert M.
Weckhuysen
 *
*
Inorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: b.m.weckhuysen@uu.nl
First published on 20th August 2020
Abstract
Metal–organic frameworks (MOFs) are a class of porous crystalline materials showing great potential for applications such as catalysis, gas storage, molecular separations, energy storage and drug delivery. The properties that render them interesting stem from their structure (e.g. morphology, porosity or metal coordination and geometry). Thus, gaining a deeper understanding strongly relies on the availability and adequate use of advanced characterization tools, which can interrogate MOFs under realistic synthesis as well as catalysis (or sorption) conditions. Herein, we present an overview of the various characterization techniques specifically suitable for the study on the underlying chemistry of the formation mechanisms and adsorption properties of three archetypal MOFs, namely MIL-100, ZIF-8 and HKUST-1. A section on using MOFs as supports for metal atoms or complexes that can be used for catalysis on the robust Zr6 nodes of UiO-66 or NU-1000, and the characterization techniques used thereof, is presented as well. In addition, we discuss recent developments on the application of nano-spectroscopic characterization for MOF thin-films and explore the potential of MOFs as model systems in catalysis. The conclusions and outlook provide future research possibilities in the field of MOF characterization.
 Miguel Rivera-Torrente | Miguel Rivera-Torrente holds a BSc in Chemistry from Universidad Autónoma de Madrid (UAM) in Spain and a MSc in Chemical Engineering from the University of Strasbourg – École de Chimie, Polymères et Matériaux (ECPM), France. After research stays at BASF and RWTH Aachen (Germany) he pursued a PhD in spectroscopy of metal–Organic frameworks at the Inorganic Chemistry and Catalysis Group of Utrecht University (The Netherlands). He currently works in different areas of catalysis, materials science and polymer chemistry as a Senior Research Specialist in Dow. |
 Laurens D. B. Mandemaker | Laurens D. B. Mandemaker received his BSc and later MSc degree in Chemistry from Utrecht University (The Netherlands) for the work performed on zeolites using in situ spectroscopic techniques in the Inorganic Chemistry and Catalysis Group. Currently, he is doing a PhD under the supervision of Prof. Bert M. Weckhuysen at Utrecht University. His research focusses on combining microscopic and spectroscopic tools to study the growth, performance and deactivation of metal–organic framework films in gas-sorption or as catalysts. |
 Matthias Filez | Matthias Filez studied Chemical Engineering and Physics at KU Leuven (Belgium). He received his PhD in 2015 at Ghent University (Belgium) with Prof. Guy B. Marin and Prof. Christophe Detavernier on the “Alternative Design of Pt-based Catalysts: An X-ray Spectroscopic View”. After his PhD, he continued as postdoctoral researcher in the group of Prof. Bert M. Weckhuysen at Utrecht University (The Netherlands), where he obtained a Marie Skłodowska-Curie fellowship. Currently, he is postdoctoral researcher in the groups of Prof. Christophe Detavernier (UGent, Belgium) and Prof. Maarten B. J. Roeffaers (KU Leuven, Belgium), where his research focusses on novel approaches in metal nanocatalysis. |
 Guusje Delen | Guusje Delen is a PhD candidate in the Inorganic Chemistry and Catalysis group at Utrecht University (The Netherlands). Prior to her PhD, she obtained her BSc and MSc degree In Nanomaterials, Chemistry and physics at Utrecht University. She obtained her MSc degree for a thesis on the synthesis of Fe based catalysts for Fischer–Tropsch synthesis. In her PhD under the supervision of Prof. Bert M. Weckhuysen, she focuses on Nano-Spectroscopy of Surface-Anchored Metal–Organic Frameworks. |
 Florian Meirer | Florian Meirer obtained his ScD degree (2008) in technical physics from the TU Wien (Vienna University of Technology) under Prof. Christina Streli. After postdoctoral stays at the Stanford Synchrotron Radiation Lightsource, USA, (Erwin Schrödinger Fellowship, FWF) and the Fondazione Bruno Kessler, Italy (Marie-Curie cofund fellowship), he moved to Utrecht to work on spectroscopic and spectromicroscopic methods for solid catalyst characterization. He is currently a tenured Assistant Professor and his fields of research include spectromicroscopy and data mining and chemometrics in the fields of heterogenous catalysis and environmental analysis. |
 Bert M. Weckhuysen | Bert Weckhuysen received his master's degree in chemical and agricultural engineering from Leuven University (Belgium) in 1991. After obtaining his PhD from Leuven University in 1995 under the supervision of Prof. Robert Schoonheydt, he has worked as a postdoc with Prof. Israel Wachs at Lehigh University (USA) and with Prof. Jack Lunsford at Texas A&M University (USA). Weckhuysen has been since 2000 a full professor of inorganic chemistry and catalysis at Utrecht University (The Netherlands). His research interests are in the development of spectroscopy and microscopy techniques for elucidating the working and deactivation principles of catalytic solids, often applied under true reaction conditions. |
1. Introduction
Since their early discovery in the 1990s, metal–organic frameworks (MOFs) have been researched with ever-increasing pace in their marked properties for gas sorption and catalysis.1–5 To unravel the chemistry of MOF formation and functioning and establish novel structure–function relationships, a toolbox of characterization techniques has been assembled and proven crucial for the knowledge build-up in the past decades. Further advances are still required though, particularly to elucidate the gaps in our understanding to better exploit MOF properties and develop protocols for obtaining application-tailored MOFs from this unexplored design space. Despite the significance of this field and the number of research reports, an overview and perspective of advanced MOF characterization tools is, to the best of our knowledge, lacking. Several review articles focused on a single or narrow selection of characterization techniques have been published, including: transmission electron microscopy (TEM),6 magnetic,7–12 X-ray13–15 and vibrational16,17 spectroscopies. One example of a broader view was provided by Howarth et al.,18 in which a selection of routinely used characterization tools was discussed, such as X-ray diffraction (XRD), thermogravimetrical analysis (TGA) and N2 physisorption, responding to the need of establishing good practices for a fair comparison of MOF reports in the field.This review article presents a critical overview of the variety of advanced spectroscopic,6,7,19–24 scattering and microscopy tools that can be applied in a state-of-the-art way to characterize the most commonly studied MOFs. This approach aims to establish a framework of thinking for moving towards a more complete set of characterization techniques for understanding synthesis, sorption and catalysis in MOFs; by expanding the number of available characterization tools and exemplifying their current and future possibilities. We present different showcases where the use of advanced characterization tools is required to decode the complexity of MOF chemistry. Specifically, the most noteworthy traits of three MOFs in their bulk and film forms, namely MIL-100/MIL-101, MAF-4/ZIF-8/ZIF-67 and Cu3BTC2/HKUST-1. These topologies have shown potential in catalytic and sorption applications, and had a huge impact on the development of the field throughout the last 20 years.25 In particular cases, relevant reports in which other topologies (or even composites) have been studied are highlighted together with the most suitable techniques used to study their properties.
2. Chemistry and characterization of archetypal metal–organic frameworks
A large number of topologies have been breakthroughs in MOF chemistry, e.g. MOF-5, UiO-66, MFM-300, MIL-53, MOF-74 or CPO-27, MIL-88, MIL-127, NH2-MIL-125, NU-1000, MOF-808, CAU-10-H, MFU-4l, DUT-5 and many others; following stability and performance criteria, as well as fundamental discovery. The reports on these structures and their properties have boosted the research on microporous crystalline materials with unprecedented properties. For instance, one important feature of MOFs, which differs from those of metal oxides or zeolites, is their flexibility, that endows them with the capacity of expanding or contracting upon gas sorption.26–28 We advise the curious reader to consult the excellent reviews on the characterization tools authored by Kitagawa et al.,29–33 Férey et al.,34,35 Fletcher et al.,36 Murdock et al.37 and Schneemann et al.26 This is also the case with the chemistry of defects and defect-engineered MOFs, recently reviewed by Fang et al.38 and Dissegna et al.39 which presents a major challenge for the spectroscopist or diffraction expert, but with the potential reward of unravelling fascinating phenomena useful for catalysis, redox chemistry or gas sorption. Other very important aspect that is inherent to the relatively dynamic nature of coordination bonds is metal exchange and mixed-metal MOFs. Again, detailed literature is available in the reviews published by Lalonde,40 Deria41 and Evans et al.,42 or Dincă et al.43 and Abedanatanzi et al.44 Another field of recent interest, sparked mainly by Bennett and others,45–50 is the development and study of disordered and amorphous MOFs. Inspired by those, we have recently shown that,51 understanding disorder may be of paramount importance for, for example, metal particle deposition. However, for a more detailed analysis and due to their higher practical advantages for technical applications, we have restricted this review article to three (MIL-100/MIL-101, ZIF-8/ZIF-67 and HKUST-1) archetypal MOF topologies for which a vast array of techniques has been used, as described hereafter. Three important aspects that have been deeply studied on these structures are: the formation mechanisms (e.g. crystallization and assembly), the formation and chemistry of coordinatively unsaturated sites (CUS) (including changes of the oxidation state of the metal center), and the diffusion of gases within the pores, as well as the interaction of those with metal and linker sites. These points will be addressed by showcasing relevant studies published for the mentioned topologies, as well as briefly outlining their implications in catalysis and gas sorption. Additionally, as highlighted by the groups of Corma52 or Gates,53 MOFs offer an excellent platform to be used as supports for metal atoms and complexes in (model) catalysis by post-synthetic modification. Thus, a section addressing the tools and hurdles of characterizing such systems is presented. In this sense, one way of minimising mass transfer issues and reducing the amount of material employed, as well as studying new fundamental properties, is by using thin-films, which are often more difficult to characterize than bulk materials. Section 5 critically discusses the necessity of using micro- and nanoscopy tools for the spatially resolved characterization of MOFs, as demonstrated for other microporous functional materials.54–56 This selection was made following a bibliometric analysis of relevant literature (ca. 2000 research articles and reviews) in the specific MOF fields in which the authors have been working on in the past years. The analysis was carried out based on the appeareance of keywords (see the ESI† for more details), and the papers grouped depending on the features that they study and the techniques used. This allowed us to pinpoint the techniques, mainly X-ray based, but also with spatially resolved tools with nanometric resolution, i.e. nanoscopy, that may be interesting to use in the coming years for the study of MOFs and will be discussed in the concluding section.2.1 Chemistry and characterization of MIL-100 and MIL-101
MIL-100 and MIL-101 have been intensively studied since they were first reported by Férey et al. in the mid-2000s as some of the most stable structures to date.57,58 They consist of 1,3,5-benzenetricarboxylate (BTC) and 1,4-benzenedicarboxylate (BDC), respectively, and μ3-O-centered trinuclear inorganic clusters [M3(μ3-O)(O2C-R)6], where M = Cr3+, Fe3+,59 Al3+,60 V3+/4+,61 Sc3+,62 Ti3+,63,64 Mn3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 65 or In3+.66 In the as-synthesized form, each metallic centre is in an octahedral environment coordinated by one μ3-O atom, shared with the other two metallic centres building the cluster, four oxygen atoms from the organic linker and one terminal ligand (Fig. 1). While two metallic octahedra per cluster are typically coordinated by terminal aqua ligands, the third terminal ligand is an anionic species, that may be either a hydroxyl ligand or a halide anion (such as F− or Cl−, depending on the synthesis conditions), needed to compensate the negative default charge per inorganic cluster of the framework. The μ3-O-centered trinuclear clusters are then linked through the organic moieties to form hybrid super tetrahedra, which further assemble into a 3D porous framework having the zeolite MTN topology. This MOF topology possesses two different types of mesoporous cages of 25 Å and 29 Å (MIL-100) and 29 Å and 34 Å (MIL-101), delimited by microporous pentagonal and hexagonal windows of 5 Å and 8.6 Å (MIL-100) and 12 Å and 14.7 Å (MIL-101). Depending on the metal and the activation procedure, MIL-100 and MIL-101 have been reported to possess BET areas up to 2300 and 4100 m2 g−1, respectively, exceptional hydrothermal stability and a relatively high thermal stability. This set of properties, together with the presence of coordinatively unsaturated metal sites (CUS) in the inorganic secondary building units (SBU) upon activation and the presence of mesoporous cages, makes these frameworks one of the topologies of choice for study in different fields.67
65 or In3+.66 In the as-synthesized form, each metallic centre is in an octahedral environment coordinated by one μ3-O atom, shared with the other two metallic centres building the cluster, four oxygen atoms from the organic linker and one terminal ligand (Fig. 1). While two metallic octahedra per cluster are typically coordinated by terminal aqua ligands, the third terminal ligand is an anionic species, that may be either a hydroxyl ligand or a halide anion (such as F− or Cl−, depending on the synthesis conditions), needed to compensate the negative default charge per inorganic cluster of the framework. The μ3-O-centered trinuclear clusters are then linked through the organic moieties to form hybrid super tetrahedra, which further assemble into a 3D porous framework having the zeolite MTN topology. This MOF topology possesses two different types of mesoporous cages of 25 Å and 29 Å (MIL-100) and 29 Å and 34 Å (MIL-101), delimited by microporous pentagonal and hexagonal windows of 5 Å and 8.6 Å (MIL-100) and 12 Å and 14.7 Å (MIL-101). Depending on the metal and the activation procedure, MIL-100 and MIL-101 have been reported to possess BET areas up to 2300 and 4100 m2 g−1, respectively, exceptional hydrothermal stability and a relatively high thermal stability. This set of properties, together with the presence of coordinatively unsaturated metal sites (CUS) in the inorganic secondary building units (SBU) upon activation and the presence of mesoporous cages, makes these frameworks one of the topologies of choice for study in different fields.67
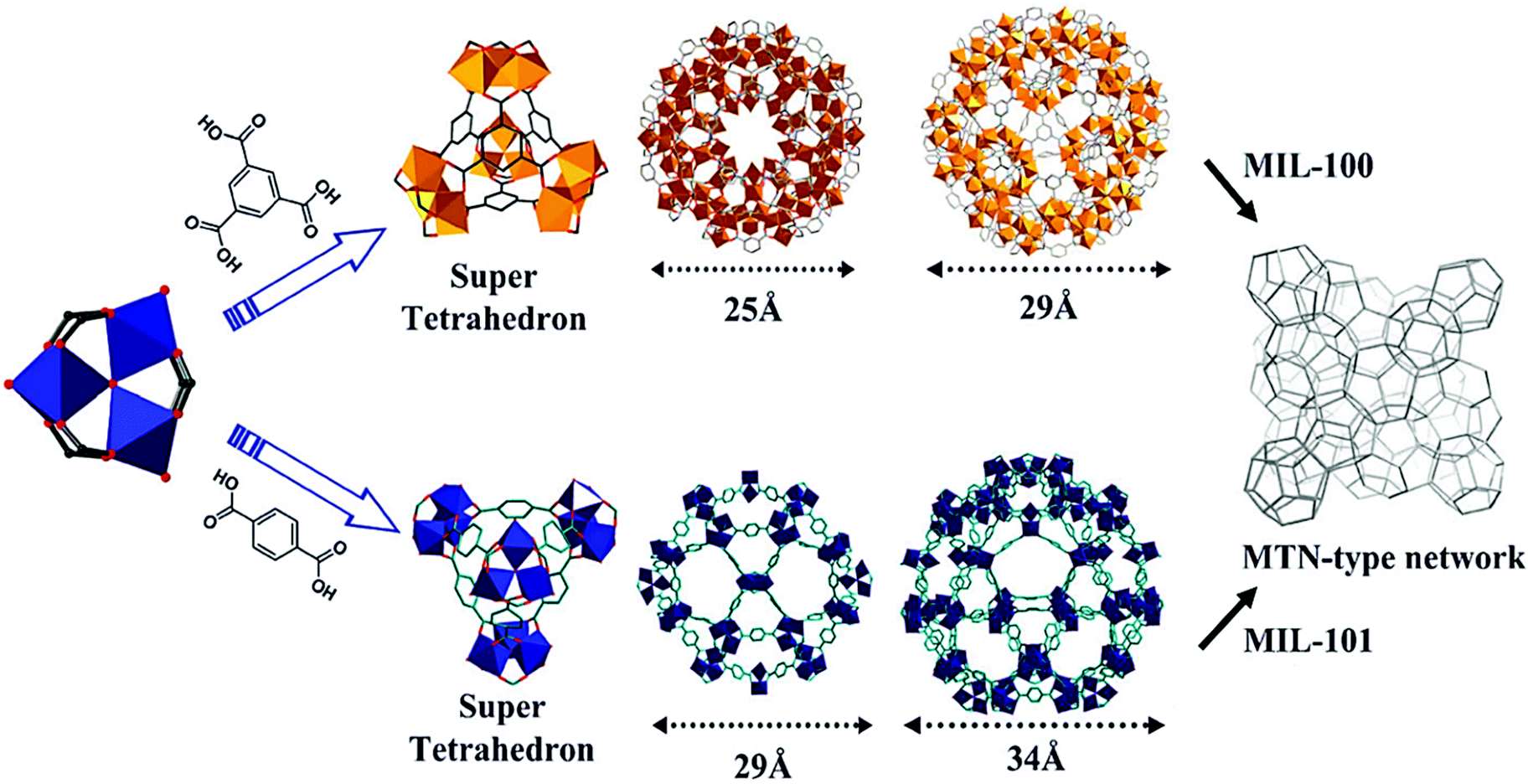 | ||
| Fig. 1 Structure of MIL-100 and 101 formed by μ3-O-centered trinuclear metallic clusters linked through the organic linker into a supertetrahedron, which further assembles into a porous framework having the MTN topology. Reproduced from ref. 84. Copyright©2012 Royal Society of Chemistry. | ||
In particular, the authors used Fe K-edge EXAFS and observed the presence of trimeric iron oxide SBUs at the different stages of the MIL-89 synthesis. XAS is indeed a very powerful technique to study the MOF formation mechanism at early stages of crystallization, providing information concerning the type, number and distance of neighbouring atoms for a specific element.86 Further insight into the stability of the trimeric iron oxide units upon MIL synthesis was obtained by Birsa Čeliĉ et al.87 The authors studied the synthesis of MIL-45(Fe) and MIL-100(Fe) from the same precursors using different solvent compositions (water or water/acetone) by XAS and Mößbauer spectroscopy at several reaction times. Interestingly, upon acetone addition a change in the iron oxidation state from Fe3+ to Fe2+ was observed upon heating, leading to the dissolution of the previously formed amorphous iron complexes. The instability of this SBU in certain solvent compositions has a pronounced effect: acetone cannot stabilize the trimeric cluster and leads to the formation of MIL-45(Fe), containing undulating chains of FeIIO6 octahedra. In water, MIL-100(Fe) is obtained where the Fe3+ based trimeric units remain unaltered throughout the crystallization process.
These examples clearly illustrate the significance of understanding the local environment of the different species present at different stages of the synthesis. However, in addition to this, it is also important to examine crystal growth over length scales of several orders of magnitude in order to build up a complete picture of crystallization. Moreover, although ex situ and pseudo in situ experiments, as those describe above, provide critical information, the interpretation of the results must be done carefully, since the delicate balance of species in solution might be altered prior to the data acquisition. In this sense, in situ SAXS/WAXS are a powerful tool, with SAXS providing information regarding the size, shape and surface of the particles and WAXS about their crystalline properties, both in a time-resolved fashion.
Further, the use of synchrotron radiation allows the study of the synthesis under harsh conditions, what facilitates the study of reactions in situ. Stavitski et al. and Goesten et al.85,88 combined in situ SAXS and WAXS to gain insight into the NH2-MIL-53(Al) and NH2-MIL-101(Al) crystallization process. In line with previous observations by Millange et al. by energy-dispersive XRD,75,89 the authors observed the formation of the NH2-MOF-235(Al) intermediate (Bragg peak observed at Q = 6.3 nm−1, Fig. 2) prior to the appearance of NH2-MIL-53(Al) and NH2-MIL-101(Al). Moreover, the study of the influence of different synthesis parameters, i.e. solvent composition (H2O/DMF ratio), temperature and concentration of precursors, on the MOF crystallization enabled the evaluation of the different factors governing the NH2-MIL-53(Al) and NH2-MIL-101(Al) formation. Specifically, the solvent composition was proven to play a key role, not only on the kinetics, but also on the sequence of events taking place during MOF synthesis. On the one hand, DMF increases the linker solubility compared to water, being the dissolution of the organic linker a rate-limiting step of the crystallization process at high H2O/DMF ratios. On the other hand, DMF seems to stabilize the intermediate phase NH2-MOF-235(Al), and the use of pure DMF is required in order to obtain NH2-MIL-101(Al).
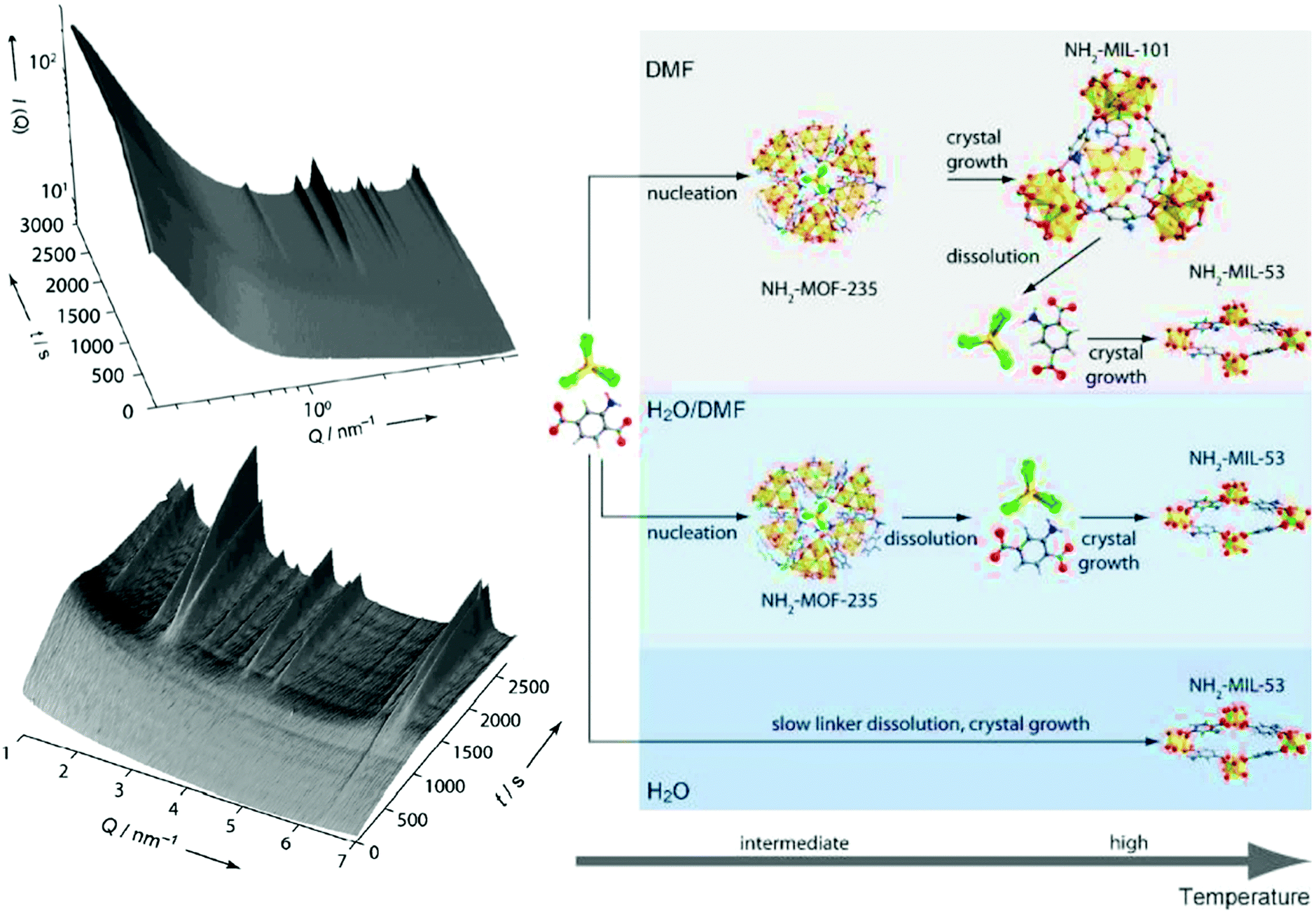 | ||
| Fig. 2 Left: In situ 3D Small Angle X-ray Scattering obtained during the crystallization of NH2-MIL-101(Al) at 403 K using DMF as solvent. Right: Sequence of events taking place during the NH2-MIL-53(Al) and NH2-MIL-101(Al) crystallization processes under different synthesis conditions. C: grey, H: white, N: blue, O: red, Al: yellow, Cl: green. Reprinted with permission from ref. 85. Copyright©2011 John Wiley & Sons, Inc. | ||
So as to gain further understanding into the role DMF plays, Goesten et al.90 applied in situ1H and 27Al NMR91 to the NH2-MIL-101(Al) synthesis and supported their observations by DFT calculations. While DFT modelling shows that DMF stabilizes the μ3-O-centered inorganic cluster present in MOF-235 and MIL-101,90,92 in line with previous observations by X-ray scattering (vide supra), in situ NMR indicates a more complex role of the solvent. In particular, the results pointed to the formation of a stable H–Cl–DMF complex that molecularly promotes the NH2-MOF-235(Al) to NH2-MIL-101(Al) transformation. The authors proposed that this complex supplies the required hydroxido ligands, which are not present in the NH2-MOF-235(Al) inorganic cluster, but that are present in that of NH2-MIL-101(Al), through a water dissociative mechanism (Fig. 3).
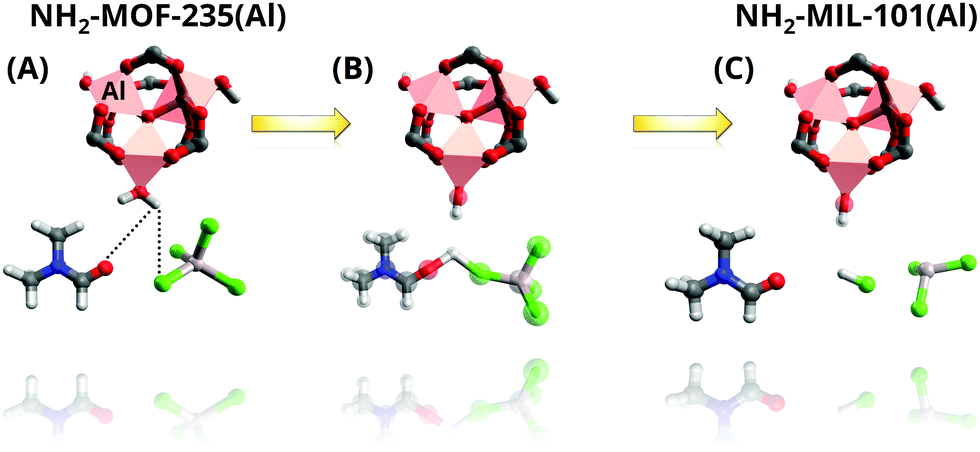 | ||
| Fig. 3 Representation of the NH2-MOF-235 to NH2-MIL-101 transformation mechanism promoted by the complexation of axial H2O coordinated to octahedral Al, DMF with Cl− anions as proposed by Goesten et al. (A) Interaction of O in DMF and Cl− in AlCl4− abstract a proton from H2O in axial position (B), leading to the formation of (C) OH hydroxy species as ligand. Colour code: pink, octahedra, Al; grey, C; red, O; white, H; blue, N; green, Cl. Adapted from ref. 90. Copyright©2014 American Chemical Society. | ||
Furthermore, with this information at hand, the authors refuted their previous assumption of a dissolution–recrystallization mechanism for the NH2-MOF-235(Al) to NH2-MIL-101(Al) transformation and proposed that the transition occurs in the solid state instead. This conclusion is further supported by the previously obtained SAXS data, from which a constant scatterer volume, morphology and intensity were observed. The data points to the formation of amorphous 25 nm scattering entities prior to the measurements, whose long-range order continue to evolve at constant volume so that Bragg peaks corresponding to NH2-MIL-235(Al) and NH2-MIL-101(Al) started appearing at 500 and 1500 s, respectively.
Although this section is not intended as a survey of all the different in situ methods available to study the formation of crystalline materials, nor as an exhaustive overview on the current insight into the formation of MOFs (for those we refer the reader to the works authored by Pienack,86 Walton93,94 and Attfield et al.),68 the examples above demonstrate that combining various synchrotron and non-synchrotron-based techniques is essential if a complete picture of the MOF crystallization is desired. Indeed, given the challenge of fully understanding the different chemical and physical events taking place during the synthesis, at all relevant length-scales.95,96 The combination of simultaneously operating, complementary techniques would allow to study the formation mechanism in one single experiment, avoiding possible reproducibility issues and facilitating the correlation of the different data. These types of systems have been already successfully used to study the synthesis of traditional porous materials,96 but they remain largely unexplored in the case of MOFs.97
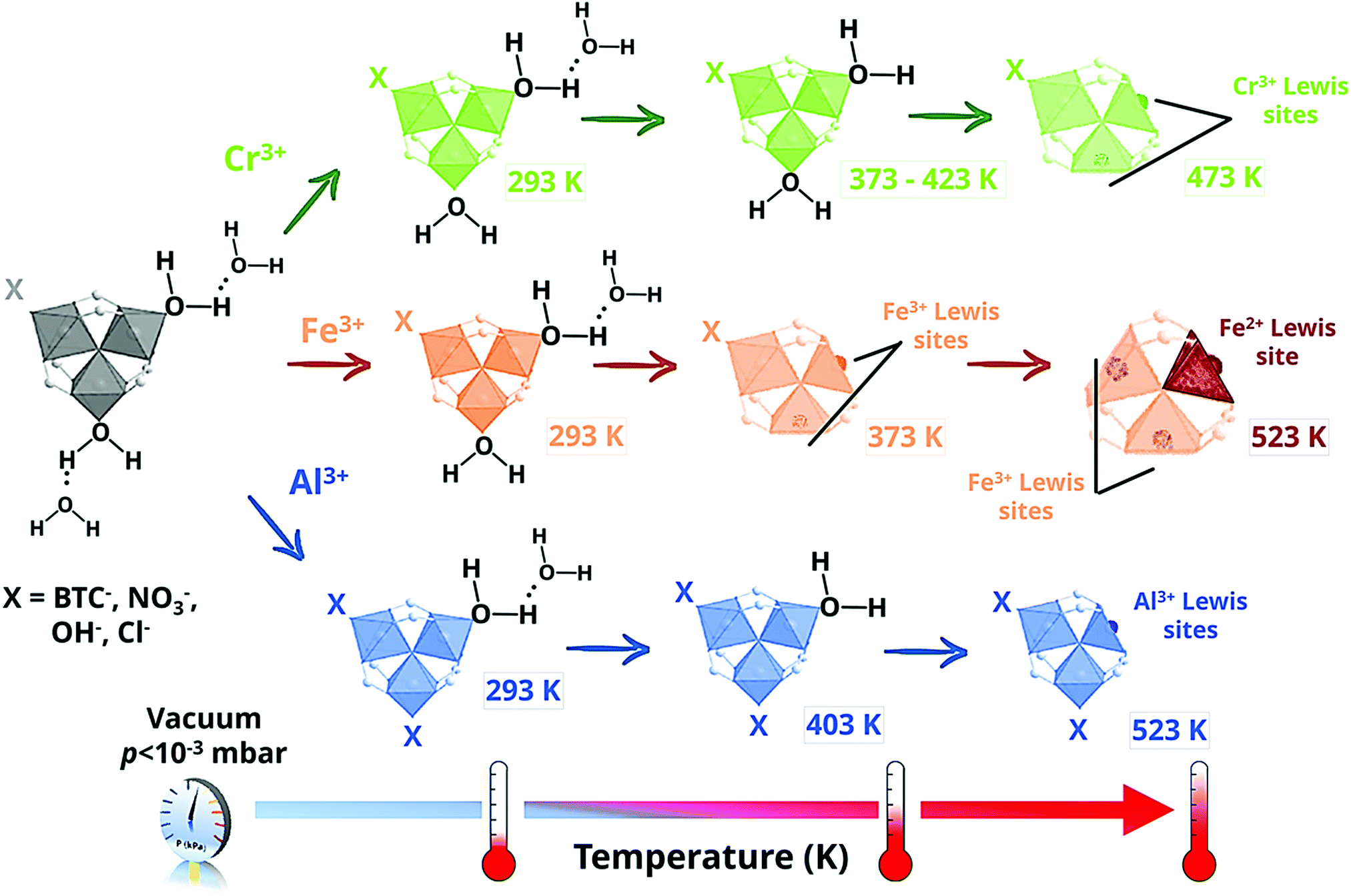 | ||
| Scheme 1 Location and interaction of water molecules and formation of the open metal sites present in the metal–oxo trimers of MIL-100(M); where M = Cr (top, green), Fe (middle, orange) and Al (bottom, blue), as a function of the MOF outgassing temperature for different for different MIL-100 analogues. | ||
| Water species | ν 3(νas) [cm−1] | ν 1(νs) [cm−1] | ν 2 + ν3 [cm−1] | ν 2(δH2O) [cm−1] |
|---|---|---|---|---|
| H2O → Cr3+ (species 1) | 3700 | 3608 | 5274 | 1596 |
| H2O → Cr3+ (species 1′) | 3683 | 3595 | 5265 | 1604 |
| H2O⋯H2O → Cr (species 2) | 3680 | 3588 | 5252 | 1603 |
| H2O⋯H2O → Cr | 3670 | 2950 | 5320 | 1650 |
In contrast to MIL-100(Cr), for which CUS with one single oxidation state were observed, i.e. Cr3+, the IR spectra acquired for MIL-100(Fe) showed two different bands at 618 and 597 cm−1, corresponding to the νas(Fe3+3O) and νas(Fe2+3O) vibrations modes, respectively.100 This partial reduction of iron at higher outgassing temperatures was further confirmed by 57Fe Mössbauer spectroscopy and it was related to the partial removal of terminal anionic ligands at 523 K (Scheme 1). In the case of MIL-100(Al), IR spectroscopy and solid-state NMR studies pointed to a different scenario, in which only one of the two terminal aqua ligands per trinuclear cluster can be removed.101,102 This different behaviour was related to the presence of different impurities, in particular NO3− from the metal salt used for the MOF synthesis and H3BTC, probably coordinated to the aluminium open metal sites.
In a further step, several research groups evaluated the Brønsted and Lewis acid sites (BAS and LAS, respectively) of different MIL-100 analogues by infrared spectroscopy using different basic probe molecules. To this end, carbon monoxide (CO) is one the most widely used probe molecules to evaluate protonic acidity. When probing BAS, the lone pair of CO interacts via σ-donation with OH groups, leading to shifts in both ν(OH) and ν(CO) IR bands, so that the larger the shift, the stronger the acidity. M–OH species (only observed for Al and Cr-based analogues, but not in the case of MIL-100(Fe) and MIL-100(V)) gave rise to Δν(OH) = 90 and 86 cm−1 for MIL-100(Cr) and MIL-100(Al), respectively, showing a fairly weak acidity, close to that of silanol groups in silicalite (Δν(OH) = 100 cm−1), regardless of the metal used. Among the different hydroxyl groups (either hydroxyl groups belonging to the framework, i.e. M–OH, or to water molecules, i.e. M–H2O or M–H2O/H2O species), the highest Brønsted acidity was observed for the M–H2O species, where the water molecules were located in the cation coordination sphere.103 In this case, the Δν(OH) value was 160 cm−1, close to that of alkali-exchanged faujasite (Δν(OH) = 160 cm−1) or P–OH groups of phosphated silica (Δν(OH) = 180 cm−1). Interestingly, as demonstrated by Vimont and co-workers, this Brønsted acidity can be modified by substituting the coordinated water by different OH-containing organic molecules, paving the way to the fine tuning of the acidic properties of MOFs.104 Furthermore, BAS can be converted into LAS upon framework dehydration, which can be also probed with different probe molecules (Table 2). A thorough study has been recently published by Hall and Bollini, in which they demonstrated that M3+⋯OH Brønsted sites should be more carefully studied.105 They used pyridine (Py) and 2,6-di-tert-butylpyridine (DBTPy) sequentially to distinguish between how Lewis or Brønsted sites play a role in acetalization reactions. This type of studies should be used as a model for other MOFs, such as Zr6 containing topologies, in which acidity is also a recurrent topic.106–111
| MIL-100 analogue | M2+ CUS [μmol g−1] | M3+ CUS [μmol g−1] | Total M CUS [μmol g−1] |
|---|---|---|---|
| MIL-100(Cr)523K | — | 3500 | 3500 |
| MIL-100(Fe)423K | 45 | 1895 | 1940 |
| MIL-100(Fe)523K | 850 | 2810 | 3660 |
| MIL-100(Al)523K | — | 1800 | 1800 |
| M3+ cation in MIL-100 | ν(CO) [cm−1] | CD3CN ν(CN) [cm−1] | C5H5N ν18 [cm−1] |
|---|---|---|---|
| Cr | 2207, 2200, 2196 | 2305 | 1015 |
| Fe | 2192–2173 | 2304 | 1014 |
| Al | 2195–2184 | 2326–2321 | 1018 |
In the case of MIL-100(Fe) two different ν(CO) bands were observed in the 2170–2166 cm−1 and 2192–2173 cm−1 ranges, which characterized CO adsorbed on Fe2+ and Fe3+ CUS, respectively.112 Moreover, for MIL-100(Al) half of the CUS calculated for MIL-100(Cr) and MIL-100(Fe) were obtained, in line with the presence of different impurities previously observed by infrared and NMR spectroscopy (see above).102
In another study, Gómez-Pozuelo et al. quantified the number of Lewis sites of the isoreticular materials MIL-100 by means of CD3CN.113 The number was lower than those reported in Table 2 although similar activation procedures were used, the reason for this discrepancy remaining unknown. This series of works highlight the importance of a thorough characterization of MOFs’ open metal sites since the different activation conditions, coordinated molecules or chemical composition influence the nature, concentration and strength of the framework acid sites. IR spectroscopy is a widely available experimental tool that, upon adsorption of different probe molecules, has been intensively used to evaluate the strength and quantify the amount of acid sites in different traditional porous materials.114 As described above, this approach has also been successfully applied to MOFs, yet some differences should be borne in mind. Although some strong bases have been commonly used to assess the acidity of many solid materials, its use to study MOF acidity can be problematic. For instance, in the case of pyridine, vibration modes sensitive to interaction with acid sites often overlap with those of the framework.102,103 Moreover, the preparation of self-supporting discs often needed to perform the experimental measurements might not be trivial for some of the aforementioned materials, given the pressure-induced amorphization reported for different MOFs.115 Although this issue is commonly overlooked, and the preparation of self-supported discs is often found in literature, the deposition of the sample by drop casting on silicon wafers is recommended for those cases in which the stability of the MOF may be a problem.
In addition to its use for evaluating the acidity of MOFs, infrared spectroscopy has also been proven as a powerful tool to study the interaction of CUS with different adsorbates. For instance, Leclerc et al.100 examined the influence of the different open metal sites present in MIL-100(Fe), i.e. Fe2+ and Fe3+, on the adsorption of several probe molecules, particularly CO, propene and propyne, able to interact through π-backdonation, and CO2, pyridine and propane, not able to provide such interaction. IR spectroscopy pointed to a more important role of Fe2+ CUS, despite their weaker acidity, due to their ability to strongly interact with CO, propene and propyne via π-backdonation, as indicated by the redshifts observed in the ν(C–O), ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), ν(C
C), ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) and ν(CH) bands. Indeed, the additional electron in Fe2+ d-orbitals enables the formation of more stable metal–adsorbate complexes by the transfer of electron density from the d orbitals of the Fe2+ metal centres to the π* antibonding orbitals of certain molecules. Yoon et al.116 and Wuttke et al.112 took advantage of the stronger interaction of Fe2+ CUS with unsaturated gas molecules, and studied the separation of propene from propane through breakthrough experiments using equimolar propene:propane mixtures. Specifically, Wuttke et al. applied in situ IR spectroscopy and studied the propene:propane separation performance with and without selectively poisoning the Fe2+ CUS. In line with previous studies, the adsorption capacity of C3H6 is markedly higher without blocking the Fe2+ CUS, leading to separation factors over 100, which drop to 5.3 upon Fe2+ NO poisoning.
C) and ν(CH) bands. Indeed, the additional electron in Fe2+ d-orbitals enables the formation of more stable metal–adsorbate complexes by the transfer of electron density from the d orbitals of the Fe2+ metal centres to the π* antibonding orbitals of certain molecules. Yoon et al.116 and Wuttke et al.112 took advantage of the stronger interaction of Fe2+ CUS with unsaturated gas molecules, and studied the separation of propene from propane through breakthrough experiments using equimolar propene:propane mixtures. Specifically, Wuttke et al. applied in situ IR spectroscopy and studied the propene:propane separation performance with and without selectively poisoning the Fe2+ CUS. In line with previous studies, the adsorption capacity of C3H6 is markedly higher without blocking the Fe2+ CUS, leading to separation factors over 100, which drop to 5.3 upon Fe2+ NO poisoning.
Further insight into the adsorption of different molecules can be obtained through variable-temperature infrared (VTIR) spectroscopy, which, as shown by Palomino et al.117 for H2 adsorption on MIL-100(Cr) and MIL-101(Cr), allows for the determination of the corresponding values of standard adsorption enthalpy (ΔH0) and entropy (ΔS0).
Two special cases for understanding formation of CUS and binding properties in the MIL-100 series are the Sc and the Al analogue, in which solid-state NMR was particularly useful. Resonance-Echo Double-Resonance (REDOR) NMR techniques allow to calculate internuclear distances and dipolar coupling of different nuclei based on the difference in dephasing radiofrequency of a given pair of nuclei, e.g.13C–15N or 29Si–27Al. It allowed Giovine et al.118 to calculate the effect a number of parameters affected on both H and C when degassing the Sc3+ centres in MIL-100(Sc) and prove the formation of penta-coordinated sites upon dehydration. A comparison with Sc3BTB2 (BTB = 1,3,5-tris(4-carboxyphenyl)benzene), which shows similar Sc sites, was established, and combining DFT calculations and multinuclear techniques (namely, Resonance-Echo Saturation-Pulse Double-Resonance (RESPDOR 13C–{45Sc}), 2- and 3-dimensional Multi-Quantum Magic Angle Spinning (2-3D MQ-MAS 45Sc–{1H}) and Cross-Polarization (CP MAS)), they were able to quantify the number of Sc3+ CUS based on their coordination numbers and the asymmetric polarization of the Sc3(μ3-O) clusters. In some cases, additional techniques such as EPR have been combined with NMR for characterizing CUS in MIL-100 materials, allowing the authors to reach conclusions that could not be drawn from vibrational techniques. For instance, Barth, Hartman and others119 studied the adsorption of NO in MIL-100(Al) by means of DRIFTS, multinuclear NMR and EPR spectroscopies. In a fist instance, they observed NO interacting weakly with the π-electrons of the aromatic linkers (band at 1861 cm−1); and physisorbed and gaseous NO (1854 and 1874 cm−1, respectively). However, while no evident bands pointing to [Al3+⋯NO] adducts were observed in the DRIFTS spectra, EPR showed that electrophilic Al3+ can polarize NO and transform it into NO+. Moreover, evolution of adsorbed species could be studied as a function of temperature to corroborate that hypothesis. Such type of weak ionic interactions cannot be detected efficiently by FT-IR, but coupling EPR and DFT calculations allowed for an accurate description of this issue.120 In order to avoid any confusion with different Al adsorption sites, 27Al{1H} heteronuclear correlation (HETCOR) upon dehydration of the MOF was used in another work,121 showing the presence of Al(OH)3 deposits that play a minor role in NO adsorption. In brief, even for a complex case of weak, ionic interactions, the authors were able to obtain an accurate description by adding magnetic spectroscopy to vibrational techniques. Further, not only bonding but also the dynamics of the adsorbed probe can be studied by means of 2D NMR, e.g. rotational motions of pyridine on Al3+ or Al–OH sites or the effects of adsorbates on the backbone.122,123 This has been also successfully employed in different frameworks,124–126 sometimes being able to discriminate between competing adsorbing molecules of very similar nature.127
Thus, beyond the use of well-known basic probe molecules to study MOF acidity (e.g. pyridine, CO, CH3CN), the use of other types sensitive to redox or ionic sites (e.g. NO, TEMPO) as well as the implementation of more sophisticated approaches, such as operando studies, provides mechanistic insight into catalytic, adsorption or separation processes, allow for the identification active species and even enable the evaluation of their thermodynamics. The development of cells that can combine different techniques, e.g. in situ XAS, XRD and FT-IR is currently under investigation by a number of groups and will certainly become more important in the coming years.128 Rivera-Torrente et al. and others have shown the importance of studying CUS formation and the fate of the network upon activation for catalytic purposes in situ spectroscopy tools.129,130 This approach will increasingly become important as MOFs are used as platforms and precursors of active catalysts.
2.2 Chemistry and characterization of MAF-4 or ZIF-8
Zeolitic imidazolate frameworks (ZIFs) are a subfamily of MOFs characterized by the use of imidazolate linkers as organic ligands so that the M–Im–M (M stands for the metal, such as Zn or Co, and Im for the imidazolate linker) angle is close to 145°, similar to the Si–O–Si angle commonly found in zeolites. This feature leads to the formation of MOFs with zeolite-like topologies, including not only those observed in zeolites, but also predicted structures not yet experimentally realized.131–138 Among the different ZIFs, ZIF-8139 (also known as metal azolate framework 4, or MAF-4)134 consists of Zn2+ (Co2+ in the case of its analogue ZIF-67) and 2-methylimidazole (MeIm), where the metallic centres are tetrahedrally coordinated to four imidazolate N atoms, giving rise to an open framework with an augmented sodalite zeolite-like topology. As shown in Fig. 4, this topology possesses sodalite-like β-cages of 11.6 Å interconnected by hexagonal pore apertures of 3.4 Å, in principle able to screen between different molecules.139–141 This feature, together with the easy synthesis of ZIF-8, its good adsorption properties and its remarkable chemical and thermal stabilities, has drawn the attention of the scientific community, being ZIF-8 one of the most studied MOFs.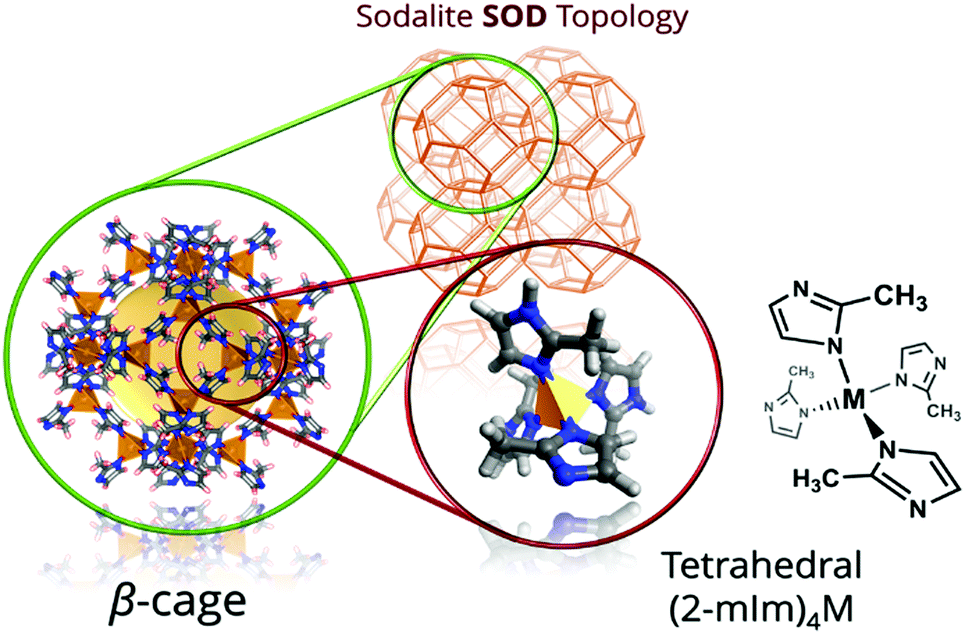 | ||
| Fig. 4 Structure of ZIF-8 sodalite (SOD) net. On the left, the sodalite-like β-cage (11.6 Å) with the yellow sphere representing the van der Waals surface within the pore, and with the ZnN4 tetrahedra highlighted on the right. | ||
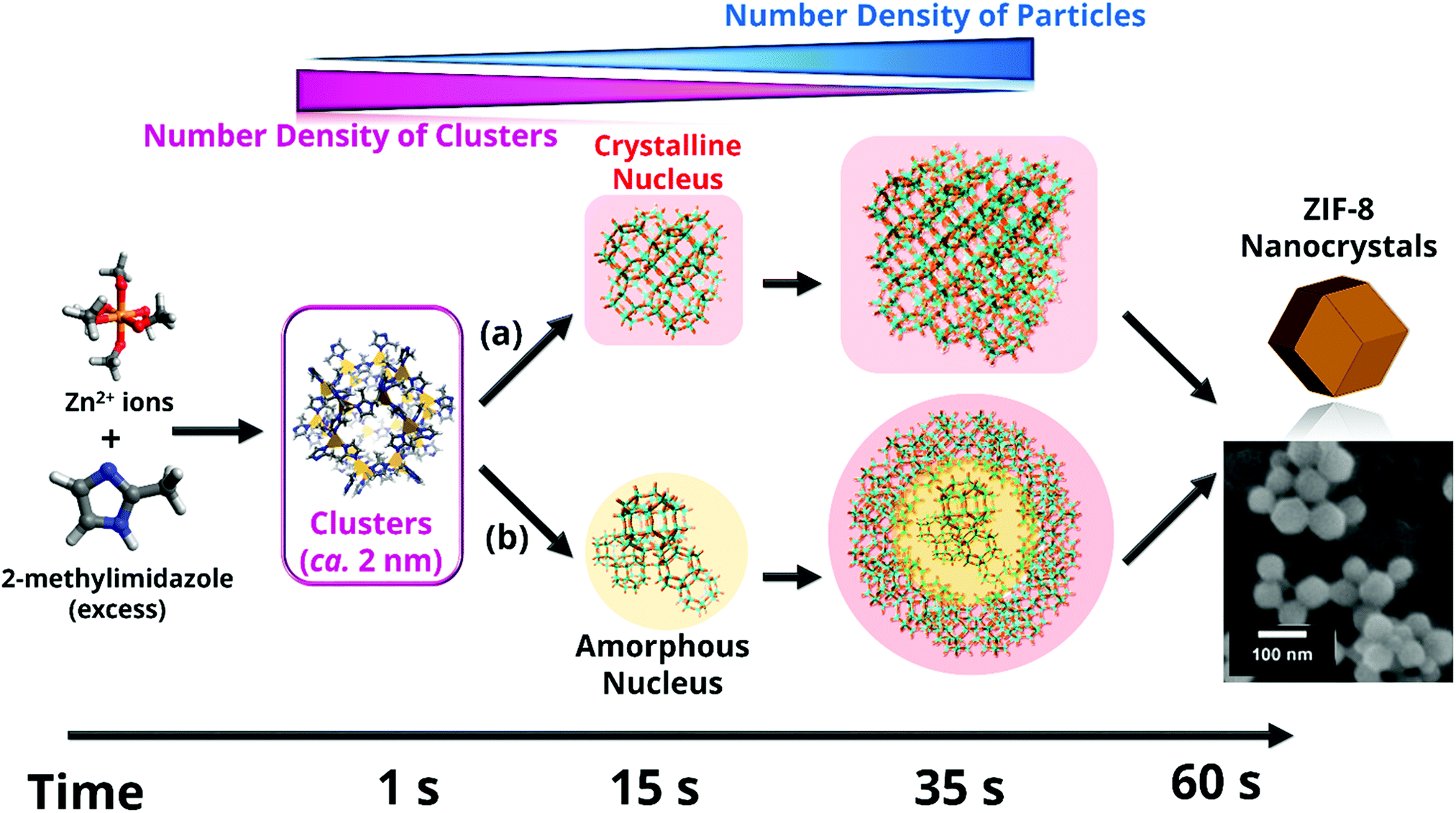 | ||
| Fig. 5 Different species formed during ZIF-8 nucleation and growth. Results point to the initial formation of very small clusters prior to the ZIF-8 nanoparticle formation, as well as to a continuous slow nucleation, which takes place simultaneously to the fast crystal growth over a period of time. Crystallinity of the first nuclei cannot be assessed given the lower sensitive of wide-angle X-ray scattering compared to small-angle X-ray scattering. Thus, two possible alternative crystallization pathways (a) and (b) are considered. Reprinted with permission from ref. 157. Copyright©2011 John Wiley & Sons, Inc. | ||
Several prominent examples have been already discussed within this review (MIL-100 and MIL-101 section) to illustrate the need of combining different techniques to unveil the MOF formation mechanism at all relevant length scales as well as the important role of in situ methods to avoid quenching, which may result in changes to the system. In this section, we aim to highlight the use of different microscopy techniques as powerful complementary analytical tools for studying MOF crystallization. Next to the use of TEM for ex situ measurements, Patterson et al.156 used liquid cell transmission electron microscopy (LCTEM) for the real-time monitoring of MOF synthesis for the first time. In particular, the authors studied the synthesis of ZIF-8, proving LCTEM is a suitable technique to gain insight into the MOF synthesis kinetics, the underlying MOF formation mechanism and the influence of the synthesis conditions on the particle size and morphology. From the LCTEM ZIF-8 synthesis direct observation (Fig. 6), it became evident that particle growth does not take place through particle coalescence, in line with previous SAXS/WAXS results by Cravillon et al.157
 | ||
| Fig. 6 (a–d) Micrographs acquired by liquid cell TEM at different synthesis times during the real-time monitoring of ZIF-8 formation, (e) image acquired after ZIF-8 growth, (f) micrograph of the same area after the cell was dried, (g) diffraction pattern obtained from the particles grown in the cell after drying and (h) mean growth kinetics of individual particles. Reprinted with permission from ref. 156. Copyright©2015 American Chemical Society. | ||
Furthermore, the growth exponents, calculated from the evolution of the particle size with time, point to the attachment of monomeric species or small cluster as the rate determining step and not the diffusion of both nutrients to the crystal nucleus. Similar conclusions were already drawn by in situ EDXRD studies,148 in which the calculated Avrami exponent n was close to 1, indicating the crystallization process is rate-limited by surface reaction. The similar results obtained by spectroscopic techniques and this LCTEM study proved the suitability of this technique for the direct observation of MOF synthesis, the electron beam having a negligible effect on the particle assembly and growth under the studied conditions.
At the molecular level, in situ atomic force microscopy (AFM) has been applied by Attfield et al.158 to study the surface crystal growth of ZIF-8 from essentially methanolic solutions. Fig. 7a shows the in situ AFM deflection images acquired for the (110) face of ZIF-8, which demonstrate that crystal growth takes place by both, spiral growth and “birth and spread” mechanisms simultaneously.158–160 Moreover, from cross-sectional analysis it was observed that most of the stable growth steps have heights of 1.2 ± 0.1 nm, indicating a strongly preferred surface termination. Even more interestingly, the real-time monitoring of a growing 2D surface nucleus (Fig. 19c) allowed for the identification of different metastable sub-steps with heights of 0.4, 0.6, 0.8, 0.9, 1.1 and 1.2 nm after 0, 2.9, 4.9, 12.8, 15.6 and 40 min, respectively.
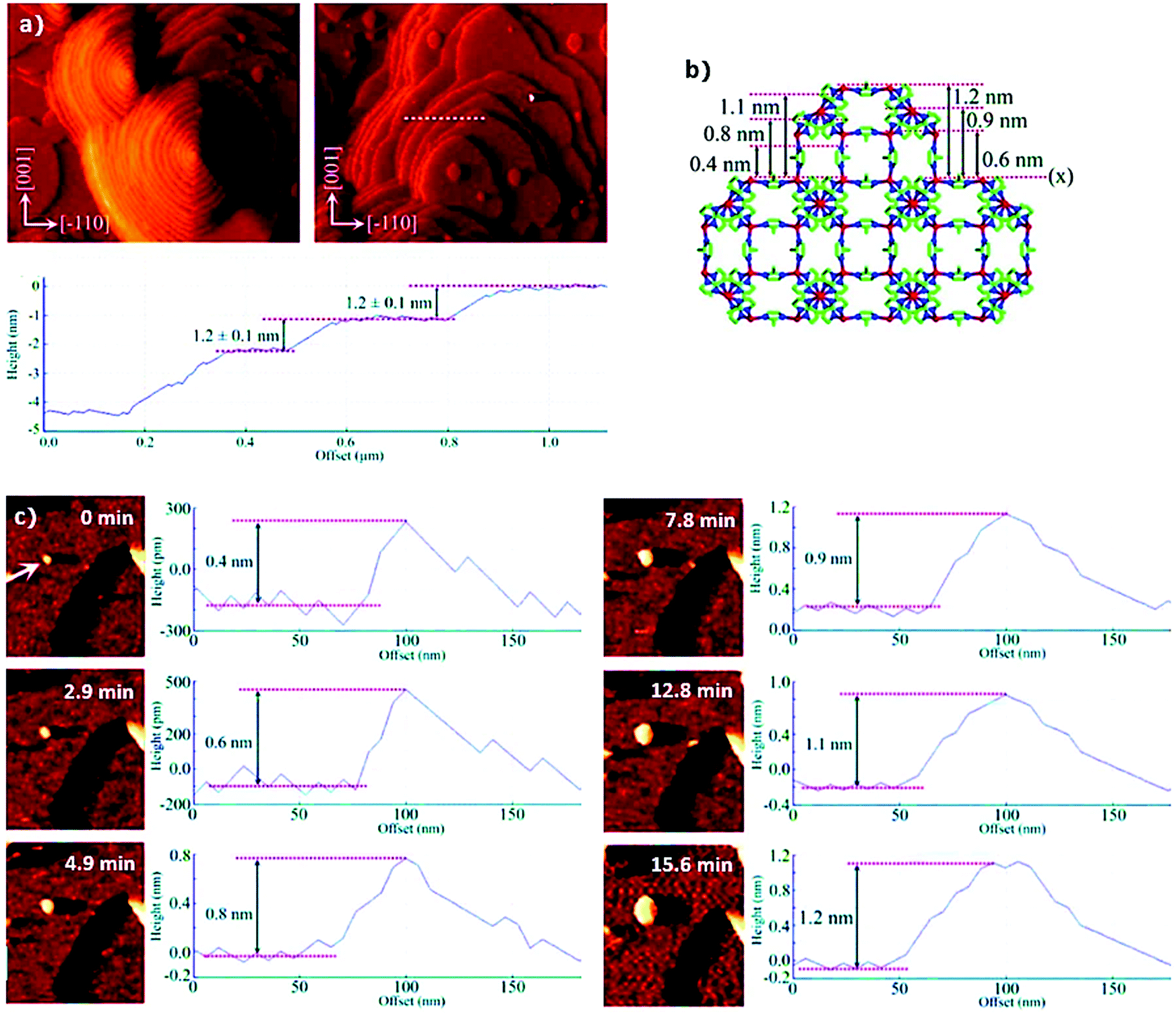 | ||
| Fig. 7 (a) In situ atomic force microscopy deflection images of the (110) face of ZIF-8 showing growth steps formed by spiral growth mechanism (left) and “birth and spread” mechanism (right) together with the cross-sectional analysis of some growth steps (dashed white line) revealing the step heights corresponding to the d110 crystal spacing of the material, (b) ZIF-8 structure viewed along the [100] direction and (c) real time AFM deflection images and cross-sectional analyses of developing growth step at 0, 2.9, 4.9, 7.8, 12.8 and 15.6 min after first observation of the MOF nucleus (highlighted with the white arrow). Reprinted with permission from ref. 158. Copyright©2011 American Chemical Society. | ||
The authors related these metastable substeps to the ZIF-8 crystal structure (Fig. 7b) obtaining further insight into the growth process. Specifically, the results point to crystal growth by addition of monomeric MeIm− and Zn2+ ion species, and not larger clusters or SBUs, until stable growth steps of 1.2 nm were formed. Similar AFM studies (both ex situ and in situ) have also been performed for other intensively studied MOFs, such as HKUST-1161–163 and MOF-5,164–166 proving the applicability of AFM to different MOF systems as well as its potential to study MOF crystal growth.
Neutron and X-ray diffraction can provide very valuable information regarding the location of the guests, yielding insight into the MOF preferential adsorption sites, one of the most important features when considering the interaction between guest molecules and MOFs.167 A recent, ground-breaking study by Hobday et al.168 showed that the arrangement of gases, such as CH4, O2, N2 or Ar, at very high pressures (up to 1.5–3 GPa) and their interactions with ZIF-8 can be studied by high-pressure crystallography combined with DFT and Grand-Canonical Monte-Carlo (GCMC) calculations. The interest of this study lies in the possibility of studying changes in the framework both from the structural and the energetic point of view. Nonetheless, in the case of molecules containing light elements, such as H, neutron diffraction is especially attractive and well-established in the community. The reason is the generally large neutron scattering cross-scattering of light elements, in contrast to the X-ray scattering cross-section which increases as a function of Z2 (where Z is the atomic number).
Many examples can be found in the field of MOFs where the adsorption of different guest molecules, such as CO2, CH4, H2 or NH3 have been studied by neutron powder diffraction (NPD).169–174 Newly developed techniques based on dielectric constant values have been recently used to study the interaction of sorbates with the framework.175 However, NPD has been the technique of choice for studying the location of gas molecules. In the particular case of ZIF-8, Wu et al.176 performed NPD studies for D2-loaded ZIF-8 and observed that the primary adsorption site for D2 was on top of the imidazolate moieties, specifically close to the CC bond (Fig. 8). Likewise, a later work by the same authors point to a similar behaviour for CD4, which was preferentially adsorbed on top of the linker with one D atom oriented towards the CC double bond.177 These results are in contrast with those commonly encountered for other MOFs, for which a preferentially adsorption on the metal nodes has often been reported. The preferential adsorption close to the organic linker further guides the possible optimization of ZIF-8 as sorbents for gas storage, where the modification of the imidazolate moieties rather than the metal sites might lead to an improved sorbent performance.
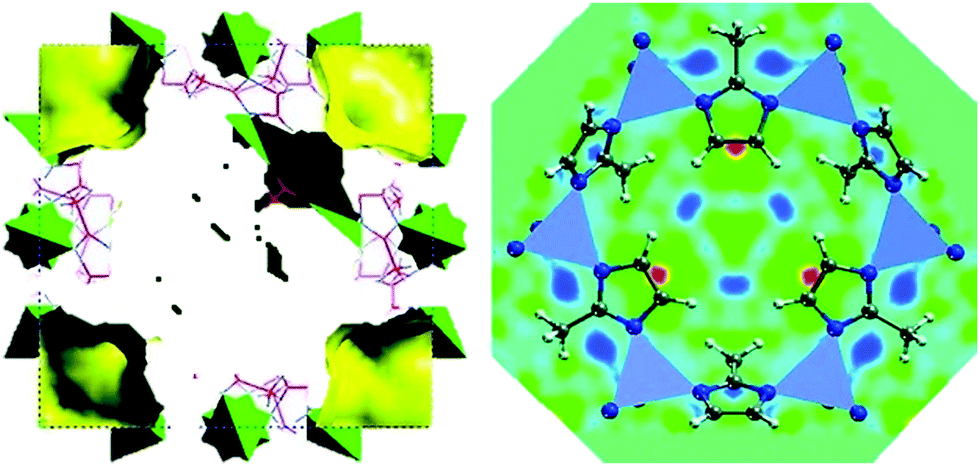 | ||
| Fig. 8 Left: (001) view of the refined crystal structure of ZIF-8 from neutron powder diffraction together with the available free space for H2 occupation. Right: (111) view of the real-space Fourier-difference scattering-length density superimposed with six-ring pore aperture of ZIF-8, indicating the location of the most favourable adsorption sites (red-yellow regions). Reprinted with permission of ref. 176. Copyright © American Chemical Society 2007. | ||
Interestingly, next to the location of preferential D2 and CD4 adsorption sites, Wu et al. observed a CD4-induced fully reversible ZIF-8 structural transition at 60 K triggered by the rearrangement of CD4 molecules at high adsorbate loadings. All in all, these works by Wu et al.170,176 illustrate the wealth of information that can be obtained by NPD, especially well-suited for hydrogen and other light elements. Monitoring the changes taking place in the atomic and molecular motions upon adsorption provides crucial insight into the nature and strength of the framework–guest interactions as well. Several spectroscopic techniques, such as infrared spectroscopy, have therefore been used to this end. Easun et al.167 recently highlighted in their review the suitability of inelastic neutron scattering (INS) to perform such a type of studies. INS provides valuable information about molecular and atomic motion modes, especially for light elements (vide supra). In the case of ZIF-8, however INS has been used to study the linker rotation upon N2 adsorption, rather than gaining insights into the adsorption sites.178 In particular, the free rotation of the methyl groups of the 2-methylimidazolate linker of ZIF-8 was observed to be hindered upon N2 adsorption. This was attributed to a change in the chemical environment of methyl groups upon swinging of the linkers, leading to a larger steric hindrance of their rotation. Furthermore, besides the location of the preferential adsorption sites and the determination of the nature and strength of the host–guest interactions, the diffusion rate should also be borne in mind when considering different applications of porous materials. Indeed, in many cases the performance of nanoporous compounds is controlled by their transport properties, playing a critical role when considering them as, for instance, catalysts or adsorbents. Hence, different techniques, a summary of which can be found in a review by Kärger et al.,179,180 have been used to study guest diffusivities as well as possible molecular transport resistances in porous materials. Among them, in this review we highlight quasi-elastic neutron scattering (QENS),181–198 pulsed field gradient (PFG)-NMR179,199–207 as well as interference208 and IR microscopy.179,208–221
QENS allows to determine the diffusion of guest molecules under equilibrium conditions on a timescale for which the guest molecules remain inside the MOF crystals, providing the intracrystalline self- and/or transport diffusivities190,222 for the incoherent and coherent neutron scattering signals, respectively. Pantatosaki et al.191 performed QENS experiments to calculate the self-diffusivity of H2 adsorbed on deuterated ZIF-8. The authors compared the experimentally obtained H2 self-diffusivities with those calculated with molecular dynamics computer simulations and observed a reasonable agreement between the experimental and simulation studies for ZIF-8, being the calculated self-diffusivity values very sensitive to the framework dynamics. In another study, Jobic et al.192 further studied the self-diffusion of CH4 by QENS in the same MOF, i.e. ZIF-8, and compared their results with those previously reported for different experimental techniques, namely PFG NMR203,206 and IR microscopy,206,211 and computational methods.223–225 Interestingly, the self-diffusivities obtained from QENS by Jobic et al.192 are in agreement, especially at low CH4 loadings, with those previously determined by PFG NMR and with the corrected diffusivities from IR microscopy. This contrasts with what has been often reported for other traditional porous materials, for which differences in the diffusivities measured with different techniques have often been encountered. These differences have been commonly attributed to the different diffusion lengths probed by the different experimental techniques together with the presence of structural defects. In this way, for example, the diffusivity values measured by QENS, for which the diffusion path covered is typically of tens of nanometres, are less affected by the possible presence of defects than in the case of PFG NMR, whose effective length scale is of several micrometres. The results reported by Jobic et al.192 point therefore to the presence of only few defects in the crystalline framework and/or to their negligible influence on CH4 diffusion. QENS and NMR have been combined in a series of recent studies in benzene motion within UiO-66(Zr) as well.226 Furthermore, a unique advantage of QENS compared to PFG NMR and interference and IR microscopy is that it also provides information of the geometry of the diffusional process by analysing the elastic component of the incoherent scattering function with different proposed models. In the particular case of the work reported by Jobic et al.,192 the spectra obtained suggest a restricted mobility of CH4 through the ZIF-8 hexagonal windows, with shorter residence times within the sodalite cages than the characteristic time for intercage hopping. Thus, despite the framework flexibility, which leads to an enlargement of the pore aperture allowing for the adsorption of CH4, the CH4 mobility is still restricted at the pore windows, being the CH4 diffusion in ZIF-8 smaller than those commonly reported for other porous materials. This ability of QENS to provide understanding into the diffusion mechanism and rate has been further exploited to shed light into the proton conductivity mechanism and proton diffusion coefficients. This is the case of the works recently reported by Pili et al.227 and Damasceno Borges et al.228 who used XRD together with QENS and molecular dynamics simulations to shed light into proton conduction on MOF.
PFG NMR provides the self-diffusivity of guest molecules adsorbed on porous materials, i.e. under equilibrium conditions, as it is the case for the incoherent contribution in QENS experiments. For PFG NMR, however, the measurement time scale allows for different diffusion path scenarios (within and outside the crystals, see review by Chmelik et al.),179 and only for sufficient large crystals, together with short observation times and low diffusivities, intracrystalline self-diffusivities can be obtained. PFG NMR has been used to determine the intracrystalline self-diffusivities of different guest molecules in several well-known MOFs,199,201,202,205,229 and in the particular case of ZIF-8, self-diffusivities of different hydrocarbons,204 small alcohols,200 CO2207 and CH4203,206 have been reported. A recent study combining XRD, Raman, FT-IR and diffuse reflectance UV-vis spectroscopies revealed that SH2 partially modified ZIF-8, although they were unable to describe the exact effect on the pores and surface.230 Very recently, Dutta et al. showed by means of PFG NMR that SH2 creates surface defects, but does not affect transport properties within the pores.231 This is in line with previous studies describing surface defects that do not alter significantly ZIF-8's internal structure.232–234 Moreover, PFG NMR, being nucleus specific, has the potential to simultaneously determine different self-diffusivities. In this sense, Chmelik et al.204 studied the diffusion of 1:1 ethane and ethane mixtures in ZIF-8 by 1H PFG MAS NMR, obtaining a diffusion selectivity towards ethene of ca. 6 at 283 K for ZIF-8 loaded with 4 or 8 molecules per cavity. Diffusion of the same mixture but including methane, and at a T = 273 K, was recently studied by a different group,235 showing again 6 molecules per cavity and a ethene:ethane diffusion selectivity of 5.8. This also proves the reliability and reproducibility of PFG NMR as a tool for studying gas diffusion within MOFs.
Finally, besides QENS and PFG-NMR, interference and IR microscopy have also been applied in the field of MOFs to study the mobility of adsorbed molecules. A recent study used time-resolved FT-IR spectroscopy upon dosing aromatic hydrocarbons i.e. benzene, toluene and xylene (BTX), in other frameworks to calculate Fick diffusion coefficients, although little information on pore location was obtained.236 For a comprehensive overview on the application of interference microscopy and IR micro-imaging to study the molecular diffusion on porous materials we refer the reader to the reviews by Kärger et al.179,216 In short, these techniques provide the transport diffusivity (i.e. under non-equilibrium conditions) calculated from the monitored evolution of the concentration profiles within the crystals, as well as the sticking probability219 and the surface permeability,208,210,218,220,221,237 the latter being inaccessible by the aforementioned experimental techniques (i.e. QENS and PFG NMR).216–218,220 While interference microscopy possesses a better spatial resolution, IR microscopy allows for the simultaneous monitoring of different species, being suitable for the study of individual transport diffusivities in multicomponent mixtures. Furthermore, this ability to track different species allows for the simultaneous study of labelled molecules (isotopes) and their respective self-diffusivity coefficients. Thus, IR microscopy provides both the transport diffusivity and the self-diffusivity depending on the conditions (non-equilibrium and equilibrium, respectively) under which the experiment is performed.215,218
2.3 Chemistry and characterization of HKUST-1 or Cu2BTC3
The topology known as HKUST-1 (Hong-Kong University of Science and Technology-1)238 or MOF-199239 has been one of the most studied MOF structures in the past 20 years. HKUST-1 is constructed of well-defined paddle-wheel secondary building units (SBU), which exhibit CUS for sorption and catalysis upon solvent removal. The framework contains four carboxylate-terminated linkers connected to dinuclear cationic clusters (Fig. 9) with either counter anions (X− or NO3−) or solvent molecules (H2O) on the axial position. The crystallographic structure relies on the tbo topology in space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. Other properties that promoted the study of HKUST-1 are their relative robustness (stable in air for several days or weeks)240 and the myriad of metals that can be introduced into the HKUST-1 nodes, including Zn,241 Mo,242 Ru,243 Ni,244 Cr245 or Fe.246 In addition, mixed oxidation states247 and radicals248 have been observed under specific conditions, rendering this MOF very interesting for applications involving redox chemistry. Recently, several groups have reported the on-purpose introduction of defective, non-coordinating linkers into the HKUST-1 structure.249 The metal sites exhibiting increased undercoordination show increased Lewis acidity,250 enhancing their potential for adsorptive and catalytic applications. As described below, spectroscopic tools have proven crucial for unravelling the physicochemical properties of these material and their potential applications.
m. Other properties that promoted the study of HKUST-1 are their relative robustness (stable in air for several days or weeks)240 and the myriad of metals that can be introduced into the HKUST-1 nodes, including Zn,241 Mo,242 Ru,243 Ni,244 Cr245 or Fe.246 In addition, mixed oxidation states247 and radicals248 have been observed under specific conditions, rendering this MOF very interesting for applications involving redox chemistry. Recently, several groups have reported the on-purpose introduction of defective, non-coordinating linkers into the HKUST-1 structure.249 The metal sites exhibiting increased undercoordination show increased Lewis acidity,250 enhancing their potential for adsorptive and catalytic applications. As described below, spectroscopic tools have proven crucial for unravelling the physicochemical properties of these material and their potential applications.
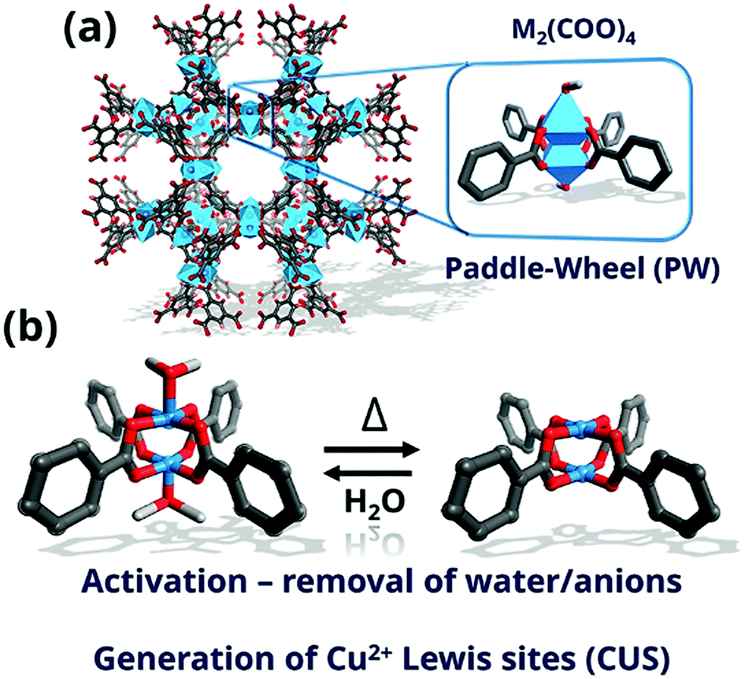 | ||
| Fig. 9 (a) Square-shaped pores of 0.9 nm × 0.9 nm dimensions of HKUST-1 formed viewed along the [100] axis; inset, highlight of the paddle-wheel unit (PW) with the metal atoms coordinated in octahedral geometry and; (b) scheme of the formation of coordinatively unsaturated sites (CUS) by desolvation by release of solvent molecules (C, grey; O, red; blue, metal). | ||
:1 mol Cu:NH3, room temperature) is adsorbed after water sorption on Cu sites.257 In the case of dry ammonia, however, the crystallinity is preserved, allowing the introduction of water even after evacuation of NH3 without amorphization or phase change. This suggests an important role of water in the degradation mechanism of the framework. DR UV-vis spectra of the dry samples (Fig. 10) with and without NH3 adsorbed showed a slight red shift of the Cu2+ d–d transition peak from 585 nm (activated) to 745 nm (+NH3), which is similar to the peak of the hydrated sample at 725 nm, as expected for these two σ-donors (H2O and NH3). Far-IR spectroscopy (Fig. 10) showed that dehydration blue-shifted all the bands corresponding to Cu–O and Cu–Cu, due to the increased donation of the carboxylate ligands. Introduction of NH3 restored the IR spectrum to that of the hydrated one. These spectral features indicate that NH3 is bonded to the open Cu2+ sites via the lone pair in a similar fashion to H2O molecules, thus, decreasing the Cu–O bond strength with the carboxylates due to increased electron density around the Cu centre. This was further corroborated by XANES which indicates an effective interaction of Cu centres with NH3 molecules derived from two pre-edge features that change when a 50 mbar equilibrium pressure of NH3 is introduced. Surprisingly, EXAFS fitting indicated a Cu–O carboxylate bond stretch of 0.025 Å and 0.065 Å for water and ammonia, respectively, compared to activated HKUST-1. This difference likely originates from a higher Lewis basicity of ammonia (pKa = 9.24) compared to water, decreasing the Cu–O bond strength as well as a degradation-free framework (XRD). It was thus possible to obtain a chemical and electronic description of the metal centres and the framework by this array of techniques (Fig. 10c). In general, no structural changes are observed upon adsorption and desorption of NH3 under dry conditions, while only hydrated HKUST-1 degrades upon NH3 dosing. Such structural disintegration of the MOF framework is likely initiated by metal-linker defect formation. In the case of HKUST-1, degradation is suggested to initiate by Cu–O carboxylate bond scission, which might result in Cu2+ to Cu+ reduction. In contrast to the work by Borfecchia et al. where no Cu2+ reduction was observed, Nijem et al.258 estimated that ca. 3.5 at% of the total Cu is Cu+ (which increased to ca. 10 at% with higher H2O pressure), being the reduction attributed to missing-linker defects. In their report, the degradation mechanism was investigated in more detail by studying a HKUST-1 thin-film by ambient pressure XPS and XANES under dry and humid NH3 adsorption conditions.
| Metal cation | Spectroscopic tools used for the study of: | Conditions (T and gas) | Ref. | ||
|---|---|---|---|---|---|
| Crystal lattice | Metal sites | Linker/gas molecules | |||
| Inelastic neutron scattering (INS), neutron diffraction (ND), thermal desorption spectroscopy (TDS), micro X-ray fluorescence (μ-XRF), pair distribution function (PDF), continuous wave electron paramagnetic resonance (cw-EPR), hyperfine sub-level correlation (HYSCORE), X-ray photoelectron spectroscopy (XPS), near-edge X-ray absorption fine structure (NEXAFS), ambient photoelectron spectroscopy (APS), neutron powder diffraction (NPD). | |||||
| Cu | XRD | EXAFS, XANES, UV-vis-NIR | Raman, IR | Thermal activation (CO and H2 probed IR at 77 K) | 254 |
| XRD, Raman, UV-vis-NIR, NMR | Raman | Raman | Chemical activation | 259 | |
| — | — | INS | CD4 (at 77 K) | 172 | |
| — | INS | CO2 (at 20 K) | 260 | ||
| ND | — | INS | H2 (at 4–5 K) | 189 and 261 | |
| ND | — | — | D2 (at 4–5 K) | 169 | |
| — | Low-temperature TDS | — | H2/D2 | 262 | |
| μ-XRF, synchrotron-based XRD, PDF | IR | I2 and H2O (at 348 K) | 263 | ||
| — | In situ IR | In situ IR | CO, CO2, NO, N2, H2 (20–77 K) | 264 | |
| XRD | XANES, UV-vis-NIR | IR | SH2 (298 K) | 230 | |
| — | XPS, NEXAFS | — | H2O, NO (at 298 K) | 265 | |
| XRD | XANES, EXAFS, UV-vis-NIR | cw EPR, HYSCORE EPR, IR | H2O, NH3 (298 K) | 257 | |
| — | Synchrotron XPS, NEXAFS, APS | — | H2O, NH3 (298 K) | 258 | |
| — | — | Solid-state NMR, IR | NO (298 K) | 266 and 267 | |
| — | — | ENDOR, HYSCORE, cw EPR | HD, D2, H2 and 13CO2/13CO | 268 and 269 | |
| — | — | Solid-state NMR | 13CO, CO2 (213–353 K) | 270 | |
| XRD | — | cw EPR and DFT | Ethene, 1-butene, ethane, and butane | 271 and 272 | |
| XRD | XPS, XANES | — | C3H6 | 273 | |
| Cr | XRD, NPD | UV-vis-NIR, XANES | In situ IR | Thermal activation and O2 sorption (variable temperature) | 256 |
| NPD | — | INS, in situ IR | H2, D2 (30–80 K) | 194 | |
| Ru | XRD | XPS | C16O/C18O-probed FT-IR | Activation | 274 |
| XRD | DRIFTS, XPS | DRIFTS | Activation and H2 (forms carbonyls and hydrides) | 275 | |
| XRD | UV/vis-, XPS | NMR, IR, Raman | — | 276 | |
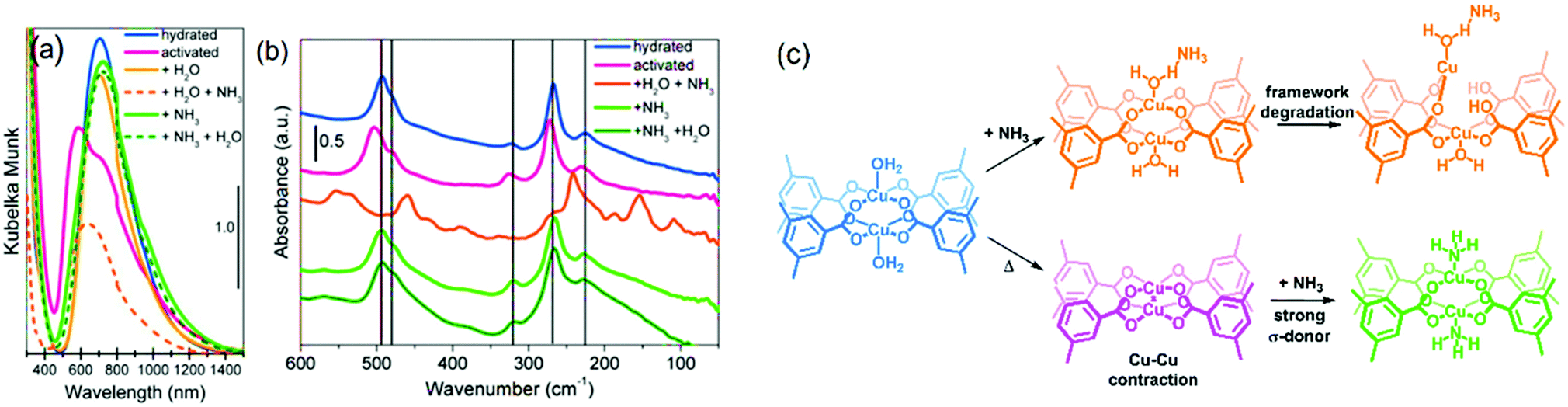 | ||
| Fig. 10 Spectroscopic analysis of HKUST-1 in hydrated and activated form, as well as upon introduction of ammonia of the hydrated and activated states. (a) Diffuse reflectance UV-vis spectroscopy and (b) far-IR spectroscopy under different conditions. (c) Different states of the Cu paddlewheel units under the different conditions reported. Adapted and modified from ref. 257. Copyright©American Chemical Society 2012. | ||
It is worth mentioning, different spots of the sample were measured to avoid beam-induced reduction of the sample. They observed the presence of the L3 Cu+ peak at ca. 935.3 eV in the XANES spectra (see Fig. 11a), indicating Cu+ in the material. Besides the mixed Cu oxidation states, experiments revealed a complex interplay between water and ammonia molecules in their coordination to Cu2+ clusters. Introduction of 0.26 Torr H2O to open Cu2+ sites results in a shoulder in the AP-XPS O 1s signal at 533.7 eV, indicating adsorbed Cu2+⋯OH2. Adding 0.03 Torr NH3 induces a shift of +0.6 eV, which is related to the formation of [O2H⋯H3N⋯Cu2+]. Therefore, NH3 replaces H2O at the Cu2+ centre due to its higher basicity.
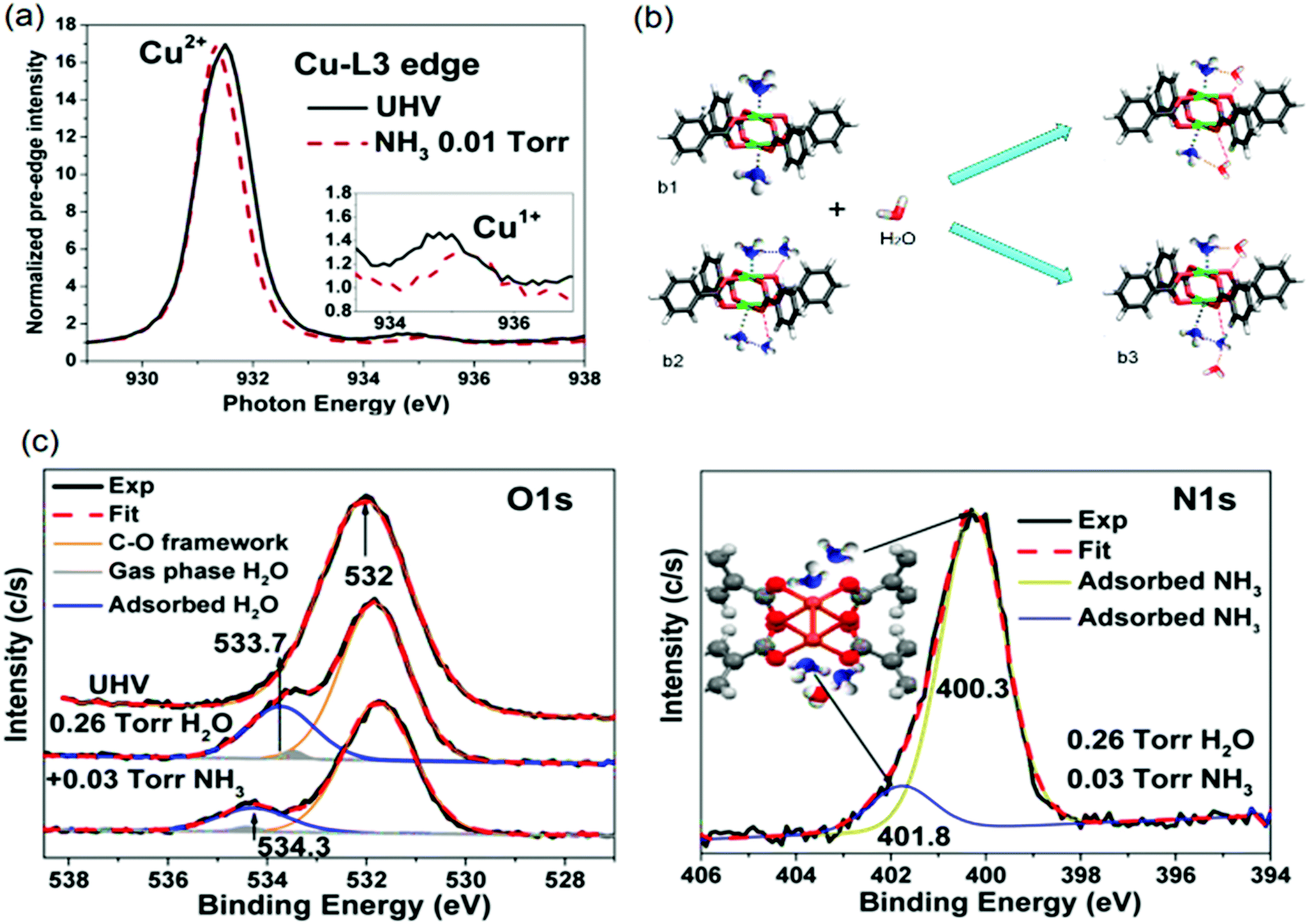 | ||
| Fig. 11 (a) X-ray absorption bands at the L3 Cu2+ edge (inset shows L3 Cu+) of the HKUST-1 film without and with 0.01 Torr of NH3 adsorbed. (b) Possible cooperative interactions between adsorbed water and ammonia molecules (C, grey; O, red; H, white; N, blue; and Cu, green). (c) O 1s and N 1s ambient pressure XPS peaks of the humid NH3 sorption experiments of HKUST-1 film. Adapted and reprinted from ref. 258. Copyright©American Chemical Society 2015. | ||
However, hydrogen bonding between water and ammonia strengthens metal–ammonia interactions due to cooperativity, resulting in a reduction of the metal oxidation state. Such Cu2+ reduction facilitates linker replacement by gas-phase (NH3–H2O) species, explaining the origin of structural degradation. The shoulder around 401.8 eV in the AP-XPS N 1s signal further corroborated the existence of such cooperative interactions, as described in the model in the inset of Fig. 10c. The structural configuration of [O2H⋯H3N(⋯H3N)⋯Cu2+] species (Fig. 11b3) involved in the degradation mechanism are displayed in Fig. 11b, established by H2O addition to NH3-preadsorbed paddlewheels at NH3:Cu 1:1 (Fig. 11b1) and 2:1 (Fig. 11b2) ratios. These in situ studies, and the characterization palette explored therein, showcase how complementary spectroscopic tools can reveal detailed molecular insight in metal–host interactions. Also, the influence of experimental conditions, such as pressure, the nature of gas phase species, to even the order of gas phase exposure, can be interrogated under in situ conditions.
In addition, the integral under the curves yielded a 49:51 ratio, indicating 1:1 ratio of each oxidation state. Based on these results, we expect possible fascinating chemistry to unveil for Fe-containing paddlewheels in MOFs. By careful design, Fe–Fe distances and coordination environments can be similar to those in enzymes, as shown for other topologies.277 More effort are being made to develop Fe containing HKUST-1 materials, mainly using the mentioned Mössbauer,278 as well as XAS and XRD tools for the purpose of studying these metal sites.279 Ru3+/Ru2+ mixed-valence states have been also observed in the HKUST-1(Ru) analogue, reported by Fischer et al. by XPS (Fig. 12a).243 Two different Ru species were identified in the mentioned study: Ru3+ and Ru2+, as confirmed by XANES analysis of both the precursors and the obtained materials.280 Similar materials were further investigated by CO-probe molecule FT-IR spectroscopy and the experimental data compared to DFT calculations.274
CO-probe FT-IR spectroscopy points to both Ru2+ and Ru3+ oxidation states since two features are observed both for labelled C16O (2171 and 2137 cm−1) and C18O (2120 and 2085 cm−1). However, DFT calculations cleared out that the presence of Cl− anions can lead to more complex scenarios in the interpretation of IR spectroscopy, for example, formation of chloroformyl and CO–Cl species, rather than purely CO on mixed-valence nodes. Partially reduced Ru paddlewheels were also observed in linker defect-engineered HKUST-1(Ru) prepared by Baiker's mixed-linker method (Fig. 12b).247,280,281 XPS of HKUST-1(Ru) with increasing amounts of low-coordinating defect linker Pydc (3,5-pyridine-dicarboxylic acid) show an emerging peak doublet at 280.0 and 284.2 eV. These peaks are characteristic for the formation of Ruδ+ besides Ru2+ (281.5 and 285.7 eV) and Ru3+ (282.5 and 286.7 eV). This peak appearance is accompanied by a decrease in the intensity of the Cl 2p peak (not shown), reinforcing their hypothesis given the proposed structural formulas in Fig. 12b. This report shows that, even in clusters containing metals with multiple oxidation states, the introduction of defect ligands can further reduce the oxidation state of the metal cations in the paddlewheel. Similar to Ru, partially reduced Cu+/Cu2+ has been observed in defective HKUST-1 by XPS and CO-probed FTIR.249 But in the last case, ambiguities exist on the cause of Cu2+ reduction being thermal activation or the inherent presence of cluster defects. A broader discussion has been established during the past years on the exact nature of Cu+ species in HKUST-1 in general, i.e. not only considering defective HKUST-1. Two possibilities are proposed: Cu+ originates from (1) extra-framework cations leading to Cu2O impurities from synthesis, (2) mixed valence Cu+/Cu2+ dimeric paddlewheels originating from (2a) defective clusters, or (2b) reversibly reducible/oxidizable Cu2+ atoms in perfectly coordinated paddlewheels. The presence of Cu+ was first reported by de Vos et al.282 by assigning the IR band at 2123 cm−1 to Cu+–CO, situated ca. 50 cm−1 below the typical Cu2+–CO band of low CO coverage at 2179 cm−1. Quantification of the Cu+/Cu2+ ratio was not possible, since the extinction coefficients for Cu+–CO is much higher compared to Cu2+–CO due to enhanced Cu+(σ) → CO(π*) back-bonding. Cu+ was presumed to originate from Cu2O agglomerates invisible to electron microscopy and/or CO-induced Cu2+ reduction into Cu+.
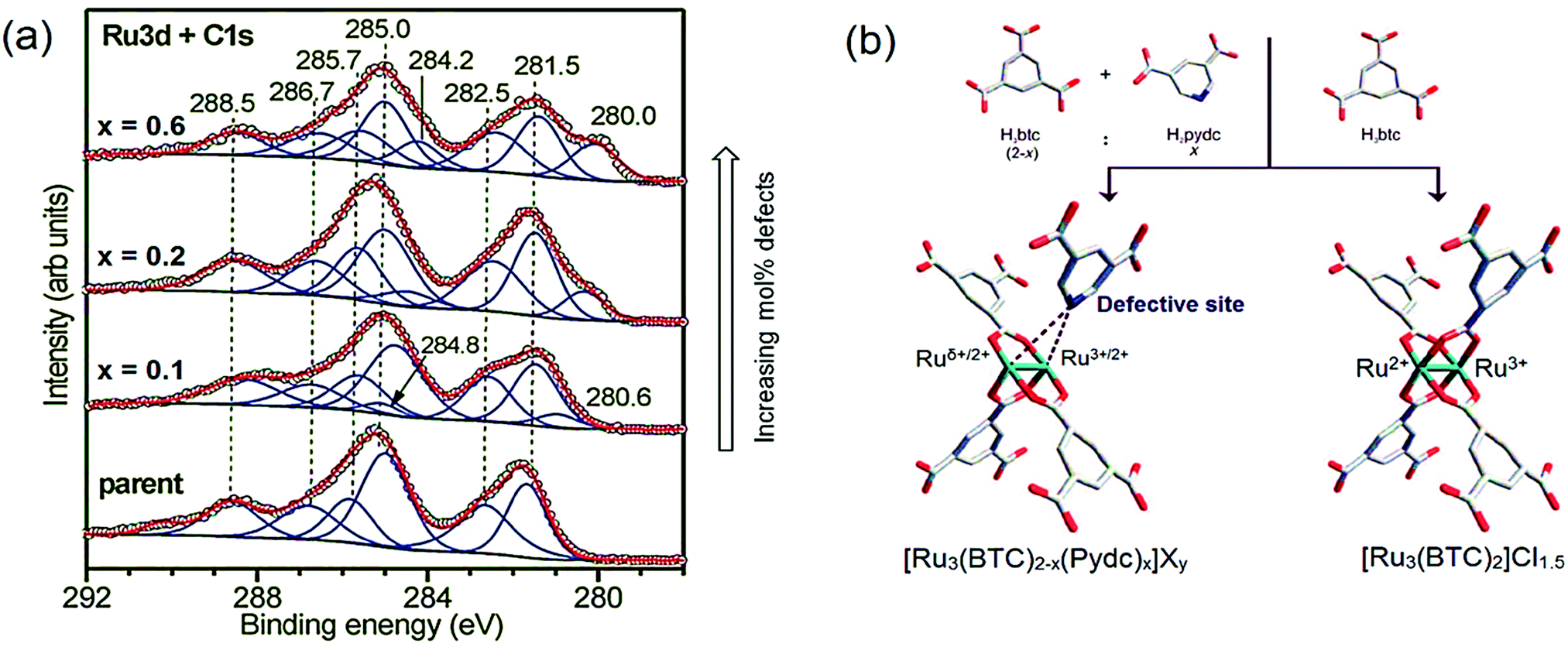 | ||
| Fig. 12 (a) X-ray photoelectron spectra of defect-engineered Ru3BTC2 with increasing defect molar content (x indicates the targeted fraction of Pydc, where Pydc = 3,5-pyrdinedicarboxylate) showing the peaks of Ruδ+ at 280.0 and 284.2 eV, Ru2+ at 281.5 and 285.7 eV and Ru3+ at 282.5 and 286.7 eV. (b) Model of parent and defective clusters including mixed-valence Ru3+/2+/δ+ species. Adapted and reprinted with permission from ref. 247 and 404. Copyright©2014, John Wiley & Sons, Inc. | ||
In a later study, Bordiga et al. observed similar CO-probed IR spectra, but rejected the CO-induced Cu2+ → Cu+ reduction mechanism proposed by de Vos et al. Instead, they ascribe the 2127 cm−1 band to minor fractions (≪1%) of Cu2O, possibly amorphous in nature as suggested by broadness of the Cu+–CO peak and its absence in XRD.264 This Cu2O phase is proposed to form during activation/heating, resulting in the desorption of solvent molecules (i.e. the aim of the treatment) which is accompanied by Cu2+ reduction into Cu+, as in Cu-exchanged zeolites. Wöll and co-workers283 later stated that they can rule out the hypothesis proposed by Bordiga et al. since their Cu3BTC2 film, grown at room temperature, is not heated yet it still showed the Cu2O peak of the 2p3/2 line in XPS (Fig. 13a) experiments and the band corresponding to Cu+–CO (Fig. 13b) adducts was present in the IR spectrum. Therefore, Wöll et al. propose Cu2O impurities are either: (i) contained in the Cu-acetate solution prior to synthesis, or (ii) formed by oxidation when exposing the MOF thin film to ambient air. Also, they confirm that the Cu2O fraction increases to as high as ca. 42 at% of all the Cu in the MOF (Fig. 13c) upon heating to 420 K. In short, XPS data showed increasing amounts of Cu2O upon heating the film. However, a fraction of Cu+ is already present in pristine Cu3BTC2, due to synthesis impurities or exposure to ambient air.
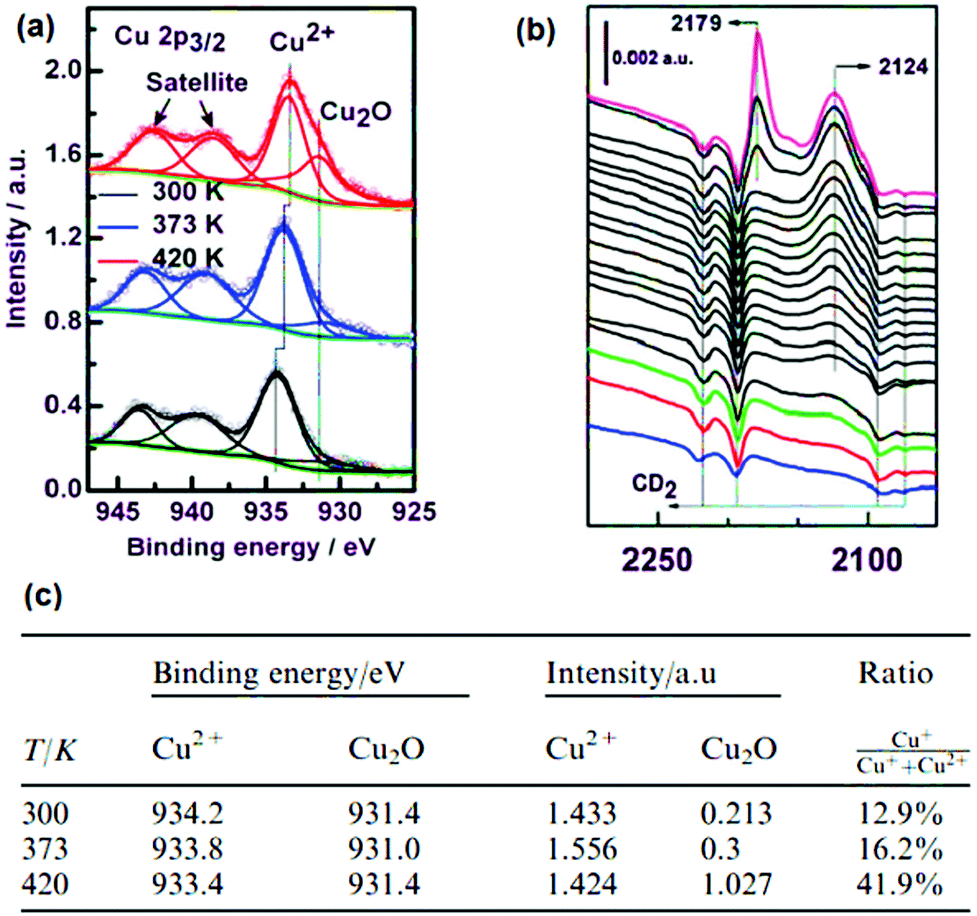 | ||
| Fig. 13 (a) XPS spectra of a HKUST-1/SAM thin film activated at different temperatures, (b) CO-probed FT-IR and (c) Cu2+/Cu+ ratios calculated from the XPS data. Adapted and reprinted from ref. 283. Copyright©Royal Society of Chemistry 2011. | ||
The studies by de Vos,282 Bordiga264 and Wöll et al.283 assumed the Cu+ signal contributions in XPS and IR mainly originate from Cu2O impurities. However, Szanyi, Daturi and co-workers284 claimed that the Cu+–CO band in their IR spectra cannot be attributed to Cu2O impurities alone, given its high relative intensity compared to the Cu2+–CO peak. Instead, they claimed to observe reversible redox properties of Basolite® C300 as a whole and, alternatively, the (Cu+/Cu2+)2(BTC)4 paddlewheel as representative redox unit. St. Petkov et al.285 confirmed the existence of mixed valence Cu+/Cu2+ dimeric paddlewheels. They elegantly combine XPS, XRD and IRRAS on model HKUST-1 surface mounted (SUR)MOFs with limited Cu2O impurities and high crystallinity on the one hand and DFT calculations on the other hand. St. Petkov et al. prove that defective (Cu+/Cu2+)3(BTC)2 units can cause the 2122 cm−1 CO-IR peak, the latter being observed even in model Cu3BTC2 SURMOFs were a relatively high concentration (ca. 5%) of the paddlewheels has missing linkers. Importantly, they show that the adsorption strength of CO on defective paddlewheels is much higher, showing their distinct chemical interaction and nature relative to non-defective paddlewheels.265 The concept of defective Cu-MOF thin films has been further extended to HKUST-1-type SURMOFs286 or paddlewheel Cu-MOFs.287 Not only synthesis conditions, thermal treatments, but also e− beams, X-rays and (long-term) exposure to ambient conditions can reduce Cu2+ to Cu+ in HKUST-1, for example by creating Cu-linker bond scission.288 Todaro et al. monitored the evolution of Cu paddlewheels over ambient exposure time by XRD, SEM, Raman and EPR.289,290 Exposure for more than 20 days results in hydrolysis of Cu–O bonds by destructive interactions with H2O that generate EPR-silent Cu+ sites and a decrease in the Raman Cu–O bond peak. This has been further corroborated by XAS,291 and surface studies combining infrared reflection absorption spectroscopy (IRRAS) and XPS.292 This dynamic character of the Cu+/2+ sites is still the subject of research and seems to strongly depend on many parameters.293,294 The presence of guest molecules, solvent, thermal treatment, presence of defects, incident beam and others, have an impact on the redox properties of PWs. This also holds true for other analogues, e.g. Ru, and strengthens the message that this material still deserves the attention is given.275
In brief, there is a long history of research on the underlying causes and nature of mixed valence metal nodes in HKUST-1, and MOFs in general, for which full agreement is not yet reached. However, opportunities lie ahead for tuning the oxidation states of these metal centres by targeted treatments which tailor their chemical properties, thus, gas sorption and catalysis abilities. As exemplified in this section, in situ spectroscopies are valuable tools to unveil the nature of redox-active clusters and can an undoubtedly will contribute to the development of more complex materials including mixed-valence metals.
3. Characterization of metalated nodes and grafted coordination complexes
One of the predicted key advantages of MOFs is the tunability of their function by hosting adsorptive or catalytic centres on their organic backbone and metal nodes.3 Among the different approaches reported to alter MOF properties, their post-synthetic modification is a very powerful method to tailor their functionality beyond the restrictions imposed by direct synthesis.295,296 This approach enables the introduction of functional groups that are unstable under the conditions for de novo synthesis, or the modification of specific sites in the framework that are otherwise impossible to be carefully tuned.In general, post-synthetic modification (PSM) has been developed in order to modify the structure in four different ways: (1) exchange of the metal cations from the framework, (2) introduction of organic functionalities into the organic ligand backbone, (3) metalation of the oxide-like cluster structure to yield supported single-metal atoms,238,297 (4) reacting metal salts with the added functionalities to create coordination complexes grafted to the organic struts.298 Given the importance of these metal sites in catalysis, gas sorption and sensing, and the outstanding examples that have been reported, a detailed analysis of the latter two approaches will be presented below. For a detailed insight on the former two, excellent literature is available on those topics.43,44,295,299–302
3.1 Metalation of the nodes
Some MOFs contain metal oxide nodes which exhibit OH groups after solvent removal. In 2009, Meilikhov et al.303 reported the first functionalization of metal oxide nodes, by reacting the μ2-OH bridging groups of two AlO6 octahedra in MIL-53(Al) with silylated ferrocene derivatives using gas phase loading, as confirmed by 29Si, 13C and 2H MAS NMR spectroscopy. Larabi et al.304 later used a similar approach to graft AuMe(PMe3) onto the nodes of UiO-67(Zr) and performed characterization using 31P, 13C, 1H MAS NMR and DRIFTS. More recently, studies reporting the introduction of metals bound to the O atoms of Zr6 clusters either via Solvothermal Deposition in a MOF (SIM) or Atomic Layer Deposition In MOFs (AIM) are listed in Table 4 for different topologies (UiO-66, UiO-67, NU-1000). Besides the techniques mentioned in this paragraph, Farha, Hupp and others have introduced XAS, EDX as well as more advanced spectroscopies to characterize the coordination geometry, oxidation state and distribution of metalated nodes. An interesting approach to understand the nature of metalated nodes was to use X-ray PDF, as reported by Kim et al.,305 after functionalizing the NU-1000 nodes with In3+ atoms by AIM. Differential PDF revealed two peaks at 2.12 and 3.33 Å corresponding to In–O nearest and In⋯Zr next-nearest neighbour distances, respectively. A broader feature at ∼4 Å reflects long range In⋯Zr neighbours within the Zr-node, exhibiting a higher degree of structural disorder. No In⋯In features were observed, suggesting that In atoms do not order relative to each other. However, the absence of In⋯In bonds could – for example – also imply that not all node sites available for metalation are occupied by In. Therefore, the application of complementary characterization methods to corroborate such finding is important to properly assess all aspects of metalation. For example, Li et al.306 deposited Co atoms on the nodes of NU-1000 both by SIM and AIM and provided a full description of the metal species with a combination of X-ray and vibrational spectroscopy. DRIFTS of the functionalized material revealed lower intensity of the OH vibrations (ca. 3800–3600 cm−1) after compared to before loading, suggesting Co2+ binding to the node-O atoms. A XANES pre-edge feature at 7709.2 eV (1s → 3d) on Co-metalated NU-1000 short after deposition indicated Co2+ species, further corroborated by XPS via a satellite 2p3/2 peak of CoO at 786 eV. Activation in O2 yielded Co3+ species as indicated by a small increase in the pre-edge feature together with a decrease in the white-line intensity, implying a mixture of Co2+ and Co3+ oxides is obtained (Fig. 14). A spinel structure was suggested from EXAFS analysis of the oxidized material which showed tetrahedral Co centres after loss of the OH groups and reduced coordination numbers of 0.8 for Co-SIM, 0.7 for Co-AIM and a decrease in Co–O neighbour distance to ∼1.96 Å. Two new peaks at ca. 2.4 and 3.1 Å matching with the Co3O4 structure, strengthened the hypothesis of such structure on the nodes. However, it should be clearly noted that drawing the latter conclusion based on the Fourier transformed EXAFS signal peaks may imply a certain risk given that signal interference in k-space can induce significant R-space artefacts which are not directly related to the physical presence of neighbours, thus leading to inconsistent interpretations.| Metal or metal complex | Topology | Spectroscopic characterization of PSM | Ref. |
|---|---|---|---|
| X-ray pair distribution function (XPDF), scanning electron microscopy energy dispersive X-ray spectroscopy (SEM-EDX), X-ray photoelectron spectroscopy (XPS), diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), difference envelope density (DED), scanning transmission electron microscopy (STEM), extended X-ray absorption fine structure (EXAFS), X-ray absorption near edge spectroscopy (XANES). | |||
| In3+ atoms | NU-1000(Zr) | XPDF, IR | 305 and 311 |
| Al3+, Zn2+ atoms | SEM-EDX, DRIFTS | 312 and 313 | |
| Co2+, Cu2+, Ni2+ atoms | 314 | ||
| Nb5+ atoms | DED | 315 | |
| CoSx | XPS, DRIFTS | 316 | |
| ZnOx species | Synchrotron XRD | 317 | |
| NiS2-like species | UV-vis, Raman, XPS | 318 | |
| Co2+–Al3+ sites | STEM-EDX, DED, DRIFTS | 319 | |
| Ir(CO)2 | UiO-66(Zr) and NU-1000(Zr) | 12CO/13CO-probe IR, EXAFS | 320 and 321 |
| Ni2+ atoms | NU-1000(Zr) | XANES, EXAFS, DRIFTS, SEM-EDX, XPS | 322 |
| V4+ and VOx species | UiO-66(Zr) | XPS, Raman, DRIFTS | 323 |
| Zr0-Benzyl sites | NU-1000(Hf) | XANES, DRIFTS, solid-state NMR, SEM-EDX | 324 and 325 |
| Ni2+(bpy)Cl2 | NU-1000(Zr) | SEM, DRIFTS, UV-vis | 326 |
| Ir3+(PtBu)Cl(res) pincer | Solid-state NMR, SEM-EDX | 327 | |
| Co2+ atoms/CoOx clusters | XANES, EXAFS, XPS, SEM-EDX, DRIFTS, DED | 328 | |
| γ-Al4 and γ-Al8 clusters | XANES, EXAFS, DED, synchrotron XRD, SEM-EDX, solid-state NMR | 329 | |
| Co–Cl, Fe–Br and Co–H | UiO-66/UiO-67(Zr) and UiO-type MOF | TEM-EDX, EXAFS, XANES, IR | 330 and 331 |
| Ir(COD)(phen) | UiO-67(Zr) | XRD, XPDF, DED, DRIFTS, XPS, XANES, EXAFS | 332 |
| Rh(C2H4)2(acac) | DRIFTS, EXAFS | 333 | |
| Al2O3 and Ir(C2H4)2(acac) | NU-1000(Zr) | FT-IR | 334 |
| Pt atoms/clusters | IR, DED, XRD, XPDF, XANES, EXAFS | 335 and 336 | |
| Cu–Oxo | XRD, XPS, DED | 337 | |
| Ni(acac)/Ni(Facac) | XANES, EXAFS, XPS, SEM-EDX | 338 | |
| Rh–Ga | STEM-EDX, DED, XANES, EXAFS | 339 | |
| Cr2+/3+ atoms | SEM-EDX, XPS, single-crystal XRD | 340 | |
| Ce4+/3+xOyHz | XPDF, DED, DRIFTS, XPS | 341 | |
| TiOx/NbOx | XRD, XPS, DRIFTS | 342 | |
| Organometallic complexes | Various | 309 | |
| Cu NPs | NU-901(Zr) and NU-907(Zr) | Synchrotron XRD, XPDF, DFT calculations | 343 |
 | ||
| Fig. 14 k 2-Weighted magnitude of the Fourier Transform of the X-ray absorption spectra of Co atoms metalated to Zr6 nodes of NU-1000 from the (a) liquid (SIM) and (b) gas (AIM) phase deposition, before and after activation in a O2 at 503 K for 4 h. (c) X-ray absorption spectra at the Co Kα edge of the different Co-NU-1000 as-synthesized and oxidized (inset shows pre-edge). (d) A proposed structural change of Co-AIM + NU-1000 upon activation. This tetranuclear cobalt cluster can be related to local structural features in spinel Co3O4. Difference envelope density of Co introduced via (e) liquid-phase and (f) gas-phase Co clusters deposited within the pores, in close vicinity of the nodes. Adapted and reprinted from ref. 306. Copyright©American Chemical Society 2017. | ||
Besides DRIFTS, XAS and XPS, a recently developed technique termed difference envelope density (DED), which can be derived from XRD data, was used.307,308 Subtraction of the electron density corresponding to the crystalline sections, i.e. the framework itself, from the electron density measured after introduction of Co generates 3D coarse maps such as the ones in Fig. 14e and f. Therein, the Co is found at ca. 8 Å along the c-axis. Computational support was given by calculating density functionals for different Co species and correlating them to the experimental data. This article provides an excellent example of the complexes approach necessary for describing this type of materials in a consistent manner, even more so when metal atoms have multiple stable oxidic structures. They have continued to extend the library of precursors and complexes that can be deposited from the gas-phase, truly making a widely applicable methodology out of this protocol.309
3.2 Grafted coordination complexes
As very recently highlighted by Drake et al.,310 MOFs offer a particularly suited scaffold for preparing homogeneous catalytic complex replicates anchored to their ligands. Several ligand-containing linkers have been used (e.g. phosphines, bipyridyl, β-diketimine, salicylaldehyde) along with different metals, for different applications. However, the most studied system has been the partially substituted UiO-67-bpy, where the 4,4′-bpdc (4,4′-biphenyl dicarboxylate) linker is partially replaced 5–10% bpydc (2,2′-bipyridine-5,5′-di-carboxylate). Platero-Prats et al. first reported a XAS-XRD study on the one-pot synthesis of homogeneous-like UiO-67-anchored Pd, Ir and Rh complexes.344,345 Øien et al. used (in situ) XANES and EXAFS to monitor the oxidation state and ligand exchange reactions in UiO-67-bpydcPt2+Cl2, respectively, during (i) high-temperature H2 reduction of Pt2+ to Pt0, (ii) oxidation to Pt4+ with liquid Br2 and (iii) ligand exchange with thiol-containing molecules (Fig. 15).346 Analysis of the coordination geometry and oxidation state of the ligated metal provided real-time insight in the nature of the resulting complexes. In addition, resonant-inelastic X-ray scattering (RIXS) on both UiO-67-bpydcPt2+Cl2 and the complex PtCl2(H2bpydc) allowed to probe the difference in electronic states when Pt is incorporated in the framework or not. Besides X-ray based characterization, CO-probe molecule IR was used to monitor the strong undercoordination of Pt0 sites via the detection of bpydcPt0(CO)2 dicarbonyl complexes as well as to exclude the formation of Pt–H hydride-type species. This study shows the potential of operando and in situ XAS for studying single-site metal-loaded (4,4′-bpy)UiO-67 or other MOFs and has been further extended for other processes: reduction of Pt2+ sites in either H2 or He to form Pt NPs,347 CO2 hydrogenation with Pt NPs,348operando hydrogenation of olefins with Pd349 and incorporation of CuCl2 and its redox-chemistry.350,351 Yet another example was reported by Van Velthoven et al. have reported this approach to graft S,O-containing ligands to the Zr6 nodes of MOF-808 then treated with Pd salts. Again, combining NMR to prove the successful grafting of the organic ligands, and HAADF-STEM together with XAS, they ruled out the formation of Pd NPs before and after catalysis. We can only stress how important is to use all these tools to rule out any extraneous species resulting in catalytic acivity.352 | ||
| Fig. 15 (4,4′-bpy)UiO-67 functionalized with Pt and the different treatments that were followed by X-ray absorption spectroscopy, as well as their observed reaction mechanism. Adapted and reprinted from ref. 346. Copyright® American Chemical Society 2015. | ||
Other complementary techniques such as UV-vis, as in the case of Cu, or XPS are useful tools for collecting corroborating evidence. Indeed, incorporation of CuBr2 in the same UiO-67 scaffold has been studied by Toyao et al.353 and the nature of the bonded (4,4′-bpy)UiO-67-CuBr2 species has been studied by UV-vis in addition to XAS. In the case of metal ripening that leads to the formation of NP@MOF composites, it is necessary to note the importance of in situ XAS for studying the evolution of metals under relevant conditions as well as TEM to disclose the formation of clusters.354 In summary, the use of complementary techniques is necessary to fully understand the nature and functioning of the post-synthetically introduced species on/in MOFs. On the one hand, when adding metals to the material, X-ray based (XAS, XRF, XPS, EM-EDX), electronic (UV-vis-NIR) and magnetic (EPR, Mößbauer) techniques are invaluable for assessing their properties. On the other hand, in the case of organic functionalities, solid state and liquid NMR, as well as FT-IR and Raman spectroscopy are more suitable for characterization. The possible non-periodicity of the post-synthesis modified functionality (see example of the non-ordered In3+ atoms in Kim et al.305 above), limits the power of the much more used single-crystal XRD. This suggests great opportunities for sensitive characterization tools that do not require long-range order, and potentially, micro-spectroscopic techniques too.
4. Metal–organic framework thin-films
So far, we have described spectroscopy and microscopy case studies using MOF powders and/or MOF single crystals. However, MOFs grown as films onto various substrates are increasingly relevant for applications such as sensors, opto-electronic devices or even catalytic membranes.283,355–361 As an added benefit, MOF thin films are highly interesting model systems for understand physicochemical processes in e.g. synthesis or catalysis. As opposed to MOF powders, thin films can be challenging to routinely characterize with conventional spectroscopic techniques, such as transmission IR spectroscopy or XRD, due to the low amount of material and high spatial resolution needed for thin film features. Therefore, other less commonly used and/or novel methods, such as IRRAS, AFM-IR or AFM combined with Tip-Enhanced Raman Spectroscopy (TERS) (vide supra), respectively, have been used.In the following paragraphs, we will present the use of these techniques through different examples published over the last few years. In particular, we will focus the discussion on surface-mounted metal–organic frameworks (SURMOFs), a subclass of thin films first described in 2007 by Shekhah et al.362 The authors showed the now widely used technique of SURMOF fabrication through liquid-phase epitaxy (LPE).264,363 This technique allows not only for a certain degree of control of the crystal growth orientation, but also for the deposition of metastable MOFs unable to form through conventional solvothermal synthesis, such as MOFs with larger pores without the presence of an additional interpenetrating lattice.364,365 Within this context, the selected examples showcase the relevance of novel tools to study SURMOFs as a system to understand physicochemical processes in MOF optimization.
4.1 Liquid-phase epitaxy synthesis
Several extensive reviews on layer-by-layer (LbL) synthesis of SURMOFs have been published.363,366–368 However, with the main focus on synthesis, an overall view on the use of spectroscopy to characterize SURMOFs is still missing, and will be precisely the focus of the following sub-sections. Using Cu2BTC3 SURMOF, Shekhah et al. showed the LbL synthesis of highly crystalline, homogeneous thin films, as well as the directing effect of self-assembled monolayers (SAMs).369,370 When using carboxylic acid terminated thiols, mimicking the functionality of the BTC linkers, the XRD diffractogram merely showed reflections for the [100] direction. Alternatively, a pure [111] orientation was achieved by using hydroxyl terminated SAMs, resembling the axial position in the Cu paddlewheel. In both cases, a smooth (crystalline) structure was confirmed by electron microscopy.However, in contrast to the seemingly defect-free crystal structure of Cu3BTC2 SURMOF showed by electron microscopy, several reports point towards the presence of defects with spectroscopy. Gu et al. synthesized HKUST-1 on quartz sheets using a LbL with and without ultrasonication step. They showed that the ultrasonication step lead to lower surface roughness and used UV-vis spectroscopy to point out that these SURMOFs have lower amounts of defects, together with their more transparent colour. The IR spectroscopy results published by St. Petkov et al.285 also highlight the presence of defects. The authors found a second absorption band in CO-adsorbed IR spectra, next to the typical band of CO adsorbed in the apical positions of the paddlewheel (2179 cm−1). This smaller second band at 2121 cm−1 points to the presence of CO adsorbed on a minority of Cu+ species of non-coordinated metal ions.285 This result was further confirmed by XPS, which corroborated the presence of up to 8% of Cu-coordination defects. CO adsorption on thin films was also studied by Mandemaker et al. using IR spectroscopy to directly compare thin films, made using the LbL synthesis, to bulk HKUST-1 crystals.371 To this means, calcium fluoride windows were functionalized with the proper SAM, which turned out to be trifluoromethyl-terminated benzene carboxylic acid or alcohol, after an essential UV-ozone treatment of the CaF2 surface. Both wafers of bulk HKUST-1 and the CaF2-mounted SURMOF were measured using FT-IR while CO or NO were dosed. Resulting from this, it was concluded that the MOF film contained 1.9 times less Cu+ species than the commercially available powder. Furthermore, the low amount of Cu+ species present was in paddlewheel conformation. To gain further insight into the nature and origin of these defects formed in the LbL synthesis, Delen et al.372 used both far-field (IRRAS) and near-field infrared spectroscopy (AFM-IR). Specifically, the authors measured IRRAS spectra during-, and post-synthesis, using commonly used Cu3BTC2 SURMOF synthesis protocols. An IR band at 1675 cm−1 was found to belong to the copper(II) acetate precursor, rather than the previously assumed fully coordinated MOF material. Nano-spectroscopic AFM-IR, combining the high spatial resolution and sensitivity of an AFM-tip as a detector while the underlying sample surface is irradiated using IR-lasers, was then further able to pinpoint individual vibrations corresponding to varying degrees of Cu-paddlewheel substitution.
This insight into the HKUST-1 synthesized allowed the authors for a larger control on the SURMOF synthesis. Particularly, the authors found that by tuning certain synthesis parameters, they could largely influence crystallinity and defect concentrations. The relevance of nano-spectroscopy techniques study these defects, such as AFM-IR, was also highlighted by Mandemaker et al.373 Using the high-spatial resolution of AFM-IR (Fig. 16a), the authors observed the desorption of the self-assembled monolayer (SAM) layer upon HKUST-1 synthesis at elevated temperatures (Fig. 16a). The strong band at 1700–1800 cm−1, attributed to the SAM carboxylates CO stretching vibration, was non-present for the areas indicated in blue (Fig. 16b, blue regions and blue spectra). This explained a quenching of nucleation observed by using in situ liquid-phase AFM during this synthesis at 323 K. Furthermore, the authors showed that a layer was formed as a ‘carpet’ surrounding the bigger crystals, debatably not as crystalline but as defect-rich Cu3BTC2.
 | ||
| Fig. 16 (a) 3D representation of an (b) atom force microscopy micrograph and the corresponding infrared spectra (right), labelled by numbers and sorted on morphological feature (red = grain, green = circle/patch, blue = in-between patches), highlighting the removal of self-assembled monolayer (CO stretch) in the blue areas. Scale bar in (b) represents 1 μm. Reproduced from ref. 373 with permission from John Wiley & Sons, copyright 2017. Note that the 3D representation was originally not published in the work, but directly obtained from the data represented in (b). (c) 3D representation of an AFM micrograph showing height segmentation, which are used as (d) regions to average Raman spectra over the given region to give chemical information using spectroscopic fingerprints correlated to topography. Colour code of the Raman relates to the colour code of each region of the map on the left. Reproduced from ref. 374 with permission from John Wiley & Sons, copyright 2019. | ||
In another recent instance, micro-spectroscopy was utilized to understand the topology-defect relation in SURZIF-8(Zn) thin films. Combining AFM, Raman, post-processing of the data using PCA and DFT to further identify chemical fingerprints, Weckhuysen et al. pinpointed the inter-grown nature of the SURZIFs (Fig. 16c). Through different spectral regions they were able to quantify the defect-richness: a sharp band at 284 cm−1, high 675/686 cm−1 ratio, low intensity for the 1180 cm−1 band (representing Zn2+ with a single linker), a low 1135/1144 cm−1 band ratio and a low 1458/1498 cm−1 band ratio (quantifying the amount of free linkers) is representative for a defect-poor material.
Using these fingerprints, the work revealed the existence of phase boundaries within 20-cycles thin-films, while applying more layers (50-cycles) yielded more (chemically) homogeneous films.374 Not only for the case in which the MOF grows directly on the substrate, but also for mixed nucleation and growth processes AFM can be used to determine film morphology. For instance, Öztürk et al. studied the influence of different parameters, such as reagent ratio or temperature, on the crystallization of ZIF-8 and ZIF-67 deposited on Au substrates.375 Moreover, film quality (i.e. density and packing onto the substrate), can be carefully tuned by choosing the appropriate number of deposition steps (Fig. 17). This has been taken to the next level by Mandemaker et al., who was able to tune the morphology showing a direct impact on the catalytic properties of Cu-BTC nanowebs.376 By tuning the deposition rate by spin-coating, the aspect-ratio and crystalline phase of the so-called nanowebs was modified, and thus, the amount of Cu+ and Brønsted sites on surface. This work further corroborated the necessity of using nanoscale tools to study film morphology, coupled with vibrational spectroscopy that provides chemical information on the materials. This, in term, allows for an extremely detailed characterization of the films.
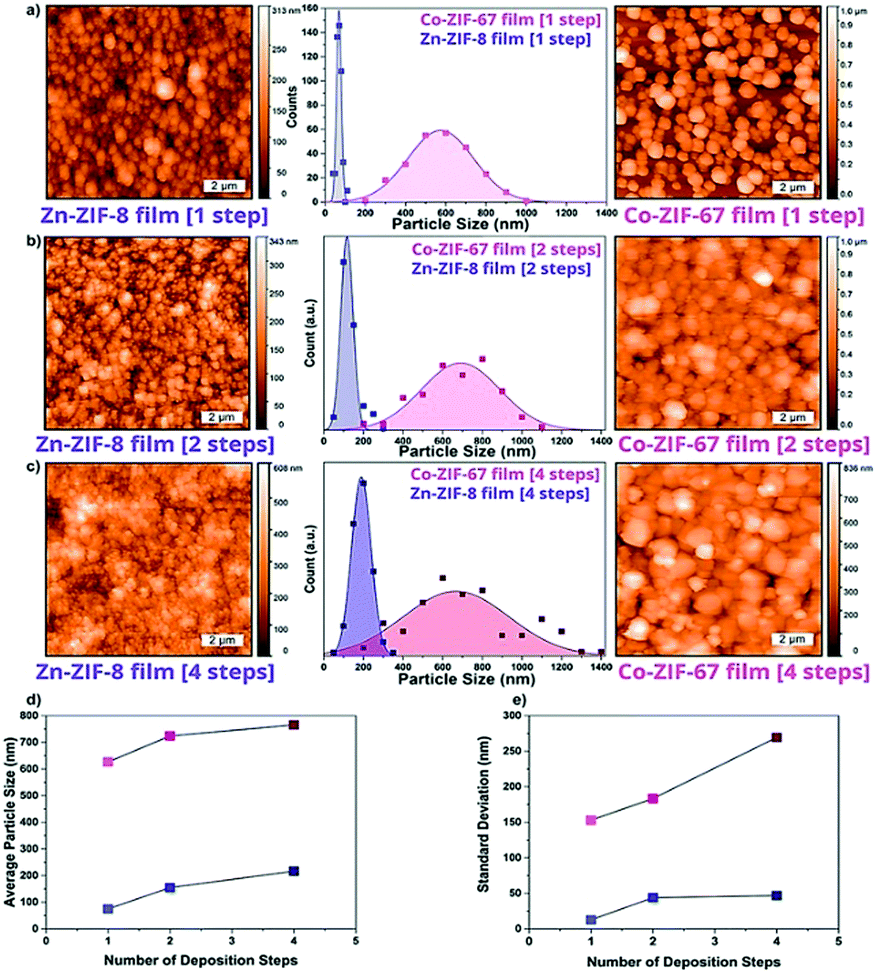 | ||
| Fig. 17 AFM topographical maps and particle size distributions of ZIF-8(Zn) (purple) and ZIF-67(Co) (pink) films prepared by direct synthesis of (a) 1 step, (b) 2 steps and (c) 4 steps. All the scalebars represent 2 μm. The variation of number of particles per mm2 and average particle size through multiple deposition steps (d), and (e) standard deviation of the given particle size distributions (metal/linker: 1 : 6.6, 20 °C, each deposition step with a time of 10 min). Adapted and reprinted with permission from ref. 375. Copyright 2017 John Wiley & Sons, Inc. | ||
4.2 Heteroepitaxial SURMOF hybrids
The fabrication of core–shell type MOFs opens the gateway to more advanced functional materials for different applications, such as selective adsorption of gaseous compounds or tandem catalysis, in the electrochemical reduction of CO2, for example.377–380 To develop such heterogeneous systems, SURMOFs provide an ideal model system, as the vertical composition can be easily altered during synthesis. The successful creation of a hetero-MOF crucially depends on the lattice matching of the stacking compounds. Studies on isoreticular SURMOF hybrids with varying degrees of strain have been published. Examples are top-layers with different metal ions, additional linker functionalization (e.g. amine groups), and varied linker length.378,380–382 For all SURMOFs it was shown with grazing incidence (GI-)XRD that the orienting effects exerted by the SAMs traverse to the shell layer(s). More specifically, Wang et al. highlighted the versatility of this approach in a trilayer SURMOF-2 thin film.382 In a hierarchal fashion, they grew a SURMOF with linkers of 1.12, 1.34, and 1.55 nm. Out-of-plane XRD showed that despite the large lattice mismatches of up to 20%, highly crystalline and oriented Cu-BPDC/Cu-NDC/Cu-BDC (where BPDC = 4,4′-biphenyl dicarboxylate and; NDC = 1,4-naphthalene dicarboxylate) SURMOF was made. The stratified arrangement was corroborated by SEM of the films stained with an Eu contrast agent, fitted only to the larger pores. Quantum chemical calculations on this system explained this unusual phenomenon in which the lattice strained is distributed onto the whole multi-linker film. The energy cost for this effect is lower than that of creating a defect site (i.e. dangling bond). In summary, through a combination of GI-XRD, SEM and theory, the authors could explain the complexity of defect formation in films. Compared to zeolites, MOFs are often reported to possess low (hydro-)thermal stability, making them a less obvious choice for industrial catalysis. However, apart from a number of XRD studies on bulk MOF at ambient temperature and pressure,383 few details on the stability of SURMOFs under elevated temperature and pressure are known. To this end, Brand et al. developed an in situ AFM utilizing a high temperature/pressure autoclave cell and combined it with ex situ IRRAS analysis.384 They used SURMOF substrates with individual grains of HKUST-1 and reported a limited thermal stability of HKUST-1 (up to 333 K), but a high mechanical stability against high pressures (up to at least 20 bar). This was concluded from both the disappearance of grains in the AFM micrographs and a decrease in HKUST-1 band intensities in the IR spectra when the sample was put at 333 K. Furthermore, they showed the morphological changes to be mostly reversible.Although not specifically mentioned in this work yet, as the focus is on spectroscopic, microscopic and diffraction-based techniques, it should be noted that for the characterization of SURMOFs the application of the quartz-crystal microbalance (QCM) has shown to be a strong toolbox. Given examples are found in the work from Stavila et al., that applied QCM to study growth kinetics, Heinke et al., which used QCM to measure diffusion coefficients and Wannapaiboon et al. which studied the loading of a SURMOF with guests.
5. Characterization of catalytic processes
Owing to the compositional flexibility of MOFs, meticulously selected metal and linker types can yield tailored functionalities and pore space topologies after their de novo synthesis. In addition, MOFs can be employed as scaffold (or equivalently catalyst support) for anchoring catalytically active sites by post-synthesis modification.53,385 These active sites range from (i) metal atoms or NPs on MOF nodes, mimicking metal atoms or NP on metal oxide supports, to (ii) metals bound to functionalized linkers, resembling anchored homogeneous catalyst complexes, and (iii) node-linker units functioning as repetitively arrayed synthetic enzyme biocatalysts. In this respect, MOFs can expose a broader class of functionalities to anchor groups and their pores more easily tuned than in the case of zeolites. Therefore, MOFs are endowed with great potential to provide a diverse platform for catalytic model studies.5.1 In situ and operando spectroscopy of metal–organic frameworks
Diffraction techniques, and obviously, single-crystal diffraction, are well suited when large crystals of a specific topology can be grown. Since MOFs offer the advantage of having repetitively ordered unit cells with well-defined, isolated sites, its structural periodicity allows for real space reconstruction of the lattice with high precision.52,385,386 MOFs can thus serve as model systems for real catalysts, such as single-site or single-atom heterogeneous catalysts or homogeneous catalyst complexes, by synthetically anchoring them in these periodic structures. As an example, Burgun et al. have reported the possibility of studying rhodium catalysed acetaldehyde synthesis with single-crystal XRD.387 A Mn(II)-based MOF with pyrazolyl moieties embedded in the linker was post-metalated with [Rh(CO)2][Rh(CO)2Cl2] to create the environment of classical hydroformylation catalysts. After installing this complex, the MOF crystals were treated with methyl bromide and were subsequently CO infused (Fig. 18). Single-crystal XRD of the material was obtained for each step, allowing the authors to obtain a definitive snapshot of each step. Although the mechanism of this reaction is long known, the interest lies in locating every atom during the catalytic cycle, which may be of interest for other reactions (e.g. chiral catalysts). This work shows the power of single-crystal XRD, in a quasi in situ approach, to unravel the catalytic reaction cycle in MOF model systems. This has been exploited in the past years, the potential of this strategy being huge as reviewed by Doonan and Sumby.388–390 However, whilst diffraction is heavily used for MOF characterization and spectroscopies become more popular, MOFs in the form of nanocrystalline powders or with heterogeneous compositions are typically used for real application studies. This question has been raised by some, and in fact some structures are not available yet as single-crystals, e.g. MIL-53(Cr) or MIL-100(Cr), and are extremely difficult, if possible, at all, to be obtained as single-crystals.391 This limits the use of single-crystal XRD for model studies, indicating the need of other bulk techniques. Thus, besides, techniques under operation conditions, i.e. in situ or operando, need to be used for the study of catalytic or gas sorption processes.392 X-ray spectroscopies, such as XAS, XES, XPS, EDX, electronic spectroscopies, such as UV-vis, and magnetic techniques, such as EPR, Mößbauer are highly sensitive to most metals with which MOFs are formed. Additionally, Raman and (probe-molecule) IR spectroscopy can provide insight into the nature of the metal phase. Notable examples of this strategy for the separation of propylene and propane with MIL-100(Fe) have been detailed in Section 2.2.2, and more recently, the speculative formation of high spin FeIVO species upon oxidation of MIL-100(Fe) sites with N2O, as suggested by Simons et al. by means of extensive in situ XAS, Mössbauer spectroscopy and DFT simulations.393 On the other hand, NMR, Raman and IR spectroscopy can characterize the linker properties and how they evolved during operation. It is then necessary for the MOF community to stir their studies towards the use of this set of techniques in operando mode.
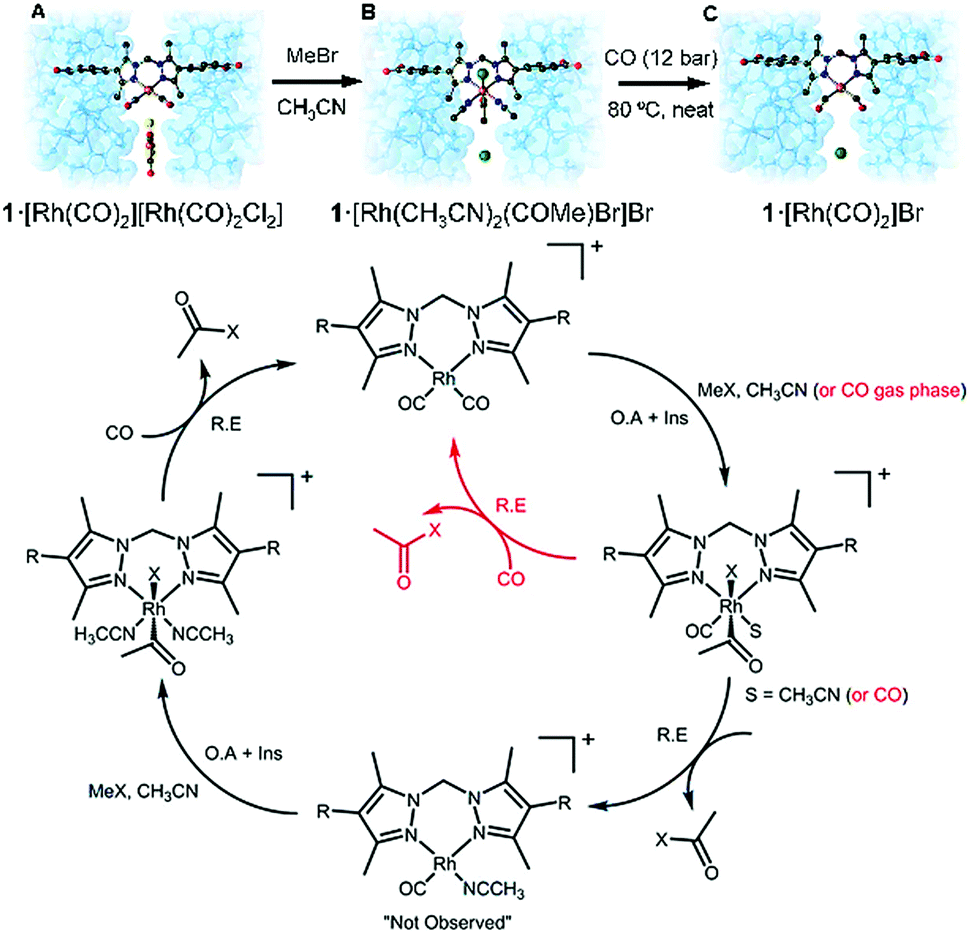 | ||
| Fig. 18 Single-crystal XRD representations of MOF 1, after (A) installing the Rh complex, (B) treatment with methyl bromide and (C) carbonylation of the methyl group releasing Br− anions. The MOF backbone is shown in blue with a transparent van der Waals surface; C black; N dark blue; O red; Rh orange, Cl yellow; Br green. The catalytic cycle shown below the crystal structures corresponds to the different steps observed by SCXRD. Adapted and reprinted with permission from ref. 387. Copyright © John Wiley & Sons, Inc. 2017. | ||
Concerning the use of MOFs as model systems (or performance materials), the weakness is still their stability under relevant conditions, namely high-temperature, pressure or extreme pH. Thus, as recently highlighted by Gates et al.,53 MOF catalysts should be directed to appropriate reactions that require mild conditions. These cases will require (already do) very advanced and sensitive techniques, owing to the fact that they may appear only under dynamic conditions and/or in very small detectable concentrations.39,394 Moreover, apparently simple processes, such as pore degassing, may reveal much more complex scenarios than initially hypothesized, as recently demonstrated by Dodson et al.395 Another possible pitfall resides in the sensitivity of spectroscopic techniques to the detection of reaction intermediates within catalytic reaction cycles. More particularly, the strong spectroscopic fingerprint of the MOF framework itself could dominate the spectroscopic contribution of the investigated reaction intermediate of catalytic MOFs. Finally, stability under the measuring beam, being electrons, neutrons, X-rays or even light, could potentially perturb or degrade the framework structure and measured properties. To prevent artefacts and false conclusions, degradation testing should be performed prior to experimentation. Despite these potential drawbacks, we foresee that MOFs will be used as model catalysts in the upcoming years. Researchers in the field need to approach most of aspects with a wide range of characterization techniques (holistic approach) to obtain a good understanding of MOF materials. Several other species that may appear during synthesis, activation or operation (e.g. lattice interpenetration, unreacted bonded molecules, defect linkers, amorphous regions or changes in the structure) must be considered.
6. Challenges and opportunities in spectroscopy of metal–organic frameworks
Besides the physicochemical phenomena discussed above, a vast number of other features (e.g. photon absorption and non-linear optics,396,397 drug release,398 photocatalytic processes,399 heat absorption/release,400 electro-conductivity401–403 or chemical sensing23,404) that lay beyond the scope of our review article may become grand challenges for characterization, let it be spectroscopy, scattering or diffraction. All these will require the use of ad hoc developed techniques, both bulk and spatially resolved.6.1 Chemical imaging and spatially resolved studies of bulk MOFs
Spatially resolved characterization of different features, e.g. chemical composition, at the nanoscale is and will be one of the major challenges in MOF chemistry. Given the heterogeneous composition in terms of elements, oxidation states, geometric configurations, etc. present in such complex materials, very advanced techniques are necessary to tackle the lack of insight.For instance, the problem of metallic spatial has not been yet thoroughly studied. Some works that make use of bulk (e.g. powder diffraction) techniques show indirect proof of metal mixing in some topologies other than MOF-5.405–411 For example, very recently, solid-solutions in the x = 0 to 1 compositional range of Zn1−xCdx in ZIF-8 were studied by high-temperature XRD,412 with the subsequent structure refinement, together with non-negative matrix factorisation (NMF) analysis of the IR spectrum. The authors were able to correlate the thermal expansion coefficient α to the arrangement of cations in the lattice. Although some degree of detail can be obtained by widely available SEM/TEM-EDX as a routine tool,413 fundamental studies in direct imaging with atomic resolution are still lacking. In spite of all these advances and other impressive reports in which high-resolution (HR)-TEM (which is limited by beam damage) has been used,414–416 there is still a lack of spatially resolved studies on metal distribution in MTV-MOFs.
Besides multi-metallic MOFs, linker-distribution in multi-variate (MTV)-MOFs, which are becoming an important area of research.417 Although indirect using NMR certainly represented a step forward in the characterization of MTV-MOFs,418 they do not provide imaging, direct evidence of the spatial linker distribution. Seminal works of this type have been previously published,234,250 but the field remains far from explored. In this sense, Katzenmeyer et al.419 applied photothermal induced resonance (PTIR) to study the chemical complexity of MTV-MIL-68(In) crystals. This technique combines the lateral resolution of AFM with the chemical specificity of IR, obtaining chemical information with nanoscale resolution (100 nm). MTV-MIL-68(In) was prepared from an equimolar mixture of NH2- and non-functionalized terephthalic acid and the resulting crystals were characterized by PTIR at different locations. In line with previous results (see above), the similar spectra obtained pointed to a homogeneous linker distribution down to ca. 100 nm (Fig. 5).
This work showcases the suitability of PTIR to characterize MOFs and nicely proves the MTV-MIL-68(In) linker homogeneity and the heterogeneous domains of crystals prepared by sequential growth (Fig. 19). In this respect, different techniques have been developed to chemically image solid materials with nanoscale resolution and may be of great use in such cases.420 An overview of the different methods available for chemical imaging can be found in the review authored by Buurmans and Weckhuysen,56,421 but due to their suitability, we would like to highlight AFM coupled with Tip-Enhanced Raman Spectroscopy (TERS) and element specific X-ray nanotomography. These techniques have been successfully applied to different heterogeneous catalysts in order to obtain chemical maps with 20 nm and sub-30 nm resolution, respectively.422–424 Further, our group has recently used atom probe tomography (ATP) to examine the atomic-scale distribution of Si, O and Al in zeolite crystals. Although extremely challenging, this technique could provide crucial information on atomic distribution in MTV-MOF materials.425
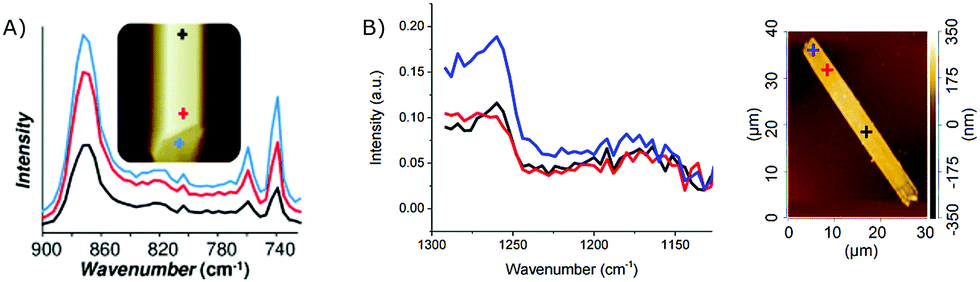 | ||
| Fig. 19 Photothermal induced resonance (PTIR) spectra acquired at different spots of (A) MTV-MIL-68(In) crystals (bands at 742 cm−1 and 762 cm−1 correspond to the C–H out-of-plane bending vibration mode for MIL-68(In) and NH2-MIL-68(In), respectively); and (B) crystals prepared by sequential growth (band at 1260 cm−1 corresponds to the C–N stretch vibration) showing a higher intensity on the blue spot (edge). Reprinted from ref. 419 Copyright©2014 John Wiley & Sons, Inc. | ||
Especially with respect to the most recent developments such as scanning near-field optical nanoscopic techniques and X-ray free electron lasers, micro- and nanoscopic tools have been developed with high speed during the past years to deliver unparalleled spatio-temporal resolution.54,55 Recently, a fist-of-its-kind study was published by Jayachandrababu et al.,426 wherein confocal fluorescence microscopy (CFM) and 1H NMR CRAMPS showed core–shell structures for post-synthetically linker exchanged ZIFs by exciting with different laser wavelengths. More recently, Schrimpf et al.427 have made use of Förster resonance energy transfer (FRET) and Fluorescence Lifetime Imaging (FLIM) to study the distribution and the presence of nanoscopic defects. These techniques are based on the energy transfer from one fluorophore donor to an acceptor, depending the efficiency of the transfer on the distance between molecules or groups. Thus, the emission can be correlated to the space between one dye linker and the other. In their study, they grafted fluorescein isothiocyanate (FITC) or rhodamine B isothiocyanate (RITC) with different concentrations to several 2-NH2-BPDC (BPDC) groups in UiO-67(Zr) crystals. In Fig. 20a, a phasor plot shows that fluorescence decay depends on the crystal size, and the exchange time with FITC. Different spatial distributions of FTIC (Fig. 20b–d) are obtained for different crystals, indicating the heterogeneity of the process at the single-particle level as well. In our opinion, this study represents a breakthrough in studying spatial distribution of mixed-linker MOFs and the presence of defects. Another example of a spatially resolved study was authored by Ghosh et al.,428 who observed local differences in solvent concentration within the pores of a HKUST-1 material as revealed by IR micro-spectroscopy in combination with NMF analysis. This shows that traditional synthesis protocols, even for archetypal MOFs, lead to heterogeneous materials at the molecular, nano- and microscale (both in composition and presence of residual molecules). Beyond chemical information, structural information may be obtained by other types of microscopy, e.g. SEM and TEM. MOFs may exhibit relatively limited stability under the electron beam, as we have previously discussed. Nevertheless, a number of high-resolution studies in which pore structures or polymorph transformations were visualized by means of aberration-corrected (AC)-TEM, have been published.429–433 Besides spatially resolved characterization, MOFs may be used as platforms for developing other advanced spectroscopic techniques.
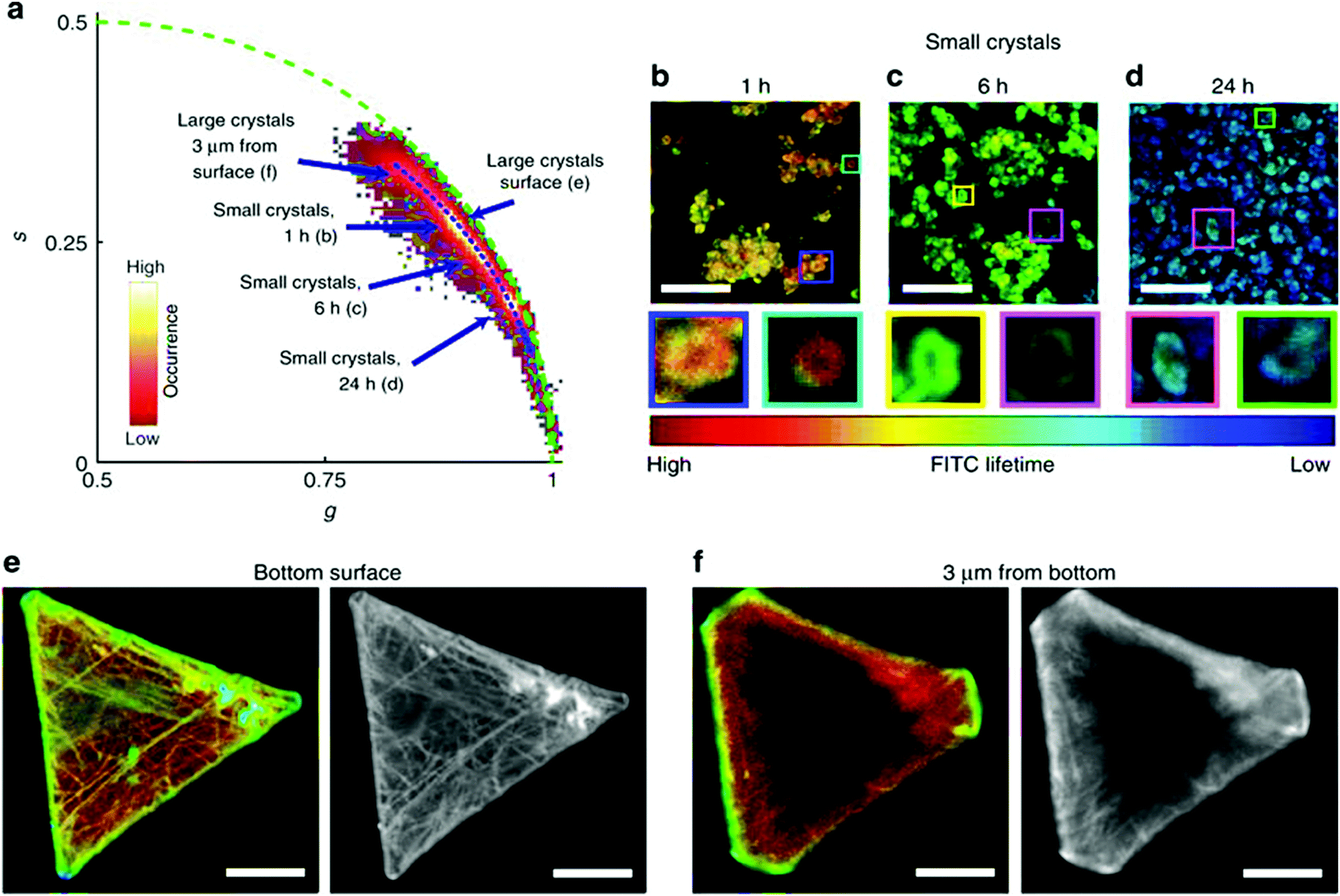 | ||
| Fig. 20 Fluorescence Lifetime Imaging (FLIM) phasor data of UiO-67 samples functionalized with fluorescein isothiocyanate (FITC) using linker exchange. (a) A phasor plot of the images shown in (b–f). The blue arrows point at the average phasor positions of the different samples. (b–d) FLIM images of small crystal UiO-67 samples subjected to linker exchange with FITC-modified linkers at 338 K for 1 h (b), 6 h (b), and 24 h (c). The inserts below are magnifications of the coloured squares. (e and f) FLIM (in colour) and fluorescence intensity (in grey scale) images of large crystal UiO-67 samples subjected to linker exchange with FITC-modified linkers for 24 h. (e) An image recorded at the bottom surface of a crystal. (f) An image recorded inside the crystal, 3 μm up from the bottom surface. The colour coding of the images is based on the blue dotted line in panel a using the colour table shown in b. The scale bar of all images is 10 μm. Adapted and reprinted from ref. 427. Copyright © 2018 Nature Publishing Group. | ||
6.2 Use of metal@MOF composites as spectroscopic tools
Although metal@MOF composites lay beyond the scope of this review, the combination of MOFs with the enhancement of Raman effects on metal surfaces has been of particular interest.434 For instance, embedded Au,435–438 Ag439–441 and even Au@Ag442 nanostructures (e.g. rods, cubes, spherical crystals, stars) have been used to as SERS substrates to study different phenomena. For instance, Zhao et al. used plasmonic Ag octahedral nanocrystals coated with a layer of Al-porphyrin MOF, to study the metalation process of the porphyrin centres by means of enhanced Raman spectroscopy. This allowed them to map the enhancement effect for individual Ag ⊂ MOF particles.443 In another example, Sim et al.444 embedded Ag nanocrystals with which they could follow the carboxylation of aniline by enhanced Raman spectroscopy. It is interesting to highlight that a number of authors have reported SERS effect in the surfaces of MOFs without the use of metal nanostructures, purely by charge-transfer resonance effects with the MOF backbone, as demonstrated by theoretical calculations.445,446 This opens the opportunity to directly study the formation of intermediates in catalysis or the interactions with liquid or gas adsorbates, on the MOF surface during sensing or gas sorption without the side effects of metal nanostructures. Not only for SERS, but also for catalysis and gas sorption charge transfer can be of high relevance. In fact, in their latest report, Olsbye and others offered a full picture of the interaction between Pt NPs and the framework during CO2 hydrogenation is given.447The key in this study is combining DFT simulations with operando IR and TEM tools to study reaction intermediates, to define what kind of products are formed and the role that the MOF plays as a support. Alternatively to IR or Raman, other techniques, such as hard XPS and NEXAFS have been used for studying orbital overlapping between Pd nanocubes and the HKUST-1(Cu) matrix and why this has impact in enhanced H2 storage, showing the necessity of using very particular techniques in certain cases.448
6.3 Latest trends for in situ and spatially resolved studies
Apart from sensing, new techniques for studying in situ the crystallization of MOFs are being developed. As described in the previous sections, much research has been devoted to the in situ study of MOF formation. In this line, Stock and co-workers have developed the SynRAC cell for crystallization studies, which provides low signal-to-noise ratios and improved time-resolution to previous set-ups.373 In showcase studies of a selected Bi-based coordination polymer, the presence of short-lived (previously undetectable) intermediates was observed.449,450 Just as shown in Sections 2.1.2 and 5.1, this kind of mechanistic studies will become more important in the future if one wants to control the different phases present during crystallization. An example beyond synchrotron technique was reported by Van Cleuvenbergen et al.451 The crystallization of ZIF-8 was followed by dynamic Angle-Resolved second harmonic scattering (dAR-SHS) shows an alternative way of obtaining relevant information about size, shape and concentration of MOFs during their formation. This summary show that MOFs are fostering progress in the development of techniques that could be used for many other types of materials. On the side of spatially resolved tools, the group of Kitagawa recently reported a study in which AFM under dynamic conditions showed layer restructuring in [(Zn2(1,4-ndc)2(dabco))n] as a response to interactions with solvent molecules.452 As seen here, already known and newly developed micro- and nanoscopic (X-ray, IR and Raman based) techniques will play a big role in the coming few years. For instance, chemical imaging by AFM-IR spectroscopy would probe dangling bonds or different linkers with resolution down to ca. 10 nm (Fig. 21a). | ||
| Fig. 21 Outlook of the challenges to be addressed in the future of metal–organic frameworks characterization at different lengthscales. (a) Microscopy at the (sub)nanometer scale by spatially resolved tools (e.g. AFM-IR spectroscopy); (b) diffusion and mass transport phenomena, tomographic reconstruction of pore networks by fluorescence and contrast-based techniques; (c) intra- and inter-particle hetereogeneities (orange and blue indicate distinct chemical compositions) studied by chemical mapping and (d) in situ/operando studies at the lab- and pilot-plant scale. A holistic approach will result in advanced understanding, thus, improved design of MOFs’ properties, leading to optimized materials for large scale applications. | ||
Another example would be the use of fluorescent probes and single-molecule fluorescence (the former already pioneered by Ameloot et al.250 in 2009 for MOF materials) for imaging pore cavities,453–457 or single-reaction (catalytic) events,458,459 respectively; polarized Raman microscopy with probe molecules460 or tomography based on X-ray contrast423,461 are a wide-open playground for the study of issues such as pore connectivity, gas and liquid diffusion barriers or separation properties in MOF crystals (Fig. 21b and c), which are crucial for regulating their performance in catalysis or gas sorption or separation.25 Examples of such strategies, such as the one reported by the group of Caro, are starting to be reported.462 These techniques have the potential to unravel physico-chemical phenomena to an unprecedented level of detail and will become more and more important. The importance of using microscopy has been recently highlighted by Rivera-Torrente et al. in the HKUST-1 system for two different applications. First, defect distribution was proven to be unequal at the inter-crystal level when 5-cyano-1,3-benzenedicarboxylic acid and 1,3,5-benzenetricarboxylic acid were mixed in the hydrothermal solution.432 In this case, the use of Raman microscopy was invaluable due to its high spatial resolution and chemical specificity for each linker. Further, in another study, a combination of Raman microscopy with advanced data analysis shed light on the distribution of a guest molecule (7,7,8,8-tetracyanoquinodimethane) at the intracrystal level.433 These two studies show the importance of using spatially resolved tools as a routine when complex composites are built as reported in the latest strategies. Eventually, the use of these, together with more traditional bulk techniques in operando modes will result in an improved design of MOFs, rendering them suitable for large-scale or commercial applications (Fig. 21). We refer the reader to recent literature on this type of techniques to understand the potential of microscopy for understanding crystallization of MOFs.420,463–465
7. Conclusions and outlook
This review article provides an overview of the use of different advanced characterization tools in the field of metal–organic frameworks (MOFs) with their potential being illustrated by using well-known, robust topologies. Characteristic traits or phenomena related to MIL-100, MAF-4/ZIF-8 and HKUST-1 were highlighted with the main (nano/micro)-spectroscopy, diffraction and scattering techniques used to uncover their nature, emphasizing potential pitfalls and constraints. Although very different in their chemical and compositional nature, the MOF field could benefit from the tools used in the past for other functional porous materials, such as zeolites or porous metal oxides.From our bibliometric analysis, it is clear that there is still plenty of room for advanced techniques, such as X-ray reflectivity (XRR), grazing incidence X-ray absorption spectroscopy (GIXAS) or electrical scanning probe microscopy techniques such as Kelvin probe force microscopy (KPFM) could provide critically relevant information on surface morphology, electronic structure or magnetic properties, respectively, of MOF surfaces. In addition, more spatially resolved studies are needed to uncover the underlying effects of atomic compositional heterogeneities, inter/intraparticle gradients and their effect on the chemistry of MOFs. They are inherently complex materials, with multiple components that can be present in different forms (i.e., geometry, configurations and oxidation state). Several fundamental questions involving crystal heterogeneities (e.g. direct imaging of atomic scale mixing, chemical nature of defects, generality of metal exchange mechanisms, pore network permeability, surface energies) are still open. These issues remain a challenge for the characterization community, in which only a few selected electron microscopy studies and the use of AFM (mainly related to the synthesis of MOF thin-films) for specific SURMOFS, have been able to provide some insight. The advent of non-diffraction limited techniques, such as AFM-IR micro-spectroscopy, together with a more widespread use of operando studies (let it be catalysis, gas sorption or molecular separations), will take understanding of MOFs to the next step.
The ultimate level of fundamental understanding will be achieved by the combination of all these features, i.e. the development of characterization tools that can provide chemical information of these materials under dynamic conditions (in situ or operando) at the nano- (and eventually atomic) scale. As a result, we foresee that MOFs will be an excellent field for the development and adaptation of (new) micro- and nano-spectroscopic methods. The use and application of these techniques have the potential to benefit the MOF community greatly in the long term, and we expect they will play a pivotal role as they already do in e.g. the fields of biology and opto-electronics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement 641887 (DEFNET). B. S. gratefully acknowledges the Dutch National Science Foundation (NWO), The Netherlands, for her personal VENI grant 722.015.007. M. F. acknowledges the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement (No. 748563). This work is supported by a Netherlands Organization of Scientific Research (NWO) Gravitation program (Netherlands Center for Multiscale Catalytic Energy Conversion, MCEC).References
- Y.-S. Ho and H.-Z. Fu, Inorg. Chem. Commun., 2016, 73, 174–182 CrossRef.
- C.-C. Wang and Y.-S. Ho, Scientometrics, 2016, 109, 481–513 CrossRef.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef CAS.
- S. R. Batten, B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1995, 117, 5385–5386 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef.
- C. Wiktor, M. Meledina, S. Turner, O. I. Lebedev and R. A. Fischer, J. Mater. Chem. A, 2017, 5, 14969–14989 RSC.
- C. Martineau-Corcos, Curr. Opin. Colloid Interface Sci., 2018, 33, 35–43 CrossRef.
- B. E. G. Lucier, S. Chen and Y. Huang, Acc. Chem. Res., 2018, 51, 319–330 CrossRef PubMed.
- P. He, B. E. G. Lucier, V. V. Terskikh, Q. Shi, J. Dong, Y. Chu, A. Zheng, A. Sutrisno and Y. Huang, J. Phys. Chem. C, 2014, 118, 23728–23744 CrossRef.
- Y. Huang, J. Xu, F. Gul-E-Noor and P. He, Metal–Organic Frameworks: NMR Studies of Quadrupolar Nuclei, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, 2014, DOI:10.1002/9781119951438.eibc2225.
- A. Krajnc, T. Kos, N. Zabukovec Logar and G. Mali, Angew. Chem., Int. Ed., 2015, 54, 10535–10538 CrossRef CAS PubMed.
- M. Haouas, Materials, 2018, 11(8), 1416 CrossRef PubMed.
- M. A. Soldatov, A. Martini, A. L. Bugaev, I. Pankin, P. V. Medvedev, A. A. Guda, A. M. Aboraia, Y. S. Podkovyrina, A. P. Budnyk, A. A. Soldatov and C. Lamberti, Polyhedron, 2018, 155, 232–253 CrossRef CAS.
- E. Borfecchia, L. Braglia, F. Bonino, S. Bordiga, S. Øien, U. Olsbye, K. P. Lillerud, J. A. van Bokhoven, K. A. Lomachenko, A. A. Guda, M. A. Soldatov and C. Lamberti, in XAFS Techniques for Catalysts, Nanomaterials, and Surfaces, ed. Y. Iwasawa, K. Asakura and M. Tada, Springer International Publishing, Cham, 2017, pp. 397–430 DOI:10.1007/978-3-319-43866-5_26.
- C. Garino, E. Borfecchia, R. Gobetto, J. A. van Bokhoven and C. Lamberti, Coord. Chem. Rev., 2014, 277-278, 130–186 CrossRef CAS.
- F. Bonino, C. Lamberti and S. Bordiga, The Chemistry of Metal–Organic Frameworks: Synthesis, Characterization, and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, pp. 657–690 DOI:10.1002/9783527693078.ch22.
- F. Bonino, C. Lamberti, S. Chavan, J. G. Vitillo and S. Bordiga, Metal Organic Frameworks as Heterogeneous Catalysts, The Royal Society of Chemistry, 2013, pp. 76–142 10.1039/9781849737586-00076.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef CAS.
- S. Bordiga, F. Bonino, K. P. Lillerud and C. Lamberti, Chem. Soc. Rev., 2010, 39, 4885–4927 RSC.
- H. C. Hoffmann, M. Debowski, P. Müller, S. Paasch, I. Senkovska, S. Kaskel and E. Brunner, Materials, 2012, 5(12), 2537–2572 CrossRef CAS.
- A. Sutrisno, V. V. Terskikh, Q. Shi, Z. Song, J. Dong, S. Y. Ding, W. Wang, B. R. Provost, T. D. Daff, T. K. Woo and Y. Huang, Chem. – Eur. J., 2012, 18, 12251–12259 CrossRef PubMed.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef.
- T. L. Easun, F. Moreau, Y. Yan, S. Yang and M. Schroder, Chem. Soc. Rev., 2017, 46, 239–274 RSC.
- M. J. Van Vleet, T. Weng, X. Li and J. R. Schmidt, Chem. Rev., 2018, 118, 3681–3721 CrossRef PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- V. Kapko, C. Dawson, M. M. J. Treacy and M. F. Thorpe, Phys. Chem. Chem. Phys., 2010, 12, 8531–8541 RSC.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357–361 CrossRef CAS PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- S. Bureekaew, S. Shimomura and S. Kitagawa, Sci. Technol. Adv. Mater., 2008, 9, 014108 CrossRef PubMed.
- K. Uemura, R. Matsuda and S. Kitagawa, J. Solid State Chem., 2005, 178, 2420–2429 CrossRef CAS.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC.
- G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- G. Férey, Dalton Trans., 2009, 4400–4415, 10.1039/B817360P.
- A. J. Fletcher, K. M. Thomas and M. J. Rosseinsky, J. Solid State Chem., 2005, 178, 2491–2510 CrossRef.
- C. R. Murdock, B. C. Hughes, Z. Lu and D. M. Jenkins, Coord. Chem. Rev., 2014, 258–259, 119–136 CrossRef.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef PubMed.
- S. Dissegna, K. Epp, W. R. Heinz, G. Kieslich and R. A. Fischer, Adv. Mater., 2018, 30, 1704501 CrossRef PubMed.
- M. Lalonde, W. Bury, O. Karagiaridi, Z. Brown, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2013, 1, 5453–5468 RSC.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- C. K. Brozek and M. Dinca, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- S. Abednatanzi, P. Gohari Derakhshandeh, H. Depauw, F.-X. Coudert, H. Vrielinck, P. Van Der Voort and K. Leus, Chem. Soc. Rev., 2019, 48, 2535–2565 RSC.
- T. D. Bennett, A. K. Cheetham, A. H. Fuchs and F.-X. Coudert, Nat. Chem., 2017, 9, 11–16 CrossRef CAS PubMed.
- T. D. Bennett, A. H. Fuchs, A. K. Cheetham and F.-X. Coudert, Dalton Trans., 2016, 45, 4058–4059 RSC.
- T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47, 1555–1562 CrossRef CAS PubMed.
- J. Hou, C. W. Ashling, S. M. Collins, A. Krajnc, C. Zhou, L. Longley, D. N. Johnstone, P. A. Chater, S. Li, M.-V. Coulet, P. L. Llewellyn, F.-X. Coudert, D. A. Keen, P. A. Midgley, G. Mali, V. Chen and T. D. Bennett, Nat. Commun., 2019, 10, 2580 CrossRef PubMed.
- R. S. K. Madsen, A. Qiao, J. Sen, I. Hung, K. Chen, Z. Gan, S. Sen and Y. Yue, Science, 2020, 367, 1473–1476 CrossRef CAS PubMed.
- J. Hou, M. L. Rios Gomez, A. Krajnc, A. McCaul, S. Li, A. M. Bumstead, A. F. Sapnik, Z. Deng, R. Lin, P. A. Chater, D. S. Keeble, D. A. Keen, D. Appadoo, B. Chan, V. Chen, G. Mali and T. D. Bennett, J. Am. Chem. Soc., 2020, 142, 3880–3890 CrossRef CAS PubMed.
- M. Rivera-Torrente, M. Filez, R. Hardian, E. Reynolds, B. Seoane, M.-V. Coulet, F. E. Oropeza Palacio, J. P. Hofmann, R. A. Fischer, A. L. Goodwin, P. L. Llewellyn and B. M. Weckhuysen, Chem. – Eur. J., 2018, 24, 7498–7506 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- M. Filez, Z. Ristanović and B. M. Weckhuysen, in Encyclopedia of Interfacial Chemistry, ed. K. Wandelt, Elsevier, Oxford, 2018, pp. 304–320 DOI:10.1016/b978-0-12-409547-2.13744-8.
- F. Meirer and B. M. Weckhuysen, Nat. Rev. Mater., 2018, 3, 324–340 CrossRef.
- A. M. Beale, S. D. M. Jacques and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4656–4672 RSC.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- P. Horcajada, S. Surble, C. Serre, D.-Y. Hong, Y.-K. Seo, J.-S. Chang, J.-M. Greneche, I. Margiolaki and G. Ferey, Chem. Commun., 2007, 2820–2822, 10.1039/B704325B.
- C. Volkringer, D. Popov, T. Loiseau, G. R. Férey, M. Burghammer, C. Riekel, M. Haouas and F. Taulelle, Chem. Mater., 2009, 21, 5695–5697 CrossRef CAS.
- A. Lieb, H. Leclerc, T. Devic, C. Serre, I. Margiolaki, F. Mahjoubi, J. S. Lee, A. Vimont, M. Daturi and J.-S. Chang, Microporous Mesoporous Mater., 2012, 157, 18–23 CrossRef CAS.
- L. Mitchell, B. Gonzalez-Santiago, J. P. S. Mowat, M. E. Gunn, P. Williamson, N. Acerbi, M. L. Clarke and P. A. Wright, Catal. Sci. Technol., 2013, 3, 606–617 RSC.
- J. A. Mason, L. E. Darago, W. W. Lukens and J. R. Long, Inorg. Chem., 2015, 54, 10096–10104 CrossRef PubMed.
- J. Castells-Gil, N. M. Padial, N. Almora-Barrios, I. da Silva, D. Mateo, J. Albero, H. Garcia and C. Marti-Gastaldo, Chem. Sci., 2019, 10, 4313–4321 RSC.
- H. Reinsch and N. Stock, CrystEngComm, 2013, 15, 544–550 RSC.
- F. Zhang, X. Zou, W. Feng, X. Zhao, X. Jing, F. Sun, H. Ren and G. Zhu, J. Mater. Chem., 2012, 22, 25019–25026 RSC.
- S. Bhattacharjee, C. Chen and W.-S. Ahn, RSC Adv., 2014, 4, 52500–52525 RSC.
- M. P. Attfield and P. Cubillas, Dalton Trans., 2012, 41, 3869–3878 RSC.
- T. Ahnfeldt, J. Moellmer, V. Guillerm, R. Staudt, C. Serre and N. Stock, Chem. – Eur. J., 2011, 17, 6462–6468 CrossRef CAS PubMed.
- F. Millange, R. El Osta, M. E. Medina and R. I. Walton, CrystEngComm, 2011, 13, 103–108 RSC.
- F. Niekiel, M. Ackermann, P. Guerrier, A. Rothkirch and N. Stock, Inorg. Chem., 2013, 52, 8699–8705 CrossRef CAS PubMed.
- F. Ragon, H. Chevreau, T. Devic, C. Serre and P. Horcajada, Chem. – Eur. J., 2015, 21, 7135–7143 CrossRef CAS PubMed.
- Y. Wu, M. I. Breeze, G. J. Clarkson, F. Millange, D. O'Hare and R. I. Walton, Angew. Chem., Int. Ed., 2016, 55, 4992–4996 CrossRef CAS PubMed.
- F. Ragon, P. Horcajada, H. Chevreau, Y. K. Hwang, U. H. Lee, S. R. Miller, T. Devic, J.-S. Chang and C. Serre, Inorg. Chem., 2014, 53, 2491–2500 CrossRef CAS PubMed.
- F. Millange, M. I. Medina, N. Guillou, G. Férey, K. M. Golden and R. I. Walton, Angew. Chem., Int. Ed., 2010, 49, 763–766 CrossRef CAS PubMed.
- M. G. Goesten, F. Kapteijn and J. Gascon, CrystEngComm, 2013, 15, 9249–9257 RSC.
- M. Haouas, C. Volkringer, T. Loiseau, G. Férey and F. Taulelle, Chem. Mater., 2012, 24, 2462–2471 CrossRef.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- C. Serre, F. Millange, S. Surblé and G. Férey, Angew. Chem., Int. Ed., 2004, 43, 6285–6289 CrossRef PubMed.
- P. Horcajada, H. Chevreau, D. Heurtaux, F. Benyettou, F. Salles, T. Devic, A. Garcia-Marquez, C. Yu, H. Lavrard, C. L. Dutson, E. Magnier, G. Maurin, E. Elkaim and C. Serre, Chem. Commun., 2014, 50, 6872–6874 RSC.
- S. Surble, C. Serre, C. Mellot-Draznieks, F. Millange and G. Ferey, Chem. Commun., 2006, 284–286, 10.1039/B512169H.
- S. Surble, F. Millange, C. Serre, G. Ferey and R. I. Walton, Chem. Commun., 2006, 1518–1520, 10.1039/B600709K.
- W. Xuan, C. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677–1695 RSC.
- M. G. Goesten, E. Stavitski, J. Juan-Alcañiz, A. Martiñez-Joaristi, A. V. Petukhov, F. Kapteijn and J. Gascon, Catal. Today, 2013, 205, 120–127 CrossRef CAS.
- N. Pienack and W. Bensch, Angew. Chem., Int. Ed., 2011, 50, 2014–2034 CrossRef CAS PubMed.
- T. Birsa Čelič, M. Rangus, K. Lázár, V. Kaučič and N. Zabukovec Logar, Angew. Chem., Int. Ed., 2012, 51, 12490–12494 CrossRef PubMed.
- E. Stavitski, M. Goesten, J. Juan-Alcañiz, A. Martinez-Joaristi, P. Serra-Crespo, A. V. Petukhov, J. Gascon and F. Kapteijn, Angew. Chem., Int. Ed., 2011, 50, 9624–9628 CrossRef CAS PubMed.
- A. C. Sudik, A. P. Côté and O. M. Yaghi, Inorg. Chem., 2005, 44, 2998–3000 CrossRef CAS PubMed.
- M. G. Goesten, P. C. M. M. Magusin, E. A. Pidko, B. Mezari, E. J. M. Hensen, F. Kapteijn and J. Gascon, Inorg. Chem., 2014, 53, 882–887 CrossRef CAS PubMed.
- G. Férey, M. Haouas, T. Loiseau and F. Taulelle, Chem. Mater., 2014, 26, 299–309 CrossRef.
- X. Yang and A. E. Clark, Inorg. Chem., 2014, 53, 8930–8940 CrossRef CAS PubMed.
- R. I. Walton and F. Millange, The Chemistry of Metal–Organic Frameworks: Synthesis, Characterization, and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, pp. 729–764 DOI:10.1002/9783527693078.ch24.
- Y. Wu, M. I. Breeze, D. O'Hare and R. I. Walton, Microporous Mesoporous Mater., 2017, 254, 178–183 CrossRef CAS.
- A. M. Beale, A. M. J. van der Eerden, S. D. M. Jacques, O. Leynaud, M. G. O'Brien, F. Meneau, S. Nikitenko, W. Bras and B. M. Weckhuysen, J. Am. Chem. Soc., 2006, 128, 12386–12387 CrossRef CAS PubMed.
- M. G. O'Brien, A. M. Beale and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4767–4782 RSC.
- D. Grandjean, A. M. Beale, A. V. Petukhov and B. M. Weckhuysen, J. Am. Chem. Soc., 2005, 127, 14454–14465 CrossRef CAS PubMed.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J.-S. Chang, D.-Y. Hong, Y. Kyu Hwang, S. Hwa Jhung and G. Férey, Langmuir, 2008, 24, 7245–7250 CrossRef CAS PubMed.
- A. Vimont, J.-M. Goupil, J.-C. Lavalley, M. Daturi, S. Surblé, C. Serre, F. Millange, G. Férey and N. Audebrand, J. Am. Chem. Soc., 2006, 128, 3218–3227 CrossRef CAS PubMed.
- H. Leclerc, A. Vimont, J.-C. Lavalley, M. Daturi, A. D. Wiersum, P. L. Llwellyn, P. Horcajada, G. Ferey and C. Serre, Phys. Chem. Chem. Phys., 2011, 13, 11748–11756 RSC.
- M. Haouas, C. Volkringer, T. Loiseau, G. Férey and F. Taulelle, J. Phys. Chem. C, 2011, 115, 17934–17944 CrossRef CAS.
- C. Volkringer, H. Leclerc, J.-C. Lavalley, T. Loiseau, G. Férey, M. Daturi and A. Vimont, J. Phys. Chem. C, 2012, 116, 5710–5719 CrossRef CAS.
- J. Jiang and O. M. Yaghi, Chem. Rev., 2015, 115, 6966–6997 CrossRef CAS PubMed.
- A. Vimont, H. Leclerc, F. Maugé, M. Daturi, J.-C. Lavalley, S. Surblé, C. Serre and G. Férey, J. Phys. Chem. C, 2007, 111, 383–388 CrossRef CAS.
- J. N. Hall and P. Bollini, ACS Catal., 2020, 10, 3750–3763 CrossRef CAS.
- J. Hajek, C. Caratelli, R. Demuynck, K. De Wispelaere, L. Vanduyfhuys, M. Waroquier and V. Van Speybroeck, Chem. Sci., 2018, 9, 2723–2732 RSC.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- R. Wei, C. A. Gaggioli, G. Li, T. Islamoglu, Z. Zhang, P. Yu, O. K. Farha, C. J. Cramer, L. Gagliardi, D. Yang and B. C. Gates, Chem. Mater., 2019, 31, 1655–1663 CrossRef CAS.
- X. Feng, J. Hajek, H. S. Jena, G. Wang, S. K. P. Veerapandian, R. Morent, N. De Geyter, K. Leyssens, A. E. J. Hoffman, V. Meynen, C. Marquez, D. E. De Vos, V. Van Speybroeck, K. Leus and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 3174–3183 CrossRef CAS PubMed.
- C. Caratelli, J. Hajek, F. G. Cirujano, M. Waroquier, F. X. Llabrés i Xamena and V. Van Speybroeck, J. Catal., 2017, 352, 401–414 CrossRef CAS.
- M. Taddei, Coord. Chem. Rev., 2017, 343, 1–24 CrossRef CAS.
- S. Wuttke, P. Bazin, A. Vimont, C. Serre, Y.-K. Seo, Y. K. Hwang, J.-S. Chang, G. Férey and M. Daturi, Chem. – Eur. J., 2012, 18, 11959–11967 CrossRef CAS PubMed.
- G. Gómez-Pozuelo, C. P. Cabello, M. Opanasenko, M. Horáček and J. Čejka, ChemPlusChem, 2016, 82, 152–159 CrossRef PubMed.
- S. Mitchell, A. B. Pinar, J. Kenvin, P. Crivelli, J. Karger and J. Perez-Ramirez, Nat. Commun., 2015, 6, 8633 CrossRef CAS PubMed.
- K. W. Chapman, G. J. Halder and P. J. Chupas, J. Am. Chem. Soc., 2009, 131, 17546–17547 CrossRef CAS PubMed.
- J. W. Yoon, Y.-K. Seo, Y. K. Hwang, J.-S. Chang, H. Leclerc, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, E. Bloch, P. L. Llewellyn, C. Serre, P. Horcajada, J.-M. Grenèche, A. E. Rodrigues and G. Férey, Angew. Chem., Int. Ed., 2010, 49, 5949–5952 CrossRef CAS PubMed.
- G. T. Palomino, C. P. Cabello and C. O. Areán, Eur. J. Inorg. Chem., 2011, 1703–1708 CrossRef CAS.
- R. Giovine, C. Volkringer, S. E. Ashbrook, J. Trébosc, D. McKay, T. Loiseau, J.-P. Amoureux, O. Lafon and F. Pourpoint, Chem. – Eur. J., 2017, 23, 9525–9534 CrossRef CAS PubMed.
- B. Barth, M. Mendt, A. Pöppl and M. Hartmann, Microporous Mesoporous Mater., 2015, 216, 97–110 CrossRef CAS.
- M. Mendt, B. Barth, M. Hartmann and A. Pöppl, J. Chem. Phys., 2017, 147, 224701 CrossRef PubMed.
- A. H. Khan, B. Barth, M. Hartmann, J. Haase and M. Bertmer, J. Phys. Chem. C, 2018, 122, 12723–12730 CrossRef CAS.
- A. E. Khudozhitkov, A. V. Toktarev, S. S. Arzumanov, A. A. Gabrienko, D. I. Kolokolov and A. G. Stepanov, Chem. – Eur. J., 2019, 25, 10808–10812 CrossRef CAS PubMed.
- A. E. Khudozhitkov, S. S. Arzumanov, D. I. Kolokolov, O. A. Kholdeeva, D. Freude and A. G. Stepanov, Chem., 2019, 25, 5163–5168 CrossRef CAS PubMed.
- A. Nandy, A. C. Forse, V. J. Witherspoon and J. A. Reimer, J. Phys. Chem. C, 2018, 122, 8295–8305 CrossRef CAS.
- C. Zhao, H. Meng, M. Nie, L. Jiang, C. Wang and T. Wang, J. Phys. Chem. C, 2018, 122, 4635–4640 CrossRef CAS.
- A. M. Sheveleva, A. V. Anikeenko, A. S. Poryvaev, D. L. Kuzmina, I. K. Shundrina, D. I. Kolokolov, A. G. Stepanov and M. V. Fedin, J. Phys. Chem. C, 2017, 121, 19880–19886 CrossRef CAS.
- T. Wittmann, A. Mondal, C. B. L. Tschense, J. J. Wittmann, O. Klimm, R. Siegel, B. Corzilius, B. Weber, M. Kaupp and J. Senker, J. Am. Chem. Soc., 2018, 140, 2135–2144 CrossRef CAS PubMed.
- S. Hinokuma, G. Wiker, T. Suganuma, A. Bansode, D. Stoian, S. C. Huertas, S. Molina, A. Shafir, M. Rønning, W. van Beek and A. Urakawa, Eur. J. Inorg. Chem., 2018, 1847–1853 CrossRef CAS.
- M. Rivera-Torrente, C. Hernández Mejía, T. Hartman, K. P. de Jong and B. M. Weckhuysen, Catal. Lett., 2019, 149, 3279–3286 CrossRef CAS.
- M. Rivera-Torrente, P. D. Pletcher, M. K. Jongkind, N. Nikolopoulos and B. M. Weckhuysen, ACS Catal., 2019, 9, 3059–3069 CrossRef CAS.
- Y.-Q. Tian, C.-X. Cai, Y. Ji, X.-Z. You, S.-M. Peng and G.-H. Lee, Angew. Chem., Int. Ed., 2002, 41, 1384–1386 CrossRef CAS PubMed.
- X. Huang, J. Zhang and X. Chen, Chin. Sci. Bull., 2013, 48, 1531–1534 Search PubMed.
- N. Masciocchi, S. Bruni, E. Cariati, F. Cariati, S. Galli and A. Sironi, Inorg. Chem., 2001, 40, 5897–5905 CrossRef CAS PubMed.
- X.-C. Huang, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS PubMed.
- Y.-Q. Tian, C.-X. Cai, X.-M. Ren, C.-Y. Duan, Y. Xu, S. Gao and X.-Z. You, Chem. – Eur. J., 2003, 9, 5673–5685 CrossRef CAS PubMed.
- Y.-Q. Tian, Z.-X. Chen, L.-H. Weng, H.-B. Guo, S. Gao and D. Y. Zhao, Inorg. Chem., 2004, 43, 4631–4635 CrossRef CAS PubMed.
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- Y. Pan, Y. Liu, G. Zeng, L. Zhao and Z. Lai, Chem. Commun., 2011, 47, 2071–2073 RSC.
- J. Cravillon, S. Münzer, S.-J. Lohmeier, A. Feldhoff, K. Huber and M. Wiebcke, Chem. Mater., 2009, 21, 1410–1412 CrossRef CAS.
- H. Reinsch, Eur. J. Inorg. Chem., 2016, 4290–4299 CrossRef CAS.
- S. Bhattacharjee, M.-S. Jang, H.-J. Kwon and W.-S. Ahn, Catal. Surv. Asia, 2014, 18, 101–127 CrossRef CAS.
- Y.-R. Lee, J. Kim and W.-S. Ahn, Korean J. Chem. Eng., 2013, 30, 1667–1680 CrossRef CAS.
- B. R. Pimentel, A. Parulkar, E.-K. Zhou, N. A. Brunelli and R. P. Lively, ChemSusChem, 2014, 7, 3202–3240 CrossRef CAS PubMed.
- J. Cravillon, R. Nayuk, S. Springer, A. Feldhoff, K. Huber and M. Wiebcke, Chem. Mater., 2011, 23, 2130–2141 CrossRef CAS.
- S. Tanaka, K. Kida, M. Okita, Y. Ito and Y. Miyake, Chem. Lett., 2012, 41, 1337–1339 CrossRef CAS.
- J. Cravillon, C. A. Schröder, H. Bux, A. Rothkirch, J. Caro and M. Wiebcke, CrystEngComm, 2012, 14, 492–498 RSC.
- M. Goesten, E. Stavitski, E. A. Pidko, C. Gücüyener, B. Boshuizen, S. N. Ehrlich, E. J. M. Hensen, F. Kapteijn and J. Gascon, Chem. – Eur. J., 2013, 19, 7809–7816 CrossRef CAS PubMed.
- L. Batzdorf, F. Fischer, M. Wilke, K.-J. Wenzel and F. Emmerling, Angew. Chem., Int. Ed., 2015, 54, 1799–1802 CrossRef CAS PubMed.
- C. A. Schröder, I. A. Baburin, L. van Wüllen, M. Wiebcke and S. Leoni, CrystEngComm, 2013, 15, 4036–4040 RSC.
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66–73 CrossRef PubMed.
- I. Halasz, S. A. J. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimäki, R. C. Nightingale, R. E. Dinnebier and T. Friščić, Nat. Protoc., 2013, 8, 1718–1729 CrossRef PubMed.
- I. Halasz, T. Friscic, S. A. J. Kimber, K. Uzarevic, A. Puskaric, C. Mottillo, P. Julien, V. Strukil, V. Honkimaki and R. E. Dinnebier, Faraday Discuss., 2014, 170, 203–221 RSC.
- I. H. Lim, W. Schrader and F. Schüth, Chem. Mater., 2015, 27, 3088–3095 CrossRef CAS.
- J. P. Patterson, P. Abellan, M. S. Denny, C. Park, N. D. Browning, S. M. Cohen, J. E. Evans and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137, 7322–7328 CrossRef CAS PubMed.
- J. Cravillon, C. A. Schröder, R. Nayuk, J. Gummel, K. Huber and M. Wiebcke, Angew. Chem., Int. Ed., 2011, 50, 8067–8071 CrossRef CAS PubMed.
- P. Y. Moh, P. Cubillas, M. W. Anderson and M. P. Attfield, J. Am. Chem. Soc., 2011, 133, 13304–13307 CrossRef CAS PubMed.
- B. Seoane, S. Castellanos, A. Dikhtiarenko, F. Kapteijn and J. Gascon, Coord. Chem. Rev., 2016, 307(Part 2), 147–187 CrossRef CAS.
- P. Cubillas, M. W. Anderson and M. P. Attfield, Chem. – Eur. J., 2013, 19, 8236–8243 CrossRef CAS PubMed.
- M. Shöâeè, J. R. Agger, M. W. Anderson and M. P. Attfield, CrystEngComm, 2008, 10, 646–648 RSC.
- M. Shoaee, M. W. Anderson and M. P. Attfield, Angew. Chem., Int. Ed., 2008, 47, 8525–8528 CrossRef CAS PubMed.
- N. S. John, C. Scherb, M. Shoaee, M. W. Anderson, M. P. Attfield and T. Bein, Chem. Commun., 2009, 6294–6296, 10.1039/B908299A.
- P. Cubillas, K. Etherington, M. W. Anderson and M. P. Attfield, CrystEngComm, 2014, 16, 9834–9841 RSC.
- P. Cubillas, M. W. Anderson and M. P. Attfield, Cryst. Growth Des., 2013, 13, 4526–4532 CrossRef CAS.
- P. Cubillas, M. W. Anderson and M. P. Attfield, Chem. – Eur. J., 2012, 18, 15406–15415 CrossRef CAS PubMed.
- T. L. Easun, F. Moreau, Y. Yan, S. H. Yang and M. Schroder, Chem. Soc. Rev., 2017, 46, 239–274 RSC.
- C. L. Hobday, C. H. Woodall, M. J. Lennox, M. Frost, K. Kamenev, T. Düren, C. A. Morrison and S. A. Moggach, Nat. Commun., 2018, 9, 1429 CrossRef PubMed.
- V. K. Peterson, Y. Liu, C. M. Brown and C. J. Kepert, J. Am. Chem. Soc., 2006, 128, 15578–15579 CrossRef CAS PubMed.
- T. Yildirim and M. R. Hartman, Phys. Rev. Lett., 2005, 95, 215504 CrossRef CAS PubMed.
- M. R. Hartman, V. K. Peterson, Y. Liu, S. S. Kaye and J. R. Long, Chem. Mater., 2006, 18, 3221–3224 CrossRef CAS.
- J. Getzschmann, I. Senkovska, D. Wallacher, M. Tovar, D. Fairen-Jimenez, T. Düren, J. M. van Baten, R. Krishna and S. Kaskel, Microporous Mesoporous Mater., 2010, 136, 50–58 CrossRef CAS.
- Y. Zhao, H. Xu, L. L. Daemen, K. Lokshin, K. T. Tait, W. L. Mao, J. Luo, R. P. Currier and D. D. Hickmott, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5727–5731 CrossRef CAS PubMed.
- X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159–2171 CrossRef CAS PubMed.
- M. Barter, J. Hartley, F.-J. Yazigi, R. J. Marshall, R. S. Forgan, A. Porch and M. O. Jones, Phys. Chem. Chem. Phys., 2018, 20, 10460–10469 RSC.
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2007, 129, 5314–5315 CrossRef CAS PubMed.
- H. Wu, W. Zhou and T. Yildirim, J. Phys. Chem. C, 2009, 113, 3029–3035 CrossRef CAS.
- M. E. Casco, Y. Q. Cheng, L. L. Daemen, D. Fairen-Jimenez, E. V. Ramos-Fernandez, A. J. Ramirez-Cuesta and J. Silvestre-Albero, Chem. Commun., 2016, 52, 3639–3642 RSC.
- C. Chmelik and J. Karger, Chem. Soc. Rev., 2010, 39, 4864–4884 RSC.
- J. Kärger and D. Freude, Chem. Eng. Technol., 2002, 25, 769–778 CrossRef.
- N. Rosenbach, H. Jobic, A. Ghoufi, F. Salles, G. Maurin, S. Bourrelly, P. L. Llewellyn, T. Devic, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 6611–6615 CrossRef CAS PubMed.
- Q. Yang, A. D. Wiersum, H. Jobic, V. Guillerm, C. Serre, P. L. Llewellyn and G. Maurin, J. Phys. Chem. C, 2011, 115, 13768–13774 CrossRef CAS.
- F. Salles, D. I. Kolokolov, H. Jobic, G. Maurin, P. L. Llewellyn, T. Devic, C. Serre and G. Ferey, J. Phys. Chem. C, 2009, 113, 7802–7812 CrossRef CAS.
- F. Salles, H. Jobic, A. Ghoufi, P. L. Llewellyn, C. Serre, S. Bourrelly, G. Férey and G. Maurin, Angew. Chem., 2009, 121, 8485–8489 CrossRef.
- F. Salles, H. Jobic, T. Devic, P. L. Llewellyn, C. Serre, G. Férey and G. Maurin, ACS Nano, 2010, 4, 143–152 CrossRef CAS PubMed.
- D. I. Kolokolov, H. Jobic, A. G. Stepanov, J. Ollivier, S. Rives, G. Maurin, T. Devic, C. Serre and G. Férey, J. Phys. Chem. C, 2012, 116, 15093–15098 CrossRef CAS.
- S. Rives, H. Jobic, F. Ragon, T. Devic, C. Serre, G. Férey, J. Ollivier and G. Maurin, Microporous Mesoporous Mater., 2012, 164, 259–265 CrossRef CAS.
- H. Jobic and D. N. Theodorou, Microporous Mesoporous Mater., 2007, 102, 21–50 CrossRef CAS.
- Y. Liu, C. M. Brown, D. A. Neumann, V. K. Peterson and C. J. Kepert, J. Alloys Compd., 2007, 446–447, 385–388 CrossRef CAS.
- Q. Yang, H. Jobic, F. Salles, D. Kolokolov, V. Guillerm, C. Serre and G. Maurin, Chem. – Eur. J., 2011, 17, 8882–8889 CrossRef CAS PubMed.
- E. Pantatosaki, H. Jobic, D. I. Kolokolov, S. Karmakar, R. Biniwale and G. K. Papadopoulos, J. Chem. Phys., 2013, 138, 034706 CrossRef PubMed.
- H. Jobic, D. I. Kolokolov, A. G. Stepanov, M. M. Koza and J. Ollivier, J. Phys. Chem. C, 2015, 119, 16115–16120 CrossRef CAS.
- H. Jobic, Phys. Chem. Chem. Phys., 2016, 18, 17190–17195 RSC.
- K. Sumida, J.-H. Her, M. Dincă, L. J. Murray, J. M. Schloss, C. J. Pierce, B. A. Thompson, S. A. FitzGerald, C. M. Brown and J. R. Long, J. Phys. Chem. C, 2011, 115, 8414–8421 CrossRef CAS.
- K. Sumida, C. M. Brown, Z. R. Herm, S. Chavan, S. Bordiga and J. R. Long, Chem. Commun., 2011, 47, 1157–1159 RSC.
- S. Yang, A. J. Ramirez-Cuesta, R. Newby, V. Garcia-Sakai, P. Manuel, S. K. Callear, S. I. Campbell, C. C. Tang and M. Schröder, Nat. Chem., 2015, 7, 121–129 CrossRef CAS PubMed.
- N. A. Ramsahye, J. Gao, H. Jobic, P. L. Llewellyn, Q. Yang, A. D. Wiersum, M. M. Koza, V. Guillerm, C. Serre, C. L. Zhong and G. Maurin, J. Phys. Chem. C, 2014, 118, 27470–27482 CrossRef CAS.
- M. E. Casco, F. Rey, J. L. Jordá, S. Rudić, F. Fauth, M. Martínez-Escandell, F. Rodríguez-Reinoso, E. V. Ramos-Fernández and J. Silvestre-Albero, Chem. Sci., 2016, 7, 3658–3666 RSC.
- F. Stallmach, S. Gröger, V. Künzel, J. Kärger, O. M. Yaghi, M. Hesse and U. Müller, Angew. Chem., Int. Ed., 2006, 45, 2123–2126 CrossRef CAS PubMed.
- J. A. Gee, J. Chung, S. Nair and D. S. Sholl, J. Phys. Chem. C, 2013, 117, 3169–3176 CrossRef CAS.
- S. Hertel, M. Wehring, S. Amirjalayer, M. Gratz, J. Lincke, H. Krautscheid, R. Schmid and F. Stallmach, Eur. Phys. J.: Appl. Phys., 2011, 55, 20702 CrossRef.
- S. Schlayer, A.-K. Pusch, F. Pielenz, S. Beckert, M. Peksa, C. Horch, L. Moschkowitz, W.-D. Einicke and F. Stallmach, Materials, 2012, 5(4), 617–633 CrossRef CAS PubMed.
- A.-K. Pusch, T. Splith, L. Moschkowitz, S. Karmakar, R. Biniwale, M. Sant, G. B. Suffritti, P. Demontis, J. Cravillon, E. Pantatosaki and F. Stallmach, Adsorption, 2012, 18, 359–366 CrossRef CAS.
- C. Chmelik, D. Freude, H. Bux and J. Haase, Microporous Mesoporous Mater., 2012, 147, 135–141 CrossRef.
- M. Wehring, J. Gascon, D. Dubbeldam, F. Kapteijn, R. Q. Snurr and F. Stallmach, J. Phys. Chem. C, 2010, 114, 10527–10534 CrossRef CAS.
- F. Stallmach, A.-K. Pusch, T. Splith, C. Horch and S. Merker, Microporous Mesoporous Mater., 2015, 205, 36–39 CrossRef CAS.
- K. Díaz, L. Garrido, M. López-González, L. F. del Castillo and E. Riande, Macromolecules, 2010, 43, 316–325 CrossRef.
- P. V. Kortunov, L. Heinke, M. Arnold, Y. Nedellec, D. J. Jones, J. Caro and J. Kärger, J. Am. Chem. Soc., 2007, 129, 8041–8047 CrossRef CAS PubMed.
- C. Chmelik, J. Kärger, M. Wiebcke, J. Caro, J. M. van Baten and R. Krishna, Microporous Mesoporous Mater., 2009, 117, 22–32 CrossRef CAS.
- C. Chmelik, F. Hibbe, D. Tzoulaki, L. Heinke, J. Caro, J. Li and J. Kärger, Microporous Mesoporous Mater., 2010, 129, 340–344 CrossRef CAS.
- H. Bux, C. Chmelik, J. M. van Baten, R. Krishna and J. Caro, Adv. Mater., 2010, 22, 4741–4743 CrossRef CAS PubMed.
- C. Chmelik, J. van Baten and R. Krishna, J. Membr. Sci., 2012, 397–398, 87–91 CrossRef CAS.
- H. Bux, C. Chmelik, R. Krishna and J. Caro, J. Membr. Sci., 2011, 369, 284–289 CrossRef CAS.
- L. Hertäg, H. Bux, J. Caro, C. Chmelik, T. Remsungnen, M. Knauth and S. Fritzsche, J. Membr. Sci., 2011, 377, 36–41 CrossRef.
- C. Chmelik, H. Bux, J. Caro, L. Heinke, F. Hibbe, T. Titze and J. Kärger, Phys. Rev. Lett., 2010, 104, 085902 CrossRef PubMed.
- J. Karger, T. Binder, C. Chmelik, F. Hibbe, H. Krautscheid, R. Krishna and J. Weitkamp, Nat. Mater., 2014, 13, 333–343 CrossRef PubMed.
- D. S. Sholl, Nat. Chem., 2011, 3, 429–430 CrossRef CAS PubMed.
- D. Tzoulaki, L. Heinke, H. Lim, J. Li, D. Olson, J. Caro, R. Krishna, C. Chmelik and J. Kärger, Angew. Chem., Int. Ed., 2009, 48, 3525–3528 CrossRef CAS PubMed.
- C. Chmelik, L. Heinke, P. Kortunov, J. Li, D. Olson, D. Tzoulaki, J. Weitkamp and J. Kärger, ChemPhysChem, 2009, 10, 2623–2627 CrossRef CAS PubMed.
- L. Heinke and J. Kärger, Phys. Rev. Lett., 2011, 106, 074501 CrossRef PubMed.
- F. Hibbe, C. Chmelik, L. Heinke, S. Pramanik, J. Li, D. M. Ruthven, D. Tzoulaki and J. Kärger, J. Am. Chem. Soc., 2011, 133, 2804–2807 CrossRef CAS PubMed.
- H. Jobic, J. Kärger and M. Bée, Phys. Rev. Lett., 1999, 82, 4260–4263 CrossRef CAS.
- E. Pantatosaki, G. Megariotis, A.-K. Pusch, C. Chmelik, F. Stallmach and G. K. Papadopoulos, J. Phys. Chem. C, 2012, 116, 201–207 CrossRef CAS.
- L. Zhang, G. Wu and J. Jiang, J. Phys. Chem. C, 2014, 118, 8788–8794 CrossRef CAS.
- X. Wu, J. Huang, W. Cai and M. Jaroniec, RSC Adv., 2014, 4, 16503–16511 RSC.
- D. I. Kolokolov, A. G. Maryasov, J. Ollivier, D. Freude, J. Haase, A. G. Stepanov and H. Jobic, J. Phys. Chem. C, 2017, 121, 2844–2857 CrossRef CAS.
- S. Pili, S. P. Argent, C. G. Morris, P. Rought, V. García-Sakai, I. P. Silverwood, T. L. Easun, M. Li, M. R. Warren, C. A. Murray, C. C. Tang, S. Yang and M. Schröder, J. Am. Chem. Soc., 2016, 138, 6352–6355 CrossRef CAS PubMed.
- D. D. Borges, S. Devautour-Vinot, H. Jobic, J. Ollivier, F. Nouar, R. Semino, T. Devic, C. Serre, F. Paesani and G. Maurin, Angew. Chem., Int. Ed., 2016, 55, 3919–3924 CrossRef CAS PubMed.
- T. Splith, E. Pantatosaki, P. D. Kolokathis, D. Fröhlich, K. Zhang, G. Füldner, C. Chmelik, J. Jiang, S. K. Henninger, F. Stallmach and G. K. Papadopoulos, J. Phys. Chem. C, 2017, 121, 18065–18074 CrossRef CAS.
- J. Ethiraj, F. Bonino, C. Lamberti and S. Bordiga, Microporous Mesoporous Mater., 2015, 207, 90–94 CrossRef CAS.
- A. Dutta, N. Tymińska, G. Zhu, J. Collins, R. P. Lively, J. R. Schmidt and S. Vasenkov, J. Phys. Chem. C, 2018, 122, 7278–7287 CrossRef CAS.
- C. Chizallet, S. Lazare, D. Bazer-Bachi, F. Bonnier, V. Lecocq, E. Soyer, A.-A. Quoineaud and N. Bats, J. Am. Chem. Soc., 2010, 132, 12365–12377 CrossRef CAS PubMed.
- C. Chizallet and N. Bats, J. Phys. Chem. Lett., 2010, 1, 349–353 CrossRef CAS.
- A. V. Kubarev and M. B. J. Roeffaers, CrystEngComm, 2017, 19, 4162–4165 RSC.
- N. Dvoyashkina, D. Freude, S. S. Arzumanov and A. G. Stepanov, J. Phys. Chem. C, 2017, 121, 25372–25376 CrossRef CAS.
- T. G. Grissom, C. H. Sharp, P. M. Usov, D. Troya, A. J. Morris and J. R. Morris, J. Phys. Chem. C, 2018, 122, 16060–16069 CrossRef CAS.
- S. Berens, C. Chmelik, F. Hillman, J. Kärger, H.-K. Jeong and S. Vasenkov, Phys. Chem. Chem. Phys., 2018, 20, 23967–23975 RSC.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- D. J. Tranchemontagne, J. R. Hunt and O. M. Yaghi, Tetrahedron, 2008, 64, 8553–8557 CrossRef CAS.
- S. P. Gavrish and Y. D. Lampeka, Theor. Exp. Chem., 2013, 49, 130–134 CrossRef CAS.
- Z. G.-S. F. Qian-Rong, X. Ming-Hong, Z. Da-Liang, S. Xin, W. Gang, T. Ge, T. Lu-Lu, X. Ming and Q. Shi-Lun, Chem. J. Chin. Univ., 2004, 25, 1016–1018 Search PubMed.
- M. Kramer, U. Schwarz and S. Kaskel, J. Mater. Chem., 2006, 16, 2245–2248 RSC.
- O. Kozachuk, K. Yusenko, H. Noei, Y. Wang, S. Walleck, T. Glaser and R. A. Fischer, Chem. Commun., 2011, 47, 8509–8511 RSC.
- P. Maniam and N. Stock, Inorg. Chem., 2011, 50, 5085–5097 CrossRef CAS PubMed.
- C. R. Wade and M. Dinca, Dalton Trans., 2012, 41, 7931–7938 RSC.
- L. Xie, S. Liu, C. Gao, R. Cao, J. Cao, C. Sun and Z. Su, Inorg. Chem., 2007, 46, 7782–7788 CrossRef CAS PubMed.
- O. Kozachuk, I. Luz, F. X. Llabrés i Xamena, H. Noei, M. Kauer, H. B. Albada, E. D. Bloch, B. Marler, Y. Wang, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7058–7062 CrossRef CAS PubMed.
- C. H. Hendon and A. Walsh, Chem. Sci., 2015, 6, 3674–3683 RSC.
- Z. Fang, J. P. Dürholt, M. Kauer, W. Zhang, C. Lochenie, B. Jee, B. Albada, N. Metzler-Nolte, A. Pöppl, B. Weber, M. Muhler, Y. Wang, R. Schmid and R. A. Fischer, J. Am. Chem. Soc., 2014, 136, 9627–9636 CrossRef CAS PubMed.
- R. Ameloot, F. Vermoortele, J. Hofkens, F. C. De Schryver, D. E. De Vos and M. B. J. Roeffaers, Angew. Chem., Int. Ed., 2013, 52, 401–405 CrossRef CAS PubMed.
- I. Senkovska and V. Bon, The Chemistry of Metal–Organic Frameworks: Synthesis, Characterization, and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, pp. 691–727 DOI:10.1002/9783527693078.ch23.
- M. Mendt, M. Šimėnas and A. Pöppl, The Chemistry of Metal–Organic Frameworks: Synthesis, Characterization, and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, pp. 629–656 DOI:10.1002/9783527693078.ch21.
- E. Borfecchia, D. Gianolio, G. Agostini, S. Bordiga and C. Lamberti, Metal Organic Frameworks as Heterogeneous Catalysts, The Royal Society of Chemistry, 2013, pp. 143–208 10.1039/9781849737586-00143.
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337–1346 CrossRef CAS.
- K.-S. Lin, A. K. Adhikari, C.-N. Ku, C.-L. Chiang and H. Kuo, Int. J. Hydrogen Energy, 2012, 37, 13865–13871 CrossRef CAS.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856–7857 CrossRef CAS PubMed.
- E. Borfecchia, S. Maurelli, D. Gianolio, E. Groppo, M. Chiesa, F. Bonino and C. Lamberti, J. Phys. Chem. C, 2012, 116, 19839–19850 CrossRef CAS.
- N. Nijem, K. Fürsich, H. Bluhm, S. R. Leone and M. K. Gilles, J. Phys. Chem. C, 2015, 119, 24781–24788 CrossRef CAS.
- H. K. Kim, W. S. Yun, M.-B. Kim, J. Y. Kim, Y.-S. Bae, J. Lee and N. C. Jeong, J. Am. Chem. Soc., 2015, 137, 10009–10015 CrossRef CAS PubMed.
- H. Wu, J. M. Simmons, G. Srinivas, W. Zhou and T. Yildirim, J. Phys. Chem. Lett., 2010, 1, 1946–1951 CrossRef CAS.
- M. C. Brown, Y. Liu, T. Yildirim, V. K. Peterson and C. J. Kepert, Nanotechnology, 2009, 20, 204025 CrossRef PubMed.
- I. Krkljus and M. Hirscher, Microporous Mesoporous Mater., 2011, 142, 725–729 CrossRef CAS.
- D. F. Sava, K. W. Chapman, M. A. Rodriguez, J. A. Greathouse, P. S. Crozier, H. Zhao, P. J. Chupas and T. M. Nenoff, Chem. Mater., 2013, 25, 2591–2596 CrossRef CAS.
- S. Bordiga, L. Regli, F. Bonino, E. Groppo, C. Lamberti, B. Xiao, P. S. Wheatley, R. E. Morris and A. Zecchina, Phys. Chem. Chem. Phys., 2007, 9, 2676–2685 RSC.
- N. Nijem, H. Bluhm, M. L. Ng, M. Kunz, S. R. Leone and M. K. Gilles, Chem. Commun., 2014, 50, 10144–10147 RSC.
- A. H. Khan, K. Peikert, M. Fröba and M. Bertmer, Microporous Mesoporous Mater., 2015, 216, 111–117 CrossRef CAS.
- K. Peikert, L. J. McCormick, D. Cattaneo, M. J. Duncan, F. Hoffmann, A. H. Khan, M. Bertmer, R. E. Morris and M. Fröba, Microporous Mesoporous Mater., 2015, 216, 118–126 CrossRef CAS.
- B. Jee, M. Hartmann and A. Pöppl, Mol. Phys., 2013, 111, 2950–2966 CrossRef CAS.
- B. Jee, P. St. Petkov, G. N. Vayssilov, T. Heine, M. Hartmann and A. Pöppl, J. Phys. Chem. C, 2013, 117, 8231–8240 CrossRef CAS.
- F. Gul-E-Noor, M. Mendt, D. Michel, A. Pöppl, H. Krautscheid, J. Haase and M. Bertmer, J. Phys. Chem. C, 2013, 117, 7703–7712 CrossRef CAS.
- A. Kultaeva, W. Böhlmann, M. Hartmann, T. Biktagirov and A. Pöppl, J. Phys. Chem. C, 2019, 123, 26877–26887 CrossRef CAS.
- A. Kultaeva, W. Böhlmann, M. Hartmann, T. Biktagirov and A. Pöppl, J. Phys. Chem. Lett., 2019, 10, 7657–7664 CrossRef CAS PubMed.
- D. M. Shakya, O. A. Ejegbavwo, T. Rajeshkumar, S. D. Senanayake, A. J. Brandt, S. Farzandh, N. Acharya, A. M. Ebrahim, A. I. Frenkel, N. Rui, G. L. Tate, J. R. Monnier, K. D. Vogiatzis, N. B. Shustova and D. A. Chen, Angew. Chem., Int. Ed., 2019, 58, 16533 CrossRef CAS PubMed.
- H. Noei, O. Kozachuk, S. Amirjalayer, S. Bureekaew, M. Kauer, R. Schmid, B. Marler, M. Muhler, R. A. Fischer and Y. Wang, J. Phys. Chem. C, 2013, 117, 5658–5666 CrossRef CAS.
- I. Agirrezabal-Telleria, I. Luz, M. A. Ortuño, M. Oregui-Bengoechea, I. Gandarias, N. López, M. A. Lail and M. Soukri, Nat. Commun., 2019, 10, 2076 CrossRef PubMed.
- W. R. Heinz, T. Kratky, M. Drees, A. Wimmer, O. Tomanec, S. Günther, M. Schuster and R. A. Fischer, Dalton Trans., 2019, 48, 12031–12039 RSC.
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocellà, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590–595 CrossRef CAS PubMed.
- Y. Yue, H. Arman, Z. J. Tonzetich and B. Chen, Z. Anorg. Allg. Chem., 2019, 645, 797–800 CrossRef CAS.
- J. Bitzer, S. Otterbach, K. Thangavel, A. Kultaeva, R. Schmid, A. Poppl and W. Kleist, Chem., 2020, 26, 5667–5675 CrossRef CAS PubMed.
- W. Zhang, M. Kauer, O. Halbherr, K. Epp, P. Guo, M. I. Gonzalez, D. J. Xiao, C. Wiktor, F. X. Liabrés i Xamena, C. Wöll, Y. Wang, M. Muhler and R. A. Fischer, Chem. – Eur. J., 2016, 22, 14297–14307 CrossRef CAS PubMed.
- S. Marx, W. Kleist and A. Baiker, J. Catal., 2011, 281, 76–87 CrossRef CAS.
- L. Alaerts, E. Séguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem. – Eur. J., 2006, 12, 7353–7363 CrossRef CAS PubMed.
- O. Shekhah, J. Liu, R. A. Fischer and C. Woll, Chem. Soc. Rev., 2011, 40, 1081–1106 RSC.
- J. Szanyi, M. Daturi, G. Clet, D. R. Baer and C. H. F. Peden, Phys. Chem. Chem. Phys., 2012, 14, 4383–4390 RSC.
- P. St. Petkov, G. N. Vayssilov, J. Liu, O. Shekhah, Y. Wang, C. Wöll and T. Heine, ChemPhysChem, 2012, 13, 2025–2029 CrossRef CAS PubMed.
- Z. Wang, H. Sezen, J. Liu, C. Yang, S. E. Roggenbuck, K. Peikert, M. Fröba, A. Mavrandonakis, B. Supronowicz, T. Heine, H. Gliemann and C. Wöll, Microporous Mesoporous Mater., 2015, 207, 53–60 CrossRef CAS.
- C.-Y. Wang, P. Ray, Q. Gong, Y. Zhao, J. Li and A. D. Lueking, Phys. Chem. Chem. Phys., 2015, 17, 26766–26776 RSC.
- A. S. Duke, E. A. Dolgopolova, R. P. Galhenage, S. C. Ammal, A. Heyden, M. D. Smith, D. A. Chen and N. B. Shustova, J. Phys. Chem. C, 2015, 119, 27457–27466 CrossRef CAS.
- M. Todaro, G. Buscarino, L. Sciortino, A. Alessi, F. Messina, M. Taddei, M. Ranocchiari, M. Cannas and F. M. Gelardi, J. Phys. Chem. C, 2016, 120, 12879–12889 CrossRef CAS.
- M. Todaro, A. Alessi, L. Sciortino, S. Agnello, M. Cannas, F. M. Gelardi and G. Buscarino, J. Spectrosc., 2016, 2016, 7 Search PubMed.
- M. Todaro, L. Sciortino, F. M. Gelardi and G. Buscarino, J. Phys. Chem. C, 2017, 121, 24853–24860 CrossRef.
- K. Müller, N. Vankova, L. Schöttner, T. Heine and L. Heinke, Chem. Sci., 2019, 10, 153–160 RSC.
- C. Schneider, M. Mendt, A. Pöppl, V. Crocellà and R. A. Fischer, ACS Appl. Mater. Interfaces, 2020, 12, 1024–1035 CrossRef PubMed.
- A. Terracina, L. N. McHugh, M. Todaro, S. Agnello, P. S. Wheatley, F. M. Gelardi, R. E. Morris and G. Buscarino, J. Phys. Chem. C, 2019, 123, 28219–28232 CrossRef.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, S. H. Jhung, Y.-K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef PubMed.
- K. K. Tanabe and S. M. Cohen, Angew. Chem., Int. Ed., 2009, 48, 7424–7427 CrossRef PubMed.
- D. Farrusseng, J. Canivet and A. Quadrelli, Metal-Organic Frameworks, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 23–48 DOI:10.1002/9783527635856.ch2.
- S. J. Garibay, Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 7341–7349 CrossRef PubMed.
- K. K. Tanabe, Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2008, 130, 8508–8517 CrossRef PubMed.
- Z. Yin, S. Wan, J. Yang, M. Kurmoo and M.-H. Zeng, Coord. Chem. Rev., 2019, 378, 500–512 CrossRef.
- M. Meilikhov, K. Yusenko and R. A. Fischer, J. Am. Chem. Soc., 2009, 131, 9644–9645 CrossRef PubMed.
- C. Larabi and E. A. Quadrelli, Eur. J. Inorg. Chem., 2012, 3014–3022 CrossRef.
- I. S. Kim, J. Borycz, A. E. Platero-Prats, S. Tussupbayev, T. C. Wang, O. K. Farha, J. T. Hupp, L. Gagliardi, K. W. Chapman, C. J. Cramer and A. B. F. Martinson, Chem. Mater., 2015, 27, 4772–4778 CrossRef.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3(1), 31–38 CrossRef PubMed.
- A. A. Yakovenko, J. H. Reibenspies, N. Bhuvanesh and H.-C. Zhou, J. Appl. Crystallogr., 2013, 46, 346–353 CrossRef.
- A. A. Yakovenko, Z. Wei, M. Wriedt, J.-R. Li, G. J. Halder and H.-C. Zhou, Cryst. Growth Des., 2014, 14, 5397–5407 CrossRef.
- I. S. Kim, S. Ahn, N. A. Vermeulen, T. E. Webber, L. C. Gallington, K. W. Chapman, R. L. Penn, J. T. Hupp, O. K. Farha, J. M. Notestein and A. B. F. Martinson, J. Am. Chem. Soc., 2020, 142, 242–250 CrossRef PubMed.
- T. Drake, P. Ji and W. Lin, Acc. Chem. Res., 2018, 51, 2129–2138 CrossRef PubMed.
- I. S. Kim, O. K. Farha, J. T. Hupp, L. Gagliardi, K. W. Chapman, C. Cramer and A. B. F. Martinson, ECS Trans., 2016, 75, 93–99 CrossRef.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed.
- W. Bury, O. K. Farha, J. T. Hupp and J. E. Mondloch, US Pat., 20150031908A1, 2015 Search PubMed.
- R. C. Klet, T. C. Wang, L. E. Fernandez, D. G. Truhlar, J. T. Hupp and O. K. Farha, Chem. Mater., 2016, 28, 1213–1219 CrossRef CAS.
- S. Ahn, N. E. Thornburg, Z. Li, T. C. Wang, L. C. Gallington, K. W. Chapman, J. M. Notestein, J. T. Hupp and O. K. Farha, Inorg. Chem., 2016, 55, 11954–11961 CrossRef CAS PubMed.
- A. W. Peters, Z. Li, O. K. Farha and J. T. Hupp, ACS Nano, 2015, 9, 8484–8490 CrossRef CAS PubMed.
- L. C. Gallington, I. S. Kim, W.-G. Liu, A. A. Yakovenko, A. E. Platero-Prats, Z. Li, T. C. Wang, J. T. Hupp, O. K. Farha, D. G. Truhlar, A. B. F. Martinson and K. W. Chapman, J. Am. Chem. Soc., 2016, 138, 13513–13516 CrossRef CAS.
- A. W. Peters, Z. Li, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2016, 8, 20675–20681 CrossRef CAS.
- A. B. Thompson, D. R. Pahls, V. Bernales, L. C. Gallington, C. D. Malonzo, T. Webber, S. J. Tereniak, T. C. Wang, S. P. Desai, Z. Li, I. S. Kim, L. Gagliardi, R. L. Penn, K. W. Chapman, A. Stein, O. K. Farha, J. T. Hupp, A. B. F. Martinson and C. C. Lu, Chem. Mater., 2016, 28, 6753–6762 CrossRef.
- D. Yang, S. O. Odoh, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2015, 137, 7391–7396 CrossRef.
- D. Yang, S. O. Odoh, J. Borycz, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, ACS Catal., 2016, 6, 235–247 CrossRef.
- Z. Li, N. M. Schweitzer, A. B. League, V. Bernales, A. W. Peters, A. B. Getsoian, T. C. Wang, J. T. Miller, A. Vjunov, J. L. Fulton, J. A. Lercher, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 1977–1982 CrossRef.
- H. G. T. Nguyen, N. M. Schweitzer, C.-Y. Chang, T. L. Drake, M. C. So, P. C. Stair, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2014, 4, 2496–2500 CrossRef.
- R. C. Klet, S. Tussupbayev, J. Borycz, J. R. Gallagher, M. M. Stalzer, J. T. Miller, L. Gagliardi, J. T. Hupp, T. J. Marks, C. J. Cramer, M. Delferro and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 15680–15683 CrossRef PubMed.
- O. K. Farha, J. T. Hupp, M. Delferro and R. C. Klet, US Pat., 20160046738A1, 2016 Search PubMed.
- S. T. Madrahimov, J. R. Gallagher, G. Zhang, Z. Meinhart, S. J. Garibay, M. Delferro, J. T. Miller, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2015, 5, 6713–6718 CrossRef.
- M. Rimoldi, A. Nakamura, N. A. Vermeulen, J. J. Henkelis, A. K. Blackburn, J. T. Hupp, J. F. Stoddart and O. K. Farha, Chem. Sci., 2016, 7, 4980–4984 RSC.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef PubMed.
- M. Rimoldi, V. Bernales, J. Borycz, A. Vjunov, L. C. Gallington, A. E. Platero-Prats, I. S. Kim, J. L. Fulton, A. B. F. Martinson, J. A. Lercher, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 1058–1068 CrossRef.
- K. Manna, P. Ji, Z. Lin, F. X. Greene, A. Urban, N. C. Thacker and W. Lin, Nat. Commun., 2016, 7, 12610 CrossRef CAS.
- P. Ji, K. Manna, Z. Lin, A. Urban, F. X. Greene, G. Lan and W. Lin, J. Am. Chem. Soc., 2016, 138, 12234–12242 CrossRef CAS PubMed.
- Z. H. Syed, Z. Chen, K. B. Idrees, T. A. Goetjen, E. C. Wegener, X. Zhang, K. W. Chapman, D. M. Kaphan, M. Delferro and O. K. Farha, Organometallics, 2020, 39, 1123–1133 CrossRef.
- V. Bernales, D. Yang, J. Yu, G. Gümüşlü, C. J. Cramer, B. C. Gates and L. Gagliardi, ACS Appl. Mater. Interfaces, 2017, 9, 33511–33520 CrossRef CAS PubMed.
- D. Yang, M. R. Momeni, H. Demir, D. R. Pahls, M. Rimoldi, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, B. C. Gates and L. Gagliardi, Faraday Discuss., 2017, 201, 195–206 RSC.
- S. Kim In, Z. Li, J. Zheng, E. Platero-Prats Ana, A. Mavrandonakis, S. Pellizzeri, M. Ferrandon, A. Vjunov, C. Gallington Leighanne, E. Webber Thomas, A. Vermeulen Nicolaas, R. L. Penn, B. Getman Rachel, J. Cramer Christopher, W. Chapman Karena, M. Camaioni Donald, L. Fulton John, A. Lercher Johannes, K. Farha Omar, T. Hupp Joseph and B. F. Martinson Alex, Angew. Chem., Int. Ed., 2018, 57, 909–913 CrossRef PubMed.
- L. R. Redfern, Z. Li, X. Zhang and O. K. Farha, ACS Appl. Nano Mater., 2018, 1, 4413–4417 CrossRef CAS.
- A. E. Platero-Prats, Z. Li, L. C. Gallington, A. W. Peters, J. T. Hupp, O. K. Farha and K. W. Chapman, Faraday Discuss., 2017, 201, 337–350 RSC.
- J. Liu, J. Ye, Z. Li, K.-I. Otake, Y. Liao, A. W. Peters, H. Noh, D. G. Truhlar, L. Gagliardi, C. J. Cramer, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2018, 140, 11174–11178 CrossRef CAS PubMed.
- S. P. Desai, J. Ye, J. Zheng, M. S. Ferrandon, T. E. Webber, A. E. Platero-Prats, J. Duan, P. Garcia-Holley, D. M. Camaioni, K. W. Chapman, M. Delferro, O. K. Farha, J. L. Fulton, L. Gagliardi, J. A. Lercher, R. L. Penn, A. Stein and C. C. Lu, J. Am. Chem. Soc., 2018, 140, 15309–15318 CrossRef CAS PubMed.
- T. A. Goetjen, X. Zhang, J. Liu, J. T. Hupp and O. K. Farha, ACS Sustainable Chem. Eng., 2019, 7, 2553–2557 CrossRef CAS.
- J. Liu, L. R. Redfern, Y. Liao, T. Islamoglu, A. Atilgan, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2019, 11, 47822–47829 CrossRef CAS PubMed.
- T. C. Wang, N. A. Vermeulen, I. S. Kim, A. B. F. Martinson, J. F. Stoddart, J. T. Hupp and O. K. Farha, Nat. Protoc., 2016, 11, 149–162 CrossRef CAS PubMed.
- M. R. Mian, L. R. Redfern, S. M. Pratik, D. Ray, J. Liu, K. B. Idrees, T. Islamoglu, L. Gagliardi and O. K. Farha, Chem. Mater., 2020, 32, 3078–3086 CrossRef CAS.
- A. E. Platero-Prats, A. Bermejo Gómez, L. Samain, X. Zou and B. Martín-Matute, Chem. – Eur. J., 2014, 21, 861–866 CrossRef PubMed.
- A. E. Platero-Prats, A. Bermejo Gómez, K. W. Chapman, B. Martín-Matute and X. Zou, CrystEngComm, 2015, 17, 7632–7635 RSC.
- S. Øien, G. Agostini, S. Svelle, E. Borfecchia, K. A. Lomachenko, L. Mino, E. Gallo, S. Bordiga, U. Olsbye, K. P. Lillerud and C. Lamberti, Chem. Mater., 2015, 27, 1042–1056 CrossRef.
- L. Braglia, E. Borfecchia, A. Martini, A. L. Bugaev, A. V. Soldatov, S. Øien-Ødegaard, B. T. Lønstad-Bleken, U. Olsbye, K. P. Lillerud, K. A. Lomachenko, G. Agostini, M. Manzoli and C. Lamberti, Phys. Chem. Chem. Phys., 2017, 19, 27489–27507 RSC.
- E. S. Gutterød, S. Øien-Ødegaard, K. Bossers, A.-E. Nieuwelink, M. Manzoli, L. Braglia, A. Lazzarini, E. Borfecchia, S. Ahmadigoltapeh, B. Bouchevreau, B. T. Lønstad-Bleken, R. Henry, C. Lamberti, S. Bordiga, B. M. Weckhuysen, K. P. Lillerud and U. Olsbye, Ind. Eng. Chem. Res., 2017, 56, 13206–13218 CrossRef.
- A. L. Bugaev, A. A. Guda, K. A. Lomachenko, E. G. Kamyshova, M. A. Soldatov, G. Kaur, S. Øien-Ødegaard, L. Braglia, A. Lazzarini, M. Manzoli, S. Bordiga, U. Olsbye, K. P. Lillerud, A. V. Soldatov and C. Lamberti, Faraday Discuss., 2018, 208, 287–306 RSC.
- L. Braglia, E. Borfecchia, L. Maddalena, S. Øien, K. A. Lomachenko, A. L. Bugaev, S. Bordiga, A. V. Soldatov, K. P. Lillerud and C. Lamberti, Catal. Today, 2017, 283, 89–103 CrossRef CAS.
- L. Braglia, E. Borfecchia, K. A. Lomachenko, A. L. Bugaev, A. A. Guda, A. V. Soldatov, B. T. L. Bleken, S. Øien-Ødegaard, U. Olsbye, K. P. Lillerud, S. Bordiga, G. Agostini, M. Manzoli and C. Lamberti, Faraday Discuss., 2017, 201, 265–286 RSC.
- N. Van Velthoven, M. Henrion, J. Dallenes, A. Krajnc, A. L. Bugaev, P. Liu, S. Bals, A. V. Soldatov, G. Mali and D. E. De Vos, ACS Catal., 2020, 10, 5077–5085 CrossRef CAS.
- T. Toyao, K. Miyahara, M. Fujiwaki, T.-H. Kim, S. Dohshi, Y. Horiuchi and M. Matsuoka, J. Phys. Chem. C, 2015, 119, 8131–8137 CrossRef CAS.
- N. Yuan, V. Pascanu, Z. Huang, A. Valiente, N. Heidenreich, S. Leubner, A. K. Inge, J. Gaar, N. Stock, I. Persson, B. Martín-Matute and X. Zou, J. Am. Chem. Soc., 2018, 140, 8206–8217 CrossRef CAS PubMed.
- J. Liu and C. Wöll, Chem. Soc. Rev., 2017, 46, 5730–5770 RSC.
- A. Betard and R. A. Fischer, Chem. Rev., 2012, 112, 1055–1083 CrossRef CAS.
- M. Tu, S. Wannapaiboon and R. A. Fischer, Inorg. Chem. Front., 2014, 1, 442–463 RSC.
- W. J. Li, M. Tu, R. Cao and R. A. Fischer, J. Mater. Chem. A, 2016, 4, 12356–12369 RSC.
- D. Bradshaw, A. Garai and J. Huo, Chem. Soc. Rev., 2012, 41, 2344–2381 RSC.
- Z. G. Gu, A. Pfriem, S. Hamsch, H. Breitwieser, J. Wohlgemuth, L. Heinke, H. Gliemann and C. Woll, Microporous Mesoporous Mater., 2015, 211, 82–87 CrossRef CAS.
- L. Heinke, M. Tu, S. Wannapaiboon, R. A. Fischer and C. Woll, Microporous Mesoporous Mater., 2015, 216, 200–215 CrossRef CAS.
- O. Shekhah, H. Wang, S. Kowarik, F. Schreiber, M. Paulus, M. Tolan, C. Sternemann, F. Evers, D. Zacher, R. A. Fischer and C. Woll, J. Am. Chem. Soc., 2007, 129, 15118–15119 CrossRef CAS.
- H. Gliemann and C. Woll, Mater. Today, 2012, 15, 110–116 CrossRef CAS.
- J. X. Liu, O. Shekhah, X. Stammer, H. K. Arslan, B. Liu, B. Schupbach, A. Terfort and C. Woll, Materials, 2012, 5, 1581–1592 CrossRef CAS.
- O. Shekhah, H. Wang, M. Paradinas, C. Ocal, B. Schupbach, A. Terfort, D. Zacher, R. A. Fischer and C. Woll, Nat. Mater., 2009, 8, 481–484 CrossRef CAS PubMed.
- D. Zacher, R. Schmid, C. Woll and R. A. Fischer, Angew. Chem., Int. Ed., 2011, 50, 176–199 CrossRef CAS.
- J.-L. Zhuang, A. Terfort and C. Wöll, Coord. Chem. Rev., 2016, 307, 391–424 CrossRef CAS.
- B. Liu and R. A. Fischer, Sci. China: Chem., 2011, 54, 1851–1866 CrossRef CAS.
- O. Shekhah, H. Wang, T. Strunskus, P. Cyganik, D. Zacher, R. Fischer and C. Woll, Langmuir, 2007, 23, 7440–7442 CrossRef CAS PubMed.
- O. Shekhah, Materials, 2010, 3, 1302–1315 CrossRef CAS.
- L. D. B. Mandemaker, M. Rivera-Torrente, R. Geitner, C. M. Vis and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006347.
- G. Delen, Z. Ristanovic, L. D. B. Mandemaker and B. M. Weckhuysen, Chem. – Eur. J., 2018, 24, 187–195 CrossRef CAS PubMed.
- L. D. B. Mandemaker, M. Filez, G. Delen, H. S. Tan, X. H. Zhang, D. Lohse and B. M. Weckhuysen, J. Phys. Chem. Lett., 2018, 9, 1838–1844 CrossRef CAS PubMed.
- B. M. Weckhuysen, Z. Öztürk, R. P. Brand, J. M. Boereboom and F. Meirer, Chem. – Eur. J., 2019, 25, 8070–8084 CrossRef CAS PubMed.
- Z. Öztürk, M. Filez and M. Weckhuysen Bert, Chem. – Eur. J., 2017, 23, 10915–10924 CrossRef PubMed.
- L. D. B. Mandemaker, M. Rivera-Torrente, G. Delen, J. P. Hofmann, M. Lorenz, A. Belianinov and B. M. Weckhuysen, Chem. – Eur. J., 2020, 26, 691–698 CrossRef CAS PubMed.
- M. Tu and R. A. Fischer, J. Mater. Chem. A, 2014, 2, 2018–2022 RSC.
- S. Wannapaiboon, M. Tu and R. A. Fischer, Adv. Funct. Mater., 2014, 24, 2696–2705 CrossRef CAS.
- P. De Luna, W. B. Liang, A. Mallick, O. Shekhah, F. P. G. de Arquer, A. H. Proppe, P. Todorovic, S. O. Kelley, E. H. Sargent and M. Eddaoudi, ACS Appl. Mater. Interfaces, 2018, 10, 31225–31232 CrossRef CAS PubMed.
- O. Shekhah, K. Hirai, H. Wang, H. Uehara, M. Kondo, S. Diring, D. Zacher, R. A. Fischer, O. Sakata, S. Kitagawa, S. Furukawa and C. Woll, Dalton Trans., 2011, 40, 4954–4958 RSC.
- Y. Yoo and H. K. Jeong, Cryst. Growth Des., 2010, 10, 1283–1288 CrossRef CAS.
- Z. B. Wang, J. X. Liu, B. Lukose, Z. G. Gu, P. G. Weidler, H. Gliemann, T. Heine and C. Woll, Nano Lett., 2014, 14, 1526–1529 CrossRef CAS PubMed.
- K. L. Mulfort, O. K. Farha, C. D. Malliakas, M. G. Kanatzidis and J. T. Hupp, Chem. – Eur. J., 2010, 16, 276–281 CrossRef CAS PubMed.
- R. P. Brand, L. D. B. Mandemaker, G. Delen, N. Rijnveld and B. M. Weckhuysen, ChemPhysChem, 2018, 19, 2397–2404 CrossRef CAS PubMed.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepulveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. Llabres i Xamena, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- C. J. Doonan and C. J. Sumby, CrystEngComm, 2017, 19, 4044–4048 RSC.
- A. Burgun, C. J. Coghlan, D. M. Huang, W. Chen, S. Horike, S. Kitagawa, J. F. Alvino, G. F. Metha, C. J. Sumby and C. J. Doonan, Angew. Chem., Int. Ed., 2017, 56, 8412–8416 CrossRef CAS PubMed.
- R. Peralta, M. Huxley, R. Young, O. M. Linder-Patton, J. D. Evans, C. J. Doonan and C. J. Sumby, Faraday Discuss., 2020 10.1039/D0FD00012D.
- R. J. Young, M. T. Huxley, E. Pardo, N. R. Champness, C. J. Sumby and C. J. Doonan, Chem. Sci., 2020, 11, 4031–4050 RSC.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- Z. Huang, M. Ge, F. Carraro, C. J. Doonan, P. Falcaro and X. Zou, Faraday Discuss., 2020 10.1039/D0FD00015A.
- V. Bon, E. Brunner, A. Pöppl and S. Kaskel, Adv. Funct. Mater., 2020, 1907847 CrossRef.
- M. C. Simons, J. G. Vitillo, M. Babucci, A. S. Hoffman, A. Boubnov, M. L. Beauvais, Z. Chen, C. J. Cramer, K. W. Chapman, S. R. Bare, B. C. Gates, C. C. Lu, L. Gagliardi and A. Bhan, J. Am. Chem. Soc., 2019, 141, 18142–18151 CrossRef CAS PubMed.
- Z. L. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- R. A. Dodson, A. G. Wong-Foy and A. J. Matzger, Chem. Mater., 2018, 30, 6559–6565 CrossRef CAS.
- R. Medishetty, J. K. Zaręba, D. Mayer, M. Samoć and R. A. Fischer, Chem. Soc. Rev., 2017, 46, 4976–5004 RSC.
- L. R. Mingabudinova, V. V. Vinogradov, V. A. Milichko, E. Hey-Hawkins and A. V. Vinogradov, Chem. Soc. Rev., 2016, 45, 5408–5431 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- M. F. de Lange, K. J. F. M. Verouden, T. J. H. Vlugt, J. Gascon and F. Kapteijn, Chem. Rev., 2015, 115, 12205–12250 CrossRef CAS PubMed.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66 CrossRef CAS PubMed.
- L. Sun, G. Campbell Michael and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- C. Schneider, D. Ukaj, R. Koerver, A. A. Talin, G. Kieslich, S. P. Pujari, H. Zuilhof, J. Janek, M. D. Allendorf and R. A. Fischer, Chem. Sci., 2018, 9, 7405–7412 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- F. Nouar, T. Devic, H. Chevreau, N. Guillou, E. Gibson, G. Clet, M. Daturi, A. Vimont, J. M. Grenèche, M. I. Breeze, R. I. Walton, P. L. Llewellyn and C. Serre, Chem. Commun., 2012, 48, 10237–10239 RSC.
- M. I. Breeze, G. Clet, B. C. Campo, A. Vimont, M. Daturi, J.-M. Grenèche, A. J. Dent, F. Millange and R. I. Walton, Inorg. Chem., 2013, 52, 8171–8182 CrossRef CAS PubMed.
- L. Mitchell, P. Williamson, B. Ehrlichová, A. E. Anderson, V. R. Seymour, S. E. Ashbrook, N. Acerbi, L. M. Daniels, R. I. Walton, M. L. Clarke and P. A. Wright, Chem. – Eur. J., 2014, 20, 17185–17197 CrossRef CAS PubMed.
- L. M. Aguirre-Díaz, F. Gándara, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. Á. Monge, J. Am. Chem. Soc., 2015, 137, 6132–6135 CrossRef PubMed.
- C. Castillo-Blas and F. Gándara, Isr. J. Chem., 2018, 58, 1036–1043 CrossRef CAS.
- A. D. Burrows, CrystEngComm, 2011, 13, 3623–3642 RSC.
- M. Giménez-Marqués, A. Santiago-Portillo, S. Navalón, M. Álvaro, V. Briois, F. Nouar, H. Garcia and C. Serre, J. Mater. Chem. A, 2019, 7, 20285–20292 RSC.
- A. F. Sapnik, H. S. Geddes, E. M. Reynolds, H. H. M. Yeung and A. L. Goodwin, Chem. Commun., 2018, 54, 9651–9654 RSC.
- C. Castillo-Blas, N. López-Salas, M. C. Gutiérrez, I. Puente-Orench, E. Gutiérrez-Puebla, M. L. Ferrer, M. Á. Monge and F. Gándara, J. Am. Chem. Soc., 2019, 141, 1766–1774 CrossRef CAS PubMed.
- D. Zhang, Y. Zhu, L. Liu, X. Ying, C.-E. Hsiung, R. Sougrat, K. Li and Y. Han, Science, 2018, 359, 675 CrossRef CAS PubMed.
- L. Zhu, D. Zhang, M. Xue, H. Li and S. Qiu, CrystEngComm, 2013, 15, 9356–9359 RSC.
- Q. Chen, C. Dwyer, G. Sheng, C. Zhu, X. Li, C. Zheng and Y. Zhu, Adv. Mater., 2020, 32, e1907619 CrossRef PubMed.
- T.-Y. Luo, C. Liu, X. Y. Gan, P. F. Muldoon, N. A. Diemler, J. E. Millstone and N. L. Rosi, J. Am. Chem. Soc., 2019, 141, 2161–2168 CrossRef CAS PubMed.
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2013, 341, 882–885 CrossRef CAS PubMed.
- A. M. Katzenmeyer, J. Canivet, G. Holland, D. Farrusseng and A. Centrone, Angew. Chem., Int. Ed., 2014, 53, 2852–2856 CrossRef CAS PubMed.
- J. Lyu, X. Gong, S.-J. Lee, K. Gnanasekaran, X. Zhang, M. C. Wasson, X. Wang, P. Bai, X. Guo, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 4609–4615 CrossRef CAS PubMed.
- I. L. C. Buurmans and B. M. Weckhuysen, Nat. Chem., 2012, 4, 873–886 CrossRef CAS PubMed.
- N. Kumar, B. Stephanidis, R. Zenobi, A. J. Wain and D. Roy, Nanoscale, 2015, 7, 7133–7137 RSC.
- F. Meirer, D. T. Morris, S. Kalirai, Y. Liu, J. C. Andrews and B. M. Weckhuysen, J. Am. Chem. Soc., 2015, 137, 102–105 CrossRef CAS PubMed.
- T. Hartman, C. S. Wondergem, N. Kumar, A. van den Berg and B. M. Weckhuysen, J. Phys. Chem. Lett., 2016, 7, 1570–1584 CrossRef CAS PubMed.
- D. E. Perea, I. Arslan, J. Liu, Z. Ristanovic, L. Kovarik, B. W. Arey, J. A. Lercher, S. R. Bare and B. M. Weckhuysen, Nat. Commun., 2015, 6, 7589 CrossRef PubMed.
- K. C. Jayachandrababu, D. S. Sholl and S. Nair, J. Am. Chem. Soc., 2017, 139, 5906–5915 CrossRef CAS PubMed.
- W. Schrimpf, J. Jiang, Z. Ji, P. Hirschle, D. C. Lamb, O. M. Yaghi and S. Wuttke, Nat. Commun., 2018, 9, 1647 CrossRef PubMed.
- A. Ghosh, P. Mukherjee, S. Deb and R. Bhargava, J. Phys. Chem. Lett., 2017, 8, 5325–5330 CrossRef CAS PubMed.
- O. I. Lebedev, F. Millange, C. Serre, G. Van Tendeloo and G. Férey, Chem. Mater., 2005, 17, 6525–6527 CrossRef CAS.
- A. Mayoral, M. Sanchez-Sanchez, A. Alfayate, J. Perez-Pariente and I. Diaz, ChemCatChem, 2015, 7, 3719–3724 CrossRef CAS.
- A. Mayoral, R. Mahugo, M. Sánchez-Sánchez and I. Díaz, ChemCatChem, 2017, 9, 3497–3502 CrossRef CAS.
- M. Rivera-Torrente, M. Filez, F. Meirer and B. M. Weckhuysen, Chem. – Eur. J., 2020, 26, 3614–3625 CrossRef CAS PubMed.
- M. Rivera-Torrente, M. Filez, C. Schneider, E. C. van der Feltz, K. Wolkersdörfer, D. H. Taffa, M. Wark, R. A. Fischer and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2019, 21, 25678–25689 RSC.
- X. Cao, S. Hong, Z. Jiang, Y. She, S. Wang, C. Zhang, H. Li, F. Jin, M. Jin and J. Wang, Analyst, 2017, 142, 2640–2647 RSC.
- K. Sugikawa, Y. Furukawa and K. Sada, Chem. Mater., 2011, 23, 3132–3134 CrossRef CAS.
- X. Ma, S. Wen, X. Xue, Y. Guo, J. Jin, W. Song and B. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 25726–25736 CrossRef CAS PubMed.
- Y. Cai, Y. Wu, T. Xuan, X. Guo, Y. Wen and H. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 15412–15417 CrossRef CAS PubMed.
- X. Qiao, B. Su, C. Liu, Q. Song, D. Luo, G. Mo and T. Wang, Adv. Mater., 2018, 30, 1702275 CrossRef PubMed.
- Z. Jiang, P. Gao, L. Yang, C. Huang and Y. Li, Anal. Chem., 2015, 87, 12177–12182 CrossRef CAS PubMed.
- C. S. L. Koh, H. K. Lee, X. Han, H. Y. F. Sim and X. Y. Ling, Chem. Commun., 2018, 54, 2546–2549 RSC.
- X. Kuang, S. Ye, X. Li, Y. Ma, C. Zhang and B. Tang, Chem. Commun., 2016, 52, 5432–5435 RSC.
- G. Zheng, S. de Marchi, V. López-Puente, K. Sentosun, L. Polavarapu, I. Pérez-Juste, E. H. Hill, S. Bals, L. M. Liz-Marzán, I. Pastoriza-Santos and J. Pérez-Juste, Small, 2016, 12, 3935–3943 CrossRef CAS PubMed.
- Y. Zhao, N. Kornienko, Z. Liu, C. Zhu, S. Asahina, T.-R. Kuo, W. Bao, C. Xie, A. Hexemer, O. Terasaki, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2199–2202 CrossRef CAS PubMed.
- H. Y. F. Sim, H. K. Lee, X. Han, C. S. L. Koh, G. C. Phan-Quang, C. L. Lay, Y.-C. Kao, I. Y. Phang, E. K. L. Yeow and X. Y. Ling, Angew. Chem., Int. Ed., 2018, 57, 17058–17062 CrossRef CAS PubMed.
- T.-H. Yu, C.-H. Ho, C.-Y. Wu, C.-H. Chien, C.-H. Lin and S. Lee, J. Raman Spectrosc., 2013, 44, 1506–1511 CrossRef CAS.
- H. Sun, S. Cong, Z. Zheng, Z. Wang, Z. Chen and Z. Zhao, J. Am. Chem. Soc., 2019, 141, 870–878 CrossRef CAS PubMed.
- E. S. Gutterød, A. Lazzarini, T. Fjermestad, G. Kaur, M. Manzoli, S. Bordiga, S. Svelle, K. P. Lillerud, E. Skúlason, S. Øien-Ødegaard, A. Nova and U. Olsbye, J. Am. Chem. Soc., 2020, 142, 999–1009 CrossRef PubMed.
- Y. Chen, O. Sakata, Y. Nanba, L. S. R. Kumara, A. Yang, C. Song, M. Koyama, G. Li, H. Kobayashi and H. Kitagawa, Commun. Chem., 2018, 1, 61 CrossRef.
- M. Köppen, A. Dhakshinamoorthy, A. K. Inge, O. Cheung, J. Ångström, P. Mayer and N. Stock, Eur. J. Inorg. Chem., 2018, 3496–3503 CrossRef.
- N. Heidenreich, S. Waitschat and H. Reinsch, Z. Anorg. Allg. Chem., 2018, 644, 1826–1831 CrossRef CAS.
- S. Van Cleuvenbergen, Z. J. Smith, O. Deschaume, C. Bartic, S. Wachsmann-Hogiu, T. Verbiest and M. A. van der Veen, Nat. Commun., 2018, 9, 3418 CrossRef PubMed.
- N. Hosono, A. Terashima, S. Kusaka, R. Matsuda and S. Kitagawa, Nat. Chem., 2019, 11, 109–116 CrossRef CAS PubMed.
- F. C. Hendriks, D. Valencia, P. C. A. Bruijnincx and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2017, 19, 1857–1867 RSC.
- F. C. Hendriks, J. E. Schmidt, J. A. Rombouts, K. Lammertsma, P. C. A. Bruijnincx and B. M. Weckhuysen, Chem. – Eur. J., 2017, 23, 6305–6314 CrossRef CAS PubMed.
- F. C. Hendriks, F. Meirer, A. V. Kubarev, Z. Ristanović, M. B. J. Roeffaers, E. T. C. Vogt, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Am. Chem. Soc., 2017, 139, 13632–13635 CrossRef CAS PubMed.
- F. C. Hendriks, S. Mohammadian, Z. Ristanović, S. Kalirai, F. Meirer, E. T. C. Vogt, P. C. A. Bruijnincx, H. C. Gerritsen and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2018, 57, 257–261 CrossRef CAS PubMed.
- G. T. Whiting, N. Nikolopoulos, I. Nikolopoulos, A. D. Chowdhury and B. M. Weckhuysen, Nat. Chem., 2019, 11, 23–31 CrossRef CAS PubMed.
- Z. Ristanović, A. V. Kubarev, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, J. Am. Chem. Soc., 2016, 138, 13586–13596 CrossRef PubMed.
- Z. Ristanović, M. M. Kerssens, A. V. Kubarev, F. C. Hendriks, P. Dedecker, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2015, 54, 1836–1840 CrossRef PubMed.
- G. Fleury, J. A. Steele, I. C. Gerber, F. Jolibois, P. Puech, K. Muraoka, S. H. Keoh, W. Chaikittisilp, T. Okubo and M. B. J. Roeffaers, J. Phys. Chem. Lett., 2018, 9, 1778–1782 CrossRef CAS PubMed.
- F. Meirer, S. Kalirai, D. Morris, S. Soparawalla, Y. Liu, G. Mesu, J. C. Andrews and B. M. Weckhuysen, Sci. Adv., 2015, 1, e1400199 CrossRef PubMed.
- I. Strauss, A. Mundstock, D. Hinrichs, R. Himstedt, A. Knebel, C. Reinhardt, D. Dorfs and J. Caro, Angew. Chem., Int. Ed., 2018, 57, 7434–7439 CrossRef CAS PubMed.
- X. Teng, F. Li and C. Lu, Chem. Soc. Rev., 2020, 49, 2408–2425 RSC.
- Y. Shen, B. Shan, B. Mu and S. Tongay, Angew. Chem., Int. Ed., 2020, 59, 9408 CrossRef CAS PubMed.
- D. N. Johnstone, F. C. N. Firth, C. P. Grey, P. A. Midgley, M. J. Cliffe and S. M. Collins, J. Am. Chem. Soc., 2020, 142(30), 13081–13089 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the bibliometric study on tools for MOF characterization. See DOI: 10.1039/d0cs00635a |
| This journal is © The Royal Society of Chemistry 2020 |