
Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes
Zhong-Liang
Li†
ab,
Gui-Chun
Fang†
a,
Qiang-Shuai
Gu†
 ab and
Xin-Yuan
Liu
*a
ab and
Xin-Yuan
Liu
*a
aShenzhen Grubbs Institute and Department of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: liuxy3@sustech.edu.cn
bAcademy for Advanced Interdisciplinary Studies, Southern University of Science and Technology, Shenzhen, 518055, China
First published on 5th December 2019
Abstract
The radical-involved 1,2-difunctionalization of alkenes has developed into a robust tool for preparation of complex organic molecules. Despite significant advances in this area, the catalytic asymmetric version still remains a challenging task mainly due to the difficulty in the stereocontrol of the highly reactive radical intermediates. Recently, owing to the good single-electron transfer ability and coordination with chiral ligands of copper catalysts, remarkable achievements in radical-involved asymmetric alkene difunctionalization have been made via synergistic combination of copper and chiral ligands. This tutorial review highlights the recent progress in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes and the mechanistic scenarios governing the stereocontrol, with an emphasis on utilization of chiral ligands.
 Zhong-Liang Li | Zhong-Liang Li obtained his BSc degree from the University of Science and Technology of China in 2009 and PhD degree from The University of Hong Kong in 2014 under the supervision of Professor Dan Yang. In 2015, he did his postdoctoral research with Professor Xin-Yuan Liu and then worked as a research assistant professor in Southern University of Science and Technology. Now his work focuses on radical-involved remote C–C bond cleavage and asymmetric radical-involved reactions. |
 Gui-Chun Fang | Gui-Chun Fang received her BS degree in chemistry in 2012 and her PhD degree in 2018 in organic chemistry from Northeast Normal University, P. R. China, under the joint supervision of Professors X. Bi and Q. Liu. Her current research interest involves transition metal-catalyzed radical transformations and asymmetric synthesis. |
 Qiang-Shuai Gu | Qiang-Shuai Gu completed his BSc studies in 2008 from the University of Science and Technology of China (USTC) and received his PhD degree in 2013 from The University of Hong Kong (HKU) under the guidance of Professor Dan Yang. He continued his research career first in Professor Yang's group at HKU and later in Professor Xin-Yuan Liu's group at Southern University of Science and Technology (SUSTech). Now, he is a research assistant professor in Academy for Advanced Interdisciplinary Studies at SUSTech. His research interests include asymmetric catalysis and asymmetric synthesis of bioactive small molecules. |
 Xin-Yuan Liu | Xin-Yuan Liu obtained his BS degree from Anhui Normal University (AHNU) and Master's degree from Shanghai Institute of Organic Chemistry, CAS and AHNU under the joint supervision of Prof. Shizheng Zhu and Prof. Shaowu Wang. He earned his PhD degree from The University of Hong Kong under the supervision of Prof. Chi-Ming Che in 2010. He continued postdoctoral study in the University of Hong Kong and The Scripps Research Institute. In 2012, he began his academic career in Southern University of Science and Technology and was promoted to tenured Full Professor in 2018. His research interest is radical-involved asymmetric chemistry. |
Key learning points(1) General reaction pathway for radical-involved alkene difunctionalization.(2) The role of copper catalysis in asymmetric radical-involved alkene difunctionalization. (3) Strategy in stereocontrol of the highly reactive radical intermediates. (4) Stereocontrol of alkene nucleocupration reaction. |
1. Introduction
Alkenes are ubiquitous in all facets of chemistry and thus are privileged building blocks in organic synthesis. In recent years, radical reactions are experiencing a renaissance due to their high reactivity, chemoselectivity and functional group tolerance.1 Thus the radical-involved 1,2-difunctionalization of alkenes, simultaneously installing two functional groups at vicinal positions, has emerged as an elegant and versatile strategy that enables straightforward construction of complex molecules from simple alkene feedstocks.2 However, the catalytic asymmetric version for expeditious assembly of enantioenriched complex molecules remains a formidable challenge, mainly due to the difficulty related to the stereocontrol of the highly reactive radical species.To solve the challenges in radical-involved asymmetric control, efficient protocols have been established during the past few decades, such as the utilization of chiral Lewis acid,3 photoredox and organocatalysis,4 and transition metal catalysis.5 In particular, pioneered by Fu, Chemler and others, significant efforts have been made by merging transition metal catalysis and radical reactions, thus bringing the radical chemistry into the realm of organometallic chemistry to achieve asymmetric control.6,7 In this scenario, the utilization of chiral transition metal catalysis to realize the asymmetric radical-involved 1,2-difunctionalizaiton of alkenes has been a hot topic in the past decade, wherein the chiral transition metal complex serves mainly as the redox catalyst.5,7 Amongst others, copper-catalysed radical-involved 1,2-difunctionalization of alkenes exhibits unique advantages: (1) copper is inexpensive, readily available and easily handled. (2) Copper is a good single-electron transfer (SET) catalyst to initiate the generation of radicals from a wide range of radical precursors. (3) Copper can efficiently trap the prochiral alkyl radical intermediate to constitute a high-valent CuIII complex, thus facilitating bond formation via reductive elimination.8 (4) As a good Lewis acid, copper can coordinate with diverse kinds of chiral ligands and has a high affinity for alkenes and other functional groups, thus promoting the stereochemical control of radical alkene 1,2-functionalization reactions.7 Therefore, an important breakthrough in copper-catalysed radical-involved asymmetric alkene difunctionalization has been made in recent years.
The aim of this review is to unveil the progress in copper-catalysed asymmetric radical difunctionalization of alkenes in the past decade, covering copper-catalysed radical addition to alkenes and alkene nucleocupration followed by radical trapping reactions (Scheme 1). The reactions are mainly classified by the chiral ligands (L*), such as chiral bisoxazoline (Box), diphosphine, chiral phosphoric acid (CPA), and cinchona alkaloid-derived sulfonamide. We will highlight the reaction design and the mechanism, providing readers with invaluable knowledge for designing novel synthetic methods in the future. We regret that due to the limited space we could not include the asymmetric synthesis of three-membered rings from alkenes which has been well documented in the literature.9
 | ||
| Scheme 1 Copper-catalysed radical-involved asymmetric alkene difunctionalization. | ||
2. Copper(I)-catalysed radical addition to alkenes
Copper (3d104s1) has four oxidation states (Cu0, CuI, CuII and CuIII) and thus is involved in a number of redox reactions either in a one-electron or two-electron manner.10 It often serves as the single-electron reduction catalyst with diverse oxidative radical precursors to initiate the radical addition to alkenes. The general catalytic cycle for this type of reactions is shown in Scheme 2.8,11 The CuIL* species first reacts with the oxidative radical precursor A–Y in a SET process to generate the corresponding radical A and CuIIL*. While A selectively adds to an alkene moiety to deliver a new carbon-centered radical I, CuIIL* undergoes transmetalation with a trapping reagent (nucleophilic reagent, [Nu]) to afford L*CuIINu. The resulting alkyl radical I reacts with L*CuIINu (II) to deliver the difunctionalization product and release the CuIL* catalyst. Thus, the catalytic cycle is closed, meanwhile achieving stereocontrol. It should be noted that in some cases of radical alkene difunctionalization, the transmetalation between CuI and the trapping reagent might occur firstly to generate a CuINu species for the subsequent catalytic cycle.12 The new chiral C–Nu bond formation may proceed via a CuIII species and subsequent reductive elimination (pathway A in Scheme 2). On the other hand, trapping of L*CuIINu by alternative outer-sphere radical substitution (pathway B) and oxidation of the intermediate II to a carbocation intermediate followed by nucleophilic trapping (pathway C) are also possible.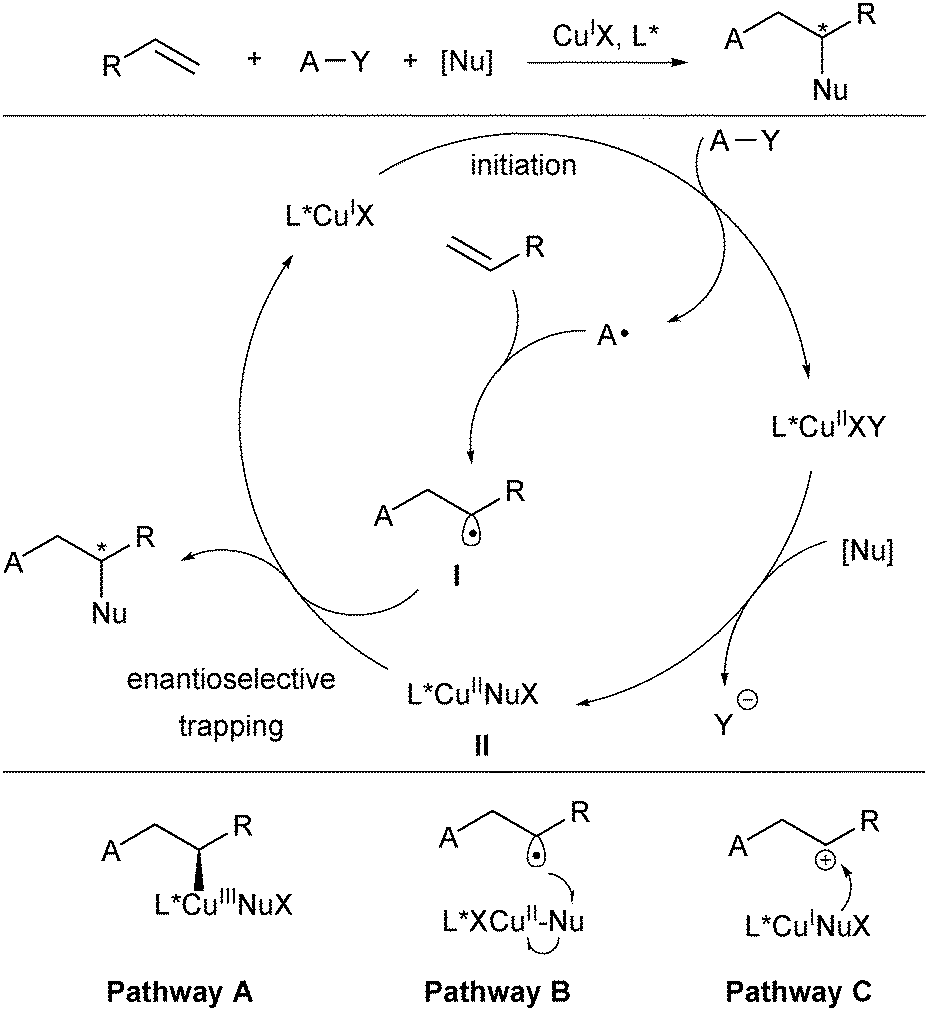 | ||
| Scheme 2 Plausible mechanism of copper(I)-catalysed radical addition to alkenes. | ||
2.1. Copper(I)/chiral Box catalysis
In 2012, Buchwald and co-workers reported a copper(I)-catalysed oxytrifluoromethylation of unactivated alkenes.13 In their study, they found that a bidentate pyridine-based ligand could assist the C–O bond formation between an oxygen-based nucleophile and the α-CF3-alkyl radical intermediate.13 This ligand effect prompted them to test the asymmetric version through the addition of a chiral Box-based ligand. Afterwards, they accomplished the copper-catalysed asymmetric oxytrifluoromethylation of alkenes with chiral Box ligands in 2013.14 They found that in the presence of [Cu(CH3CN)4]PF6 and Box ligand L1, arylated or heteroarylated alkenes 1 and Togni's reagent 2a reacted smoothly to deliver the trifluoromethylated lactones 3 in good enantioselectivity (Scheme 3). Both 5- and 6-member-ring lactones can be obtained. Based on their mechanistic study, the authors proposed a possible mechanism (Scheme 3). A SET process between L1CuI and 2a generated a CF3 radical and L1CuII. The in situ generated CF3 radical selectively added to the alkene of 1 to provide a transient α-CF3 alkyl radical intermediate I, which underwent the enantioselective C–O bond formation with the assistance of L1CuII and released L1CuI possibly via a CuIII intermediate.15 | ||
| Scheme 3 Buchwald's asymmetric oxytrifluoromethylation of alkenes. | ||
In 2015, the same group extended this protocol to a diverse radical enantioselective oxyfunctionalization of alkenes 1 under slightly modified reaction conditions (Scheme 4).15 For example, utilizing TMSN3 as the azidyl radical precursor and PhI(OAc)2 as an oxidant, the asymmetric oxyazidation products 5 were afforded in up to 92% ee. When sulfonyl chloride 6 was employed as the radical precursor and silver carbonate as both a base and a chloride scavenger, the oxysulfonylation products 7 were generated in up to 80% ee. Further, using aryl diazonium salt 8 as the radical precursor and 2,6-di-tert-butylpyridine as a base, the reaction provided products 9 in up to 76% ee. A trial with benzoyl peroxide 10, in which Mn was used to control the concentration of active radicals, allowed the formation of 11 and 12 in moderate enantioselectivity. Based on their mechanistic study, the authors assumed that the CuII carboxylate might combine with the prochiral benzyl radical intermediate I to form a CuIII complex II, followed by reductive elimination to construct the C–O bond (Scheme 4b). Since the reductive elimination step is rapid, it is likely that the combination of CuII with the prochiral benzyl radical is the enantiodetermining step. Notably, the authors cannot exclude the involvement of a tertiary carbocation intermediate although a Hammett study showed that a fully developed benzylic carbocation is unlikely.
 | ||
| Scheme 4 Radical-involved asymmetric oxyfunctionalization of alkenes with diverse radical precursors. | ||
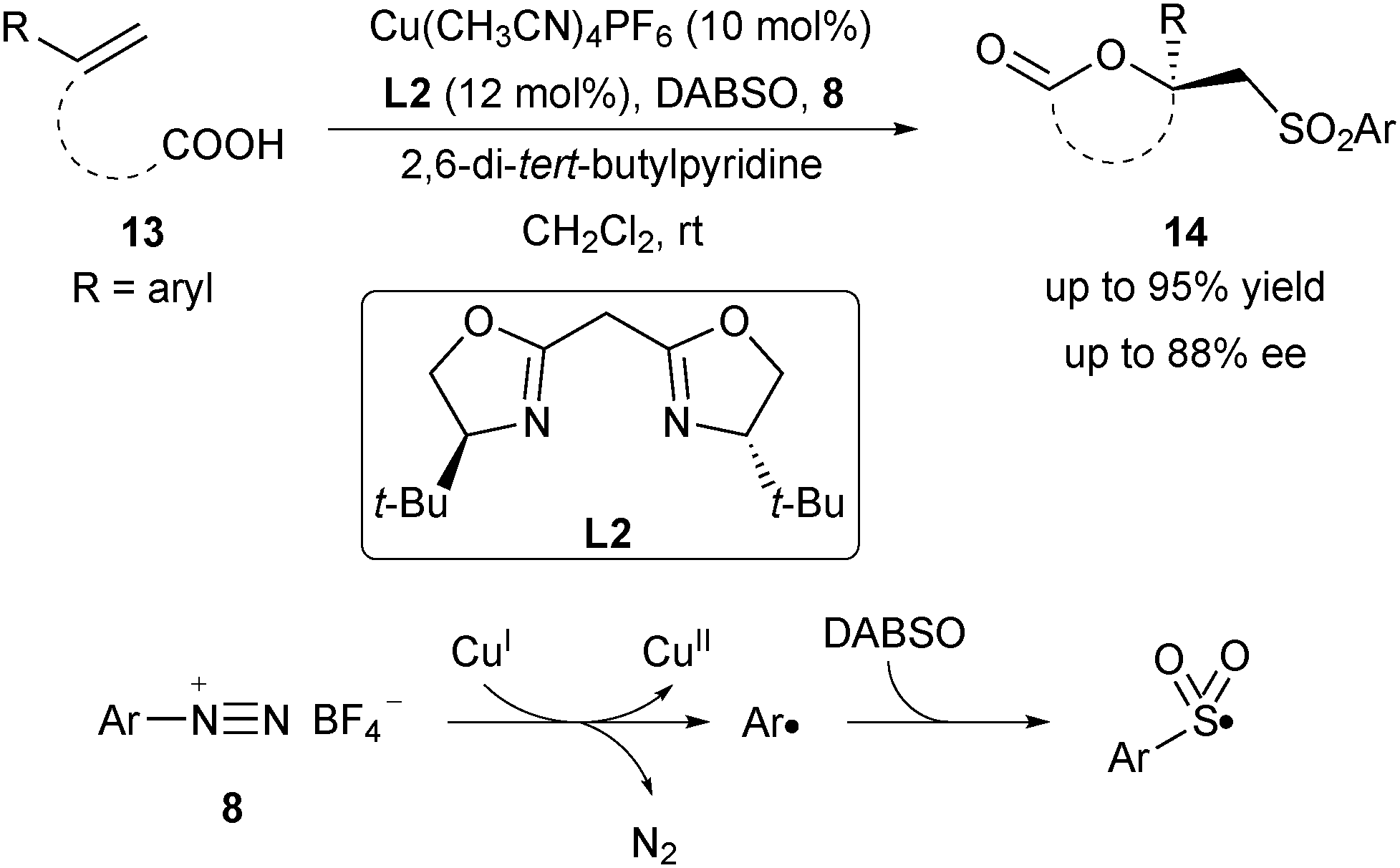
Following Buchwald's oxysulfonylation, Han and co-workers reported an asymmetric oxysulfonylation of unsaturated carboxylic acids 13via a multi-component cascade reaction in 2018 (Scheme 5).16 The combination of aryl diazonium salt 8 and DABCO·(SO2)2 (DABSO) was employed as the sulfonyl radical precursor. This new type of oxysulfonylation reaction exhibited a broad substrate scope to furnish products 14 in up to 88% ee. Mechanistically, aryl diazonium salt 8 underwent a SET process with CuI to generate an aryl radical and the CuII species via the release of nitrogen gas. The aryl radical was trapped by DABSO and generated the aryl sulfonyl radical, which then took part in the asymmetric oxysulfonylation reaction.
 | ||
| Scheme 5 Han's asymmetric oxysulfonylation of alkenes. | ||
From 2014 to 2015, Liu and co-workers reported a series of copper-catalysed intermolecular trifluoromethylation reactions of alkenes to install a trifluoromethyl group and an additional nucleophile to the alkene moiety.8 In their systematic study, they found that the radical intermediate generated from the addition of a trifluoromethyl radical to alkene can be selectively trapped by the nucleophile with the assistance of copper catalyst.8,11 These results encouraged them to explore the possibility of asymmetric radical transformation by the combination of copper catalysis and a chiral ligand.
After extensive screening of reaction parameters, Liu reported an asymmetric radical-involved trifluoromethylcyanation of alkenes under the CuI/chiral Box catalytic system in 2016 (Scheme 6).17 Utilizing Togni's reagent 2b as the radical source, the reaction of arylated or heteroarylated alkenes and TMSCN 16 provided enantioenriched benzyl nitriles in good enantioselectivity in the presence of Cu(CH3CN)4PF6 and L3. This reaction exhibited a broad substrate scope and functional group compatibility. A mechanistic study revealed that the reaction exhibited an induction period and a low concentration of the CN anion was crucial to the success of the reaction. Consequently, they proposed a plausible mechanism shown in Scheme 6: TMSCN combined with 2b to provide a mutual activation complex I. The formed L3CuICN in the induction period acted as the active catalyst, which underwent a SET process with the complex I to generate a CF3 radical and L3CuII(CN)2. The CF3 radical attacked styrene 15a rapidly to provide a prochiral benzylic radical II, followed by subsequent trapping with L3CuII(CN)2 to afford a CuIII species III. Eventually, the reductive elimination of the CuIII species III led to the desired C–CN bond-forming product 17a and released L3CuICN.
 | ||
| Scheme 6 Liu's asymmetric trifluoromethylcyanation of alkenes. | ||
In 2017, the same group realized an asymmetric aminocyanation of styrenes using N-fluorobenzenesulfonimide (NFSI) 18 as the radical precursor.18 Ligand L4 proved to be the best one under this catalytic system to deliver a wide range of chiral benzyl nitriles 19 (Scheme 7). Chiral β-amino nitriles are important both as natural products and bioactive compounds and as vital intermediates in the synthesis of chiral 1,3-diamines. However, it is difficult to obtain free amine derivatives from 19 due to the harsh conditions to remove the sulfonyl group. Thus, they developed an asymmetric azidocyanation reaction to successfully provide the azidocyanation products 20 in the presence of CuCl2 and ligand L5, with TMSN34 as the radical precursor and PhI(O2CEt)2 as the oxidant (Scheme 7). Similar to the trifluoromethylcyanation reaction, both amino- and azidocyanation reaction proceeded via a possible CuIII complex followed by a reductive elimination step to construct the chiral C–CN bond.17
 | ||
| Scheme 7 Liu and Lin's asymmetric aminocyanation and azidocyanation of alkenes. | ||
In 2018, Han and Mei reported an asymmetric radical cyanoalkylation of styrenes by merging photoredox and copper catalysis (Scheme 8).19 They used the alkyl N-hydroxyphthalimide ester (NHP ester) 21 derived from the inexpensive and abundant carboxylic acid as the radical precursor. Thus, in the presence of Ir(ppy)3, CuBr and L6, the visible light-induced cyanoalkylation of styrenes 15 was performed to provide alkylated benzyl nitriles 22 in good yield and enantioselectivity. One representative feature of this protocol was that diverse alkyl carboxylic acids were applicable, thus significantly expanding the scope of radical precursors. Under irradiation, Ir(ppy)3 was irradiated to the activated state, which underwent a SET process with the radical precursor 21 to form an alkyl radical upon release of CO2 and phthalimide anion I and concurrently generate oxidative IrIV (Scheme 8). Meanwhile, L6CuICN was oxidized by IrIV to L6CuIICN to finish the Ir catalytic cycle for generation of the alkyl radical.
 | ||
| Scheme 8 Han and Mei's photoredox-promoted asymmetric cyanoalkylation of alkenes. | ||
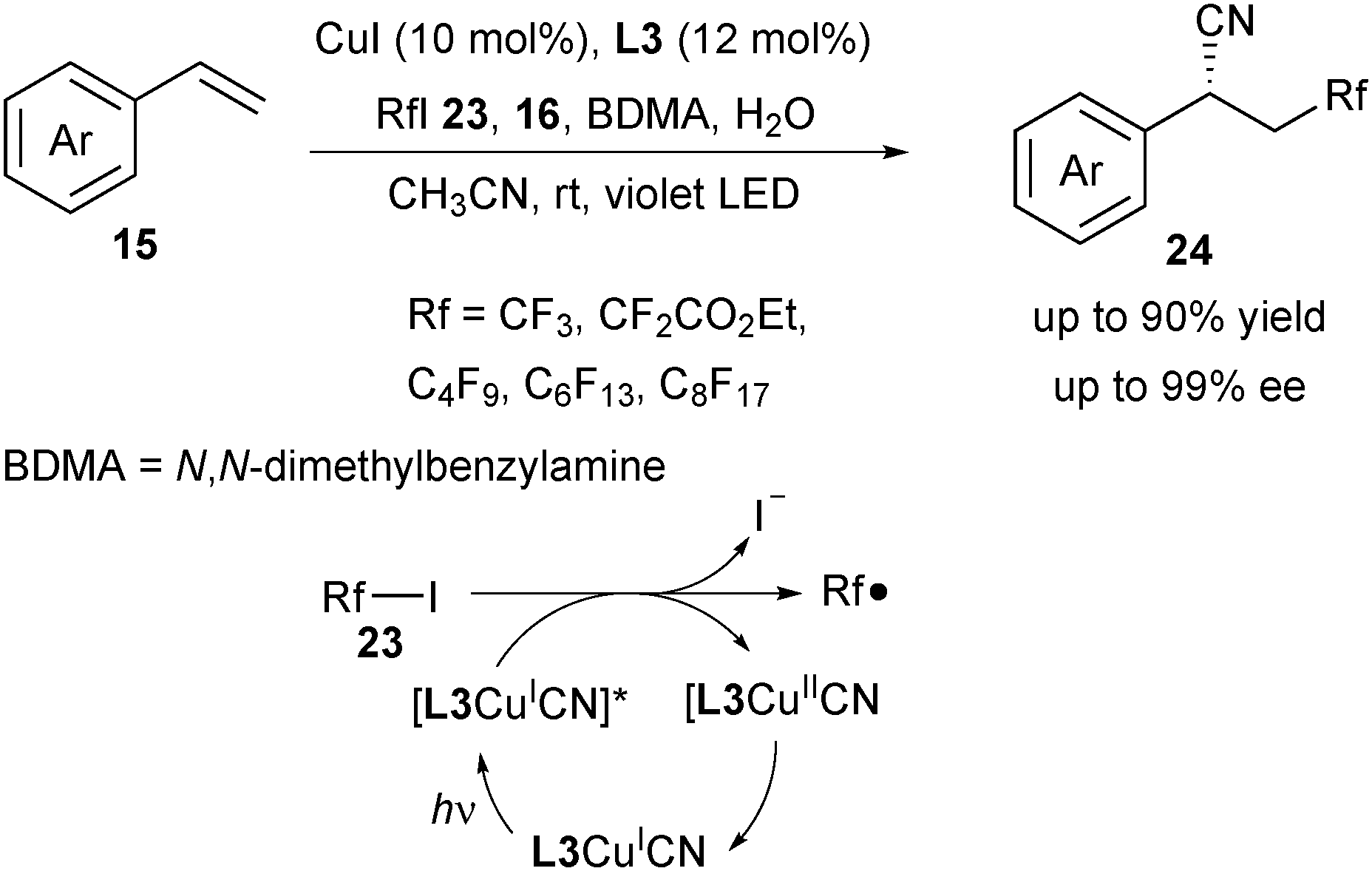
Compared with Togni's reagent 2a and 2b, fluoroalkyl iodide 23 is inexpensive and readily available, which can work as an ideal precursor for diverse fluoroalkylation reactions. Wang and Xu recently reported a copper-catalysed photoredox enantioselective cyanofluoroalkylation of styrenes to deliver diverse β-fluoroalkylated benzylic nitriles 24 (Scheme 9).20 In this strategy, copper played a dual role, both as the photoredox catalyst to undergo a SET process with fluoroalkyl iodides and as the cross-coupling catalyst for C–CN bond formation. Instead of a blue or green LED, a violet LED was employed to irradiate the copper complex. Based on their mechanistic study, the authors proposed that it was the L3CuICN species which was irradiated to its excited state [L3CuICN]* with the violet LED. The following oxidative quenching step between [L3CuICN]* and 23 generated a Rf radical and L3CuIICN. They assumed that the addition of H2O might promote the homocleavage of TMSCN and N,N-dimethylbenzylamine (BDMA) might act as a base to neutralize the acidic byproducts.
 | ||
| Scheme 9 Wang and Xu's photoredox asymmetric cyanofluoroalkylation of alkenes. | ||
Very recently, Liu and Zou described an asymmetric copper-catalysed phosphinoylcyanation of styrenes, offering a direct route to various chiral phosphine-containing alkylnitriles 26 (Scheme 10a).21 In this process, t-BuOOH and TMSCN reacted quickly to generate t-BuOOTMS, which underwent SET with L7CuICN to afford the t-butoxy radical and L7CuIICN in the presence of HCN via a proton-coupled-electron transfer (PCET) pathway (complex I in Scheme 10). The t-butoxy radical then abstracted the hydrogen of Ph2P(O)H to provide a phosphinoyl radical to initiate the subsequent catalytic cycle.17 Almost at the same time, Lin's group developed an electrochemical strategy to accomplish the asymmetric phosphinoylcyanation reaction (Scheme 10b). They further applied the protocol to asymmetric cyanosulfinylation of styrenes using sulfinic acids as radical precursors.22
 | ||
| Scheme 10 Liu's and Lin's asymmetric cyanofunctionalization of styrenes. | ||
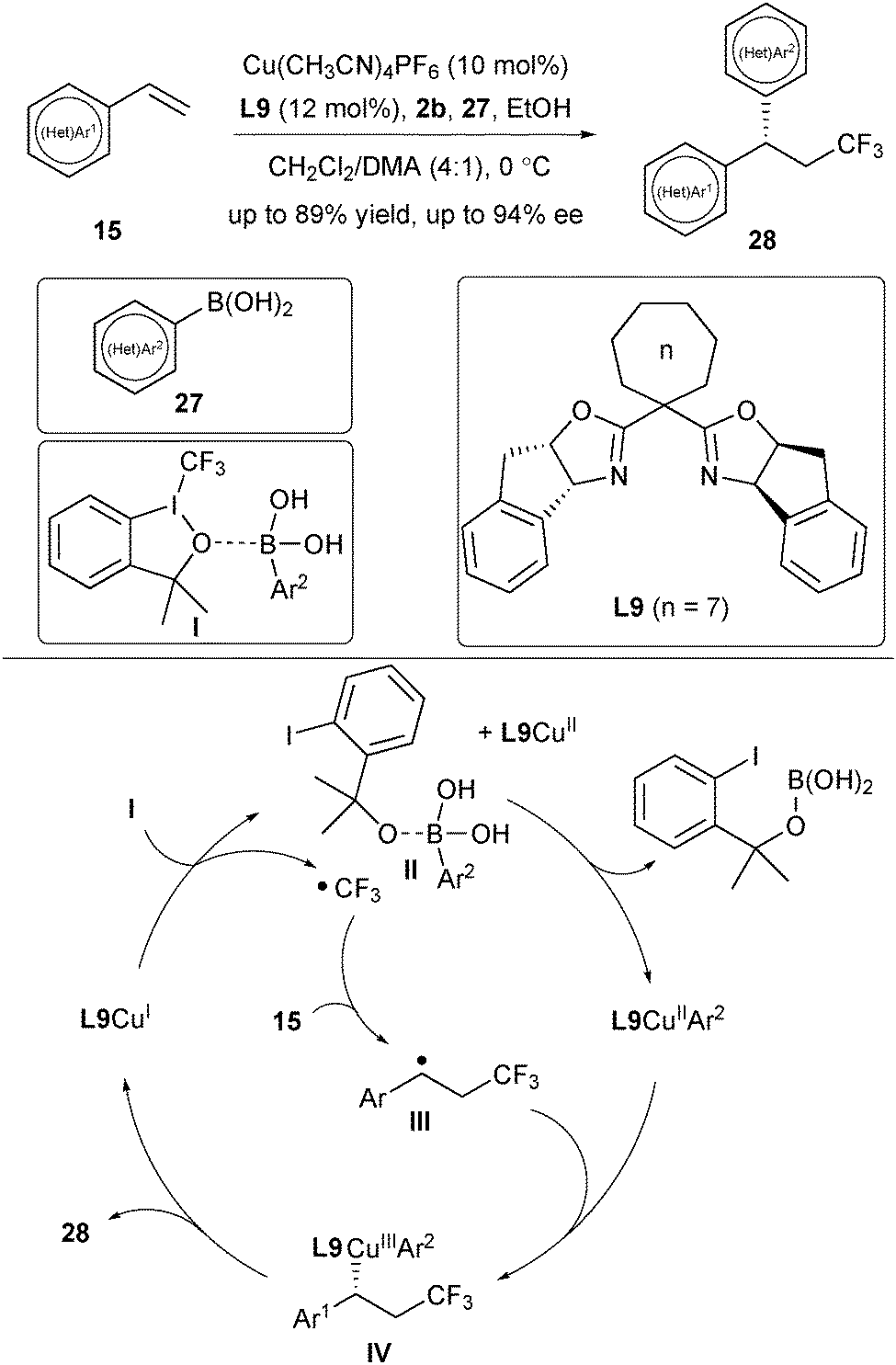
Inspired by the success of asymmetric control via the combination of a benzyl radical intermediate with L*CuIICN, Liu's group then focused on the utilization of boronic acids as a nucleophile for asymmetric radical arylation reactions. In 2017, they reported an asymmetric trifluoromethylarylation of styrenes for the assembly of chiral CF3-containing 1,1-diarylmethane derivatives (Scheme 11).23 Screening of ligands disclosed a correlation between the enantiomeric excess of the products 28 and the bite angle of the Box ligand. Increasing the ring size of the Box ligand (n = 3–7) is beneficial to the enantioselectivity, and ligand L9 (n = 7) with a small bite angle provided the best enantiomeric excess. The substrate scope is quite broad and not only styrenes and heteroarylated alkenes are suitable for the reaction, but the aryl boronic acids are also variable, ranging from various functionalized aryl boronic acids to heteroaryl boronic acids. Analogously, a SET process between the activated complex I and L9CuI would generate a CF3 radical (Scheme 11). Meanwhile, the in situ generated alkoxy anion from 2b acted as a strong base to activate Ar2B(OH)2 to form an ate complex II, which promoted the transmetalation process to deliver the L9CuIIAr2 species. The benzylic radical III derived from the addition of a CF3 radical to alkene would be trapped by L9CuIIAr2 to afford a CuIII species IV. A final reductive elimination step delivered the desired products 28, while releasing the L9CuI species.
 | ||
| Scheme 11 Liu's asymmetric trifluoromethylarylation of styrenes. | ||
Based on the asymmetric arylation strategy, Liu and Lin further described an asymmetric aminoarylation of styrenes for the synthesis of chiral 2,2-diarylethylamines by an applicable N-based radical precursor (Scheme 12).24 In their initial study, they used NFSI as the N-centered radical precursor. However, after extensive screening of reaction conditions, the aminoarylation product was obtained only in 35% yield and 84% ee with almost complete consumption of 15b (Scheme 12). They assumed that the generation of L9CuIIAr via transmetalation of arylboronic acid with CuII is too slow to match the rate of benzylic radical generation. Density functional theory (DFT) study revealed that the N-alkylsulfonamidyl radical was less electrophilic and exhibited a higher energy barrier toward radical addition to styrenes compared with that of the bisulfonimidyl radical. The reaction rate of this kind of radicals with alkenes is capable of matching the transmetalation step. Following this concept, they designed a series of N-centered radical precursors 29 and found that the N-methylsulfonamidyl radical provided the best yield and ee. The addition of LiOt-Bu and lowering down the reaction temperature to −10 °C afforded the aminoarylation product in 81% yield and 93% ee.
 | ||
| Scheme 12 Liu and Lin's asymmetric aminoarylation of styrenes. | ||
Recently, Liu's group discovered an asymmetric trifluoromethylarylation of alkenes for the construction of quaternary all-carbon stereocenters.25 Notably, the construction of quaternary all-carbon centered products has been quite difficult in previous intermolecular asymmetric radical alkene difunctionalization. In this strategy, α-substituted acrylamides 31 were utilized for the trifluoromethylarylation with boronic acids 27 and Togni's reagent 2b, leading to the corresponding CF3-containing products 32 with excellent enantioselectivity (Scheme 13). When screening the ligands, the authors identified that the gem-dibenzyl group in the Box ligand (L5–L10) is crucial for enantioselectivity, probably attributing to the side arm effect.
 | ||
| Scheme 13 Liu's asymmetric trifluoromethylarylation of acrylamides. | ||
The key to the success of copper-catalysed asymmetric cyanation and arylation lies in that TMSCN and ArB(OH)2 serve as ideal trapping reagents to form L*CuIINu. The in situ generated complexes react with the benzylic radical intermediate to accomplish the enantioselective control. Liu further reasoned that if suitable alkynylating reagents were used via a similar strategy for stereocontrol (Scheme 14), the asymmetric alkynylfunctionalization of alkenes would be feasible. Following this concept, they tested alkynylating reagents 33a–33e and found that in the presence of Cu(CH3CN)4PF6 and ligand L5, only 33a can react with alkene 15c and Togni's reagent 2b to provide the desired product 34 in 89% yield, albeit with 33% ee (Scheme 14).26 Further screening of ligands revealed that L9 gave the trifluoromethylalkynylation product 34 with the best result (83% yield and 92% ee).
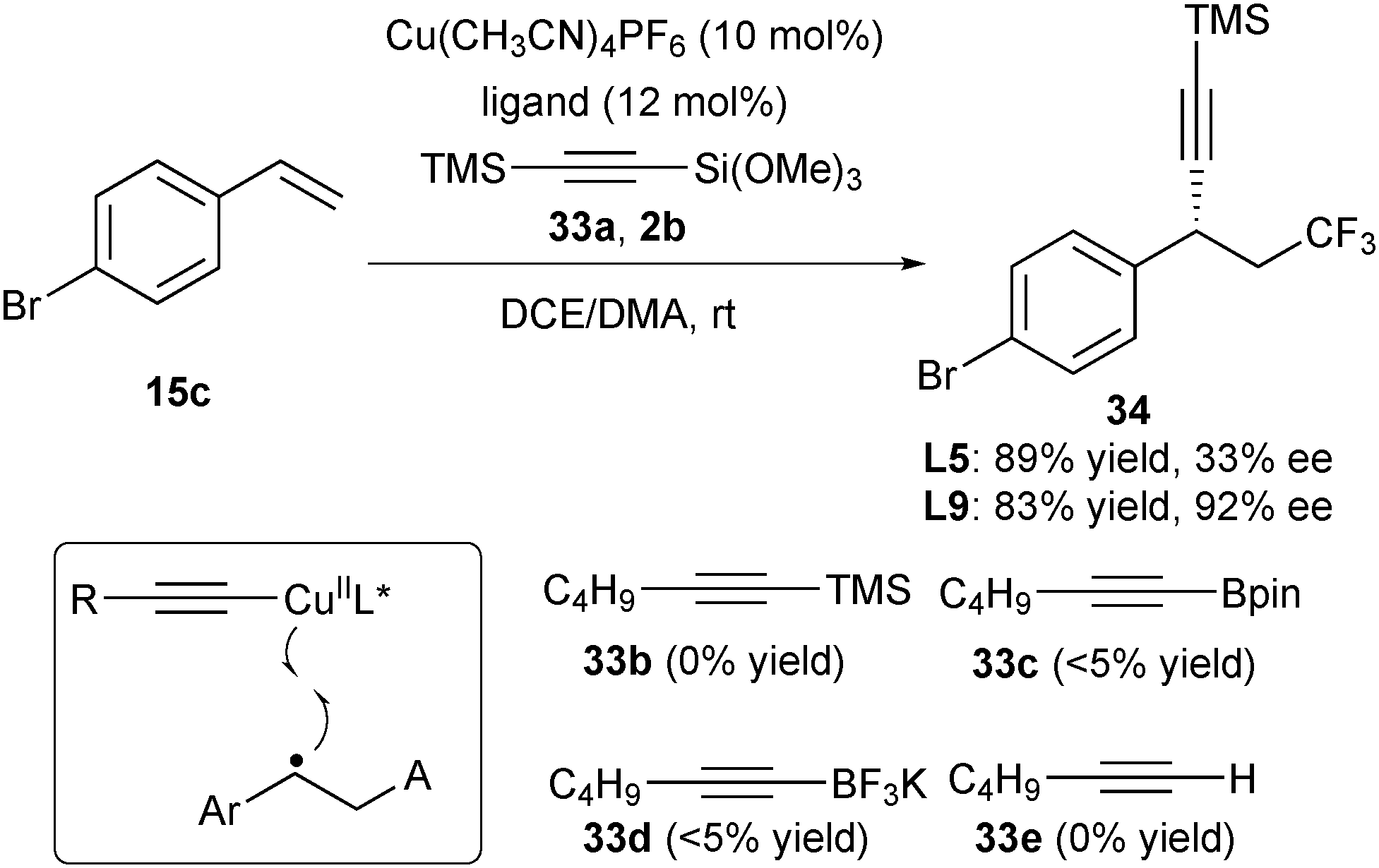 | ||
| Scheme 14 Liu's asymmetric trifluoromethylalkynylation of alkenes. | ||
Very recently, Chen and He extended the asymmetric radical alkene difunctionalization reaction to an intramolecular aminotrifluoromethylation of unactivated alkenes with 2b as the CF3 radical source (Scheme 15).27 Remarkably, their strategy could be applied to the mono-substituted unactivated alkene substrates 35, produced from readily available homoallylic alcohols and aryl nitriles. Treating alkenes 35 in the presence of Cu(CH3CN)4PF6 and L11 enabled the asymmetric aminotrifluoromethylation reaction with excellent enantioselectivity. The reaction may also proceed via a CuIII complex followed by reductive elimination to produce the chiral 1,3-oxazines 37.
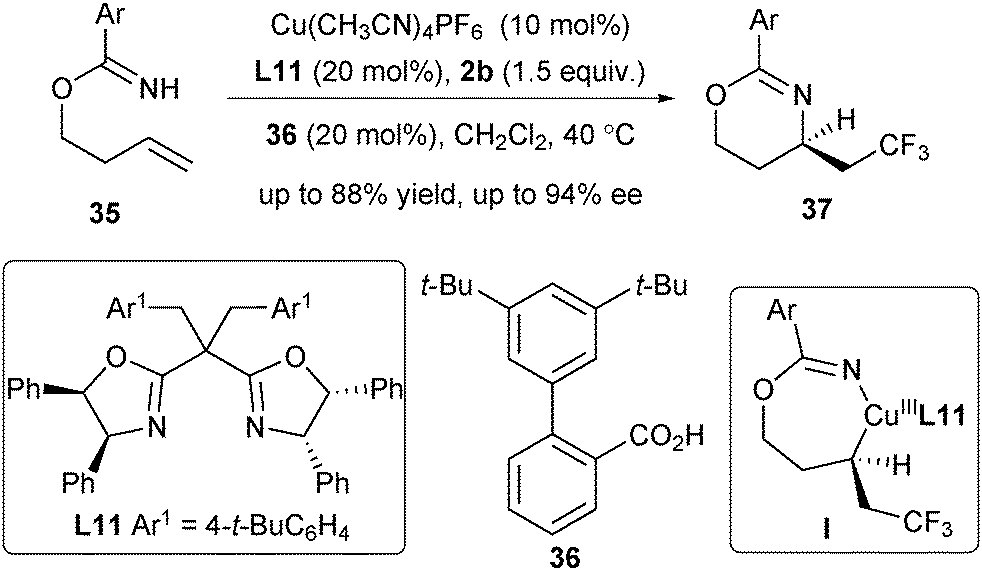 | ||
| Scheme 15 Chen and He's intramolecular aminotrifluoromethylation of unactivated alkenes. | ||
In 2009, Yoon and co-workers described an asymmetric aminohydroxylation of styrenes with oxaziridine to produce enantioenriched vicinal amino alcohols in the presence of CuII/chiral Box catalysis (Scheme 16).28 This environmentally benign aminohydroxylation under osmium-free conditions offers an attractive alternative to the well-established Sharpless asymmetric aminohydroxylation. The authors proposed a radical mechanism in which the key aminocopper(III) species II might be generated from the reaction of 15a and complex I (Scheme 16).
 | ||
| Scheme 16 Yoon's copper(II)-catalysed aminohydroxylation of styrenes. | ||
2.2. Copper(I)/chiral diphosphine catalysis
In 2007, Shi and co-workers achieved a regioselective copper(I)-catalysed diamination of conjugated alkenes.29 Using di-tert-butyldiaziridinone 41 as the diamine reagent, the reaction occurs selectively on terminal alkenes and they assume that the reaction probably proceeds via a radical mechanism (Scheme 17). Firstly, CuI promoted a reductive cleavage of the N–N bond of 41 to form an N-centered radical I, which added to the terminal alkene of 40 to provide the intermediate II. The possible pathway for the formation of the C–N bond is that the carbon-centered radical of II coordinated with the CuII center to generate a CuIII complex III and subsequent reductive elimination afforded the final products 42 and regenerated the CuI catalyst. | ||
| Scheme 17 Shi's copper-catalysed diamination of dienes. | ||
Afterwards, they were encouraged to develop the more challenging asymmetric diamination reaction by screening diverse chiral ligands. Among the screened neutral chiral ligands, only chiral diphosphine ligand (R)-DTBM-SEGPHOS L13 provided a promising enantioselectivity (up to 74% ee) (Scheme 18).30
 | ||
| Scheme 18 Shi's copper/diphosphine-catalysed asymmetric diamination of dienes. | ||
2.3. Copper(I)/CPA catalysis
In 2009, Shi and co-workers explored the asymmetric diamination of conjugated alkenes by utilizing the CuI/chiral anion derived from mesitylcopper(I) and chiral phosphoric acid (CPA) L14 (Scheme 19).31 Under optimal conditions, aryl, heteroaryl-conjugated diene and triene 40 are applicable to provide the diamination products 42 in promising enantioselectivity (up to 61% ee).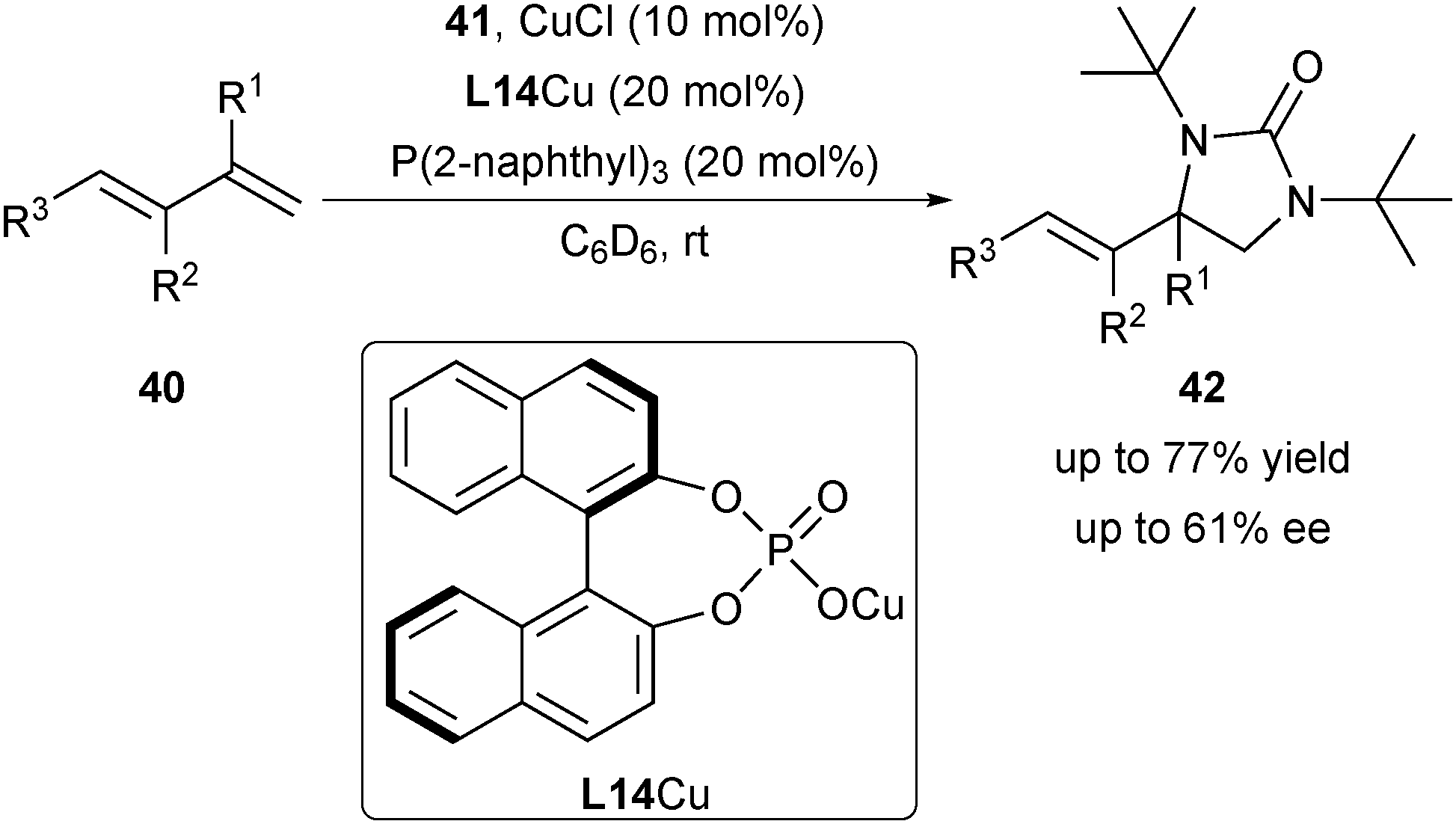 | ||
| Scheme 19 Shi's copper/CPA-catalysed asymmetric diamination of dienes. | ||
Liu's group has been dedicated to the design of a new catalytic system to solve the challenging asymmetric radical-involved alkene difunctionalization reactions since 2014. In 2014, Liu and co-workers disclosed a copper(I)/CPA-catalysed radical-involved remote asymmetric C–H functionalization.32 Their results proved that the copper(I)/CPA catalytic system was compatible with radical-involved reactions. This encouraged them to exploit the radical-involved asymmetric alkene difunctionalization using the copper(I)/CPA catalytic system.
In 2016, Liu and co-workers designed Cu(I)/CPA as a novel SET catalyst to establish an asymmetric radical-involved intramolecular aminotrifluoromethylation of arylated alkenes (Scheme 20).33 At the outset, they screened a range of protecting groups of amine and found that only substrates with tosyl and urea protecting groups provided promising enantioselectivity under the Cu(I)/CPA catalytic system (Scheme 20). After screening of reaction conditions, they identified that in the presence of CuCl and L15, alkenes 43 and 2b reacted smoothly in ethyl isobutyrate to provide 44 in excellent enantioselectivity, providing a straightforward access to diversely substituted CF3-containing pyrrolidines bearing an α-tertiary stereocenter. The key to success is not only the introduction of Cu(I)/CPA as a novel SET catalyst, but also the use of urea with two acidic N–H as both the nucleophile and directing group.
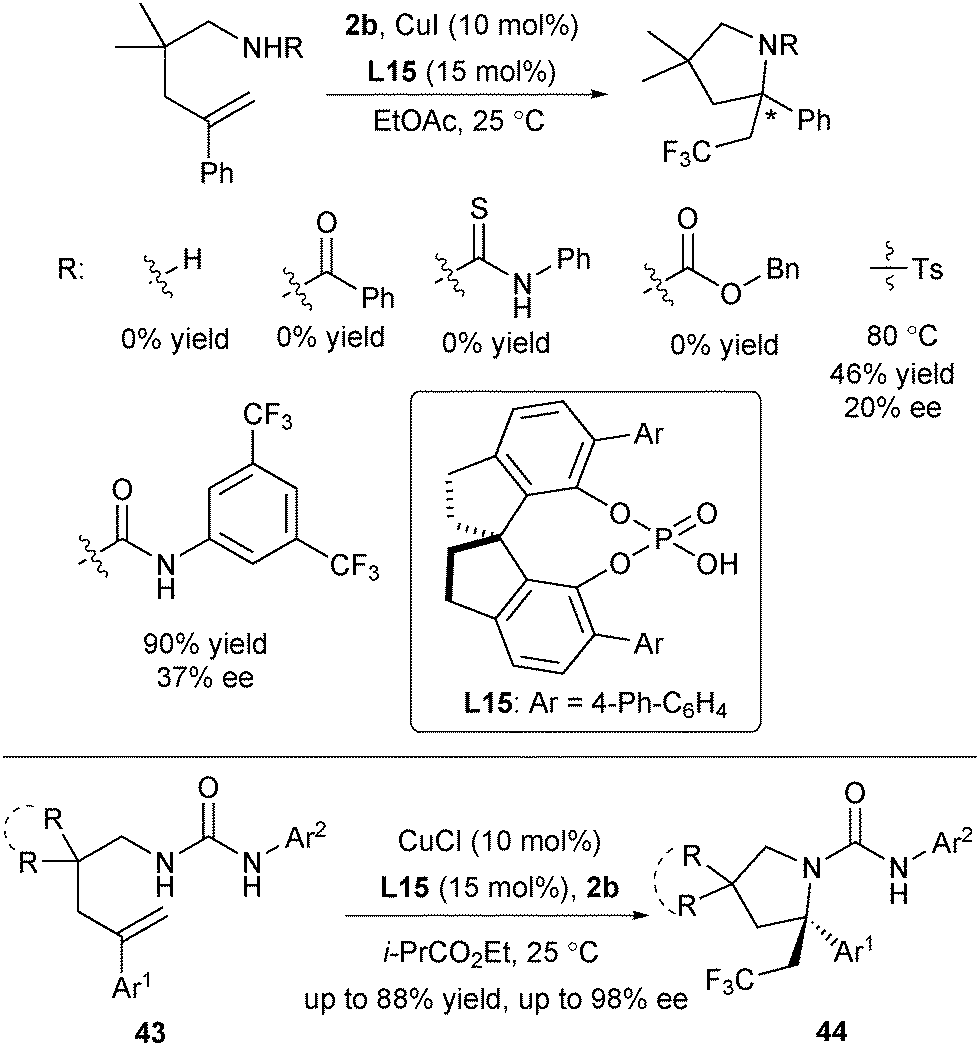 | ||
| Scheme 20 Liu's copper/CPA-catalysed asymmetric aminotrifluoromethylation. | ||
Based on their mechanistic study, Liu proposed a plausible mechanism as shown in Scheme 21.33 In the first step, CuI would undergo a single-electron transfer with the CPA-activated Togni's reagent 2b to generate a CF3 radical and chiral CuII phosphate (Scheme 21). The CF3 radical selectively added to the terminal alkene of substrates 43 to afford a tertiary benzylic radical intermediate I, which would be trapped by CuII/phosphate to either generate a CuIII complex II or form a carbocation intermediate III. The chiral phosphate could control the selectivity via both hydrogen-bonding interactions with the N–H bond adjacent to the aryl group and ion-pairing interactions in a concerted transition state. The subsequent C–N bond formation delivered the final products 44 while releasing CuI and CPA.
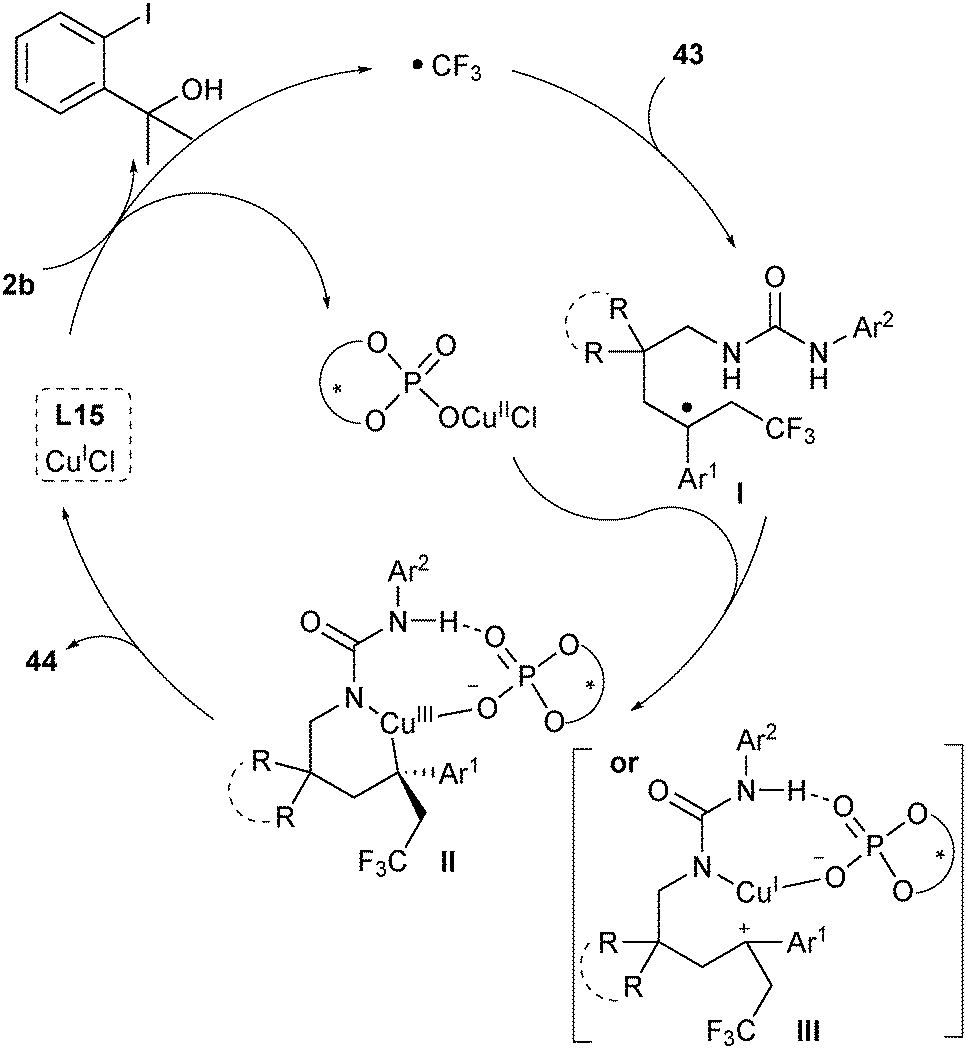 | ||
| Scheme 21 Mechanistic proposal for Liu's asymmetric aminotrifluoromethylation. | ||
While Liu's aminotrifluoromethylation reaction with 2b successfully generated chiral β-trifluoromethylated amines, it proved unfeasible to access more chiral β-fluoroalkyl amines due to the lack of the analogous reagents. Therefore, Liu and co-workers subsequently developed a radical-involved asymmetric aminofluoroalkylation of arylated alkenes utilizing sulfonyl chloride 45 as the fluoroalkyl radical source with CuI/CPA as the SET catalyst (Scheme 22).34 This strategy provides a platform to access a diverse array of β-perfluorobutanyl, trifluoromethyl-, difluoroacetyl-, and difluoromethyl chiral amines in excellent enantioselectivity. The reaction proceeded via a similar pathway as in Scheme 21. Unlike the reaction with Togni's reagent 2b, in the initiation step to generate a fluoroalkyl radical from fluoroalkylsulfonyl chloride, a strong HCl acid was also generated. The HCl acid would result in strong background reactions against the CPA-catalysed process and hydroamination of alkene. Thus, the use of silver carbonate is crucial to suppress the strong background and side hydroamination reactions.
 | ||
| Scheme 22 Liu's asymmetric aminofluoroalkylation of alkenes. | ||
Chiral vicinal diamines are not only key structural motifs in natural products and pharmaceutical reagents but also versatile platforms for development of chiral ligands, organocatalysts and auxiliaries in asymmetric synthesis. Inspired by their asymmetric aminofluoroalkylation of arylated alkenes with chiral CuI/phosphate catalysis, Liu's group utilized the N-centered radical to facilitate the synthesis of chiral vicinal diamines via asymmetric diamination of arylated alkenes (Scheme 23).35 One unique feature of the strategy is the dual role of O-acylhydroxylamine 47 as both an alkylamine source and an oxidant in this process. This protocol prevented the catalyst poisoning associated with the use of a free alkylamine and meanwhile avoided the use of an external oxidant required in oxidative diamination reactions. Further expanding the N-centered radical to the azidyl radical produced from hypervalent iodine reagent 49 delivered the desired chiral azidoamination products 50 (Scheme 23).35
 | ||
| Scheme 23 Liu's asymmetric diamination of alkenes. | ||
In 2018, Liu and co-workers disclosed an asymmetric aminoarylation of arylated alkenes by utilizing aryl diazonium salt 8 as the radical precursor and CuI/phosphate as the SET catalyst (Scheme 24).36 A broad scope of substrates worked well to afford the expected pyrrolidines 51 in good enantioselectivity. This reaction undergoes a similar CuII/phosphate-mediated enantioselective radical trapping process (via the intermediate I in Scheme 24), which is similar to that depicted in Scheme 21. It affords a mechanistically distinct and complementary approach for the traditional asymmetric alkene aminoarylation invoking a key nucleophilic aminopalladation step (via the intermediate II, Scheme 24).
 | ||
| Scheme 24 Liu's asymmetric aminoarylation of alkenes. | ||
Very recently, Liu and co-workers discovered an asymmetric radical aminosilylation of arylated alkenes with hydrosilane 52 as the silyl radical source, affording diverse N-heterocycles 55 including pyrrolidine, indoline as well as isoindoline (Scheme 25).37 Unlike the generation of radicals from oxidative radical precursors,33–36 Cu(I) reacts with CPA-activated LPO 53 followed by the loss of CO2 to afford a highly reactive alkyl radical accompanied by the generation of the crucial L17CuIITc complex. Hydrogen abstraction from 52 by the alkyl radical gives the silicon-centered radical. A mechanistic study indicated that the formation of the chiral C–N bond is via the carbocation intermediate I rather than a possible CuIII elimination pathway.37
 | ||
| Scheme 25 Liu's asymmetric aminosilylation of alkenes. | ||
Very recently, Liu's group disclosed a copper(I)/CPA-catalysed asymmetric radical-involved intermolecular dicarbofunctionalization of 1,1-diarylalkenes (Scheme 26).38 This three-component radical reaction mainly focused on the utilization of sulfonyl chloride 45 (or 2b) as the radical precursor and indole (or pyrrole) 57 as the nucleophile, providing a direct access to synthetically challenging chiral triarylmethanes 58. Similarly, the reaction firstly generated the radical addition intermediate I. In this case, the intermediate I underwent a SET process with CuII to provide the cation intermediate II (or III), followed by a nucleophilic attack of 57 to produce desired products 58. DFT study disclosed that the critical chiral environment was created by the hydrogen-bonding and ion-pair interactions across L18 (the intermediate II and III with the N–H and O–H moiety in Scheme 26).
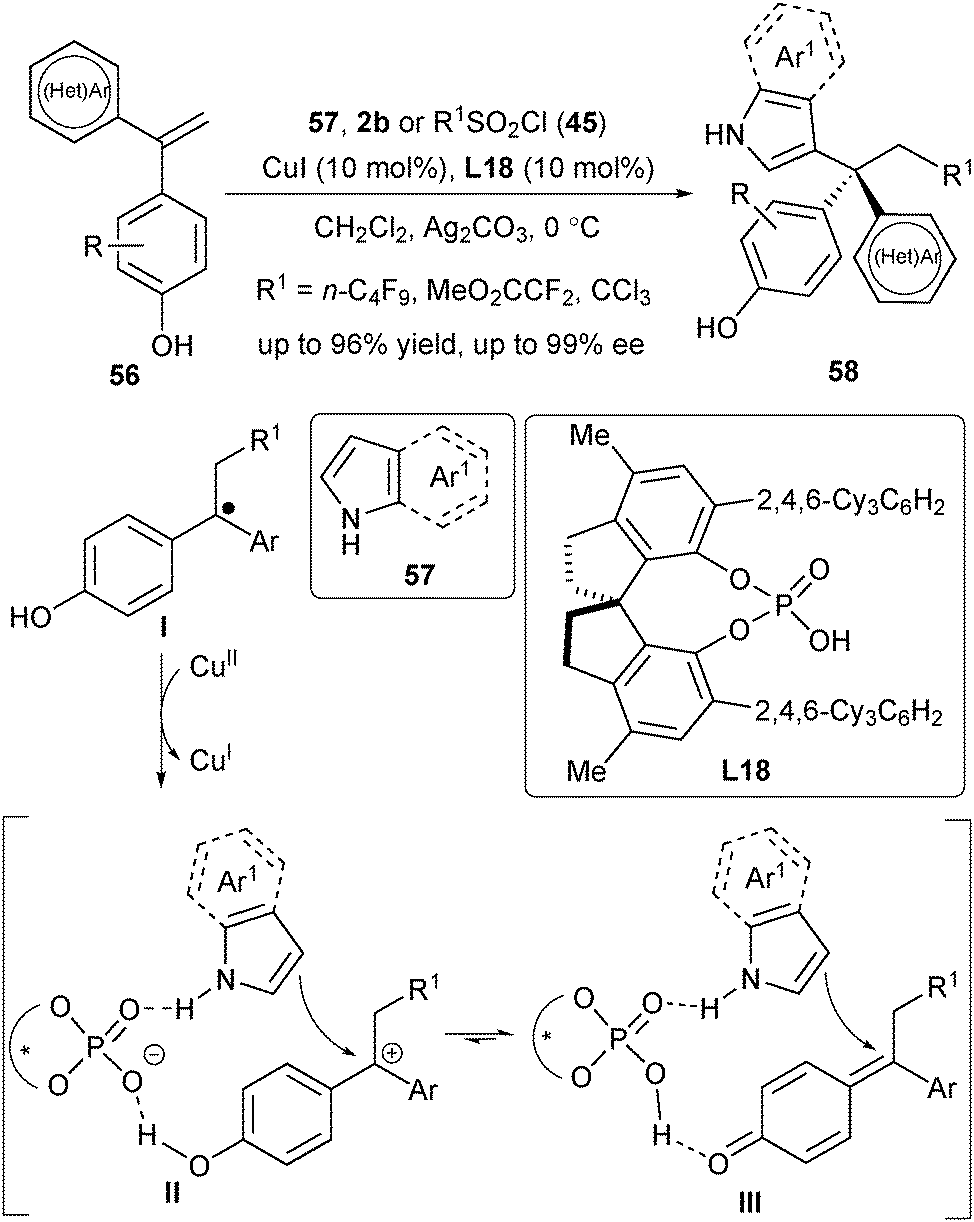 | ||
| Scheme 26 Asymmetric radical-involved intermolecular alkene dicarbofunctionalization. | ||
In Buchwald's work to achieve the asymmetric oxytrifluoromethylation of alkenes, the copper(I)/Box catalytic system is found to be incompatible with alcohol-based nucleophiles probably due to the low binding affinity of alcohol to the chiral copper complex.14 To solve this challenge, Liu and co-workers utilized the CuI/CPA catalyst to achieve the asymmetric radical oxytrifluoromethylation of alkenes with alcohols (Scheme 27).39 A wide range of CF3-substituted tetrahydrofurans 61 bearing an α-tertiary stereocenter was obtained. Analogously, the intermediate I was generated via addition of the CF3 radical to alkenes (Scheme 27).33 Although they could not exclude the carbocation pathway, they suggested that the enantioselective trapping by the alcohol nucleophile provided the final product 61via a CuIII intermediate and the subsequent reductive elimination process.33 One unique feature of the strategy is that the achiral pyridine 60 may act as a coordinative ligand on the copper metal to stabilize the high-valent Cu species. They further assumed that this stabilization effect might make the more enantioselective CuIII pathway favourable compared with other competing radical substitution or carbocation pathways (Scheme 2).
 | ||
| Scheme 27 Liu's asymmetric radical oxytrifluoromethylation of alkenyl alcohols. | ||
2.4. Copper(I)/cinchona alkaloid-derived sulfonamide catalysis
During Liu's study of asymmetric oxytrifluoromethylation of alkenyl alcohols,39 they realized that the addition of an achiral neutral ligand could stabilize the high-valent Cu species, thus tuning the chemo- and enantioselectivity of the radical-involved reactions. This phenomenon inspired them to exploit a multidentate anionic–neutral hybrid ligand in order to realize not only the mechanistic chemocontrol by forging the CuIII pathway but also the stereocontrol by offering a more rigid chiral environment. Following this concept, they designed a cinchona alkaloid-derived sulfonamide to accomplish the copper-catalysed radical-involved asymmetric oxytrifluoromethylation of alkenyl oximes (Scheme 28).40 With this strategy, a wide range of CF3-containing isoxazolines 63 bearing an α-tertiary stereocenter were generated using L20 as the ligand. Based on their mechanistic study, they proposed a mechanistic pathway as depicted in Scheme 28. The L20CuI species underwent a SET process with 2b to afford a CF3 radical and L20CuII. The CF3 radical added to the alkene of 62 to deliver the intermediate I, which would react with L20CuII to give the complex II, followed by the formation of the chiral C–O bond to furnish the desired products 63. | ||
| Scheme 28 Liu's asymmetric oxytrifluoromethylation of alkenyl oximes. | ||
3. Copper(II)/Box-catalysed nucleocupration of alkenes
Since 2004, Chemler's group has developed a family of intramolecular 1,2-amino-/oxyfunctionalization of unactivated terminal alkenes that enables the installation of an amine or alcohol into alkenes, directly providing valuable pyrrolidines, indolines as well as other heterocyles.7,41 Since the cyclization of the amine or alcohol moiety onto unactivated alkenes will not occur easily, copper(II) catalysis is used to activate the inert alkene electrophile to initiate the nucleocupration process in Chemler's chemistry. The general catalytic cycle is shown in Scheme 29. The amine/alcohol group of the substrate coordinates with the CuII species to generate the complex I. A subsequent Lewis acid-promoted cis-nucleocupration across the alkene occurs to provide a cyclized intermediate II. Homolysis of the C–CuII bond in the intermediate II results in an alkyl radical III, which is rapidly trapped with various radical trapping reagents to deliver the amino- or oxyfunctionalization products and regenerate CuI, which is then oxidized to CuII for the next catalytic cycle. Based on this concept, Chemler's group has accomplished a series of asymmetric versions by utilizing chiral Box ligands for the assembly of a library of enantioenriched pyrrolidines, indolines and other heterocycle scaffolds. A mechanistic study indicated that the cis-nucleocupration of I towards II is the rate-determining and enantiodetermining step (Scheme 29).42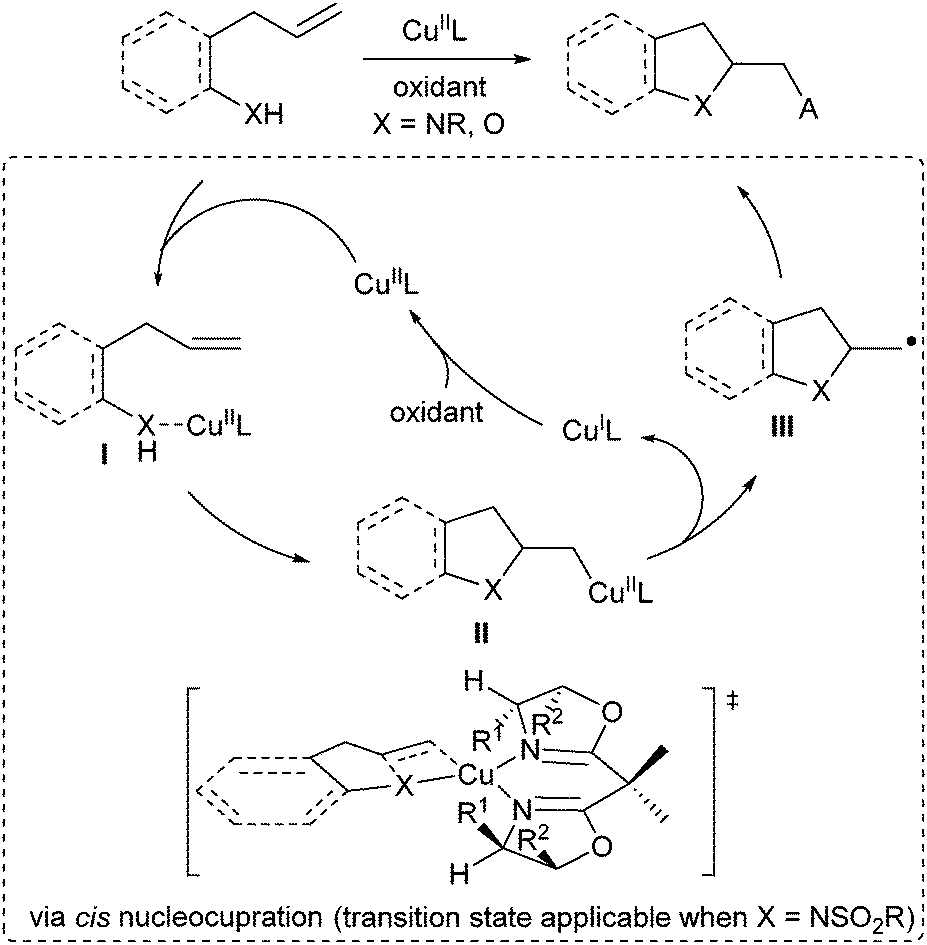 | ||
| Scheme 29 Chemler's nucleocupration for radical-involved alkene difunctionalization. | ||
In 2007, Chemler and co-workers developed the first example of enantioselective intramolecular carboamination of alkenes for expeditious synthesis of chiral sultams using the CuII/Box catalytic system (Scheme 30).43 In the presence of Cu(OTf)2, L12, K2CO3, and oxidant MnO2, the reaction of alkenyl arylsulfonamides 64 afforded sultams 65 in excellent yield and enantioselectivity (up to 94% ee). Analogous to the general mechanistic proposal in Scheme 29, the reaction firstly underwent a cis aminocupration to deliver the intermediate II, followed by homolysis of the Cu–C bond to provide CuI and the alkyl radical intermediate III. The radical addition of the alkyl radical to an intramolecular aryl or heteroaryl ring and subsequent rearomatization afforded the desired products 65. The oxidant is required to convert CuI back to CuII and may participate in the oxidation of the aryl radical intermediate IV.
 | ||
| Scheme 30 Chemler's enantioselective intramolecular alkene carboamination. | ||
Based on this concept, Chemler and co-workers designed another two classes of substrates, which are applicable to the copper-catalysed enantioselective intramolecular carboamination of unactivated terminal alkenes. Alkenylsulfonamides 66 bearing γ-substituted groups underwent this reaction smoothly for synthesis of hexahydro-1H-benz[f]indoles 67 in excellent yield and selectivity, bearing vicinal tertiary and quaternary carbon stereocenters (Scheme 31a).44 When using alkenylsulfonamides 68 bearing β-substituted groups as the substrates, the enantioselective alkene carboamination provided the bridged heterocycles 69 (Scheme 31b).45
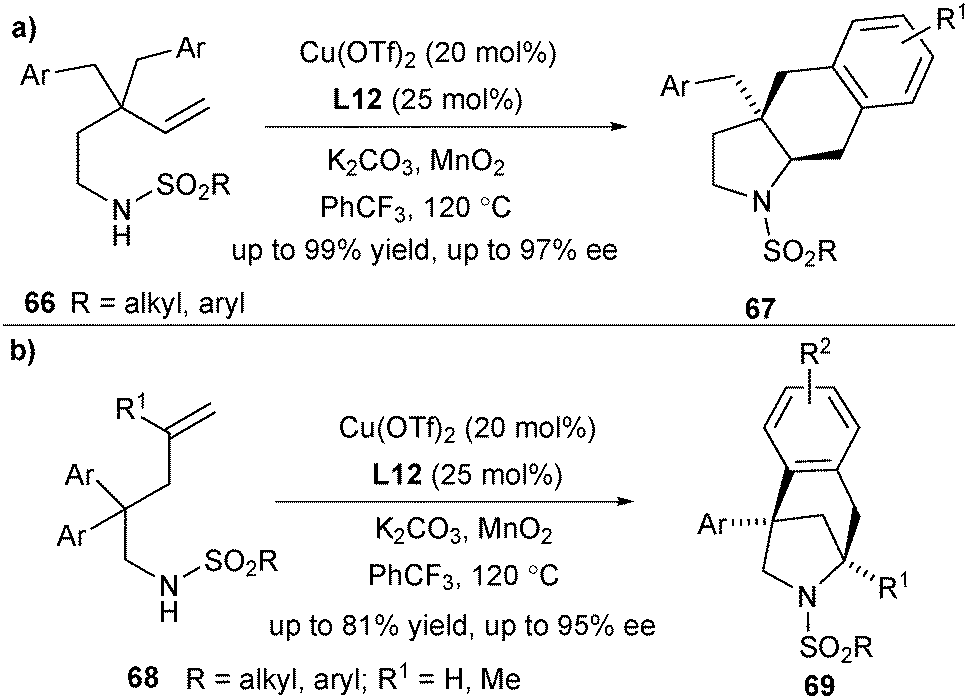 | ||
| Scheme 31 Enantioselective synthesis of hexahydro-1H-benz[f]indoles and 6-azabicyclo[3.2.1]octanes via carboamination. | ||
Compared with intramolecular carboamination, the intermolecular carboamination version is more challenging, since the intramolecular version to generate sultam will compete with the desired process.43 Thus, the employment of good radical acceptors is critical to the success of the intermolecular carboamination. To their delight, utilizing 1,1-diphenylethylene and related styrenes 70 as the radical acceptors, Chemler and co-workers achieved a formal Heck-type coupling reaction via enantioselective intermolecular carboamination to synthesize a variety of enantioenriched indolines, pyrrolidines, and tetrahydroisoquinolines 71 (Scheme 32).46 During their investigation of the radical acceptors, they found that the less reactive alkene benzofuran 70a required the removal of the aryl ring in the substrate for avoidance of intramolecular reaction and caused the decrease of the ee value. They proposed that in case that the rate of the radical trapping step is slow and the generated aminyl radical is relatively stable, the radical intermediate I may undergo a ring-opening process, generating the aminyl radical II and the subsequent unselective ring-closing process will reduce the enantioselectivity (Scheme 32).
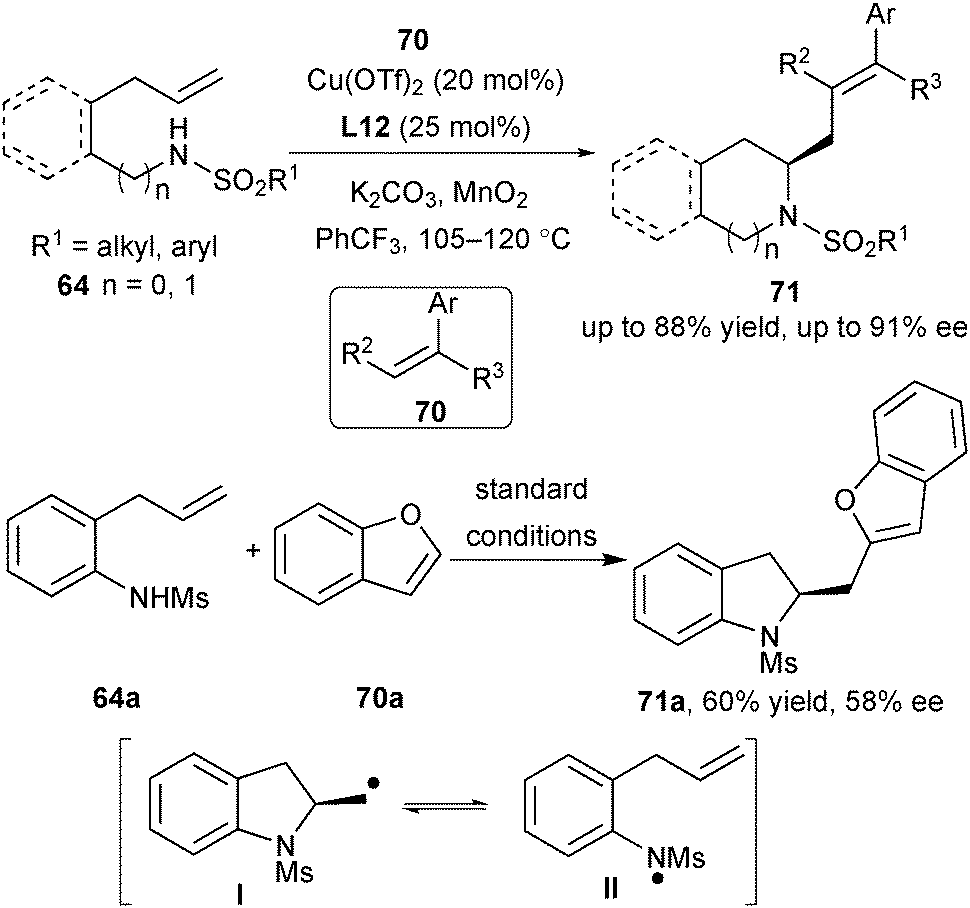 | ||
| Scheme 32 Chemler's enantioselective intra/intermolecular carboamination of alkenes. | ||
In 2008, Chemler established the first enantioselective alkene aminooxygenation reaction, in which tetramethylaminopyridyl radical (TEMPO) serves as both the trapping reagent and an oxidant (Scheme 33).47 This reaction also supported the generation of a carbon radical species upon the homolysis of the Cu–C bond.42 Under the modification of reaction conditions, they found that with L21 as the chiral ligand, alkenyl sulfonamides 64 reacted smoothly with TEMPO, resulting in chiral indolines and pyrrolidines 72 in high enantioselectivity. The use of environmentally benign O2 as a co-oxidant was necessary to achieve a full conversion of some substrates. The TEMPO adduct 72a could be efficiently converted to the corresponding chiral amino alcohol 73 and aldehyde 74 without significantly diminished enantioselectivity (Scheme 33).
 | ||
| Scheme 33 Chemler's enantioselective alkene aminooxygenation with TEMPO. | ||
The importance of chiral vicinal diamines prompted Chemler's group to extend their aminocupration strategy for the realization of enantioselective diamination of unactivated terminal alkenes for straightforward access to a range of chiral vicinal diamines. In 2014, they realized such transformations by utilizing sulfonamides as the trapping reagent (Scheme 34).48 One unique feature of this strategy is the introduction of a catalytic amount of KMnO4 to assist in the oxidation phase of the catalytic cycle. Although they were not certain of the exact role of KMnO4, they speculated that it might serve to regenerate the poorly insoluble MnO2. The reaction was suitable for a wide range of aryl and alkyl sulfonamides 75, providing indolines and pyrrolidines 77 in excellent enantioselectivity (up to >95% ee) (Scheme 34). Mechanistically, the in situ generated alkyl radical Ivia homolysis of the C–Cu bond may combine with CuIINHSO2R to form the CuIII species II and subsequent reductive elimination leads to the construction of the second C–N bond of 77.
 | ||
| Scheme 34 Chemler's enantioselective diamination of unactivated alkenes. | ||
In 2015, Zeng and co-workers reported a copper-catalysed enantioselective intramolecular diamination of unactivated terminal alkenyl ureas (Scheme 35).49 This double intramolecular diamination offered a protocol for concise and enantioselective synthesis of chiral vicinal bicyclic heterocycles 79. One difference between this protocol and Chemler's enantioselective aminocupration lies in the protecting group of amine. While Chemler used the sulfonyl group in most of their asymmetric aminocupration reactions, Zeng designed a urea group as the diamine source in this work.
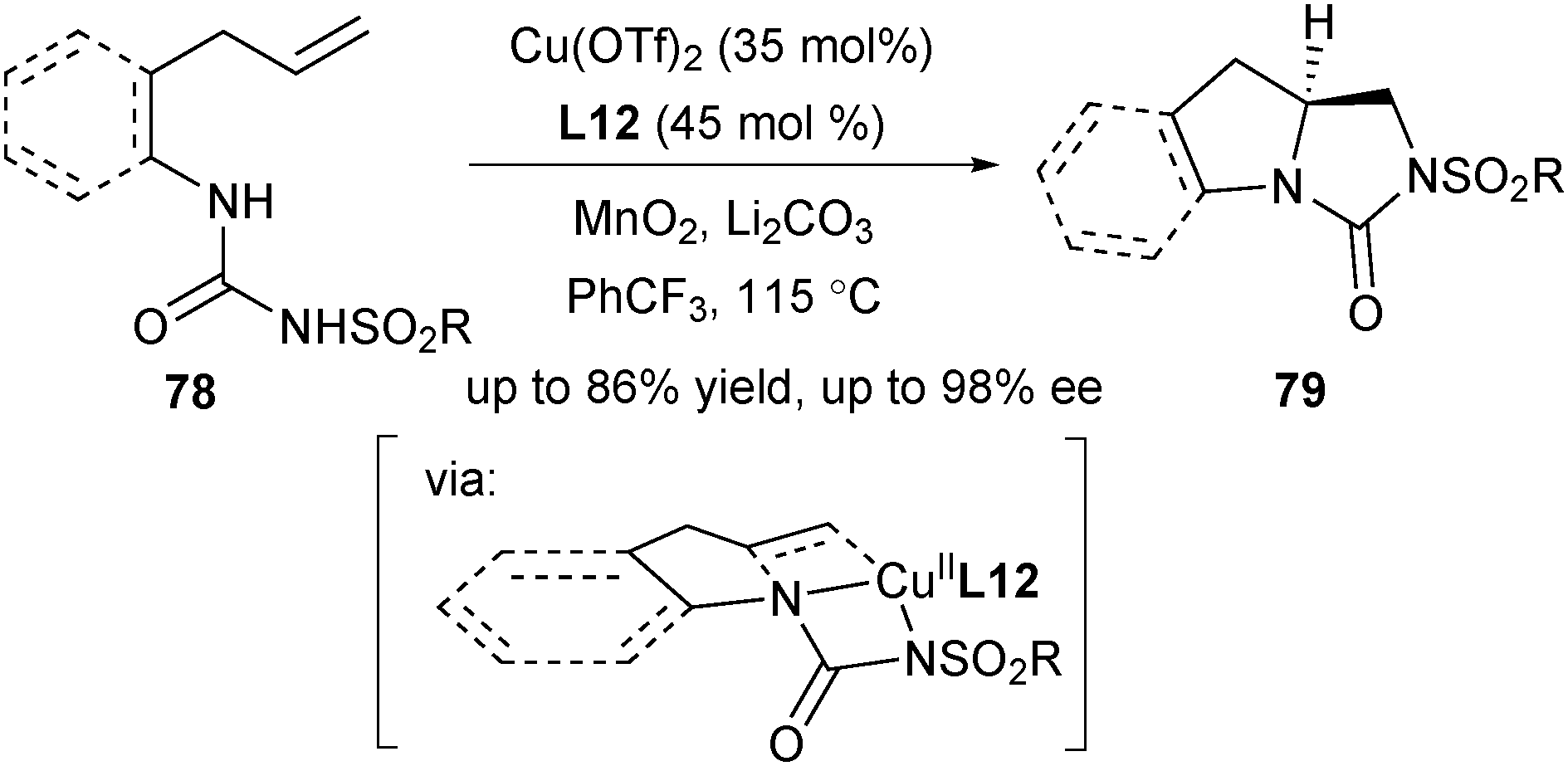 | ||
| Scheme 35 Zeng's intramolecular enantioselective diamination of unactivated alkenes. | ||
Chemler's group envisioned that if the carbon radical Iin situ generated from the homolysis of the Cu–C bond underwent a halogen atom transfer with the corresponding halogen atom donor, an aminohalogenation of unactivated terminal alkenes would be realized (Scheme 36). With this in mind, after screening of several halogen sources, Chemler and co-workers demonstrated a copper-catalysed enantioselective alkene aminohalogenation reaction, with 2-iodopropane 80a, 1,1-dichloroethene 80b and 80c as the corresponding halogen atom source (Scheme 36).50 Moreover, the aminohalogenation products 81 are crystalline and easily subjected to chiral amplification by recrystallization.
 | ||
| Scheme 36 Chemler's enantioselective alkene aminohalogenation. | ||
Inspired by the aminocupration process, Chemler's group developed a copper-catalysed intramolecular carboetherification of unactivated terminal alkenes in 2012, providing fused and bridged-ring tetrahydrofurans from alkenyl alcohols. One representative transformation to heterocycles 83 is shown in Scheme 37a.51 Based on their detailed mechanistic study, Chemler and co-workers proposed a plausible mechanism analogous to aminocupration. Alkenyl alcohol 82 coordinated with CuIIL and the subsequent cis oxycupration resulted in an intermediate I. Homolysis of the C–Cu bond of this intermediate provided the alkyl radical species II, followed by an intramolecular radical addition to arene and oxidative rearomatization afforded the final product 83. In their initial study of enantioselective alkene carboetherification, they found that using the best ligand L12 in previous aminocupration, substrate 82a generated almost a racemic product 83a, while substrate 82b delivered 83b in estimated 75% ee (Scheme 37b). They envisioned that the steric effect of the substrate near the alcohol region is critical to the enantioselectivity of the oxycupration step and increasing the steric effect would be beneficial to enantioselectivity (transition state IV in Scheme 37b). They also observed that the same ligand L12 would favour the (S)-carboamination product and (R)-carboetherification product, respectively. They assumed that it is because the large group of the substrates has switched in these two reactions. For the sulfonamide, the NSO2R group is larger than the alkene. However, for the alcohol, the alkene is larger than the alkoxyl group (IV and V in Scheme 37b).
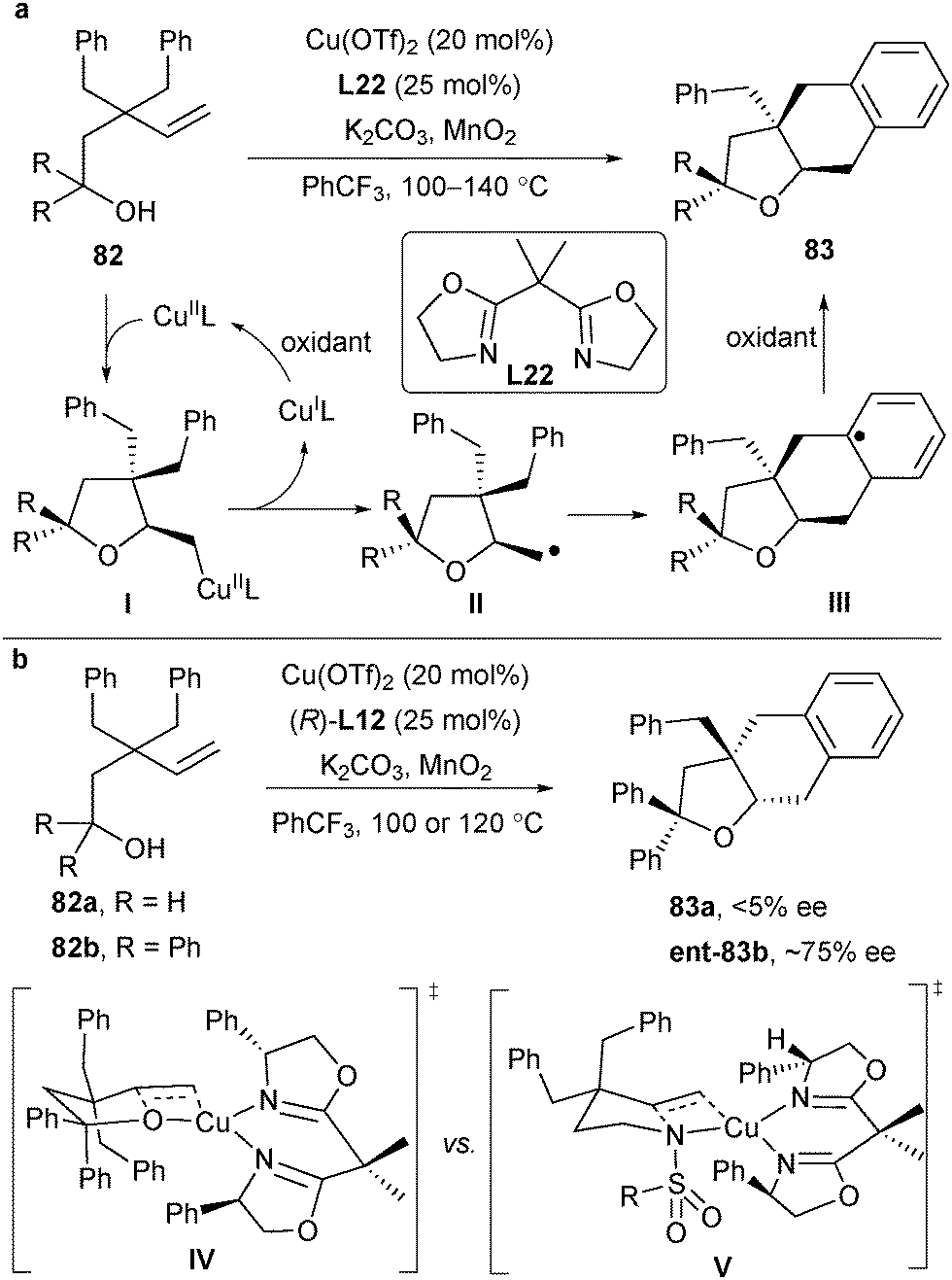 | ||
| Scheme 37 Chemler's intramolecular alkene carboetherification. | ||
The initial promising enantioselectivity experiment prompted Chemler to study the asymmetric carboetherification systematically. Thus changing the substituents of the box ligand from the Ph (L12) to t-butyl group (L1) led to a great increase of the ee value (Scheme 38).52 Screening of substrate scope revealed that the enantioselective carboetherification could yield a range of chiral fused and bridged-tetrahydrofurans in excellent enantioselectivity. Moreover, the carboetherification reaction was also applicable to the intermolecular version with styrenes 70 as the radical acceptors,46 thus allowing the assembly of skeletally diverse chiral tetrahydrofurans (Scheme 38).52
 | ||
| Scheme 38 Chemler's enantioselective carboetherification of unactivated alkenes. | ||
The enantioselective synthesis of spirocyclic ethers utilizing strategies involving the construction of both rings in one-step with concomitant stereocontrol at the quaternary carbon center is quite rare. To address this issue, Chemler and co-workers applied their enantioselective carboetherification of unactivated alkenes for the synthesis of diverse spirocyclic ethers in one step in 2018 (Scheme 39).53 Investigation of the substrate scope demonstrated that aryl-substituted alkenols 86 favour an endo addition to give 5,6-spirocycles 87 and 6,6-spirocycles (not shown here), while alkenyl- and alkynyl-substituted alkenols 88 undergo an exo addition to provide the 5,5-spirocycles 89.
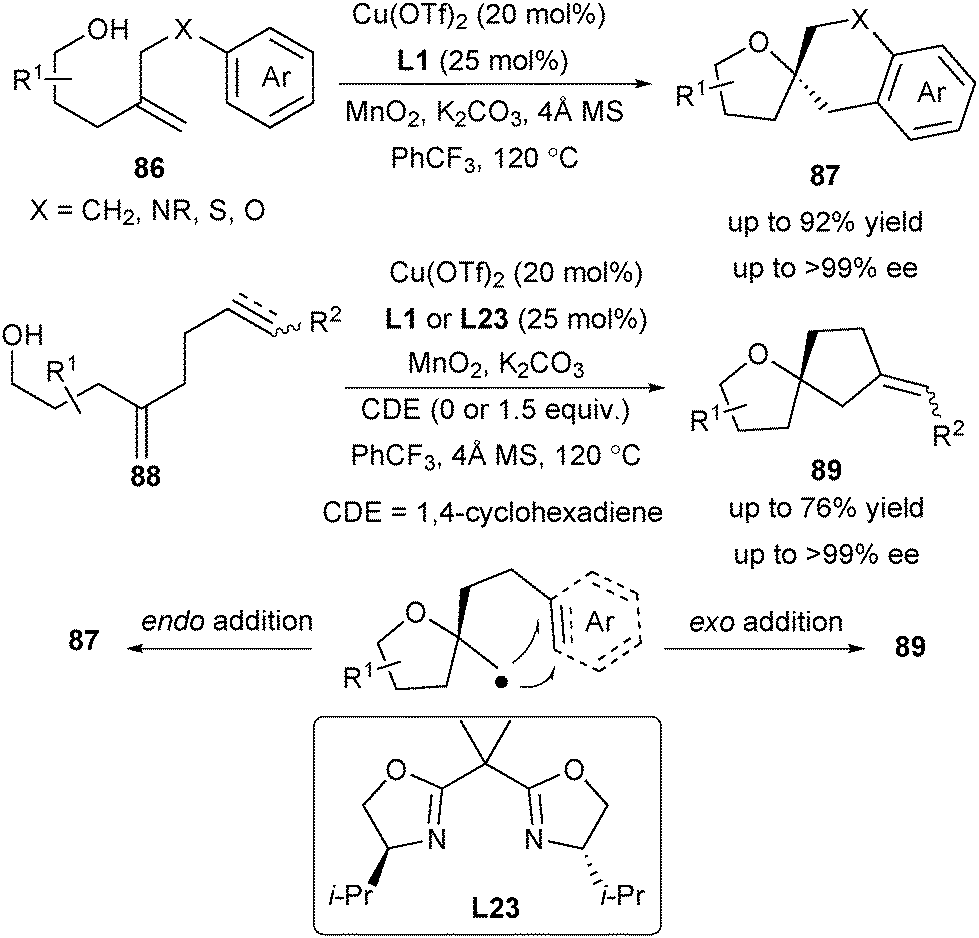 | ||
| Scheme 39 Enantioselective alkene carboetherification towards spirocyclic ethers. | ||
Recently Chemler's group applied the enantioselective alkene carboetherification strategy for the synthesis of enantioenriched phthalans from 2-vinyl benzyl alcohols (Scheme 40).54 Vinylarenes 91 were used as the radical acceptors in the enantioselective cyclization/coupling sequences of unsaturated alcohols 90. The authors further demonstrated its synthetic utility in the enantioselective synthesis of (R)-fluspidine 93, a σ1 receptor ligand.
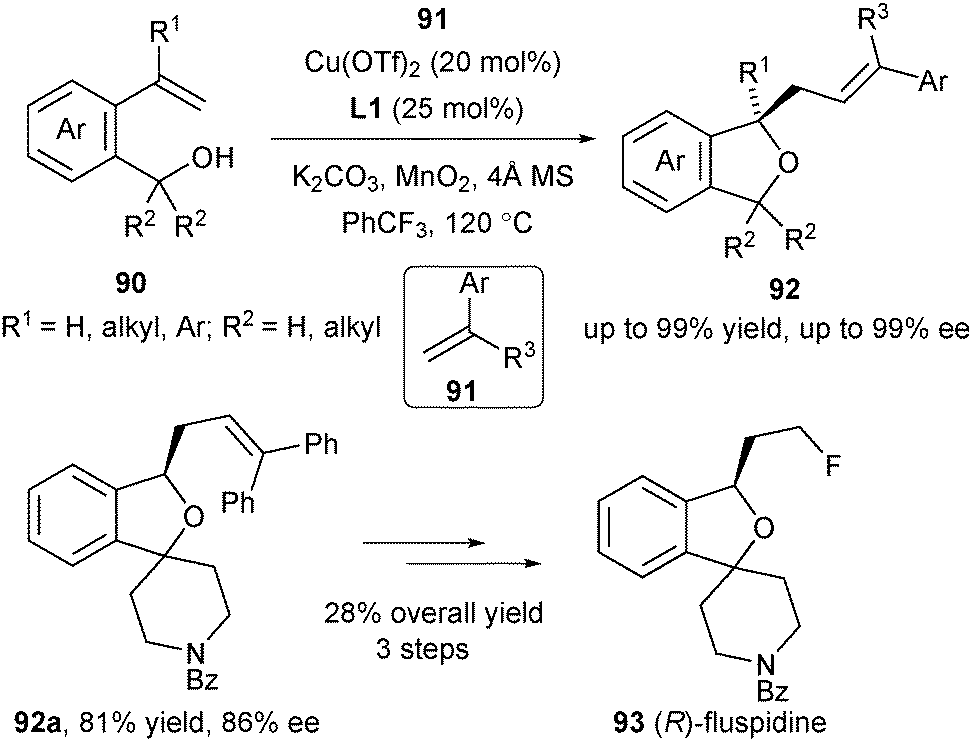 | ||
| Scheme 40 Enantioselective carboetherification for synthesis of phthalans. | ||
4. Conclusion and outlook
The radical-involved asymmetric 1,2-difunctionalization of alkenes has appeared as an elegant strategy for the rapid assembly of enantioenriched complex molecules from simple alkene feedstocks. This review highlights the recent advances in radical-involved asymmetric 1,2-difunctionalization of alkenes by merging chiral copper catalysis and radical chemistry. In this aspect, the key issue ultimately focuses on the design of chiral ligands to coordinate with copper salt to provide a chiral environment for asymmetric introduction. Fortunately, copper is not only a good SET catalyst to generate radicals with radical precursors, but also a good Lewis acid, coordinating with diverse kinds of chiral ligands, such as Box, diphosphine, CPA and cinchona alkaloid-derived sulfonamide to offer a chiral environment for asymmetric control. Conceptually, two distinct strategies have been developed for the asymmetric control of alkene difunctionalization. The first one mainly deals with the trapping of the prochiral alkyl intermediate generated from the addition of the radical to alkene with CuII/L*, followed by a reductive elimination or nucleophilic addition to carbocation. The key to the success of this strategy lies in the stereocontrol of the highly reactive alkyl radical intermediate. On the other hand, the second strategy focuses on the CuII/L*-mediated enantiodetermining nucleocupration and a sequential homolysis of the Cu–C bond/radical trapping process.Despite the enormous efforts in the field, several challenges have yet to be addressed: (1) almost all the reactions covering the trapping of the prochiral alkyl radical intermediate with CuII/L* illustrated above deal with styrene type alkenes. It is highly desirable to extend the approach to unactivated aliphatic alkenes. (2) In the nucleocupration reaction, only intramolecular amino- and oxycupration reactions are unveiled, and the realization of intermolecular amino- and oxycupration reactions remains a significant challenge. (3) Until now, we have witnessed the versatile application of neutral Box and anionic CPA ligands for construction of the chiral C–C(N,O) bond in the radical-involved alkene difunctionalization. While these ligands can provide a good chiral environment for stereocontrol, they are limited for tuning the redox potential of copper catalysts and only highly oxidative radical precursors are applicable in copper-catalysed radical addition to alkenes. Therefore, the design of powerful multidentate anionic–neutral hybrid ligands is urgent not only for offering a rigid chiral environment but also for tuning the redox potential of copper catalysts to be compatible with more radical precursors and for realizing more radical-involved asymmetric reactions. We hope that the concept introduced in this review will stimulate further research in this field to exploit new and efficient methodology in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (No. 21722203, 21831002, and 21801116), Shenzhen Special Funds for the Development of Biomedicine, Internet, New Energy, and New Material Industries (JCYJ20170412152435366, JCYJ20180302180235837), Guangdong Natural Science Foundation (2018A030310083), and Shenzhen Nobel Prize Scientists Laboratory Project (C17783101). The authors gratefully acknowledge Professor Sherry Chemler (The State University of New York at Buffalo) for her valuable suggestions on the manuscript.Notes and references
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC.
- M. P. Sibi, S. Manyem and J. Zimmerman, Chem. Rev., 2003, 103, 3263–3296 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- K. Wang and W. Kong, Chin. J. Chem., 2018, 36, 247–256 CrossRef CAS.
- J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed.
- S. R. Chemler, S. D. Karyakarte and Z. M. Khoder, J. Org. Chem., 2017, 82, 11311–11325 CrossRef CAS PubMed.
- F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS PubMed.
- A. B. Charette, H. Lebel and M. Roy, Asymmetric Cyclopropanation and Aziridination Reactions, in Copper-Catalyzed Asymmetric Synthesis, ed. A. Alexakis, N. Krause and S. Woodward, Wiley-VCH, Weinheim, 5th edn, 2014, pp. 203–238 Search PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed , and references cited therein.
- F. Wang, D. Wang, X. Mu, P. Chen and G. Liu, J. Am. Chem. Soc., 2014, 136, 10202–10205 CrossRef CAS PubMed.
- J. He, C. Chen, G. C. Fu and J. C. Peters, ACS Catal., 2018, 8, 11741–11748 CrossRef CAS PubMed.
- R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 12462–12465 CrossRef CAS PubMed.
- R. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 12655–12658 CrossRef CAS PubMed.
- R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 8069–8077 CrossRef CAS PubMed.
- Y. Wang, L. Deng, J. Zhou, X. Wang, H. Mei, J. Han and Y. Pan, Adv. Synth. Catal., 2018, 360, 1060–1065 CrossRef CAS.
- F. Wang, D. Wang, X. Wan, L. Wu, P. Chen and G. Liu, J. Am. Chem. Soc., 2016, 138, 15547–15550 CrossRef CAS PubMed.
- D. Wang, F. Wang, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2017, 56, 2054–2058 CrossRef CAS PubMed.
- W. Sha, L. Deng, S. Ni, H. Mei, J. Han and Y. Pan, ACS Catal., 2018, 8, 7489–7494 CrossRef CAS.
- Q. Guo, M. Wang, Q. Peng, Y. Huo, Q. Liu, R. Wang and Z. Xu, ACS Catal., 2019, 9, 4470–4476 CrossRef CAS.
- G. Zhang, L. Fu, P. Chen, J. Zou and G. Liu, Org. Lett., 2019, 21, 5015–5020 CrossRef CAS PubMed.
- N. Fu, L. Song, J. Liu, Y. Shen, J. C. Siu and S. Lin, J. Am. Chem. Soc., 2019, 141, 14480–14485 CrossRef CAS PubMed.
- L. Wu, F. Wang, X. Wan, D. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 2904–2907 CrossRef CAS PubMed.
- D. Wang, L. Wu, F. Wang, X. Wan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 6811–6814 CrossRef CAS PubMed.
- L. Wu, F. Wang, P. Chen and G. Liu, J. Am. Chem. Soc., 2019, 141, 1887–1892 CrossRef CAS PubMed.
- L. Fu, S. Zhou, X. Wan, P. Chen and G. Liu, J. Am. Chem. Soc., 2018, 140, 10965–10969 CrossRef CAS PubMed.
- X.-Q. Mou, F.-M. Rong, H. Zhang, G. Chen and G. He, Org. Lett., 2019, 21, 4657–4661 CrossRef CAS PubMed.
- D. J. Michaelis, K. S. Williamson and T. P. Yoon, Tetrahedron, 2009, 65, 5118–5124 CrossRef CAS PubMed.
- Y. Zhu, R. G. Cornwall, H. Du, B. Zhao and Y. Shi, Acc. Chem. Res., 2014, 47, 3665–3678 CrossRef CAS PubMed.
- H. Du, B. Zhao, W. Yuan and Y. Shi, Org. Lett., 2008, 10, 4231–4234 CrossRef CAS PubMed.
- B. Zhao, H. Du and Y. Shi, J. Org. Chem., 2009, 74, 8392–8395 CrossRef CAS PubMed.
- P. Yu, J.-S. Lin, L. Li, S.-C. Zheng, Y.-P. Xiong, L.-J. Zhao, B. Tan and X.-Y. Liu, Angew. Chem., Int. Ed., 2014, 53, 11890–11894 CrossRef CAS PubMed.
- J.-S. Lin, X.-Y. Dong, T.-T. Li, N.-C. Jiang, B. Tan and X.-Y. Liu, J. Am. Chem. Soc., 2016, 138, 9357–9360 CrossRef CAS PubMed.
- J.-S. Lin, F.-L. Wang, X.-Y. Dong, W.-W. He, Y. Yuan, S. Chen and X.-Y. Liu, Nat. Commun., 2017, 8, 14841 CrossRef PubMed.
- F.-L. Wang, X.-Y. Dong, J.-S. Lin, Y. Zeng, G.-Y. Jiao, Q.-S. Gu, X.-Q. Guo, C.-L. Ma and X.-Y. Liu, Chem, 2017, 3, 979–990 CAS.
- X.-F. Li, J.-S. Lin, J. Wang, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, Acta Chim. Sin., 2018, 76, 878–882 CrossRef CAS.
- Y. Zeng, X.-D. Liu, X.-Q. Guo, Q.-S. Gu, Z.-L. Li, X.-Y. Chang and X.-Y. Liu, Sci. China: Chem., 2019, 62, 1529–1536 CrossRef CAS.
- J.-S. Lin, T.-T. Li, J.-R. Liu, G.-Y. Jiao, Q.-S. Gu, J.-T. Cheng, Y.-L. Guo, X. Hong and X.-Y. Liu, J. Am. Chem. Soc., 2019, 141, 1074–1083 CrossRef CAS PubMed.
- Y.-F. Cheng, X.-Y. Dong, Q.-S. Gu, Z.-L. Yu and X.-Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 8883–8886 CrossRef CAS PubMed.
- X.-T. Li, Q.-S. Gu, X.-Y. Dong, X. Meng and X.-Y. Liu, Angew. Chem., Int. Ed., 2018, 57, 7668–7672 CrossRef CAS PubMed.
- S. R. Chemler, Science, 2013, 341, 624 CrossRef CAS PubMed.
- M. C. Paderes, J. B. Keister and S. R. Chemler, J. Org. Chem., 2013, 78, 506–515 CrossRef CAS PubMed.
- W. Zeng and S. R. Chemler, J. Am. Chem. Soc., 2007, 129, 12948–12949 CrossRef CAS PubMed.
- L. Miao, I. Haque, M. R. Manzoni, W. S. Tham and S. R. Chemler, Org. Lett., 2010, 12, 4739–4741 CrossRef CAS PubMed.
- B. J. Casavant, A. S. Hosseini and S. R. Chemler, Adv. Synth. Catal., 2014, 356, 2697–2702 CrossRef CAS PubMed.
- T. W. Liwosz and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 2020–2023 CrossRef CAS PubMed.
- P. H. Fuller, J.-W. Kim and S. R. Chemler, J. Am. Chem. Soc., 2008, 130, 17638–17639 CrossRef CAS PubMed.
- B. W. Turnpenny and S. R. Chemler, Chem. Sci., 2014, 5, 1786–1793 RSC.
- S. Fu, H. Yang, G. Li, Y. Deng, H. Jiang and W. Zeng, Org. Lett., 2015, 17, 1018–1021 CrossRef CAS PubMed.
- M. T. Bovino and S. R. Chemler, Angew. Chem., Int. Ed., 2012, 51, 3923–3927 CrossRef CAS PubMed.
- Y. Miller, L. Miao, A. S. Hosseini and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 12149–12156 CrossRef CAS PubMed.
- M. T. Bovino, T. W. Liwosz, N. E. Kendel, Y. Miller, N. Tyminska, E. Zurek and S. R. Chemler, Angew. Chem., Int. Ed., 2014, 53, 6383–6387 CrossRef CAS PubMed.
- S. D. Karyakarte, C. Um, I. A. Berhane and S. R. Chemler, Angew. Chem., Int. Ed., 2018, 57, 12921–12924 CrossRef CAS PubMed.
- D. Chen and S. R. Chemler, Org. Lett., 2018, 20, 6453–6456 CrossRef CAS PubMed.
Footnote |
| † These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |