
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormazanate coordination compounds: synthesis, reactivity, and applications
Joe B.
Gilroy
 *a and
Edwin
Otten
*b
*a and
Edwin
Otten
*b
aDepartment of Chemistry and The Centre for Advanced Materials and Biomaterials Research (CAMBR), The University of Western Ontario, London, ON, Canada N6A 5B7. E-mail: joe.gilroy@uwo.ca
bStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: edwin.otten@rug.nl
First published on 5th December 2019
Abstract
Formazans (Ar1-NH-N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR3-NN-Ar5), a class of nitrogen-rich and highly colored compounds, have been known since the late 1800s and studied more closely since the early 1940s. Their intense color has led to their widespread use as dyes, especially in cell biology where they are most often used to quantitatively assess cell-viability. Despite structural similarities to well-known ligand classes such as β-diketiminates, the deprotonated form of formazans, formazanates, have received relatively little attention in the transition metal and main group coordination chemistry arenas. Formazanate ligands benefit from tunable properties via structural variation, rich optoelectronic properties owing to their highly delocalized π-systems, low-lying frontier orbitals that stabilize otherwise highly reactive species such as radicals, and redox activity and coordinative flexibility that may have significant implications in their future use in catalysis. Here, we review progress in the coordination chemistry of formazanate ligands over the past two decades, with emphasis on the reactivity and applications of the subsequent complexes.
CR3-NN-Ar5), a class of nitrogen-rich and highly colored compounds, have been known since the late 1800s and studied more closely since the early 1940s. Their intense color has led to their widespread use as dyes, especially in cell biology where they are most often used to quantitatively assess cell-viability. Despite structural similarities to well-known ligand classes such as β-diketiminates, the deprotonated form of formazans, formazanates, have received relatively little attention in the transition metal and main group coordination chemistry arenas. Formazanate ligands benefit from tunable properties via structural variation, rich optoelectronic properties owing to their highly delocalized π-systems, low-lying frontier orbitals that stabilize otherwise highly reactive species such as radicals, and redox activity and coordinative flexibility that may have significant implications in their future use in catalysis. Here, we review progress in the coordination chemistry of formazanate ligands over the past two decades, with emphasis on the reactivity and applications of the subsequent complexes.
 Joe B. Gilroy | Joe Gilroy is an Associate Professor in the Department of Chemistry at The University of Western Ontario (aka Western University). Originally from the West Coast of Canada, he completed both his BSc and PhD at the University of Victoria where he conducted research in stable radical chemistry under the supervision of Prof. Robin Hicks. He moved to the University of Bristol for his postdoctoral studies where he worked in various areas of metallopolymer chemistry with Prof. Ian Manners as an NSERC and EU Marie Curie PDF. At Western, Joe leads a talented team of students and postdocs who work in many different areas of synthetic materials chemistry, with major focuses including the development of luminescent and semiconducting molecular and polymeric materials. He has received several awards, including the Thieme Chemistry Journal Award, Western's Petro-Canada Young Innovator and Faculty Scholar Awards, the CNC-IUPAC travel award, and an Ontario Early Researcher Award. |
 Edwin Otten | Edwin Otten is an Associate Professor in the Stratingh Institute for Chemistry at the University of Groningen. He obtained a PhD in chemistry in 2008 (supervisor: Prof. Bart Hessen, University of Groningen). He subsequently moved to the University of Toronto as a NWO Rubicon postdoctoral fellow, where he worked on small-molecule activation by Frustrated Lewis Pairs with Prof. Doug Stephan. After a short period in industry (SABIC), Edwin was appointed as tenure-track Assistant Professor of Molecular Inorganic Chemistry at the University of Groningen in 2011. In the same year he was awarded a Veni grant, and in 2015 he received a Vidi grant from NWO. His research focuses on the development of new (catalytic) reactions using metal complexes with ligands that incorporate reactive (‘non-innocent’) sites. In addition, he is interested in ligand design for manipulating the electronic structure of main group and transition metal complexes, and how this impacts reactivity. |
1. Introduction
Formazans 1 were first reported in the 1890s, although they have only been studied extensively since the early 1940s.1–3 The intense color and redox-chemistry that originates from their nitrogen-rich backbone has led to their widespread use as dyes, mainly in chemical biology as the colored component of cell-viability assays,4,5 and as precursors to a family of stable radicals known as verdazyls 2 (Chart 1).6–8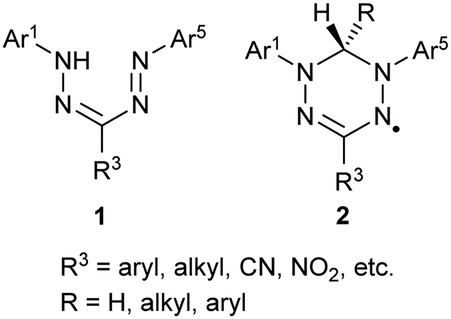 | ||
| Chart 1 | ||
The Ar1-NH-NCR3-NN-Ar5 backbone of formazans provides a platform for structural isomerization (Fig. 1). Bulky alkyl or aryl R3 substituents tend to favor the ‘closed’ (trans–syn, s-cis) isomer, while relatively small R3 substituents (e.g., H, Me, SMe, CN) allow for the ‘open’ (trans–syn, s-trans) and ‘linear’ (trans–anti, s-trans) geometry to be adopted. There is a strong correlation between the identity of the isomer adopted and the color of formazans, with the ‘closed’ isomer exhibiting a characteristic blood red color, while the ‘open’ and ‘linear’ forms taking on orange and yellow colors, respectively.
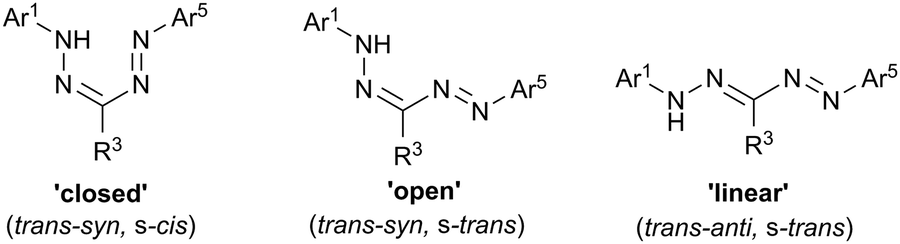 | ||
| Fig. 1 Structural isomers of formazans. | ||
One of the most intriguing features of formazan chemistry is their facile synthesis that allows for libraries of compounds to be prepared and facilitates property modulation through structural variation. While numerous synthetic routes to formazans exist,1 the most widely employed methods rely on the reaction of aryldiazonium salts with substrates possessing activated carbon functionalities. For example, triarylformazans 3 can be prepared by coupling one equivalent of aryl diazonium salt with hydrazones (Scheme 1a).1,9 This method is modular and provides access to asymmetric formazans with different Ar1 and Ar5 substituents, and can also be employed to prepare 3-alkyl formazans 4 (Scheme 1b).1,10–12 Alternatively, coupling two equiv. of aryldiazonium salts with compounds containing activated methylene groups yields symmetric formazans (Ar1 = Ar5). Thus, two equiv. of aryldiazonium salts react with the activated methylene group of phenylpyruvic acid derivatives to give triarylformazans 3 (Scheme 1c).1,13 Similar reactions lead to the formation of 3-cyanoformazans 5 (Scheme 1d)1,14 and 3-nitroformazans 6 (Scheme 1e).14,15 Although the yield of formazan products using these procedures is often moderate to good (typically 40–80%), the stability of the requisite diazonium salts may present problems, in particular when sterically demanding substituents are introduced.
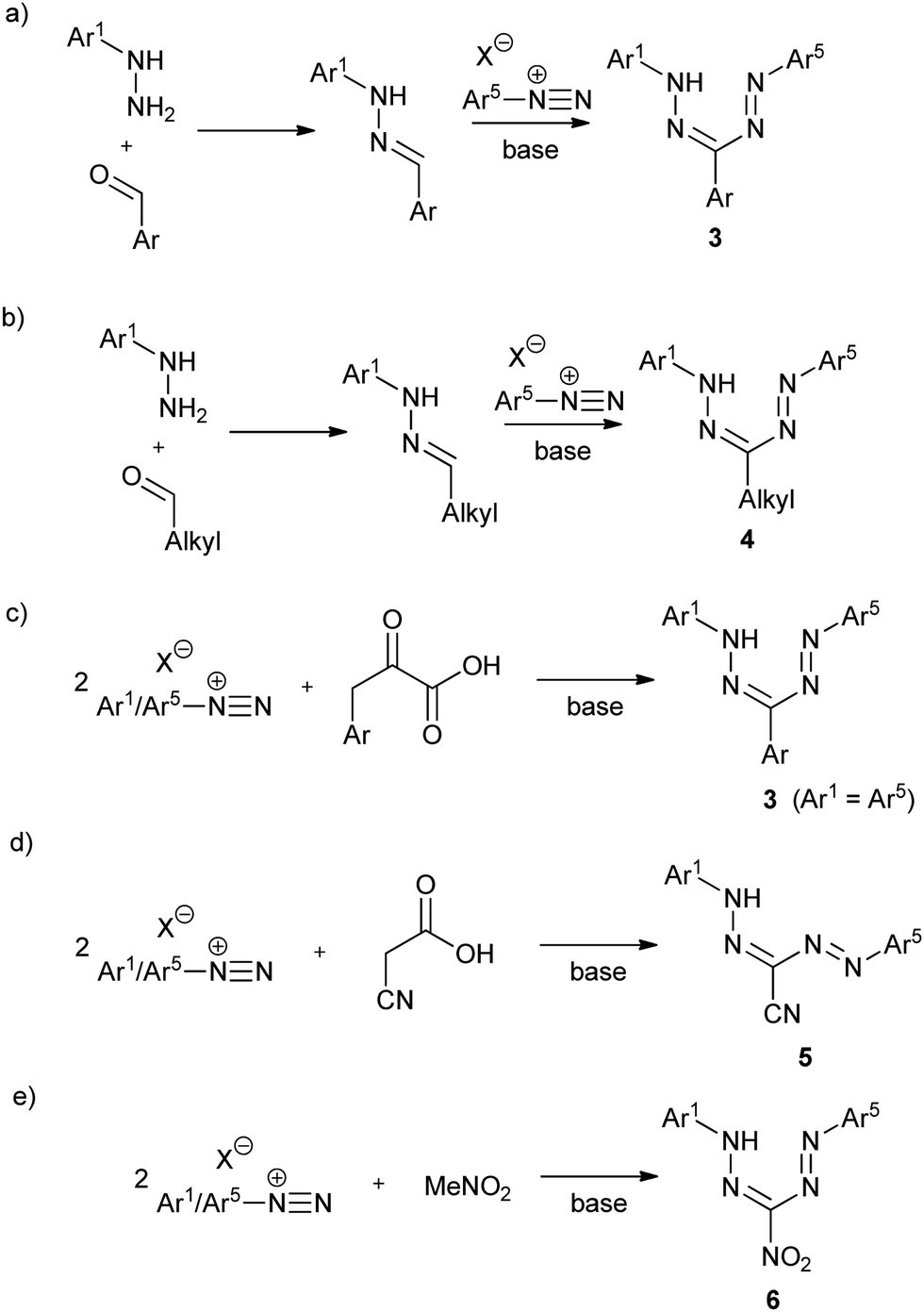 | ||
| Scheme 1 Common routes for the synthesis of formazans. | ||
Despite their structural similarity to other families of chelating N-donor ligands (e.g., β-diketiminates 7),16,17 it may be somewhat surprising that the coordination chemistry of the anionic form of formazans, from this point forward referred to as formazanates 8 (Chart 2), has not been studied to the same extent.3,18
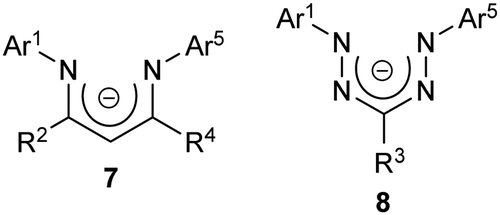 | ||
| Chart 2 | ||
Formazanate ligands offer several unique and potentially useful traits when compared to related families of ligands. One important feature is their ability to engage in both oxidative and reductive redox chemistry due to the presence of high-lying filled (e− donor) and low-lying empty (e− acceptor) orbitals of π-symmetry (Fig. 2A). The ready availability of formazans with diverse substitution patterns (Scheme 1) allows rational tuning of the energies of these frontier orbitals. Both the HOMO and LUMO are mainly composed of orbitals centered on the NNCNN backbone, with additional contributions from the π-conjugated aromatic N-Ar1/Ar5 groups. Thus, the energies of both these orbitals should be sensitive to substituents at the ortho- and para-position of the N-aryl rings, primarily via resonance effects. The LUMO is fully π-anti-bonding between the four nitrogen atoms in the formazanate framework and presents a nodal plane that runs through the C-R3 fragment. As a result, structural variation at this position only has an inductive effect on the energy of the LUMO. In comparison to β-diketiminates and other related chelating N-donor ligands, the LUMO is low in energy, and compounds containing formazanate ligands may therefore be expected to readily engage in ligand-based reduction reactions (i.e., formazanates are redox-active ligands).19–21 Moreover, the low-lying π*-orbital gives rise to electronic transitions in the visible range of the spectrum, and both the absorption and emission properties of formazanate complexes are readily tunable by modifying the ligand structure. Finally, the coordinative flexibility that results from the four nitrogen atoms in the backbone allows for formation of four-, five- and six-membered chelates (Fig. 2B). In the symmetrical coordination modes (i.e., with four- and six-membered chelate rings) the formazanate ligand backbones tend to exhibit bond lengths intermediate between single and double bonds of the respective atoms, indicating a high degree of electronic delocalization within the π-electron system. In the case of five-membered chelate rings, the metrical parameters in the formazanate backbone are often indicative of a more localized bonding picture although this appears to be dependent on the ligand substitution pattern and the nature of the central inorganic element.
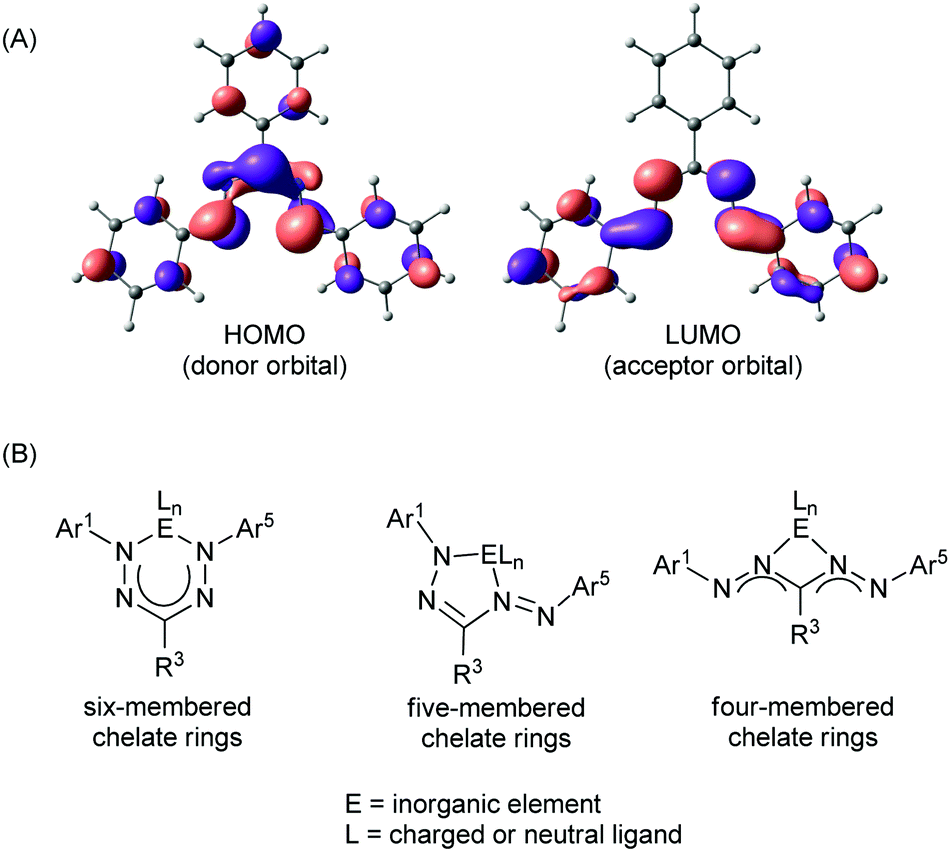 | ||
| Fig. 2 (A) Frontier (Kohn–Sham) orbitals of the triphenylformazanate anion in the commonly observed ‘closed’ form, calculated using density functional theory (B3LYP/6-31G(d)). (B) Common coordination modes of formazanate ligands in inorganic complexes. | ||
The facile synthesis and unique attributes of formazans and formazanates has led to rejuvenated interest in their coordination chemistry over the past two decades. Herein, we provide a review of recent developments with focus on the reactivity and applications facilitated by this unique class of ligands. We have chosen to organize our review by group in the periodic table and begin with alkali metal complexes.
2. Formazanate coordination chemistry
2.1 Alkali metals (Na, K)
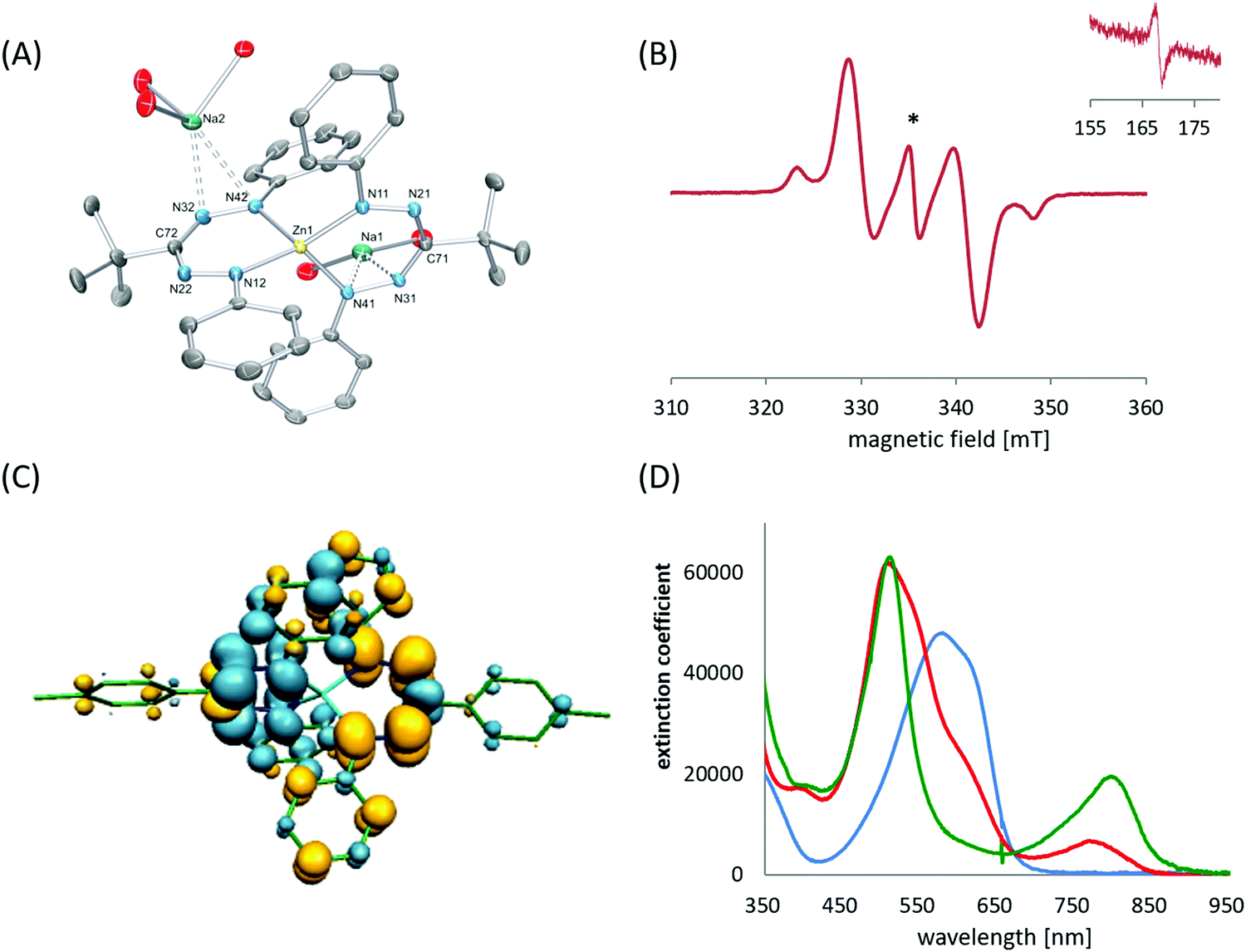
The deprotonation of formazans with strong alkali metal bases such as NaH or KH has been shown to cleanly generate the corresponding alkali metal formazanate salts (Scheme 2).22 Three different formazans were studied, all with aromatic N-groups (Ar1/5 = Ph, Mes) and substituents at the central C-atom (R3) that were either a p-tolyl (3a), t-butyl (4a) or cyano moiety (5a). The molecular structures in the solid state, obtained by X-ray crystallography, demonstrate the flexibility of these ligands in their coordination behavior: both 4- and 5-membered chelate rings are accessible due to the presence of the four nitrogen atoms in the formazanate NNCNN backbone which allows the terminal as well as the internal N-atoms to interact with the metal center in a bidentate binding mode. This leads to solid state structures that range from dimeric (9) to hexameric (10) and polymeric (11) (Scheme 2, dashed lines indicate bonds that cause aggregation). In these ionic compounds, a high degree of π-delocalization is indicated by the equivalent N–N and C–N bond lengths within the ligand core, regardless of the chelate ring size. Cation exchange of the potassium salt 9 with [Bu4N][Br] afforded the ion pair 12. The crystal structure of 12 shows that, in the absence of a coordinating cation, the triarylformazanate anion adopts a linear arrangement. The solution structure of 9 was examined by variable-temperature NMR spectroscopy, which indicated that the dimeric structure is retained even in a donor solvent such as THF. These alkali metal salts are potentially useful reagents for the transfer of formazanate ligands to transition metals via salt metathesis reactions.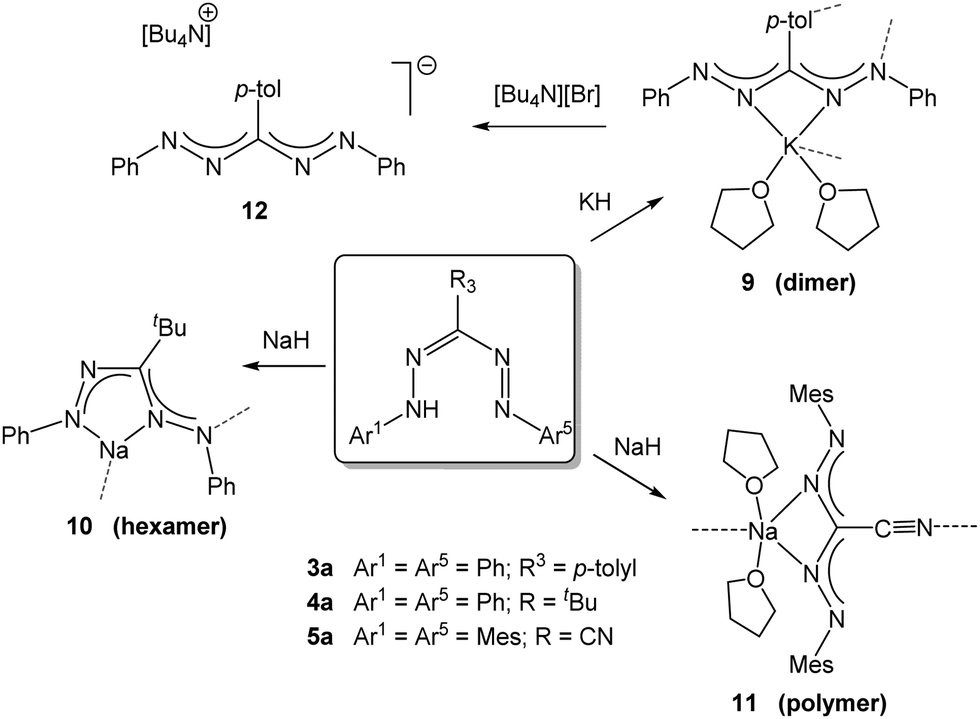 | ||
| Scheme 2 Synthesis of alkali metal salts of formazanate ligands.22 | ||
While alkali metal formazanate salts are stable when non-functionalized alkyl or aryl substituents are present, introduction of a C6F5 group as the R3 substituent does not allow the synthesis of the corresponding formazanate salt by deprotonation. Instead, the nucleophilic formazanate anion 13− evolves by nucleophilic aromatic substitution (SNAr) onto the ortho-position of the C6F5 ring to give cyclization product 14 as a mixture of regioisomers (Scheme 3). The arylazoindazole 14 presents a novel heteroaromatic motif for azo photoswitches, and it was shown to have good photoconversion, thermal stability as well as fatigue resistance.23
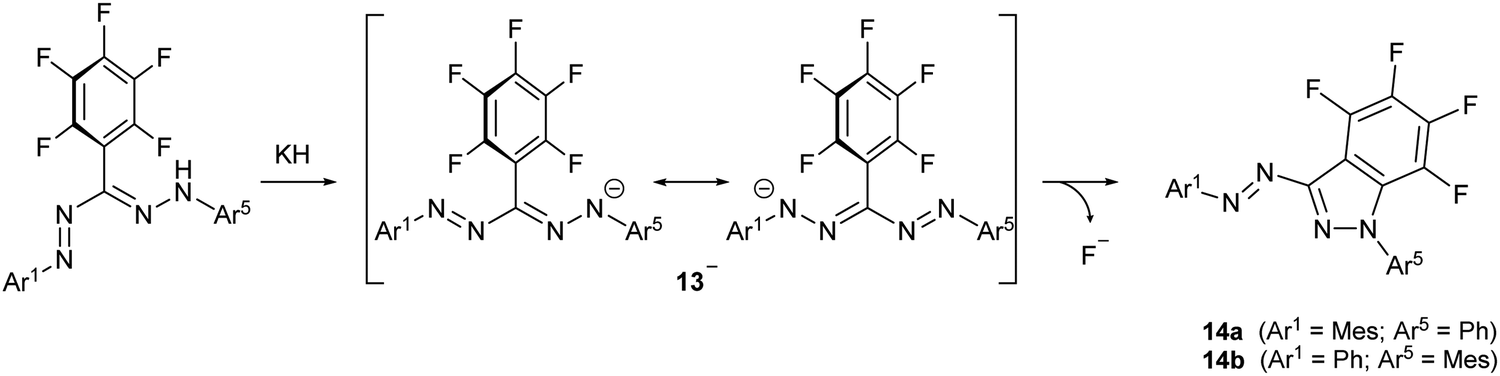 | ||
| Scheme 3 SNAr cyclization upon deprotonation of a 3-perfluorophenylformazan to give arylazoindazoles 14.23 | ||
2.2 Group 7 (Mn)
Formazanate complexes of group 7 metals have been reported only once in a 1998 report by the group of Brown with the preparation of the bis(benzothiazolylformazanate) manganese(II) complex 15.24 Compound 15 was obtained from the manganese(III) precursor Mn(acac)3 by refluxing with formazan in EtOH under N2 atmosphere (Scheme 4). The Mn(II) product likely arises from reduction by excess formazan, which in this process is oxidized to the corresponding tetrazolium salt. In the low-spin Mn(II) complex, the formazanate ligands are bound to the metal center in a N,N′,N′′-tridentate fashion involving the benzothiazolyl substituent to give an octahedral coordination geometry. Formazanate complexes with the heavier congeners in group 7 have not been reported to the best of our knowledge.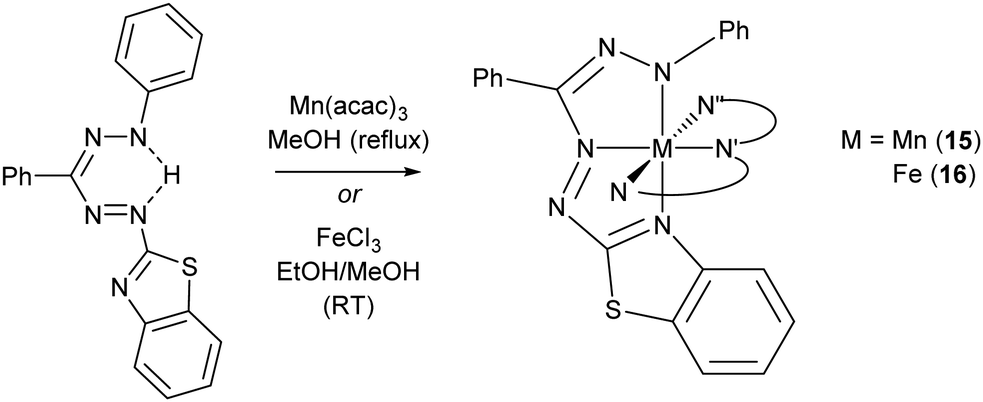 | ||
| Scheme 4 Synthesis of homoleptic manganese(II) and iron(II) complexes with benzothiazolyl-substituted formazanate ligands (only one ligand shown in full, other abbreviated (N,N′,N′′) for clarity).24–26 | ||
2.3 Group 8 (Fe, Ru, Os)
Early work on Fe complexes with benzothiazolylformazanate ligands mirrors the results described above for Mn: treatment of Fe(III) salts with formazans leads to formation of low-spin octahedral Fe(II) complexes (16, Scheme 4), some of which were crystallographically characterized.24–26 More recently, Hicks and co-workers reported an Fe(III) complex with a trianionic N2O2 ligand based on the formazanate scaffold, in which the N-aryl groups have an additional phenoxide O-donor moiety.27 The low-spin Fe(III) complex 17 was obtained by salt metathesis from in situ generated Na-salt of the tetradentate N2O2 ligand with FeCl3 (Scheme 5). Two additional pyridine ligands are bound trans to each other to give a pseudo-octahedral coordination geometry in 17. Apparently, reduction to Fe(II) as discussed above does not occur in the synthesis of 17,24 which may be due to the presence of the additional anionic O-donor groups.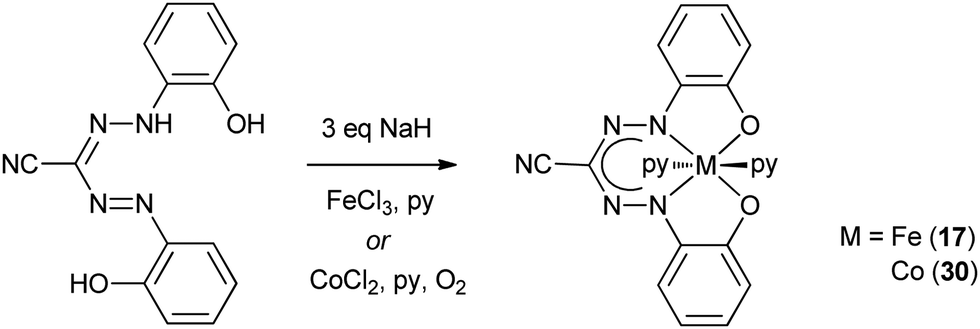 | ||
| Scheme 5 Synthesis of Fe(III) and Co(III) complexes with trianionic N2O2 ligands based on the formazanate scaffold.27 | ||
In 2016, the synthesis of the Fe(II) complex 18 with two simple, monoanionic triarylformazanate ligands was reported via salt metathesis using the potassium formazanate 9 (Scheme 6).28 The absence of additional coordinating groups (e.g., the benzothiazolyl substituent in compounds 16) results in a four-coordinate environment around the iron center with the formazanate ligands bound via the terminal N-atoms. In contrast to the benzothiazolylformazanate complex 16, the coordination mode of the formazanate ligands in compound 18 gives rise to six-membered chelate rings. The crystallographically determined solid-state structure of 18 showed a ‘flattened’ tetrahedral structure with very short Fe–N bond lengths of 1.8174(16)–1.8330(16) Å. Other remarkable features of this compound included its NMR and UV/vis spectra, which indicated a temperature-dependent equilibrium between a diamagnetic state (S = 0) at low temperature and a paramagnet (S = 2) at high temperature.
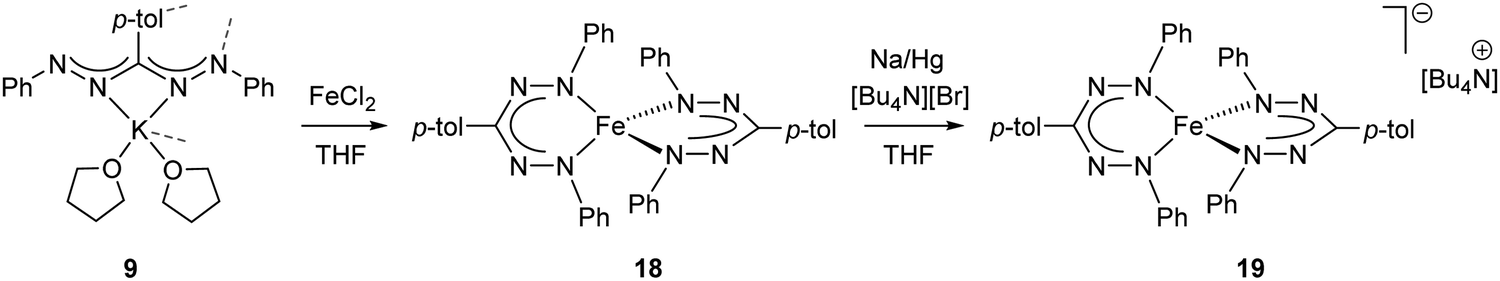 | ||
| Scheme 6 Synthesis of bis(formazanate)iron(II) complex 18 and the corresponding one-electron reduction product 19.28 | ||
The unusual occurrence of spin-crossover in a four-coordinate complex was attributed to π-backdonation from the d6 metal center to a low-lying formazanate π*-orbital, which stabilizes one of the high-energy orbitals and leads to an energy-ordering of the d-orbital manifold that has a 2-over-3 splitting (Fig. 3) that is reminiscent of that of six-coordinate, octahedral complexes (ligand-field ‘inversion’).29 Cyclic voltammetry measurements for 18 showed quasi-reversible redox-events at −1.21 and −2.01 V (vs. Fc0/+), which prompted attempts to synthesize and characterize these reduced species on a preparative scale. One-electron reduction of the Fe(II) complex 18 using one equiv. of Na/Hg as the reducing agent allowed the high-yield synthesis of the anionic complex 19. Analysis of structural (X-ray diffraction), spectroscopic (EPR, Mössbauer) and computational (DFT) data showed that 19 is best formulated as a low-spin (S = 1/2) Fe(I) complex containing closed-shell, monoanionic formazanate ligands. An alternative description in which reduction takes place at the ligand (with the additional electron in the low-energy formazanate π*-orbital) is not supported by the empirical data, despite the prominence of ligand-based reductions for these ‘redox-active’ ligands (see right).
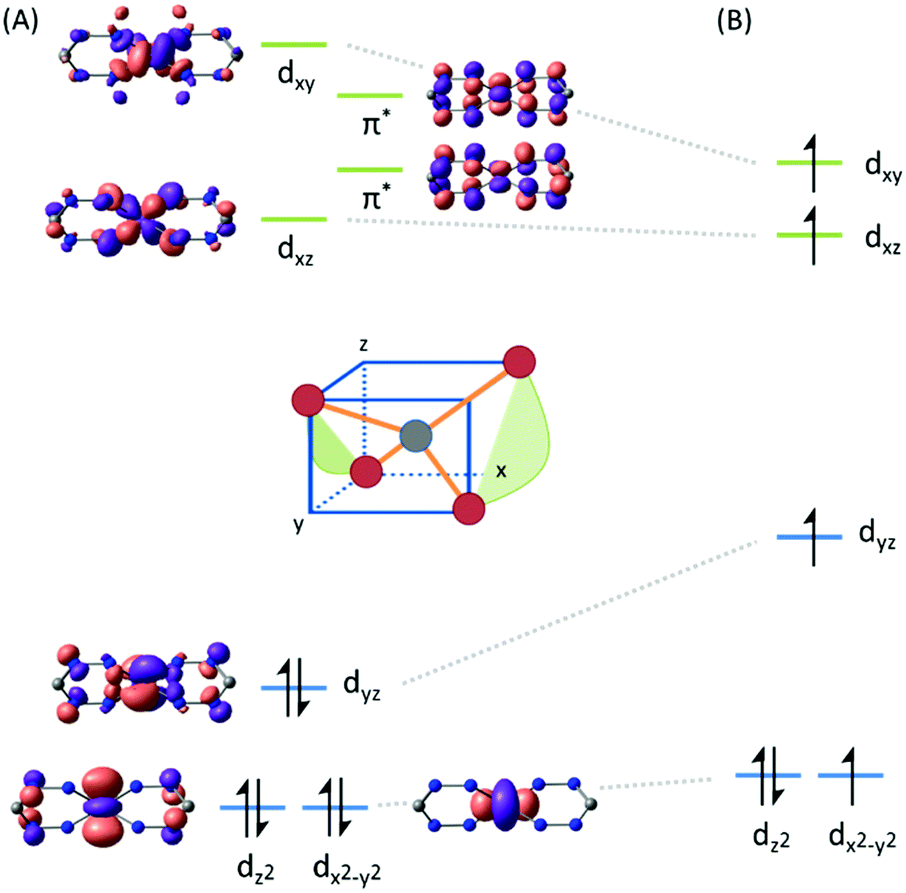 | ||
| Fig. 3 DFT calculated frontier molecular orbitals in the low-spin (A) and high-spin state (B) of compound 18. Reproduced from ref. 28 (https://pubs.acs.org/doi/10.1021/jacs.6b01552) with permission from the American Chemical Society. Further permissions related to the material excerpted should be directed to the ACS. | ||
The redox-active nature of the formazanate ligand in Fe(II) complexes was explored by the Holland group. Mono-formazanate iron amide 20 or its THF adduct 20-THF were used to prepare the one-electron reduction product 21 (Scheme 7), which was studied using a variety of spectroscopic and computational techniques.30 A multi-configurational quartet ground state was calculated for 21 using SORCI, which reproduces the empirical spectroscopic data. The calculations suggest two configurations to be dominant (ca. 25% contribution each). One of those represents a high-spin Fe(II) center that is anti-ferromagnetically coupled to a singly occupied ligand π*-orbital, whereas the other is best described as high-spin Fe(I) without ligand participation.
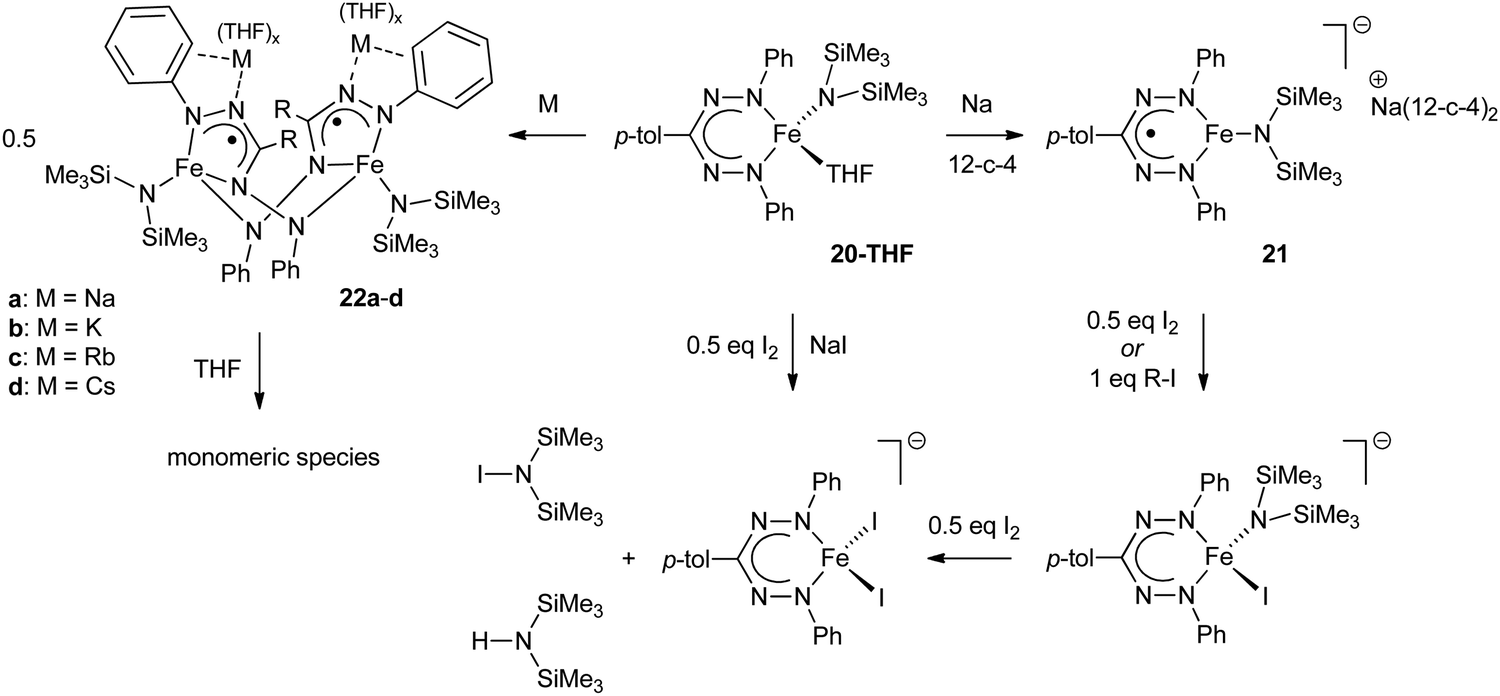 | ||
| Scheme 7 Conversion of mono(formazanate)iron(II) amide complex 20-THF to the one-electron reduction products 21 and 22.30,31 | ||
The reactivity of 21 towards alkyl iodides and I2 was investigated, which suggested that formation of the reductive elimination product IN(SiMe3)2 occurred (at least formally) via a pathway that involves redox-reactions in the ligand (Scheme 7). IN(SiMe3) was also obtained directly from 20-THF and I2/NaI.
Another study from these authors examined the effect of the countercation on the structures and reactivity of a series of derivatives of 21.31 It was shown that when the countercation is one of the alkali metals (Na+, K+, Rb+ or Cs+), the compounds are dimeric in the solid state (22a–d, Scheme 7). In these dimers, the formazanate ligands coordinate in the ‘open’ form to give five-membered metallacycles, in which the ‘pendant’ terminal N-atom bridges to another Fe center. In contrast, sequestering the alkali cation with crown ether or allowing the dimers to equilibrate in THF solution results in monomeric species in solution similar to 21 in which the formazanate ligand gives rise to a six-membered metallacycle.
The reactivity of the neutral complex 20 (or the THF-adduct 20-THF) towards CO2 was examined and shown to cleanly form isocyanate and the dimeric iron siloxide product 23via the carbamate intermediate 24, which was isolated from a low-temperature reaction and crystallographically characterized (Scheme 8).32
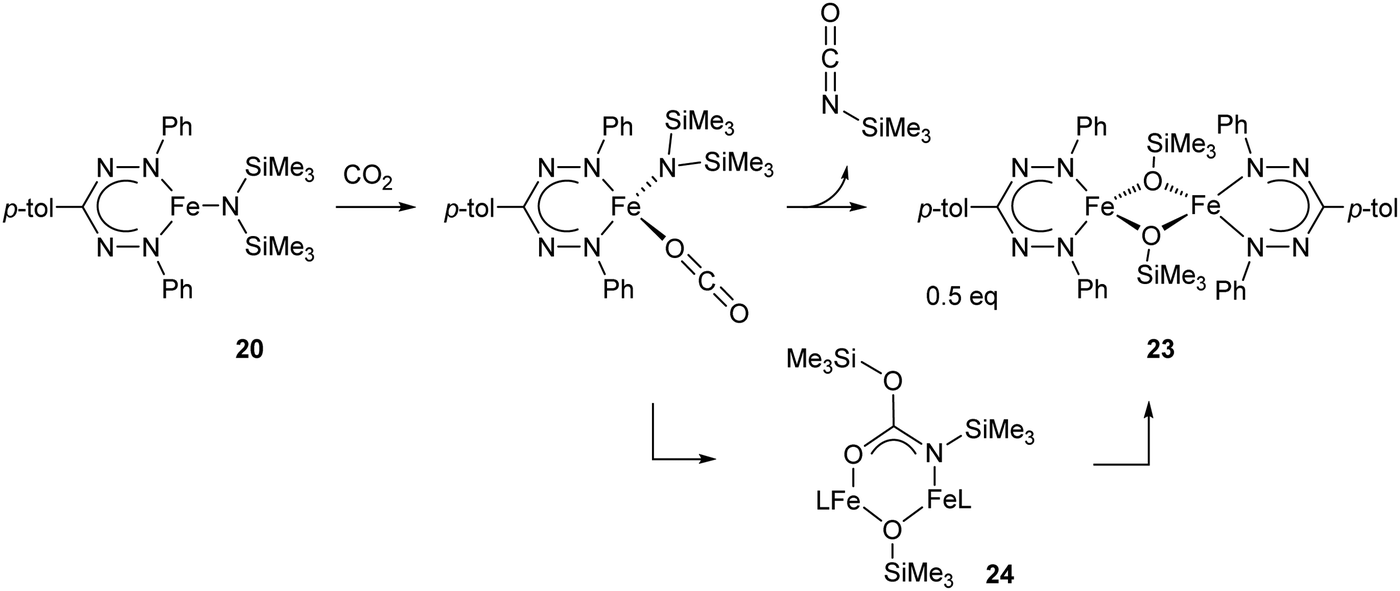 | ||
| Scheme 8 Reaction of CO2 and mono(formazanate)iron(II) amide complex 20 to form isocyanate.32 | ||
Mono(formazanate)iron(II) complexes with halide co-ligands are prone to ligand exchange reactions to the thermodynamically favored bis(formazanate) complexes (e.g., 19), but carrying out salt metathesis reactions in the presence of an additional equivalent of halide (such as [Bu4N][X]) allows high-yield synthesis of four-coordinate ferrate complexes 25.33 The halide ligands in these complexes are labile, as demonstrated by the formation of octahedral, cationic complexes 26 upon treatment with isocyanide (Scheme 9a). Making use of its labile nature, 25 can be used as a source of three-coordinate Fe(II) and was shown to be an active catalyst for the synthesis of cyclic organic carbonates from CO2 and epoxides, even in the absence of an external nucleophilic co-catalyst (Scheme 9b). It was proposed that the lability of a halide ligand in 25 allows binding of epoxide to the Lewis acidic Fe center, and the halide that is liberated acts as a nucleophile for the ring-opening of the epoxide to initiate the reaction.34
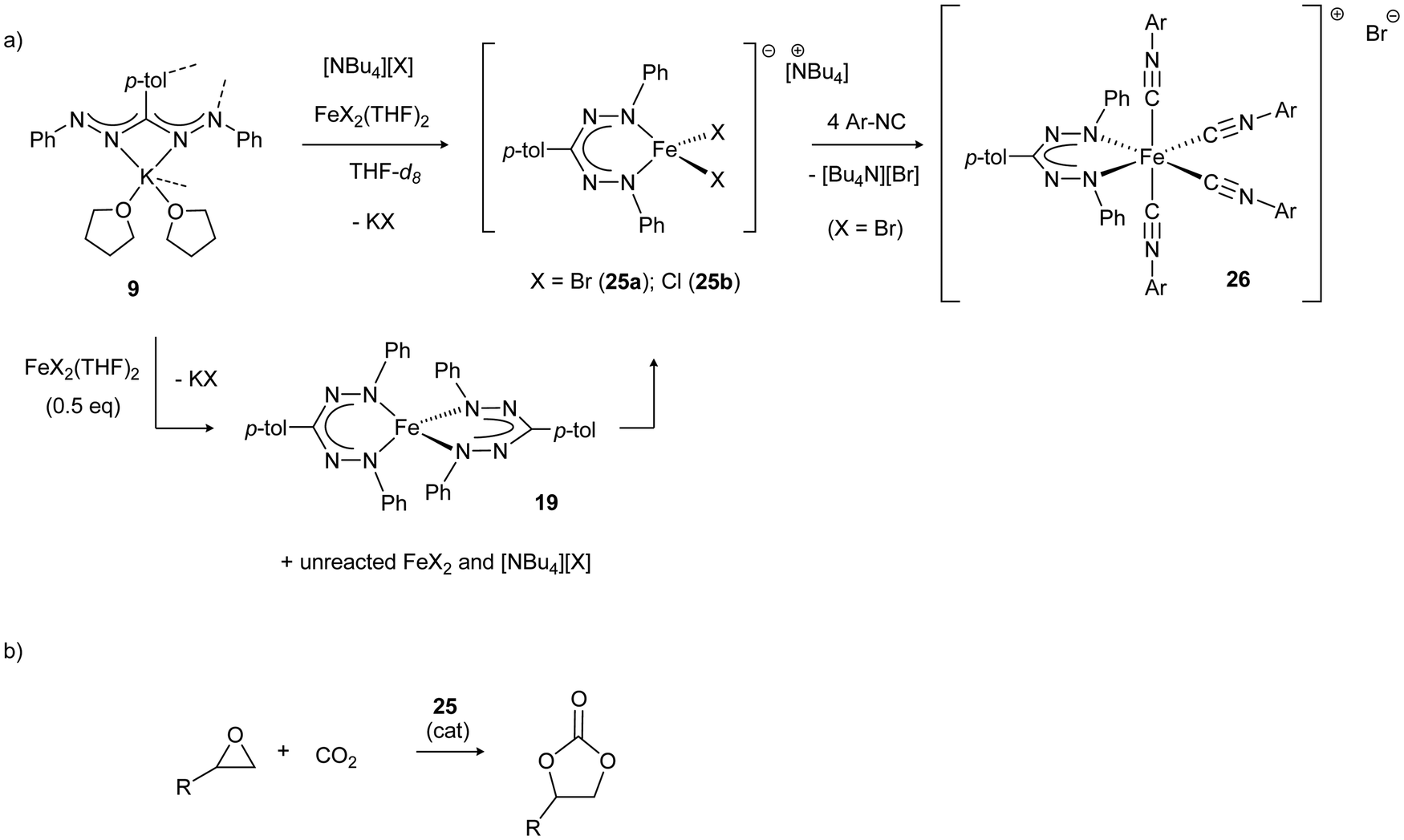 | ||
| Scheme 9 (a) Synthesis of ferrate complexes 25 and subsequent halide exchange for isocyanide to give cationic complex 26. (b) Application of 25 in the catalytic conversion of CO2/epoxide to cyclic carbonates.33,34 | ||
Formazanate complexes with the heavier elements in group 8 (Ru, Os) have only been sporadically investigated. Early work by Ibers and co-workers described the synthesis of complexes in which C–H activation of the NPh group took place to afford cyclometallated derivatives.35 More recently, Lahiri et al. reported Ru complexes with a formazanate ligand and acetylacetonate, bipyridine or 2-phenylazopyridine co-ligands to give neutral complex 27 (Scheme 10a) and the cations 28+ and 29+, respectively (Scheme 10b). These studies established bidirectional redox-noninnocence for the formazanate ligand: starting from complexes with a closed-shell monoanionic form of the ligand, both oxidation and reduction reactions were shown by spectroelectrochemisty and computational studies to occur in the formazanate moiety. This study provides the first evidence that the formazanate ligand can also bind in the neutral formazanyl radical form (L˙ in the dicationic complex 292+), thus extending the range of stable ligand oxidation states (Scheme 10c).36
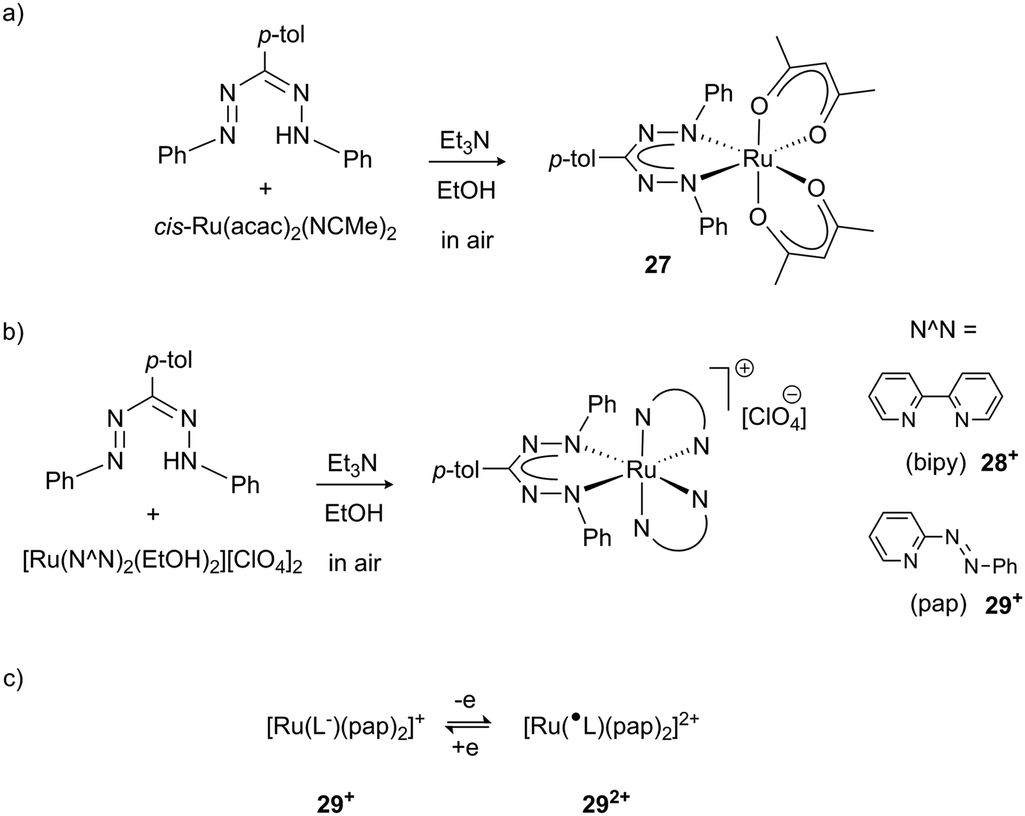 | ||
| Scheme 10 Ruthenium complexes 27 and 28+/29+ that show formazanate-based reduction and oxidation.36 | ||
2.4 Group 9 (Co, Ir)
In 2008, the Hicks group reported the preparation of the Co(III) complex 30 with a trianionic, tetradentate N2O2 cyano-formazanate ligand.27 Compound 30 was obtained via salt metathesis with Co(II), followed by air oxidation, and was shown to be isostructural with Fe(III) complex 17 by X-ray diffraction (Scheme 5). Poddel'sky and co-workers reported triphenylformazanate cobalt complexes with one or two semiquinonate (SQ) ligands (31/32, Scheme 11).37 The crystallographic, magnetic and spectroscopic data for these compounds indicates that the formazanate is bound as the closed-shell, monoanionic form of the ligand. The bidentate oxygen-ligands derived from 3,6-di-tert-butyl-o-benzoquinone (3,6-Q) are present as monoanionic semiquinonate radicals. Antiferromagnetic coupling, either between the S = 1/2 Co(II) center and a semiquinonate radical anion in 31 or between the two ligand radicals in the Co(III) complex 32, leads to diamagnetic ground states for both compounds. In subsequent work, the authors examined the reduction chemistry of both cobalt complexes. These data suggest that in these compounds, reductions are ligand-based but occur in the quinone-derived ligand rather than in the formazanate to give a Co(II) product with closed-shell, dianionic catecholate ligand (31−).38 The corresponding one-electron reduction product 32− is suggested to feature valence tautomerism (redox-isomerism) between Co(II) and Co(III) complexes with corresponding changes in oxidation state of the ligand (semiquinonate (SQ) and catecholate (Cat), respectively, see Scheme 11).38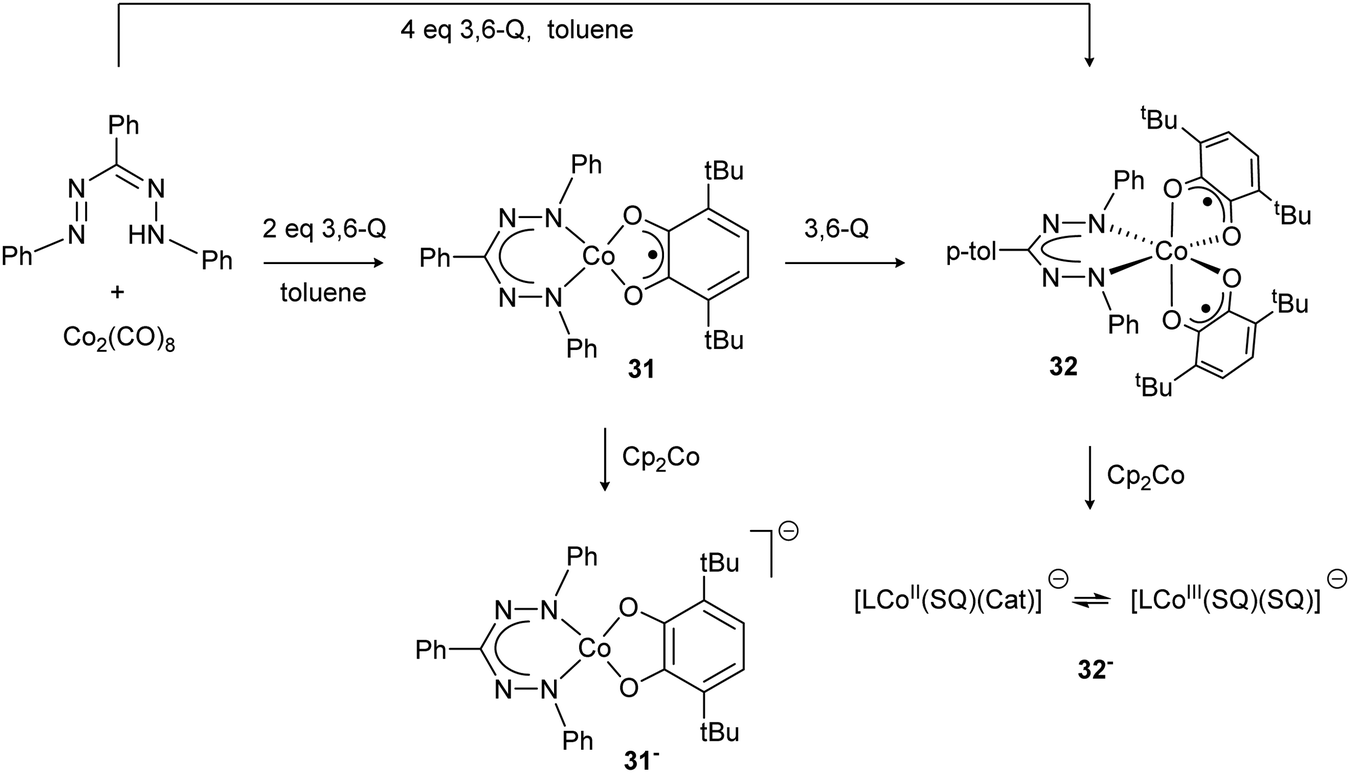 | ||
| Scheme 11 Formazanate cobalt complexes with o-quinone-based co-ligands and their reduction chemistry.37,38 | ||
Recent work from the Teets group has investigated a series of formazanate iridium(III) complexes with cyclometalated (C^N) ligands.39 Both the substituent pattern on the formazanate as well as the nature of the C^N ligand were systematically varied to allow rational tuning of the electrochemical and optical properties of these octahedral Ir complexes. Stirring the dimeric bis-cyclometallated iridium chloride precursors with formazans at 80–85 °C in EtOH in the presence of NEt3 afforded the formazanate complexes 33–36 in moderate to good yields (Scheme 12). The crude products are obtained as mixture of two isomeric products which differ in the coordination mode of the formazanate ligand. The metal center is coordinated either in a five- or six-membered chelate ring, which gives rise to complexes with C1- or C2-symmetry, respectively. The majority of these compounds could be obtained as a single isomer by recrystallization, and were shown by X-ray crystallography and NMR spectroscopy to have a five-membered chelate structure (“open” form of the ligand). Only compounds 33b′ and 33c′ crystallized as the C2-symmetric (“closed”) isomer, whereas the isomers for compound 33a could not be separated by crystallization or other means. Pure samples were shown to be kinetically stable: the other isomer that was present in the crude reaction mixture does not form upon heating CDCl3 solutions to 60 °C. The different binding modes lead to a different degree of π-delocalization: the “open” isomers show alternating short/long bond lengths in the NNCNN core due to localized bonding, whereas the pairs of N–N and N–C distances are similar in the “closed” form. The preference for the “open” form of the ligand in these compounds stands in marked contrast to the symmetrical (“closed”) binding mode that is commonly observed in formazanate transition metal complexes, and was attributed to a release of steric hindrance in the former. Analysis of the UV/vis absorption spectra for these compounds showed that there is little difference between the “open” and “closed” isomers, but that their intense, lowest-energy absorption (520–677 nm) due to a formazanate-based π→π* transition is sensitive to ligand substituent effects. Moreover, the absorption bands at higher energy (near-UV-visible) depend on the nature of the cyclometalated ligand, and are ascribed to metal-to-ligand charge transfer (Ir(d) → C^N(π*)). Cyclic voltammetry showed formazanate-centered reduction waves between −1.22 and −1.98 V vs. Fc0/+ that were ascribed to reduction of the formazanate ligand. A second formazanate-based reduction was observed for several compounds, but this was generally not reversible and led to the appearance of additional oxidation waves upon the return scan. At more positive potentials (>−0.10 V vs. Fc0/+), an oxidation wave was observed that is also quite sensitive to the formazanate substituents. Thus, while this may formally be assigned to a Ir(III)/Ir(IV) redox couple, there is also a significant formazanate-contribution to the HOMO in these compounds.
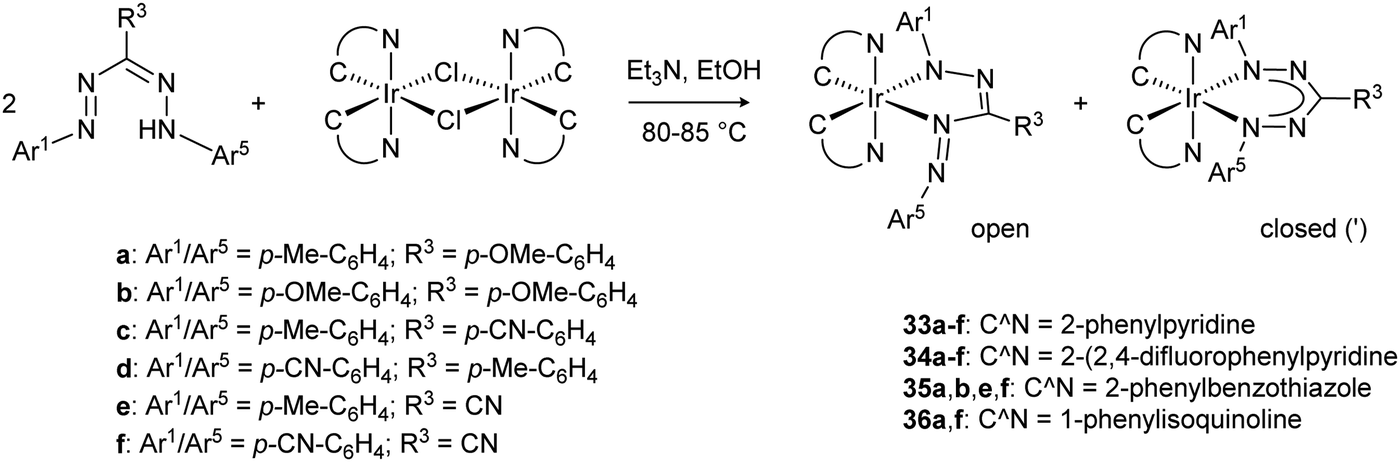 | ||
| Scheme 12 Synthesis of iridium(III) formazanate complexes with cyclometalated C^N co-ligands.39 | ||
2.5 Group 10 (Ni, Pd, Pt)
Reports published since 2000 on nickel complexes with formazanate ligands have established detailed insight in the structures and properties of these compounds, and applications are starting to emerge. In 2006, the group of Vatsadze unambiguously confirmed the symmetrical, six-membered chelate structure for the bis(triphenylformazanate)nickel complex 37a,40 which had been known since 194141 (the crystal structure of a related 3-Me substituted derivative was reported in 1967).42 Bis(formazanate)nickel(II) complexes have square planar coordination geometries and thus are diamagnetic. The metal ion is displaced out of the ligand plane to minimize steric interactions between the N-Ar groups. Derivatives with pyridine substituents on the formazanate backbone were prepared as potential metalloligands in supramolecular chemistry (37b–d, Scheme 13a).40 Tezcan and co-workers reported related Ni complexes, and studied the effect of ligand substituents on the spectroscopic and electrochemical properties.43 Zaidman et al. prepared a series of bis(formazanate) nickel complexes from formazans with various (heteroaromatic) substituents or linked bis-formazan scaffolds, and studied their catalytic activity in ethylene oligomerization (for representative structures 38/39, see Scheme 13b).44 Upon treatment with AlEtCl2, the complexes showed some catalytic activity towards the formation of a mixture of butenes, hexenes and octenes, but also resulted in substantial amounts of (poly)ethyltoluenes by Friedel–Crafts alkylation of the toluene solvent with ethylene.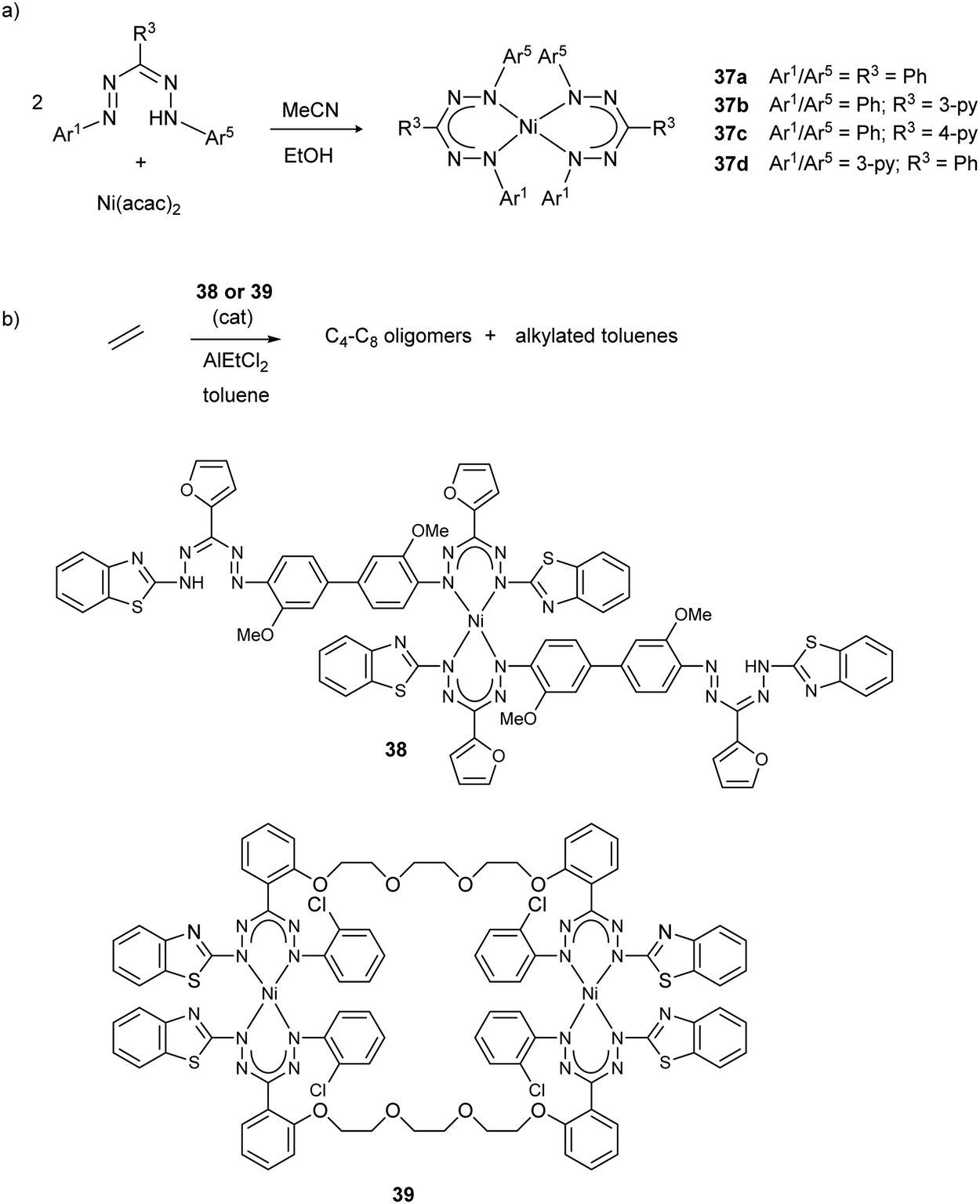 | ||
| Scheme 13 (a) Synthesis of square planar nickel complexes with triarylformazanate ligands. (b) Representative structures of nickel complexes described by Zaidman et al., and their application as catalysts for ethylene oligomerization.40–44 | ||
Nitro- or cyanoformazans were used by Hicks et al. to prepare the nickel complexes 40 and 41 (Scheme 14).27 While cyanoformazans reacted with Ni(OAc)2 to produce ill-defined (oligomeric/polymeric) products due to coordination of the cyano group, nitroformazans led to bis(formazanate)Ni complexes when small N-aryl substituents are used (e.g., Ar1/Ar5 = p-tolyl). On the other hand, sterically more demanding substituents (2,6-Me2 substituted aromatic rings) prevented formation of homoleptic complexes and instead led to mono(formazanate) nickel hydroxides, which are dimeric in the solid state (Scheme 14b).
 | ||
| Scheme 14 (a) Synthesis of homoleptic bis(formazanate) nickel complex 40. (b) Synthesis of heteroleptic cyano/nitro-formazanate nickel hydroxide complexes 41 with larger N-aryl groups.27 | ||
Palladium complexes 42 with nitroformazanate and fluorinated acetylacetonate ligands (Scheme 15a) were prepared and their electrochemical properties evaluated.45 The irreversible reductions observed in the cyclic voltammograms led the authors to conclude that the radical dianionic ligand that is generated is not stable in these ‘metalaverdazyl’-type palladium complexes.
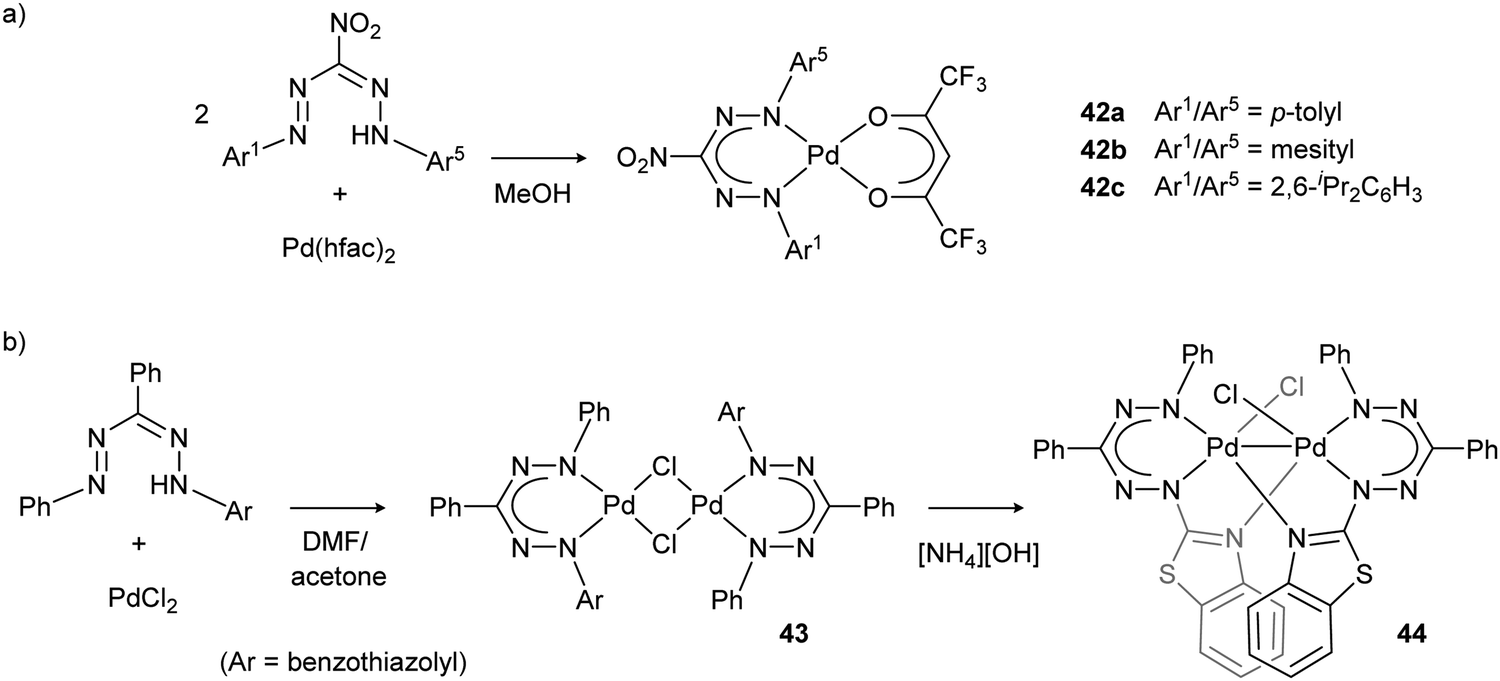 | ||
| Scheme 15 (a) Synthesis of nitroformazanate palladium complex 42 with fluorinated acetylacetonate ligands. (b) Synthesis of binuclear Pd–Pd bonded complex 44via the chloride-bridged dimer 43.45,46 | ||
Formazanate palladium complexes 43 with heteroaromatic substituents were reported by Lipunova et al. (Scheme 15b).46 Starting from PdCl2, several complexes were obtained with the empirical formula (formazanate)PdCl that show intense absorption maxima in the near-IR range (820–1020 nm). Although the structure of these compounds remains unknown, a Cl-bridged dimeric structure was suggested based on the mass spectrum. Addition of amine bases results in loss of the near-IR band and appearance of a new absorption in the visible range (630 nm). The product after treatment with [NH4][OH] was identified as a dimeric palladium species (44) by using single-crystal X-ray diffraction.
The synthesis of organometallic mono(formazanate) palladium complexes with a Pd–Me bond was described by the groups of Otten and Milani (Scheme 16).47 Salt metathesis between potassium formazanate and the palladium precursor (COD)Pd(CH3)Cl in THF was unsuccessful due to the poor stability of the putative three-coordinate complex (formazanate)Pd(CH3). From these reactions, the corresponding homoleptic bis(formazanate) palladium complex was invariably obtained (a closely related compound was described previously by Siedle).48 However, addition of an equivalent of [Bu4N][Cl] to the reaction mixture afforded the four-coordinate palladate complex 45, in which the halide ligand is labile and thus allows binding of unsaturated substrates. Insertion of CO, isocyanide and methyl acrylate into the Pd–CH3 bond was demonstrated, but less reactive olefins (ethylene, styrene) did not react.
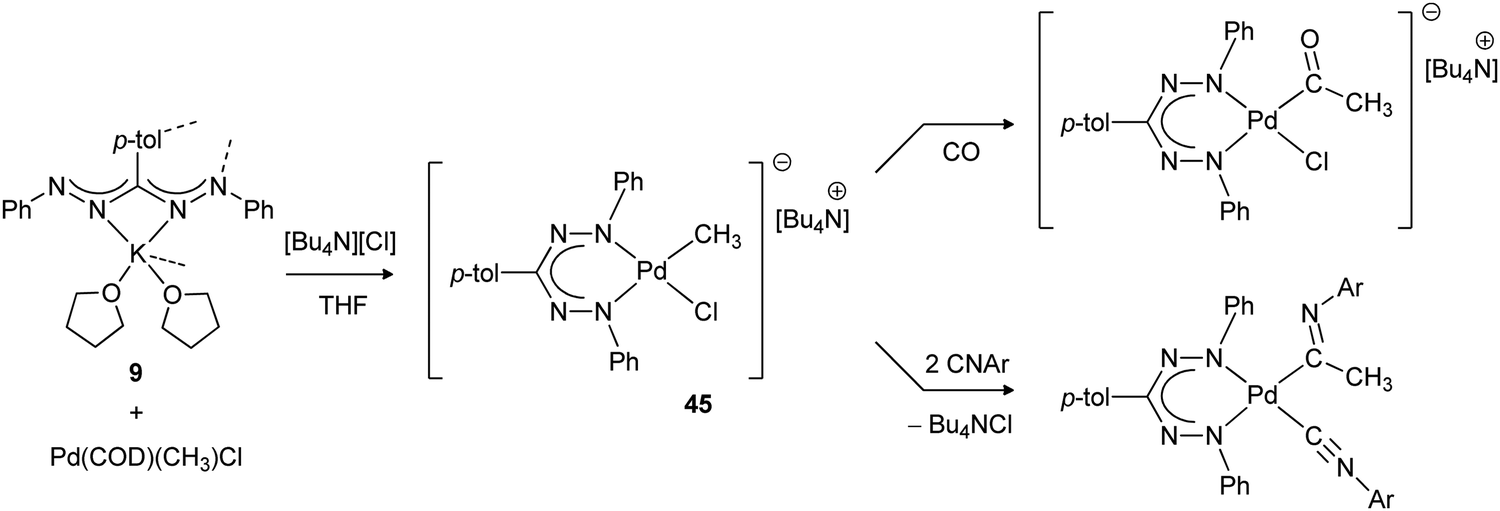 | ||
| Scheme 16 Synthesis of organometallic mono(formazanate) palladium complex 45 and subsequent insertion reactions with unsaturated substrates.47 | ||
The properties of platinum formazanate complexes 46–49 with cyclometallated ligands were studied by Teets and co-workers with a wide range of ligand substituents (Scheme 17a).49,50 Formazanate coordination to the platinum center was shown by a combination of spectroscopic, electrochemical and computational studies to lead to substantial changes in comparison to the free ligands, with absorption maxima that are red-shifted to ca. 660 nm. In addition to the typical ligand π → π* transition, a (minor) contribution of Pt(d) → formazanate charge transfer character is observed in the excited state. Computational data indicate that both the HOMO and LUMO are primarily ligand-based, and spectroelectrochemical data support the notion that ligand-based one-electron reductions take place at relatively accessible potentials between −1.2 and −1.6 V vs. Fc0/+. Compounds 46e/47e (with highly electron-withdrawing ligands) are exceptions and show even more facile reductions at −0.84 and −0.80 V vs. Fc0/+, respectively. A second reduction occurs at more negative potentials (∼−2 V vs. Fc0/+), and both these reduction waves were assigned to formazanate-based processes. Both reduction waves respond in a similar manner to the presence of electron-donating/withdrawing substituents on the formazanate ligands, resulting in a relatively constant separation between these two of ca. 0.6–0.7 V. A third cathodic wave (<−2.3 V) was assigned to reduction of the Pt–C^N fragment. Moreover, irreversible oxidations were also observed at mild potentials (around +0.1 V vs. Fc0/+). Despite the rather large variation of the LUMO energy levels, with first reduction potentials differing by as much as 0.79 V across the series, the electronic spectra show rather similar absorption maxima in the visible range (λmax = 600–665 nm, a range of ca. 0.2 eV). Apparently, both the HOMO and LUMO energy are affected to approximately the same extent by substituent effects, a feature that is in line with the computed frontier orbital compositions of formazanate ligands (Fig. 2, see also Section 2.8.1).
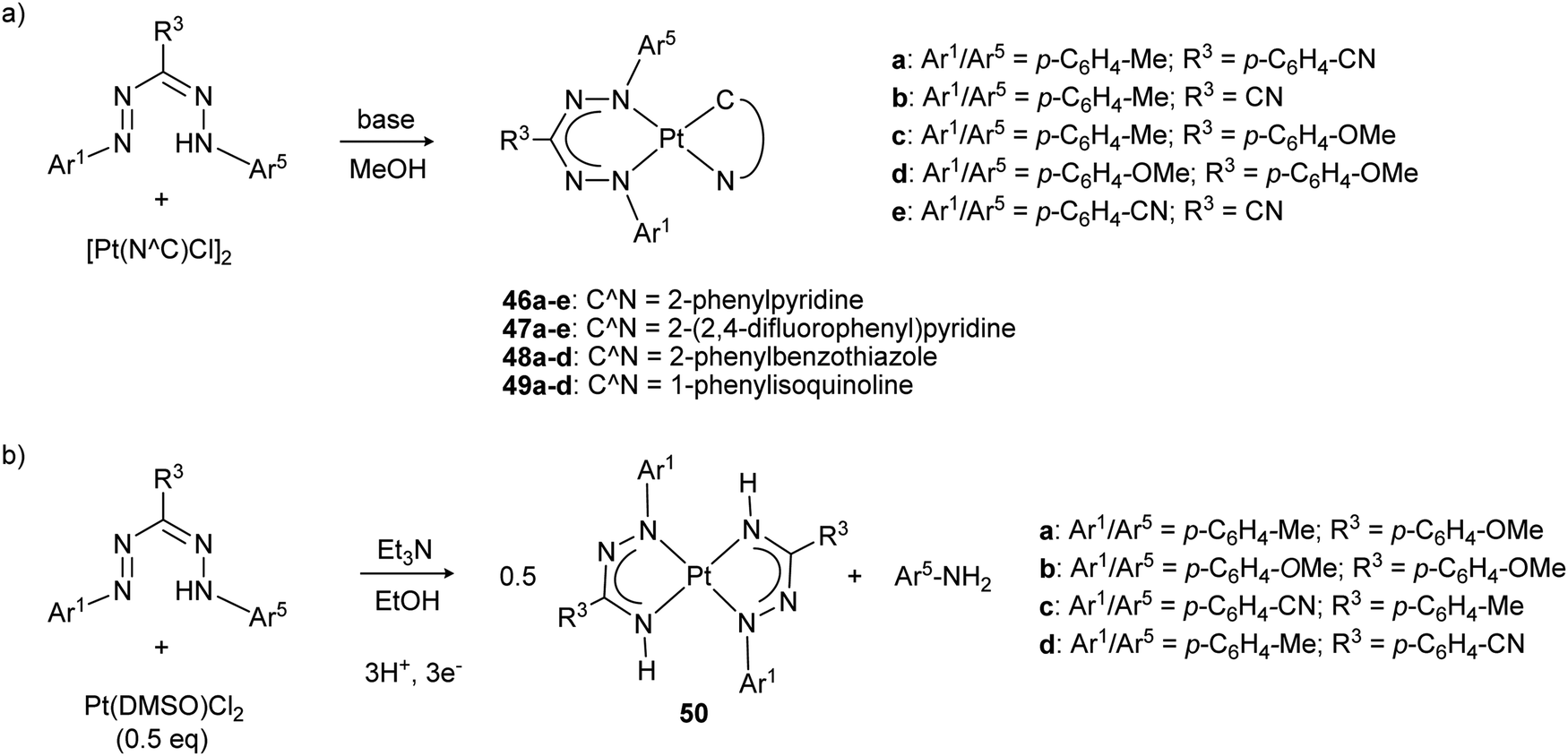 | ||
| Scheme 17 (a) Synthesis of square planar platinum complexes with formazanate and cyclometallated C^N ligands. (b) Synthesis of homoleptic platinum azoiminate complexes via reductive N–N bond cleavage.49–51 | ||
Changing the platinum precursor from a cyclometallated species (e.g., [Pt(C^N)Cl]2) to Pt(DMSO)Cl2 under similar reaction conditions to those used for the synthesis of 46–49 led to reductive cleavage of the formazan N–N bond to give azo-iminate complexes 50 (Scheme 17b).51 Although the mechanism for this overall 3H+/3e− transformation is not fully understood, control experiments indicated that the solvent (MeOH, EtOH) is likely involved as the source of protons and electrons. On the one hand, this study highlights that the reactivity of formazans can be utilized to form organic ligands that are otherwise difficult to access (e.g., azo-iminates). On the other hand, it shows that reductive N–N bond cleavage is a potential decomposition reaction for formazanate complexes in reduced states, albeit that a general understanding of this type of reactivity remains to be established.
2.6 Group 11 (Cu)
The Hicks group reported the copper formazanate complex 51 in which both N-aryl substituents carry an additional –OMe donor functionality (Scheme 18).27 The solid state structure of the complex was determined by X-ray crystallography, which showed a pseudo-five-coordinate geometry around the Cu center. One of the OMe groups is clearly bonded with a Cu–O distance of 2.068(2) Å, whereas the other is significantly further away at 2.479(2) Å. Magnetic and spectroscopic characterization confirmed the presence of a S = 1/2 Cu(II) center with the unpaired electron in the dx2−y2 orbital (g‖ = 2.174 and g⊥ = 2.064).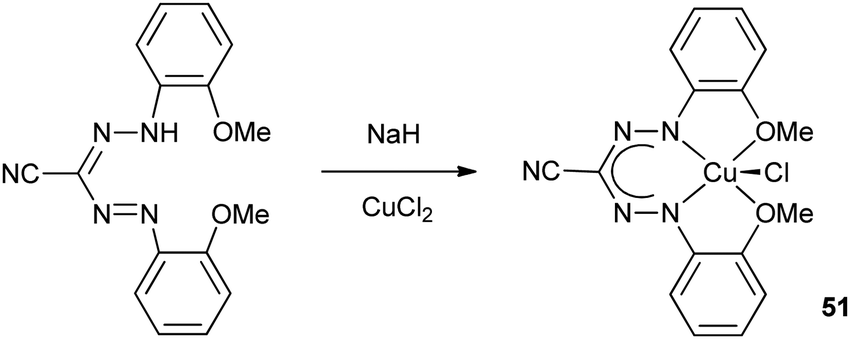 | ||
| Scheme 18 Synthesis of Cu(II) complex 51 with tetradentate, monoanionic N2O2 formazanate ligand.27 | ||
The unusual electronic features of formazanate ligands were explored in the context of Cu(I)-mediated dioxygen activation. Hicks, Tolman and co-workers evaluated the reactivity of a mononuclear Cu(I) complex 52, which is coordinated by a sterically demanding nitroformazanate ligand with N-2,6-diisopropylphenyl substitution pattern (Scheme 19).52,53 The ligand was shown to be similar in its electron-donating properties to electron-poor CF3-substituted β-diketiminate ligands by comparing oxidation potentials and CO stretching frequencies. Reaction of 52 with O2 at room temperature afforded the bis(μ-hydroxo)dicopper(II) complex 53. Performing the reaction at −80 °C, however, allowed identification of an intermediate that was assigned as the bis(μ-oxo)dicopper complex 54-i. Characterization of this intermediate by UV/vis spectroscopy showed that the nature of the electronic transitions in the formazanate and β-diketiminate complexes are markedly different. The low-energy transition (λmax = 525 nm) observed for 54-i was shown by DFT calculations to have substantial contribution from orbitals on the formazanate framework (Scheme 19). In contrast to the β-diketiminate analogues, decay of the initial oxygenation product 54-i at temperatures above −50 °C showed the formation of a second intermediate (54-ii) en route to the bis(μ-hydroxo)dicopper(II) end-product 53. The spectroscopic features of 54-ii (UV/vis: λmax = 724 nm; X-band EPR at 4 K: isotropic signal at g ∼ 2.0) led the authors to suggest that this species is formed by hydrogen-atom abstraction from the solvent by the Cu(III)2O2 core and contains an organic (ligand) radical. The product of this H˙ transfer, formally containing a Cu(II)Cu(III)(OH)(O) moiety, presumably undergoes electron-transfer from Cu(II) to the formazanate ligand and forms the dicopper(III) complex 54-ii with a radical dianionic formazanate ligand. An alternative description based on a coordinated neutral formazanyl radical was considered less likely due to the similarity of the spectroscopic features of 54-ii with those observed for other compounds containing radical dianionic formazanate ligands.
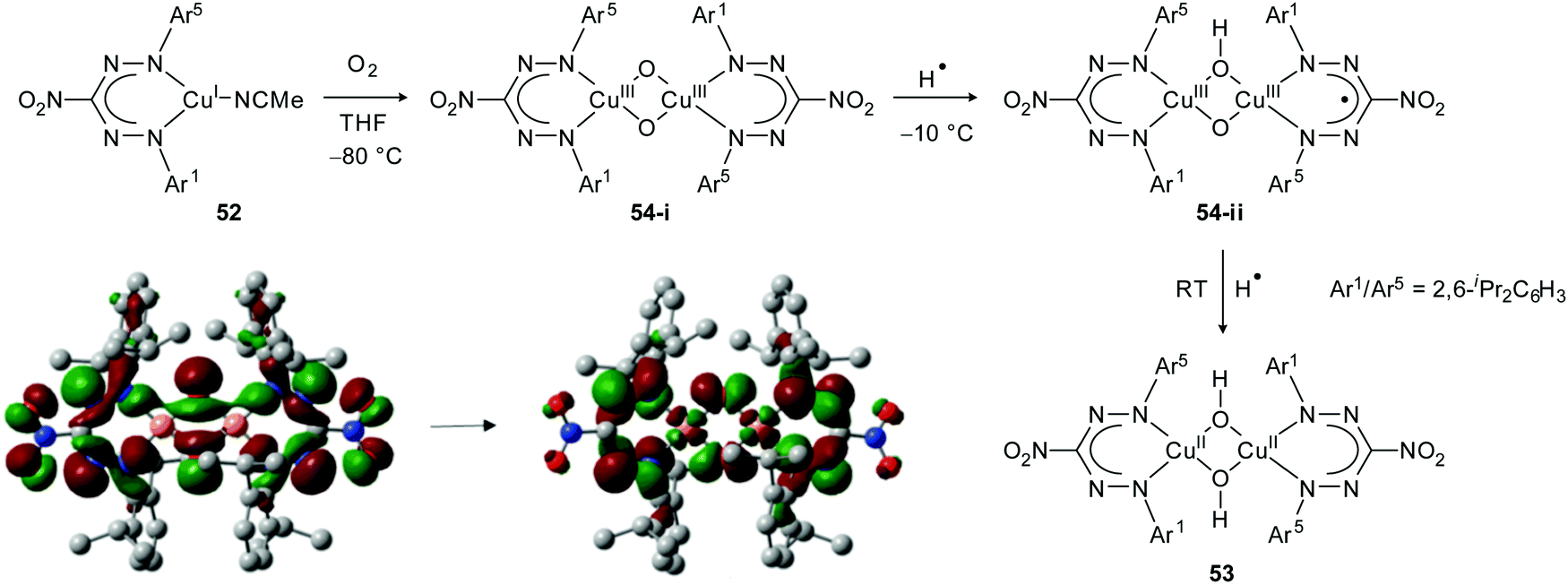 | ||
| Scheme 19 Reaction of formazanate Cu(I) complex 52 with O2. Donor and acceptor Kohn–Sham orbitals involved in the lowest-energy electronic transition in intermediate 54-i are adapted from ref. 52 with permission from the American Chemical Society. | ||
2.7 Group 12 (Zn)
Although ligands based on the formazanate NNCNN framework have a long history in the spectrophotometric quantification of zinc and copper ions (Zincon),54 reports from recent years have provided a better understanding of the properties of formazanate zinc complexes, with a specific focus on the ability of formazanates to function as electron-reservoirs (i.e., as redox-active ligands). In 2014, the synthesis of bis(formazanate) zinc complexes 55a/b was described via protonolysis using ZnMe2 (Scheme 20), and the properties of these compounds were explored by cyclic voltammetry and chemical synthesis.55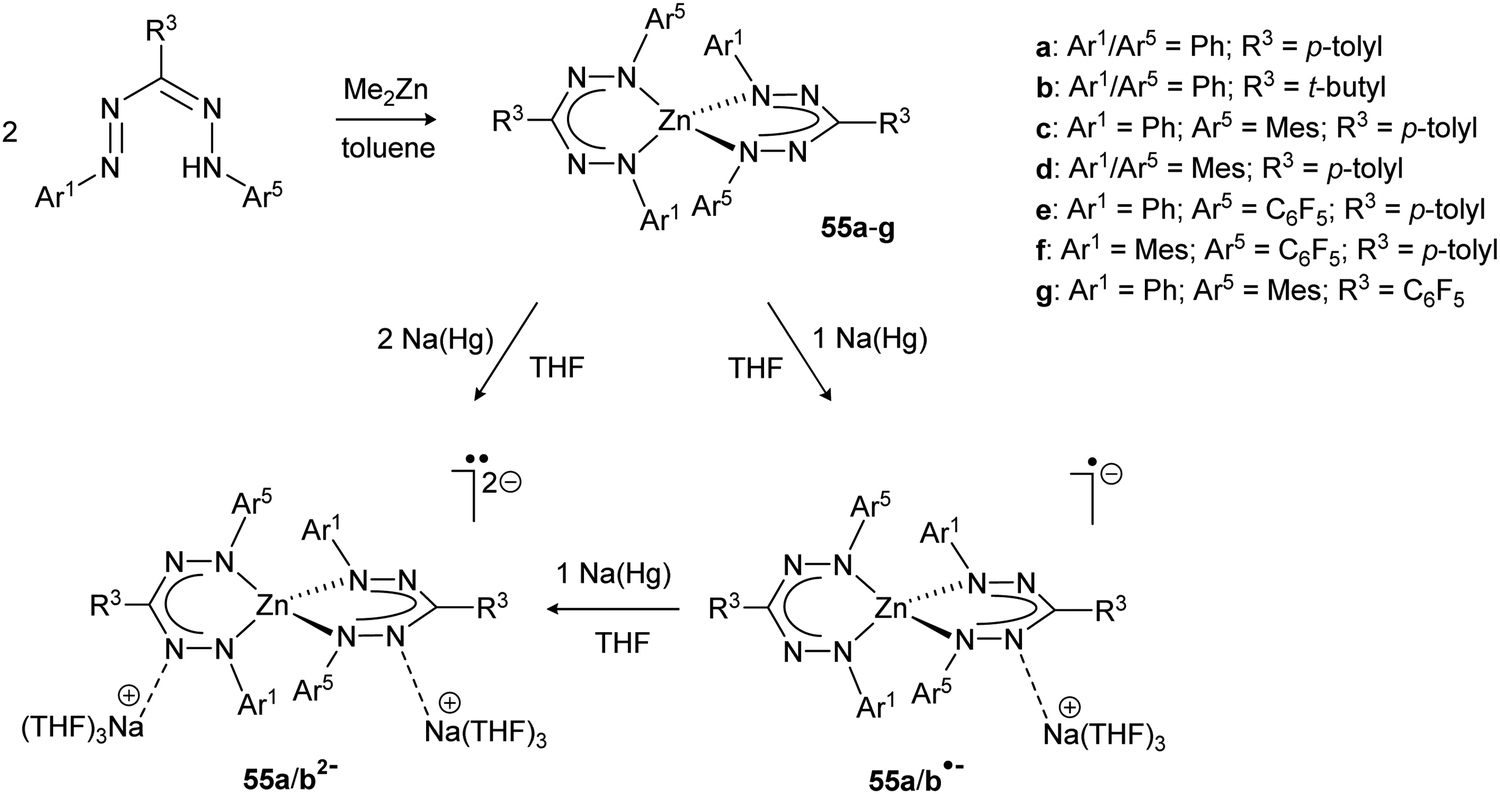 | ||
| Scheme 20 Synthesis of bis(formazanate) zinc complexes 55, and subsequent reduction to the corresponding radical anions 55˙− and dianions 552−.55,56 | ||
Two quasi-reversible, single-electron redox processes were observed in the voltammogram of 55a at −1.31 and −1.55 V vs. Fc0/+ that correspond to the redox-couples 55a0/− and 55a−/2−, respectively. Qualitatively similar data were obtained for the 3-tert-butyl formazanate complex 55b, but a cathodic shift (to −1.57/−1.85 V vs. Fc0/+) was observed due to the presence of the electron-donating tBu substituent. The stability of the one- and two-electron reduced species on the electrochemical timescale prompted the chemical synthesis of these compounds. Addition of one equivalent of Na/Hg reducing agent to THF solutions of 55a/b afforded the corresponding radical anions 55a/b˙−, respectively, which were characterized by X-ray crystallography. In these radical anionic complexes, the Na+ countercation interacts with the nitrogen atoms in the ligand backbone. The two formazanate ligands around the zinc center were shown to be different, with one having intraligand metrical parameters that are very similar to the neutral starting materials whereas the other ligand (where the cation is bound) shows substantially elongated N–N bonds (avg. 1.304 Å in 55avs. 1.363 Å in 55a˙−). It was concluded that the latter ligand is present as a radical dianion, i.e., in the metallaverdazyl form. Treatment of 55a/b with 2 equiv. of Na/Hg was shown to form the corresponding dianionic complexes (55a/b2−), in which both ligands are present as radical dianions bonded to a Zn2+ center, as demonstrated by the crystal structure for 55b2− (Fig. 4A), which shows all N–N bonds being elongated (>1.355 Å). The EPR features of 552− at 77 K in frozen THF solution are consistent with a triplet organic diradical, with g = 2.0028 and a zero-field splitting parameter D ≈ 11.5 × 10−3 (Fig. 4B). A variable-temperature EPR study suggested that although the triplet state is apparently significantly populated at 77 K, 55a2− has an unusual singlet biradical electronic ground state. This was confirmed by broken-symmetry DFT calculations (Fig. 4C). The presence of radical dianionic ligands in the reduced compounds was furthermore confirmed by UV/vis spectroscopy, which showed features typical for verdazyl-type radicals (low-energy absorptions at λ > 750 nm, Fig. 4D).
 | ||
| Fig. 4 Molecular structure of 55b2−, with hydrogen atoms and coordinated THF molecules (except the O-atoms) omitted for clarity (A). EPR spectrum of 55a2− recorded at 77 K in frozen THF solution (B); asterisk denotes doublet impurity, inset shows half-field region). Spin density plot for the broken-symmetry DFT calculations on 55a2− (C). UV-vis spectra for 55a (blue), 55a˙− (red) and 55a2− (green) in THF solution (D). Adapted from ref. 55 with permission from John Wiley and Sons. | ||
The influence of formazanate substitution pattern on the structure and electronic properties of the resulting bis(formazanate) zinc complexes was investigated by X-ray crystallography, cyclic voltammetry and UV/vis spectroscopy.56 The majority of the compounds 55a–g have the formazanate ligands bound in a six-membered chelate ring, but when the N-aryl substituents are electronically dissimilar the π-delocalization is less pronounced and five-membered chelate rings become energetically accessible. For example, compound 55f (Ar1 = Mes; Ar5 = C6F5) was found to have one ligand bound in the unusual ‘open’ geometry (via both a terminal and internal N-atom) whereas the other is present in the more common ‘closed’ form. Solution NMR studies indicated a dynamic equilibrium between these isomers. It was found that the redox-potentials vary in a predictable manner based on the electron-donating/withdrawing ability of the substituents, and one-electron reduction potentials that span a wide range (between −1.17 to −1.86 V vs. Fc0/+) were demonstrated. Gilroy and co-workers have examined in more detail these substituent effects, and correlated the optoelectronic properties of formazanate boron difluoride compounds with computed frontier molecular orbitals (see Section 2.8.1). In addition to reduction waves corresponding to the formation of the radical anions 55˙− and diradical dianions 552−, the voltammograms indicated that these compounds can be further reduced at more negative potentials (<−2.5 V vs. Fc0/+). Although the stability of these highly reduced species is limited, the observation that five different oxidation states are accessible in these simple compounds (i.e., ranging from neutral 55 to the tetranion 554−) is noteworthy and suggests that each formazanate ligand can be reduced by up to two electrons. This study furthermore demonstrated that bis(formazanate) zinc complexes with two different formazanate ligands are accessible in a stepwise manner (e.g., 56, Scheme 21a), and provided the first example of a zinc complex with a parent (neutral) formazan ligand when the less basic reagent Zn(C6F5)2 was used (57, Scheme 21b).
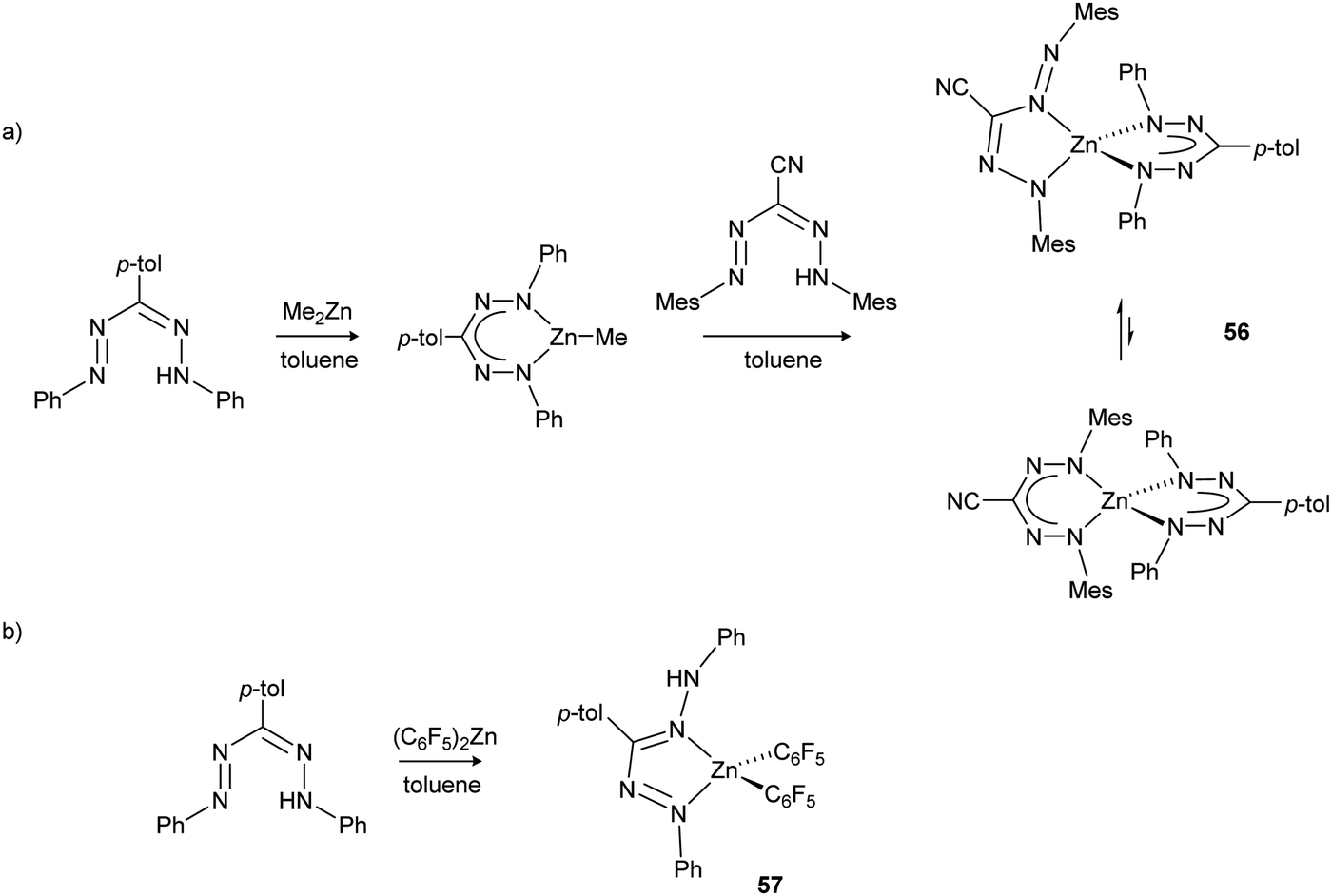 | ||
| Scheme 21 (a) Synthesis of bis(formazanate) zinc complexes 56 with two different formazanate ligands. (b) Synthesis of formazan complex 57 using the less basic reagent Zn(C6F5)2.56 | ||
2.8 Group 13 (B, Al, Ga, In)
 | ||
| Scheme 22 Synthesis of boron diacetate complexes of formazanate ligands and their corresponding radical anions.57 | ||
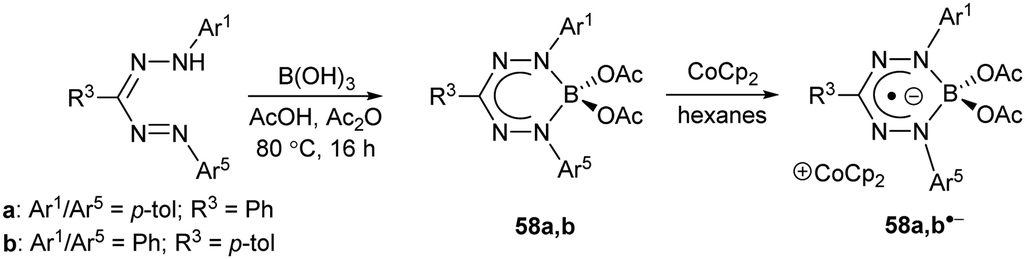
Inspired by the rich chemistry of boron difluoride (BF2) adducts of chelating N-donor ligands,58 including boron dipyrromethenes (BODIPYs),59,60 the first examples of BF2 formazanates 59 were synthesized from the corresponding homoleptic Zn(II) complexes 55 (Scheme 23a).61 The resulting complexes, which benefited from structural rigidity and stability associated with the ‘BF2+’ fragment, could be electrochemically reduced in two steps to the corresponding radical anions and dianions. Chemical reduction with cobaltocene (CoCp2) afforded the first structurally characterized examples of radical anions supported by boron adducts of formazanates, which showed characteristic elongation of the formazanate N–N bonds from an average of ca. 1.309 Å for 59a to 1.362 Å for 59a˙− due to the population of the LUMO, which possesses N–N antibonding character (Scheme 24).61 An alternative synthesis involving the conversion of 3-cyanoformazans 5 to BF2 complexes 60 by heating toluene solutions containing excess NEt3 and BF3·OEt2 at 80 °C overnight was published62 soon after the initial report (Scheme 23b) and was later extended to 3-aryl (3)63 and 3-nitroformazans (6)64 to produce BF2 complexes 59 and 61 (Scheme 23a and c). The 3-cyano and 3-nitro derivatives could also be electrochemically reduced in two steps, and were also the first examples of BF2 formazanate dyes to exhibit appreciable photoluminescence.
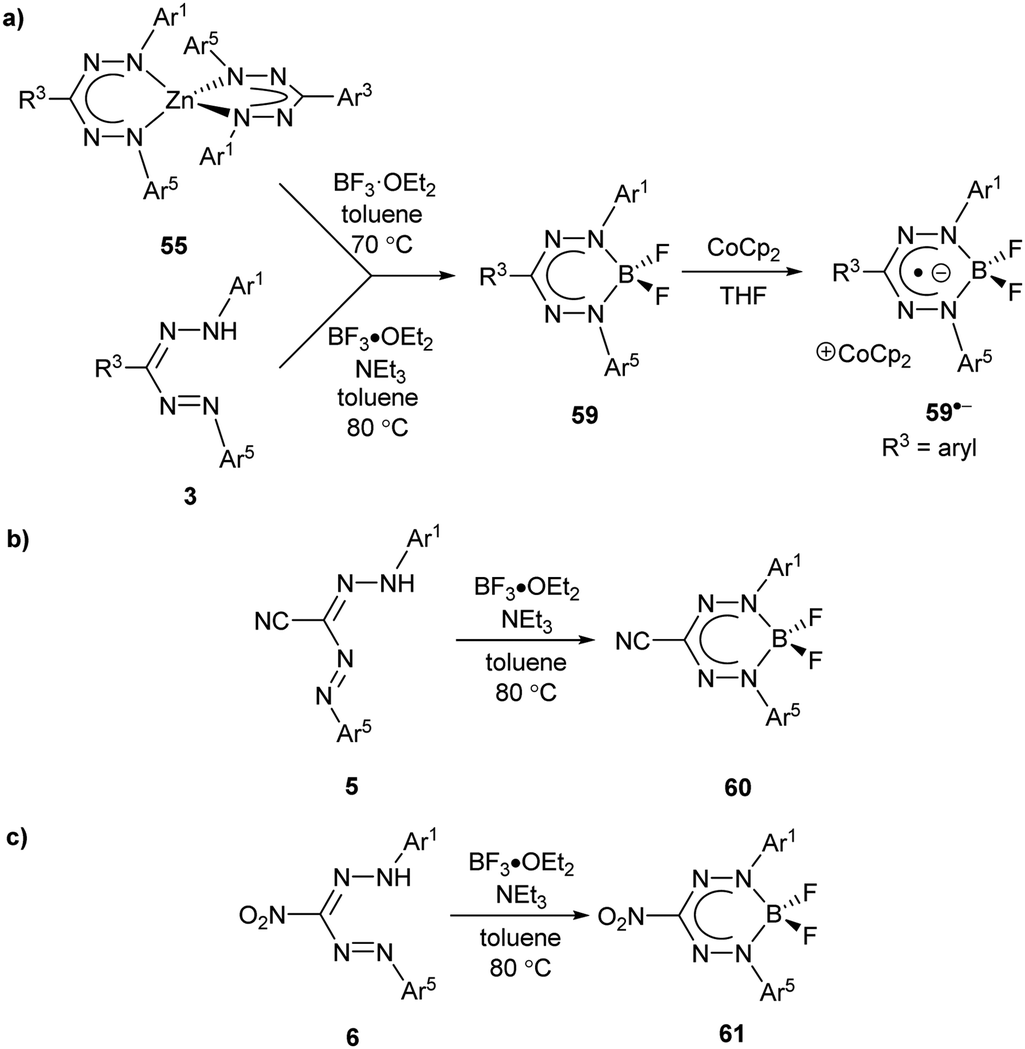 | ||
| Scheme 23 Synthetic routes to boron difluoride complexes of (a) triarylformazanate ligands (and corresponding radical anion), (b) 3-cyanoformazanate ligands, and (c) 3-nitroformazanate ligands.61–64 | ||
As noted earlier, BF2 formazanate complexes can be electrochemically reduced in two one-electron steps. These reduction events occur at Ered1 ∼ −0.8 and Ered2 ∼ −1.9 V relative to the Fc0/+ redox couple for adducts of triarylformazanates (59),63,65Ered1 ∼ −0.6 and Ered2 ∼ −1.7 V for 3-cyanoformazanates (60),62 and Ered1 ∼ −0.5 and Ered2 ∼ −1.6 V for 3-nitroformazanates (61)64 bearing phenyl substituents at the 1,5 position of the ligand backbone. There are a number of reagents that could potentially reduce BF2 formazanates to their radical anion forms, including CoCp2 and its permethylated analogue (CoCp*2), which are both 19-electron metal complexes.66 However, relatively few reducing agents are strong enough to generate the dianion form of BF2 formazanates.66 The isolation of the dianion form of BF2 formazanates was attempted using a Na/Hg amalgam as a chemical reductant (Scheme 24).67 Treatment of complex 59a with two equiv. of Na/Hg resulted in the production of novel, crystallographically-characterized BN heterocycles 62–65, whose formation was driven by the production of NaF and likely implicates B(I) carbenoid intermediates 66 and 66′. Treatment of heterocycles 62 and 63 with XeF2 resulted in the regeneration of BF2 complex 59a in high yield.
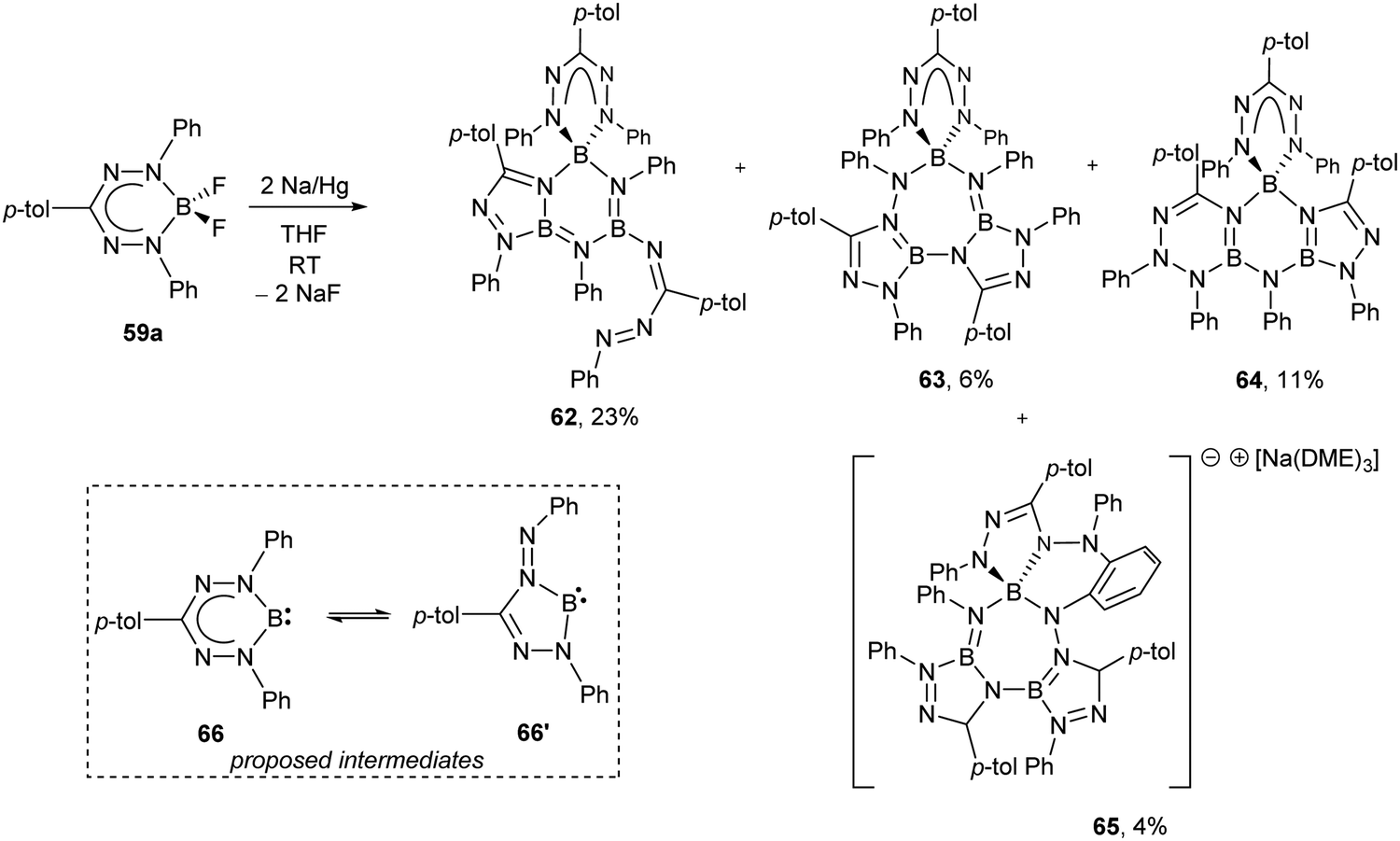 | ||
| Scheme 24 Synthesis of novel BN heterocycles 62–65via treatment of BF2 formazanate 59a with Na/Hg amalgam and proposed B(I) carbenoid intermediates 66 and 66′.67 | ||
Attempts to exploit the chelate effect toward the isolation of complex 67 from the potentially tetradentate formazanate ligand 1,5-bis(2-hydroxyphenyl)-3-cyanoformazan resulted in the production of a number of unprecedented BN heterocycles 68–72 (Scheme 25).68 The absence of 67 in the resulting reaction mixtures can be attributed to ring strain associated with the 5-membered BOCCN chelate rings that would need to form during its synthesis. This hypothesis was supported by the fact that switching from phenol to benzyl alcohol N-aryl substituents resulted in the clean formation of 73, which involved only the formation of 6-membered BOCCCN chelate rings, which impose a lesser degree of ring strain. Compounds 68–72 were isolated from crude reaction mixtures through the use of column chromatography and all but 68 were crystallographically characterized. Complex 68, which includes a formazanate ligand bound to tetrahedral boron in a tridentate fashion is unstable in solution and spontaneously converts to dimers 69 and 71. Compounds 69 and 70 are structural isomers that differ in the structure of the 10-membered BNCCOBNCCO rings that form their cores (pseudo chair and boat conformations for 69 and 70, respectively). Each formazanate ligand in complex 69 could be chemically reduced in a stepwise fashion to yield the corresponding radical anion (69˙−) and diradical dianion (69˙˙2−). BN heterocycles 71 and 72 form by the Lewis-acid-assisted hydrolysis of the nitrile group in the ligand (Scheme 25). Complex 71 spontaneously converts to complex 72 in solution, and the latter can be converted to its corresponding anion (72−) upon reduction with CoCp2 and loss of H˙.
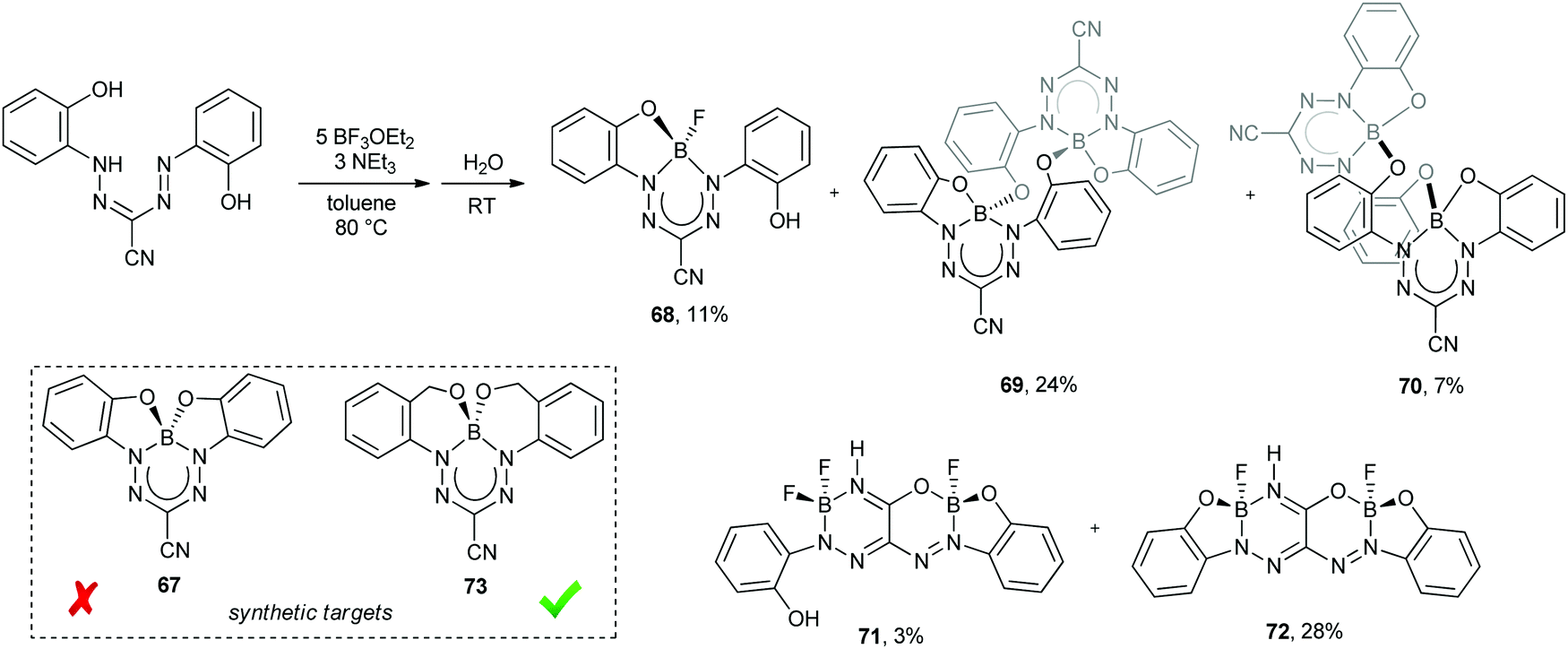 | ||
| Scheme 25 Synthesis of unusual BN heterocycles 68–72 from a potentially tetradentate N2O23− formazanate ligand and synthetic targets 67 and 73.68 | ||
The optoelectronic properties of BF2 formazanates have been systematically explored to probe the effect of variation of the R1, R3, and R5 substituents.61–63,65,69–71 In order to understand trends in these properties, it is vital to first examine the frontier molecular orbitals of BF2 formazanates as they are directly implicated in their reduction chemistry and low-energy absorption/photoluminescence properties. Fig. 5 shows the HOMO and LUMO isosurfaces calculated using DFT.64 Both orbitals are highly delocalized over the formazanate backbone and the N-aryl substituents for each series of compounds. However, only the HOMOs have significant contribution from the R3 substituents due to the presence of a nodal plane in the LUMOs. Thus, properties such as electrochemical reduction that implicate only the LUMO are generally affected to a greater extent by structural variation at the N-aryl substituents than the R3 substituents. Properties such as absorption and photoluminescence, which implicate both the HOMO and LUMO (as confirmed by TDDFT64) are expected to be affected in a similar fashion, although structural variation at the R3 substituent cannot be ignored in this context as the HOMOs possess significant contribution from those substituents. Additionally, there is significant orbital density at the para-carbons of each aryl ring, indicating that substitution at the para-position is likely to have a significant impact on the properties of BF2 formazanates. General trends have been demonstrated experimentally through systematic studies of BF2 triarylformazanates.63
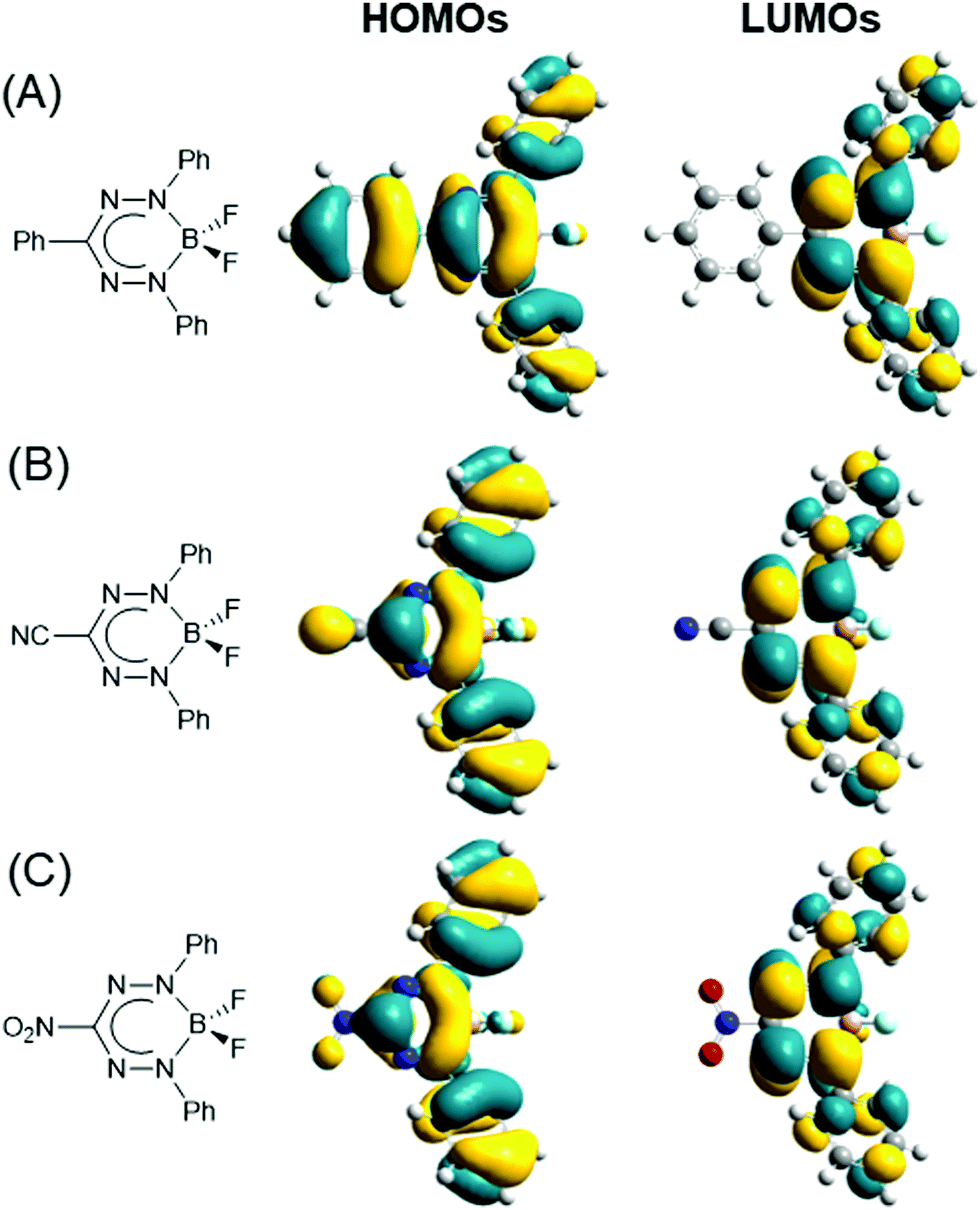 | ||
| Fig. 5 HOMOs and LUMOs calculated (DFT: M06/6-311+G*) for toluene solutions of BF2 complexes of (A) triarylformazanate ligands, (B) 3-cyanoformazanate ligands, and (C) 3-nitroformazanate ligands. In all cases Ar1/Ar5 = Ph. Adapted from ref. 64 with permission from the American Chemical Society. | ||
For brevity, we will discuss the structure–property relationships reported for BF2 complexes of 3-cyanoformazanate ligands (60).62,64,65,70 Representative data are presented in Fig. 6 and summarized in Table 1. As mentioned above, BF2 formazanates possess highly delocalized π-electron systems that give rise to low-energy λmax and λPL values and large Stokes shifts (νST). The relative strength of photon absorption can be compared using molar extinction coefficients (ε) while photoluminescence quantum yields (ΦPL) are used to quantify and compare the efficiency of photoluminescence.
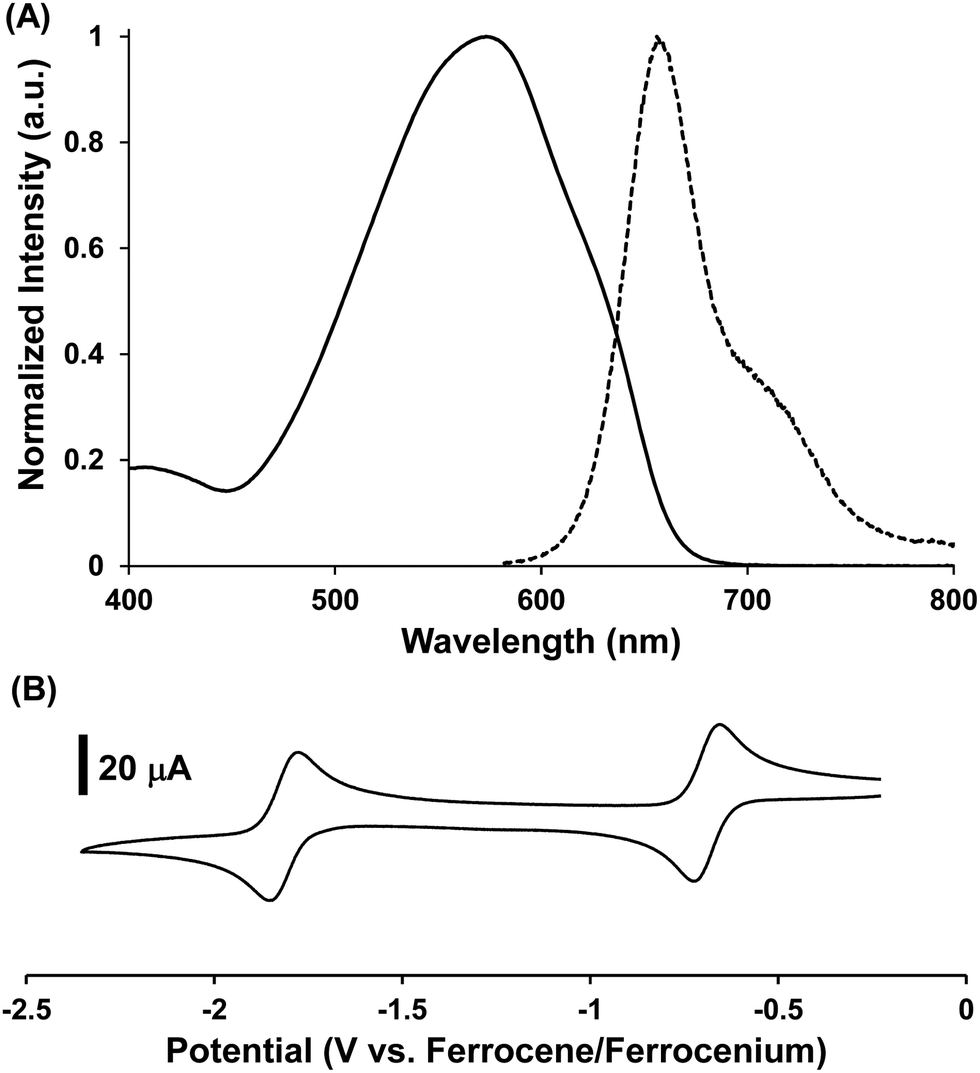 | ||
| Fig. 6 Representative (A) normalized UV-vis absorption (solid line) and photoluminescence (dashed line) spectra collected in toluene and (B) CV collected at 0.25 V s−1 in CH3CN for 60d. Adapted from ref. 62 with permission from John Wiley and Sons. | ||
| Compound | Ar1/Ar5 | λ max (nm) | ε (M−1 cm−1) | λ PL (nm) | Φ PL (%) | ν ST (nm) | ν ST (cm−1) | E red1 (V vs. Fc0/+) | E red2 (V vs. Fc0/+) |
|---|---|---|---|---|---|---|---|---|---|
| a Recorded in toluene. b Recorded in CH3CN. c Recorded in dichloroethane. | |||||||||
| 60a | Ph | 502 | 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
586 | 15 | 84 | 2855 | −0.53 | −1.68 |
| 60a·B(C6F5)3 | Ph | 502 | 20600 |
632 | <1 | 130 | 4098 | −0.67c | −1.75c |
| 60b | Naphthyl | 581 | 25700 |
670 | 39 | 89 | 2286 | −0.49 | −1.54 |
| 60c | p-C6H4CN | 515 | 35000 |
598 | 14 | 83 | 2695 | −0.21 | −1.25 |
| 60d | p-C6H4OMe | 572 | 42700 |
656 | 77 | 84 | 2239 | −0.68 | −1.82 |
| 60e | o-C6H4OMe | 467 | 16000 |
592 | 5 | 125 | 4521 | −0.73 | −1.88 |
| 60f | m-C6H4OMe | 525 | 21100 |
635 | 13 | 110 | 3300 | −0.50 | −1.62 |
| 60g | p-C6H4NMe2 | 728 | 47800 |
834 | 8 | 106 | 1746 | −1.02 | −2.05 |
Spectroscopic analysis of toluene solutions of 60a (Ar1/Ar5 = Ph) yielded λmax = 502 nm (ε = 30400 M−1 cm−1) and λPL = 586 nm (ΦPL = 15%) and cyclic voltammetry revealed that this compound can be electrochemically reduced in two one-electron steps at potentials of Ered1 = −0.53 V and Ered2 = −1.68 V relative to the Fc0/+ redox couple.62 Coordination of the Lewis acid B(C6F5)3 to the nitrogen atom of the 3-cyano substituent in 60a·B(C6F5)3 had little effect on the absorption maxima, although the intensity of the absorption was reduced by approximately one third. While the photoluminescence maximum is unaffected, coordination of B(C6F5)3 significantly decreased the quantum yield (ΦPL < 1%) providing an indication that Lewis acid coordination has a substantial effect (i.e., quenching) on the excited state.65 Increasing the size of the π-electron system by varying the Ar1/Ar5 substituents from phenyl to naphthyl in 60b resulted in a red-shift in λmax by 79 nm (2708 cm−1) and λPL by 84 nm (2140 cm−1). This extended electronic delocalization also resulted in stabilization of the LUMO orbital, rendering 60b easier to reduce electrochemically than 60a.64 The introduction of p-cyanophenyl N-aryl substituents in 60c rendered the complex much easier to reduce than 60a, as would be expected for an electron-withdrawing substituent. However, perhaps counterintuitively, this structural modification red-shifted the absorption and photoluminescence spectra of 60c relative to 60a as a result of the modest increase in the size of the π-electron system when CN groups were introduced.
Complex 60d, which possesses electron-donating p-methoxyphenyl N-aryl substituents, yielded absorption and photoluminescence spectra that were dramatically red-shifted and intensified (λmax = 572 nm, ε = 42700 M−1 cm−1; λPL = 656 nm, ΦPL = 77%) compared to most other BF2 formazanate complexes (Fig. 6). The dramatic red-shift may be attributed to the donor (p-methoxyphenyl) – acceptor (BF2 formazanate) electronic structure of 60d and the fact that the oxygen lone pairs can be delocalized into the π-electron system to promote a planar structure. The structural planarity also has implications on the aggregation characteristics of 60d in THF solutions containing various concentrations of H2O, where aggregation-caused quenching (ACQ) was observed due to π–π stacking.71 Detailed studies of the photoexcitation of 60d revealed that excitation to a high energy, bent excited state with complex vibrational fine structure occurs on the microsecond timescale. This non-emissive state relaxes to the planar, emissive excited state mentioned previously before radiative decay occurs.72 This behaviour leads to the large Stokes shifts observed for this family of compounds. Complex 60d was more difficult to reduce to its radical anion and dianion forms when compared to 60a due to the presence of the electron-donating methoxy substituents. Changing the N-aryl substituent from p-methoxyphenyl in 60d to o-methoxyphenyl in 60e resulted in a dramatic change in both the photophysical properties and aggregation behavior due to sterically-driven twisting of the N-aryl substituents out of the plane of the BF2 formazanate ligand. This twisting results in the observation of aggregation-induced emission enhancement (AIEE) upon the addition of H2O to THF solutions as the twisted o-methoxyphenyl substituents prevent π–π stacking, and thus ACQ.71 The m-substituted complex 60f had properties intermediate of the o- and p-derivatives.70
Finally, the introduction of strongly electron-donating p-dimethylaminophenyl substituents in 60g resulted in photophysical properties that extend into the near-infrared region of the electromagnetic spectrum and dramatically shifted reduction potentials (λmax = 728 nm, ε = 47800 M−1 cm−1; λPL = 834 nm, ΦPL = 8%; Ered1 = −1.02 V and Ered2 = −2.05 V). Donation of a nitrogen lone pair appears to be the origin of the property changes, and evidence of quinoidal character (i.e., bond alternation within the phenyl ring and N–C bonds with significant double bond character) within the N-aryl substituents were observed using X-ray crystallography.73
The optoelectronic properties of BF2 complexes of 3-nitroformazanates (61)64 and triarylformazanates (59)63,65 are qualitatively similar to those described above. The 3-substituent has a dramatic influence on electrochemical reduction potentials with nitroformazanate complexes reduced more easily than analogous compounds based on 3-cyanoformazantes due to the greater electron-withdrawing character of NO2 relative to CN. The opposite is true for triarylformazante complexes as the C-aryl substituents are relatively weakly electron-withdrawing or electron-donating. The most dramatic difference found within this series is the fact that ΦPL values for BF2 triarylformazanates are generally very low and often <1% and Stokes shifts for the same species are generally larger than those of 3-cyano and 3-nitro derivatives. The latter trait can be linked to the strictly planar structure adopted by these species in the excited state, as discussed above,65,74,75 while the former has been attributed to enhanced probability of non-radiative decay associated with vibration and/or free rotation of the 3-aryl substituents. This hypothesis was tested by examining the protonation of compound 59Py to form 59Py·H+, which possesses a 2-pyridyl substituent at the 3-position of the formazanate backbone.76 Upon protonation, it is thought that rotation and vibration associated with the 2-pyridyl substituent are dramatically attenuated resulting in enhanced photoluminescence intensity that varied linearly with decreasing pH (Fig. 7).
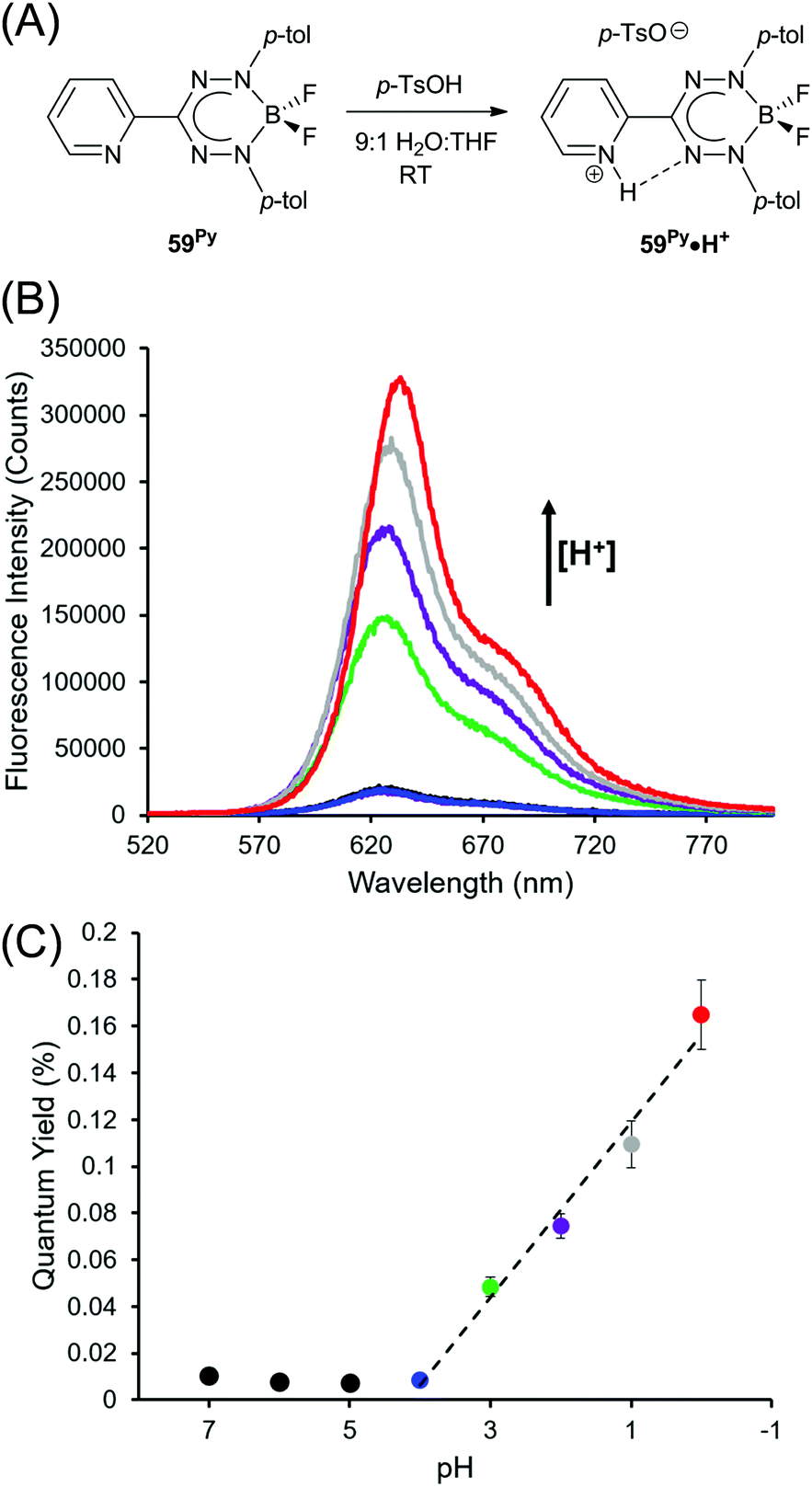 | ||
| Fig. 7 (A) Protonation of complex 59Py. (B) pH-Dependent photoluminescence spectra. (C) pH-Dependent photoluminescence quantum yields. Adapted from ref. 76 with permission from the American Chemical Society. | ||
A second non-radiative decay pathway was uncovered when the Lewis-acid-supported oxoborane (BO) complex 64 was isolated (Scheme 26).77 Access to this species required a halide exchange reaction between BF2 triarylformazanate 59b (Ar1/Ar5 = p-tolyl; R3 = Ph) and BCl3 to generate BCl2 complex 63. The BCl2 unit in compound 63 was subsequently converted to the oxoborane by treatment with one equivalent of AlCl3 and H2O. As mentioned earlier, the structural reorganization associated with electronic excitation observed for BF2 triarylformazanates has been linked to the large Stokes shifts observed for these species. It is also feasible that this structural reorganization is a potential pathway for non-radiative decay upon photoexcitation that contributes to the low ΦPL values observed for this subclass of dyes. The photoluminescence characteristics of oxoborane 64 support this hypothesis, as the formation of the BO unit turns on photoluminescence (λPL = 636 nm, ΦPL = 36%).77 The origin of this behavior lies in the fact that both the ground and excited states of compound 64 adopt a planar structure as the result of sp2 hybridization at boron, thus limiting structural reorganization upon photoexcitation and attenuating non-radiative decay. Based on these studies, it is reasonable to assume that maximum photoluminescence from boron triarylformazanate complexes will be achieved with immobilized C-bound substituents and sp2 hybridized boron units. This remains an area for exploration in the future.
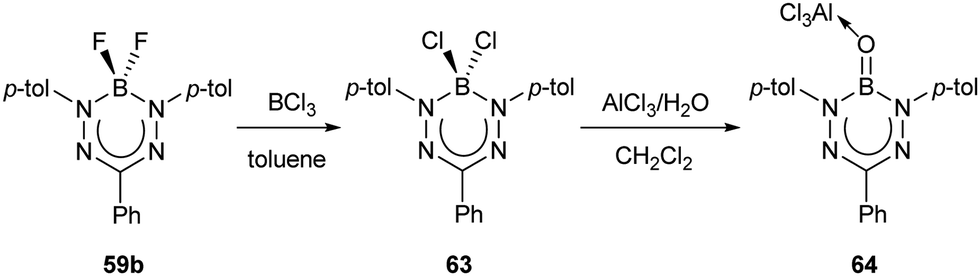 | ||
| Scheme 26 Synthesis of oxoborane formazanate complex 64.77 | ||
 | ||
| Chart 3 | ||
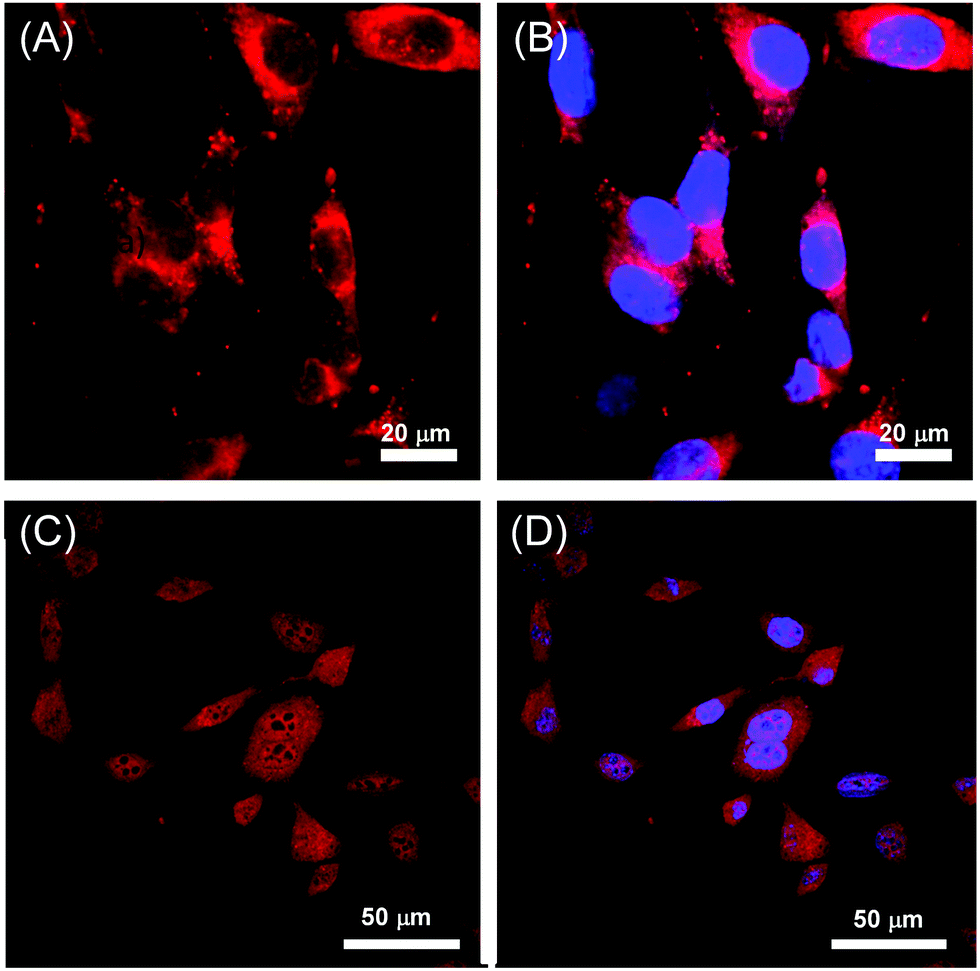 | ||
| Fig. 8 Confocal fluorescence microscopy images of mouse fibroblast cells with filters applied for the visualization of (A) 60d, (B) 60d + DAPI, (C) 60h, and (D) 60h + DAPI. Adapted from ref. 70 and 79 with permission from John Wiley and Sons and the Royal Society of Chemistry. | ||
Electrogenerated luminescence or electrochemiluminescence (ECL)80 is a phenomenon that results in the emission of light from an excited state produced by the reaction of electrogenerated species. Ideally, these species will exist at similar oxidation/reduction potentials and are highly reactive. BF2 formazanates are strong candidates for ECL as a result of their photoluminescence and redox activity. However, it is their oxidation that has proved most useful for the generation of ECL when combined with the coreactant tri-n-propylamine (TPrA) (e.g., Fig. 9).73,75,81
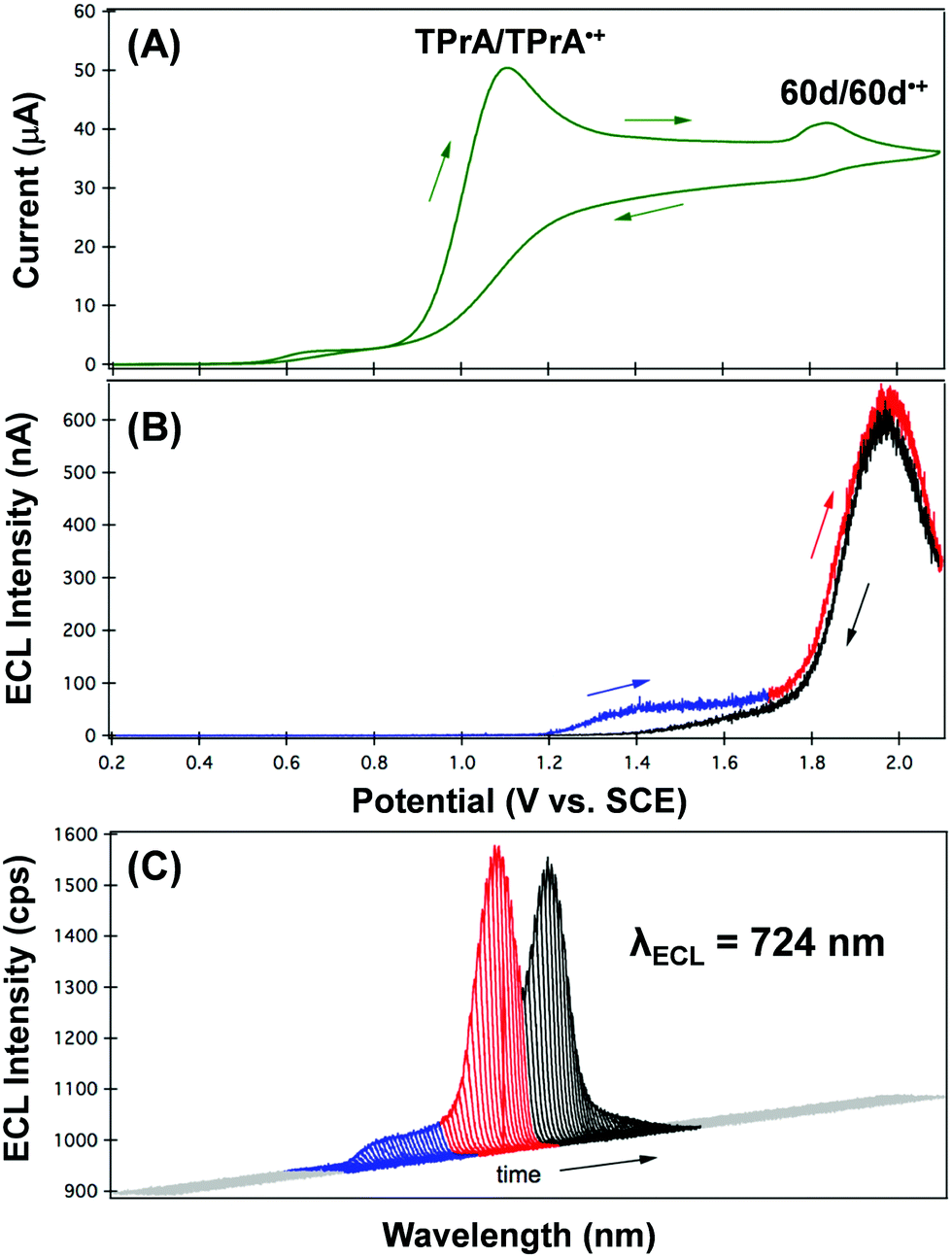 | ||
| Fig. 9 (A) CV, (B) ECL–voltage curve, and (C) spooling ECL spectra acquired for a 0.1 mM CH3CN solution of 60d in the presence of 20 mM TPrA and 0.1 M [Bu4N][PF6] at a scan rate of 0.020 V s−1. The wavelength of maximum ECL intensity was 724 nm in all of the spooling ECL spectra reported in panel (C). Adapted from ref. 81 with permission from the Royal Society of Chemistry. | ||
The CV of a solution containing BF2 formazanate 60d and an excess of TPrA is shown in Fig. 9A. Upon scanning to positive potentials, TPrA is first oxidized to its radical cation form (TPrA˙+), which loses a proton to generate the reducing agent TPrA˙ (eqn (1)).82Fig. 9B shows the intensity of ECL generated during the CV scan as an ECL–voltage curve, and reveals relatively low-intensity ECL centered at ca. 1.4 V vs. SCE arising from chemical reactions implicating 60d, TPrA˙+, and TPrA˙.81 At higher potentials, 60d is oxidized to 60d˙+ and 60d2+ (eqn (2)) and it was shown computationally that the comproportionation reaction involving 60d2+ and 60d generating two equiv. of 60d˙+ was energetically favorable (eqn (3)). Thus, the concentration of 60d˙+ is relatively high above potentials of ca. 1.8 V vs. SCE. In these potential regions, 60d˙+ can react with TPrA˙ to produce the excited state 60d* (eqn (4)), which radiatively relaxes to its grounds state by ECL with maximum intensity at a wavelength (λECL) of 724 nm (eqn (5)). This process shows little dependence on the scan direction (red and blue regions of Fig. 9B), and can be monitored in real time using spooling ECL spectroscopy. Crucially, the spooling spectra (color coded in Fig. 9C) all have ECL maxima centered at 724 nm, indicating a common excited state (60d*) throughout the entire scan window.
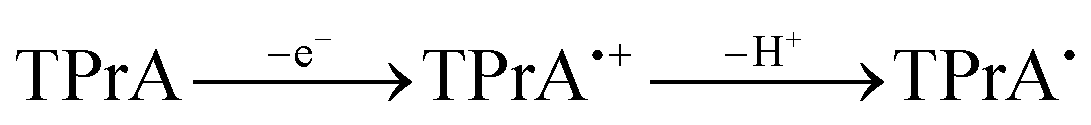 | (1) |
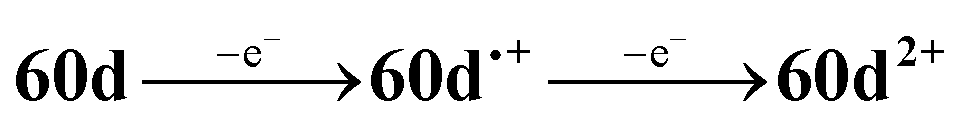 | (2) |
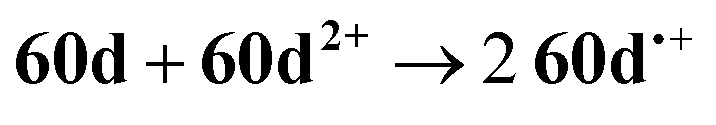 | (3) |
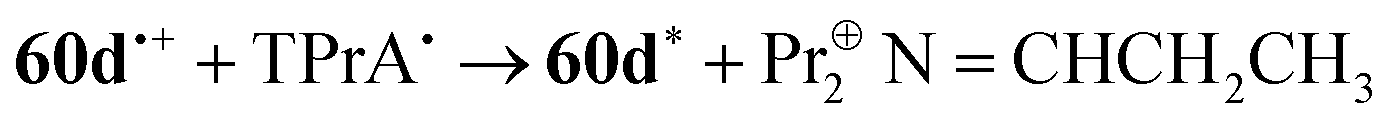 | (4) |
 | (5) |
The ECL efficiency of BF2 formazanates is dependent on a number of factors, including: match with the oxidation potential of TPrA, the stability/instability of the radical cation and dication forms, and the balance between radiative and non-radiative decay pathways associated with the excited states involved in ECL. The efficiency (ΦECL) of these processes was quantified by relative comparison with the [Ru(bipy)3][PF6]2/TPrA system under identical conditions. Complex 60d had a maximum ECL efficiency of 450%, which remains the highest reported to date for BF2 formazanates.81 The λECL and ΦECL values reported for related compounds are summarized in Table 2.
The attractive optoelectronic properties of BF2 formazanates make them excellent candidates for incorporation into functional polymers, whereby film-forming properties may facilitate their incorporation into various organic electronics. One of the primary challenges in their polymerization is the discovery of reaction conditions that are compatible with BF2 formazanates. Ring-opening metathesis polymerization (ROMP)83 has been used to produce side-chain polymers comprised with pendant BF2 triarylformazanates 6684 and 3-cyanoformazanates 67 (Chart 4).85 Polymer 66 retained many of the traits of molecular BF2 triarylformazanates, including intense absorption in the red region (λmax = 518 nm), weak photoluminescence (λPL = 645 nm, ΦPL = 2.5%) and reversible reduction to its poly(radical anion) form (Ered1 = −0.95 V vs. Fc0/+).84 In an effort to enhance photoluminescence intensity, (co)polymers 67 containing BF2 3-cyanoformazanate complexes were prepared through variation of the mole fraction (fBF2) of BF2 formazanate monomer incorporated.85 Despite the monomer exhibiting respectable photoluminescence (λPL = 561 nm, ΦPL = 30%), the photoluminescence intensity of solutions of homopolymer 67 (fBF2 = 1) were reduced (λPL = 559 nm, ΦPL = 11%), likely as a result of ACQ. To circumvent this issue, random (co)polymers with cis-dimethyl-5-norbornene-exo-2,3-dicarboxylate were produced in order to increase the average distance between the BF2 formazanates. This strategy resulted in reduced quenching and enhanced photoluminescence up to a maximum of ΦPL = 24% when fBF2 was 0.08. These findings may provide opportunity for side-chain BF2 formazanate polymers to be used as the active layer in electroluminescent devices in the future.
 | ||
| Chart 4 | ||
While side-chain polymers of BF2 formazanates allow for their desirable characteristics to be combined with advantageous film-forming properties, main-chain polymer architectures offer the latter in combination with opportunities to tune electronic structure via extended π-electron conjugation. For example, CuAAC chemistry was used to produce polymer 68 (Chart 4), which gave rise to λmax = 557 nm (ε = 25700 M−1 cm−1) and λPL = 696 nm (ΦPL = 1%) in DMF.86 Polymer 68 also exhibited a reversible wave associated with its electrochemical reduction to its poly(radical anion) form (Ered1 = −0.73 V vs. Fc0/+). Thorough examination of the properties of 68 compared to model compounds constructed from various combinations of the building blocks of the polymer later revealed that π-electron conjugation involving the BF2 formazanate units did not extend beyond the triazole rings formed during polymerization. Despite the relatively localized electronic structure, polymer 68 exhibited a narrow thin-film optical band gap (Eg) of 1.7 eV. The combination of BF2 formazanates and Pt(II) acetylides in polymer 69 (Chart 4) resulted in further narrowing of the band gap (Eg = 1.4 eV), which could be readily tuned via the stepwise reduction of the BF2 formazanate repeating units, providing an entry point for future studies of their redox-tunable semiconductivity.87 The red-shifted absorption maxima (λmax = 661 nm, ε = 45500 M−1 cm−1) exhibited by polymer 69 relative to its parent BF2 formazanate unit arises due to extended π-conjugation through the Pt(II) acetylides units that implicates backbonding-type interactions between the π* orbitals of the alkyne units and the dxy and dxz orbitals of the Pt(II) center. The extended electronic conjugation mentioned above was verified by comparison with model compounds constructed from the various polymer building blocks, which in all cases absorbed at shorter wavelengths than polymer 69. A follow-up study focused on examining the photoluminescence properties of molecular analogues of 69 revealed that conjugation of BF2 formazanates with Pt(II) acetylides resulted in fluorescence quenching with no signs of phosphorescence observed, even at 77 K.88
 | ||
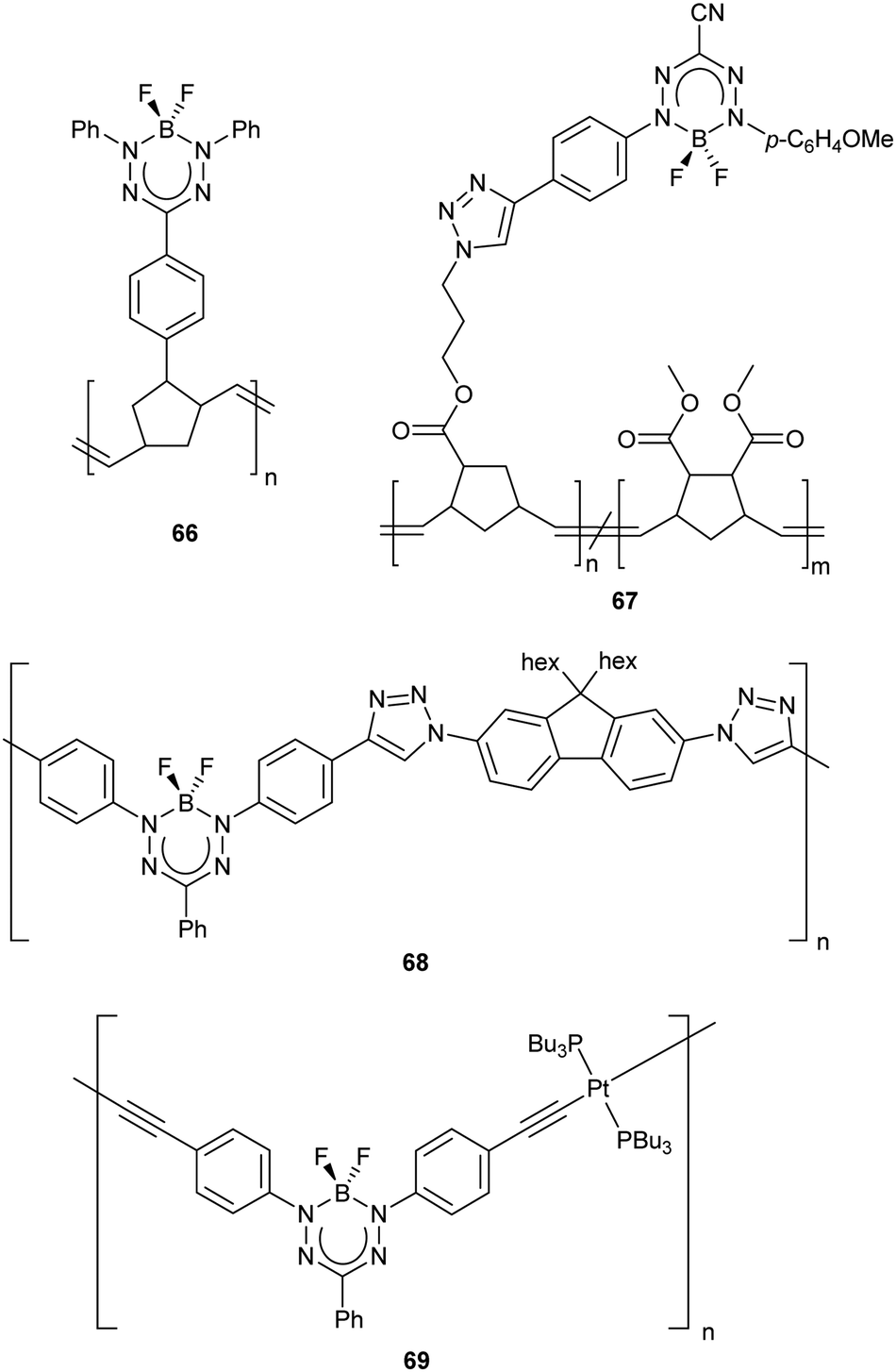
| Scheme 27 (a) Representative synthesis of BH2 formazanate complex 70 and subsequent thermally-driven hydride transfer reactions.89 (b) Proposed mechanism of thermally-induced hydride transfer reactions of BH2 formazanate complex 70.89 | ||
The mechanism of these unusual reactions warrants further discussion, and is proposed in Scheme 27b. Step 1 involves the isomerization of complex 70 to form complex 73, which possesses a 5-member formazanate chelate ring. Similar isomerization behavior has previously been observed for Zn adducts of formazanate ligands.56 Step 2 involves hydride transfer to the N-aryl substituent, exclusively at the ortho position, to generate 74. Step 3 results in hydrogen migration from the cyclohexadiene group to the terminal nitrogen atom, resulting in rearomatization of the N-aryl substituent in 71-ii. Step 4 generates a formally low-valent boron center via transfer of the remaining borohydride to the internal nitrogen atom in 75, which allows for reductive N–N bond cleavage to form triamidoborane 71-ii. DFT calculations (B3LYP/6-31G(d)) indicated that the isomerization step was energetically uphill (5.0 kcal mol−1 for 70 → 73) and that all other steps are energetically downhill.89 These studies provided significant insight into the use of formazanate ligands as redox-active ligands as each hydride transfer corresponds to the transfer of two electrons and a proton and served as a starting point for further reactivity studies.
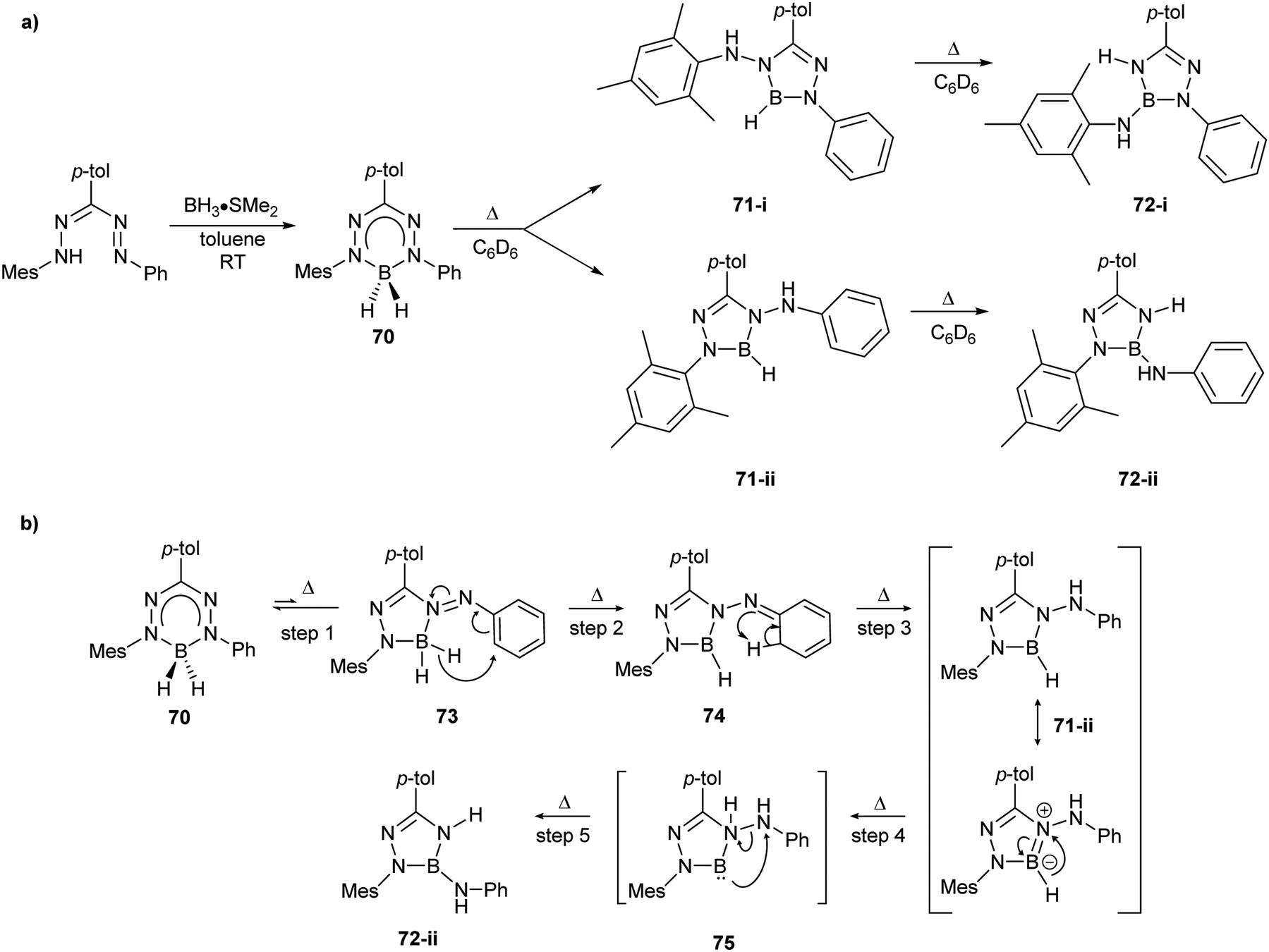
As noted earlier (Scheme 24), a significant hurdle in the development of the two-electron reactivity of complexes of redox-active formazanate ligands is the realization of methods for the production of their dianion form. Boron diphenyl (BPh2) formazanates are excellent candidates for this purpose as the absence of boron–halide bonds in their frameworks should circumvent the formation of salts (e.g., LiF in Scheme 24) that complicated the redox reactivity of BF2 formazanates.67 BPh2 formazanate 76 was generated by refluxing BPh3 with the appropriate formazan in toluene (Scheme 28a).90 The radical anion 76˙− and dianion 762− were subsequently generated by treatment with 1 equiv. of CoCp*2 and 2 equiv. of Na/C10H8, respectively. Their solid-state structures were confirmed crystallographically. Crucially, for the first time, the dianion of a formazanate complex was produced, setting the stage for further reactivity studies designed to take advantage of the unique two-electron redox chemistry of the formazanate ligand scaffold.
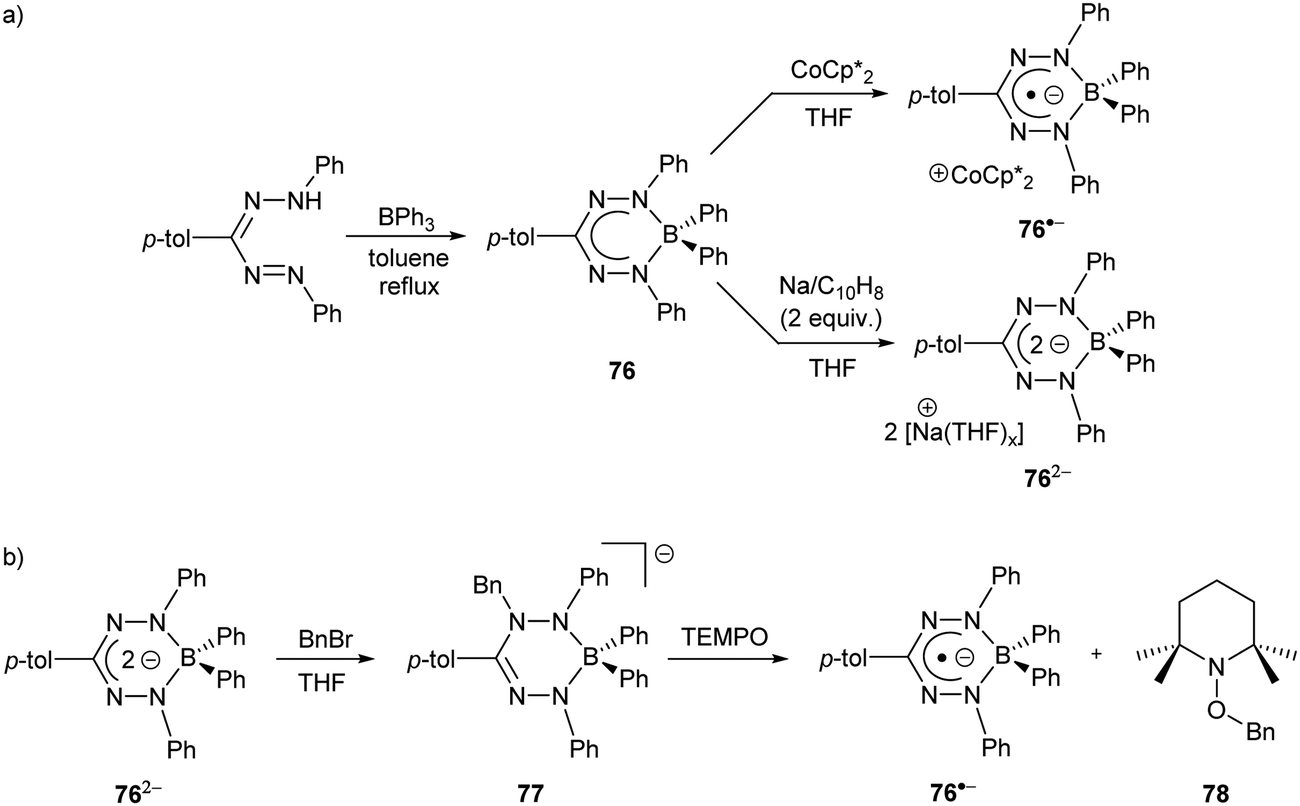 | ||
| Scheme 28 Representative (a) synthesis and chemical reduction chemistry of BPh2 formazanate complexes90 and (b) bond formation/homolysis reactions involving 762− and BnBr.91 | ||
Reaction of dianion 762− with benzyl bromide (BnBr) or H2O resulted in the production of novel anionic analogues of leucoverdazyls (e.g., 77) that demonstrated the ability of the formazanate ligand framework to store two electrons and an electrophile (Scheme 28b).91 The N–Bn or N–H bonds formed were highly reactive, and favored homolytic cleavage pathways due to the inherent stability of the relevant radical anions (e.g., 76˙−). This was experimentally demonstrated, for example, by reacting 77 with the (2,2,6,6-tetramethylpiperidin-1-yl)oxyl radical (TEMPO) in THF, resulting in the regeneration of 76˙− and the formation of adduct 78. This reactivity clearly demonstrated that the unique redox chemistry of formazanate ligands can be used to transfer electrons to electrophiles, in these cases resulting in the formation of Bn˙ and H˙.
BF2 formazanate 59e was converted to the corresponding dialkynylborane complexes 79 upon treatment with a slight excess of alkynyllithium reagents in yields ranging from 17–35%,92 which were lower than related dipyrrin complexes93 as a result of steric crowding imposed by the N-aryl substituents (Scheme 29a). These complexes provided an opportunity to study the relationship between structure and properties via variation of boron-bound substituents (Table 3). The λmax values obtained for toluene solutions of dialkynylborane complexes 79a–c were red-shifted by ca. 10 nm compared to the parent BF2 complex 59e and were relatively insensitive to the nature of the alkyne substituents employed. This insensitivity arises as a result of the tetrahedral geometry at boron, which prevents π-orbital overlap between the formazanate and substituted alkynes. The dialkynylborane complexes could be electrochemically reduced in two steps (79 → 79˙− and 79˙− → 792−), and also exhibited reversible oxidation (79 → 79˙+) waves in CH2Cl2. The potentials at which these events occurred were dependent on the nature of the alkyne substituents, due to inductive effects, with electron-donating substituents (e.g., 79b, R = OMe) rendering the complexes easier to oxidize and harder to reduce than the corresponding phenylacetylene complex 79a. The opposite trend was observed when electron-withdrawing substituents were introduced in complex 79c (R = CF3). Chemical reduction with CoCp2 or CoCp*2 resulted in the formation of stable radical anions (79˙−). Future opportunities in this area should focus on energy/electron transfer between the formazanate ligand backbone and alkynyl substituents bearing π-conjugated functional groups for light-harvesting and charge-transport applications.
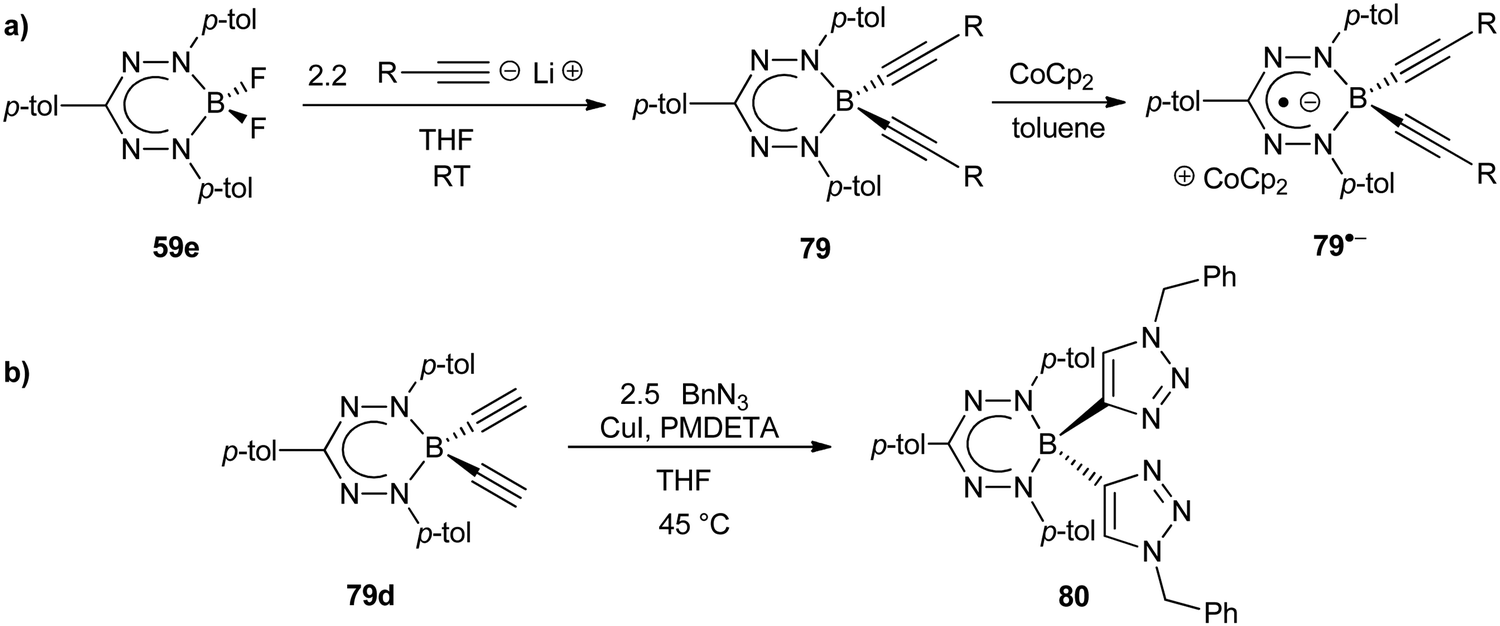 | ||
| Scheme 29 (a) Synthesis of dialkynylborane formazanate complexes 79. (b) Proof of concept CuAAC chemistry to produce bis(triazolyl)borane complex 80.92 | ||
| Compound | R | λ max (nm) | ε (M−1 cm−1) | E red2 (V vs. Fc0/+) | E red1 (V vs. Fc0/+) | E ox1 (V vs. Fc0/+) |
|---|---|---|---|---|---|---|
| a Recorded in toluene. b Recorded in CH2Cl2. c Irreversible peak. Potential at maximum cathodic current reported. d Irreversible peak. Potential at maximum anodic current reported. | ||||||
| 59e | — | 532 | 20200 |
−1.99c | −1.04 | 1.03 |
| 79a | H | 521 | 12000 |
−2.03c | −1.18 | 0.83 |
| 79b | OMe | 519 | 12500 |
−2.12c | −1.21 | 0.86d |
| 79c | CF3 | 521 | 13700 |
−2.01c | −1.14 | 0.89 |
Dialkynylborane complexes 79 also provided an opportunity for the elaboration of the structural diversity of boron formazanate complexes using CuAAC chemistry. For example, the reaction of complex 79d (R = H) with benzyl azide (BnN3) resulted in triazole formation at the boron-bound alkyne and demonstrated that, despite the steric congestion at boron, this transformation is feasible and complex 80 could be formed (Scheme 29b). This reactivity could be used to leverage the applications described above or even to produce polymeric structures if bifunctional dialkynylborane formazanate complexes and azides were employed.
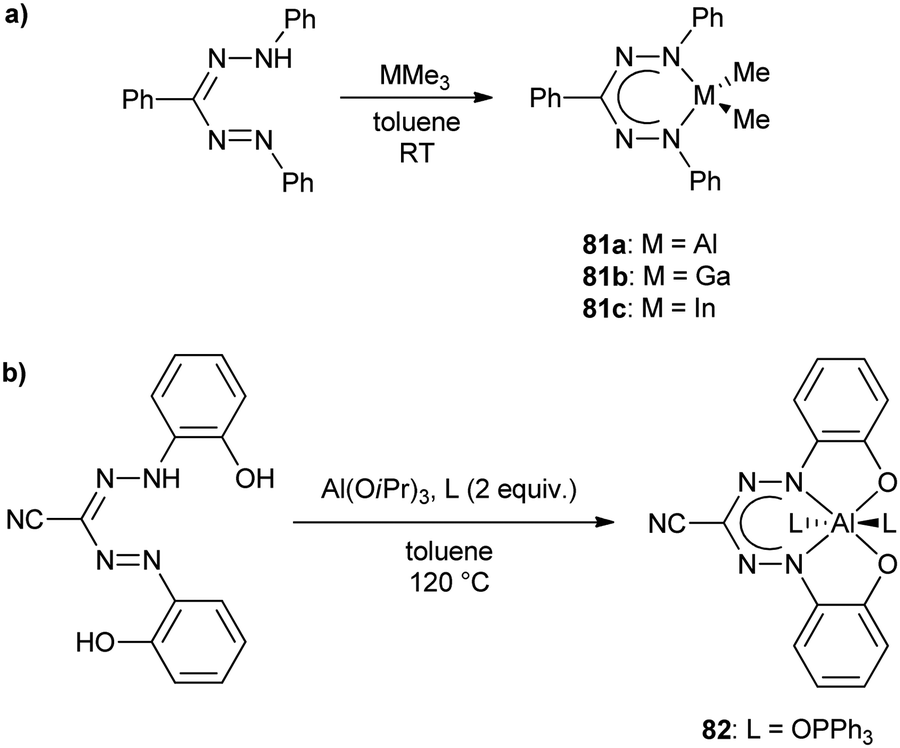
210 M−1 cm−1; 81b: λmax = 576 nm, ε = 20960 M−1 cm−1; 81c: λmax = 591 nm, ε = N/A).94 Preliminary CV studies indicated that the complexes could not be reversibly oxidized or reduced.
 | ||
| Scheme 30 (a) Synthesis of heavy group 13 complexes of formazanates 81.94 (b) Representative synthesis of octahedral aluminum formazanate complexes with phosphine oxide substituents 82.95 | ||
Six-coordinate aluminum complexes of N2O23− formazanate ligands (e.g., 82) were prepared by heating N-(ortho-hydroxyphenyl) substituted cyanoformazan with Al(OiPr)3 and two equiv. of phosphine oxide ligand in hot toluene solution resulting in their production in 52–86% yield (Scheme 30b).95 The solid-state structures of these complexes confirmed an octahedral geometry at aluminum with the formazanate adopting a planar structure and a tetradentate binding mode. DFT calculations (M06/6-311+G(d,p)) indicated that the frontier orbitals were centered on the formazanate ligand, consistent with the fact that variation of the phosphine oxide ligands involved had little effect on the spectroscopic characteristics of the resulting complexes. In the case of complex 82, its photophysical properties were examined in detail revealing the following data: λmax = 629 nm, ε = 24700 M−1 cm−1; λPL = 690 nm, ΦPL = 2%. It is worth noting that the low-energy maximum was accompanied by related peaks associated with the vibrational fine structure of the rigid complex (82) and that detailed studies of these properties indicated that the phosphine oxide ligands were labile in solution. Examination of the same properties in the presence of 50 equiv. of phosphine oxide ligand confirmed that the emissive species was indeed the octahedral complex 82. The CV of 82 both in the absence and presence of excess triphenyl phosphine revealed irreversible oxidation and reduction events, with the latter giving rise to the following potentials at peak anodic and cathodic currents: Epc = −1.34 V and Epa = 0.29 V. The combined photoluminescence and oxidation behavior of 82 prompted the examination of its ECL in the presence of TPrA and excess phosphine oxide. These studies gave rise to λECL = 735 nm and ΦECL = 7%, demonstrating for the first time that aluminum formazanate complexes may offer utility in light-emitting technologies.
More recently, Mondol and Otten conducted a study that led to the dramatic expansion of the structural diversity of aluminum formazanate complexes, including the first isolated examples of ligand-supported radical anions and dianions derived from aluminum formazanates.96 Their work began with the synthesis of tetrahedral AlR2 complexes (R = Me, Et) that were similar to those reported by Sundermeyer and co-workers.94 Treatment with one equivalent or excess I2 resulted in the asymmetrically substituted AlMeI and AlEtI complexes, respectively.
The reaction of potassium salt 9 with AlCl3 in THF yielded a distorted trigonal bipyramidal (τ = 0.64) aluminum formazanate complex 83 (Scheme 31a), which yielded a 27Al NMR resonance at 43.0 ppm. In contrast to most group 13 adducts of formazanate ligands, the low-energy absorption maxima observed for 83 was blue-shifted by ca. 10 nm compared to the parent formazan. This behavior was attributed to a significant structural distortion associated with the Al atom being displaced by more than 1 Å from the plane of the formazanate ligand that causes the ligand backbone to deviate from planarity. Complex 83 exhibited two quasi-reversible reduction waves at potentials of −1.36 V and −1.67 V relative to the Fc0/+ redox couple when studied by cyclic voltammetry in THF and was cleanly converted to its radical anion form 83˙− by treatment in the same solvent with CoCp2. Density functional calculations indicated that the unpaired electron density associated with this radical anion was primarily located on the nitrogen atoms of both formazanate ligands.
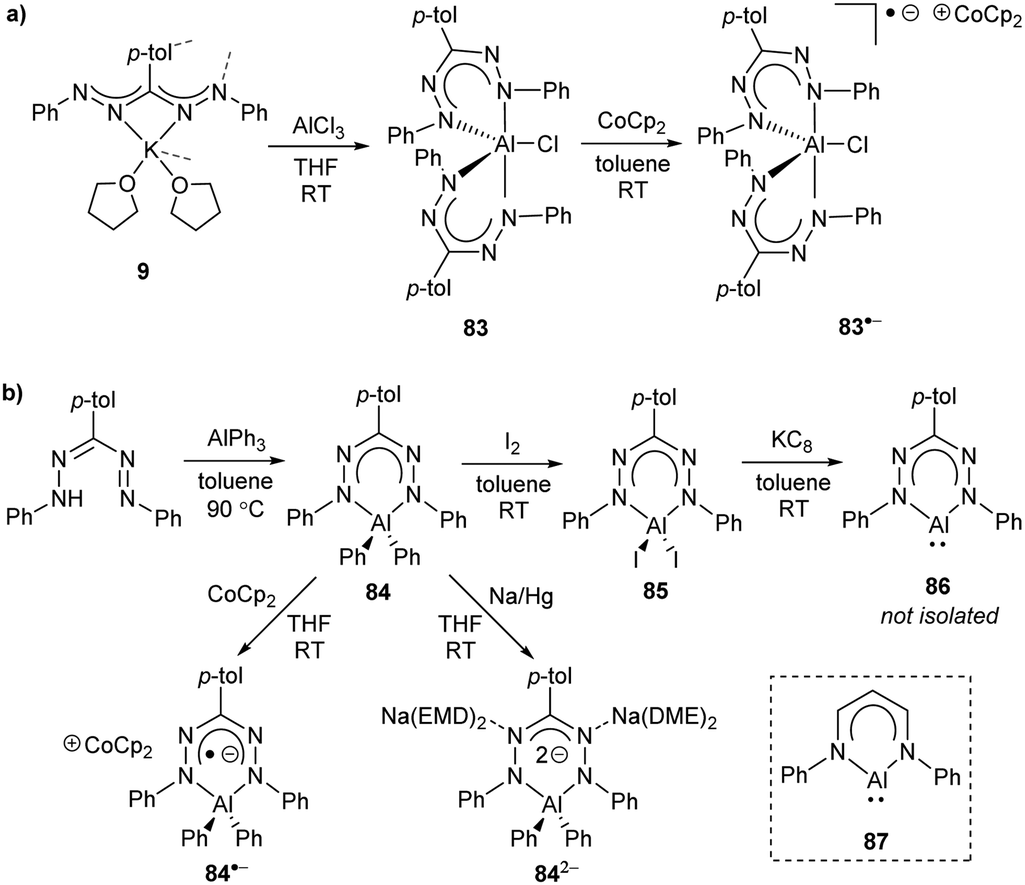 | ||
| Scheme 31 (a) Synthesis of aluminum formazanate complex 83 and its corresponding radical anion. (b) Synthesis of diphenyl aluminum complex 84 and its conversion to radical anion 84˙−, dianion 842−, and aluminum diodide complex 85. The attempted conversion of compound 85 to Al(I) carbenoid 86 and the structure of β-diketiminate complex 87 are also included to supplement the discussion above.96 | ||
The reaction of triarylformazan with AlPh3 afforded the tetrahedral diphenyl aluminum complex 84 and created several new synthetic opportunities (Scheme 31b). Firstly, isolable radical anion 84˙− and dianion 842− were prepared by treatment with CoCp2 or Na/Hg amalgam, respectively. Ligand exchange resulting from the treatment of complex 84 with I2 yielded complex 85. Inspired by the work of Roesky on low-valent Al chemistry supported by β-diketiminate ligands,97 complex 85 was treated with two equiv. of KC8 with the goal of performing a two-electron reduction. The observed color changes were indicative of radical anion formation, but the Al(I) carbenoid 86 was not isolated from this reaction. This prompted a detailed investigation of the electronic structures of proposed complex 86 and the related β-diketiminate complex 87 using DFT calculations. As expected, the presence of additional nitrogen atoms in the ligand backbone of 86 resulted in the stabilization of the HOMO, LUMO, and LUMO+1 orbitals relative to those of 87 (Table 4). However, this did not explain the apparent difference in stability between the two families of compounds. Examination of the singlet–triplet energy gaps for each species provided further insight. The calculated values were 11.5 kcal mol−1 for the formazanate complex 86 and 30.7 kcal mol−1 for β-diketiminate complex 87, with the marked difference potentially accounting for the differences in observed reactivity.
| −(ES − ET) (kcal mol−1) | HOMO (eV) | LUMO (eV) | LUMO+1 (eV) |
|---|---|---|---|
| 11.5 | −5.25 | −3.00 | −1.43 |
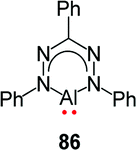
|
|
|
|
| 30.7 | −4.77 | −1.79 | −0.80 |
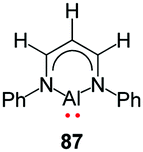
|
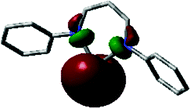
|
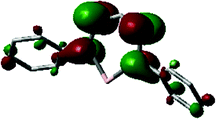
|
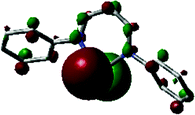
|
Comparison of structure, bonding, and reactivity of AlPh2 complex 842− with its BPh2 analogue 762−, both possessing formazanate ligands in their trianionic form, led to several important conclusions.98 (i) The Al–N bonds have primarily ionic character and B–N bonds have primarily covalent character. (ii) The introduction of an Al ion resulted in N–Caryl bonds with significant π character; a structural feature that appears to have been recently exploited to induce near-IR absorption and emission in similar compounds.73 (iii) Coordination of Al significantly alters the reactivity at the unsubstituted nitrogen atoms, weakening the N–Bn bonds in trapped species (e.g., 77) and setting the stage for two-electron redox chemistry to be exploited.
2.9 Group 14 (Si, Ge, Sn)
The first examples of group 14 formazanate complexes were reported in 2018.99 After deprotonating 1,5-bis(2-hydroxyphenyl)-3-phenylformazan with NaH to generate its trianion, the formazanate was combined with the respective phenyl trichloride of Si, Ge, and Sn (EPhCl3) in THF to form complexes 88a–c (Scheme 32), which contain hypervalent (5-coordinate) group 14 atoms. Unlike closely related hypervalent group 14 complexes of dipyrrin N2O23− ligands that adopted a trigonal bipyramidal geometry,10088a–c adopt a distorted square pyramidal geometry. This is particularly relevant when considering the potential conversion of these complexes to their corresponding radical anions 88a–c˙−, where π-electron delocalization is required to enhance radical stability. The formazanate ligand occupies the base of the distorted square pyramid formed by the respective ligands, imparting planarity. This planar configuration led to a delocalized structure and low-energy absorption maxima of 662 nm (ε = 16800 M−1 cm−1) for 88a, 681 nm (ε = 21200 M−1 cm−1) for 88b, and 681 nm (ε = 22200 M−1 cm−1) for 88c in CH2Cl2 (Fig. 10A–C), and varied based on the group 14 element incorporated. The planarity of the formazanate ligand was quantified by assessing the displacement of the group 14 atom from the N4O2 plane (88a: 0.467 Å; 88b: 0.593 Å; 88c: 0.739 Å) and the average angle between the N4O2 plane and the plane defined by the N-aryl substituents (88a: 22.44°; 88b: 22.63°; 88c: 26.81°) in the solid state. When the relatively large Sn atom was incorporated in 88c, the solid-state structure deviated from planarity significantly. This influenced the electronic properties of the resulting complex, and rendered 88c more difficult to reduce than 88a and 88b as a result of a destabilized LUMO orbital that results from disruption of the π-electron system. Furthermore, the reduction events associated with radical anion and dianion formation that were reversible in the CVs collected for 88a and 88b became irreversible for 88c (Fig. 10F). This was reflected during efforts to chemically reduce 88a–c to 88a–c˙−, with the Sn-containing compound 88c˙− being the only unstable compound in the series as a result of the poorer electron-accepting ability of its relatively less planar formazanate ligand. Radicals 88a˙− and 88b˙− were structurally characterized, once again revealing square pyramidal geometries at Si and Ge, reduced separation between the main group atoms and the N4O2 plane, and an overall greater degree of formazanate ligand planarity. In solution, these species gave rise to new absorption bands centered at ca. 800 nm (consistent with verdazyl-type radicals) and complex EPR spectra implicating hyperfine coupling to the ortho- and para-protons of the N-aryl substituents and two sets of equivalent nitrogen atoms of the formazanate backbone. Simulation of the spectra, yielded the following g-factors and hyperfine coupling constants (a) for 88a˙−: g = 2.0037, aN(bottom) = 5.05 G, aN(top) = 3.37 G, aH(ortho) = 1.61 G, aH(para) = 1.58 G and 88b˙−: g = 2.0035, aN(bottom) = 4.94 G, aN(top) = 3.75 G, aH(ortho) = 1.56 G, aH(para) = 1.55 G (Fig. 10D and E). These stable radicals are rare examples that do not require the presence of significant steric bulk as a stabilizing factor and demonstrate the importance of structural planarity and π-electron delocalization for the stabilization of radical species.
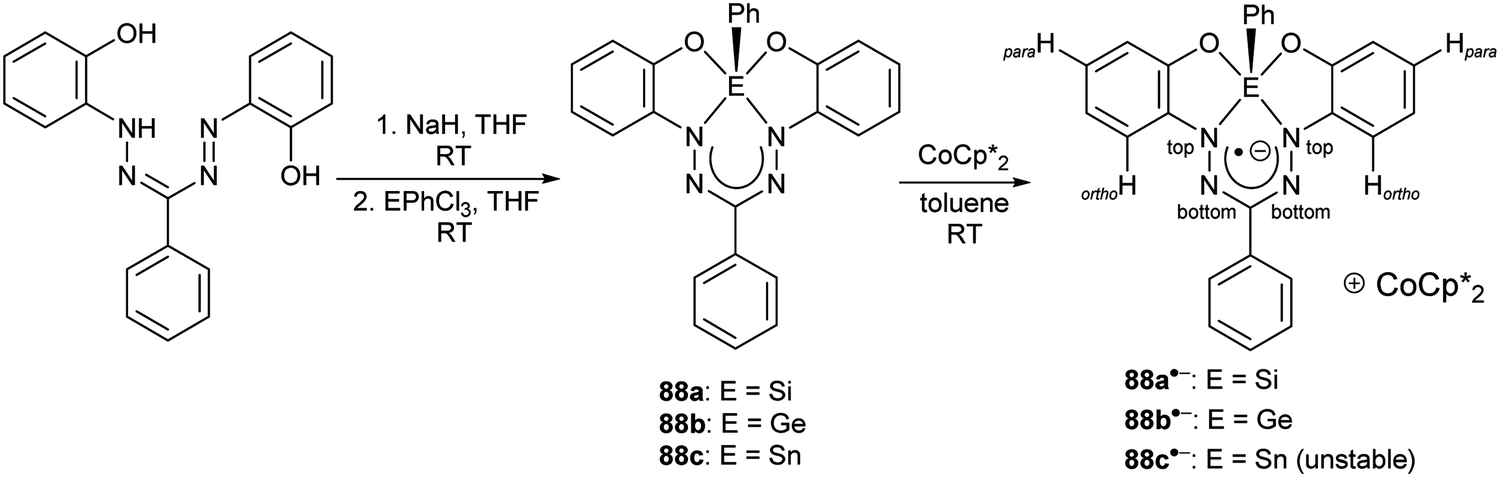 | ||
| Scheme 32 Synthesis of group 14 complexes of formazanate ligands 88a–c and their corresponding radical anions 88a–c˙−.99 | ||
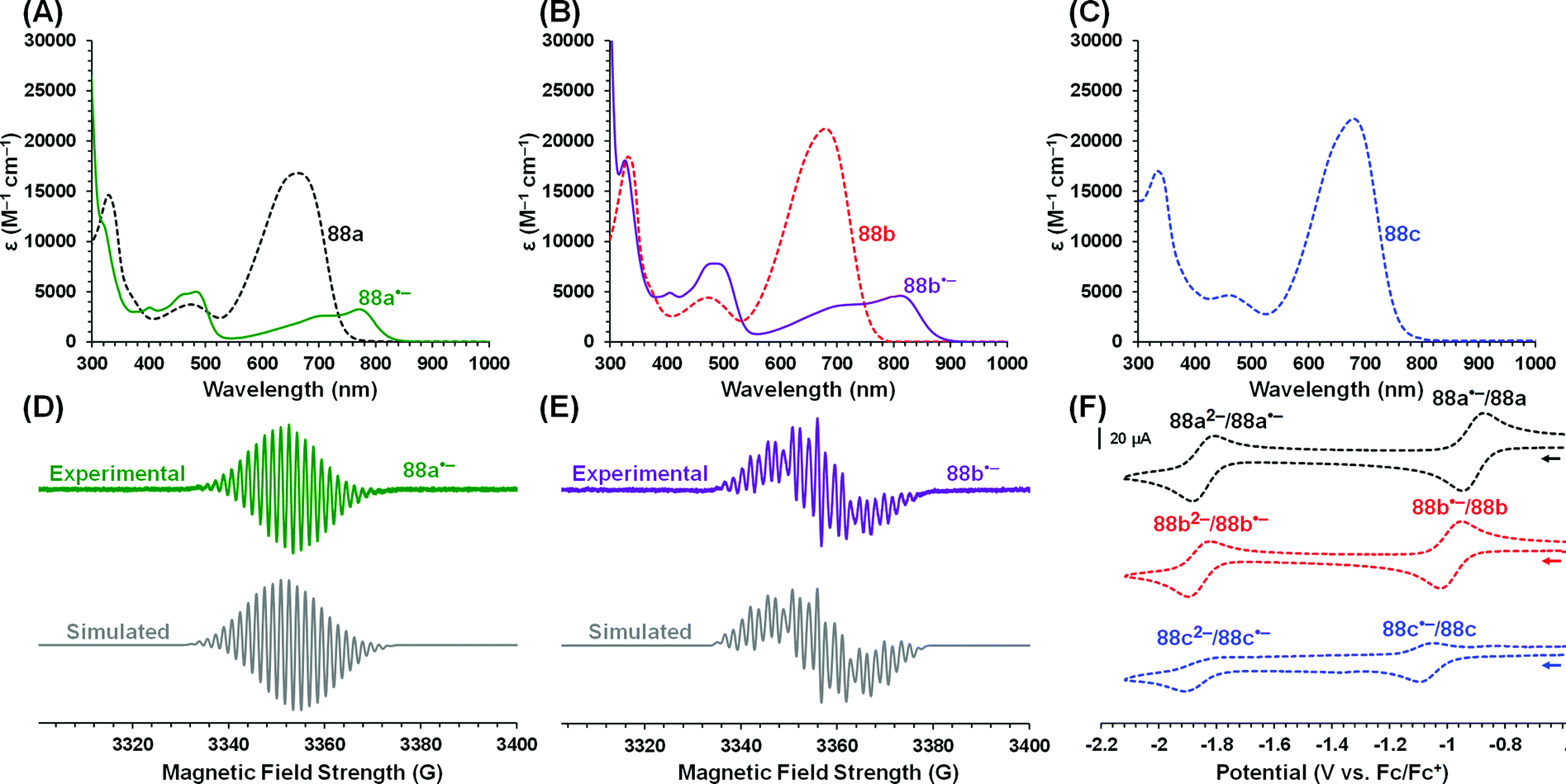 | ||
| Fig. 10 (A–C) UV-vis absorption spectra of complexes 88a–c and radicals 88a˙− and 88b˙−, (D and E) EPR spectra of radicals 88a˙− and 88b˙−, and (F) CVs of complexes 88a–c (0.25 V s−1) recorded in dry, degassed CH2Cl2 containing ca. 1 mM analyte and 0.1 M [Bu4N][PF6]. The arrows denote the scan direction.99 Reproduced from ref. 99 with permission from John Wiley and Sons. | ||
2.10 Group 15 (P)
To the best of our knowledge, there have been no reports of compounds prepared by the direct coordination of formazanate ligands to group 15 atoms. However, the Hicks group has reported the synthesis of several examples of phosphaverdazyl radicals via indirect routes (Scheme 33).101–103 Bis(hydrazide)s 89a–c were reacted with trimethyl orthobenzoate to form phosphaleucoverdazyls 90a–c. Like their purely organic analogs, the leucoverdazyls were readily oxidized to their radical forms by treatment with NaH/[Bu4N][IO4] or benzoquinone to form phosphaverdazyls 91a˙–91c˙. These compounds are effectively phosphorus(V) adducts of the radical dianion form of formazanate ligands. | ||
| Scheme 33 Synthesis of phosphaverdazyls 91a˙–91c˙.101–103 | ||
EPR spectroscopic analysis of 91a˙ revealed hyperfine coupling constants to two pairs of equivalent nitrogen atoms that were typical of other verdazyl radicals. However, perhaps surprisingly given the non-planar structure of the heterocyclic core, an appreciable hyperfine constant attributed to phosphorus (aP = 5.2 G) was observed, presumably as a result of spin leakage via spin polarization. This observation prompted the study of phosphaverdazyl 91b˙, which possesses an additional p orbital on the Me2N group that could allow for a spiroconjugation pathway for radical transfer from the π-system of the phosphaverdazyl core to the Me2N group. EPR analysis again revealed typical coupling to two pairs of equivalent nitrogen atoms and an appreciable hyperfine coupling to the exocyclic nitrogen of 4.6 G. To further probe this effect, phosphaverdazyl 91c˙ was prepared and potential radical transfer between the phosphaverdazyl and phosphazene ring was examined. Hyperfine coupling constants <1 G were obtained for the phosphazene nitrogen atoms, indicating inefficient radical transfer despite the seemingly ideal orbital arrangement for electronic communication via spiroconjugation of the heterocycle π-systems. The authors therefore concluded that the most likely coupling pathway involved spin polarization through the σ bond framework of the molecules.
3. Conclusions
Formazans (organic compounds with the Ar1-NH-NCR3-NN-Ar5 structure) have a venerable history in chemistry, with the development of synthetic procedures dating back to the end of the 1800s. Deprotonation gives rise to monoanionic chelating N-donor ligands (‘formazanates’) that are related to the well-known β-diketiminate and dipyrrinate classes of ligands, but with distinct electronic features. From the studies of the last two decades discussed in this review it is clear that the relatively straightforward, modular synthesis of the parent formazans allows a facile entry into formazanate complexes with tunable steric/electronic properties. Coordination compounds with formazanate ligands are now known in both the main group as well as d-block of the periodic table. Despite the advances made in recent years, the overview presented here also clearly demonstrates that formazanate complexes of the most electropositive elements (e.g., early transition metals and lanthanides) have not yet been reported. Thus, substantial gaps remain in our understanding of the fundamental coordination chemistry of these ligands, which means that this research area is still very much amenable to further growth.
A feature of formazanate ligands that sets these apart from β-diketiminates/dipyrrinates and other bidentate N-donor ligands is the presence of a delocalized, low-lying LUMO, which is π-anti-bonding between the four nitrogen atoms in the ligand backbone. Complexes with a coordinated formazanate ligand thus generally have a small HOMO–LUMO gap, which results in the intense coloration of these compounds due to symmetry-allowed (π→π*) electronic transitions in the visible range. Moreover, the energetically accessible π*-orbital renders formazanate complexes redox-active, and allows reversible ligand-based storage of electrons in these compounds. In transition metal formazanate complexes, the interaction of the d-orbitals with the close-lying ligand π*-orbital has been shown to lead a high degree of metal–ligand bond covalency (π-backdonation) and unusual electronic/magnetic properties (e.g., spin-crossover, valence tautomerism). With the vast majority of studies on transition metal formazanate compounds focussing on synthetic aspects, not much is known yet on leveraging the distinctive structural, magnetic and spectral properties of these compounds in applications. The same holds true for the reactivity of transition metal formazanate complexes, and the use of formazanates as supporting ligands in organometallic chemistry and homogeneous catalysis is currently little explored. Nevertheless, it is clear that the incorporation of redox-active formazanate ligands leads to reactivity that does not simply mirror that of complexes with related (redox-inert) anionic bidentate N-donor ligands: the possibility to access reduced and/or oxidized states of the formazanate ligand enables new modes of reactivity. An additional aspect that requires further study is the ability of formazanates to access different coordination modes, which potentially allows complexes with these ligands to adapt the coordination sphere around the central element in response to varying steric/electronic demands throughout a sequence of (catalytic) reaction steps.
Perhaps the most well-established area of application is based on the optoelectronic properties of boron difluoride adducts of the formazanate anion. The absorption and emission spectra of these dyes can be rationally tuned by substituent effects at the formazanate N-Ar1/5 positions, in particular via the extent of π-conjugation. Boron difluoride compounds containing 3-cyanoformazanate ligands combine high photoluminescence quantum yields with large Stokes shifts that render them effective cell-imaging agents. This class of compounds was also used for efficient electrochemiluminescence at potentials >1.8 V vs. SCE in the presence of tri-n-propylamine. The high stability of formazanate boron compounds bodes well for applications in materials, especially considering that incorporation of redox-active formazanate units in polymers leads to a low band-gap that may be further modified by (partial) reduction.
Studies related to the reduction chemistry of boron and aluminum complexes with formazanate ligands have provided detailed insight in the structure/reactivity of these compounds, demonstrating that ligand-based ‘storage’ of up to two electrons in a single formazanate ligand is feasible and allows the development of (stoichiometric) multi-electron chemical transformations that are purely ligand-based. In the course of these studies, decomposition pathways have been identified that indicate that cleavage of an N–N bond in the backbone is potentially deleterious.
The parallels that exist between the chemistry of formazanate coordination complexes and that of stable organic verdazyl radicals is perhaps most clearly demonstrated with the recent synthesis of Si, Ge and Sn compounds with a tetradentate formazanate-based N2O2 ligand: hypervalent heavier group 14 analogues of verdazyl radicals were obtained that are exceptionally stable despite the absence of sterically demanding substituents or other stabilizing effects.
To conclude, we believe that the fundamental insight that has been accumulated over the last two decades on the synthesis, properties and reactivity of formazanate coordination complexes provides a solid basis for the development of new applications that make use of the distinctive optoelectronic features and versatile electrochemistry that formazanate ligands offer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the University of Western Ontario and the University of Groningen for financial support. Our own work in this area is supported by funding from the NSERC of Canada, the Canadian Foundation for Innovation, the Ontario Ministry of Research and Innovation (JBG) and the Netherlands Organisation for Scientific Research (NWO) via VENI and VIDI grants (EO). We also thank Dr. Ryan R. Maar and Francesca Milocco for valuable discussions and proofreading the document.Notes and references
- A. W. Nineham, Chem. Rev., 1955, 55, 355–483 CrossRef CAS.
- A. S. Shawali and N. A. Samy, J. Adv. Res., 2015, 6, 241–254 CrossRef CAS PubMed.
- G. N. Lipunova, T. G. Fedorchenko and O. N. Chupakhin, Russ. J. Gen. Chem., 2019, 89, 1225–1245 CrossRef CAS.
- E. Grela, J. Kozlowska and A. Grabowiecka, Acta Histochem., 2018, 120, 303–311 CrossRef CAS PubMed.
- J. C. Stockert, R. W. Horobin, L. L. Colombo and A. Blázquez-Castro, Acta Histochem., 2018, 120, 159–167 CrossRef CAS PubMed.
- B. D. Koivisto and R. G. Hicks, Coord. Chem. Rev., 2005, 249, 2612–2630 CrossRef CAS.
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC.
- D. J. R. Brook, Comments Inorg. Chem., 2015, 35, 1–17 CrossRef CAS.
- A. R. Katritzky, S. A. Belyakov, D. Cheng and H. D. Durst, Synthesis, 1995, 577–581 CrossRef CAS.
- E. Bamberger and J. Müller, Ber. Dtsch. Chem. Ges., 1894, 27, 147–155 CrossRef.
- E. Wedekind, Ber. Dtsch. Chem. Ges., 1897, 30, 2993–2999 CrossRef CAS.
- F. A. Neugebauer and H. Trischmann, Justus Liebigs Ann. Chem., 1967, 706, 107–111 CrossRef CAS.
- Y. A. Ibrahim, A. H. M. Elwahy and A. A. Abbas, Tetrahedron, 1994, 50, 11489–11498 CrossRef CAS.
- J. B. Gilroy, P. O. Otieno, M. J. Ferguson, R. McDonald and R. G. Hicks, Inorg. Chem., 2008, 47, 1279–1286 CrossRef CAS PubMed.
- D. M. Hubbard and E. W. Scott, J. Am. Chem. Soc., 1943, 65, 2390–2393 CrossRef CAS.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3065 CrossRef CAS PubMed.
- R. L. Webster, Dalton Trans., 2017, 46, 4483–4498 RSC.
- G. I. Sigeikin, G. N. Lipunova and I. G. Pervova, Russ. Chem. Rev., 2006, 75, 885–900 CrossRef CAS.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- B. de Bruin, P. Gualco and N. D. Paul, in Ligand Design in Metal Chemistry, ed. M. Stradiotto and R. J. Lundgren, Wiley, 2016, ch. 7, pp. 176–204 Search PubMed.
- R. Travieso-Puente, M.-C. Chang and E. Otten, Dalton Trans., 2014, 43, 18035–18041 RSC.
- R. Travieso-Puente, S. Budzak, J. Chen, P. Stacko, J. T. B. H. Jastrzebski, D. Jacquemin and E. Otten, J. Am. Chem. Soc., 2017, 139, 3328–3331 CrossRef CAS PubMed.
- D. A. Brown, H. Bögge, G. N. Lipunova, A. Müller, W. Plass and K. G. Walsh, Inorg. Chim. Acta, 1998, 280, 30–38 CrossRef CAS.
- A. Müller, H. Bögge, E. Diemann, D. Brown, S. O'Shea and G. Lipunova, Naturwissenschaften, 1994, 81, 136–137 Search PubMed.
- Y. A. Gorbatenko, Z. G. Rezinskikh, G. N. Lipunova, I. G. Pervova, T. I. Maslakova, P. A. Slepukhin and I. N. Lipunov, Russ. J. Appl. Chem., 2008, 81, 2127–2131 CrossRef CAS.
- J. B. Gilroy, B. O. Patrick, R. McDonald and R. G. Hicks, Inorg. Chem., 2008, 47, 1287–1294 CrossRef CAS PubMed.
- R. Travieso-Puente, J. O. P. Broekman, M.-C. Chang, S. Demeshko, F. Meyer and E. Otten, J. Am. Chem. Soc., 2016, 138, 5503–5506 CrossRef CAS PubMed.
- R. Hoffmann, S. Alvarez, C. Mealli, A. Falceto, T. J. Cahill, T. Zeng and G. Manca, Chem. Rev., 2016, 116, 8173–8192 CrossRef CAS PubMed.
- D. L. J. Broere, B. Q. Mercado, J. T. Lukens, A. C. Vilbert, G. Banerjee, H. M. C. Lant, S. H. Lee, E. Bill, S. Sproules, K. M. Lancaster and P. L. Holland, Chem. – Eur. J., 2018, 24, 9417–9425 CrossRef CAS PubMed.
- D. L. J. Broere, B. Q. Mercado, E. Bill, K. M. Lancaster, S. Sproules and P. L. Holland, Inorg. Chem., 2018, 57, 9580–9591 CrossRef CAS PubMed.
- D. L. J. Broere, B. Q. Mercado and P. L. Holland, Angew. Chem., Int. Ed., 2018, 57, 6507–6511 CrossRef CAS PubMed.
- F. Milocco, S. Demeshko, F. Meyer and E. Otten, Dalton Trans., 2018, 47, 8817–8823 RSC.
- A. J. Kamphuis, F. Milocco, L. Koiter, P. P. Pescarmona and E. Otten, ChemSusChem, 2019, 12, 3635–3641 CAS.
- G. B. Jameson, A. Muster, S. D. Robinson, J. N. Wingfield and J. A. Ibers, Inorg. Chem., 1981, 20, 2448–2456 CrossRef CAS.
- A. Mandal, B. Schwederski, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 8126–8135 CrossRef CAS PubMed.
- N. A. Protasenko, A. I. Poddel'sky, A. S. Bogomyakov, G. K. Fukin and V. K. Cherkasov, Inorg. Chem., 2015, 54, 6078–6080 CrossRef CAS PubMed.
- N. A. Protasenko, A. I. Poddel'sky, A. S. Bogomyakov, A. G. Starikov, I. V. Smolyaninov, N. T. Berberova, G. K. Fukin and V. K. Cherkasov, Inorg. Chim. Acta, 2019, 489, 1–7 CrossRef CAS.
- E. Kabir, G. Mu, D. A. Momtaz, N. A. Bryce and T. S. Teets, Inorg. Chem., 2019, 58, 11677–11683 CrossRef PubMed.
- N. A. Frolova, S. Z. Vatsadze, V. E. Zavodnik, R. D. Rakhimov and N. V. Zyk, Russ. Chem. Bull., 2006, 55, 1810–1818 CrossRef CAS.
- L. Hunter and C. B. Roberts, J. Chem. Soc., 1941, 823–826 RSC.
- D. Dale, J. Chem. Soc. A, 1967, 278–287 RSC.
- H. Tezcan, E. Uzluk and M. L. Aksu, Electrochim. Acta, 2008, 53, 5597–5607 CrossRef CAS.
- A. V. Zaidman, I. G. Pervova, A. I. Vilms, G. P. Belov, R. R. Kayumov, P. A. Slepukhin and I. N. Lipunov, Inorg. Chim. Acta, 2011, 367, 29–34 CrossRef CAS.
- J. B. Gilroy, M. J. Ferguson, R. McDonald and R. G. Hicks, Inorg. Chim. Acta, 2008, 361, 3388–3393 CrossRef CAS.
- G. N. Lipunova, Z. G. Rezinskikh, T. I. Maslakova, P. A. Slepukhin, I. G. Pervova, I. N. Lipunov and G. I. Sigeikin, Russ. J. Coord. Chem., 2009, 35, 215–221 CrossRef CAS.
- F. Milocco, F. de Vries, A. Dall'Anese, V. Rosar, E. Zangrando, E. Otten and B. Milani, Dalton Trans., 2018, 47, 14445–14451 RSC.
- A. R. Siedle and L. H. Pignolet, Inorg. Chem., 1980, 19, 2052–2056 CrossRef CAS.
- E. Kabir, C.-H. Wu, J. I. C. Wu and T. S. Teets, Inorg. Chem., 2016, 55, 956–963 CrossRef CAS PubMed.
- E. Kabir, D. Patel, K. Clark and T. S. Teets, Inorg. Chem., 2018, 57, 10906–10917 CrossRef CAS PubMed.
- G. Mu, L. Cong, Z. Wen, J. I. C. Wu, K. M. Kadish and T. S. Teets, Inorg. Chem., 2018, 57, 9468–9477 CrossRef CAS PubMed.
- S. Hong, L. M. R. Hill, A. K. Gupta, B. D. Naab, J. B. Gilroy, R. G. Hicks, C. J. Cramer and W. B. Tolman, Inorg. Chem., 2009, 48, 4514–4523 CrossRef CAS PubMed.
- S. J. Hong, A. K. Gupta and W. B. Tolman, Inorg. Chem., 2009, 48, 6323–6325 CrossRef CAS PubMed.
- R. M. Rush and J. H. Yoe, Anal. Chem., 1954, 26, 1345–1347 CrossRef CAS.
- M.-C. Chang, T. Dann, D. P. Day, M. Lutz, G. G. Wildgoose and E. Otten, Angew. Chem., Int. Ed., 2014, 53, 4118–4122 CrossRef CAS PubMed.
- M.-C. Chang, P. Roewen, R. Travieso-Puente, M. Lutz and E. Otten, Inorg. Chem., 2015, 54, 379–388 CrossRef CAS PubMed.
- J. B. Gilroy, M. J. Ferguson, R. McDonald, B. O. Patrick and R. G. Hicks, Chem. Commun., 2007, 126–128 RSC.
- D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- Y. Ni and J. Wu, Org. Biomol. Chem., 2014, 12, 3774–3791 RSC.
- M.-C. Chang and E. Otten, Chem. Commun., 2014, 50, 7431–7433 RSC.
- S. M. Barbon, P. A. Reinkeluers, J. T. Price, V. N. Staroverov and J. B. Gilroy, Chem. – Eur. J., 2014, 20, 11340–11344 CrossRef CAS PubMed.
- S. M. Barbon, J. T. Price, P. A. Reinkeluers and J. B. Gilroy, Inorg. Chem., 2014, 53, 10585–10593 CrossRef CAS PubMed.
- S. M. Barbon, V. N. Staroverov and J. B. Gilroy, J. Org. Chem., 2015, 80, 5226–5235 CrossRef CAS PubMed.
- M.-C. Chang, A. Chantzis, D. Jacquemin and E. Otten, Dalton Trans., 2016, 45, 9477–9484 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- M.-C. Chang and E. Otten, Inorg. Chem., 2015, 54, 8656–8664 CrossRef CAS PubMed.
- S. M. Barbon, V. N. Staroverov and J. B. Gilroy, Angew. Chem., Int. Ed., 2017, 56, 8173–8177 CrossRef CAS PubMed.
- S. M. Barbon, J. T. Price, U. Yogarajah and J. B. Gilroy, RSC Adv., 2015, 5, 56316–56324 RSC.
- R. R. Maar, S. M. Barbon, N. Sharma, H. Groom, L. G. Luyt and J. B. Gilroy, Chem. – Eur. J., 2015, 21, 15589–15599 CrossRef CAS PubMed.
- R. R. Maar and J. B. Gilroy, J. Mater. Chem. C, 2016, 4, 6478–6482 RSC.
- A. Melenbacher, J. S. Dhindsa, J. B. Gilroy and M. J. Stillman, Angew. Chem., Int. Ed., 2019, 58, 15339–15343 CrossRef CAS PubMed.
- R. R. Maar, R. Zhang, D. G. Stephens, Z. Ding and J. B. Gilroy, Angew. Chem., Int. Ed., 2019, 58, 1052–1056 CrossRef CAS PubMed.
- A. D. Laurent, E. Otten, B. Le Guennic and D. Jacquemin, J. Mol. Model., 2016, 22, 263 CrossRef PubMed.
- M. Hesari, S. M. Barbon, R. B. Mendes, V. N. Staroverov, Z. Ding and J. B. Gilroy, J. Phys. Chem. C, 2018, 122, 1258–1266 CrossRef CAS.
- S. M. Barbon, J. V. Buddingh, R. R. Maar and J. B. Gilroy, Inorg. Chem., 2017, 56, 12003–12011 CrossRef CAS PubMed.
- R. R. Maar, N. A. Hoffman, V. N. Staroverov and J. B. Gilroy, Chem. – Eur. J., 2019, 25, 11015–11019 CrossRef CAS PubMed.
- M. V. Berridge, P. M. Herst and A. S. Tan, Biotechnol. Annu. Rev., 2005, 11, 127–152 CAS.
- S. M. Barbon, S. Novoa, D. Bender, H. Groom, L. G. Luyt and J. B. Gilroy, Org. Chem. Front., 2017, 4, 178–190 RSC.
- A. J. Bard, Electrogenerated Chemiluminescence, Marcel Dekker Inc., 2004 Search PubMed.
- M. Hesari, S. M. Barbon, V. N. Staroverov, Z. F. Ding and J. B. Gilroy, Chem. Commun., 2015, 51, 3766–3769 RSC.
- R. Y. Lai and A. J. Bard, J. Phys. Chem. A, 2003, 107, 3335–3340 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- S. Novoa, J. A. Paquette, S. M. Barbon, R. R. Maar and J. B. Gilroy, J. Mater. Chem. C, 2016, 4, 3987–3994 RSC.
- S. Novoa and J. B. Gilroy, Polym. Chem., 2017, 8, 5388–5395 RSC.
- S. M. Barbon and J. B. Gilroy, Polym. Chem., 2016, 7, 3589–3598 RSC.
- J. S. Dhindsa, R. R. Maar, S. M. Barbon, M. O. Avilés, Z. K. Powell, F. Lagugné-Labarthet and J. B. Gilroy, Chem. Commun., 2018, 54, 6899–6902 RSC.
- J. S. Dhindsa, A. Melenbacher, S. M. Barbon, M. J. Stillman and J. B. Gilroy, Dalton Trans., 2020, 49 10.1039/C9DT03417J.
- M.-C. Chang and E. Otten, Organometallics, 2016, 35, 534–542 CrossRef CAS.
- R. Mondol, D. A. Snoeken, M.-C. Chang and E. Otten, Chem. Commun., 2017, 53, 513–516 RSC.
- R. Mondol and E. Otten, Inorg. Chem., 2018, 57, 9720–9727 CrossRef CAS PubMed.
- A. Van Belois, R. R. Maar, M. S. Workentin and J. B. Gilroy, Inorg. Chem., 2019, 58, 834–843 CrossRef CAS PubMed.
- C. Goze, G. Ulrich and R. Ziessel, J. Org. Chem., 2007, 72, 313–322 CrossRef CAS PubMed.
- W. Schorn, D. Grosse-Hagenbrock, B. Oelkers and J. Sundermeyer, Dalton Trans., 2016, 45, 1201–1207 RSC.
- R. R. Maar, A. Rabiee Kenaree, R. Zhang, Y. Tao, B. D. Katzman, V. N. Staroverov, Z. Ding and J. B. Gilroy, Inorg. Chem., 2017, 56, 12436–12447 CrossRef CAS PubMed.
- R. Mondol and E. Otten, Inorg. Chem., 2019, 58, 6344–6355 CrossRef CAS PubMed.
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed.
- R. Mondol and E. Otten, Dalton Trans., 2019, 48, 13981–13988 RSC.
- R. R. Maar, S. D. Catingan, V. N. Staroverov and J. B. Gilroy, Angew. Chem., Int. Ed., 2018, 57, 9870–9874 CrossRef CAS PubMed.
- M. Yamamura, M. Albrecht, M. Albrecht, Y. Nishimura, T. Arai and T. Nabeshima, Inorg. Chem., 2014, 53, 1355–1360 CrossRef CAS PubMed.
- R. G. Hicks and R. Hooper, Inorg. Chem., 1999, 38, 284–286 CrossRef CAS.
- R. G. Hicks, L. Öhrström and G. W. Patenaude, Inorg. Chem., 2001, 40, 1865–1870 CrossRef CAS PubMed.
- T. M. Barclay, R. G. Hicks, A. S. Ichimura and G. W. Patenaude, Can. J. Chem., 2002, 80, 1501–1506 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |