
Voltage issue of aqueous rechargeable metal-ion batteries
Zhuoxin
Liu†
ab,
Yan
Huang†
c,
Yang
Huang†
 a,
Qi
Yang
b,
Xinliang
Li
b,
Zhaodong
Huang
b and
Chunyi
Zhi
*bd
a,
Qi
Yang
b,
Xinliang
Li
b,
Zhaodong
Huang
b and
Chunyi
Zhi
*bd
aCollege of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, China
bDepartment of Materials Science and Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Hong Kong 999077, China. E-mail: cy.zhi@cityu.edu.hk
cState Key Laboratory of Advanced Welding and Joining, Harbin Institute of Technology (Shenzhen), Shenzhen 518055, China
dShenzhen Research Institute, City University of Hong Kong, Shenzhen 518057, China
First published on 29th November 2019
Abstract
Over the past two decades, a series of aqueous rechargeable metal-ion batteries (ARMBs) have been developed, aiming at improving safety, environmental friendliness and cost-efficiency in fields of consumer electronics, electric vehicles and grid-scale energy storage. However, the notable gap between ARMBs and their organic counterparts in energy density directly hinders their practical applications, making it difficult to replace current widely-used organic lithium-ion batteries. Basically, this huge gap in energy density originates from cell voltage, as the narrow electrochemical stability window of aqueous electrolytes substantially confines the choice of electrode materials. This review highlights various ARMBs with focuses on their voltage characteristics and strategies that can effectively raise battery voltage. It begins with the discussion on the fundamental factor that limits the voltage of ARMBs, i.e., electrochemical stability window of aqueous electrolytes, which decides the maximum-allowed potential difference between cathode and anode. The following section introduces various ARMB systems and compares their voltage characteristics in midpoint voltage and plateau voltage, in relation to respective electrode materials. Subsequently, various strategies paving the way to high-voltage ARMBs are summarized, with corresponding advancements highlighted. The final section presents potential directions for further improvements and future perspectives of this thriving field.
 Zhuoxin Liu | Zhuoxin Liu received his PhD degree from City University of Hong Kong in 2019. Later, he joined Collage of Materials Science and Engineering, Shenzhen University as an assistant professor. His research mainly focuses on flexible aqueous energy storage devices, including supercapacitors and aqueous metal-ion batteries. |
 Yan Huang | Yan Huang received her PhD degree in University of Rochester in 2013. Then she moved to City University of Hong Kong as a post-doctoral fellow, followed by a research fellow. She has been a professor in School of Materials Science and Engineering at Harbin Institute of Technology, Shenzhen since September 2017. Her research interests include flexible and wearable electronic devices, mainly energy storage (batteries, supercapacitors) and conversion (fuel cells). |
 Yang Huang | Yang Huang was awarded his PhD degree from City University of Hong Kong in 2016. He is currently an assistant professor in the College of Materials Science and Engineering, Shenzhen University. His research mainly focuses on low-dimensional materials (e.g. transition metal carbides and nitrides, known as MXenes), and their applications in advanced micro-/nano-electronic devices (e.g. battery and supercapacitor). |
 Chunyi Zhi | Chunyi Zhi got his PhD degree in physics from Institute of Physics, Chinese Academy of Sciences. After that, he started to work as a postdoctoral researcher in National Institute for Materials Science (NIMS) in Japan, followed by a research fellow in International Center for Young Scientists in NIMS and a permanent position in NIMS as a senior researcher. He is currently an associate professor in Department of Materials Science and Engineering, City University of Hong Kong. His research focuses on flexible/wearable energy storage devices, etc. |
1. Introduction
Human activities of economical production and consumption require the use of energy, which affects the environment in aspects of land use, water use, water/air pollution, harm to public health, endangerment to wildlife and loss of their habitat, greenhouse gas emissions, etc.1,2 With our planet facing such energy and environmental challenges, there has been a growing requirement for high-energy energy storage systems (ESSs) in applications of consumer electronics and electrical vehicles where long-lasting power supply is needed, as well as in connection with renewable power plants where clean and sustainable energy resources such as solar, wind, tide and geothermal heat can be penetrated for large-scale stationary energy storage.3–5 Electrochemical EESs, especially rechargeable batteries, possessing a variety of advantageous characteristics including high round-trip efficiency, eco-friendly operation, tunable power and energy output, and simple maintenance, have been regarded as the most competitive candidates for power supply in terms of either portable or large-scale stationary applications.4,6,7Current market of rechargeable batteries is undoubtedly dominated by lithium-ion batteries (LIBs), as they show relatively high energy density, decent cycle stability and good energy efficiency among various battery systems,8 thus they have been serving many applications ranging from small electronics to automotive vehicles. Despite the tremendous success in commercialization, conventional LIBs suffer from issues of cost and safety, where the former mainly attributes to the scarcity of lithium resources and the strict requirement of environment during manufacturing, while the latter originates from the flammable organic electrolyte and the thermal runaway induced by the reactivity of electrodes with electrolytes.9–12 P. Rüetschi proposed the “three E” criteria for the development of battery system for sustainable applications,13i.e., energy (high gravimetric and volumetric energy density), economics (low cost, low maintenance need, long service life), and environment (non-toxic, safe, reliable, recyclable). Apparently, conventional LIBs are no longer an ideal candidate for the blooming flexible and wearable electronics as the involvement of mechanical stresses and deformations during use arouse higher requirement for safety.14–17 Meanwhile, temporally intermittent and spatially dispersed renewable energy sources call for high reliability, absolute safety and cost efficiency, which makes conventional LIBs less feasible for the corresponding large-scale energy storage.5,18
Aqueous rechargeable metal-ion batteries (ARMBs) are promising alternatives that can resolve the major challenges facing conventional LIBs, as water is an ideal electrolyte solvent considering the following aspects: (i) the safety issue of flammable organic electrolytes that cause fire and explosions can be fundamentally avoided, (ii) the strict manufacturing requirements can be exempted, (iii) the cost of water and water-soluble electrolyte salts is relatively low, (iv) the ionic conductivity of aqueous electrolytes can be 2 orders of magnitude higher than that of organic electrolytes, resulting in better round-trip efficiency and rate capability, and (v) water solvent is totally non-toxic and environmentally benign.6,19,20 In these regards, the suitability of ARMBs for portable/wearable applications and for large-scale grid-storage has been revisited.
Dahn and colleagues first proposed an aqueous rechargeable lithium-ion battery (ARLB) where the flammable organic solvent was replaced by water.21 Various ARLBs have been thoroughly studied and substantial improvements in electrochemical performances have been achieved ever since, which has attracted considerable attention of more researchers to aqueous energy storage systems.14,22–27 To date, a variety of aqueous rechargeable batteries based on many other metal ions including naturally-abundant alkali metal ions (Na+ and K+) and multivalent charge carriers (Zn2+, Mg2+, Al3+, etc.) has also been investigated.6,18,28–31 On one hand, the monovalent alkali metal ions of Na and K are more cost-effective in developing low-cost energy storage systems regarding their abundant reserves in Earth's crust. On the other hand, multivalent metal cations can, theoretically, transfer more than one electron and thereby excel in achieving higher specific capacity and energy density. Basically, these ARMBs share similar electrochemistry with their counterparts based on organic electrolytes, where metal ions could intercalate from/into electrode hosts through aqueous media.6,32 Besides, in contrast with traditional aqueous lead acid (Pb–acid), nickel–cadmium (Ni–Cd) and nickel–metal hydride (Ni–MH) batteries, these intercalation-based ARMBs possess the merits of better cycle stability arising from reversible intercalation reactions, and higher power density originating from rapid ion-diffusion pathways offered by the intercalative electrode structures, without the liability of memory effect, poor energy efficiency and environmental pollution.18,33
However, the major obstacle that hampers the practical applications of ARMBs is energy density. Fig. 1 compares the energy density between typical ARMBs and conventional LIBs. It is highly possible that ARMBs replace LIBs in many fields if their energy density can be raised to a considerably high level. There are two factors directly responsible for the energy density of a given battery system, i.e., specific capacity and cell voltage.34 Generally, specific capacity of most active materials in aqueous electrolytes is almost the same as it in organic electrolytes,6,35 while cell voltage is absolutely confined by electrochemical stability window of electrolytes, as the operating voltage of an ARMB cell is decided by the potential difference of redox reactions between cathode and anode, which should lie within the electrochemical stability window of aqueous electrolytes. Whereas, the stable voltage window of aqueous electrolytes is much narrower than that of current organic electrolytes, which is only approximately 1.23 V. Beyond this window, decomposition (electrolysis) of water occurs with the evolution of H2 and O2, disabling the possibility of redox reactions of electrode materials. It should be noted that in practice this voltage window can be wider due to kinetic effects, including overpotentials for H2/O2 evolution at electrode surfaces and the interactions between ions and solvents that increase the difficulty for water decomposition.36,37 Therefore, the choice of electrode materials in aqueous electrolytes is substantially limited, especially when taking side reactions with water or/and oxygen, proton co-insertion, and dissolution of electrode materials into account,6,35,38 thus resulting in a relatively-low cell operating voltage and an inferior energy density.
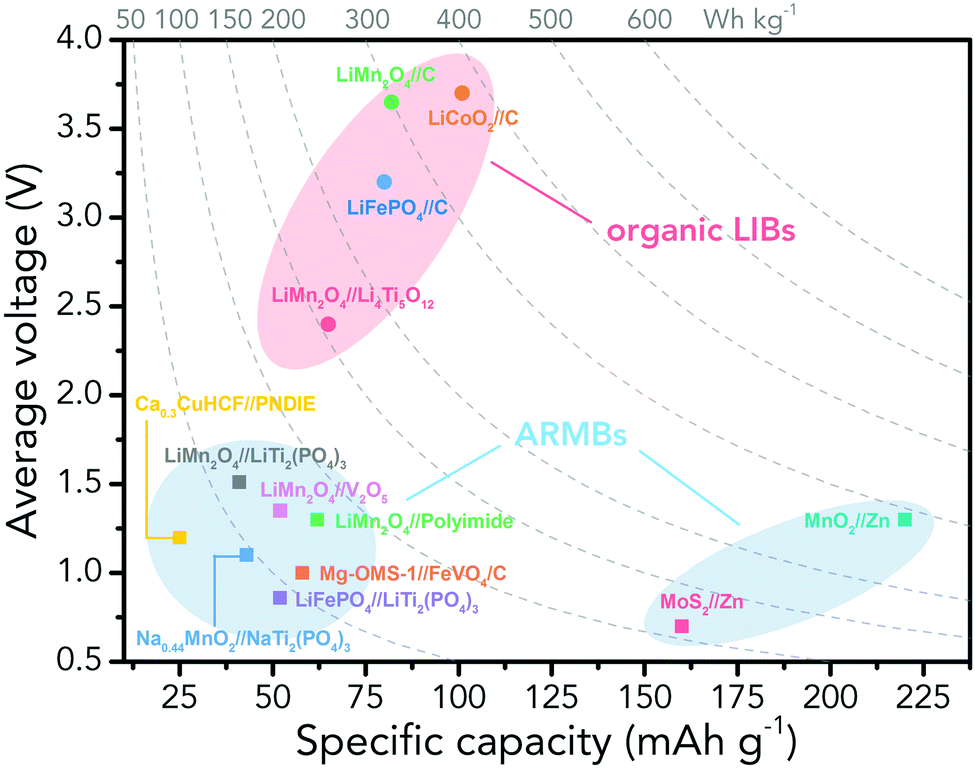 | ||
| Fig. 1 Energy comparison between typical ARMBs and conventional organic LIBs. | ||
Albeit lower in energy density, various ARMB systems have grown into a vigorous force in the field of energy storage due to their irreplaceable characteristics. Normally, ARMBs employ metal-ion intercalative cathodes and aqueous electrolytes with shuttling metal ions.39,40 As for anodes, most pure metals cannot be directly used. Fig. 2 compares different metal anodes in terms of their standard electrode potentials and specific capacities. Obviously, although all these metals are light enough to deliver high capacity, the potentials of most metals (except for zinc) are too low to allow reversible redox reactions before hydrogen evolution, meaning alternative metal-ion intercalative anodes are also needed for these ARMB systems. Therefore, regarding cell voltage, the selection of suitable cathodes and anodes together with electrolyte voltage window are critical in determining battery energy density.
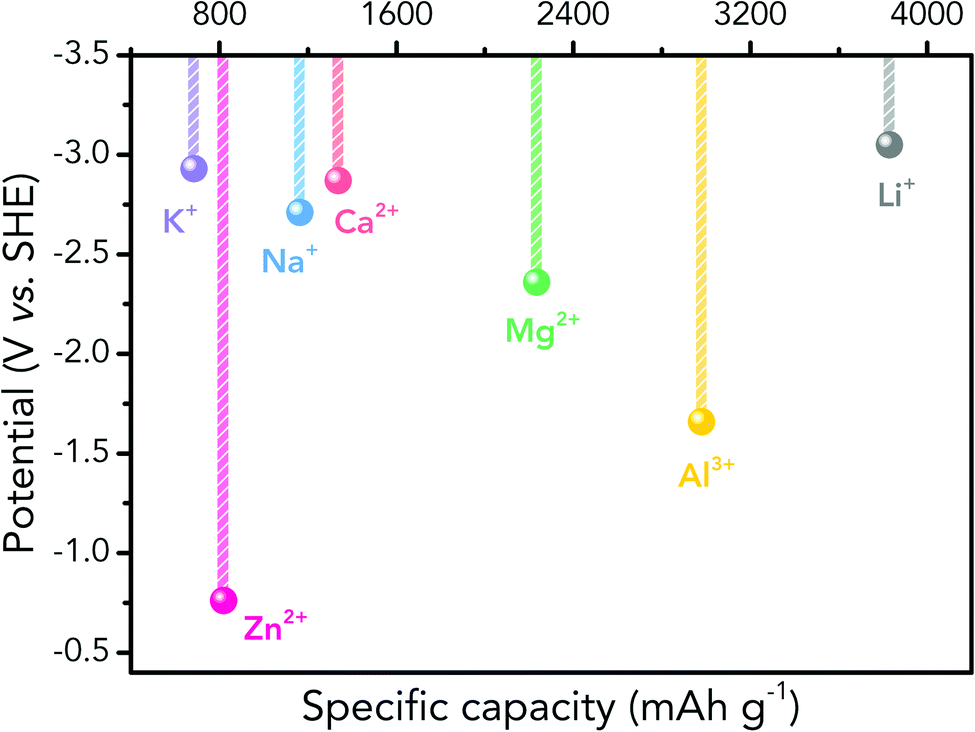 | ||
| Fig. 2 Comparison of standard electrode potentials and theoretical capacities of various metal anodes. | ||
Currently, there are no reviews that have specifically focused on, and comprehensively analysed and summarized, the voltage issue of ARMBs under the purpose of attaining high energy density. We consider it timely and necessary to present a review on this research frontier, to provide recent advances and potential guidance for improving the overall performances of ARMBs. This review article first introduces the fundamental basics of electrochemical stability window of aqueous electrolytes, as well as how it can be further manipulated to benefit the voltage of ARMBs. The following section covers the development of various ARMB systems and compares their voltage characteristics in midpoint voltage and plateau voltage, in relation to respective electrode materials. Then, we go into details of various strategies that pave the way to high-voltage ARMBs, with corresponding advancements highlighted. The final section discusses the challenges and perspectives of this blooming field so that insights for future development of high-energy ARMBs can be rendered.
2. Electrochemical stability window of aqueous electrolytes
In principle, most ARMBs discussed in this article share a “rocking chair” type of working mechanism, where metal ions shuttle between cathode and anode via electrolyte. These metal ions (or together with hydrogen ions) can reversibly intercalate into/de-intercalate from cathode and anode, resulting in a rechargeable energy storage system, as illustrated in Fig. 3a.18 While zinc-ion battery is an exception, as Zn/Zn2+ electrode gives a standard electrode potential of E = −0.76 V, which allows its stripping/plating in many aqueous electrolytes without significant hydrogen evolution. Therefore, another mechanism involving metal stripping/plating at anode side and ion intercalation at cathode side is commonly seen for zinc-ion batteries (Fig. 3b).19,30,31 Moreover, due to the limited choice of electrode materials in aqueous electrolytes, some representative battery systems employing conversion-type electrodes involving metal ion shuttling and conversion reactions without metal ion intercalation/de-intercalation, and polymer electrodes involving doping/de-doping or enolization/de-enolization, are also discussed. It should be also noted that the redox mechanisms of some electrode materials are still under debate, meaning their category as intercalation-type or conversion-type electrode is not clearly identified yet.41–44 Several traditional alkaline battery systems are also involved for comparison.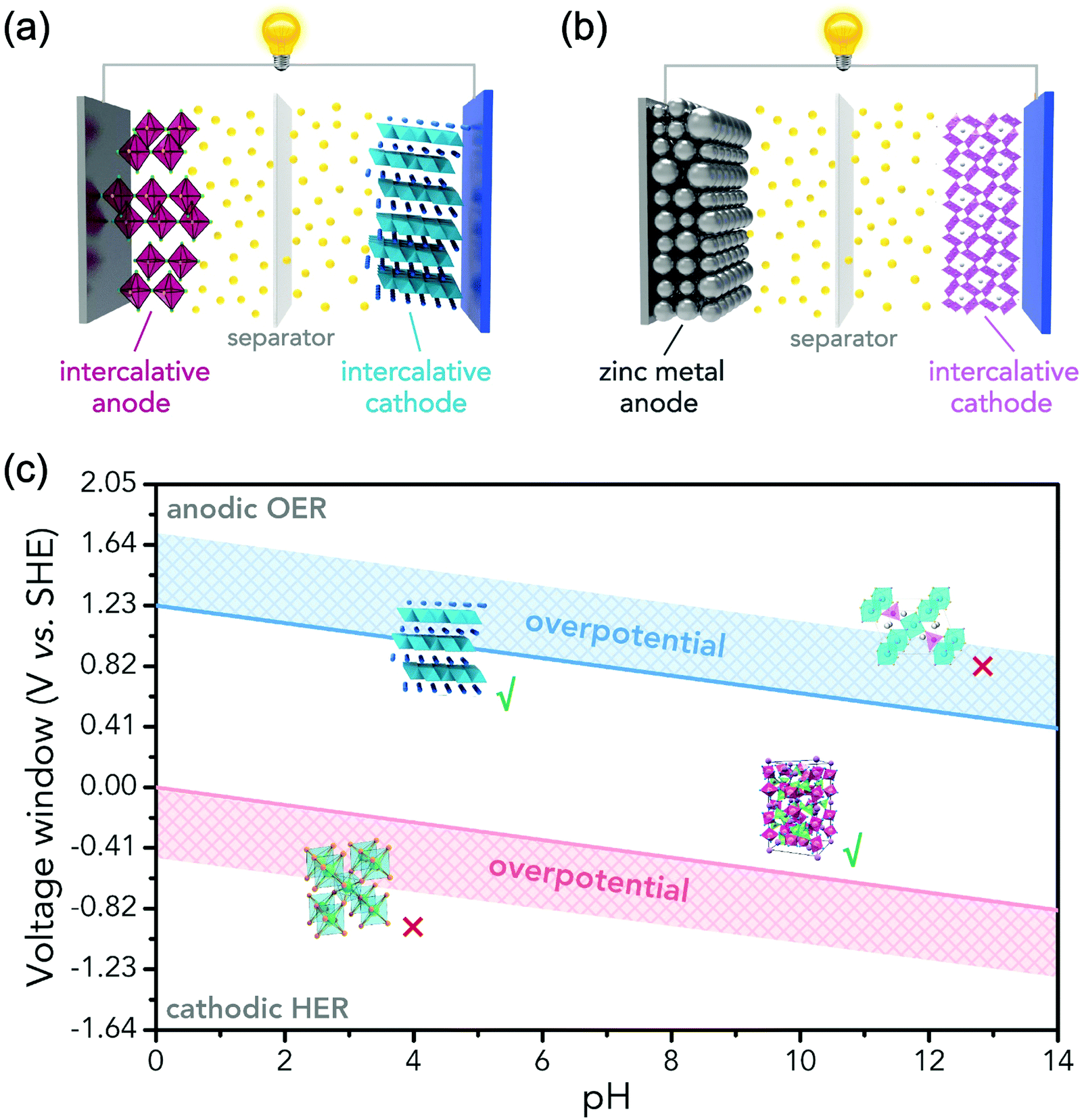 | ||
| Fig. 3 (a) Illustration of the configuration of a battery using metal ion-intercalative cathode and anode. (b) Illustration of the configuration of a zinc-ion battery with zinc stripping/plating at anode and zinc-ion intercalation at cathode. It should be noted that some conversion-type materials and organic compounds have also been utilized to construct high-performance aqueous battery systems. (c) Pourbaix diagram showing the voltage window of aqueous electrolytes. To initiate a reversible ARMB system, working potentials of electrode materials should locate within the window of OER and HER potentials. | ||
To initiate intercalation/de-intercalation of metal ions, or stripping/plating of metal anodes, the electrochemical stability of solvent water should be carefully considered. When applied potential is lower than that of hydrogen evolution reaction (HER), or higher than that of the oxygen evolution reaction (OER), water molecules would be decomposed into hydrogen and oxygen, that's what we call the electrolysis of water. Water decomposition is an irreversible process, which can be depicted by the following equation:
| H2O → H2 + ½O2 |
In a battery system, this process is separated into two parts occurring at cathode and anode, respectively. At cathode side, oxygen evolution reaction (OER) occurs:
| 2OH− → H2O + ½O2 + 2e− (neutral/alkaline) |
| H2O → 2H+ + ½O2 + 2e− (acidic) |
| 2H2O + 2e− → H2 + 2OH− (neutral/alkaline) |
| 2H+ + 2e− → H2 (acidic) |
The thermodynamic potential of water decomposition varies with temperature. The potential difference ΔE0 that triggers the decomposition of water can be thermodynamically determined by the following calculation:
| ΔG = −nFΔE0 |
Water decomposition is pH-dependant, and the potentials where OER and HER should occur as a function of pH value are shown in the Pourbaix diagram (Fig. 3c). The practical electrochemical processes in aqueous electrolytes are very complicated, and actual water decomposition usually requires higher potential than theoretical values. The excess potential needed is termed as overpotential (η). Take HER as an example, which is considered to involve three steps in acidic condition.45 The first step is H+ + e− → Hads, it is called Volmer step where a proton combines with an electron to yield a hydrogen atom absorbed at electrode surface. HER is subsequently facilitated by the following reactions of Tafel step (2Hads → H2) or Heyrovsky step (Hads + H+ + e− → H2), or both. No matter what steps occur, Hads is always involved. The Gibbs free energy of the absorption of hydrogen atom on electrode surface ΔGH should be overcome by applying excess potential, i.e., overpotential. If Hads is strongly bound to electrode, suggesting large and positive ΔGH, the corresponding overpotential to initiate HER is also large. Overpotentials that overcome intrinsic activation barriers at cathode and anode, as well as solution resistance and contact resistance, etc., vary with electrode materials, electrolytes and pH values.6,37,49 Thus, the practical electrochemical stability of water in terms of potential difference is described as:50
| ΔE = ΔE0 + ηc + ηa + ηother |
Therefore, as illustrated in Fig. 3c, only electrode materials with working potentials located within the window of OER and HER potentials are useable in constructing a reversible ARMB system. Electrode materials with working potentials beyond this window are not suitable in aqueous electrolytes, unless special precautions are taken to prevent OER and/or HER, which will be discussed in Section 4. In addition, chemical stability and solubility in water at certain pH values should also be considered when screening electrode materials.6
Albeit wider than 1.23 V, the electrochemical stability window of water is still not comparable to that of organic electrolytes (>4 V),51 which substantially confines the choice of electrode materials. As mentioned before, two factors directly decide the energy density of a battery system: specific capacity and cell voltage. While the specific capacity of many electrode materials changes little in aqueous electrolytes compared to it in organic electrolytes, it is of great significance to increase the cell voltage of ARMBs if targeting at higher energy density for practical applications. Wider electrochemical stability window of water can enable electrode materials with higher or lower working potentials, thus resulting in a larger cell voltage. In the following parts we will discuss the factors that influence the voltage window of water, and how this window can be further manipulated to benefit the voltage of ARMBs.
2.1 pH of electrolytes and local pH change
As illustrated in Fig. 3c, OER and HER potentials are substantially affected by electrolyte pH, thus the voltage window can be manually adjusted to cover higher or lower working potentials of electrode materials, allowing for wider cell voltage and more stable charging/discharging. Literatures have reported the deliberate choosing of acidic or alkaline electrolytes for better battery performances. For example, OER easily occurs for LiMn2O4 cathode for aqueous lithium-ion batteries (ALIBs) when working at high pH values, because OER potential shifts to lower potential in alkaline condition, thus leading to inferior cycle performance.52 LiNi1/3Co1/3Mn1/3O2 (NCM), known as a promising high-voltage cathode for LIBs, should be used in acidic to mild alkaline aqueous electrolytes. If using at the pH of 13, OER occurs at a lower potential (0.75 V vs. SCE, saturated calomel electrode) that overlaps with the working potential of NCM, leading to the insufficient utilization of NCM capacity of lithium storage.53 Wang et al. suggested that the pH of electrolytes for FePO4·2H2O anode in ALIBs should not be too low in order to fully utilize its capacity, otherwise HER cannot be excluded during electrochemical processes.54Regardless of the special purposes to adjust electrolyte pH, the widest voltage window of an aqueous electrolyte is usually achieved at a neutral pH. Yokayama et al. compared the voltage windows of 0.5 M H2SO4, 1.0 M HClO4, 0.33 M H3PO4 and 1.0 M NaOH solutions and found that all the acidic solutions exhibited similar voltage windows except for NaOH, mainly due to the shifting of OER to a lower potential in alkaline solutions. Moreover, all these electrolytes yielded narrower voltage windows compared to 1 M neutral electrolyte.55 Similarly, Wessells et al. reported the electrochemical stability window of 1 M LiNO3 electrolyte gradually decreased with the increase of pH value, which reached the widest at a neutral pH.56 For the anode material of Na2V6O16·0.14H2O used in ALIBs, the pH of Li2SO4 electrolyte was tuned by LiOH. As the pH increases from 7 to 12, the voltage window of electrolyte decreased correspondingly.57 In addition, electrode materials may exhibit different redox reactions that occur at different potentials depending on electrolyte pH. For example, the Co3O4 cathode we developed for zinc-based battery underwent significantly different reactions in mild electrolyte compared to it in alkaline condition, which led to a much higher cut-off voltage of 2.2 V, as well as largely enhanced cycle stability.58 Although Co3O4 is not a intercalative cathode for zinc ions, its pH-dependant behaviour can be a good reference in developing high-voltage ARMBs.
Generally, the pH of aqueous electrolytes adjacent to electrodes will change due to the ions generated or consumed during electrochemical processes. Thus, if cathode and anode are well partitioned in anolyte and catholyte by a separator (which is a commonly adopted configuration in battery devices), the corresponding acidity and alkalinity are both expected to rise more than the case of free mixing of electrolyte without separation, which is ascribed to the increased concentration of H+ and OH− in respective compartment. Such pH change only induces a slight change to the potential of OER and HER, however, if we examine a rather small area that surrounds electrodes in neutral electrolytes, where the proton concentration is very low (ca. 10−7 M at pH = 7), the local pH in the vicinity of electrodes would change drastically due to the produced H+/OH− by OER/HER. As a result, the voltage window of neutral electrolytes is expanded via the shifting of OER and HER occurring upon drastic local pH change. Such local pH change is effectively suppressed by the buffering of H+/OH− in acidic/alkaline electrolytes, therefore acidic/alkaline electrolytes usually exhibit narrower voltage window compared to neutral electrolytes.55
2.2 Electrolyte salts and concentration
At similar concentration and pH, the type of electrolyte salt has moderate influences on the electrochemical stability window of aqueous electrolytes. In the study of Wessells and co-workers, albeit petty, the electrolytes of 1 M Li2SO4, 1 M LiNO3, 1 M NaNO3, and 1 M Mg(NO3)2 exhibit varied voltage windows.56 Similar phenomenon was observed for NaClO4, NaNO3, LiNO3 and lithium bis(trifluorosulfonyl)imide (LiTFSI) solutions with 1 M concentration.55 Whereas, the discovered influences of salt type on electrolyte voltage window is not significant so far, and there’re no developed guidelines in determining the voltage window range of a given salt unless further experimental studies are conducted.Besides the type of electrolyte salt, one effective approach to expand electrochemical stability window of aqueous electrolytes is to increase the concentration of electrolyte salts. This approach can be divided into two strategies, one is to increase salt concentration but still keep water as the major constituent of electrolyte. Wessells et al. found that the concentration of Li2SO4 and LiNO3 has similar influences on both cathodic and anodic processes, leading to a wider full cell voltage with increasing salt concentration.56 A distinct correlation between water concentration and electrolyte voltage window was also observed for a series of aqueous electrolytes,55 of which voltage windows were significantly expanded with the decrease of water concentration (the increase of salt concentration), as reduced water concentration would lead to reduced water activity, resulting in positively-shifted/negatively-shifted potentials of OER/HER and correspondingly decreased reaction rate, although the influences on OER and HER are asymmetrical. Moreover, a saturated aqueous solution of sodium perchlorate was reported to yield a remarkable voltage window as wide as 3.2 V. The authors ascribed the wide voltage window to the increased difficulty in water electrolysis,59 where the O–H bond of water was enhanced due to the loss of hydrogen bond in concentrated electrolytes with increased ions. In addition, concentrated electrolytes may affect the working voltage of certain battery systems. In a study of Zhao and co-workers,60 LiFePO4//LiV3O8 full cell with varied LiNO3 electrolyte concentrations (2 M, 5 M and 9 M) showed different working voltages. The voltage plateau was raised with the increase of LiNO3 concentration, despite the little change in electrode polarization and voltage lag.
The other strategy is to utilize “water-in-salt”-like electrolytes, which was developed by dissolving ultra-soluble salts into water to obtain solutions containing more salt than water in both volume and weight ratio. Due to the absence of free water molecules, “water-in-salt” electrolytes hinder the direct interaction of water molecules with electrode materials. Besides, a protective layer analogous to solid–electrolyte interphase (SEI) in organic electrolytes could form at electrode surface due to the reduction of salt anions. “Water-in-salt” electrolyte was first proposed by Suo et al., the 21 M LiTFSI electrolyte they developed enabled a stable voltage window as wide as 3.0 V, allowing for an ALIB with a cell voltage over 2.3 V.61 Having inherited the foundation of “water-in-salt”, hydrate-melt and “water-in-bisalt” electrolytes were subsequently developed,62,63 targeting at even higher voltage windows. This concept was further extended to other types of ARMBs including sodium-ion battery, potassium-ion battery, magnesium-ion battery, etc.64–67
Increasing the concentration of electrolytes could also extend the working temperature of batteries, as the freezing point of electrolytes could be effectively depressed at high salt concentration.68,69 However, lower temperature may decrease the solubility of electrolyte salts, resulting in salt deposition and reduced concentration. Therefore, the solubility of electrolyte salts at low temperatures should be thoroughly considered. It should also be noted that ionic conductivity of electrolytes usually increases with the increase of salt concentration and reaches its maximum value at optimum salt concentration (Kohlrausch law).70 When salt concentration continues rising, ionic conductivity will pass its peak value and gradually decrease, affecting the overall battery performances. Thus, it is important to avoid unnecessary high salt concentration when expanding electrolyte voltage window.
2.3 Electrolyte additives
It is well established that the decomposition of organic electrolyte components in conventional LIBs result in the formation of a passivation layer at electrode surface, which is called SEI.71–75 This protective SEI extends the voltage window of organic electrolytes and allows for the stable cycling of LIBs over a broad voltage range. Whereas, aqueous electrolytes lack of such organic species essential to the formation of SEI, their voltage window is thus restricted by the thermodynamic potentials of OER/HER. One possible method to stabilize and/or expand their voltage window is to use additives that are capable of forming a SEI-like protective layer on electrode surface. For example, vinylene carbonate (VC) that has been widely employed in organic electrolytes to yield a stable SEI was proved to be an effective additive in aqueous electrolytes.76 The protective film derived from VC to some extent prevented the penetration of water molecules into electrode materials, thus alleviating water decomposition and resulting in better coulombic efficiency and battery cycle stability. It is also reported that OER was effectively suppressed in the aqueous electrolyte containing disodium propane-1,3-disulfonate (PDSS), leading to a wide electrochemical stability window that was positively extended to 1.6 V (vs. Ag/AgCl).77 Although the mechanism remains unclear, the authors suggested that the bulky anions of PDSS served as a barrier between electrolyte and electrode, which played a similar role as SEI and increased the resistance of oxidative decomposition of water. Hou et al. further reported that the addition of sodium dodecyl sulfate (SDS) into aqueous electrolyte resulted in the expanding of voltage window to around 2.5 V, as the hydrophobic layer formed via SDS being electrostatically adsorbed onto electrode surface can effectively suppress OER/HER.782.4 Dissolved oxygen
In a study of Xia and co-workers, dissolved oxygen in aqueous electrolytes was found to be a nonnegligible factor causing electrochemical instability of ALIBs.79 Their investigations revealed that discharged state of ALIB anodes would react with water and oxygen, regardless of pH and electrolyte salt, leading to capacity decay during cycling. Therefore, eliminating the oxygen dissolved in electrolytes as well as sealing cells in the absence of oxygen can notably stabilize and widen voltage window of electrolytes. Many studies reported the improvements in cycle stability by bubbling nitrogen or argon into cells to exclude oxygen before electrochemical tests.60,78,80–82 Chen et al. further demonstrated an ALIB with oxygen self-elimination capability, where the lithiated polyimide anode could be oxidized by the generated oxygen to its pristine state. The sealed battery that overcharged to 2.2 V can still restore its initial capacity and high coulombic efficiency, showing remarkable stability.83 Nevertheless, the strategy of oxygen elimination is only effective in aspect of preventing the shrinkage of voltage window caused by oxygen-involved side reactions, instead of expanding the intrinsic voltage window of a given electrolyte. Besides, data of long-term cycling test indicate that the removal of dissolved oxygen does not completely address the issue of electrochemical instability in aqueous electrolytes.80,842.5 Overpotentials
As aforementioned, overpotentials to overcome intrinsic activation barriers of water decomposition push OER and HER potentials apart, resulting in wider voltage window of aqueous electrolytes. Overpotentials usually vary with a few factors including pH values, electrolytes, electrode materials, etc. Generally, these factors work synergistically to affect overpotentials. In neutral electrolytes, the overpotential of HER could readily reach 200 mV, and the overpotential for OER could extend to over 1.8 V (vs. SHE, standard hydrogen electrode).85–87The electrocatalytic activity of electrode materials on water decomposition is one major concern for constructing high-voltage ARMBs. To ensure high overpotentials, highly catalytic electrode materials should be avoided. MoS2, for example, a material from the transition metal dichalcogenide family, possesses high specific capacity.88,89 While the overpotential of MoS2 in acid electrolytes is only 40–50 mV, leading to a narrow operational voltage window.90,91 When used in neutral or alkaline electrolytes, MoS2 could work at a higher operating voltage as electrode for supercapacitors.92,93 Similar cases are IrO2 and RuO2 electrodes.94 In the cathodes of ALIBs, de-lithiated spinel LiMn2O4 and Li2MnP2O7 are proved to be catalytic on OER, meaning they facilitate oxygen evolution and lower upper voltage limit of electrolyte, thus deteriorating battery performances.95,96
In practice, current collectors are essential components of electrodes, their electrochemical stability should also be thoroughly considered. Among the various current collectors employed in ARMBs, stainless steel was found to be more electrochemically stable than carbon nanotubes (CNTs)-based current collectors,97 and nickel mesh was demonstrated to yield a 2.8 V-wide voltage window.98 As the widely-adopted current collectors for conventional LIB cathodes in industry is aluminium, Kühnel et al. successfully applied aluminium current collectors in concentrated LiTFSI electrolyte, where the aluminium dissolution in aqueous solution was substantially suppressed.99 The high overpotential of aluminium with oxidized passivation layer promised a voltage window of 4.2 V, highlighting the possibility of using a cost-effective and light-weight current collector in ARMBs.
The above parts introduce the fundamental basics of water decomposition, summarize and highlight the factors that affect the electrochemical stability window of aqueous electrolytes. Put together, precise control of electrolyte pH, careful selecting of electrolyte salt and tuning of its concentration, introducing of additives, elimination of dissolved oxygen, and consideration of overpotentials are crucial for stabilizing and expanding voltage window of aqueous electrolytes. Meanwhile, wide voltage window does not directly promise high-voltage ARMBs, it only offers a possibility for pairing more distant cathode and anode regarding their redox potentials. Therefore, the choosing of suitable cathode and anode is the final step that decides the operating voltage of a battery system.
3. Marching of aqueous rechargeable metal-ion batteries: performances, voltage characteristics and comparison
Holding the merits of intrinsic safety, cost efficiency and eco-friendliness, various ARMBs have grown into a vigorous force in the field of energy storage. Regarding cell voltage, some claimed high voltage values actually refer to open circuit voltage, which cannot reflect the real working voltage of an ARMB. Besides, some ARMBs exhibit clear discharge plateau, which extends from the initial voltage drop at the beginning of discharging to the knee of the discharging curve, while some only display capacitor-like sloping discharging profiles. Moreover, it is commonly seen that ARMBs are discharged to a low cut-off voltage to struggle for more capacity, thereby their capacity/energy mainly comes from low-voltage zone, which shows little practicability. To fully reveal voltage characteristics, in this article, we employ the concepts of midpoint voltage and plateau voltage to depict ARMBs. Midpoint voltage is defined as the battery voltage when 50% of the discharging capacity is delivered, and plateau voltage is defined as the voltage at the midpoint of a discharge plateau. As discharge profiles vary with discharge rate, these two voltage parameters are both picked from the lowest reported discharge rate for a certain study. In this section, a series of ARMB systems are introduced in aspects of their electrochemical properties (e.g. specific capacity, cycling stability, energy and power densities, etc.), together with a focus on their voltage characteristics (e.g. midpoint voltage, plateau voltage, and open circuit voltage). Meanwhile, upon a rational comparison within a certain ARMB system, proposed strategies including materials optimization and parameter regulation to further improve their energy storage performances are discussed in detail.3.1 Aqueous lithium-ion batteries
Aqueous rechargeable lithium-ion batteries (ALIBs) were first launched in 1994, which could operate reversibly with a midpoint voltage of around 1.5 V and simultaneously provide an energy density of 75 W h kg−1 based on the mass of both active materials, comparable to both Pb–acid and Ni–Cd batteries.21 As pointed out in this conceptual and pioneering study, the potentials of hydrogen and oxygen evolution should be taken into account as an important factor when selecting both of the electrode materials for ALIBs. In other words, the working potential of electrode materials should be located between OER and HER to avoid the electrolysis of H2O in electrolyte.100 So far, a variety of electrode materials have been developed for high-performance ALIBs. Representative cathode and anode materials with the consideration of their redox potentials are discussed as follows.In order to maximize energy density, working potential of cathode materials should approach OER potential. Therefore, LiMn2O4 is one of the most promising cathode materials in consideration of its suitable Li+ intercalation potential. Typically, it displays two redox pairs at around 0.75 V/0.89 V and 0.86 V/1.05 V (vs. SCE), respectively (Fig. 4a), resulting in a midpoint voltage of 0.77 V (vs. SCE).115 Nanostructures have been introduced to improve its electrochemical performances. For example, Wu's group reported a porous LiMn2O4 synthesized by utilizing polystyrene as sacrificial template (Fig. 4b).25 It exhibited excellent rate performances, maintaining 95% of its initial capacity (118 mA h g−1) at the high current density of 10 A g−1. Its capacity retention reached 93% after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 charge/discharge cycles at 1.0 A g−1, suggesting a lifetime usability without maintenance. The porous nanomorphology and high crystallinity should account for such high performances. They further developed LiMn2O4 nanotubes via a two-step method by employing multiwall CNTs (MWCNTs) as template (Fig. 4c).116 This LiMn2O4 nanotube cathode can be operated at ultrafast second-level charge/discharge rates, and even retained 53.9% of its capacity at 600C. As revealed by literatures, the long-term cycling issue of LiMn2O4 cathode could be well addressed by several effective strategies, including Al doping to suppress Jahn–Teller distortion and stabilize octahedral sites,117,118 designing porous structure to ensure high crystallinity and alleviate Jahn–Teller distortion strain,119 and introducing tubular structure to buffer the strain and stress from Jahn–Teller effects.120 In addition to LiMn2O4, layered lithium oxides, including LiCoO2 and LiNi1/3Co1/3Mn1/3O2 (NCM), which are commonly used in traditional organic LIBs, have also been investigated as cathode materials for ALIBs due to their relatively high redox potentials.70,98,121–125 However, H+ insertion occurs preferentially over Li+ insertion in this type of materials when using LiOH electrolyte.126,127 As indicated by later studies, reversible Li+ insertion into and extraction from LiCoO2 is possible in LiNO3 and Li2SO4 electrolyte.70,98,121,122 It was found that the redox potential of LiCoO2 increased linearly with the electrolyte concentration, suggesting reversible Li+ intercalation instead of H+ insertion in high concentration electrolytes (Fig. 4d). As expected, LiCoO2 cathode can be cycled with relative low polarization in a potential range of 0.55–1.15 V (vs. SHE) and deliver a capacity of 105 mA h g−1 in 5 M LiNO3 with a midpoint voltage of 0.93 V (vs. SHE).121 Raising upper potential limit to 1.4 V, LiCoO2 cathode yielded a capacity of 135 mA h g−1 with a deteriorated coulombic efficiency of 98.5%.70 The decreased reversibility of LiCoO2 above 1.3 V should be attributed to water electrolysis. Besides, with optimized morphology, nano-LiCoO2 achieved a charge/discharge capacity of 147 mA h g−1 at 1000 mA g−1 (7C rate) in 0.5 M Li2SO4 electrolyte.128 As an alternative to LiCoO2 in organic LIBs, NCM has also been studied in aqueous electrolytes. Its electrochemical stability was seriously affective by electrolyte pH, as its oxidation peak shifted obviously to higher potentials in repeated cyclic voltammetry (CV) cycles at the pH of 7 and 9.123,124 Literatures have reported that NCM electrode has relatively low stability under low-pH conditions due to H+ exchange/insertion, which could be further stabilized by polypyrrole (PPy) coating and nano-structuring that favour Li+ intercalation/de-intercalation reversibility and charge transfer kinetics.125,129,130 It could be operated in the potential range of 0–1.05 V (vs. SCE) in 0.5 M Li2SO4 with a midpoint voltage of 0.65 V (vs. SCE). All the above-discussed cathode materials exhibit tilting plateau upon discharging.
000 charge/discharge cycles at 1.0 A g−1, suggesting a lifetime usability without maintenance. The porous nanomorphology and high crystallinity should account for such high performances. They further developed LiMn2O4 nanotubes via a two-step method by employing multiwall CNTs (MWCNTs) as template (Fig. 4c).116 This LiMn2O4 nanotube cathode can be operated at ultrafast second-level charge/discharge rates, and even retained 53.9% of its capacity at 600C. As revealed by literatures, the long-term cycling issue of LiMn2O4 cathode could be well addressed by several effective strategies, including Al doping to suppress Jahn–Teller distortion and stabilize octahedral sites,117,118 designing porous structure to ensure high crystallinity and alleviate Jahn–Teller distortion strain,119 and introducing tubular structure to buffer the strain and stress from Jahn–Teller effects.120 In addition to LiMn2O4, layered lithium oxides, including LiCoO2 and LiNi1/3Co1/3Mn1/3O2 (NCM), which are commonly used in traditional organic LIBs, have also been investigated as cathode materials for ALIBs due to their relatively high redox potentials.70,98,121–125 However, H+ insertion occurs preferentially over Li+ insertion in this type of materials when using LiOH electrolyte.126,127 As indicated by later studies, reversible Li+ insertion into and extraction from LiCoO2 is possible in LiNO3 and Li2SO4 electrolyte.70,98,121,122 It was found that the redox potential of LiCoO2 increased linearly with the electrolyte concentration, suggesting reversible Li+ intercalation instead of H+ insertion in high concentration electrolytes (Fig. 4d). As expected, LiCoO2 cathode can be cycled with relative low polarization in a potential range of 0.55–1.15 V (vs. SHE) and deliver a capacity of 105 mA h g−1 in 5 M LiNO3 with a midpoint voltage of 0.93 V (vs. SHE).121 Raising upper potential limit to 1.4 V, LiCoO2 cathode yielded a capacity of 135 mA h g−1 with a deteriorated coulombic efficiency of 98.5%.70 The decreased reversibility of LiCoO2 above 1.3 V should be attributed to water electrolysis. Besides, with optimized morphology, nano-LiCoO2 achieved a charge/discharge capacity of 147 mA h g−1 at 1000 mA g−1 (7C rate) in 0.5 M Li2SO4 electrolyte.128 As an alternative to LiCoO2 in organic LIBs, NCM has also been studied in aqueous electrolytes. Its electrochemical stability was seriously affective by electrolyte pH, as its oxidation peak shifted obviously to higher potentials in repeated cyclic voltammetry (CV) cycles at the pH of 7 and 9.123,124 Literatures have reported that NCM electrode has relatively low stability under low-pH conditions due to H+ exchange/insertion, which could be further stabilized by polypyrrole (PPy) coating and nano-structuring that favour Li+ intercalation/de-intercalation reversibility and charge transfer kinetics.125,129,130 It could be operated in the potential range of 0–1.05 V (vs. SCE) in 0.5 M Li2SO4 with a midpoint voltage of 0.65 V (vs. SCE). All the above-discussed cathode materials exhibit tilting plateau upon discharging.
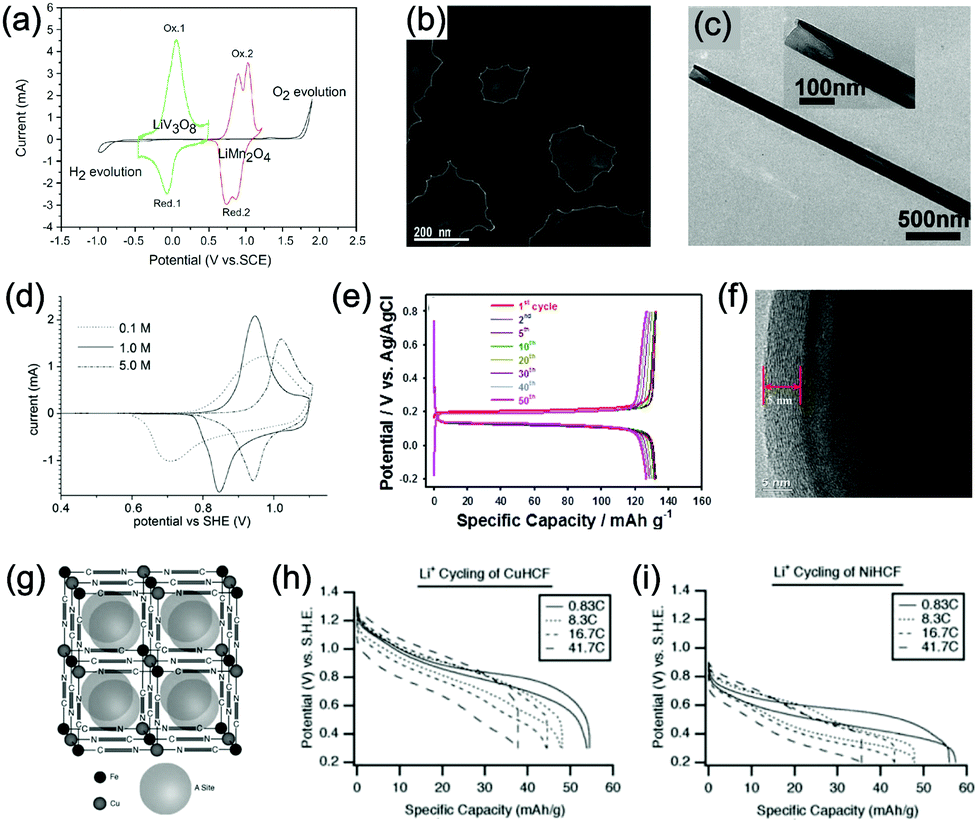 | ||
| Fig. 4 (a) CV curves of LiMn2O4 cathode and LiV3O8 anode in 2 M Li2SO4 aqueous electrolyte at 0.2 mV s−1. Reproduced with permission from ref. 115, Copyright 2007 Elsevier. (b) TEM image of the synthesized porous LiMn2O4. Reproduced with permission from ref. 25, Copyright 2011 Royal Society of Chemistry. (c) TEM image of the synthesized LiMn2O4 nanotubes. Reproduced with permission from ref. 116, Copyright 2013 American Chemical Society. (d) CV curves of LiCoO2 cathode at 0.1 mV s−1 in aqueous LiNO3 electrolytes with different concentrations. Reproduced with permission from ref. 121, Copyright 2009 Elsevier. (e) GCD curves of carbon coated LiFePO4 of different cycles. Reproduced with permission from ref. 131, Copyright 2017 American Chemical Society. (f) TEM image showing the coated carbon layer on LiFePO4. Reproduced with permission from ref. 132, Copyright 2011 Elsevier. (g) The unit cell of Prussian blue crystal structure. Each of the eight sub-cells within the unit cell contains an open “A site”. (h) GCD curves of CuHCF as cathode for ALIB at various rates. (i) GCD curves of NiHCF as cathode for ALIB at various rates. Reproduced with permission from ref. 114, Copyright 2011 The Electrochemical Society. | ||
Another group of promising cathode material is polyanionic compounds. LiFePO4 with olivine structure was reported as the first polyanionic cathode for ALIBs.108 Though Li+ intercalation/de-intercalation in LiFePO4 was proved to be feasible by electrochemical analysis and ex situ characterizations, the capacity of LiFePO4 was far lower than its theoretical value (70 mA h g−1, corresponding to a utilization rate of 41%), and faded rapidly after several charge/discharge cycles. The oxidation of LiFePO4 to FePO4 was not fully reversible, and subsequent reduction resulted in the mixture of LiFePO4 and Fe3O4 that led to poor cyclability.108 LiFePO4 showed a sharp redox pair at 0.12 V/0.32 V (vs. Ag/AgCl), yielding a flat discharge plateau at around 0.12 V (vs. Ag/AgCl) (Fig. 4e).131 It was found out that O2 and OH− in aqueous electrolytes significantly deteriorated the cycling stability of LiFePO4.132 For example, when using 0.5 M Li2SO4 (pH = 7) electrolyte in the presence of O2, obvious capacity degradation was observed during cycling. Transmission electron microscopy (TEM) results further revealed that floccules formed on the surface of LiFePO4 with the existence of O2, which were identified as Fe(OH)2 and Fe2O3. Whereas these impurities were not detected in the absence of O2. Thus, side reactions originated from O2 and OH− should account for the cycling instability of LiFePO4. With a thin carbon coating (about 5 nm) on LiFePO4 (Fig. 4f), the attack from dissolved O2 and OH− could be eased and cycling stability could thus be enhanced. Besides, metal ion doping is an effective strategy to adjust working potentials and to improve the overall electrochemical performances of polyanionic compounds, this includes Mn/Ni co-doping, Fe/Mn co-doping, Ni/Co doping/co-doping, etc.112,113,133–135
PBAs that possess a structure of archetypal hexacyanometalate framework are described in a general formula of AxPR(CN)6, in which nitrogen-coordinated transition metal cations (P) and hexacyanometalate complexes (R(CN)6) form a face-centred cubic framework with large interstitial A sites (Fig. 4g).136–140 The ionic occupancy in A sites varies from 0 to 2 depending on valence changes in one or both of the P and R species.141 These materials have been proposed as potential cathodes for aqueous battery systems as they are able to accommodate a variety of metal ions, including Li+, Na+, K+, and NH4+ in aqueous solutions. In a representative work, copper hexacyanoferrate (CuHCF) and nickel hexacyanoferrate (NiHCF) were prepared via a co-precipitation method and exhibited certain electrochemical activity in 1 M LiNO3 electrolyte (pH = 2).114 Both CuHCF and NiHCF were able to provide a specific capacity of around 56 mA h g−1 at 0.83C, and retained 65% and 58% of the capacity, respectively, at the high current density of 2500 mA g−1 (41.7C), as shown in Fig. 4h and i. However, cycling stability of CuHCF and NiHCF was poor, only 40% of their initial capacities could be maintained. This was due to the dissolution of active materials, which may result from the larger Stokes radius of Li+ (2.4 Å) compared with the radius of connected channels between A sites (1.6 Å) in Prussian blue structure.142,143 The cathodes of CuHCF and NiHCF were operated in the potential range of 0.4–1.3 V and 0.2–0.9 V, with a midpoint voltage of 0.83 V and 0.59 V, respectively (vs. SHE).
Monoclinic structured VO2(B) was used as anode material for the first proposed ALIB in 1994, as it possesses suitable tunnel structure that allows for the rapid intercalation/de-intercalation of Li+,164 and its redox potential is moderately higher than that of HER in an alkaline electrolyte, excluding the potential hydrogen insertion or the involvement of H+/OH− during electrochemical processes.144,165 Whereas, it was revealed that VO2(B) suffered from poor cycling stability in a strong basic electrolyte (at the pH of 11.3) due to its spontaneous dissolution. Though decreasing pH values from 11.3 to 6.2 can increase its discharge capacity and coulombic efficiency, HER occurs when pH goes below 7.165 In one study, it was suggested that the highly concentrated LiTFSI aqueous electrolyte could improve the electrochemical properties of monoclinic VO2, which resulted in an ALIB with a plateau voltage of around 1.35 V (Fig. 5a) and a good capacity retention of 84% after 1000 cycles at 100 mA g−1 when coupling with LiVOPO4 cathode.166 Detailed discussion on concentrated LiTFSI electrolyte is given in Section 4.4.2. Another method to enhance the electrochemical properties of VO2(B) is morphological optimization. Through a hydrothermal route using polyvinyl pyrrolidone as capping reagent, flowerlike VO2(B) assembled by singly-crystalline nanosheets was synthesized (Fig. 5b), which was paired with a LiMn2O4 cathode to yield an ALIB operating in the voltage range of 0.5–1.65 V with a specific capacity of 74.9 mA h g−1 at 60 mA g−1.146 Its midpoint voltage was located at 1.10 V. The high aspect ratio and surface areas derived from the flowerlike nanostructures favour Li+ migration, leading to improved electrochemical kinetics.
 | ||
| Fig. 5 (a) GCD curves of the VO2//LiVOPO4 full cell in the initial cycles at 50 mA g−1. Reproduced with permission from ref. 166, Copyright 2019 Elsevier. (b) TEM image of an individual loose flowerlike VO2 sphere. Reproduced with permission from ref. 146, Copyright 2009 American Chemical Society. (c) GCD curves of the LiNi0.81Co0.19O2 cathode, the LiV3O8 anode, and the corresponding full cell in 1 M Li2SO4 aqueous electrolyte. Reproduced with permission from ref. 167, Copyright 2000 Elsevier. (d) Schematic illustration of the PPy coating ontoLiV3O8 particles. Reproduced with permission from ref. 169, Copyright 2018 Elsevier. (e) CV curves of TiP2O7 at 0.2 mV s−1 in 5 M LiNO3 aqueous electrolyte. (f) CV curves of LiTi2(PO4)3 at 0.2 mV s−1 in 5 M LiNO3 aqueous electrolyte. Reproduced with permission from ref. 170, Copyright 2007 Elsevier. (g) Working mechanism of the ALIB with doping/de-doping anode and intercalation/de-intercalation cathode. Reproduced with permission from ref. 156, Copyright 2008 Wiley-VCH. (h) The cell configuration based on polyimide anode with enolization redox mechanism and an inorganic intercalative cathode. (i) GCD curves of polyimide//LiCoO2 full cell in 5 M LiNO3 aqueous electrolyte at the current density of 100 mA g−1. Reproduced with permission from ref. 157, Copyright 2014 Elsevier. | ||
γ-LiV3O8 was fist investigated by Kohler et al. using 1 M Li2SO4 or LiCl electrolyte.167 The full cell based on γ-LiV3O8 anode and LiNi0.81Co0.19O2 cathode showed sloping discharge profiles, realizing a midpoint voltage of 0.87 V and a total capacity of 45 mA h g−1 (based on the mass of both active materials), as shown in Fig. 5c. However, its cycling stability was far from satisfactory. Further ex situ XRD analysis uncovered the distortion of crystal structure, the additional peaks emerged after 100 cycles suggested the formation of new compounds including LiV2O5 and V2O5. Besides, the decreased (100) peak indicated a deteriorated layered structure of γ-LiV3O8. These irreversible changes in crystal structure impeded Li+ intercalation and accounted for the inferior cycling stability. By modifying morphology, electrochemical performances of γ-LiV3O8 anode could be enhanced. For example, a macaroni-like Li1.2V3O8 nanomaterial with high discharge capacity was fabricated by a solution process using β-cyclodextrin as template.168 The cell based on this nanostructured Li1.2V3O8 anode and a LiMn2O4 cathode could operate at a voltage range of 0.5–1.4 V in 1.0 M Li2SO4 aqueous electrolyte. It exhibited three-staged discharge profiles with a midpoint voltage located at 1.0 V. Specific capacities were 189, 140 and 101 mA h g−1 (based on the mass of anode material) at 0.1, 0.5, and 1.0C, respectively. Such increased capacities were attributed to the porous structure of Li1.2V3O8 anode that facilitated Li+ intercalation. In a more recent study from our group, PPy coating was employed to improve the cycling stability of LiV3O8 (Fig. 5d), which achieved a capacity retention of 73.2% after 100 cycles at 0.5 A g−1, whereas the pristine uncoated LiV3O8 only retained 30.6% of its original capacity.169 It was also discovered that PPy coating suppressed the hydrogen evolution at LiV3O8 side, as the conductive PPy layer facilitated ion and charge transfer, enabling the predominant reaction of Li+ intercalation over HER. Literature has also shown that increasing electrolyte concentration and eliminating dissolved oxygen resulted in a consistent capacity during 100 charge/discharge cycles for the ALIB consisting of LiV3O8 anode and LiFePO4 cathode, which operated in the narrow voltage range of 0–0.8 V with a flat but low plateau voltage at 0.23 V.60 The enhanced cycling stability should attribute to reduced electrochemical resistance and less polarization of the optimized aqueous electrolyte. In general, LiV3O8 can work as a reliable anode material for ALIBs when rational precautions are taken to tackle its problematic instability. Similarly, V2O5 showed poor cyclability in aqueous media due to dissolution issue and crystal structure changes.107,151,160 To enhance its reversibility in aqueous electrolytes, polymer coatings of polyaniline (PANI) and PPy was introduced, resulting in full cells with notably improved cycling stability, delivering midpoint voltages of 0.80 V and 1.05 V when coupled with LiNi1/3Mn1/3Co1/3O2 and LiMn2O4, respectively.107,151
In addition to oxides, polyanionic compounds have been investigated as anode materials for ALIBs due to their flat charge/discharge plateau and proper redox potentials. In 2007, the redox potentials of two polyanionic compounds, namely, pyrophosphate TiP2O7 and NASICON-type LiTi2(PO4)3, were systematically investigated in 5 M LiNO3 aqueous solution.170 CV curves of TiP2O7 and LiTi2(PO4)3 clearly indicated that they could act as anode materials for ALIBs without obvious hydrogen evolution, since their redox reactions occur at the potentials of approximately −0.35 and −0.45 V (vs. SHE), respectively, which were moderately higher than that of HER (Fig. 5e and f). The resultant TiP2O7/LiMn2O4 and LiTi2(PO4)3//LiMn2O4 full cells delivered specific capacities of around 42 and 45 mA h g−1 (based on the mass of both active materials) with high midpoint voltages of 1.38 and 1.47 V, respectively. However, both batteries decayed rapidly due to structural decomposition and morphological changes.159 Several effective approaches have been proposed to further improve the electrochemical performances of these polyanionic anodes, including porous structuring, carbon coating and electrolyte optimization (pH adjust and oxygen elimination), which realized 80–90% capacity retention after several hundreds of charge/discharge cycles or even over 1000 cycles.101,153,163
Considering their redox potential position of −0.8 to 0.3 V (vs. SHE) and the intrinsic deficiency of lithium ion, organic polymer materials mainly serve as anodes for ALIBs. For example, A cell consisting of a PPy anode and a spinel LiMn2O4 cathode could be operated reversibly within a voltage range of 0–1.6 V in a saturated Li2SO4 aqueous electrolyte (pH = 7), delivering a midpoint voltage of around 0.44 V.156 Different from other anode materials, PPy realises energy storage through a reversible doping and de-doping mechanism (Fig. 5g) at an average redox potential of −0.27 V. Moreover, unlike many vanadium-based anodes, PPy does not dissolve in aqueous solution, and its doping/de-doping mechanism is highly reversible. Therefore, it should exhibit good stability during repeated electrochemical processes. As revealed by Wang et al., the PPy//LiCoO2 cell with a midpoint voltage of 0.80 V showed no obvious degradation after 120 charge/discharge cycles.171 Another example of organic polymer anode material is polyimide with conjugated carbonyl groups based on 1,4,5,8-naphthaleneteracarboxylic dianhydride moiety.157 Polyimide undergoes an enolization process to combine with Li+ and simultaneously redistribute charges within the conjugated aromatic molecule (Fig. 5h), which is significantly distinguished from Li+ intercalation/de-intercalation mechanism or doping/de-doping mechanism. Polyimide anode could provide a high discharging/charging capacity of 192 mA h g−1/160 mA h g−1 with midpoint discharging/charging voltage of −0.50 V/−0.39 V (vs. SCE). Assembled with a LiCoO2 cathode in 5 M LiNO3, the resultant ALIB exhibited a discharge capacity of 71 mA h g−1 and a specific energy of 80 W h kg−1 at 100 mA g−1 (based on the mass of both active materials), along with plateau voltage of 1.10 V (Fig. 5i). This ALIB also displayed a good cyclability by maintaining 80% of its original capacity after 200 charge/discharge cycles, which was attributed to the unique Li+ diffusion-independent enolization mechanism that avoided strains and structure distortions during repeated electrochemical processes.
For ALIBs, the options of anode materials from traditional organic LIBs are less compared to cathode materials, as most anode materials used for traditional LIBs possess redox potentials significantly lower than that of HER. Besides, many organic compounds used in traditional LIBs cannot be directly utilized as electrodes in ALIBs because of their instability in aqueous electrolytes.172 Thereby, researchers have devoted tremendous efforts in developing anode materials with suitable redox potentials for aqueous electrolytes. Redox potentials of representative cathode and anode materials for ALIBs are summarized and compared in Fig. 6. The notably higher redox potential of LiMn2O4 makes it the most widely-adopted cathode for high-energy ALIBs. As for anode materials, attention should be paid to the low-potential NASICON-type LiTi2(PO4)3 and its fluoride LiTi2(PO4)2.88F0.12. Although some vanadium oxides show low redox potentials, their toxic synthesis and intrinsic instability in water hinder their further applications. Besides electrochemical potentials, extensive efforts are also needed to better address the issues of unsatisfactory cyclability and insufficient utilization of theoretical capacity of current electrode materials, as well as to explore new families of high-performance electrode materials.
 | ||
| Fig. 6 Comparison of redox potentials of representative electrode materials for ALIBs. Red-colour columns and blue-colour columns represent cathodes and anodes, respectively. | ||
3.2 Aqueous sodium-ion and potassium-ion batteries
Aqueous sodium-ion (ASIBs) and aqueous potassium-ion batteries (AKIBs) with similar monovalent ion intercalation/de-intercalation mechanisms to ALIBs have been developed as promising alternative energy storage systems due to the easy accessibility of sodium/potassium resources, which could be better choices for grid-scale energy storage that imposes higher requirement on cost. Their electrode materials are also quite limited considering the narrow electrochemical stability window of water, and they usually exhibit lower energy density compared to ALIBs due to the higher atomic mass of sodium/potassium. In this section, the development of ASIBs and AKIBs will be discussed in terms of their electrochemical performances and voltage characteristics based on cathode and anode materials, which mainly include spinel-structured materials, polyanionic compounds, PBAs, etc.Transition metal oxides can be readily synthesized with controllable morphology and high electrochemical activity, therefore have attracted tremendous attention in battery research.173 As a representative, MnO2 has been demonstrated the capability of hosting sodium ions.174 For example, an ASIB based on cubic spinel-type λ-MnO2 cathode delivered a high capacity up to 78 mA h g−1 and a high midpoint voltage of 1.35 V when coupled with an activated carbon anode.175 Minakshi et al. reported γ-MnO2 as cathode for Na+ intercalation/de-intercalation in 7 M NaOH aqueous electrolyte.176 A high specific capacity of 225 mA h g−1 was obtained accompanied with a midpoint voltage of 1.32 V (vs. Zn/Zn2+). They confirmed the intercalation of sodium ions into MnO2 structure via XRD and proton induced X-ray emission analysis. Qu et al. examined the electrochemical behaviour of V2O5·0.6H2O in aqueous electrolytes containing different alkali metal sulphates.177 Galvanostatic charge/discharge (GCD) curves exhibited an capacitor-like linear relationship with time between the potential range of 0–1 V (vs. SCE) with midpoint voltages at around 0.4 V (vs. SCE), and the specific capacities for Li+, Na+, and K+ storage were 37, 43 and 50 mA h g−1, respectively. Their study revealed that the intercalation/de-intercalation of K+ into/from the interlayered space of V2O5·0.6H2O was more facile compared to Li+ and Na+, which may be attributed to the moderate charge density of K+. Another attractive category of metal oxide for ASIBs and AKIBs is MxMnO2 (M = Na, K), which possesses various crystal structures and properties depending on sodium/potassium ratio. Among them, Na0.44MnO2 with three-dimensionally interconnected S-shaped Na tunnels has been widely studied, and it was found that Na+ ions could reversibly intercalate/de-intercalate into/from Na0.44MnO2 host at three redox potentials (0.05, 0.27, and 0.50 V vs. SCE) in the composition range of Na0.44−xMnO2 (0.25 < x < 0.44).178 Capacity and stability of Na0.44MnO2 can be further improved by heteroatom substitution, as reported by Wang et al.179 The Ti-substituted Na0.44MnO2 cathode exhibited a reversible capacity of 76 mA h g−1 at 2C and a high rate performance up to 8C with 60 mA h g−1 retained in a full cell coupled with NaTi2(PO4)3/C anode, which was superior to pristine Na0.44MnO2 (30–40 mA h g−1). This ASIB exhibited a good midpoint voltage of around 1.13 V. As for KxMnO2, Liu et al. discovered the charge storage mechanism involved K+ extraction in the first charge process and reversible Na+ intercalation/de-intercalation in subsequent cycles.180 The full cell assembled by coupling K0.27MnO2 cathode with NaTi2(PO4)3 anode delivered a specific capacity of 68.5 mA h g−1 at 0.2 A g−1 in the voltage range of 0–1.6 V with a midpoint voltage of 0.71 V (Fig. 7a–c). Such ASIB was further improved by designing a hollow nanostructure for K0.27MnO2 to facilitate electron/ion transport, which increased the specific capacity to 84.9 mA h g−1 at 150 mA g−1 and 56.6 mA h g−1 at 600 mA g−1 (Fig. 7d and e).181
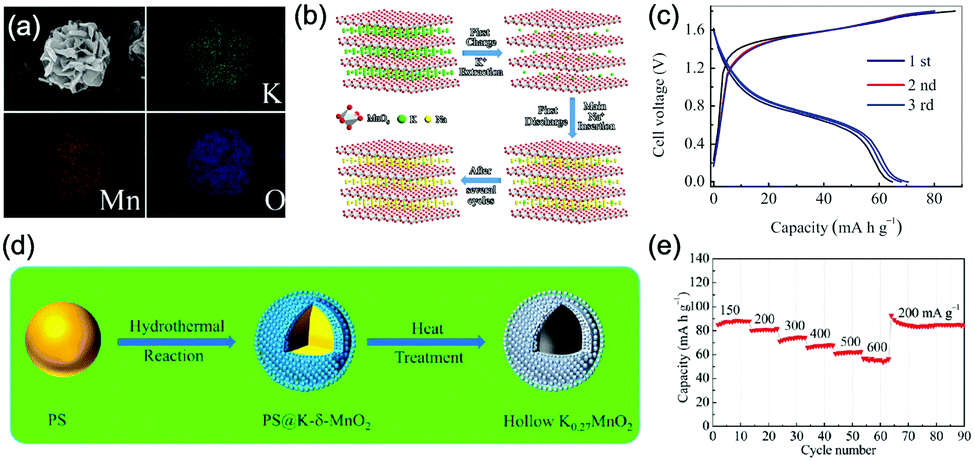 | ||
| Fig. 7 (a) EDS elemental mapping images of K0.27MnO2. (b) Schematic illustration of the electrochemical mechanisms of the layered K0.27MnO2 cathode. (c) GCD curves of the NaTi2(PO4)3//K0.27MnO2 full cell at 200 mA g−1. Reproduced with permission from ref. 180, Copyright 2014 Elsevier. (d) Schematic illustration of the synthesis of the K0.27MnO2 with hollow nanostructure. (e) Rate capacity of the NaTi2(PO4)3//K0.27MnO2 full cell at various current densities. Reproduced with permission from ref. 181, Copyright 2016 American Chemical Society. | ||
Polyanionic compounds contain a series of tetrahedron anion units (XO4)n− or their derivatives (XmO3m+1)n− (X = S, P, Si, As, Mo, or W) with strong covalent-bonded MOx polyhedra (M represents a transition metal). The (XO4)n− unit not only allows fast conduction of alkali ions in an open framework, but also stabilizes the redox potentials of transition metals. Therefore, a series of polyanionic compounds including NASICON-type material, phosphate, pyrophosphate, fluorinated pyrophosphate, olivine-type material, etc., have been developed as cathode materials for ASIBs. For example, the representative NASICON-type cathode Na3V2(PO4)3 (NVP) has large tunnels to accommodate sodium ions. Song et al. investigated the electrochemical behaviour of NVP cathode in aqueous solutions of 1 M Li2SO4, 1 M Na2SO4, and 1 M K2SO4, respectively. Its storage of Na+ was promising, which exhibited a capacity of around 50 mA h g−1 (209 F g−1) at 8.5C with a flat discharge plateau at around 0.4 V (vs. SCE) (Fig. 8a), whereas its Li+ and K+ storage capability was far from satisfactory.182 The intercalation of Li+ into NVP framework is irreversible due to the larger hydrated ionic radius of Li+, and K+ tends to form electric double-layer capacitance on NVP surface rather than intercalate into the framework because of the weak solvation and high conductivity of K+. Qin et al. reported a fluoridised NASICON-type compound of NaVPO4F (NVPF) for ASIBs, which showed shifted potentials with two pairs of redox peaks at 0.26 V/0.20 V and 0.79 V/0.77 V (vs. SCE) in 5 M NaNO3 aqueous electrolyte, promising higher cell voltage compared to NVP (Fig. 8b).157 Another widely studied NASICON-type compound is iron-based pyrophosphate, which includes Na2FeP2O7, LiFePO4, NaFePO4, etc. Their redox potentials approximately locate in the range of 0.45–0.72 V (vs. SCE), suggesting a moderate full cell working voltage of 0.95–1.22 V if coupling with a −0.5 V (vs. SCE) anode.
 | ||
| Fig. 8 (a) Initial GCD curves of Na3V2(PO4)3 cathode at various rates. Reproduced with permission from ref. 182, Copyright 2014 Wiley-VCH. (b) CV curves of polyimide anode and NaVPO4F cathode in 5 M NaNO3 aqueous electrolyte at 0.1 mV s−1. Reproduced with permission from ref. 157, Copyright 2014 Elsevier. (c) The illustrative structure of CuNiHCF cell containing a framework of hexacyanoferrate groups linked by nitrogen coordinated P site transition-metal ions of Cu and Ni. (d) TEM image revealing the 20–50 nm particles of CuNiHCF. Reproduced with permission from ref. 186, Copyright 2012 American Chemical Society. (e) Rietveld refinement patterns of X-ray diffraction data of K2FeIIFeII(CN)6·2H2O nanocubes. (f) FESEM image of K2FeIIFeII(CN)6·2H2O nanocubes. (g) The first discharge curves of K2FeIIFeII(CN)6·2H2O cathode at various current densities showing two flat plateaus. Reproduced with permission from ref. 66, Copyright 2017 American Chemical Society. (h) GCD curves of the NaTi2(PO)3/C anode, K2NiFe(CN)6·1.2H2O cathode and the corresponding full cell. Reproduced with permission from ref. 187, Copyright 2018 Wiley-VCH. | ||
As aforementioned, the ionic radii of Na+ and K+ are larger than that of Li+, the choice of host materials for their reversible intercalation/de-intercalation is thus more limited. Therefore, PBAs with open framework that are suitable to host Na+ and K+ are urgently needed. Their large ionic channels, compositional and electrochemical tenability are highly potential in both Na+ and K+ storage.183 The utilization of PBA frameworks for selective cation insertion could date back to 1970s–1980s, when a number of thin-film PBA frameworks (KM[Fe(CN)6], M = Co, Ni, Cu, Cr, and Ti) were proved electrochemically active in aqueous electrolytes.184 In recent years, a series of high performance PBA cathodes for ASIBs and AKIBs were reported by Cui's group. They first proposed a NiHCF with a chemical composition of K0.6Ni1.2Fe(CN)6·3.6H2O for reversible intercalation/de-intercalation of Na+ and K+ in mildly acidic NaNO3 or KNO3 electrolyte.137 Based on the redox couple of Fe(CN)64−/Fe(CN)63−, the NiHCF cathode delivered a specific capacity of around 60 mA h g−1 at 0.83C with midpoint voltages at 0.59 V and 0.69 V (vs. SHE) for Na+ and K+ intercalation, respectively. This cathode also exhibited an extremely stable cycle performance with charge/discharge cycles up to 5000 at 8.3C for both Na+ and K+ storage, which benefited from the intrinsic rigid metal–organic framework. They also synthesized a low-cost PBA by replacing the nitrogen-coordinated Fe3+ with Cu2+ through a controlled co-precipitation method.185 The CuHCF delivered a similar capacity for K+ storage compared to NiHCF, but achieved a higher midpoint voltage at 0.95 V (vs. SHE). The low-strain structure of CuHCF also contributed to an excellent cycling performance, which obtained 83% of the original capacity after 40000 deep discharge cycles. Furthermore, the redox potential of PBA can be precisely tuned by adjusting Cu/Ni ratio in the framework of a Cu–Ni alloyed PBA.186 As revealed, Cu and Ni could form a fully miscible solution at particular sites in the framework without perturbing the structure (Fig. 8c and d). The CuNiHCF integrated the high rate, long cycle life of both NiHCF and CuHCF cathodes with controllable redox potentials. The highest midpoint voltages achieved for K+ and Na+ storage was around 0.91 V and 0.88 V, respectively. However, the sodium-deficient nature of NiHCF and CuHCF may restrict their practical application due to the need for a sodiated anode. Thus, a cathode of high K+-content potassium iron(II) hexacyanoferrate dihydrate (K2FeIIFeII(CN)6·2H2O) nanocubes was synthesized for AKIBs (Fig. 8e and f).66 The K2FeIIFeII(CN)6·2H2O cathode can provide two-electron transfer per molecular unit, thereby delivering remarkable capacities in aqueous electrolytes (up to 120 mA h g−1). It exhibited two discharge plateaus at 0.85 V and 0.20 V (vs. Ag/AgCl), respectively (Fig. 8g). Another potassium-rich mesoporous nickel ferrocyanide(II) (K2NiFe(CN)6·1.2H2O) to host K+ was synthesized recently, which realized impressive rate performances.187 Its single charge/discharge could be completed within 4.1 s, with a capacity of 42 mA h g−1 delivered. A flat discharge plateau at 0.51 V could be observed, contributing to a plateau voltage of 1.35 V for the full cell coupled with NaTi2(PO)3/C anode (Fig. 8h). It should be mentioned that this PBA type cathode was also capable of hosting bivalent magnesium and trivalent aluminium ions, showing high versatility. In addition, many other PBAs including FeHCF (with two discharge plateaus at 0.8 V and 0.1 V (vs. Ag/AgCl)),188 NaCoHCF (with two discharge plateaus at 0.90 V and 0.38 V (vs. Ag/AgCl)),189 NaMnHCF (with two redox pairs at 0.58 V/0.39 V and 1.17–1.23 V/1.00 V (vs. Ag/AgCl)),190 VHCF (with a midpoint voltage of 0.84 V (vs. Ag/AgCl)),191etc., have been investigated for sodium storage, showing high diversity of this open-framework materials.
Compared with inorganic materials, polymers represent a promising candidate as battery electrodes because of their merits of metal-free nature, flexibility, light weight, and cost-effectiveness. More importantly, their structural diversity and capability of molecular functionalization enable subtle control of their redox properties and thus providing many opportunities in improving cell voltage and battery performances.192,193 Whereas, although organic polymer electrodes have been widely investigated in ALIBs,194 their applications in ASIBs and AKIBs are still lacking. Koshika et al. reported a poly(2,2,6,6-tetramethylpiperidinyloxy-4-yl vinylether) (PTVE) cathode for sodium storage with one-electron reaction in 0.1 M NaCl aqueous solution.195 The PTVE cathode exhibited a flat discharge plateau at 0.73 V (vs. Ag/AgCl) with a reversible capacity of 130 mA h g−1 at 60C, and maintained 75% of the initial capacity after 1000 cycles. Besides, PPy-based cathodes were also discovered for ASIBs, whose redox potential was located between 0 and 0.8 V (vs. Ag/AgCl), and maintained a high specific capacity of around 110 mA h g−1 for hundreds of cycles.196,197 It is worth mentioning that the energy storage mechanism of PTVE and PPy is based on the redox reaction of nitroxide radicals or conjugated structure, which does not involve Na+ intercalation/de-intercalation, meaning that these polymers can readily be utilized for other alkali metal-ion battery systems.
In early works, low-cost activated carbon (AC) has been utilized as anode material for ASIBs due to its excellent stability in aqueous media.175 It was demonstrated that high surface area activated carbons or surface modified carbons exhibited a pseudocapacitive and possible hydrogen storage behaviours between −1.0 and 0.3 V (vs. Hg/Hg2SO4). However, its specific capacity was just around 50 mA h g−1.
The research of metal oxides as anode has been conducted by Wu's group, where PPy-coated MoO3 nanobelts for sodium storage were developed.198 This anode showed two pairs of redox potentials located at −0.25 V/−0.08 V and −0.49 V/−0.34 V (vs. SCE) with a reversible capacity of 33 mA h g−1. When coupled with Na0.35MnO2, the full ASIB yielded sloping discharge profiles with a midpoint voltage of 0.6 V. While its capacity is still not satisfactory, which is much lower compared to many anodes of ALIBs, thus having urged researchers to pursue anodes with higher capacities for high-performance ASIBs and AKIBs. Deng et al. developed a 1D-nanostructured sodium vanadium oxide (Na2V6O16·nH2O, NVO) through a hydrothermal method (Fig. 9a).199 The layer-structured NVO anode enabled Na+ intercalation/de-intercalation with a solid-state diffusion coefficient of about 10−14 cm S−1. More importantly, it delivered an initial specific capacity of 123 mA h g−1 with a midpoint voltage at −0.41 V (vs. SCE). The major drawback of this material is that its capacity faded quickly in subsequent charge/discharge cycles due to irreversible structural change upon sodium intercalation/de-intercalation, which is a common issue for vanadium oxides.
 | ||
Fig. 9 (a) TEM image of the 1D nanostructured Na2V6O16·nH2O. Reproduced with permission from ref. 199, Copyright 2014 Elsevier. (b) GCD curves of the NaTi2(PO4)3//Na cell in non-aqueous electrolyte and the NaTi2(PO4)3//Zn cell in aqueous electrolyte at 2.0 mA cm−2. Inset shows the cycle stability in aqueous electrolyte. Reproduced with permission from ref. 201, Copyright 2011 The Electrochemical Society. (c) Working mechanism of the MnII–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–MnIII/II//CuII–NC–FeIII/II ASIB based on Na+ intercalation/de-intercalation. The anode and the cathode have the same open framework crystal structure. (d) GCD curves of the MnII–NC–MnIII/II//CuII–NC–FeIII/II full cell at various C rates. Reproduced with permission from ref. 139, Copyright 2014 Springer Nature. (e) The reduction potentials of quinone electrodes measured by CV test vs. electrolyte pH values. It can be seen that PPTO could be used as anode for various ARMB systems. Reproduced with permission from ref. 205, Copyright 2017 Springer Nature. C–MnIII/II//CuII–NC–FeIII/II ASIB based on Na+ intercalation/de-intercalation. The anode and the cathode have the same open framework crystal structure. (d) GCD curves of the MnII–NC–MnIII/II//CuII–NC–FeIII/II full cell at various C rates. Reproduced with permission from ref. 139, Copyright 2014 Springer Nature. (e) The reduction potentials of quinone electrodes measured by CV test vs. electrolyte pH values. It can be seen that PPTO could be used as anode for various ARMB systems. Reproduced with permission from ref. 205, Copyright 2017 Springer Nature. | ||
NASICON-type NaTi2(PO4)3 (NTP) is a representative phosphate anode material for ASIBs due to its open framework for sodium storage.200 Park et al. studied the electrochemical behaviour of NTP in both organic and aqueous electrolyte.201 It delivered a specific capacity of 120 mA h g−1 in organic electrolyte with a midpoint voltage of 2.1 V (vs. Na+/Na), and a specific capacity of 123 mA h g−1 in aqueous electrolyte with a midpoint voltage of −0.77 V (vs. Ag/AgCl) (Fig. 9b), corresponding to the redox conversion between Ti3+ and Ti4+. The cyclability of NTP anode was better in aqueous electrolyte, whereas, it still faded quickly within 30 charge/discharge cycles, which is similar to that of LiTi2(PO4)3. Xia's group revealed that the capacity fading of aqueous batteries should be ascribed to side reactions between discharged electrodes with water and/or oxygen in aqueous electrolytes.163 Eliminating dissolved oxygen, adjusting electrolyte pH and carbon coating are effective approaches to improve the cycling stability of NTP. For example, a microwave-assisted method was employed to synthesize carbon-coated NTP, which exhibited an improved capacity retention of 86% after 100 cycles at 1C in 1 M Na2SO4 aqueous electrolyte.202 Frogspawn-structured hierarchical porous NTP/C arrays were also reported for ASIBs with a superior cycle life up to 2000 cycles at 20C.203
As discussed in Section 3.2.1, PBAs have been used as cathode materials for ASIBs due to their relatively high redox potentials of Fe and other N-coordinated metal ions. Such unique open framework-structured materials, however, have seldom been reported as anode for sodium/potassium storage. The first study of utilizing PBA as anode for ASIB was proposed by Pasta et al.139 Aiming to develop an anode with sufficiently low potential while avoiding triggering HER, they screened a series of PBAs including CrII–NC–MnII/I (−0.312 V vs. SHE), MnII–NC–MnIII/II (0.052 V vs. SHE), FeII–NC–MnIII/II (0.075 V vs. SHE) and CrII–NC–MnIII/II (0.352 vs. SHE), and selected MnII–NC–MnIII/II in consideration of stability and crystallinity. The prepared MnHCF anode exhibited a specific capacity of 57 mA h g−1 at 50C and yielded a full cell midpoint voltage of 0.88 V when paired with CuII–NC–FeIII/II cathode (Fig. 9c and d). This full ASIB showed negligible capacity loss over 1000 cycles in 10 M NaClO4 aqueous electrolyte saturated with Mn(ClO4)2 at pH = 6.4.
In addition, as carbonyl-based organic electrodes have been widely investigated in organic rechargeable batteries, it is feasible to borrow some effective strategies from these well-studied representatives to develop high-performance ASIBs and AKIBs. In 2014, Qin et al. proposed a 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTCDA)-derived polyimide as anode for ASIB.157 It involved an enolization process during discharge/charge to reversibly combine Na+. The polyimide anode showed symmetric redox peaks at −0.34 V/−0.52 V (vs. SCE) and delivered a discharging capacity of 184 mA h g−1 and a charging capacity of 165 mA h g−1 in a 5 M NaNO3 aqueous solution. When coupled with NaVPO4F cathode, the full ASIB exhibited a midpoint voltage of 0.90 V. Meanwhile, Deng et al. reported another redox-active and water-insoluble polyimides, namely, poly-(naphthalene four formyl ethylenediamine) (PNFE).204 The reversible Na+-association/dissociation with the carbonyl groups occurred within the potential range of −1.0 to 0 V with a midpoint voltage of −0.58 V (vs. Ag/AgCl). This PNFE not only delivered a high capacity of 130 mA h g−1, but also provided a superior rate capability as well as an excellent capacity retention of 91.2% over 1000 cycles. Besides, as a widely investigated organic compound in organic batteries, quinones have been demonstrated as suitable anode materials for aqueous batteries by Yao's group. They discovered that prepared quinones were able to operate with different charge carrier species (including H+, Li+, Na+, K+, Mg2+) under different pH values (from −1 to 15), temperatures (−35 to 25 °C) and atmospheres (with/without O2).205 By rationally selecting/designing quinone molecules with desired structure, they further developed three major systems coupling with various industrially-mature cathode materials, including PbO2 (in acidic condition), LiMn2O4 (in neutral condition), and Ni(OH)2 (in alkaline condition). Among these quinone molecules, polymerized pyrene-4,5,9,10-tetraone (PPTO) was especially versatile as anode for ARMBs (Fig. 9e). Besides the capability to initiate ALIBs, it was also coupled with NVP cathode in a neutral 5 M NaNO3 electrolyte, resulting an ASIB showing a high specific capacity of 201 mA h g−1 as well as a good cyclability of 79% capacity retention over 80 charge/discharge cycles. The full ASIB exhibited sloping discharge profiles with a midpoint voltage of 0.65 V.
For ASIBs and AKIBs, representative cathode and anode materials have been discussed in this section, unveiling their inferior performances compared to ALIBs, which is mainly due to their larger ionic radius that imposes higher requirement on host materials including larger intercalative framework and better structural stability. Redox potentials of reprehensive cathode and anode materials for ASIBs and AKIBs are summarized and compared in Fig. 10. As revealed, NASICON-type materials exhibit desired potentials as either cathode or anode. Some PBA-type materials are also preferential for ASIB and AKIB cathode. It should be noted that except for the study employing a concentrated aqueous electrolyte (introduced in Section 4.4.2), no stable anode materials in normal aqueous electrolytes have been reported for AKIBs so far. To facilitate the development of ASIBs and AKIBs, more studies should be implemented to promote electrochemical kinetics and structural stability of electrode materials.
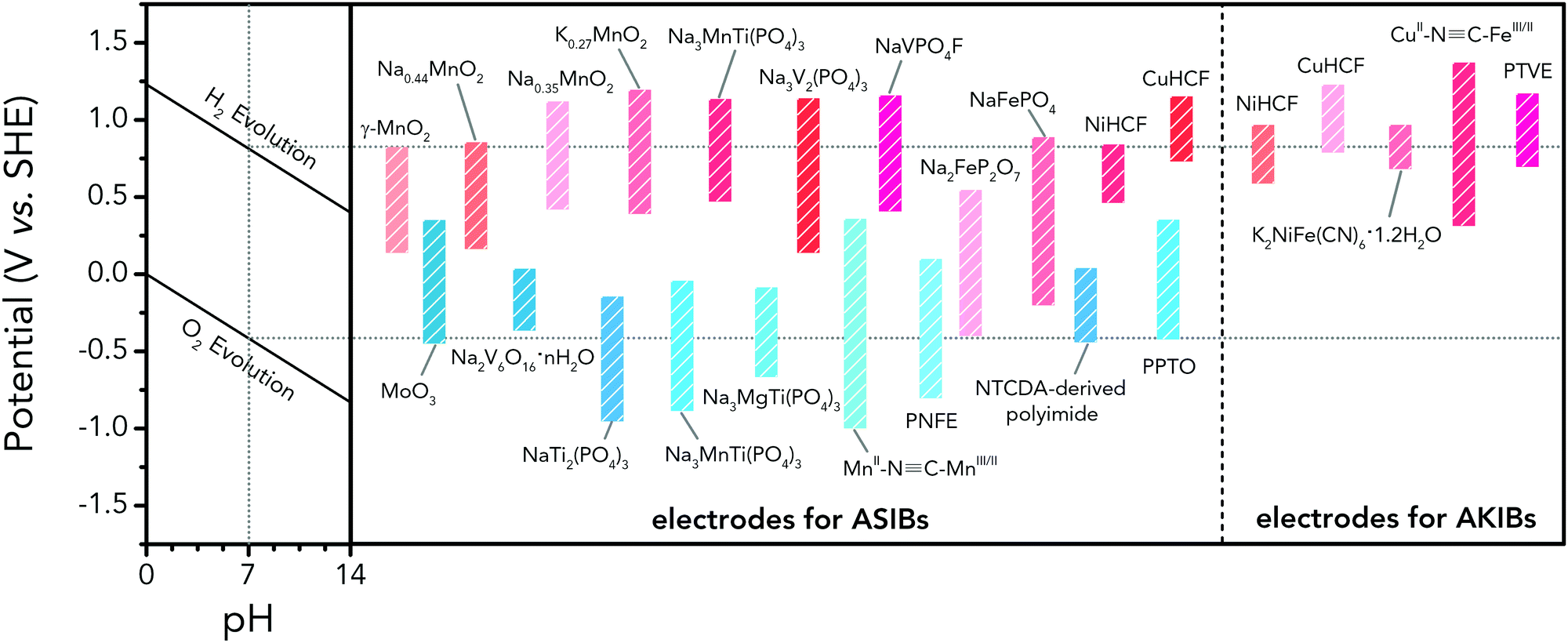 | ||
| Fig. 10 Comparison of redox potentials of representative electrode materials for ASIBs and AKIBs. Red-colour columns and blue-colour columns represent cathodes and anodes, respectively. | ||
3.3 Aqueous zinc-ion batteries
Traditional alkaline zinc-based batteries are either non-rechargeable or extremely poor in cycle stability. Zinc-ion batteries employing neutral/mild aqueous electrolytes have shown great improvements in reversibility and rate performance. Aside from the nature abundance and eco-friendliness, zinc metal is regarded as an ideal anode material for aqueous zinc-ion batteries (AZIBs) due to its high theoretical capacity (820 mA h g−1), high electrical conductivity, relatively low redox potential, and high safety.206–209 Due to the feasibility of directly utilizing zinc metal as anode in aqueous electrolytes, research efforts of AZIBs are mainly focused on developing suitable cathode materials. In this section, a variety of representative cathodes for AZIBs such as manganese-based materials, vanadium-based materials, organic materials, and other compounds including PBAs, two-dimensional (2D) materials and nickel/cobalt oxides, are thoroughly discussed in terms of their electrochemical performances and voltage characteristics. It should be noted that all the specific capacity values of reported AZIBs are based on the mass of cathode materials unless otherwise specified.In the early stage, MnO2 was the most investigated cathode material for AZIBs due to its tunnel or layered structure that enables reversible intercalation/de-intercalation of zinc ions. MnO2 exhibits different crystallographic polymorphs including α-, β-, γ-, R-type (tunnel structures), δ-type (layered structure), and λ-type (spinel structure).220 The phase structure and morphology of MnO2 can play a significant role in the electrochemical properties and voltage characteristics of AZIBs. For example, the commercial α-MnO2 with 2 × 2 tunnels shows two discharge plateaus at around 1.4 and 1.3 V (vs. Zn/Zn2+), corresponding to H+ and Zn2+ ions insertion, both of which are very tilting owing to the large α-MnO2 particle size. As a result, the AZIB based on commercial α-MnO2 could only provide a capacity of around 187 mA h g−1 at 100 mA g−1. To improve electrochemical properties, nanostructured MnO2 was further electrodeposited on carbon fibre paper (MnO2@CFP).43 The AZIB based on MnO2@CFP presented a long and flat discharge plateau at 1.3 V accompanied with a tilting plateau at 1.4 V in mild acidic ZnSO4 + MnSO4 electrolyte (Fig. 11a), which delivered a discharge capacity of 290 mA h g−1 at 90 mA g−1 and a cyclability of 10000 cycles at 6.5C with a capacity decay rate of 0.007% per cycle. The improvement of working voltage of MnO2@CFP cathode arose from the highly-porous nanoflake structure and strong interfacial adhesion that favoured the reversible insertion/extraction of H+ and Zn2+ ions during charge/discharge (Fig. 11b), which also ensured high cycling stability. Among the MnO2 polymorphs, β-MnO2 is considered to be the most thermodynamically stable one as it is composed of MnO6 octahedral unit single chains by forming a 1 × 1 tunnel along c axis. Although such narrow tunnel is not favourable for the diffusion of Zn2+,221 it was revealed in a recent study that the AZIB based on β-MnO2 nanorod cathode could deliver a high capacity of 270 mA h g−1 at 100 mA g−1 with two tilting discharge plateaus at 1.40 V and 1.24 V, and retain 75% of its original capacity after 200 charge/discharge cycles.222Ex situ characterization technologies confirmed the storage of Zn2+via the intercalating of Zn2+ into the β-MnO2, resulting in the formation of Zn-intercalated phases and the precipitation of ZnSO4·3Zn(OH)2·5H2O. γ-MnO2 with randomly arranged 1 × 2 and 1 × 1 tunnels has also demonstrated reversible zinc storage capability with a high discharge capacity of 285 mA h g−1 at 0.05 mA cm−1 and similar discharge profiles to α-MnO2 with two tilting plateaus.223 In addition, δ-MnO2 with typical layered structure was found to exhibit one discharge plateau at 1.23 V during its initial cycling, while two discharge plateaus at 1.38 V and 1.23 V were observed in the subsequent cycling (vs. Zn/Zn2+).224 Detailed investigations revealed that the intercalation of Zn2+ caused the phase transformation of layered δ-MnO2 to spinel-type ZnMn2O4 and layered-type δ-ZnxMnO2 with Mn(II) phase. In a later study, this ZnMn2O4 was turned into a viable zinc host material by introducing cation vacancies.219 The resultant AZIB with cation-defective spinel ZnMn2O4 cathode provided a specific capacity of 150 mA h g−1 at 50 mA g−1 with a midpoint voltage of 1.33 V. Facile charge transfer and Zn2+ intercalation in the robust cation-defective spinel structure were revealed to account for its good performances (Fig. 11c).
 | ||
| Fig. 11 (a) GCD curves of the Zn//MnO2@CFP full cell at 0.3C in mild acidic ZnSO4 + MnSO4 aqueous electrolyte. (b) Discharge galvanostatic intermittent titration technique (GITT) curves of the Zn//MnO2@CFP full cell at 50 mA g−1 for 2 min with 4 h rest. It is proposed that the region I voltage plateau attributes to H+ insertion and the region II voltage plateau attributes to Zn2+ insertion. Reproduced with permission from ref. 43, Copyright 2017 American Chemical Society. (c) Schematic illustration of Zn2+ intercalation/de-intercalation in the 3D framework of ZnMn2O4 (left) and hypothesized Zn2+ diffusion passage in ZnMn2O4 without and with Mn vacancies (right). Reproduced with permission from ref. 219, Copyright 2016 American Chemical Society. (d) Schematic illustration of the synthesis of PANI-intercalated MnO2 nanolayers. (e) Cycling test of the Zn//PANI-intercalated MnO2 full cell at 200 mA g−1. Reproduced with permission from ref. 234, Copyright 2018 Springer Nature. | ||
Despite its advantages, MnO2 cathode suffers from intrinsic poor electrical conductivity that hinders electrochemical kinetics. One obvious symptom is that the charging/discharging curves of pristine MnO2 display sloping voltage plateau and shorter duration period at high rates. So far, it is generally accepted that the combination of a conducting material (e.g. CNTs, graphene) with MnO2 is an effective strategy to improve its conductivity, thus bringing about better electrochemical properties.225–228 For example, Wu et al. attempted to increase the conductivity of α-MnO2 nanowire by coating graphene scroll on its surface with an average width of 5 nm. It displayed a discharge capacity of 382.2 mA h g−1 at 0.3 A g−1 and a superior cyclability of 3000 cycles with 94% capacity retention at 3.0 A g−1.229 In addition, doping metal ions (e.g. Al3+, Co2+, Ni2+) into MnO2 lattice could facilitate Zn2+ intercalation and enhance working potential.230–232 Yang et al. studied the electronic structure of Al-doped α-MnO2via first-principle calculations, and found that the substitution of Mn with Al atom caused redistribution of charge density, which provided more electrons for α-MnO2, thus contributing to an enhanced electronic conductivity.232 Another feasible approach is to engineer oxygen vacancy in MnO2, which can improve electrical conductivity by regulating surface chemistry. Owing to the presence of oxygen vacancy, the Gibbs free energy of Zn2+ absorption on the MnO2 surface can be reduced to thermoneutral value, indicating the highly reversible adsorption/desorption process of Zn2+. Besides, more electrons can be contributed into the delocalized electron cloud of oxygen-deficient MnO2, since fewer electrons are required for the formation of Zn–O bonding, which improves the attainable capacity. As a result, the oxygen-deficient MnO2 cathode achieved a high discharge capacities of 345 mA h g−1 and a remarkable stability of 84% capacity retention after 2000 cycles at 5 A g−1.233 In addition to intrinsically poor conductivity, repeated insertion of hydrated cations during electrochemical processes destroys the ordered structure of MnO2, leading to inevitable capacity fading. Introducing a polymer (e.g. polyaniline) into MnO2 layers has been proposed to effectively consolidate the latter's layered structure and avoid the phase transformation induced by hydrated H+/Zn2+ insertion, which resulted in improved cycling stability and discharge capacity (Fig. 11d and e).234 Another novel approach incorporated crystal water into the layered host structure of MnO2 and shielded the electrostatic interactions between zinc ions and host materials, revealing promoted Zn2+ diffusion. Moreover, such interlayer crystal water molecules assisted the stabilization of layered MnO2 framework by suppressing side reactions during cycling. The as-assembled AZIB with a high open-circuit voltage of 1.55 V delivered a high capacity of 350 mA h g−1 at 100 mA g−1 along with improved cycling stability and rate capability.235
V2O5 has been widely studied for AZIBs owing to its extremely high theoretical capacity of 589 mA h g−1 based on two-electron redox centres.236 Whereas, V2O5 for Zn2+ intercalation generally exhibits poor cycling stability and low rate capability because of its unstable layered structure during electrochemical processes. Similar to the strategies used for MnO2, to promote performances, structural water and hetero metal ions can be introduced into layered V2O5 as pillars that strengthen its interlayer structure.236,237 For example, Yan et al. fabricated a V2O5·nH2O/graphene (VOG) composite via a liquid phase method.238 The initial CV curve of VOG showed two pairs of redox peaks associating with Zn2+ intercalation/de-intercalation located at 0.97 V/0.91 V and 0.63 V/0.54 V, respectively. Subsequent CV curves overlapped with the initial one, indicating the solid-solution reaction of Zn2+ intercalation/de-intercalation was highly reversible (Fig. 12a). The AZIB with VOG cathode delivered a high initial capacity of 381 mA h g−1 at 60 mA g−1 and retained 71% of its initial capacity after 900 cycles. Its discharge profiles showed no obvious plateau but a midpoint voltage of 0.68 V. Ming et al. reported a layered Mg2+-intercalated V2O5 cathode with enhanced electrochemical activity for AZIBs (Fig. 12b).239 Due to the larger radius of hydrated Mg2+ (4.3 Å), the expanded interlayer spacing (13.4 Å) allowed for efficient Zn2+ intercalation/de-intercalation, resulting an AZIB with a wide working voltage range of 0.1–1.8 V (Fig. 12c). The AZIB based on Mg0.34V2O5·nH2O cathode delivered 353 mA h g−1 and 264 mA h g−1 at 100 mA g−1 and 1.0 A g−1, respectively, along with a 97% capacity retention after 2000 cycles at 5.0 A g−1. It showed no obvious discharge plateau, and the midpoint voltage located at around 0.70 V.
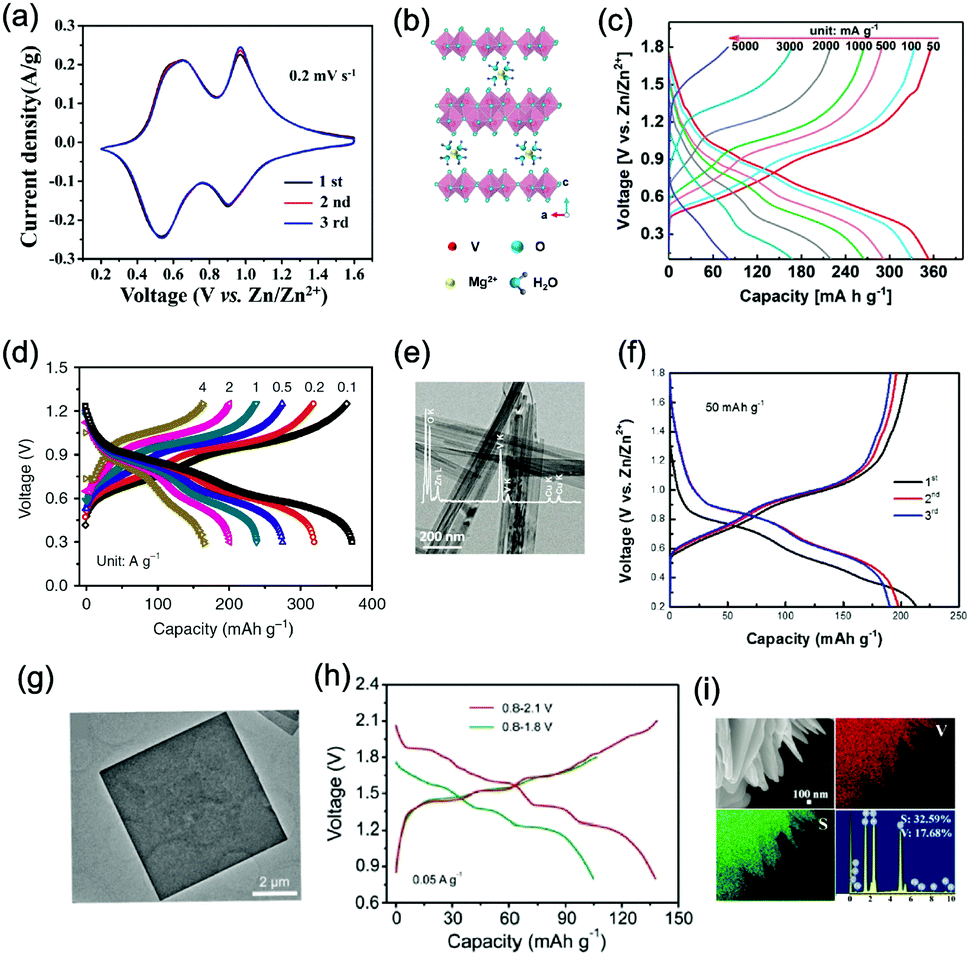 | ||
| Fig. 12 (a) CV curves of the as-prepared VOG for the initial 3 cycles. Reproduced with permission from ref. 238, Copyright 2018 Wiley-VCH. (b) Crystal structure of the layered Mg2+-intercalated V2O5. (c) GCD curves of the Mg2+-intercalated V2O5 cathode at various current densities. Reproduced with permission from ref. 239, Copyright 2018 American Chemical Society. (d) GCD curves of the Zn//NVO full cell at various current densities. Reproduced with permission from ref. 240, Copyright 2018 Springer Nature. (e) TEM image of the ZVO nanowires. Inset shows its EDS mapping, revealing the existence of Zn, O and V. (f) GCD curves of the ZVO cathode at 50 mA g−1 for the initial 3 cycles. Reproduced with permission from ref. 244, Copyright 2018 Wiley-VCH. (g) TEM image of the VOPO4 particle. (h) Comparison of the 2nd GCD curves between Zn//VOPO4 batteries in 21 M LiTFSI electrolyte and in 1 M Zn(Tr)2 electrolyte. Reproduced with permission from ref. 247, Copyright 2019 Wiley-VCH. (i) SEM–EDX of the as-prepared layered VS2. Reproduced with permission from ref. 248, Copyright 2017 Wiley-VCH. | ||
Bearing relative high oxidation states of V5+ and layered structure, MxV3O8 (M = Li, Na, H, etc.) composed of VO6 octahedra and V2O8 square pyramids have been studied as cathode candidates for Zn2+ intercalation/de-intercalation. For example, Wan et al. proposed a simple liquid–solid stirring strategy to fabricate NaV3O8·1.5H2O (NVO) nanobelts for AZIBs.240 CV profiles of the resultant AZIB exhibited two pairs of redox peaks located at 0.55 V/0.77 V and 0.85 V/1.06 v which were owing to the reversible redox reactions from NaZn0.1V3O8·1.5H2O to H2.14NaZn0.2V3O8·1.5H2O and then to H3.9NaZn0.5V3O8·1.5H2O, corresponding to the valence changes of vanadium from V5+ to V4+ and V4+ to V3+.241–243 The AZIB achieved a highly reversible capacity of 380 mA h g−1 at 0.05 A g−1 along with a midpoint voltage located 0.72 V (Fig. 12d). 1 M Na2SO4 as an additive was added into the ZnSO4 electrolyte to suppress the dissolution of NVO and the growth of Zn dendrites, which notably improved the reversibility of Zn2+ intercalation/de-intercalation, resulting in a high capacity retention of 82% after 1000 cycles at 4 A g−1. Zn3V2O7(OH)2·2H2O and Zn2V2O7 are another type of vanadium-based cathode materials for AZIBs, which have porous crystal framework and expanded interlayer space. A study showed that an AZIB employing ultralong Zn3V2O7(OH)2·2H2O (ZVO) nanowires as cathode delivered a specific capacity of 213 mA h g−1 at 50 mA g−1 along with a midpoint voltage of 0.75 V (Fig. 12e and f).244 Since the open-framework structure of ZVO cathode allows fast immigration of Zn2+, the AZIB exhibited very good rate capability, achieving 76 mA h g−1 at a high current density of 3000 mA g−1. Besides, as an alternative to Zn3V2O7(OH)2·2H2O, Zn2V2O7 nanowires were synthesized by one-step hydrothermal method recently.245 AZIBs based on Zn2V2O7 cathode could be operated in a voltage range of 0.4–1.4 V with a midpoint voltage of 0.67 V. After fully activation, the battery delivered a specific capacity of 197.4 mA h g−1 at 300 mA g−1. Benefitted from its nanowire morphology and robust structure, the AZIB exhibited high rate performance and long-term cyclability, achieving 85% capacity retention (138 mA h g−1) after 1000 cycles at 4000 mA g−1.
Other vanadium-based cathodes include vanadium-based phosphates and sulphides. For example, owing to the presence of PO43− polyanions, NASICON-typed NVP cathode exhibited a flat discharge plateau located at 1.10 V (vs. Zn/Zn2+), exceeding the midpoint voltages of most vanadium oxides.209 But it suffered serious capacity decay during long-term cycles. To address this issue, Li et al. fabricated a Na3V2(PO4)2F3 cathode for AZIBs, the full cell displayed an even higher plateau voltage of 1.63 V, as well as a high energy density of 97.5 W h kg−1 and an excellent cyclability with 95% capacity retained after 4000 cycles.246 This good electrochemical performance was due to the strong affinity of F atoms, which endowed Na3V2(PO4)2F3 with more stable structure that led to “zero-strain” change during Zn2+ intercalation/de-intercalation. In addition, the introduction of extra anionic redox reaction is another effective strategy to enhance the working voltage and energy density of AZIBs, thus a high-performance AZIB with reversible oxygen redox chemistry was developed based on layered VOPO4 cathode and “water-in-salt” electrolyte (Fig. 12g).247 Due to the contribution of oxygen redox reactions, the Zn//VOPO4 battery displayed an expanded working voltage range of 0.8–2.1 V, which was much wider than the voltage range of 0.8–1.8 V when merely relying on the redox reactions of vanadium (Fig. 12h). This broadened voltage range notably benefited battery capacity (from 109 mA h g−1 to 139 mA h g−1) and midpoint voltage (from 1.32 V to 1.44 V). Moreover, a sulphide of VS2 synthesized via a facile hydrothermal reaction (Fig. 12i) was employed as cathode for AZIBs in 2017 for the first time, the full cell exhibited a midpoint voltage of 0.63 V with two flat discharge plateaus located at 0.69 V and 0.61 V.248 This battery delivered a specific capacity of 190.3 mA h g−1 at 0.05 A g−1 and an energy density of 123 W h kg−1 (the latter was based on the mass of both active materials).
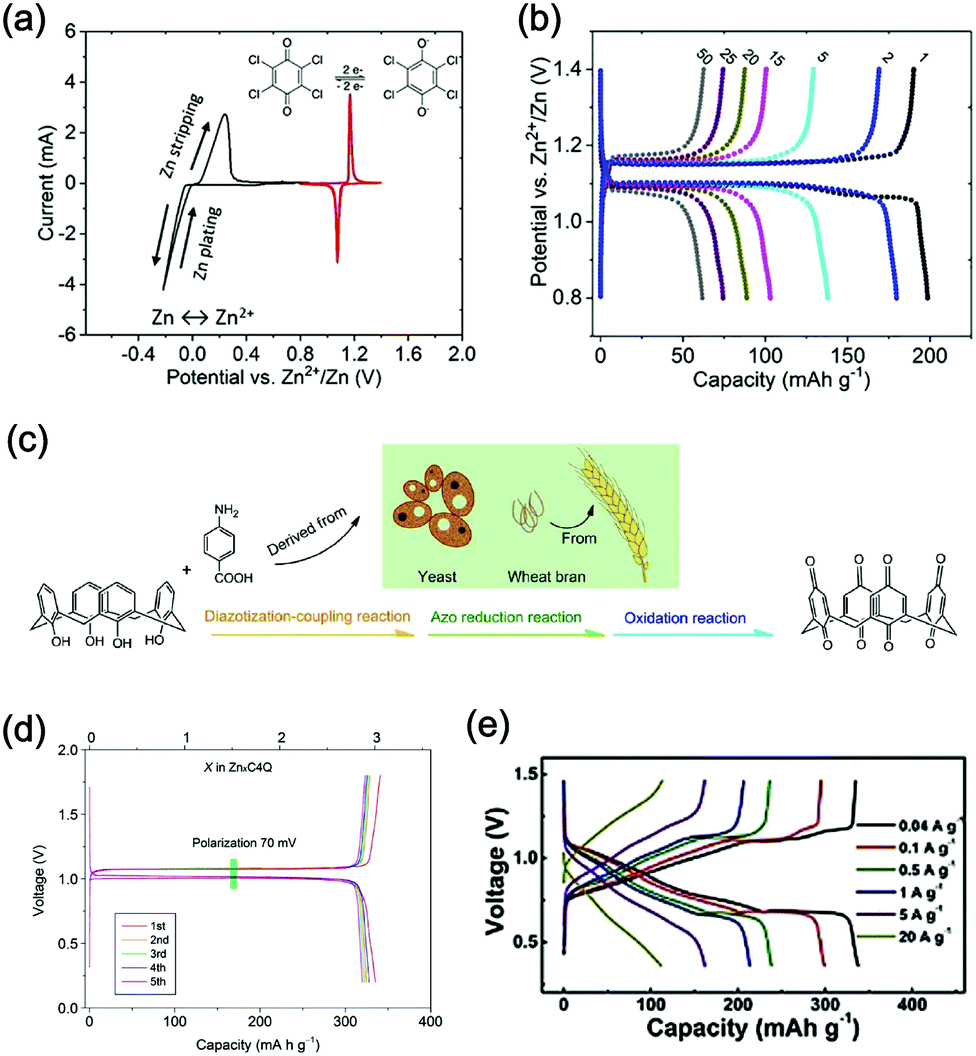 | ||
| Fig. 13 (a) CV curves of the Zn anode (black line) and the p-chloranil cathode (red line) in 1 M Zn(OTf)2–H2O electrolyte at 5 mV s−1 and 0.1 mV s−1, respectively. (b) Selected cycles of GCD curves of the p-chloranil cathode at C/5 rate. The numbers indicate cycle number. Reproduced with permission from ref. 250, Copyright 2018 American Chemical Society. (c) Schematic illustration of the synthesis of C4Q. (d) GCD curves of the Zn//C4Q full cell at 20 mA g−1. The upper X axis indicates the uptake number of zinc ions. Reproduced with permission from ref. 251, Copyright 2018 American Association for the Advancement of Science. (e) GCD curves of the Zn//PTO full cell at various current densities. Reproduced with permission from ref. 252, Copyright 2018 Wiley-VCH. | ||
Compared to metal oxides and organic carbonyl compounds, conducting polymers with long-range π-conjugation system possess higher electrical conductivity and structural stability. Recently, two typical conducting polymers of PANI and PPy have been investigated as cathode for AZIBs. Kim et al. proposed a polyaniline-coated carbon fibre (PANI/CF) cathode for flexible AZIBs.253 It was found that a thin (150 nm) and highly porous PANI layer was immobilized on carbon fibres, providing high surface area and enhanced electrical conductivity for redox reactions.254 Such PANI/CF cathode could be operated at a high rate of 600C within the voltage range of 0.7–1.7 V. In another study, PANI nanorods were deposited on lens papers with large voids that could effectively promote electrolyte infiltration and ion diffusion.255 The resultant AZIB delivered a specific capacity of 142.3 mA g g−1 at 0.2 A g−1 over the voltage range of 0.7–1.7 V. One drawback of PANI cathode is its instability in acidic electrolytes, where PANI undergoes deprotonation during repeated charge/discharge, leading to severe degradation. To solve this problem, Shi et al. reported a sulfo-self-doped PANI electrode (PANI-S) where SO3− self-dopant acted as internal proton reservoir to maintain high local proton concentration on the polymer backbone and promoted reversible redox reactions during charge/discharge. AZIB employing such cathode was operated in the voltage range of 0.5–1.6 V with a discharge capacity of 184 mA h g−1 at 0.2 A g−1.256 All these PANI-based AZIBs showed sloping discharge profiles with a midpoint voltage at around 1.09 V. Similar to PANI, PPy can also imitate reversible doping/de-doping when used as cathode for AZIBs. As reported by Wang et al., the Zn//PPy battery provided a capacity of 123 mA h g−1 and a midpoint voltage of 0.55 V within the voltage range of 0–1.2 V, retaining 41% and 38% of its initial capacity after 100 and 200 cycles, respectively.252 Li et al. further fabricated nanostructured PPy composite aerogels that allowed easy electrolyte infiltration and ion diffusion.257 The obtained AZIB delivered a higher specific capacity of 150 mA h g−1 and a higher midpoint voltage of 1.04 V within the voltage range of 0.6–1.6 V, and retained 76.7% of its initial capacity after 1000 charge/discharge cycles at 8 A g−1.
000 charge/discharge cycles (at 3 A g−1), respectively (Fig. 14a and b). Kinetic analyses revealed that such remarkable performances were attributed to the increased redox active sites and stable structure induced by the high-voltage activation. Whereas, this AZIB exhibited a midpoint voltage of around 1.25 V, which is lower compared to the AZIBs using ZnHCF and CuHCF cathodes.
 | ||
| Fig. 14 (a) GCD curves of the FeHCF cathode after various cycles at the current density of 1 A g−1, showing increasing capacity. (b) A record-high cycle stability of the Zn//FeHCF full cell with in-depth discharge/charge processes up to 10000 cycles at 3 A g−1. Reproduced with permission from ref. 261, Copyright 2019 Wiley-VCH. (c) Schematic illustration of the fabrication of E-MoS2. (d) GCD curves of Zn/E-MoS2 full cell at various current densities. Reproduced with permission from ref. 264, Copyright 2018 Elsevier. (e) Schematic illustration of the formed quasi-SEI layer between zinc metal anode and the solid-state PANa electrolyte. (f) Comparison of capacity retention with the state-of-the-art aqueous alkaline rechargeable batteries. Reproduced with permission from ref. 16, Copyright 2018 Wiley-VCH. | ||
Besides, it is well-known that 2D materials with large interlayer space could offer abundant active sites with reduced ion diffusion resistance, which is favourable for metal ion intercalation/de-intercalation.262 Chao et al. reported the first AZIB utilizing a 2D Layered cathode of zinc orthovanadate array.263 This AZIB exhibited a specific capacity of 204 mA h g−1 and sloping discharge profiles with a midpoint voltage of 0.86 V at 0.5C. Its maximum energy and power densities reached 115 W h kg−1 and 5100 W kg−1, respectively (based on the mass of both electrodes including current collectors). The good zinc ion storage capability was attributed to the ultrathin mesoporous arrays featuring high surface areas and single-crystalline layered structure with 2D facile ion transport pathways. Our group also demonstrated an AZIB utilizing a 2D layered material of MoS2 nanosheets with expanded interlayer spacing (E-MoS2) grown on carbon fibre cloth as zinc-ion intercalative cathode.264 The enlarged interlayer distance of E-MoS2 enable Zn2+ intercalation with fast reaction kinetics and low energy barrier. Thus, the Zn/E-MoS2 battery exhibited a high specific capacity (202.6 mA h g−1 at 0.1 A g−1) and an outstanding long-term cycling stability (98.6% capacity retention after 600 cycles). However, this AZIB showed sloping discharge profiles with a low midpoint voltage of 0.6 V, which impedes its energy density (Fig. 14c and d).
In addition, some high-performance AZIBs with conversion-type nickel/cobalt oxide cathodes have been developed. For example, a flexible Zn/Ni battery operated within the voltage range of 1.4–1.9 V was proposed by using a self-supported NiCo2O4 cathode, which delivered a high capacity of 183.1 mA h g−1 and an energy density of 303.8 W h kg−1.265 However, due to the insufficient active sites and intrinsic poor conductivity (semiconductor nature), the capacity and rate capability of NiCo2O4 cathodes are far from satisfactory. To tackle these issues, ultrathin NiCo2O4 nanosheets (P-NiCo2O4−x) were prepared using a facile solvothermal method with the simultaneous introduction of oxygen vacancies and phosphate ions, which exhibited improved electrochemical properties.266 It is found that oxygen vacancies not only acted as shallow donor to improve electrical conductivity of NiCo2O4, but also provided sufficient active sites for Zn2+ insertion. Besides, phosphate ion has a longer bond length and lower electronegativity compared to O2− on the surface, leading to a decreased energy of electron transport, thus improving surface activity and reaction kinetics. As a result, the P-NiCo2O4−x cathode presented a tilting high-voltage plateau at 1.68 V, which was notably superior to pristine NiCo2O4 cathode. At the high current density of 3 A g−1, the P-NiCo2O4−x cathode provided a high capacity of 361.3 mA h g−1, which was ten times higher than that of pristine NiCo2O4 cathode. It also achieved an outstanding energy density of 616.5 W h kg−1 at a power density of 5.15 kW kg−1. In a recent study, our group has demonstrated for the first time that electrolyte played a unique and important role in improving cycle stability for AZIBs based on NiCo-hydroxide cathode.16 The developed sodium polyacrylate (PANa) hydrogel held a concentrated electrolyte solution, where water was well absorbed and retained, and zinc ions were immobilized to form a quasi-SEI on the PANa network (Fig. 14e), thus facilitating ion transport and suppressing zinc dendrite formation. The resultant battery exhibited a tilting discharge plateau at 1.58 V and retained 73% and 65% of its initial capacity after 10000 and 16000 charge/discharge cycles at 96C (Fig. 14f), respectively. Although possessing attractive performances, it should be noted that these reported zinc batteries based on conversion-type nickel/cobalt oxides usually adopt alkaline electrolytes for reversible cycling, which should be further optimized in terms of corrosiveness and eco-friendliness.
Therefore, the suppression of zinc dendrites is of vital importance in achieving and maintaining satisfactory AZIB performances. A variety of studies on zinc dendrites have been conducted for alkaline zinc-based batteries (the cathodes of which are non-intercalative). For example, back to the 1990s, Kan et al. reported Triton X-100 additive as an effective inhibitor on zinc dendrite growth, which can improve the dispersion and cathodic polarization of electrolyte, resulting in finer deposited zinc particles that helped raise efficiency and prolong battery cycle life.269 Later on, other additives such as nickel triflate,270 polyethylene glycol,271 benzyltrimethylammonium hydroxide,272etc., were reported effective in suppressing zinc dendrite growth in alkaline electrolytes. These additives resulted in the formation of an interphase layer originated from either molecular absorption or chemical reactions, which affected the nucleation and growth of zinc and restricted the motion of deposition kinetics, hence preventing the runaway growth of large dendrites. Parker et al. initially proposed to suppress zinc dendrite growth by constructing three-dimensional zinc anode.273 The three-dimensional zinc sponge they demonstrated reached ca. 90% utilization (corresponding to 728 mA h gZn−1) when discharged in alkaline electrolyte, and it can be cycled in various cells without the formation of macroscale dendrites. Another method proposed by Higashi et al. employed a backside-plating configuration, where the backside plating of zinc was achieved via coating an insulation layer on the edges and the “front” side of a copper foil that faces zinc metal counter electrode.274 In this way, zinc ions have to travel over the edge and deposit on the open back side of the copper foil, enabling long-term cycling of zinc batteries without shorting.
In neutral or mildly acidic aqueous electrolytes (which are adopted by most AZIBs), two main strategies have been developed to effectively suppress zinc dendrites. One is to construct artificial SEI. As revealed by Li et al., the Zn@C anode where carbon coating layer served as an artificial SEI realized stable zinc stripping/plating and improved kinetics without obvious zinc dendrite formation, which should be attributed to the even current distribution and resultant homogeneous zinc deposition facilitated by the porous carbon coating film.275 Nanosized metal–organic frameworks and in situ reduced graphene oxide have also been utilized as artificial SEI layer to obtain dendrite-free zinc anode, which were claimed to be capable of regulating electrolyte flux on anode surface, resulting in a homogeneous plating process.276,277 Another strategy is to construct unique anode structures. A free-standing, three-dimensional layered Ti3C2Tx MXene@Zn paper anode as an alternative to bulky zinc metal was developed by Tian et al. via in situ electroplating.278 This anode can provide rapid electron transport pathways, thereby realizing uniform charge distribution and thus effectively suppressing the formation of dendrites. SEM images revealed the smooth zinc surface on Ti3C2Tx MXene in aqueous electrolyte with outstanding durability. In addition, microporous zinc-based metal–organic framework ZIF-8 treated at 500 °C and flexible three-dimensional CNT framework were both found to be attractive host matrix for zinc plating and stripping, and the resultant anodes with unique structures realized high Coulombic efficiency and dendrite-free plating attributing to decreased local current density, low zinc nucleation overpotential as well as homogeneous electric field distribution.279,280
In a study that investigates the fractal growth of zinc dendrites in alkaline electrolytes, the authors pointed out that zinc dendrite growth patterns are affected by various factors, including electrolyte concentration, electrolyte thickness, voltage variation, anion variation and solvent nature.281 Recently, our group studied the growth behaviour of zinc dendrites in neutral/mild aqueous electrolytes and found that dendrites growth is substantially affected by battery configuration, applied current density and the mass loading of cathode materials, which explained the diverse results of battery cycle life reported in literatures.282 Subsequently, we proposed an electrohealing methodology to in situ eliminate the already-formed zinc dendrites by decreasing applied current density, aiming to prolong battery lifetime or to in situ rescue in-service AZIBs.
Back to battery voltage characteristics, for AZIBs, major types of cathode materials have been discussed in this section, with their redox potentials summarized and compared in Fig. 15. As revealed, PBA-type materials could provide high redox potentials, while vanadium oxides, sulphides and polymers show relatively low redox potentials. However, discharge capacities of high-potential PBAs are rather small (<80 mA h g−1) compared to other materials (200–300 mA h g−1). Cathodes with simultaneous high redox potentials and high specific capacities are lacking. Due to the thick solvation sheath and large charge/radius ratio, the intercalation kinetics of zinc ions are usually sluggish, and their high atomic mass and strong positive polarity lead to poor kinetics of ion transport and low solid-state solubility in bulky electrodes. Therefore, in pursue of high-performance AZIBs, it is highly desired to continue discovering new cathode materials that bear high capacity, high rate capability, stable cycle performance and high redox potentials with flat and high-voltage discharge plateaus.
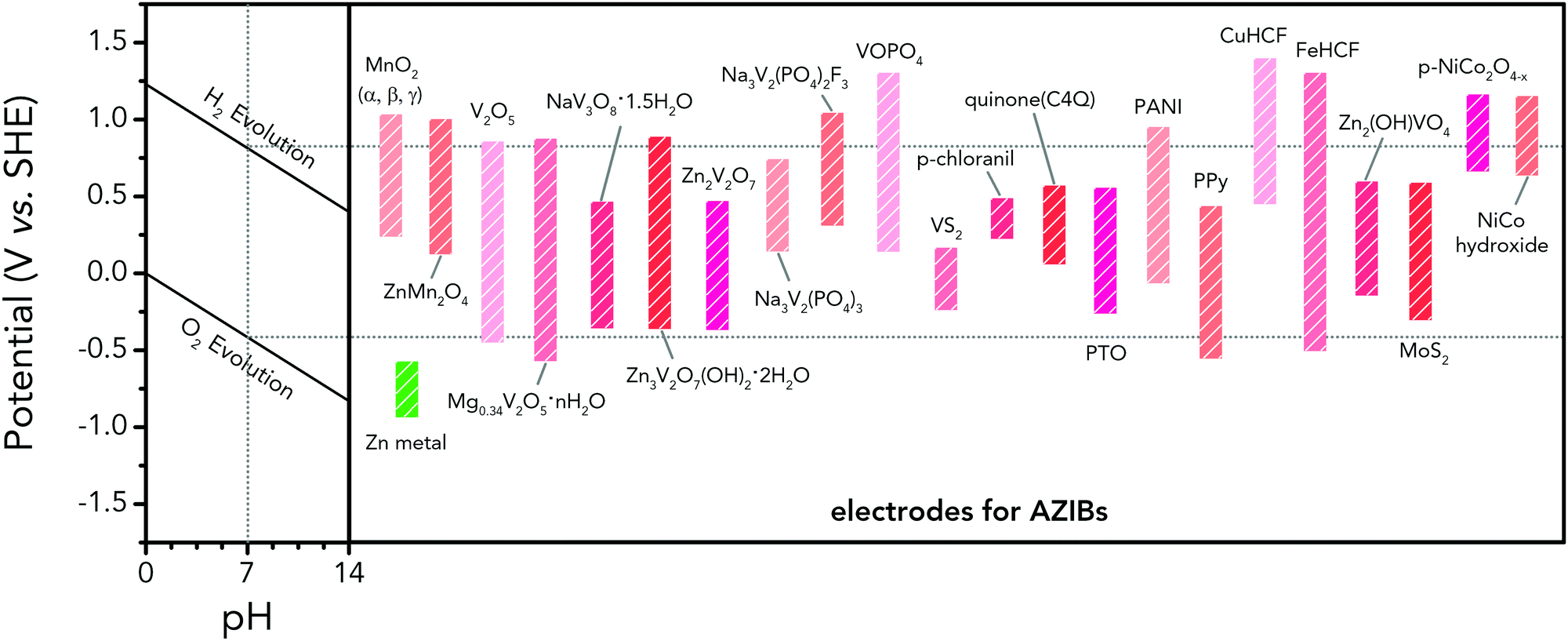 | ||
| Fig. 15 Comparison of redox potentials of representative electrode materials for AZIBs. Red-colour columns and green-colour column represent cathodes and zinc metal anode, respectively. (Some conversion-type NiCo compounds are also included.). | ||
3.4 Other aqueous multivalent metal-ion batteries
Aqueous multivalent metal-ion batteries are considered as promising alternatives to aqueous monovalent metal-ion battery systems. Besides the earth abundance of these metal elements, one major reason favours their high potential is that, multivalent metal cations can, theoretically, transfer more than one electron and thereby achieve higher specific capacity and energy density.283–289 However, multivalent cations with large charge densities diffuse much more slowly in electrode materials than monovalent cations do due to their strong cation–cation repulsive and cation–anion attractive forces.287–292 Therefore, current aqueous multivalent metal-ion batteries exhibit less ideal electrochemical performances, high-performance aqueous multivalent metal-ion battery systems have been rarely reported so far.000 cycles at 2000 mA g−1. The reversible capacity of 88.6 mA h g−1 is also remarkable as even the well-known stable PBA electrodes just deliver capacities of <80 mA h g−1 for monovalent metal-ion batteries137,185 and <50 mA h g−1 for divalent metal-ion batteries136 at similar current densities in aqueous electrolytes.
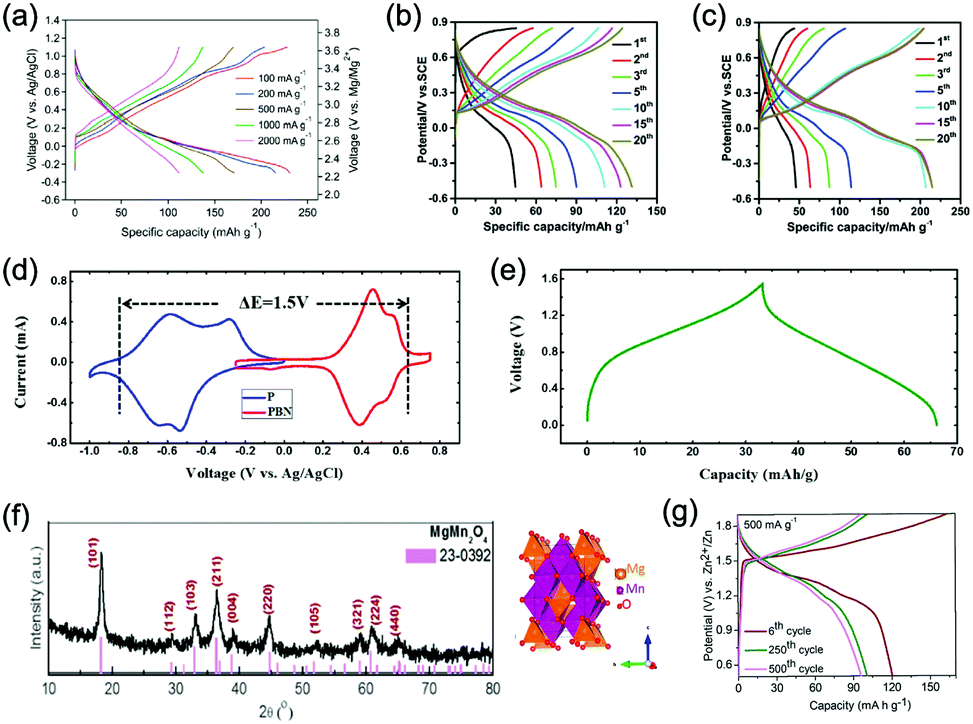 | ||
| Fig. 16 (a) GCD curves of the Mg-B cathode at various current densities. Reproduced with permission from ref. 300, Copyright 2015 American Chemical Society. (b) GCD curves of MgMn2O4 at the current density of 50 mA g−1. (c) GCD curves of MgMn2O4/rGO at the current density of 50 mA g−1. Reproduced with permission from ref. 303, Copyright 2018 Royal Society of Chemistry. (d) CV curves of PBN cathode and polyimide anode at 1 mV s−1. (e) GCD curves of the polyimide//PBN full cell at 1 A g−1. Reproduced with permission from ref. 328, Copyright 2017 American Chemical Society. (f) PXRD pattern of MgMn2O4 with an illustration of its crystal structure (right). (g) GCD curves of the MgMn2O4 cathode in 1 M “MgSO4 + ZnSO4” + 0.1 M MnSO4 aqueous electrolyte at 500 mA g−1. Reproduced with permission from ref. 322, Copyright 2018 American Chemical Society. | ||
Other types of manganese oxides have also been widely-studied for AMIBs. Cabello et al. observed that magnesium could be reversibly extracted/inserted from/into MgMn2O4 during electrochemical processes in both aqueous and non-aqueous electrolytes, although the cycling of MgMn2O4 in aqueous electrolyte was not as stable as in the non-aqueous electrolyte based on carbonate solvent. This cathode exhibited sloping discharge profiles within the potential range of 0–1.3 V (vs. Ag/AgCl).302 Actually the poor conductivity of MgMn2O4 seriously confines its electrochemical performances, therefore reduced graphene oxide (rGO) was further introduced to enhance its electron transfer.303 The as-prepared MgMn2O4/rGO nanocomposite delivered a specific capacity of 140.1 mA h g−1 at 1000 mA g−1, which was 69% higher than that of pristine MgMn2O4. While it still didn’t exhibit obvious discharge plateau when operated in the potential range of −0.45 to 0.9 V (vs. SCE) (Fig. 16b and c). Besides, one unique category of manganese dioxide minerals, manganese oxide octahedral molecular sieves (OMS), has been frequently reported in the fields of energy storage and catalysis due to its diverse tunnel structures.304–307 Zhang et al. studied Mg-OMS-1 with 3 × 3 tunnels, Mg-OMS-7 with 1 × 1 tunnels308 and Mg-OMS-2 with 2 × 2 and 1 × 1 tunnels309 in various aqueous electrolytes including 0.5 M MgCl2, 0.5 M Mg(NO3)2 and 0.5 M MgSO4. These cathodes were operated in the potential range of −0.6 to 0.8 V (vs. SCE) with sloping discharge profiles. In fact, the capacity contribution in the potential range of −0.6 to 0 V was quite small, which should be exempted unless considerably low-potential anodes could be identified to fully utilize this low discharge portion of cathodes. Their specific capacities were roughly in the range of 150–250 mA h g−1, which are much higher compared to most electrode materials employed in ALIBs. They can also be stably cycled in various aqueous electrolytes for hundreds of charge/discharge cycles, showing high potential for high-performance AMIBs. In addition, α-MnO2 nanosheets and commercial Mn3O4 have also been investigated as cathode for magnesium storage,310,311 which was operated in the potential range up to 1.50 V (vs. SCE) and 0.95 V (vs. Ag/AgCl) with discharge capacities of 145 mA h g−1 and 105 mA h g−1 at low current densities, respectively.
Regarding anode, due to the negative redox potential of magnesium metal, it is irreversible to utilize it as anode in aqueous or moisture-contaminated nonaqueous electrolytes. Similar to coated/protected lithium metal anode for ALIBs, the strategy of coating/protecting magnesium metal anode with a thin membrane should be a feasible approach to initiate full cells.312–314 Still, most reported aqueous magnesium-ion electrochemistry were only investigated in a half cell configuration due to the absence of proper electrochemical couples136,315 that originates from limited Mg2+ host materials316–323 and the narrow voltage window of aqueous electrolytes.324,325 Examples of full AMIBs are few. Zhang et al. reported the utilization of FeVO4 as anode for AMIBs, which was operated in the potential range of −1.0 to 0 V (vs. SCE) with a specific capacity over 180 mA h g−1 at 50 mA g−1.326,327 However, when coupled with Mg-OMS-1 cathode, the obtained AMIB exhibited sloping discharge profiles, whose midpoint voltage was just over 0.4 V, showing poor practicability. Chen et al. reported a full AMIB by employing a PBA type nickel hexacyanoferrate cathode (NiHCF) coupled with a polyimide anode.328 Its working principle involved Mg2+ intercalation/de-intercalation at cathode side and reversible enolization at anode side. CV profiles indicated that magnesium ions can be extracted from PBN cathode successfully prior to oxygen evolution and be inserted into polyimide anode ahead of hydrogen evolution (Fig. 16d). The AMIB delivered an initial capacity of 33 mA h g−1 (based on the mass of both active materials) and achieved 60% capacity retention after 5000 charge/discharge cycles within the voltage range of 0–1.55 V. It showed no obvious discharge plateau, and the midpoint voltage was at around 0.75 V (Fig. 16e). Furthermore, due to the shortage of AMIB anodes, Soundharrajan et al. constructed a magnesium–zinc hybrid battery by using a Mg2+ intercalative cathode of MgMn2O4 and a zinc metal anode, which benefited from the low redox potential of zinc stripping/plating and thus exhibited a high midpoint voltage of 1.35 V with a tilting plateau at 1.32 V (Fig. 16f and g).322 Nevertheless, to facilitate the development of high-performance AMIBs for practical applications, more electrode materials with high capacities and suitable redox potentials are to be explored.
It has been demonstrated that aqueous electrolytes could promote Ca2+ diffusion in electrodes, as calcium ions are hydrated by water molecules, the shielding effect of water weakens their charge density and thus enables their fast diffusion.28,329–331 Large channels allowing for Ca2+ diffusion are also necessary for electrode materials. Several research groups including Cui's group have demonstrated that calcium ions could rapidly diffuse into a PBA type electrode of NiHCF that possesses large open interstitial sites and channels within the cyano-bridged perovskite framework.136,329 This PBA cathode exhibited a tilting discharge plateau at 0.62 V (vs. SCE). However, its relatively low capacity (around 50 mA h g−1) and cycle instability during initial cycles inhibited its practical applications. The PBA cathode of K2BaFe(CN)6 was also investigated in 1 M Ca(NO3)2 aqueous electrolyte, although showing a flat plateau, its redox potential was located at around 0.15 V (vs. Ag/AgCl), which was too low for a cathode material.332 Lee et al. studied the effect of the concentration of aqueous electrolytes on electrochemical performances of CuHCF cathode for AMIBs. They found that super-concentrated electrolyte could enhance the electrochemical performances by reducing the hydration number and radius of hydrated calcium ionms.333,334 The capacity was ca. 13% higher than that in dilute electrolyte. Besides, as shown in Fig. 17a, ca. 97% of the initial capacity could be maintained after 150 cycles in the super-concentrated electrolyte. In contrast, only ca. 58% of the initial capacity was retained in the dilute electrolyte. This may be attributed to the suppressed structural collapse of CuHCF electrode in super-concentrated electrolyte.333 In addition, cyclability gradually improved with the rise of electrolyte concentration.334 The capacity retention after 1000 cycles in 1 M Ca(NO3)2 electrolyte was only 49%, whereas 89% was realized after 5000 cycles in 8.4 M Ca(NO3)2 electrolyte. Compared to the previous reported PBA cathodes,136,329 this was a huge improvement. The authors proposed two potential reasons for the well maintenance of CuHCF structure in the 8.4 M electrolyte: (1) the dissolution of transition metal ions from CuHCF into electrolyte was suppressed by the highly concentrated Ca2+, and (2) the repetitive hydrated Ca2+ intercalation/de-intercalation-induced structural stress in the CuHCF electrode was relatively weak as Ca2+ have a smaller hydration radius in the 8.4 M electrolyte than in 1 M electrolyte. Whereas, the tilting discharge plateau of CuHCF became less obvious with the increase of electrolyte concentration. The midpoint voltage was at around 0.51 V (vs. SCE) in 8.4 M Ca(NO3)2 electrolyte (Fig. 17b).
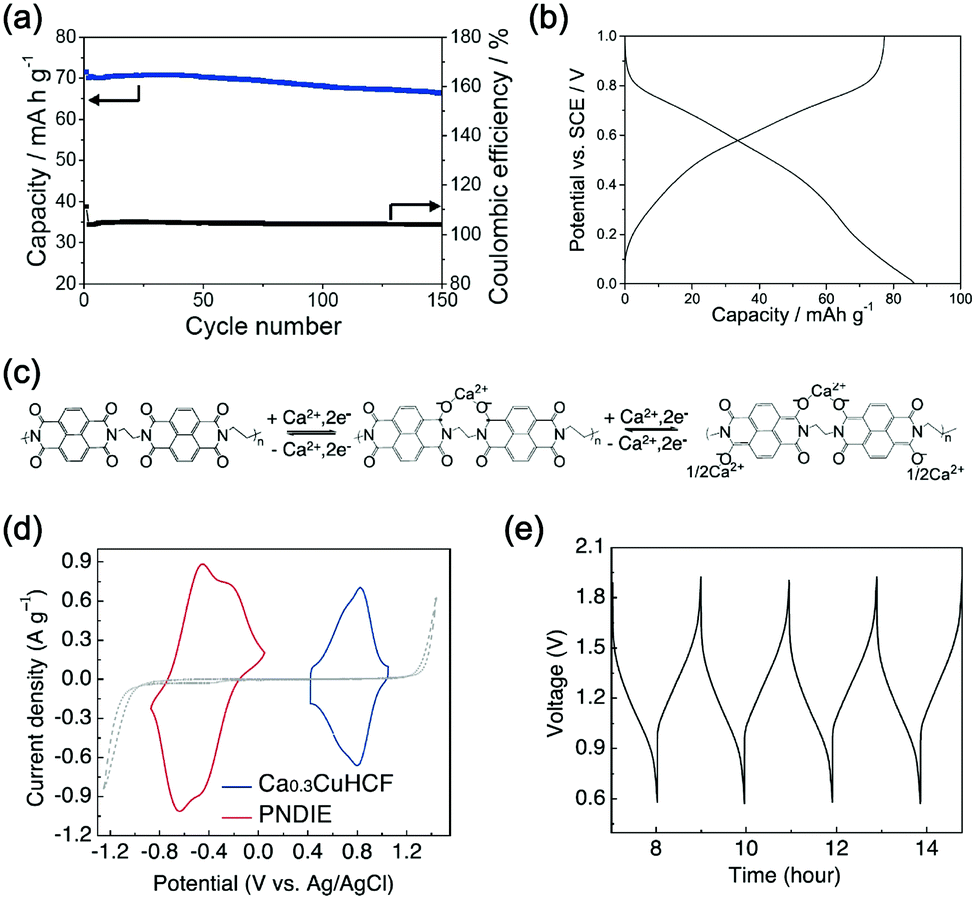 | ||
| Fig. 17 (a) Cycle test of the CuHCF cathode in 8.37 M Ca(NO3)2 aqueous electrolyte at 2C. Reproduced with permission from ref. 333, Copyright 2016 The Chemical Society of Japan. (b) GCD curves of the CuHCF cathode in 8.4 M Ca(NO3)2 aqueous electrolyte at 0.2C. Reproduced with permission from ref. 334, Copyright 2018 Elsevier. (c) Illustration of the possible electrochemical redox reactions of PNDIE anode. (d) CV curves of PNDIE anode and Ca0.3CuHCF cathode in 2.5 M Ca(NO3)2 aqueous electrolyte at 1 mV s−1. (e) GCD curves of the PNDIE//Ca0.3CuHCF full cell at 1C rate (450 mA g−1, according to the total mass of active materials). Reproduced with permission from ref. 335, Copyright 2017 Wiley-VCH. | ||
A low-cost aqueous full ACIB was fabricated based on a Ca0.3CuHCF cathode and a poly[N,N′-(ethane-1,2-diyl)-1,4,5,8-naphthalenetetracarboxiimide] (PNDIE) anode.335 As depicted in Fig. 17c, each reduction step to form radical anion and dianion was associated with Ca2+ coordination to the enolate groups of PNDIE with neglect change and damage to its framework, as charges were redistributed within the conjugated aromatic molecule.336 Typical CVs of the two electrodes are displayed in Fig. 17d, clearly showing redox potentials of both cathode and anode located within the electrochemical stability window of the 2.5 M Ca(NO3)2 aqueous electrolyte. The redox peaks of PNDIE anode were at −0.44 V/−0.19 V and −0.65 V/−0.45 V (vs. Ag/AgCl), indicating a two-step electron transfer during calciation/de-calciation. The redox peak of Ca0.3CuHCF cathode was also well-defined at 0.83 V/0.80 V (vs. Ag/AgCl), resulting in a full ACIB with a midpoint voltage of 1.13 V that showed no obvious discharge plateau (Fig. 17e). Another aqueous full ACIB was fabricated by using barium hexacyanoferrate (BaHCF), meso-carbon micro beads (MCMB) and 1 M Ca(ClO4)2 as cathode, anode and electrolyte, respectively.337 The BaHCF cathode exhibited a reduction peak at 0.28 V and an oxidation peak at 0.34 V (vs. Ag/AgCl), and the MCMB anode showed broad redox peaks at −0.6 V and −0.4 V (vs. Ag/AgCl). Consequently, the full battery delivered nearly 40 mA h g−1 capacity (based on the mass of cathode material) within the voltage range of 0–2.0 V, while it showed no discharge plateau and its midpoint voltage was just around 0.6 V. Like the case of AMIBs, the limited choices of electrode materials, especially anode materials, substantially restrict the advancing of high-performance ACIBs. Further simulations and experiments are needed to discover new electrode materials with desired electrochemical characteristics.
Currently, aluminium metal cannot be directly employed as anode in aqueous electrolytes as its electrochemical plating/stripping potentials are far beyond the stable voltage window of water. Anatase TiO2 with good chemical stability and nontoxicity has been widely studied in lithium storage,347 and its feasibility in aluminium storage has also been validated. Liu et al. first demonstrated the reversible intercalation/de-intercalation of Al3+ into/from anatase TiO2 nanotubes in 1 M AlCl3 aqueous electrolyte.348 Typically, a large redox pair can be observed at around −1.26 V/−0.84 V (vs. SCE) (Fig. 18a). Al3+ intercalation/de-intercalation occurred prior to HER (−1.43 V vs. SCE) because the strong solvation of aluminium ions in aqueous electrolytes resulted in a high overpotential for hydrogen evolution.349 Based on the analysis of electrochemical kinetics, it was found that solid phase diffusion during Al3+ intercalation/de-intercalation was dominant for the anatase TiO2 nanotube arrays. The electrode delivered a capacity of 75 mA h g−1 under the current density of 4 mA cm−2. Besides, single-crystal black anatase TiO2 nano-leaves were subsequently prepared via a solution plasma processing technique by He et al.350Fig. 18b shows a low-magnification image of the as-prepared 2D willow leaf-like TiO2 nanostructure. These TiO2 nano-leaves demonstrated better Al3+ storage performance than commercial white anatase TiO2 as the nano-leaves exhibited a more negative potential for Al3+ de-intercalation and a more positive potential for Al3+ intercalation (Fig. 18c), indicating that the electrochemical processes were more reversible. CV curves and electrolyte pH values remained almost unchanged after cycling, demonstrating that the electrochemical reactions were stable and reversible, and HER was successfully avoided. Fig. 18d reveals that the capacities of the TiO2 nano-leaves were 259.7 and 141.3 mA h g−1 at 0.1 A g−1 and 2 A g−1, respectively, significantly higher than commercial TiO2 electrodes. Such remarkable difference arose from the fast electron transport and Al3+ diffusion in TiO2 nano-leaves due to their short diffusion path. In addition, it is reported that electrically conductive agents such as CNTs and graphene are essential in facilitating Al3+ intercalation/de-intercalation in TiO2 electrode.351,352 Study estimated that graphene could remarkably enhance Al3+ ion diffusion coefficient in TiO2 by 672 times.353
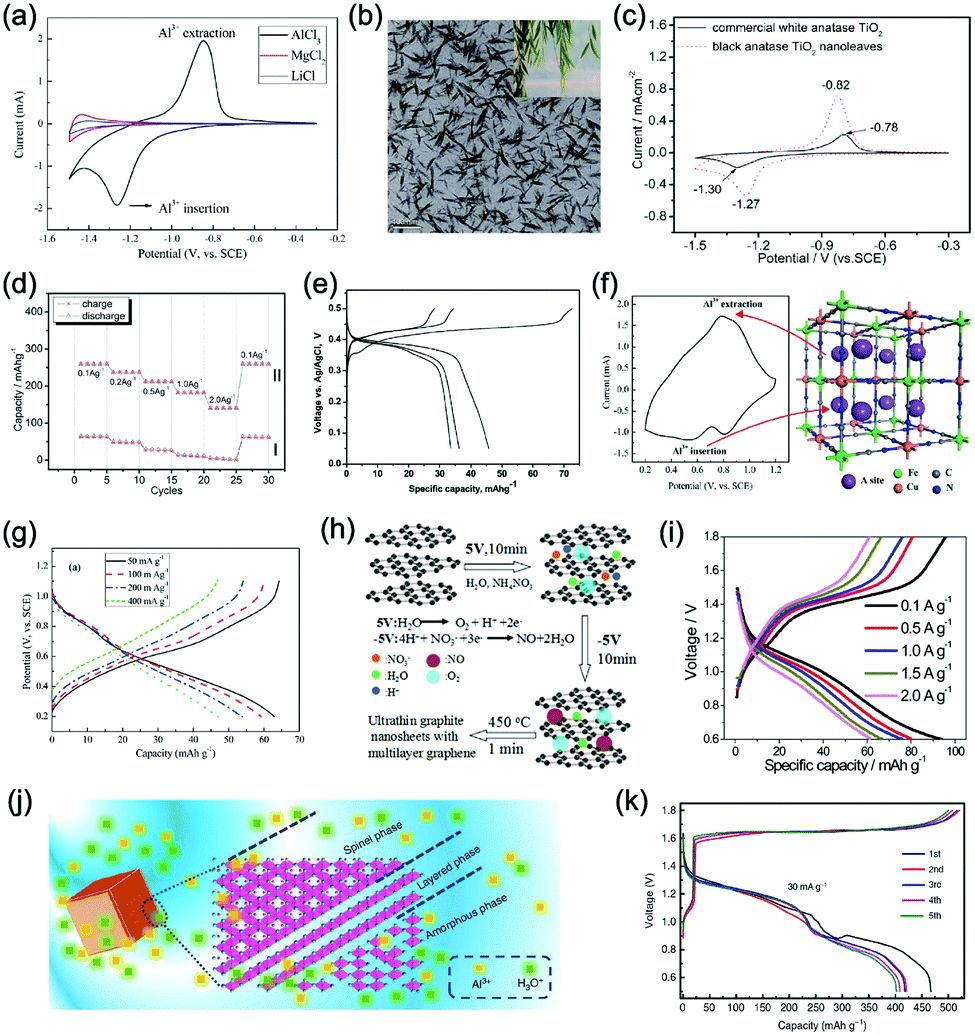 | ||
| Fig. 18 (a) CV curves of the anatase TiO2 nanotubes in 1 M AlCl3, MgCl2 and LiCl aqueous electrolytes at 20 mV s−1. Reproduced with permission from ref. 348, Copyright 2012 Royal Society of Chemistry. (b) Low magnification TEM image of the black anatase TiO2 nano-leaves. Inset is a photograph of willow leaves. (c) Comparison of CV profiles of the black anatase TiO2 nano-leaves and commercial white anatase TiO2 at 1 mV s−1. (d) Comparison of rate performance of (I) commercial white anatase TiO2 and (II) black anatase TiO2 nano-leaves. Reproduced with permission from ref. 350, Copyright 2014 Royal Society of Chemistry. (e) GCD curves of the NVP cathode in aqueous AlCl3 electrolyte of the initial several cycles at 10 mA g−1. Reproduced with permission from ref. 356, Copyright 2018 Elsevier. (f) CV curve of the CuHCF cathode in aqueous Al2(SO4)3 electrolyte with a schematic illustration showing the positions of Al3+ in CuHCF framework. (g) GCD curves of the CuHCF cathode in aqueous Al2(SO4)3 electrolyte at various current densities. Reproduced with permission from ref. 357, Copyright 2015 Royal Society of Chemistry. (h) Synthesis route of the ultrathin graphite nanosheet cathode. (i) GCD curves of the Zn//graphite nanosheet full cell at various current densities. Reproduced with permission from ref. 359, Copyright 2016 American Chemical Society. (j) Schematic illustration of the structure of AlxMnO2·nH2O. (k) GCD curves of the Al//AlxMnO2·nH2O full cell at 30 mA g−1 of the initial 5 cycles. Reproduced with permission from ref. 360, Copyright 2019 Springer Nature. | ||
Not many stable cathode materials have been discovered for AAIBs so far. Orthorhombic vanadium pentoxide (V2O5) with a layered structure built up from VO5 square pyramids could serve as cathode for ALIBs and AZIBs, and its xerogel (xero-V2O5) consisting of V2O5 bilayers with water in between intrigues significant changes in electrochemistry.354 The intercalation of aluminium ions into V2O5 xerogel in aqueous electrolytes was initially investigated by González et al.355 This xero-V2O5 exhibited a redox pair at around −0.08 V/−0.28 V (vs. mercury–mercurous sulfate electrode, MSE) with a discharge capacity of 250 mA h g−1 at the current density of 20 mA g−1 in 1 M LiCl aqueous electrolyte. While this capacity rapidly decayed to 130 mA h g−1 at the 5th cycle, showing poor cycle stability. Nacimiento et al. reported a NASICON-type NVP as cathode for AAIBs using AlCl3 dissolved in oxygen-free water as aqueous electrolyte.356 The NVP cathode showed a 47 mA h g−1 specific capacity at the current density of 10 mA g−1 with a flat discharge plateau at around 0.37 V (vs. SCE) (Fig. 18e). They proposed a mechanism wherein NVP loses Na+ during the first charging cycle, and subsequent discharging leads to both bulk intercalation and surface storage of Al3+, both of which contribute to the resultant capacity. Multiple characterization techniques have clearly demonstrated the successful Al3+ intercalation/de-intercalation, despite the poor cycle stability that needs further optimization. Moreover, although monovalent and divalent cations can readily insert into PBAs framework as demonstrated in many battery systems, the intercalation chemistry of trivalent cations that would carry more charges to PBAs has rarely been studied. Liu et al. investigated CuHCF as cathode material for AAIBs for the first time, achieving a specific capacity of 64 mA h g−1 at 50 mA g−1 with a midpoint voltage of 0.52 V (vs. SCE) in 0.5 M Al2(SO4)3 aqueous solution (Fig. 18f and g).357 Broadened CV peaks for Al3+ intercalation were observed in CuHCF, suggesting complex mechanisms and poor insertion kinetics.329,357 This CuHCF cathode sustained 1000 charge/discharge cycles, which is far superior to the above-introduced cathode materials. While the capacity retention of 54.9% is still not satisfactory compared to relatively mature ALIBs and AZIBs.
A full AAIB coupling CuHCF cathode and TiO2 anode was fabricated by Holland et al., showing a relatively high tilting discharge plateau at 1.45 V.358 While its specific capacity based on both cathodes was merely 10.6 mA h g−1, making the good capacity retention of 70% over 1750 charge/discharge cycles almost meaningless. The authors stated that the slight shift in neutron diffraction parameters was not sufficient to verify the major mechanism of charge storage for TiO2 anode, which should be via either Al3+ intercalation or surface adsorption. To achieve higher capacity, an aqueous rechargeable Al3+–Zn2+ dual-ion hybrid battery was developed based on zinc metal anode and ultrathin graphite nanosheet cathode (Fig. 18h) in aqueous Al2(SO4)3–Zn(CH3COO)2 electrolyte.359 The stripping/plating of zinc metal anode and the Al3+ intercalation/de-intercalation into/from the outer graphene layers of graphite cathode led to a discharge capacity of 94 mA h g−1 for the hybrid Zn//graphite battery with a midpoint voltage of 1.0 V (Fig. 18i). Besides, this hybrid battery could be rapidly charged within 2 min while maintaining a high capacity and demonstrated a good cycle stability with 94% capacity retention after 200 cycles. More recently, Wu et al. fabricated an AAIB by successfully utilizing an aluminium metal anode in 4 M aluminium trifluoromethanesulfonate (Al(OTF)3) aqueous electrolyte.360 In this electrolyte, passivation occurred on aluminium metal that effectively suppressed hydrogen evolution, pushing its onset potential from 1.1 V to −0.3 V (vs. Al3+/Al), allowing for the reversible stripping/plating of aluminium metal. An AlxMnO2·nH2O cathode was synthesized via incorporating water and Al3+ into spinel Mn3O4 structure through in situ electrochemical transformations (Fig. 18j). It was found that the aqueous electrolyte and crystal water in this cathode could screen the electrostatic interaction between aluminium ions and cathode frameworks, thus enabling reversible electrochemical trivalent processes. The resultant Al//AlxMnO2·nH2O full cell in 4 M Al(OTF)3 yielded an open circuit voltage over 1.6 V with a midpoint voltage at 1.05 V. Although its working voltage is not the highest among all AAIBs, it exhibited a remarkable discharge capacity of 467 mA h g−1 at 30 mA g−1, leading to a record high energy density of 481 W h kg−1 (Fig. 18k). The strategy of enabling passivation of aluminium metal in aqueous electrolyte and tailoring polarity between metal ions and host frameworks could open a new way to explore high-voltage and high-energy reversible ARMB configurations.
On top of that, the type of electrolyte has a prominent effect on Al3+ intercalation behaviour. For example, it was difficult for Al3+ to intercalate into TiO2 nanotube arrays in aqueous Al2(SO4)3 electrolyte.361 The same phenomenon was observed in an aprotic electrolyte system: no redox peaks were shown in the electrolyte of Al triflate in PC/THF from −0.75 V to 4.2 V, while an obvious redox pair was detected in the electrolyte of AlCl3 in [EMIm]Cl.340 It is also revealed that Al3+ intercalation/de-intercalation in MoO3 is favoured in specific types of aqueous electrolyte. Compared with Al2(SO4)3 and Al(NO3)3, AlCl3 electrolyte provides higher Al3+ storage capacity, superior long-term stability and less polarization. In detail, the Al3+ storage capacity of MoO3 in the first discharge cycle was found to be 680 mA h g−1 in AlCl3 electrolyte, which is the highest reported value so far for AAIBs.362 Therefore, it is anticipated that Cl− played a significant role in facilitating the reversible and effective Al3+ intercalation/de-intercalation,361 which could shed light on the research of Al3+ storage in other transition metal electrodes (e.g., TiO2, V2O5, etc.).
This section discusses aqueous multivalent metal-ion batteries including AMIBs, ACIBs and AAIBs. Redox potentials of their electrode materials are summarized and compared. As shown in Fig. 19, not many electrode materials have been developed so far, the studies of AMIBs, ACIBs and AAIBs are still in their infancy. The multivalence of their metal ions usually induces strong electrostatic interaction with anions in electrode hosts, resulting in sluggish kinetics or even inertness. It should be noted that PBA-type materials are extremely versatile in ARMBs, as they show excellent capability to host various metal ions including not only Li+, Na+, K+, and Zn2+, but also Mg2+, Ca2+, and Al3+. In-depth studies on PBAs may help facilitate the advancement of AMIBs, ACIBs and AAIBs.
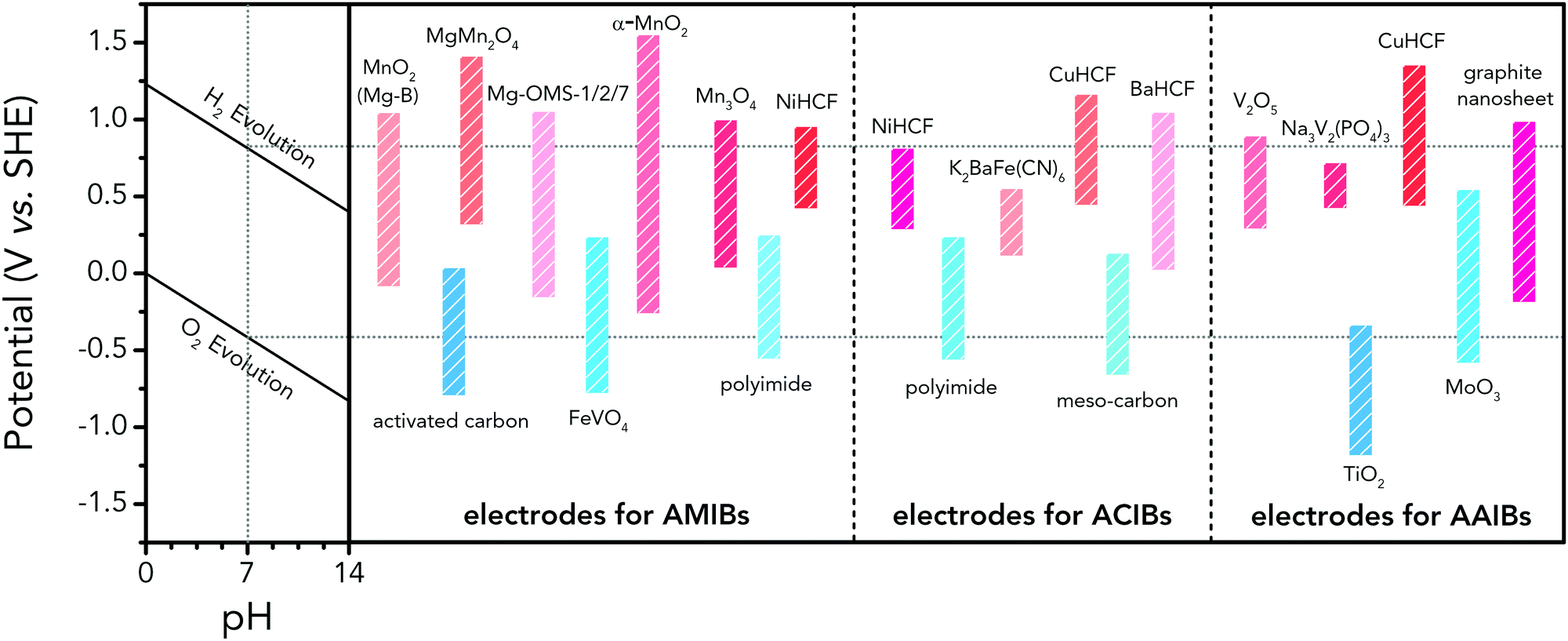 | ||
| Fig. 19 Comparison of redox potentials of representative electrode materials for AMIBs, ACIBs and AAIBs. Red-colour columns and blue-colour columns represent cathodes and anodes, respectively. | ||
We further compare the voltage characteristics including midpoint voltage and plateau voltage of various ARMB systems in Fig. 20. As revealed, most reported ARMBs exhibit a midpoint voltage less than 1.5 V. The majority of the ARMBs do not yield a flat discharge plateau, especially for AMIBs, ACIBs and AAIBs, all of which just show sloping discharge profiles. Generally speaking, the midpoint voltages of AMIBs, ACIBs and AAIBs are lower than those of ALIBs, ASIBs, AKIBs and AZIBs, except for the cell utilizing TiO2 anode, as TiO2 could provide a significant low redox potential (Fig. 19). The difference in midpoint voltage is small among all these ARMBs, which is mainly due to the narrow voltage window of aqueous electrolytes that substantially confines cell voltage. In the following sections, we will focus on the strategies that can effectively raise cell voltage.
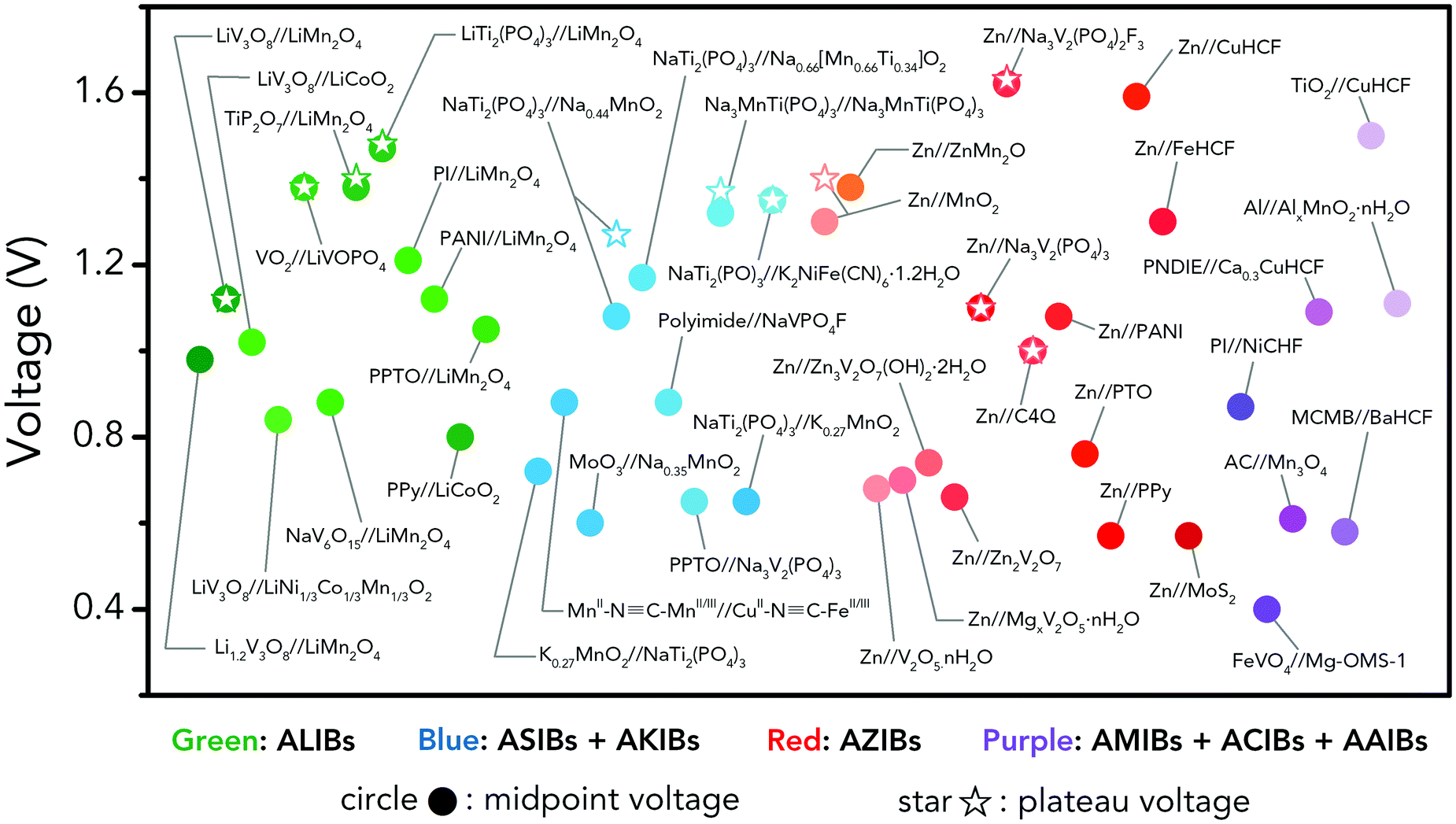 | ||
| Fig. 20 Voltage comparison among various ARMB systems. Solid circles represent midpoint voltages, and hollow stars at the same x-axis position represent corresponding plateau voltages (the voltage of the first discharge plateau is adopted if a battery yields more than one plateau). Some sodium–potassium hybrid batteries are also included. | ||
4. Towards high-voltage ARMBs: strategies and advancements
Regarding the constrained cell voltage of ARMBs in aqueous electrolytes, in this section, we discuss five major categories of strategies that have been demonstrated effective in raising cell voltage. These strategies involve the screening of suitable electrode materials to fully utilize the electrochemical stability window of normal aqueous electrolytes, the isolating of water or the avoiding of electrode-water contact, and the broadening of electrolyte voltage window. Generally, type of electrode material, electrolyte pH, electrolyte concentration, etc., all have a significant influence on the parasitic gas evolution at electrode–electrolyte interface. By adopting effective strategies and cautiously tuning parameters, ARMBs with higher operating voltage that even exceeds 2 V could thus be realized.4.1 Fully exploit electrolyte voltage window
Electrode materials play a vital role in determining cell voltage, and the voltage window of electrolytes decides the feasibility of pairing up electrodes. Thus, with a given electrolyte, the voltage of a battery can be regulated by carefully selecting electrodes whose redox potentials are well within the electrolyte voltage window. By pairing a cathode with a more positive redox potential, with an anode with a more negative redox potential, the battery voltage can be considerably raised in conventional aqueous electrolytes. Besides, in terms of electrode materials, the core of suppressing hydrogen and oxygen evolution is to utilize anode with high hydrogen evolution overpotential and cathode with high oxygen evolution overpotential. This section highlights the studies on achieving high-voltage ARMBs with conventional aqueous electrolytes via examining the redox potentials as well as the overpotentials of both cathodes and anodes.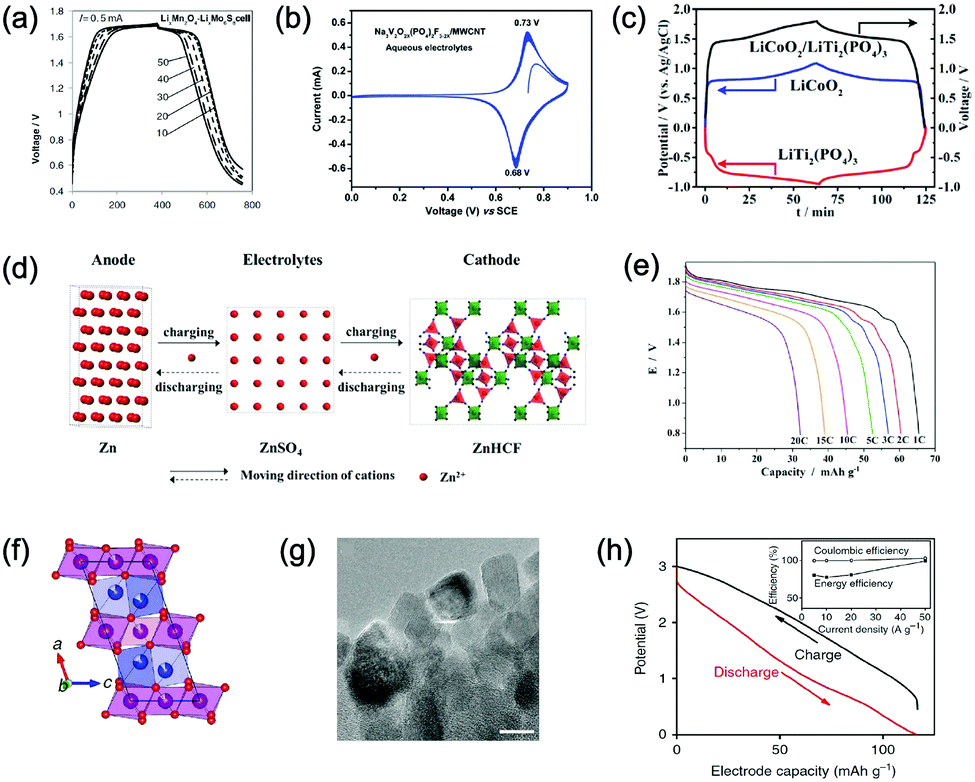 | ||
| Fig. 21 (a) GCD curves of the Mo6S8//LiMn2O4 full cell in 2 M Li2SO4 electrolyte for consecutive 50 cycles at 0.5 mA. Cycle numbers are indicated. Reproduced with permission from ref. 363, Copyright 2012 Springer Nature. (b) CV curves of the Na3V2O2x(PO4)2F3−2x/CNTs cathode at 0.2 mV s−1 in aqueous electrolyte. Reproduced with permission from ref. 367, Copyright 2015 Royal Society of Chemistry. (c) GCD curves of individual electrode and the corresponding LiTi2(PO4)3/C//LiCoO2 full cell in aqueous Li2SO4 electrolyte. Reproduced with permission from ref. 368, Copyright 2019 Elsevier. (d) A schematic of the Zn//ZnHCF battery. (e) Discharge curves of the Zn//ZnHCF battery at various rates. Reproduced with permission from ref. 258, Copyright 2015 Wiley-VCH. (f) Illustration of the lattice structure of Mn5O8. Purple represents Mn4+, blue represents Mn2+, red represents O, and white indicates the possible unoccupied sites. (g) TEM image of the Mn5O8 particles. (h) GCD curves of the Mn5O8 symmetric cell at 5 A g−1. Inset shows its energy efficiency and coulombic efficiency. Reproduced with permission from ref. 370, Copyright 2016 Springer Nature. | ||
It is most favourable to combine two electrodes with distant redox potentials that are still well within electrolyte voltage window, so that the cell voltage could be maximized. Xue et al. prepared 3D hierarchy LiCoO2 nanorod arrays as cathode and coupled it with LiTi2(PO4)3 anode. The former exhibited redox potentials at around 0.8 V and the latter exhibited redox potentials at around −0.7 V, leading to a tilting discharge plateau of 1.51 V for the resultant ALIB (Fig. 21c).368 Liu et al. utilized the high redox potential of LiMn2O4 cathode and the low redox potential of TiO2, constructed an ALIB showing a high midpoint voltage of around 2.06 V with two discharge plateaus at 2.24 V and 1.90 V.369 In this ALIB, HER was effectively suppressed in the mixed 3.5 M LiCl/0.25 M Li2SO4 electrolyte due to the high anodic hydrogen evolution overpotential, enabling the reversible lithium-ion intercalation/de-intercalation at TiO2 anode side. As the commonly employed anode for AZIBs is zinc metal, the selection of electrode materials to realize high-voltage AZIBs depends on the select of cathode materials. Compared to the widely studied MnO2 cathode, PBA cathodes are inferior in terms of specific capacity, while they offer attractive redox potentials that are usually higher than 1.5 V (vs. Zn/Zn2+). For example, Zhang et al. fabricated ZnHCF for AZIBs, and the resultant full battery exhibited a tilting discharge plateau of around 1.72 V, much higher than the 1.38 V and 1.2 V of Zn//MnO2 battery (Fig. 21d and e).258 Moreover, Shan et al. synthesized surface hydroxylated Mn5O8 electrodes to construct a symmetrical ASIB (Fig. 21f and g).370 This Mn5O8 electrode showed a wide potential window in a 0.1 M Na2SO4 electrolyte between −1.7 V (with an overpotential of 0.64 V towards HER) and 0.8 V (with an overpotential of 0.63 V towards OER), which originated from sluggish HER and OER kinetics that were verified by TAFEL plots. The interaction between hydroxylated interphase on the surface and the unique bivalence structure of Mn5O8 accounted for the suppression of HER and OER, and a two-electron charge transfer mechanism via Mn2+/Mn4+ was identified, providing facile pathway for Na+ diffusion. The as-assembled symmetrical ASIB could be charged to 3.0 V and exhibited remarkable rate capability. While its charge/discharge profiles were sloping, resulted in a midpoint voltage of around 1.0 V, which is relatively low compared to its upper cut-off voltage of 3.0 V (Fig. 21h).
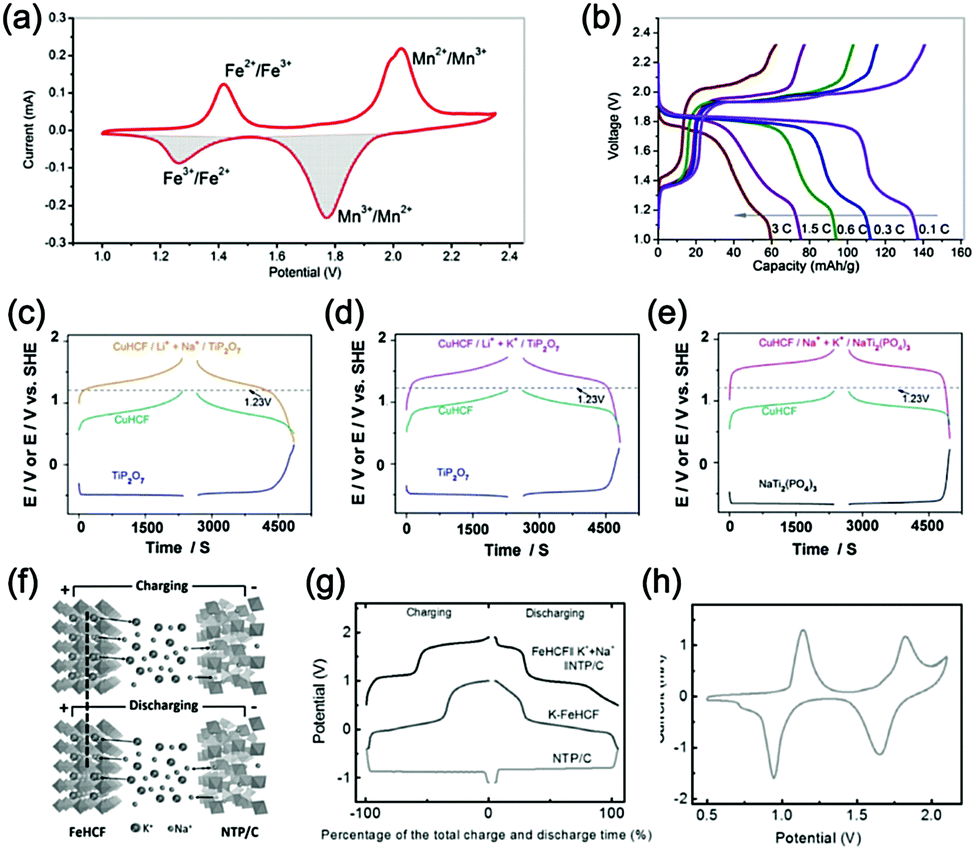 | ||
| Fig. 22 (a) CV curves of the Zn/LiMn0.8Fe0.2PO4 hybrid battery at 0.1 mV s−1 in the aqueous electrolyte of 21 M LiTFSI + 0.5 M ZnSO4. (b) GCD curves of the Zn/LiMn0.8Fe0.2PO4 hybrid battery at various rates. Reproduced with permission from ref. 376, Copyright 2016 Elsevier. (c–e) GCD curves of aqueous mixed-ion batteries of CuHCF/Li+ + Na+/TiP2O7, CuHCF/Li+ + K+/TiP2O7 and CuHCF/Na+ + K+/NaTi2(PO4)3 and their individual electrodes at the rate of 1.5C. Reproduced with permission from ref. 377, Copyright 2017 Royal Society of Chemistry. (f) Illustration of the working mechanism of the K–Na hybrid aqueous battery system. (g) GCD curves of the K–Na hybrid aqueous battery and their individual electrodes (vs. SCE) at 1C rate. (h) CV curves of the K–Na hybrid aqueous battery at 1 mV s−1. Reproduced with permission from ref. 378, Copyright 2018 Wiley-VCH. | ||
Moreover, high-voltage hybrid batteries with dual-intercalative electrodes have also been designed. In a work of Jiang et al., a variety of high-voltage hybrid batteries have been demonstrated with CuHCF as cathode and TiP2O7 or NaTi2(PO4)3 as anode.377 It was found that the intercalation voltage for the ion-selective CuHCF cathode followed the sequence of K+ > Na+ > Li+. As a result, a series of high-voltage batteries based on “M+/N+-dual shuttles” including CuHCF/Li+ + Na+/TiP2O7, CuHCF/Li+ + K+/TiP2O7 and CuHCF/Na+ + K+/NaTi2(PO4)3 were obtained with midpoint voltages at 1.36 V, 1.42 V and 1.55 V, respectively (Fig. 22c–e). These hybrid batteries relied on two electrochemical processes: co-intercalation/de-intercalation of M+ (small ionic radius) and N+ (large ionic radius) at CuHCF cathode, and release/storage of M+ at the ion-selective TiP2O7 or NaTi2(PO4)3 anode. Similarly, Liu et al. constructed a high-voltage potassium–sodium hybrid battery based on a K-FeHCF cathode and a carbon-coated NaTi2(PO4)3 (NTP/C) anode.378 Utilizing the unique cation selectivity of both electrodes, the resultant hybrid battery using mixed-ion electrolyte (containing both Na+ and K+) involved the K+ intercalation/de-intercalation at K-FeHCF cathode and Na+ intercalation/de-intercalation at NTP/C anode (Fig. 22f), which exhibited two-stage discharge profiles with two flat plateaus at 1.70 V and 0.98 V, respectively (Fig. 22g and h). It delivered a high capacity of 160 mA h g−1 at a 0.5C and retained 94.3% of its initial capacity after 1000 charge/discharge cycles at 60C. Its high energy density of 69.6 W h kg−1 (based on the mass of both active materials) is superior to that of traditional lead–acid, Ni–Cd and Ni–MH batteries.
4.2 Regulate electrolyte pH
As the potentials of OER and HER vary with pH values, theoretically, the electrochemical stability window of aqueous electrolytes can be deliberately adjusted to accommodate electrode materials with even more positive or negative redox potentials by regulating electrolyte pH. Moreover, electrode materials may exhibit different electrochemical behaviours that occur at different potentials depending on electrolyte pH. In this section, studies employing aqueous electrolytes with regulated pH values for achieving more stable cycle performance and/or higher cell voltage are discussed. This section is divided into two parts, focusing on regulating the general pH and designing multiple electrolytes with varied pH values, respectively. Although these strategies were majorly adopted in supercapacitors, alkaline batteries and metal-ion batteries with conversion-type electrodes, their potential in ARMBs should not be neglected.First, redox reactions of some electrodes that beyond electrolyte voltage window at neutral pH can be reversibly initiated under certain pH conditions. For example, Liu et al. fabricated vertically oriented anatase TiO2 nanotube arrays as lithium-ion intercalative anode to pair with H+ insertive α-NiOH cathode.379 CV curve of this anatase TiO2 in a mixed alkaline LiOH/KOH aqueous electrolyte revealed redox peaks at −1.39 V and −1.11 V (vs. Hg/HgO) attributing to lithium-ion intercalation/de-intercalation, and the HER peak was notably shifted to a more negative potential of −1.53 V (vs. Hg/HgO), suggesting that HER was substantially suppressed in this alkaline electrolyte, allowing for the prior occurrence of lithium-ion intercalation/de-intercalation in TiO2 anode. As a result, the assembled battery yielded a flat high-voltage plateau at 1.74 V.
Second, the better utilization of electrode materials with distant potentials by regulating general pH can result in more stable performances. Pei et al. first investigated the spinel LiMn2O4 cathode in aqueous electrolytes with different pH values. In their studies, OER occurred more easily with increasing pH values (Fig. 23a), which is in accordance with the Pourbaix diagram shown in Fig. 3c. Therefore, the battery employing a LiMn2O4 cathode and a zinc foil anode displayed better cycle stability at pH = 2 than at pH = 5.52 For the lithium-ion intercalative FePO4, it has long been used as a cathode for conventional LIBs due to its potential of 3.4 V vs. (Li+/Li).380 By properly tuning electrolyte pH and pairing with a suitable cathode, Wang and co-workers successfully utilized olivine FePO4 and amorphous FePO4·2H2O as anodes for ALIBs.54 Although these anodes are stable in a broad pH range, according to calculation, electrolyte pH should not be too low, which was optimized at pH = 5. Otherwise full capacity of anodes cannot be utilized due to severe HER at lower pH values. The authors further pointed out that many common cathodes of LIBs are confined to alkaline conditions because of the instability of anodes in acidic conditions. If paired with a stable anode and put in acidic an electrolyte, cathodes such as LiFePO4 and LiCoO2 should exhibit more stable performances as OER would be effectively suppressed.
 | ||
| Fig. 23 (a) CV curves of LiMn2O4 in electrolytes with varied pH values. Reproduced with permission from ref. 52, Copyright 1996 Elsevier. (b) GCD curves of the Zn//Co3O4 battery in alkaline and mild electrolytes. Reproduced with permission from ref. 58, Copyright 2018 Royal Society of Chemistry. (c) Illustration of the configuration of the Zn//KMnO4 battery using an acidic–alkaline dual-electrolyte. Reproduced with permission from ref. 382, Copyright 2013 Royal Society of Chemistry. (d) Illustrative concept of the cell with KOH and Na2SO4 as catholyte and anolyte, respectively. (e) Electrode potential limits vs. voltage measured at 40 mA g−1; (f) CV curves of the cell for various voltage windows up to 1.6 V measured at 0.4 mV s−1. Reproduced with permission from ref. 383, Copyright 2018 Wiley-VCH. | ||
Third, it is possible that the change of electrolyte pH value leads to the change of redox potential of electrode materials. Although no intercalative electrode materials of ARMBs have been reported to manifest such feature, experiences can be borrowed from other conversion-type electrodes. One example is the Zn//Co3O4 battery developed by our group.58 2 M ZnSO4 + 0.2 M CoSO4 mild electrolyte was used to replace traditional alkaline electrolyte for the first time, and the OER potential for Co(III) rich-Co3O4 cathode was found shifted to a significantly higher position of 2.26 V, compared to 1.93 V in alkaline electrolyte (vs. Zn/Zn2+). Obviously, such broadened voltage window is ascribed to the lower pH value of electrolyte, as revealed in Fig. 3c. Furthermore, we found that the redox reaction of Co3O4 in mild electrolyte was notably different from that in alkaline electrolyte, which was a highly reversible conversion reaction that yielded a higher cell voltage (Fig. 23b). The as-fabricated Zn//Co3O4 battery in mild electrolyte exhibited a high discharge voltage plateau of 1.9 V, as well as an exceptional cycling stability (92% capacity retention after 5000 cycles at 4 A g−1).
For electrodes of alkaline batteries, their redox potentials may shift with pH variation according to the Nernst equation. By utilizing this, Li et al. developed a battery with three electrolytes.381 PbO2 cathode was placed in acidic H2SO4 electrolyte, and NiMHx anode was placed in alkaline KOH electrolyte. To avoid self-neutralization, an additional neutral K2SO4 electrolyte separated by cation exchange membrane from acidic electrolyte and anion exchange membrane from alkaline electrolyte was used in between instead of applying a bipolar membrane, which was expensive and vulnerable to large current density. Due to the positive shift of PbO2 potential and negative shift of NiMHx potential, a battery with an open circuit voltage up to 2.64 V was obtained, which was remarkable compared to 1.92 V of conventional lead–acid battery in acidic electrolyte and 1.40 V of Ni–MH battery in alkaline electrolyte. The battery delivered a flat high-voltage discharge plateau at around 2.12 V.
OER and HER also vary with pH. If the high OER potential in acidic condition and the low HER potential in alkaline condition can be combined, the overall voltage window of electrolytes can be substantially expanded. To realize this, a thin film of ceramic lithium super-ionic conductor (LATSP) was employed to partition acidic and alkaline electrolytes while lithium ions can still pass through, resulting an acidic-alkaline dual-electrolyte (Fig. 23c).382 To exploit the expanded voltage window, KMnO4 with a high working potential of 1.5 V (vs. SHE) was chosen to pair with Zn anode, giving a battery with a high open circuit voltage of 2.8 V and a midpoint voltage of 2.2 V, whose theoretical energy density may be comparable to conventional LIBs. Similar studies were conducted on supercapacitors.383–385 For example, Ratajczak and co-worker experimentally verified the concept of using two aqueous electrolytes of different pH values separated by a cationic exchange membrane to enable a high-voltage electrochemical cell based on carbon materials (Fig. 23d).383 According to calculations employing the Nernst equation, the cell using 0.5 M KOH (pH = 13.2) catholyte and 1.0 M Na2SO4 (pH = 6.6) anolyte were able to operate up to 1.62 V without water decomposition (Fig. 23e and f).
Given the above facts, there is still plenty of room for facilitating the strategy of designing multiple electrolytes to high-voltage electrochemical cells, especially to high-voltage ARMBs. As more cathodes and anodes are being discovered, electrolytes with varied pH values that yield wider voltage window could better accommodate their working potentials, which is promising in developing low-cost and high-energy AMRBs.
4.3 Construct artificial interphase layers
Solid–electrolyte interphase (SEI) in non-aqueous LIBs in situ forms from the decomposition of organic electrolyte species, which can significantly alleviate parasitic reactions and stabilize/expand electrochemical stability windows. Whereas, such protective interphase layer cannot form spontaneously in conventional aqueous electrolytes. This section introduces the strategy of constructing artificial interphase layers to enable high-voltage ARMBs. Generally, two specific methods can be conceived. One is to employ organic–aqueous hybrid electrolyte, where the organic part serves as a thick interphase layer to isolate anodes from the aqueous part of electrolyte, enabling the reversible utilizing of anodes with low working potentials in organic electrolyte. The other is to construct a thin SEI-like layer directly on electrode surfaces, blocking the contact with water molecules, thus suppressing water decomposition and extending voltage window.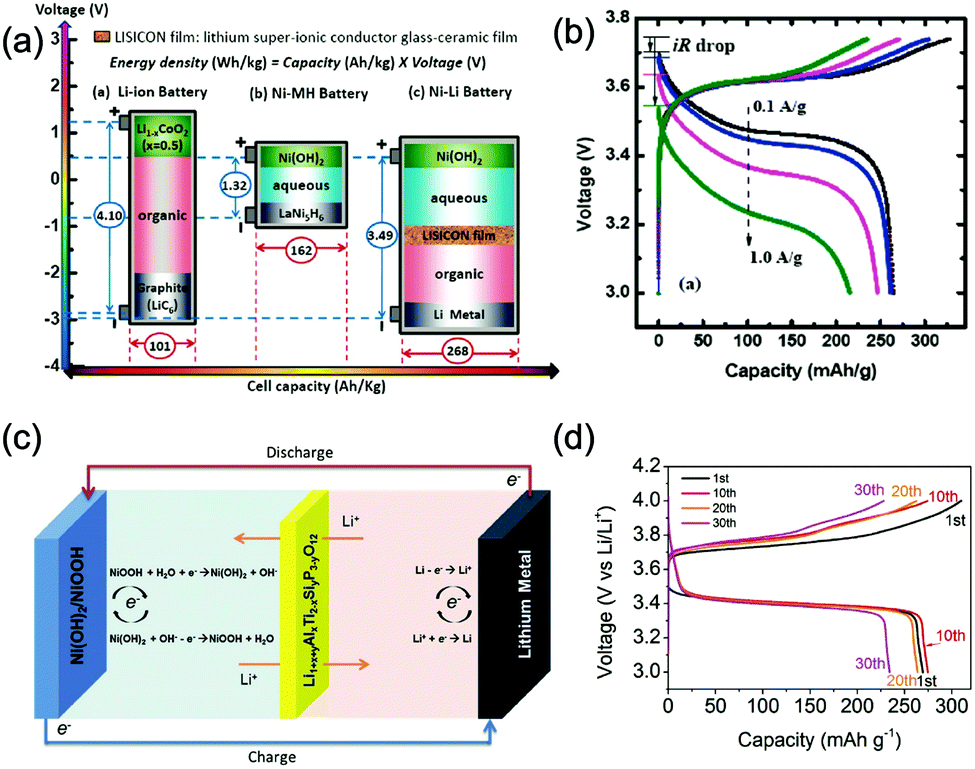 | ||
| Fig. 24 (a) Characteristics and configurations of traditional Li-ion battery, Ni–MH battery, and the proposed Ni–Li battery. (b) GCD curves of the Ni–Li battery between 3.0 and 3.75 V. The battery was discharged at 0.1, 0.2, 0.5, and 1.0 A g−1, respectively. Reproduced with permission from ref. 386, Copyright 2009 American Chemical Society. (c) Illustration of the as-fabricated Li–Ni battery with hybrid electrolyte separated by a LATP film. (d) GCD curves of the Li–Ni battery to 30th cycle at 50 mA g−1. Reproduced with permission from ref. 387, Copyright 2017 Wiley-VCH. | ||
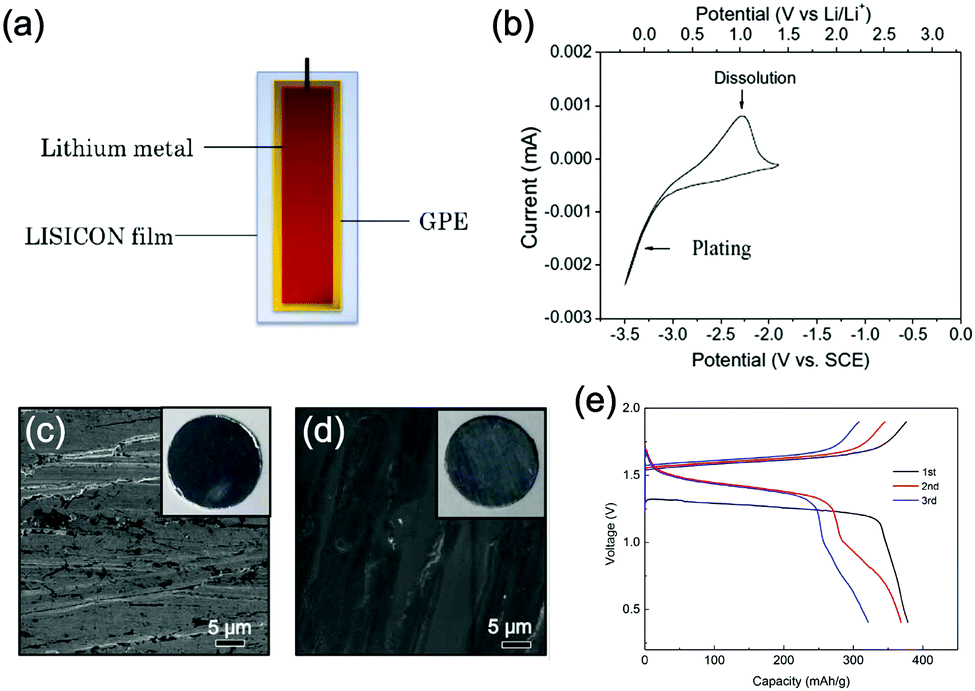 | ||
| Fig. 25 (a) The schematic illustration of the LISICON film-coated lithium metal. GPE represents a gel polymer electrolyte containing organic lithium salt solution. (b) CV curve of the LISICON film-coated lithium metal at 0.1 mV s−1 in 0.5 M Li2SO4 aqueous electrolyte. Reproduced with permission from ref. 388, Copyright 2013 Springer Nature. (c) SEM image of pristine Al metal. (d) SEM image of Al metal after treatment with AlCl3-IL electrolyte. Insets of (c and d) are digital photos of Al metals. (e) GCD curves of the aqueous Al-ion battery employing treated Al metal in 2 M Al(CF3SO3)3 electrolyte at 100 mA g−1 (current density was based on the mass of MnO2 cathode). Reproduced with permission from ref. 393, Copyright 2018 American Association for the Advancement of Science. | ||
An artificial SEI was further designed for aluminium anode to enable its consistent usability in aqueous electrolytes.393 It is known that Al readily forms a high-bandgap passivation layer of Al2O3, which turns Al to be inert in electrochemical processes. Particularly, if used in aqueous electrolytes, higher potential is needed to drive ion transport through the passivation layer, leading to severe hydrogen evolution. It was found that immersing Al in the acidic ionic liquid (IL) electrolyte of AlCl3-[EMIm]Cl for more than one day could induce a tightly bound, IL-enriched interphase on Al surface (Fig. 25c and d), which erodes the passivation coating of Al2O3 and prevents its subsequent formation. Moreover, this interphase is permanent and favourable for aluminium ion transport. By utilizing this artificial SEI, a battery based on Al anode and MnO2 cathode was constructed, which can be reversibly cycled in aqueous Al(CF3SO3)3 electrolyte without notable hydrogen evolution, delivering an energy density up to 500 W h kg−1 due to its high specific capacity and high cell voltage (with a midpoint voltage of 1.37 V and a plateau voltage of 1.40 V) (Fig. 25e).
Based on the above discussion, it is understood that constructing artificial interphase layers usually involves the addition of organic electrolyte, meaning the cost and safety concerns of conventional organic battery systems cannot be completely erased. Although some approaches have been proposed to construct artificial SEI on cathode or anode without the using of organic electrolyte, they just managed to stabilize electrode and achieve better cycle performance, rather than raise cell voltage.394,395 Therefore, the direct and effective strategy of constructing artificial interphase layers still needs further optimization.
4.4 Concentrated aqueous electrolytes
Studies have shown that concentrated aqueous electrolytes have profound influences on electrochemical stability window as well as on potentials of electrode materials. This section divides these studies into three categories with thorough discussion. The first category involves electrolytes with increased concentration of conventional salts, which alters OER/HER potentials and suppresses water decomposition rate. The second category refers to the “water-in-salt”/“water-in-bisalt” systems where the absence of free water molecules and the formation of anodic SEI originating from salt anions jointly prevents the direct interaction between water and electrode surfaces. The third category is about hydrate-melt electrolytes, where no free water molecule exists, and all water molecules participate in ion hydration, resulting in unique physicochemical behaviours. All the three categories of concentrated aqueous electrolytes feature a wider electrochemical stability window and help realize a higher battery voltage.Tomiyasu et al. proposed a novel mechanism that accounts for the concentration-dependent voltage window of electrolytes.59 In their study, they focused on hydrogen bond in aqueous electrolytes, which is a bonding between a hydrogen and an adjacent oxygen within 0.3 nm. It's like the tug-of-war, i.e., the weaker the intermolecular hydrogen bonding, the tougher the intramolecular O–H bonding, and the more difficult the occurrence of water decomposition. Therefore, by using extremely soluble sodium perchlorate (219.6 g can be dissolved in 100 g water at 25 °C), the authors obtained a saturated aqueous solution where the ratio of water molecules to sodium perchlorate molecules was only 3.3. It is comprehensible that the addition of salt disturbs hydrogen bonding between water molecules, as the electronegative oxygen atoms would be more attracted to cations and the electron poor hydrogen atoms would be more attracted to anions. In saturated sodium perchlorate solution, the hydrogen bonding may be eliminated because of the absence of neighbouring water molecules as well as their intense hydration to salt ions (Fig. 26a). Consequently, the voltage window of this solution was expanded to 3.2 V due to the loss of hydrogen bonding and the resultant enhancement of O–H bonding (Fig. 26b).
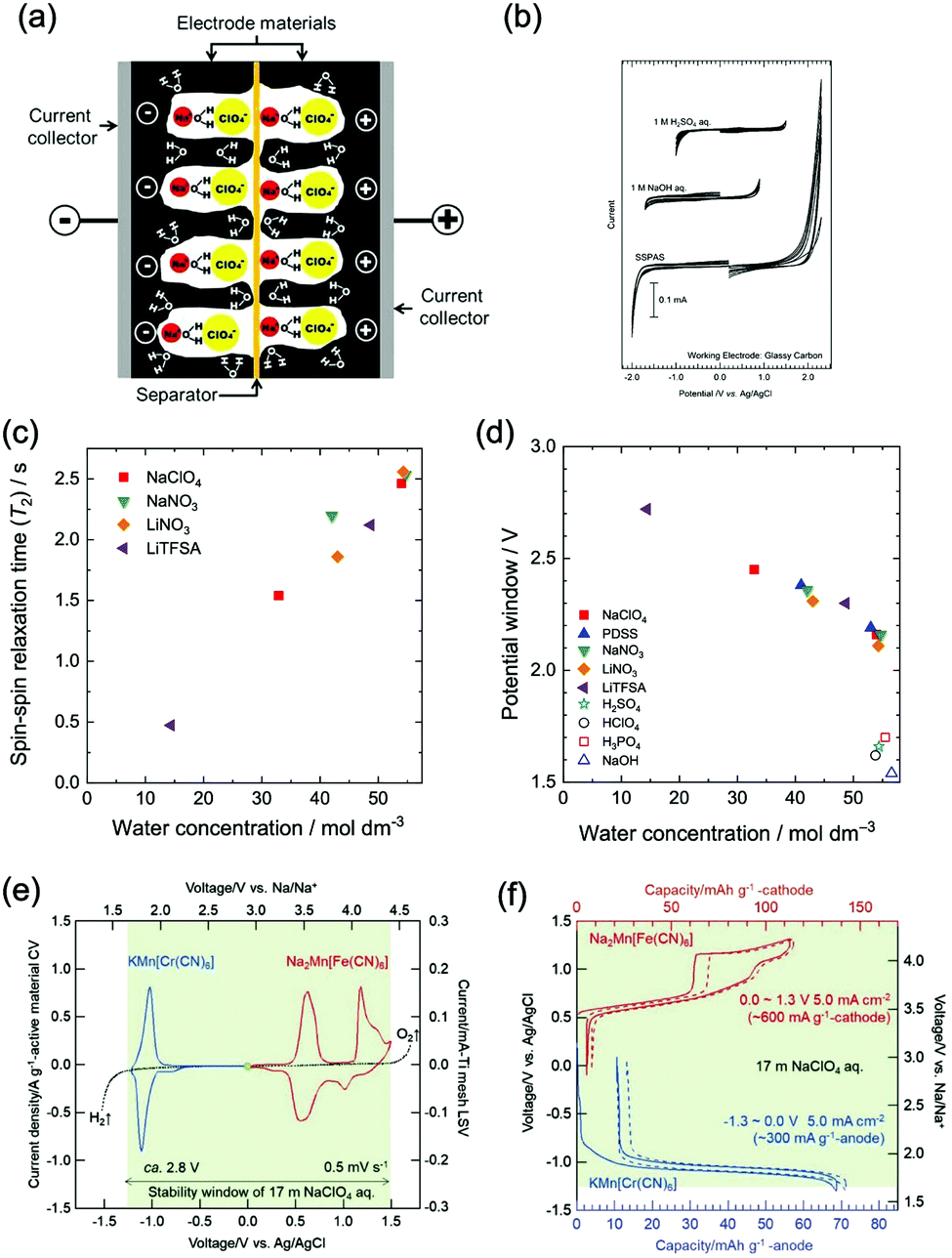 | ||
| Fig. 26 (a) Estimated structure of a supercapacitor based on saturated sodium perchlorate electrolyte. At least one water molecule should be strongly hydrated to NaClO4, and the hydrogen bonding between water molecules would be weakened. (b) Electrochemical stability windows of saturated sodium perchlorate electrolyte, 1 M H2SO4, and 1 M NaOH aqueous electrolytes using a glassy carbon as working electrode measured from 30 mV s−1 to 200 mV s−1 at 25 °C. Reproduced with permission from ref. 59, Copyright 2017 Springer Nature. (c) Spin–spin relaxation time of various electrolytes with various water concentrations. (d) Width of voltage window versus water concentration according to various electrolytes. Reproduced with permission from ref. 55, Copyright 2018 The Electrochemical Society. (e) Linear sweep voltammograms of Ti current collectors (dashed lines) and CV curves of Na2Mn[Fe(CN)6] and KMn[Cr(CN)6] electrodes in 17 M NaClO4 electrolyte. (f) GCD curves of Na2Mn[Fe(CN)6] and KMn[Cr(CN)6] electrodes in 17 M NaClO4 electrolyte. Reproduced with permission from ref. 396, Copyright 2018 Wiley-VCH. | ||
In a study of Yokoyama et al. seeking the origin of electrochemical stability of aqueous electrolytes, the authors demonstrated that water concentration was a universal feature that determines the voltage windows of aqueous electrolytes, as a clear correlation between water concentration and voltage window could be observed regardless the type of electrolyte salt (Fig. 26c and d).55 In the case of concentrated electrolytes, water activity can no longer be approximated to unity. According to Nernst equation, potentials of OER (EOER) and HER (EHER) can be thus expressed as below,

where

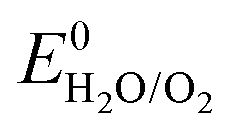 and
and  are the standard potentials of OER and HER, respectively. R is the gas constant, T is temperature, F is the Faraday constant, and K is the equilibrium constant of water ionization aH2O represents water activity. Reduced water concentration (increased salt concentration) of electrolytes decreases water activity, thus increasing OER potential. However, the term involving water activity or water concentration is not included in the equation for HER, therefore, the authors concluded that the influence of water concentration on potentials of OER and HER is asymmetrical. The widest voltage window achieved for conventional electrolyte salt in their study was around 2.45 V (33 mol dm−3 NaClO4 solution) when measured at the current density of 0.1 mA cm−2.
are the standard potentials of OER and HER, respectively. R is the gas constant, T is temperature, F is the Faraday constant, and K is the equilibrium constant of water ionization aH2O represents water activity. Reduced water concentration (increased salt concentration) of electrolytes decreases water activity, thus increasing OER potential. However, the term involving water activity or water concentration is not included in the equation for HER, therefore, the authors concluded that the influence of water concentration on potentials of OER and HER is asymmetrical. The widest voltage window achieved for conventional electrolyte salt in their study was around 2.45 V (33 mol dm−3 NaClO4 solution) when measured at the current density of 0.1 mA cm−2.
Nakamoto et al. demonstrated that the practical voltage window of a highly concentrated electrolyte of 17 M NaClO4 aqueous solution can be extended to 2.8 V.396 Based on this highly concentrated electrolyte, Na2Mn[Fe(CN)6] (NMHCF) cathode exhibited two redox pairs at 0.6 V and 1.0 V (vs. Ag/AgCl), while KMn[Cr(CN)6] (KMHCC) anode exhibited a redox pair at around −1.1 V (vs. Ag/AgCl), reflecting the intercalation/de-intercalation of sodium ions into/from the open frameworks attributing to Fe2+/Fe3+ and Mn2+/Mn3+ in NMHCF and Cr3+/Cr2+ in KMHCC, respectively (Fig. 26e and f). An ASIB that can be charged to 2.6 V with a discharge midpoint voltage of 1.61 V was thus obtained. Despite the high voltage, the specific capacity of the battery based on the mass of both active materials was less than 40 mA h g−1, which was mainly restricted by the intrinsically low sodium ion-accommodation capacity of KMHCC (which is only around 70 mA h g−1). Thus, the resultant energy density of this battery was just around 58 W h kg−1, much less than conventional LIBs. Moreover, its cycle stability (80% capacity retention after 100 cycles at 30C rate) also needs further improvement.
It should be noted that to obtain highly concentrated aqueous electrolytes, the electrolyte salt should be highly soluble in water. In these concentrated electrolytes enabled by conventional inorganic salts, water is still the major constituent, which differs from the categories of “water-in-salt” electrolytes and hydrate-melt electrolytes.
 | ||
| Fig. 27 (a) Electrochemical stability window of LiTFSI–H2O electrolytes with various concentrations measured on nonactive electrodes at 10 mV s−1. (b) GCD curves of the Mo6S8//LiMn2O4 full cell in 21 M LiTFSI “water-in-salt” electrolyte at 0.15C. (c) TEM images of initial Mo6S8 (left) and cycled Mo6S8 (right), showing the formation of the protective SEI. (d) Illustration of the evolution of Li+ primary solvation sheath in diluted and “water-in-salt” electrolytes. Reproduced with permission from ref. 61, Copyright 2015 American Association for the Advancement of Science. (e) GCD curves of the LiMn2O4//C-TiO2 full cell in “water-in-bisalt” electrolyte at the 2nd, 5th and 100th cycles. Reproduced with permission from ref. 63, Copyright 2016 Wiley-VCH. | ||
Inherited from “water-in-salt” electrolytes, a “water-in-bisalt” electrolyte was subsequently developed based on the knowledge that a saturated electrolyte could further accommodate another unhydrated salt with similar chemical structures to form a dual-salt mixture, resulting in an even higher salt ion/water ratio.63 The developed “water-in-bisalt” electrolyte consisted of 21 M LiTFSI and 7 M LiOTf (lithium trifluoromethane sulfonate), thereby its lithium ion concentration reached 28 M, and the corresponding ratio of water molecules to salt ions was approximately 2. Benefited from the higher salt concentration, a denser SEI layer could be promoted and water activity could be further decreased, allowing a wider electrochemical stability window of around 3.1 V. An ALIB with an open circuit voltage up to 2.5 V (showing a flat plateau at 2.07 V) was fabricated and reversibly cycled by coupling of LiMn2O4 cathode and TiO2 anode, delivering an energy density of 100 W h kg−1 (based on the mass of both active materials) (Fig. 27e).
Since the voltage window of “water-in-salt”/“water-in-bisalt” electrolytes exceeds 3.0 V, ARMBs based on such electrolytes could further raise their operating voltage by employing suitable electrodes with redox potentials sitting at the edge of electrolyte voltage window. For example, commercial spinel P4332 structured LiNi0.5Mn1.5O4 cathode has a considerably high potential, while the positive shift of its potential in “water-in-salt” electrolytes hinders its utilization.398 Wang et al. studied the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m type LiNi0.5Mn1.5O4 cathode, which bears two redox pairs at 4.6 V (Ni2+/Ni3+) and 4.8 V(Ni3+/Ni4+).399 The former can be fully utilized as it is completely within the voltage window of 21 M LiTFSI “water-in-salt” electrolyte even taking potential shift into account (Fig. 28a). An ALIB showing two flat discharge plateaus at 2.56 V and 2.10 V with a midpoint voltage of 2.50 V was thus obtained by coupling Fdm LiNi0.5Mn1.5O4 cathode and Mo6S8 anode, delivering an energy density of 80 W h kg−1 with a capacity decay of only 0.075% per charge/discharge cycle at 5C rate. Furthermore, by adjusting electrolyte pH, the voltage window was positively shifted by 0.1 V, allowing for the better utilization of cathode capacity. The Fdm LiNi0.5Mn1.5O4 cathode at the pH of 5 could deliver a higher capacity of 125 mA h g−1 when being charged to 5.05 V (vs. Li+/Li), almost having reached its full capacity, leading to a 126 W h kg−1 energy density for the resultant LiNi0.5Mn1.5O4//Mo6S8 ALIB with similar voltage profiles (Fig. 28b and c). Yang and co-workers further applied “water-in-salt” as gel polymer electrolyte (GPE) by dissolving 25 M LiTFSI into water at 90 °C followed by adding PVA.400 The realization of the high concentration of 25 M suggested the interaction between PVA chains with LiTFSI, which must have facilitated the latter's solubility. A symmetric ALIB employing LiVPO4F as both cathode and anode based on this GPE exhibited a flat high-voltage discharge plateau (2.44 V) (Fig. 28d), superior energy density (141 W h kg−1 based on the mass of both active materials) and remarkable cycle life (4000 charge/discharge cycles) (Fig. 28e), which could be attributed to the extremely high concentration that ensured its ultra-stable reversibility.
m type LiNi0.5Mn1.5O4 cathode, which bears two redox pairs at 4.6 V (Ni2+/Ni3+) and 4.8 V(Ni3+/Ni4+).399 The former can be fully utilized as it is completely within the voltage window of 21 M LiTFSI “water-in-salt” electrolyte even taking potential shift into account (Fig. 28a). An ALIB showing two flat discharge plateaus at 2.56 V and 2.10 V with a midpoint voltage of 2.50 V was thus obtained by coupling Fdm LiNi0.5Mn1.5O4 cathode and Mo6S8 anode, delivering an energy density of 80 W h kg−1 with a capacity decay of only 0.075% per charge/discharge cycle at 5C rate. Furthermore, by adjusting electrolyte pH, the voltage window was positively shifted by 0.1 V, allowing for the better utilization of cathode capacity. The Fdm LiNi0.5Mn1.5O4 cathode at the pH of 5 could deliver a higher capacity of 125 mA h g−1 when being charged to 5.05 V (vs. Li+/Li), almost having reached its full capacity, leading to a 126 W h kg−1 energy density for the resultant LiNi0.5Mn1.5O4//Mo6S8 ALIB with similar voltage profiles (Fig. 28b and c). Yang and co-workers further applied “water-in-salt” as gel polymer electrolyte (GPE) by dissolving 25 M LiTFSI into water at 90 °C followed by adding PVA.400 The realization of the high concentration of 25 M suggested the interaction between PVA chains with LiTFSI, which must have facilitated the latter's solubility. A symmetric ALIB employing LiVPO4F as both cathode and anode based on this GPE exhibited a flat high-voltage discharge plateau (2.44 V) (Fig. 28d), superior energy density (141 W h kg−1 based on the mass of both active materials) and remarkable cycle life (4000 charge/discharge cycles) (Fig. 28e), which could be attributed to the extremely high concentration that ensured its ultra-stable reversibility.
 | ||
| Fig. 28 (a) The electrochemical stability window of “water-in-salt” electrolyte measured on non-active current collectors at 10 mV s−1. The corresponding redox potentials of LiNi0.5Mn1.5O4 cathodes with different crystal structures and Mo6S8 anode (inset) were measured at 0.1 mV s−1. (b) GCD curves of the Fdm LiNi0.5Mn1.5O4 cathode in “water-in-salt” electrolytes before and after pH adjustment. (c) GCD curves of the LiNi0.5Mn1.5O4//Mo6S8 full cell in pH-adjusted “water-in-salt” electrolyte at 0.5C. Reproduced with permission from ref. 399, Copyright 2017 Wiley-VCH. (d) GCD curves of the symmetric LiVPO4F full cell at various C rates. (e) Cycle stability of the symmetric LiVPO4F full cell at 20C. The specific capacities were based on total electrode weight. Reproduced with permission from ref. 400, Copyright 2017 Wiley-VCH. | ||
Besides ALIBs, the concept of “water-in-salt” electrolyte has also been extended into other ARMB systems including ASIBs,64,401–403 AKIBs,65 AMIBs,66 hybrid zinc batteries,67,275,404etc. For example, in the study of Kühnel and co-workers,64 sodium bis(fluorosulfonyl)imide (NaFSI) was employed as electrolyte salt to construct “water-in-salt” electrolyte, as its melting point of 106 °C is much lower than that NaTFSI (257 °C), suggesting weaker lattice energy and higher solubility. Experimental data revealed that its solubility at room temperature can be as high as 35 M, while better ionic conductivity can be still maintained compared to LiTFSI (Fig. 29a). This “water-in-salt” electrolyte of 35 M NaFSI gave a voltage window slightly wider than that of 21 M LiTFSI (Fig. 29b), suggesting the feasibility of 2 V ASIBs and more stable cycling. Very recently, Jiang et al. extended the voltage window of aqueous potassium-ion electrolyte to 3.0 V by increasing the concentration of KCF3SO3 to 22 M (Fig. 29c).254 They synthesized a Fe-substituted Mn-rich PBA cathode (KxFeyMn1−y[Fe(CN)6]w·zH2O) and an organic 3,4,9,10-perylenetetracarboxylic diimide (PTCDI) anode, resulting in an AKIB showing a midpoint voltage of 1.50 V with a flat but short discharge plateau at 1.78 V. It also achieved a high energy density of 80 W h kg−1 and remarkable cycling stability that retained 73% capacity after 2000 charge/discharge cycles at 4C. In addition, the large organic anion of electrolyte salt is not always necessary to achieve the effect of “water-in-salt”. For example, Liu et al. used a 17 M NaClO4 aqueous electrolyte to enable an ASIB with an open circuit voltage of 1.92 V and a midpoint voltage of 1.50 V.403 Leonard et al. demonstrated a 3.2 V-wide electrochemical stability window spanning from −1.7 V to 1.5 V (vs. Ag/AgCl) by using a 30 M potassium acetate (KOAc) aqueous electrolyte, which allowed for the reversible cycling of the low-potential KTi2(PO4)3 anode (−1.4 V/−1.0 V vs. Ag/AgCl) for AKIBs (Fig. 29d and e).65 The potential hysteresis between K+ intercalation and de-intercalation was smaller in 30 M KOAc (around 0.3 V) than in nonaqueous electrolyte (around 0.5 V), suggesting higher energy efficiency. Furthermore, Lukatskaya et al. constructed a “water-in-bisalt” electrolyte by mixing inorganic salts of lithium acetate (LiOAc) and KOAc. According to MD simulation results, the ionic hydration shells of the highly concentrated 32 M KOAc + 8 M LiOAc electrolytes exhibit significantly interpenetrated feature, where every water molecule is involved in at least one hydration shell at any given time (Fig. 29f). Thus, it can be regarded as a typical “water-in-bisalt” electrolyte, where no free water exists, and the solvation sheath consists of solely shared water molecules. The molecular water-to-salt ratio was pushed to an ultralow value of 1.3. Resultantly, conventional LIB anode materials including Li4Ti5O12 and TiO2 exhibited reversible Li+ intercalation/de-intercalation in the 32 M KOAc + 8 M LiOAc “water-in-bisalt” electrolyte (Fig. 29g), and the corresponding TiO2//LiMn2O4 full ALIB yielded a flat high-voltage discharge plateau of 2.10 V.405 These organic component-free “water-in-salt”/“water-in-bisalt” electrolytes are cheap and environmentally benign, and can be readily regulated over a wide range, showing high adaptability and versatility.
 | ||
| Fig. 29 (a) Ionic conductivity of NaFSI and LiTFSI electrolytes of various concentrations at 20 °C. (b) Electrochemical stability window of various “water-in-salt” electrolytes measured on stainless steel at 0.1 mV s−1. Redox potentials of NaTi2(PO4)3 and Na3(VOPO4)2F electrodes in 35 M NaFSI at 0.05 mV s−1 were also shown. Reproduced with permission from ref. 64, Copyright 2017 American Chemical Society. (c) Electrochemical stability windows of 1 M and 22 M KCF3SO3 electrolytes measured at 1 mV s−1, which are marked with red and black, respectively. Reproduced with permission from ref. 254, Copyright 2019 Springer Nature. (d) CV curves of KTi2(PO4)3 anode in 30 M KAc and nonaqueous electrolytes at 0.5 and 0.1 mV s−1, respectively. Inset shows its charge–discharge profiles in 30 M KAc at the current density of 200 mA g−1. (e) Cycle stability of the KTi2(PO4)3 anode in 30 M KAc electrolyte. Reproduced with permission from ref. 65, Copyright 2018 American Chemical Society. (f) Visualization of the equilibrium electrolytes of 1 M KOAc, 1 M LiOAc, 27 M KOAc, and 32 M KOAc + 8 M LiOAc. Atom color: K, purple; Li, green; O, red; C, grey; H, white. (g) CV curves of various electrodes in 32 M KOAc + 8 M LiOAc electrolyte at 0.2 mV s−1. Reproduced with permission from ref. 405, Copyright 2018 Royal Society of Chemistry. | ||
 | ||
| Fig. 30 (a) Liquidus line of LiTFSI + LiBETI mixtures in water. The light blue area above represents a stable liquid phase of fully miscible salts and water, whereas the dark blue area underneath represents a partially miscible phase. (b) Stoichiometric amounts of salts and water used to prepare the hydrate melt of Li(TFSI)0.7(BETI)0.3·2H2O, resulting a transparent liquid. (c) Snapshots of equilibrium trajectories obtained from first-principles DFT-MD simulations of Li(TFSI)0.7(BETI)0.3·2H2O hydrate melt and dilute LiTFSI/H2O. Atom color: C, dark grey; H, light grey; O, red; N, blue; S, yellow; F, green; Li, purple. (d) Equilibrium potential shift of lithium ion intercalation versus Li+ molality of electrolytes. (e) Voltage windows of pure water, conventional LiTFSI/H2O electrolyte, and the hydrate-melt electrolyte with redox potentials of Li4Ti5O12, LiCoO2 and LiNi0.5Mn1.5O4 indicated. Reproduced with permission from ref. 62, Copyright 2016 Springer Nature. | ||
It should be noted that “water-in-salt” type electrolytes differ from hydrate-melt electrolytes as they still contain non-negligible amount of free water due to their off-eutectic design. For the solution with the water/anion molar ratio of 2 (Li(TFSI)0.7(BETI)0.3·2H2O), a stable state was discovered where all water molecules were coordinated with lithium ions and intramolecular hydrogen bonding was neglectable (Fig. 30c). The authors also investigated the equilibrium potential shift of intercalation/de-intercalation reaction of lithium ions, which was commonly observed in “water-in-salt” type electrolytes. The equilibrium potential shift was measured to be 0.25 V in Li(TFSI)0.7(BETI)0.3·2H2O hydrate melt, higher than that of 22 M LiTFSI (Fig. 30d). Anodic and cathodic potential limits were further pushed due to a thermodynamic mechanism (lowered HOMO level) and a kinetic mechanism (anion-based passivation), respectively, resulting in an ultra-wide voltage window of 3.8 V. Moreover, due to the positive shift of reactive potentials, this voltage window allows for reversible cycling of Li4Ti5O12 anode, whose reaction potentials are completely located within the voltage window. High-voltage LiCoO2 and LiNi0.5Mn1.5O4 cathodes can also be utilized as their upward-shifted potentials are still within the voltage window (Fig. 30e). As a result, the Li4Ti5O12//LiCoO2 and the Li4Ti5O12//LiNi0.5Mn1.5O4 full ALIBs exhibited high-voltage discharge plateaus at 2.26 V and 3.10 V, respectively.
So far, there are no other investigations employing hydrate-melt electrolytes for ARMBs, except that Wu et al. developed a Li–O2 battery based on a hydrate-melt electrolyte, which was organic solvent-free and thus was effectively exempted from side reactions regarding solvent degradation and by-products formation.408 Therefore, more studies are needed to further evaluate and improve the practicability of this novel kind of electrolytes.
4.5 Electrolyte additives with interphase-forming capability
Differing from artificial SEI, additives with interphase-forming capability tend to in situ form an extra interphase between electrode and electrolyte, via either polymerization, physical absorption, or chemical decomposition of additives during electrochemical processes. This section highlights the studies involving electrolyte additives that benefit battery voltage. The in situ-formed, additive-originated interphases hinder the transport of water molecules to electrode surfaces, suppress the evolution of hydrogen or oxygen, and thus result in stabilized cycling of high-voltage electrode materials or broadened voltage windows.In non-aqueous electrolytes, interphase-forming additives have been reported to be utilized to enhance stability of batteries, which is realized by their preferential reduction before electrolyte components during electrochemical processes that strengthens SEIs.409 The first report of the successful application of an electrolyte additive into an aqueous energy storage system was introduced by Stojković et al.76 In their work, the addition of 1% VC (vinylene carbonate) in aqueous LiNO3 electrolyte greatly boosted the capacity of Li1.05Cr0.10Mn1.85O4 cathode, with a notable increase in its coulombic efficiency and a significant reduction of its capacity decay. It is claimed that the addition of VC facilitated the formation of a thin interphase of polymeric species onto electrode surface, resulting in a protective interphase that can hinder the penetration of water molecules and alleviate the harm it does to lithium ion intercalation.410–412
Electrolyte additives have been reported to be physically absorbed onto electrode surfaces, serving as a barrier interphase to isolate water molecules, thus raising the resistance to reductive/oxidative decomposition of electrolytes, resulting a wider voltage window. For example, Miyazaki and co-workers saturated 0.5 M LiNO3 electrolyte with disodium propane-1,3-disulfonate (PDSS), enabling the utilization of the high-potential LiNi0.5Mn1.5O4 cathode.77 The authors explained that the PDSS anions were like bulky TFSI anions, which could effectively cover the surface of LiNi0.5Mn1.5O4 cathode and repel water molecules during electrochemical processes. Compared to other highly concentrated electrolytes, the addition of PDSS does not change the concentration of electrolyte salt (i.e., lithium salt for this case), therefore, the potentials for lithium ion intercalation/de-intercalation would not be affected. In addition, the dissolution of the high-potential cathode material in low-pH condition was suppressed by adding a phosphate buffer. Resultantly, a wide voltage window positively extending to 1.6 V (vs. Ag/AgCl) was obtained (Fig. 31a), allowing for the reversible redox reactions of the high-potential LiNi0.5Mn1.5O4 cathode (two redox pairs at 1.45 V/1.49 V and 1.48 V/1.52 V (vs. Ag/AgCl), respectively). Similarly, a hybrid aqueous battery system by coupling Na2MnFe(CN)6 cathode and zinc anode showing two flat discharge plateaus at 1.75 V and 1.58 V was developed.78 It was confirmed that the second redox pair of Na2MnFe(CN)6 cathode located at around 1.1 V (vs. SHE) could not be initiated in conventional 1 M Na2SO4 + 1 M ZnSO4 electrolyte due to severe OER. With the addition of sodium dodecyl sulfate (SDS), OER was effectively suppressed and the second redox reaction of cathode could thus be reversed (Fig. 31b and c). The resultant battery delivered a high energy density of 170 W h kg−1 (based on the mass of cathode material) as well as a good cycling stability. DFT calculations further revealed the existence of a high-energy barrier that blocks the transport of water molecules through the dense hydrophobic interphase of SDS additives on electrode surface, OER and HER potentials are thus increased, leading to the expanding of voltage window.
 | ||
| Fig. 31 (a) Linear sweep voltammograms of Pt electrodes in aqueous electrolyte of 0.5 M LiNO3 + saturated PDSS buffered with 0.25 M Li–PO4 (pH = 7) showing the positive expanding of voltage window to 1.6 V (vs. Ag/AgCl). Reproduced with permission from ref. 77, Copyright 2016 Royal Society of Chemistry. (b) CV curves of the Na2MnFe(CN)6 cathode in the pristine electrolyte at 5 mV s−1. (c) CV curves of the Na2MnFe(CN)6 cathode in the electrolyte containing SDS at 5 mV s−1. Reproduced with permission from ref. 78, Copyright 2017 Royal Society of Chemistry. (d) CV curves of graphite anode pre-coated with LiTFSI-HFE gel and LiVPO4F cathode in “water-in-bisalt” electrolyte coupled with lithium metal anode pre-coated with LiTFSI-HFE gel measured at 0.5 mV s−1. Insets illustrate the cell configurations. (e) GCD curves of LiVPO4F cathode coupled with Li metal anode at 0.3C. Reproduced with permission from ref. 413, Copyright 2017 Elsevier. (f) Illustration of the formation of SEI-like interphase on the anode in HANE. (g) TEM image of fully lithiated Li4Ti5O12 anode. (h) Electrochemical stability window of the HANE measured on pure current collectors and on active materials of Li4Ti5O12 and LiNi0.5Mn1.5O4. (i) Discharge profiles of the Li4Ti5O12//LiNi0.5Mn1.5O4 full cell at various C rates. Reproduced with permission from ref. 414, Copyright 2018 Elsevier. | ||
Decomposition of electrolyte additives can also in situ form a protective, SEI-like interphase. The formation of a protective interphase through additive decomposition should meet the following requirements: (1) the additive should be chemically stable in the given electrolyte solution. (2) It should be electrochemically unstable so that decomposition can occur and interphase ingredient can be released during electrochemical processes. Yang et al. introduced a highly-fluorinated ether (HFE) as an electrolyte additive, which was mixed with 0.5 M LiTFSI and 10 wt% polyethylene oxide to form a transparent gel. This gel was extremely hydrophobic and exhibited complete inertness toward lithium metal. It also remained phase separated from the 21 M LiTFSI + 7 M LiOTf “water-in-bisalt” GPE. The pre-coating of LiTFSI-HFE gel onto graphite or lithium metal electrode initiated their reversible cycling in “water-in-bisalt” GPE (Fig. 31d), leading to the feasibility of a series of high-voltage ALIBs with discharge plateaus located at around 4.0 V and energy densities approaching conventional state-of-the-art LIBs (Fig. 31e). It was discovered that the LiTFSI-HFE gel were consumed to yield a solid interphase consisting of both organic fluorinated hydrocarbon and inorganic fluorides, in accordance with previous reported organic electrolytes.413 Moreover, in the hybrid aqueous/non-aqueous electrolyte (HANE) developed by Wang et al.,414 the non-aqueous component of dimethyl carbonate (DMC) served as an additive in “water-in-salt” electrolyte, which provided a secondary ingredient of alkylcarbonate in addition to LiF originating from TFSI reduction to strengthen the SEI-like interphase formed on anode during electrochemical processes (Fig. 31f and g), extending voltage window downward to 1.0 V (vs. Li/Li+) (Fig. 31h). The electrochemical stability window of the HANE was around 4.1 V wide, enabling an ALIB with a flat discharge plateau at 3.2 V based on LiNi0.5Mn1.5O4 cathode and Li4Ti5O12 anode (Fig. 31i). This ALIB showed an energy density as high as 165 W h kg−1 and a cycle stability over 1000 charge/discharge cycles. More recently, Wang's group proposed a conversion-intercalation chemistry of the cathode containing equimolar lithium halide salts (LiBr)0.5(LiCl)0.5–graphite (LBC–G) (Fig. 32a).415 In this study, “water-in-bisalt” electrolyte was utilized to bind partially hydrated LiBr/LiCl within the cathode host, and HFE gel was employed to coat graphite anode so that a protective interphase could be formed to initiate its electrochemical processes. This new cathode chemistry combined the high energy of the conversion reaction and the good reversibility of intercalation process, resulting in a full ALIB with a midpoint voltage of 3.98 V and an ultrahigh energy density of 460 W h kg−1 (based on the mass of both electrodes) (Fig. 32b and c).
 | ||
| Fig. 32 (a) Schematic illustrations of the conversion–intercalation chemistry of the LBC–G cathode upon oxidation. The two-step reactions include the oxidation of Br− (at 4.0 V) and Cl− (at 4.2 V) and their subsequent intercalation into the graphitic host. Discharge process is a reversal. (b) GCD curves of ALIBs based on LBC–G cathodes with anhydrous LiBr/LiCl or LiBr/LiCl monohydrates and HFE–PEO gel coated graphite anodes at 0.2C. (c) Energy density of the graphite//LBC–G full cell compared with other batteries (based on the mass of both electrodes). Reproduced with permission from ref. 415, Copyright 2019 Springer Nature. (d and e) TEM images of the LiCoO2 cathode after cycling in 21 M LiTFSI “water-in-salt” electrolyte containing 0.1 wt% TMSB additive. A protective interphase can be clear observed. (f) GCD curves of the LiCoO2//Mo6S8 full cell at 0.5C. Inset shows its CV curves. Reproduced with permission from ref. 418, Copyright 2016 Royal Society of Chemistry. | ||
In addition, an electrolyte additive has been reported to stabilize high-voltage cathode material. Layered LiCoO2, when charged to 4.2 V (vs. Li/Li+), could deliver a specific capacity of 140 mA h g−1, corresponding to 50% Li extraction to Li0.5CoO2, which has been regarded as a relatively reliable cathode material. If charged to 4.5 V (vs. Li/Li+), a higher specific capacity of 180 mA h g−1 (corresponding to 70% Li extraction) could be realized in organic electrolytes. However, such high capacity decays during cycling in aqueous electrolytes due to the lack of protective SEI on cathode.121,416,417 Wang et al. introduced an additive of tris(trimethylsilyl) borate (TMSB) into “water-in-salt” electrolyte, which was subsequently oxidized to form a protective interphase on LiCoO2 cathode (Fig. 32d and e).418 At a high cut-off voltage, the interphase-protected LiCoO2 delivered a high capacity of 170 mA h g−1 with remarkable cycling stability. An ALIB coupling LiCoO2 cathode with Mo6S8 anode exhibited a 2.50 V open circuit voltage and a 1.95 V midpoint voltage (Fig. 32f), achieving an energy density as high as 120 W h kg−1 and a low capacity decay rate of 0.013%/charge–discharge cycle, as the protective interphase effectively suppressed OER and cobalt dissolution from cathode to electrolyte.
Voltage characteristics of high-voltage ARMBs employing the above-mentioned various strategies are compared in Fig. 33. As revealed, most of these ARMBs possess midpoint voltages higher than 1.5 V, and the majority of them exhibit a flat plateau upon discharge, showing high potential in providing high-energy and consistent output. Midpoint voltages of the ARMBs based on conventional aqueous electrolytes that fully exploit voltage window (introduced in Section 4.1) are basically in the range of 1.2–2.0 V. By regulating electrolyte pH, midpoint voltages can be raised to 1.8–2.3 V (ARMBs introduced in Section 4.2). Concentrated aqueous electrolytes including the novel “water-in-salt”/“water-in-bisalt” and hydrate-melt electrolytes further pushes midpoint voltages up to 2.5 V and above (ARMBs introduced in Section 4.4). Impressively, protective interphase layers either artificially formed (ARMBs introduced in Section 4.3) or in situ formed via electrolyte additives (ARMBs introduced in Section 4.5) can ultimately realize ARMBs with midpoint voltages over 4.0 V, which are even comparable to traditional organic LIBs. Still, there is a long way to go before they can be practically applied and commercialized, further researches on cost cut-down, cell assembly and encapsulation techniques are needed.
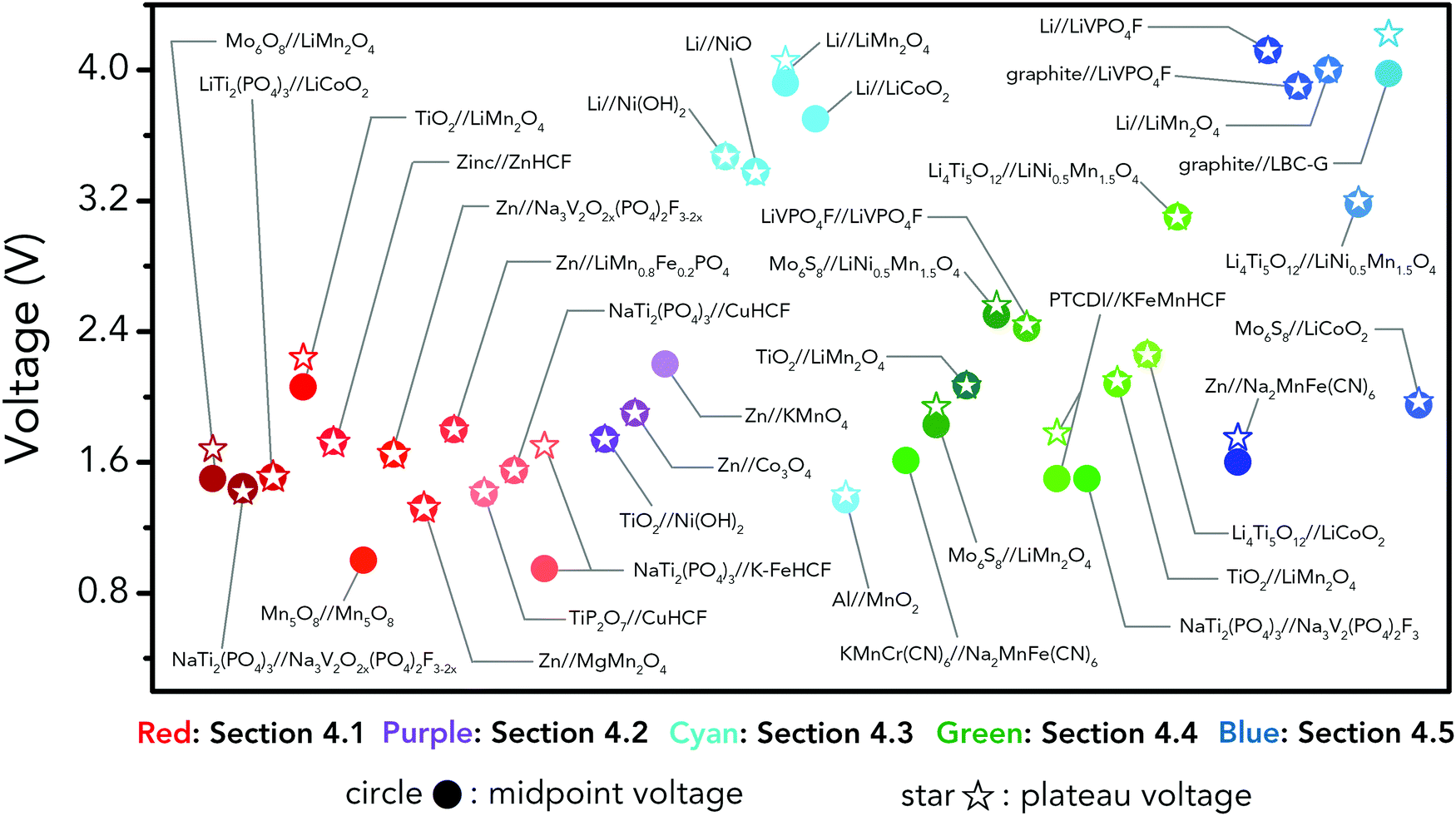 | ||
| Fig. 33 Voltage comparison among various high-voltage ARMB systems. Solid circles represent midpoint voltages, and hollow stars at the same x-axis position represent corresponding plateau voltages (the voltage of the first discharge plateau is adopted if a battery yields more than one plateau). | ||
5. Future perspectives and concluding remarks
Compared with conventional organic LIBs, ARMBs possess a number of irreplaceable merits including low cost, high power, intrinsic safety and environmental benignity, showing great potential in large-scale stationary energy storage as well as in small electronics concerning wearable applications. The introducing of aqueous electrolytes accounts for the superiority of ARMBs, which erases the usage of expensive, flammable and toxic organic electrolytes. Whereas, aqueous electrolytes inevitably bring the confinement of operating voltage for ARMBs due to the narrow electrochemical stability window of water (∼1.23 V). Consequently, batteries with aqueous electrolytes normally exhibit lower energy density compared to their counterparts based on organic electrolytes. To alleviate this problematic issue, effective strategies have been proposed to broaden electrolyte voltage window, resulting in ARMBs with higher operating voltages. Nevertheless, running high-voltage ARMBs far away from their intrinsic “safe zone” leads to other problems, including jeopardized cycle stability originating from aggravated side reactions, higher cost due to the use of expensive chemicals/components, more complicated configurations, etc.Reviewing representative ARMBs, it is delighted to see that some tough problems have been more or less addressed by applying a variety of ingenious solutions. However, ideal ARMBs prototypes possessing high energy and power density with simultaneous long-term cycling stability are rare, not to mention the concern about cost. As the ultimate goal of ARMBs is commercialization, it is an urgent need to upgrade their practicability to a new level. According to the achievements that have been made so far, potential directions for further improvements of future ARMBs are proposed in the following aspects.
5.1 Envision more cost-effective and facile strategies
Although effective, current strategies to obtain high-voltage ARMBs are usually complex and/or costly. Specifically, the effect of regulating electrolyte pH is moderate, while it may be more demanding on cell configuration, or induce side reactions of electrode materials, or cause safety concerns of erosion. Constructing artificial interphase is even more complicated in engineering, and it usually involves the use of an extra layer of lithium-ion conductor and organic electrolyte to initiate the utilization of low-potential lithium metal or carbon anode, meaning the cost and safety concern are actually not much eased. The novel “water-in-salt”/“water-in-bisalt” electrolytes and hydrate-melt electrolytes are impressive in expanding voltage window, which makes the cycling of electrode materials with higher/lower redox potentials in aqueous solutions possible. Whereas, they generally employ salts with large organic groups, which are extremely expensive, and the high concentration further pushes up their cost. As for electrolyte additives, their long-term influences on electrochemical performances should be carefully observed, and their decomposition to form SEI-like interphase usually associate with the use of organic components and “water-in-salt”/“water-in-bisalt” electrolytes.Therefore, to enable practicability, more cost-effective and facile strategies should be envisioned and implemented. For example, there are many experiences of constructing artificial SEIs for conventional LIBs,419–421 which can be translated to ARMBs with suitable adjustments. This kind of strategy should be carefully screened and optimized to cater for the original intentions of low cost and industrial simplicity. Besides, almost all works based on “water-in-salt”/“water-in-bisalt” electrolytes and hydrate-melt electrolytes are limited to several types of organic salts, there should be cheaper and more effective options for constructing high-voltage ARMBs with reasonable cost. Some ionic liquids (ILs) have been reported to be good supporting components for aqueous electrolytes, and their mixture with water mitigates the disadvantages of high viscosity and low ionic conductivity, which could also be a promising research direction of expanding electrolyte voltage window.241,422–424 Moreover, studies have shown that some gel polymer electrolytes (GPEs) have better tolerance for high voltages, which was ascribed to the mechanism of “molecular cages” where water molecular are “locked” and more interacted with polymer chains,425 or a special ionic conduction mechanism of cooperative O → B− coordination bonds in polymer.241 These gel polymers enabled high-voltage energy storage devices without severe water electrolysis. Although more comprehensive investigations are needed to further reveal their in-depth mechanisms, it would be inspiring if GPEs with stronger capability to suppress water electrolysis are discovered.
5.2 Discover new electrode materials for aqueous electrolytes
Comparative studies have demonstrated that the difference of electrochemical performances of some existed electrode materials in aqueous electrolytes and non-aqueous electrolytes is not noticeable, which means traditional methods of material optimization could be readily translated into ARMBs to enhance battery performances. Besides, due to the narrower voltage window of aqueous electrolytes, it is more important to fully exploit the voltage window by utilizing electrode materials with desired redox potentials sitting at the edge of the window. In return, their redox potentials are affected by electrolyte pH, electrolyte salt concentration, crystal orientation, etc.,58,60,61,399 which requires a more comprehensive and accurate control of the whole electrochemical system. Moreover, some reported high voltage values for ARMBs actually refer to open circuit voltage, while the corresponding batteries only yielded low discharge plateau or no plateau but capacitor-like sloping discharge profiles, which is not favourable to energy density and stable energy output. Therefore, electrode materials with high redox potentials and simultaneously fast redox kinetics are highly desired to construct ARMBs with high-voltage discharge plateaus. In addition, the seeking of more suitable electrode materials should not only be focused on redox potentials, materials with larger specific capacities are always preferential regarding to the promotion in energy density of ARMBs. For example, Kundu et al. reported a vanadium oxide bronze pillared by interlayer zinc ions and water as cathode material for AZIBs, although the average operating voltage of the resultant cell was less than 1.0 V, the energy density reached 250 W h kg−1 (based on the mass of cathode material) due to the large specific capacity (220 mA h g−1) at 15C-rate, meaning large specific capacity could help compensate low operating voltage in practical applications.426 However, when discovering new electrode materials for ARMBs, their stability in aqueous solutions should be thoroughly considered, especially at certain pH values, as literatures have demonstrated the dissolution of electrode materials or their intermediate compounds into water during electrochemical processes, which are mainly responsible for battery capacity decay.27,57,132,427,4285.3 Systematic studies of current collectors and binders
Various current collectors have been employed in ARMBs, including nickel mesh, stainless steel sheet/grid, titanium foil, graphite foil, carbon fabric, carbon nanotube fabric, etc. However, detailed studies of the influences of current collectors on battery performances in aqueous electrolytes are absent. Conventional LIBs widely adopt aluminium as current collector because of its good electrical conductivity, light weight and cost efficiency. While for practical application of ARMBs, the select of current collectors should be conducted with extra cautiousness regarding the narrower voltage window and more complicated electrochemical processes. In a study of Coustan and coworkers, voltage windows of various metallic current collectors in LiTFSI solutions with different concentrations were compared, which showed that the shift of voltages window at different LiTFSI concentrations is highly dependent on the choice of metallic electrode.429 Lahan et al. demonstrated that current collector played a unique role in determining the electrochemical performance of TiO2 anode for aluminium-ion batteries. The employed graphene current collector significantly suppressed HER on anode, allowing for reversible Al3+ intercalation/de-intercalation.430Fig. 34 gives the relationship between hydrogen evolution from the interaction of water and the strength of adsorbed metal–hydrogen (M–H) bonds on current collectors.431,432 It is clearly seen that the exchange current for HER increases to a peak and then gradually decreases with the increasing of M–H bond strength. Consideration of the interaction energy between atomic hydrogen and surface metal atoms actually results in the divide of metals into two distinct groups. The peak of the curve should correspond to Δg0 = 0, where Δg0 is the standard free energy of hydrogen adsorption. Consequently, in the left rising part of the curve in Fig. 34 there should be metals not-adsorbing hydrogen, while in the right descending part there should be metals adsorbing hydrogen. At low M–H bond strength, the trend for HER is low as hydrogen bonds weakly with metal surface. At high M–H bond strength, H–H recombination is decreased as the bonding between hydrogen and metal is so strong.433 Therefore, metals sit at either extreme of this curve are preferred for current collectors in aqueous electrolytes as their overpotential for HER is high, contributing to wider electrochemical stability window. | ||
| Fig. 34 Relationship between exchange current for HER and metal–hydrogen bond strength of various metal electrodes. Reproduced with permission from ref. 432, Copyright 2019 Wiley-VCH. | ||
Besides, the corrosion of metallic current collectors is more severe in aqueous electrolytes.434 Platinum, titanium, and nickel are demonstrated to be more resistant to corrosion, while they are more expensive than stainless steel, copper and aluminium.435 To avoid corrosion and facilitate long-term stability, the contact area between current collector and electrolyte is suggested to be minimized. This can be achieved via fully coating current collectors with electrode material on both sides, and/or protecting the bare current collector area with inert coatings.436,437 Moreover, the choice of binder for ARMBs is quite limited. The widely-adopted polyvinylidene-di-fluoride (PVDF) binder suffers from high cost and toxicity, and the solvent of N-methyl-pyrrolidone (NMP) used to dissolve PVDF has similar disadvantages.438 The gradual dissolution of PVDF into aqueous electrolytes further required the seeking for safer, more cost-effective and more stable binders for ARMBs.439 Overall, the choice of current collector and binder for ARMBs seems trivial, but it is crucial in terms of improving battery performances. Therefore, systematic and in-depth studies in these aspects are urgently needed.
5.4 Enhancement of separators/solid-state electrolytes
A separator is usually in the form of a porous membrane sandwiched between cathode and anode, permeable to electrolyte ions but preventing electric contact of electrodes. It plays a vital role in a battery system, and should meet the following requirements: (1) electric insulation; (2) low ionic resistance; (3) chemical stability against electrolyte, electrode reactants and by-products; (4) mechanically strong and stable; (5) readily wettable by electrolyte.440 It should be clarified that currently there is no single separator that can satisfy all batteries with varied chemistries, configurations and geometries. Conventional LIBs extensively adopt polyolefins (polyethylene and polypropylene) with microporous structures as separators, as they possess good mechanical properties, chemical stability and affordability. But these polyolefins are inherently non-wettable by aqueous solutions, so that aqueous electrolytes cannot penetrate these membranes and ions migration cannot occur. Recent researches of ARMBs tend to utilize cellulose-based microporous nonwoven fabrics as separators, which are chemically stable and hydrophilic. Whereas, sufficient thickness is needed for them to maintain satisfactory mechanical strength.To avoid liquid leakage and simplify battery configuration, solid-state electrolytes that can serve simultaneously as battery separators have been accordingly developed. For conventional LIBs, commonly-adopted solid-state electrolytes are polymer electrolytes, gel polymer electrolytes and inorganic solid-state electrolytes.441,442 Although attractive in operation safety and cell assembly, these solid-state electrolytes suffer either low lithium-ion conductivity, low mechanical strength or poor flexibility. For ARMBs, as water is the electrolyte solvent, it is feasible to utilize hydrogel as solid-state electrolyte. Hydrogels are typically swollen polymer networks with a large amount of absorbed water, resulting in high ionic conductivity and stability against liquid leakage. Some hydrogel electrolytes such as poly(vinyl alcohol), sodium polyacrylate and gelatin are actually not advantageous in maintaining consistent battery performance, because they would be easily affected or damaged upon high shearing force, pressure, or sharp cut. But through careful design and modification, we have demonstrated the successful applications of self-healing hydrogel electrolyte and soft-yet-tough hydrogel electrolyte in various aqueous battery systems, having achieved improved device-level usability and durability.213,443,444
Compared to the studies on electrode materials and liquid electrolytes, very little work is directed toward characterizing, evaluating and developing separators/solid-sate electrolytes for ARMBs. To score higher energy density and maintain consistent energy output, researchers should devote more into these areas. Mechanical strength, chemical stability, thermal stability, permeability, porosity, wettability, electrolyte absorption and retention, thickness, and dimensional stability should all be considered when enhancing separators/solid-sate electrolytes. It is worth bearing in mind that it is eventually the applications that decide what kind of separator/solid-sate electrolyte is most suitable for a certain ARMB system, as no single separator/solid-sate electrolyte satisfies all the needs. Thus, compromises need to be made. There has always been a growing demand for thinner separators/solid-sate electrolytes to push up battery energy density,445 and the blooming field of wearable electronics call for highly flexible, highly mechanically-durable separators/solid-sate electrolytes to ensure both comfortable wearing and stable energy supply. Researchers should work along with industrial manufacturers to create the next generation of ARMBs with enhanced separators/solid-sate electrolytes.
5.5 Thoroughly examine OER and HER behaviour in ARMBs
As researches are mainly focused on directly enhancing battery performances, the profound influences of OER and HER during the electrochemical processes of ARMBs have not been thoroughly investigated yet. For ARMBs, OER and HER together decide the electrochemical stability window of their aqueous electrolytes, thus affecting the usability of electrode materials and resultant cell voltages. Besides, OER and HER themselves as well as associated parasitic reactions significantly deteriorate the long-term stability of ARMBs, attributing to either alteration in electrolyte concentration and/or accessibility due to water consumption, or dissolution of intermediate by-products originated from electrode active materials. On top of that, even minor gas evolution could disable the sealing of ARMBs, which impedes long-term durability, torturing the encapsulation for further commercialization. Therefore, it is of tremendous importance to systematically study OER and HER during battery charge/discharge processes with respect to various battery components including electrode materials, electrolytes, binders, conductive additives, and current collectors. Due to the similarities in electrochemistry, research methods and experiences can be borrowed from the studies of metal–air batteries446–449 for OER and the studies of electrocatalytic hydrogen production37,450–452 for HER. Moreover, real-time monitoring electrolyte local pH may help quantify OER and HER processes that involve hydrogen ions and hydroxides, and pH change could in return affect the activity of OER and HER.453,454 Coulombic efficiency may also be utilized as an effective indicator for examining OER and HER, and literatures have already paid attention to its importance in galvanostatic charge/discharge cycling, self-discharge test and galvanostatic intermittent titration technique (GITT) test.63,399,400,455,456 With in-depth understanding of OER and HER behaviour in certain ARMB configurations, effective and accurate strategies can thus been envisioned to better enhance battery performances.5.6 In-depth exploration of degradation mechanisms of ARMBs
Degradation mechanisms of conventional LIBs have been thoroughly investigated, which has helped address the cycle life issue and achieve wide applications of LIBs. For ARMBs, few studies have specially focused on their degradation mechanisms during cycling. Actually, the adoption of aqueous solutions brings about more complicated chemical reactions and thus should result in more diverse degradation processes. Literatures have demonstrated that some vanadium-based compounds can be utilized as promising cathodes for various ARMB systems, while their fast dissolution into aqueous electrolytes accounts for the rapid capacity fading of ARMBs.457 Phase transformation of electrode materials is another major factor that causes battery performance decay. For example, MnO2 goes through irreversible phase transformation to spinel ZnMn2O4. The latter possesses a high negative formation energy (−1293.3 kJ mol−1) that can well stabilize this compound, leading to unfavourable zinc ion extraction during charging. Together with the loss of redox-active Mn element originating from Mn2+ dissolution, capacity fading of the Zn//MnO2 battery is thus inevitable.44,432,458 Na2/3[Ni1/3Mn2/3]O2 for sodium intercalation and LiNiO2 for lithium intercalation were also found to experience phase transformation upon cycling.459,460 Nayak et al. reported that the layered-to-spinel phase transformation of Li- and Mn-rich cathode Li1.2Ni0.16Mn0.56Co0.08O2 was responsible for its average discharge voltage decay.461 Also, the migration of some transition metal ions in electrode materials would cause concentration gradient in lattice, leading to the formation of structural reconstruction layer throughout the whole electrochemical process and the resultant performance decay.462 Besides, it is reported that structural instability of high-voltage oxygen-containing cathodes can result in the release of lattice oxygen. Extensive oxygen release causes severe cathode performance decay and triggers thermal runaway of conventional LIBs.463,464 In addition, other causes including irreversible electrochemical instabilities at electrode–electrolyte interfaces,465,466 induced microcracks within secondary particles of electrode materials,467 redox reactions between electrode and binder,468 and non-uniform coatings of electrodes469 that would deteriorate battery long-term performance have also been located.To deal with the suffering battery degradation issue, researchers have proposed corresponding solutions. Polymer coating strategy and addition of specific salt to change dissolution equilibrium were proved effective to suppress the dissolution of vanadium-based compounds.27,240 Doping electrode materials with various valence groups can affect their structures and properties in various ways, which include acting as pillars to uphold electrode structural integrity,470 increasing lattice spacing for faster ion intercalation,471 decreasing lattice distortion,472 increasing the energy barrier for transition metal migration,461 and delaying oxygen release reaction,473 thus alleviating their structural transformations. Constructing core–shell structures via modification of surface chemical composition without affecting bulk composition has also been proposed to enhance the structural stability of electrodes.474–479 Moreover, for AZIBs, Wang et al. and Wan et al. emphasized on the co-insertion process of protons and zinc ions, where the cathode lattice contraction induced by zinc ion intercalation and the expansion caused by hydronium intercalation offset each other, favouring lattice integrity and battery long-term stability.240,480
Exploring degradation mechanisms is extremely important in terms of either commercialisation of ARMBs or achieving high-voltage ARMBs. For instance, replacing oxide ion with polyanions can expand charge voltage limited by the top of the anion-p bands of cathodes, while it could result in structural instability and performance decay after cycling.481 Also, some electrodes show electrochemical instability at high applied voltage.460 Currently, during the investigations of ARMB degradation mechanisms, more attentions are devoted to electrode intrinsic properties, while their association with aqueous electrolytes has seldom been attended. Besides, the studies on the degradation of aqueous electrolyte and hydrogel electrolytes themselves are usually neglected. Therefore, researchers are suggested to conduct more comprehensive and in-depth exploration of the degradation mechanism of ARMBs.
5.7 Employment of in situ characterization techniques
Electrochemical testing approaches such as cyclic voltammetry, galvanostatic charge/discharge, electrochemical impedance spectroscopy and rate performance test are widely-adopted methods to investigate the electrochemical mechanisms and evaluate the performances of ARMBs. While these methodologies are incapable of describing the detailed reaction mechanisms and microscopic variations of electrode chemical/physical state during electrochemical processes, hence ARMBs in operation are like a “black box”. This “black box” can be unfolded via various ex situ characterization techniques. However, for ex situ measurements, the ongoing electrochemical processes have to be stopped and the electrode/electrolyte components obtained from the disassembled cells need to be carefully handled prior to the tests. In this respect, ex situ techniques cannot reveal the exact reaction routes happened in ARMBs. Instead, in situ characterization techniques can track the whole electrochemical processes without disturbing ARMBs, omitting the influence and uncertainty brought by cell disassembly and electrode/electrolyte post-treatment, thereby providing systematic, accurate and profound information of structural evolutions and reaction mechanisms.Various in situ characterization techniques including X-ray, electron, neutron, optical, and scanning probes have been applied to the studies of LIBs and lithium–sulfur batteries.482,483 Whereas, only in situ X-ray diffraction and Raman spectroscopy have been relatively widely-adopted for the studies of ARMBs. It is suggested that researchers conduct more systematic in situ characterizations with the cutting-edge techniques to detect ARMB electrode at different depths of discharge and states of charge, as well as the influences of aqueous electrolytes and the interfaces between electrolytes and electrodes, therefore building up a bridge linking the macroscopic electrochemical performances with the microscopic physical and chemical variations of ARMB components. For example, in situ optical spectroscopy characterizations such as Fourier transform infrared spectroscopy and ultraviolet-visible absorption spectroscopy can be combined with Raman spectroscopy to quantitatively or semi-quantitatively identify fingerprints of electrodes, electrolyte and reaction products under different electrochemical operating stages.484In situ transmission electron microscopy and scanning electron microscopy can reveal the morphology evolution including expansion, extraction and crack formation of electrodes, and multi-dimensional information of electrodes can be obtained via transmission X-ray microscopy together with X-ray absorption near-edge structure.483 Nuclear magnetic resonance imaging has been reported to real-time monitor the microstructure growth of lithium metal, suggesting its huge potential in the studies of zinc dendrites in AZIBs.485 Moreover, structure- and dynamic-function relations of energy materials can be unveiled by in situ neutron-based techniques, and identifying battery working mechanism at molecular and/or atomic scale is thus feasible. Specifically, in situ neutron powder diffraction can be applied to help understand the complicated ion-intercalation and phase transitions in electrode materials, and even modest dynamical information can be probed from atomic displacement parameters.486 Besides, in situ small-angle neutron scattering that probes materials at larger length scales from 1 nanometre up to several micrometres is particularly powerful for characterization of structural evolution at nanometre scales.487 Driven by these cutting-edge in situ characterization techniques, energy material optimization and battery cell design could be substantially promoted, thus advancing current ARMB technologies.
5.8 More general theory and regulation strategies
A profound understanding of energy storage mechanisms is the foundation for continuous evolution of ARMBs. To date, compared with the fast advancing of electrochemical performances (e.g. capacity), in-depth understanding of electrochemical reaction mechanisms has not been correspondingly realized, and some explanations of electrochemical processes are still under debate. For example, co-insertion of protons with guest ions play entirely different roles in different ARMB systems, and in one case, they lead to poor cyclability,163 whereas in other case they result in increased capacity.43 Thus, it is important to establish a detailed theory that uncovers the relationship between electrode composition/structure evolution and corresponding changes in electrochemical characteristics, providing convincing information for tuning dominant physical and chemical properties to further improve electrochemical performances. Considering the variety of ARMBs together with the large quantity of constituent electrode materials, it is a challenging task to summarize general rules and subsequently build a versatile theory from traditional perspectives. Common mechanisms are few among various ARMBs despite the similarity of guest ions intercalation/de-intercalation. This is the reason why most of the existing explanations or mechanisms were made on a case-by-case basis. Here, starting from voltage characteristics may provide a different perspective angle to construct a theory with high generality and practicability for electrochemical improvements disregarding battery and electrode types. For example, flat discharge plateau indicates stable energy output and high energy efficiency, which can be found in both organic and inorganic electrode materials among different ARMBs.120,251 Whereas sloping discharge profiles are also commonly seen in some systems with slow electrochemical kinetics. Therefore, by studying general strategies that can regulate discharge plateau or other voltage characteristics, it is highly feasible to develop universal approaches to control the electrochemical performance of various ARMBs. To achieve this, precise characterization technologies and convincing theoretical calculations/simulations are indispensable.Thorough studies have been made on ARMBs in the past two decades, while the attempts to achieve practically usable high-voltage and high-energy ARMBs are preliminary and deficient. Although ARMBs cannot compete with commercial LIBs yet, recent investigations and the proposed perspectives have provided insightful strategies and promising opportunities that can facilitate the practical development of high-voltage and high-energy ARMBs.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Iranpour, M. K. Stenstrom, G. Tchobanoglous, D. Miller, J. Wright and M. Vossoughi, Science, 1999, 285, 706–711 CrossRef CAS PubMed.
- C. P. Grey and J. M. Tarascon, Nat. Mater., 2016, 16, 45–56 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- H. Kim, J. Hong, K. Y. Park, H. Kim, S. W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS.
- K. S. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- O. Schmidt, A. Hawkes, A. Gambhir and I. Staffell, Nat. Energy, 2017, 2, 17110 CrossRef.
- N. Alias and A. A. Mohamad, J. Power Sources, 2015, 274, 237–251 CrossRef CAS.
- B. Li, J. Zheng, H. Zhang, L. Jin, D. Yang, H. Lv, C. Shen, A. Shellikeri, Y. Zheng, R. Gong, J. P. Zheng and C. Zhang, Adv. Mater., 2018, 30, 1705670 CrossRef.
- F. Beck and P. Rüetschi, Electrochim. Acta, 2000, 45, 2467–2482 CrossRef CAS.
- Z. Liu, F. Mo, H. Li, M. Zhu, Z. Wang, G. Liang and C. Zhi, Small Methods, 2018, 2, 1800124 CrossRef.
- Z. Wang, H. Li, Z. Tang, Z. Liu, Z. Ruan, L. Ma, Q. Yang, D. Wang and C. Zhi, Adv. Funct. Mater., 2018, 28, 1804560 CrossRef.
- Y. Huang, Z. Li, Z. Pei, Z. Liu, H. Li, M. Zhu, J. Fan, Q. Dai, M. Zhang, L. Dai and C. Zhi, Adv. Energy Mater., 2018, 8, 1802288 CrossRef.
- Q. Yang, Y. Wang, X. Li, H. Li, Z. Wang, Z. Tang, L. Ma, F. Mo and C. Zhi, Energy Environ. Mater., 2018, 1, 183–195 CrossRef.
- Z. Xing, S. Wang, A. Yu and Z. Chen, Nano Energy, 2018, 50, 229–244 CrossRef CAS.
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- W. Tang, Y. Zhu, Y. Hou, L. Liu, Y. Wu, K. P. Loh, H. Zhang and K. Zhu, Energy Environ. Sci., 2013, 6, 2093–2104 RSC.
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS.
- X. L. Dong, L. Chen, X. L. Su, Y. G. Wang and Y. Y. Xia, Angew. Chem., Int. Ed., 2016, 55, 7474–7477 CrossRef CAS.
- J. X. Zheng, Y. Y. Hou, Y. D. Duan, X. H. Song, Y. Wei, T. C. Liu, J. T. Hu, H. Guo, Z. Q. Zhuo, L. L. Liu, Z. Chang, X. W. Wang, D. Zherebetskyy, Y. Y. Fang, Y. Lin, K. Xu, L. W. Wang, Y. P. Wu and F. Pan, Nano Lett., 2015, 15, 6102–6109 CrossRef CAS.
- W. Tang, L. L. Liu, Y. S. Zhu, H. Sun, Y. P. Wu and K. Zhu, Energy Environ. Sci., 2012, 5, 6909–6913 RSC.
- Q. T. Qu, L. J. Fu, X. Y. Zhan, D. Samuelis, J. Maier, L. Li, S. Tian, Z. H. Li and Y. P. Wu, Energy Environ. Sci., 2011, 4, 3985–3990 RSC.
- Y. Zhao, Y. Zhang, H. Sun, X. L. Dong, J. Y. Cao, L. Wang, Y. F. Xu, J. Ren, Y. N. Hwang, I. H. Son, X. L. Huang, Y. G. Wang and H. S. Peng, Angew. Chem., Int. Ed., 2016, 55, 14382–14386 Search PubMed.
- Z. X. Liu, H. F. Li, M. S. Zhu, Y. Huang, Z. J. Tang, Z. X. Pei, Z. F. Wang, Z. C. Shi, J. Liu, Y. Huang and C. Y. Zhi, Nano Energy, 2018, 44, 164–173 CrossRef CAS.
- R. K. Guduru and J. C. Icaza, Nanomaterials, 2016, 6, 41 CrossRef.
- T. Liu, X. Cheng, H. Yu, H. Zhu, N. Peng, R. Zheng, J. Zhang, M. Shui, Y. Cui and J. Shu, Energy Storage Mater., 2019, 18, 68–91 CrossRef.
- Y. Li, J. Fu, C. Zhong, T. Wu, Z. Chen, W. Hu, K. Amine and J. Lu, Adv. Energy Mater., 2019, 9, 1802605 CrossRef.
- M. Song, H. Tan, D. Chao and H. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- D. Bin, F. Wang, A. Tamirat, L. Suo, Y. Wang, C. Wang and Y. Xia, Adv. Energy Mater., 2018, 8, 1703008 CrossRef.
- J. O. G. Posada, A. J. R. Rennie, S. P. Villar, V. L. Martins, J. Marinaccio, A. Barnes, C. F. Glover, D. A. Worsley and P. J. Hall, Renewable Sustainable Energy Rev., 2017, 68, 1174–1182 CrossRef CAS.
- A. Eftekhari, Adv. Energy Mater., 2018, 8, 1801156 CrossRef.
- J. Huang, Z. Guo, Y. Ma, D. Bin, Y. Wang and Y. Xia, Small Methods, 2019, 3, 1800272 CrossRef.
- E. Roduner, Catal. Today, 2018, 309, 263–268 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- Y. Wang, J. Yi and Y. Xia, Adv. Energy Mater., 2012, 2, 830–840 CrossRef CAS.
- S. Husmann and A. J. G. Zarbin, Electrochim. Acta, 2018, 283, 1339–1350 CrossRef CAS.
- W. Zuo, W. Zhu, D. Zhao, Y. Sun, Y. Li, J. Liu and X. W. Lou, Energy Environ. Sci., 2016, 9, 2881–2891 RSC.
- S. H. Yu, X. Feng, N. Zhang, J. Seok and H. D. Abruna, Acc. Chem. Res., 2018, 51, 273–281 CrossRef CAS.
- D. C. Hannah, G. Sai Gautam, P. Canepa and G. Ceder, Adv. Energy Mater., 2018, 8, 1800379 CrossRef.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods - Fundamentals and Applications, John Wiley & Sons, Inc., 2nd edn, 2001 Search PubMed.
- W. Chen, F. Ambrosio, G. Miceli and A. Pasquarello, Phys. Rev. Lett., 2016, 117, 186401 CrossRef.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 99th edn, 2018 Search PubMed.
- P. Peljo and H. H. Girault, Energy Environ. Sci., 2018, 11, 2306–2309 RSC.
- X. Zang, C. Shen, M. Sanghadasa and L. Lin, ChemElectroChem, 2019, 6, 976–988 CrossRef CAS.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS.
- R. Chen, R. Luo, Y. Huang, F. Wu and L. Li, Adv. Sci., 2016, 3, 1600051 CrossRef.
- W. Pei, Y. Hui and H. Q. Yang, J. Power Sources, 1996, 63, 275–278 CrossRef.
- Y. G. Wang, J. Y. Lou, W. Wu, C. X. Wang and Y. Y. Xia, J. Electrochem. Soc., 2007, 154, A228–A234 CrossRef CAS.
- Y. Wang, S. Z. Yang, Y. You, Z. Feng, W. Zhu, V. Gariepy, J. Xia, B. Commarieu, A. Darwiche, A. Guerfi and K. Zaghib, ACS Appl. Mater. Interfaces, 2018, 10, 7061–7068 CrossRef CAS.
- Y. Yokoyama, T. Fukutsuka, K. Miyazaki and T. Abe, J. Electrochem. Soc., 2018, 165, A3299–A3303 CrossRef CAS.
- C. Wessells, R. Ruffo, R. A. Huggins and Y. Cui, Electrochem. Solid-State Lett., 2010, 13, A59–A61 CrossRef CAS.
- D. Zhou, S. Liu, H. Wang and G. Yan, J. Power Sources, 2013, 227, 111–117 CrossRef CAS.
- L. Ma, S. Chen, H. Li, Z. Ruan, Z. Tang, Z. Liu, Z. Wang, Y. Huang, Z. Pei, J. A. Zapien and C. Zhi, Energy Environ. Sci., 2018, 11, 2521–2530 RSC.
- H. Tomiyasu, H. Shikata, K. Takao, N. Asanuma, S. Taruta and Y. Y. Park, Sci. Rep., 2017, 7, 45048 CrossRef CAS.
- M. Zhao, B. Zhang, G. Huang, H. Zhang and X. Song, J. Power Sources, 2013, 232, 181–186 CrossRef CAS.
- L. M. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. L. Fan, C. Luo, C. S. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129 CrossRef CAS.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 7136–7141 CrossRef CAS.
- R.-S. Kühnel, D. Reber and C. Battaglia, ACS Energy Lett., 2017, 2, 2005–2006 CrossRef.
- D. P. Leonard, Z. Wei, G. Chen, F. Du and X. Ji, ACS Energy Lett., 2018, 3, 373–374 CrossRef CAS.
- F. Wang, X. Fan, T. Gao, W. Sun, Z. Ma, C. Yang, F. Han, K. Xu and C. Wang, ACS Cent. Sci., 2017, 3, 1121–1128 CrossRef CAS.
- P. Hu, M. Yan, T. Zhu, X. Wang, X. Wei, J. Li, L. Zhou, Z. Li, L. Chen and L. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 42717–42722 CrossRef CAS PubMed.
- P. W. Wilson and A. D. Haymet, Phys. Chem. Chem. Phys., 2009, 11, 2679–2682 RSC.
- M. S. Wei, B. X. Li, X. N. Zang, W. S. Chen, Y. Chu, M. Sanghadasa and L. W. Lin, 2017 19th International Conference on Solid-State Sensors, Actuators and Microsystems (Transducers), 2017, 702–705.
- R. Ruffo, F. La Mantia, C. Wessells, R. A. Huggins and Y. Cui, Solid State Ionics, 2011, 192, 289–292 CrossRef CAS.
- Z. Zhu, Y. Tang, Z. Lv, J. Wei, Y. Zhang, R. Wang, W. Zhang, H. Xia, M. Ge and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 3656–3660 CrossRef CAS.
- Y. Gao, Z. Yan, J. L. Gray, X. He, D. Wang, T. Chen, Q. Huang, Y. C. Li, H. Wang, S. H. Kim, T. E. Mallouk and D. Wang, Nat. Mater., 2019, 18, 384–389 CrossRef CAS PubMed.
- A. J. Louli, L. D. Ellis and J. R. Dahn, Joule, 2019, 3, 745–761 CrossRef CAS.
- W. J. Chang, S. H. Kim, J. Hwang, J. Chang, D. W. Yang, S. S. Kwon, J. T. Kim, W. W. Lee, J. H. Lee, H. Park, T. Song, I. H. Lee, D. Whang and W. Il Park, Nat. Commun., 2018, 9, 3461 CrossRef.
- C. Cao, I. I. Abate, E. Sivonxay, B. Shyam, C. Jia, B. Moritz, T. P. Devereaux, K. A. Persson, H.-G. Steinrück and M. F. Toney, Joule, 2019, 3, 762–781 CrossRef CAS.
- I. B. Stojković, N. D. Cvjetićanin and S. V. Mentus, Electrochem. Commun., 2010, 12, 371–373 CrossRef.
- K. Miyazaki, T. Shimada, S. Ito, Y. Yokoyama, T. Fukutsuka and T. Abe, Chem. Commun., 2016, 52, 4979–4982 RSC.
- Z. Hou, X. Zhang, X. Li, Y. Zhu, J. Liang and Y. Qian, J. Mater. Chem. A, 2017, 5, 730–738 RSC.
- J. Y. Luo, W. J. Cui, P. He and Y. Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS.
- Z. Guo, Y. Zhao, Y. Ding, X. Dong, L. Chen, J. Cao, C. Wang, Y. Xia, H. Peng and Y. Wang, Chem, 2017, 3, 348–362 CAS.
- X. Zhang, Z. Hou, X. Li, J. Liang, Y. Zhu and Y. Qian, J. Mater. Chem. A, 2016, 4, 856–860 RSC.
- K. Nakamoto, Y. Kano, A. Kitajou and S. Okada, J. Power Sources, 2016, 327, 327–332 CrossRef CAS.
- L. Chen, W. Li, Z. Guo, Y. Wang, C. Wang, Y. Che and Y. Xia, J. Electrochem. Soc., 2015, 162, A1972–A1977 CrossRef CAS.
- F. Yu, S. Zhang, C. Fang, Y. Liu, S. He, J. Xia, J. Yang and N. Zhang, Ceram. Int., 2017, 43, 9960–9967 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS.
- N. Batisse and E. Raymundo-Piñero, J. Power Sources, 2017, 348, 168–174 CrossRef CAS.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 RSC.
- X. Chia, A. Y. Eng, A. Ambrosi, S. M. Tan and M. Pumera, Chem. Rev., 2015, 115, 11941–11966 CrossRef CAS.
- W. Chen, J. Gu, Q. Liu, R. Luo, L. Yao, B. Sun, W. Zhang, H. Su, B. Chen, P. Liu and D. Zhang, ACS Nano, 2018, 12, 308–316 CrossRef CAS PubMed.
- D. Kong, H. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341–1347 CrossRef CAS.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P. L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 16070 CrossRef CAS.
- Y. Yang, H. Fei, G. Ruan, C. Xiang and J. M. Tour, Adv. Mater., 2014, 26, 8163–8168 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS.
- D. M. Robinson, Y. B. Go, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2010, 132, 11467–11469 CrossRef CAS.
- J. Park, H. Kim, K. Jin, B. J. Lee, Y. S. Park, H. Kim, I. Park, K. D. Yang, H. Y. Jeong, J. Kim, K. T. Hong, H. W. Jang, K. Kang and K. T. Nam, J. Am. Chem. Soc., 2014, 136, 4201–4211 CrossRef CAS.
- J. Liu, L. Yi, L. Liu and P. Peng, Mater. Chem. Phys., 2015, 161, 211–218 CrossRef CAS.
- G. J. Wang, N. H. Zhao, L. C. Yang, Y. P. Wu, H. Q. Wu and R. Holze, Electrochim. Acta, 2007, 52, 4911–4915 CrossRef CAS.
- R. S. Kühnel, D. Reber, A. Remhof, R. Figi, D. Bleiner and C. Battaglia, Chem. Commun., 2016, 52, 10435–10438 RSC.
- W. Li, W. R. Mckinnon and J. R. Dahn, J. Electrochem. Soc., 1994, 141, 2310–2316 CrossRef CAS.
- J.-Y. Luo and Y.-Y. Xia, Adv. Funct. Mater., 2007, 17, 3877–3884 CrossRef CAS.
- A. Eftekhari, Electrochim. Acta, 2001, 47, 495–499 CrossRef CAS.
- J.-W. Lee and S.-I. Pyun, Electrochim. Acta, 2004, 49, 753–761 CrossRef CAS.
- R. L. Deutscher, T. M. Florence and R. Woods, J. Power Sources, 1995, 55, 41–46 CrossRef CAS.
- A. Yuan and Q. Zhang, Electrochem. Commun., 2006, 8, 1173–1178 CrossRef CAS.
- V. S. Nair, Y. L. Cheah and S. Madhavi, J. Electrochem. Soc., 2014, 161, A256–A263 CrossRef CAS.
- H. Wang, K. Huang, Y. Zeng, F. Zhao and L. Chen, Electrochem. Solid-State Lett., 2007, 10, A199–A203 CrossRef CAS.
- M. Manickam, P. Singh, S. Thurgate and K. Prince, J. Power Sources, 2006, 158, 646–649 CrossRef CAS.
- F. Sauvage, L. Laffont, J. M. Tarascon and E. Baudrin, J. Power Sources, 2008, 175, 495–501 CrossRef CAS.
- H. Manjunatha, T. V. Venkatesha and G. S. Suresh, Electrochim. Acta, 2011, 58, 247–257 CrossRef CAS.
- M. Minakshi, Electrochim. Acta, 2010, 55, 9174–9178 CrossRef CAS.
- M. Minakshi, P. Singh, N. Sharma, M. Blackford and M. Ionescu, Ind. Eng. Chem. Res., 2011, 50, 1899–1905 CrossRef CAS.
- M. Minakshi, P. Singh, D. Appadoo and D. E. Martin, Electrochim. Acta, 2011, 56, 4356–4360 CrossRef CAS.
- C. D. Wessells, S. V. Peddada, M. T. McDowell, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2011, 159, A98–A103 CrossRef.
- G. J. Wang, H. P. Zhang, L. J. Fu, B. Wang and Y. P. Wu, Electrochem. Commun., 2007, 9, 1873–1876 CrossRef CAS.
- W. Tang, Y. Hou, F. Wang, L. Liu, Y. Wu and K. Zhu, Nano Lett., 2013, 13, 2036–2040 CrossRef CAS.
- A. Yuan, L. Tian, W. Xu and Y. Wang, J. Power Sources, 2010, 195, 5032–5038 CrossRef CAS.
- W. Xu, A. Yuan, L. Tian and Y. Wang, J. Appl. Electrochem., 2011, 41, 453–460 CrossRef CAS.
- Q. Qu, L. Fu, X. Zhan, D. Samuelis, J. Maier, L. Li, S. Tian, Z. Li and Y. Wu, Energy Environ. Sci., 2011, 4, 3985–3990 RSC.
- W. Tang, Y. Hou, F. Wang, L. Liu, Y. Wu and K. Zhu, Nano Lett., 2013, 13, 2036–2040 CrossRef CAS.
- R. Ruffo, C. Wessells, R. A. Huggins and Y. Cui, Electrochem. Commun., 2009, 11, 247–249 CrossRef CAS.
- G. J. Wang, Q. T. Qu, B. Wang, Y. Shi, S. Tian, Y. P. Wu and R. Holze, Electrochim. Acta, 2009, 54, 1199–1203 CrossRef CAS.
- Y.-g. Wang, J.-y. Luo, C.-x. Wang and Y.-y. Xia, J. Electrochem. Soc., 2006, 153, A1425–A1431 CrossRef CAS.
- Y.-g. Wang, J.-y. Lou, W. Wu, C.-x. Wang and Y.-y. Xia, J. Electrochem. Soc., 2007, 154, A228–A234 CrossRef CAS.
- R. B. Shivashankaraiah, H. Manjunatha, K. C. Mahesh, G. S. Suresh and T. V. Venkatesha, J. Solid State Electrochem., 2012, 16, 1279–1290 CrossRef CAS.
- M. Mohan Rao, M. Jayalakshmi, O. Schäf, H. Wulff, U. Guth and F. Scholz, J. Solid State Electrochem., 2001, 5, 50–56 CrossRef.
- M. Mohan Rao, M. Jayalakshmi, O. Schäf, U. Guth, H. Wulff and F. Scholz, J. Solid State Electrochem., 1999, 4, 17–23 CrossRef.
- W. Tang, L. L. Liu, S. Tian, L. Li, Y. B. Yue, Y. P. Wu, S. Y. Guan and K. Zhu, Electrochem. Commun., 2010, 12, 1524–1526 CrossRef CAS.
- L. Liu, F. Tian, X. Wang, Z. Yang, Q. Chen and X. Wang, J. Solid State Electrochem., 2012, 16, 491–497 CrossRef CAS.
- F. Wang, S. Xiao, Z. Chang, Y. Yang and Y. Wu, Chem. Commun., 2013, 49, 9209–9211 RSC.
- A. Tron, Y. N. Jo, S. H. Oh, Y. D. Park and J. Mun, ACS Appl. Mater. Interfaces, 2017, 9, 12391–12399 CrossRef CAS.
- P. He, J.-L. Liu, W.-J. Cui, J.-Y. Luo and Y.-Y. Xia, Electrochim. Acta, 2011, 56, 2351–2357 CrossRef CAS.
- X.-H. Liu, T. Saito, T. Doi, S. Okada and J.-i. Yamaki, J. Power Sources, 2009, 189, 706–710 CrossRef CAS.
- M. Zhao, G. Huang, W. Zhang, H. Zhang and X. Song, Energy Fuels, 2013, 27, 1162–1167 CrossRef CAS.
- M. Minakshi, N. Sharma, D. Ralph, D. Appadoo and K. Nallathamby, Electrochem. Solid-State Lett., 2011, 14, A86–A89 CrossRef CAS.
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Y. Cui, Nano Lett., 2013, 13, 5748–5752 CrossRef CAS PubMed.
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421–5425 CrossRef CAS.
- M. Pasta, C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2012, 3, 1149 CrossRef PubMed.
- M. Pasta, C. D. Wessells, N. Liu, J. Nelson, M. T. McDowell, R. A. Huggins, M. F. Toney and Y. Cui, Nat. Commun., 2014, 5, 3007 CrossRef.
- Z. Wu, J. Xie, Z. J. Xu, S. Zhang and Q. Zhang, J. Mater. Chem. A, 2019, 7, 4259–4290 RSC.
- H. Kim, J. Hong, K.-Y. Park, H. Kim, S.-W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS.
- K. Itaya, T. Ataka and S. Toshima, J. Am. Chem. Soc., 1982, 104, 4767–4772 CrossRef CAS.
- P. C. F. Pau, J. O. Berg and W. G. McMillan, J. Phys. Chem., 1990, 94, 2671–2679 CrossRef CAS.
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS.
- W. Li, W. R. McKinnon and J. R. Dahn, J. Electrochem. Soc., 1994, 141, 2310–2316 CrossRef CAS.
- S. Zhang, Y. Li, C. Wu, F. Zheng and Y. Xie, J. Phys. Chem. C, 2009, 113, 15058–15067 CrossRef CAS.
- J. Ni, W. Jiang, K. Yu, Y. Gao and Z. Zhu, Electrochim. Acta, 2011, 56, 2122–2126 CrossRef CAS.
- G. Wang, L. Fu, N. Zhao, L. Yang, Y. Wu and H. Wu, Angew. Chem., 2007, 119, 299–301 CrossRef.
- H. Heli, H. Yadegari and A. Jabbari, J. Phys. Chem. C, 2011, 115, 10889–10897 CrossRef CAS.
- H. Li, T. Zhai, P. He, Y. Wang, E. Hosono and H. Zhou, J. Mater. Chem., 2011, 21, 1780–1787 RSC.
- W. Tang, X. Gao, Y. Zhu, Y. Yue, Y. Shi, Y. Wu and K. Zhu, J. Mater. Chem., 2012, 22, 20143–20145 RSC.
- K. H. Reiman, K. M. Brace, T. J. Gordon-Smith, I. Nandhakumar, G. S. Attard and J. R. Owen, Electrochem. Commun., 2006, 8, 517–522 CrossRef CAS.
- J.-Y. Luo, J.-L. Liu, P. He and Y.-Y. Xia, Electrochim. Acta, 2008, 53, 8128–8133 CrossRef CAS.
- C. Wessells, F. La Mantia, H. Deshazer, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2011, 158, A352–A355 CrossRef CAS.
- C. Wessells, R. A. Huggins and Y. Cui, J. Power Sources, 2011, 196, 2884–2888 CrossRef CAS.
- G. Wang, Q. Qu, B. Wang, Y. Shi, S. Tian and Y. Wu, ChemPhysChem, 2008, 9, 2299–2301 CrossRef CAS PubMed.
- H. Qin, Z. P. Song, H. Zhan and Y. H. Zhou, J. Power Sources, 2014, 249, 367–372 CrossRef CAS.
- A. Caballero, J. Morales and O. A. Vargas, J. Power Sources, 2010, 195, 4318–4321 CrossRef CAS.
- K. Sun, D. A. Juarez, H. Huang, E. Jung and S. J. Dillon, J. Power Sources, 2014, 248, 582–587 CrossRef CAS.
- H. Wang, Y. Zeng, K. Huang, S. Liu and L. Chen, Electrochim. Acta, 2007, 52, 5102–5107 CrossRef CAS.
- M. Manickam, P. Singh, T. B. Issa and S. Thurgate, J. Appl. Electrochem., 2006, 36, 599–602 CrossRef CAS.
- W. Li and J. R. Dahn, J. Electrochem. Soc., 1995, 142, 1742–1746 CrossRef CAS.
- J.-Y. Luo, W.-J. Cui, P. He and Y.-Y. Xia, Nat. Chem., 2010, 2, 760 CrossRef CAS.
- D. W. Murphy, P. A. Christian, F. J. DiSalvo, J. N. Carides and J. V. Waszczak, J. Electrochem. Soc., 1981, 128, 2053–2060 CrossRef CAS.
- M. Zhang and J. R. Dahn, J. Electrochem. Soc., 1996, 143, 2730–2735 CrossRef CAS.
- M. Shao, J. Deng, F. Zhong, Y. Cao, X. Ai, J. Qian and H. Yang, Energy Storage Mater., 2019, 18, 92–99 CrossRef.
- J. Köhler, H. Makihara, H. Uegaito, H. Inoue and M. Toki, Electrochim. Acta, 2000, 46, 59–65 CrossRef.
- C. Cheng, Z. H. Li, X. Y. Zhan, Q. Z. Xiao, G. T. Lei and X. D. Zhou, Electrochim. Acta, 2010, 55, 4627–4631 CrossRef CAS.
- Z. Liu, H. Li, M. Zhu, Y. Huang, Z. Tang, Z. Pei, Z. Wang, Z. Shi, J. Liu, Y. Huang and C. Zhi, Nano Energy, 2018, 44, 164–173 CrossRef CAS.
- H. Wang, K. Huang, Y. Zeng, S. Yang and L. Chen, Electrochim. Acta, 2007, 52, 3280–3285 CrossRef CAS.
- G. J. Wang, L. C. Yang, Q. T. Qu, B. Wang, Y. P. Wu and R. Holze, J. Solid State Electrochem., 2010, 14, 865–869 CrossRef CAS.
- J. Xie and Q. Zhang, J. Mater. Chem. A, 2016, 4, 7091–7106 RSC.
- S. Wang, C. Sun, N. Wang and Q. Zhang, J. Mater. Chem. A, 2019, 7, 10138–10158 RSC.
- L. Athouël, F. Moser, R. Dugas, O. Crosnier, D. Bélanger and T. Brousse, J. Phys. Chem. C, 2008, 112, 7270–7277 CrossRef.
- J. F. Whitacre, T. Wiley, S. Shanbhag, Y. Wenzhuo, A. Mohamed, S. E. Chun, E. Weber, D. Blackwood, E. Lynch-Bell, J. Gulakowski, C. Smith and D. Humphreys, J. Power Sources, 2012, 213, 255–264 CrossRef CAS.
- M. Minakshi, Mater. Sci. Eng., B, 2012, 177, 1788–1792 CrossRef CAS.
- Q. T. Qu, L. L. Liu, Y. P. Wu and R. Holze, Electrochim. Acta, 2013, 96, 8–12 CrossRef CAS.
- F. Sauvage, E. Baudrin and J. M. Tarascon, Sens. Actuators, B, 2007, 120, 638–644 CrossRef CAS.
- Y. Wang, L. Mu, J. Liu, Z. Yang, X. Yu, L. Gu, Y.-S. Hu, H. Li, X.-Q. Yang, L. Chen and X. Huang, Adv. Energy Mater., 2015, 5, 1501005 CrossRef.
- Y. Liu, Y. Qiao, W. Zhang, H. Xu, Z. Li, Y. Shen, L. Yuan, X. Hu, X. Dai and Y. Huang, Nano Energy, 2014, 5, 97–104 CrossRef CAS.
- Y. Liu, Y. Qiao, X. Lou, X. Zhang, W. Zhang and Y. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 14564–14571 CrossRef CAS.
- W. Song, X. Ji, Y. Zhu, H. Zhu, F. Li, J. Chen, F. Lu, Y. Yao and C. E. Banks, ChemElectroChem, 2014, 1, 871–876 CrossRef CAS.
- J. Qian, C. Wu, Y. Cao, Z. Ma, Y. Huang, X. Ai and H. Yang, Adv. Energy Mater., 2018, 8, 1702619 CrossRef.
- V. D. Neff, J. Electrochem. Soc., 1978, 125, 886–887 CrossRef CAS.
- C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2011, 2, 550 CrossRef.
- C. D. Wessells, M. T. McDowell, S. V. Peddada, M. Pasta, R. A. Huggins and Y. Cui, ACS Nano, 2012, 6, 1688–1694 CrossRef CAS.
- W. Ren, X. Chen and C. Zhao, Adv. Energy Mater., 2018, 8, 1801413 CrossRef.
- X. Wu, Y. Luo, M. Sun, J. Qian, Y. Cao, X. Ai and H. Yang, Nano Energy, 2015, 13, 117–123 CrossRef CAS.
- X. Wu, M. Sun, S. Guo, J. Qian, Y. Liu, Y. Cao, X. Ai and H. Yang, ChemNanoMat, 2015, 1, 188–193 CrossRef CAS.
- K. Nakamoto, R. Sakamoto, M. Ito, A. Kitajou and S. Okada, Electrochemistry, 2017, 85, 179–185 CrossRef CAS.
- J.-H. Lee, G. Ali, D. H. Kim and K. Y. Chung, Adv. Energy Mater., 2017, 7, 1601491 CrossRef.
- J. Xie, P. Gu and Q. Zhang, ACS Energy Lett., 2017, 2, 1985–1996 CrossRef CAS.
- J. Xie, C. E. Zhao, Z. Q. Lin, P. Y. Gu and Q. Zhang, Chem. – Asian J., 2016, 11, 1489–1511 CrossRef CAS.
- Y. Xu, M. Zhou and Y. Lei, Mater. Today, 2018, 21, 60–78 CrossRef CAS.
- K. Koshika, N. Sano, K. Oyaizu and H. Nishide, Chem. Commun., 2009, 836–838, 10.1039/B818087C.
- H. Long, W. Zeng, H. Wang, M. Qian, Y. Liang and Z. Wang, Adv. Sci., 2018, 5, 1700634 CrossRef.
- Y. Wei, Q. Hu, Y. Cao, D. Fang, W. Xu, M. Jiang, J. Huang, H. Liu and X. Fan, Org. Electron., 2017, 46, 211–217 CrossRef CAS.
- Y. Liu, B. H. Zhang, S. Y. Xiao, L. L. Liu, Z. B. Wen and Y. P. Wu, Electrochim. Acta, 2014, 116, 512–517 CrossRef CAS.
- C. Deng, S. Zhang, Z. Dong and Y. Shang, Nano Energy, 2014, 4, 49–55 CrossRef CAS.
- Q. Yang, S. Cui, Y. Ge, Z. Tang, Z. Liu, H. Li, N. Li, H. Zhang, J. Liang and C. Zhi, Nano Energy, 2018, 50, 623–631 CrossRef CAS.
- S. I. Park, I. Gocheva, S. Okada and J.-i. Yamaki, J. Electrochem. Soc., 2011, 158, A1067–A1070 CrossRef CAS.
- W. Wu, J. Yan, A. Wise, A. Rutt and J. F. Whitacre, J. Electrochem. Soc., 2014, 161, A561–A567 CrossRef CAS.
- B. Zhao, B. Lin, S. Zhang and C. Deng, Nanoscale, 2015, 7, 18552–18560 RSC.
- W. Deng, Y. Shen, J. Qian and H. Yang, Chem. Commun., 2015, 51, 5097–5099 RSC.
- Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841 CrossRef CAS.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- H. Zhang, K. Ye, X. Huang, X. Wang, K. Cheng, X. Xiao, G. Wang and D. Cao, J. Power Sources, 2017, 338, 136–144 CrossRef CAS.
- J. F. Parker, C. N. Chervin, I. R. Pala, M. Machler, M. F. Burz, J. W. Long and D. R. Rolison, Science, 2017, 356, 415–418 CrossRef CAS.
- G. Li, Z. Yang, Y. Jiang, C. Jin, W. Huang, X. Ding and Y. Huang, Nano Energy, 2016, 25, 211–217 CrossRef CAS.
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y.-K. Sun and S.-T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS.
- M. H. Alfaruqi, S. Islam, D. Y. Putro, V. Mathew, S. Kim, J. Jo, S. Kim, Y.-K. Sun, K. Kim and J. Kim, Electrochim. Acta, 2018, 276, 1–11 CrossRef CAS.
- M. Chamoun, W. R. Brant, C.-W. Tai, G. Karlsson and D. Noréus, Energy Storage Mater., 2018, 15, 351–360 CrossRef.
- Z. Liu, D. Wang, Z. Tang, G. Liang, Q. Yang, H. Li, L. Ma, F. Mo and C. Zhi, Energy Storage Mater., 2019, 23, 636–645 CrossRef.
- G. Wu, Y. Xue, L. Wang, X. Wang and G. Chen, J. Mater. Chem. A, 2018, 6, 3376–3380 RSC.
- M. Zhu, Z. Wang, H. Li, Y. Xiong, Z. Liu, Z. Tang, Y. Huang, A. L. Rogach and C. Zhi, Energy Environ. Sci., 2018, 11, 2414–2422 RSC.
- I. Stosevski, A. Bonakdarpour, F. Cuadra and D. P. Wilkinson, Chem. Commun., 2019, 55, 2082–2085 RSC.
- B. Jiang, C. Xu, C. Wu, L. Dong, J. Li and F. Kang, Electrochim. Acta, 2017, 229, 422–428 CrossRef CAS.
- C. Zhu, G. Fang, J. Zhou, J. Guo, Z. Wang, C. Wang, J. Li, Y. Tang and S. Liang, J. Mater. Chem. A, 2018, 6, 9677–9683 RSC.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS PubMed.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- C. Wei, C. Xu, B. Li, H. Du and F. Kang, J. Phys. Chem. Solids, 2012, 73, 1487–1491 CrossRef CAS.
- S. Islam, M. H. Alfaruqi, V. Mathew, J. Song, S. Kim, S. Kim, J. Jo, J. P. Baboo, D. T. Pham, D. Y. Putro, Y.-K. Sun and J. Kim, J. Mater. Chem. A, 2017, 5, 23299–23309 RSC.
- M. H. Alfaruqi, V. Mathew, J. Gim, S. Kim, J. Song, J. P. Baboo, S. H. Choi and J. Kim, Chem. Mater., 2015, 27, 3609–3620 CrossRef CAS.
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, D. T. Pham, J. Jo, Z. Xiu, V. Mathew and J. Kim, Electrochem. Commun., 2015, 60, 121–125 CrossRef CAS.
- S. Luo, L. Xie, F. Han, W. Wei, Y. Huang, H. Zhang, M. Zhu, O. G. Schmidt and L. Wang, Adv. Funct. Mater., 2019, 29, 1901336 CrossRef.
- D. Xu, B. Li, C. Wei, Y.-B. He, H. Du, X. Chu, X. Qin, Q.-H. Yang and F. Kang, Electrochim. Acta, 2014, 133, 254–261 CrossRef CAS.
- L. Chen, Z. Yang, H. Qin, X. Zeng and J. Meng, J. Power Sources, 2019, 425, 162–169 CrossRef CAS.
- B. Li, J. Chai, X. Ge, T. An, P.-C. Lim, Z. Liu and Y. Zong, ChemNanoMat, 2017, 3, 401–405 CrossRef CAS.
- B. Wu, G. Zhang, M. Yan, T. Xiong, P. He, L. He, X. Xu and L. Mai, Small, 2018, 14, 1703850 CrossRef.
- C. Pan, R. Zhang, R. G. Nuzzo and A. A. Gewirth, Adv. Energy Mater., 2018, 8, 1800589 CrossRef.
- C. Yuan, Y. Zhang, Y. Pan, X. Liu, G. Wang and D. Cao, Electrochim. Acta, 2014, 116, 404–412 CrossRef CAS.
- Z. Yang, X. Wang and Y. Huang, Curr. Appl. Phys., 2015, 15, 1556–1561 CrossRef.
- T. Xiong, Z. G. Yu, H. Wu, Y. Du, Q. Xie, J. Chen, Y.-W. Zhang, S. J. Pennycook, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2019, 9, 1803815 CrossRef.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef.
- K. W. Nam, H. Kim, J. H. Choi and J. W. Choi, Energy Environ. Sci., 2019, 12, 1999–2009 RSC.
- N. Zhang, Y. Dong, M. Jia, X. Bian, Y. Wang, M. Qiu, J. Xu, Y. Liu, L. Jiao and F. Cheng, ACS Energy Lett., 2018, 3, 1366–1372 CrossRef CAS.
- J. Meng, Z. Liu, C. Niu, X. Xu, X. Liu, G. Zhang, X. Wang, M. Huang, Y. Yu and L. Mai, J. Mater. Chem. A, 2016, 4, 4893–4899 RSC.
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang, Y. Shao, K. T. Mueller, L. Mai, J. Liu and J. Yang, Adv. Mater., 2018, 30, 1703725 CrossRef PubMed.
- F. Ming, H. Liang, Y. Lei, S. Kandambeth, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2018, 3, 2602–2609 CrossRef CAS.
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef.
- M. Jiang, J. Zhu, C. Chen, Y. Lu, E. S. Pampal, L. Luo, P. Zhu and X. Zhang, J. Mater. Chem. A, 2016, 4, 16588–16596 RSC.
- G. He, C. A. Bridges and A. Manthiram, Chem. Mater., 2015, 27, 6699–6707 CrossRef CAS.
- J.-H. Kim, A. Huq, M. Chi, N. P. W. Pieczonka, E. Lee, C. A. Bridges, M. M. Tessema, A. Manthiram, K. A. Persson and B. R. Powell, Chem. Mater., 2014, 26, 4377–4386 CrossRef CAS.
- C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef.
- B. Sambandam, V. Soundharrajan, S. Kim, M. H. Alfaruqi, J. Jo, S. Kim, V. Mathew, Y.-k. Sun and J. Kim, J. Mater. Chem. A, 2018, 6, 3850–3856 RSC.
- W. Li, K. Wang, S. Cheng and K. Jiang, Energy Storage Mater., 2018, 15, 14–21 CrossRef.
- F. Wan, Y. Zhang, L. Zhang, D. Liu, C. Wang, L. Song, Z. Niu and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 7062–7067 CrossRef CAS.
- P. He, M. Yan, G. Zhang, R. Sun, L. Chen, Q. An and L. Mai, Adv. Energy Mater., 2017, 7, 1601920 CrossRef.
- J. Xie and Q. Zhang, Small, 2019, 15, 1805061 CrossRef.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef.
- Z. Guo, Y. Ma, X. Dong, J. Huang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 11737–11741 CrossRef CAS PubMed.
- C. Kim, B. Y. Ahn, T.-S. Wei, Y. Jo, S. Jeong, Y. Choi, I.-D. Kim and J. A. Lewis, ACS Nano, 2018, 12, 11838–11846 CrossRef CAS PubMed.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y.-S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- Y. Ma, X. Xie, R. Lv, B. Na, J. Ouyang and H. Liu, ACS Sustainable Chem. Eng., 2018, 6, 8697–8703 CrossRef CAS.
- H. Y. Shi, Y. J. Ye, K. Liu, Y. Song and X. Sun, Angew. Chem., Int. Ed., 2018, 57, 16359–16363 CrossRef CAS.
- X. Li, X. Xie, R. Lv, B. Na, B. Wang and Y. He, Energy Technol., 2019, 7, 1801092 CrossRef.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- R. Trócoli and F. La Mantia, ChemSusChem, 2015, 8, 481–485 CrossRef.
- G. Kasiri, J. Glenneberg, A. Bani Hashemi, R. Kun and F. La Mantia, Energy Storage Mater., 2019, 19, 360–369 CrossRef.
- Q. Yang, F. Mo, Z. Liu, L. Ma, X. Li, D. Fang, S. Chen, S. Zhang and C. Zhi, Adv. Mater., 2019, 31, 1901521 CrossRef.
- X. Zhan, Z. Chen and Q. Zhang, J. Mater. Chem. A, 2017, 5, 14463–14479 RSC.
- D. Chao, C. R. Zhu, M. Song, P. Liang, X. Zhang, N. H. Tiep, H. Zhao, J. Wang, R. Wang, H. Zhang and H. J. Fan, Adv. Mater., 2018, 30, 1803181 CrossRef.
- H. Li, Q. Yang, F. Mo, G. Liang, Z. Liu, Z. Tang, L. Ma, J. Liu, Z. Shi and C. Zhi, Energy Storage Mater., 2019, 19, 94–101 CrossRef.
- H. Zhang, X. Zhang, H. Li, Y. Zhang, Y. Zeng, Y. Tong, P. Zhang and X. Lu, Green Energy Environ., 2018, 3, 56–62 CrossRef.
- Y. Zeng, Z. Lai, Y. Han, H. Zhang, S. Xie and X. Lu, Adv. Mater., 2018, 30, 1802396 CrossRef PubMed.
- F. R. Mclarnon and E. J. Cairns, J. Electrochem. Soc., 1991, 138, 645–664 CrossRef CAS.
- R. Y. Wang, D. W. Kirk and G. X. Zhang, J. Electrochem. Soc., 2006, 153, C357–C364 CrossRef CAS.
- J. Q. Kan, H. G. Xue and S. L. Mu, J. Power Sources, 1998, 74, 113–116 CrossRef CAS.
- Z. Liu, T. Cui, G. Pulletikurthi, A. Lahiri, T. Carstens, M. Olschewski and F. Endres, Angew. Chem., Int. Ed., 2016, 55, 2889–2893 CrossRef CAS PubMed.
- A. Mitha, A. Z. Yazdi, M. Ahmed and P. Chen, ChemElectroChem, 2018, 5, 2409–2418 CrossRef CAS.
- X. Liu, O. Bolton and R. Akolkar, J. Electrochem. Soc., 2019, 166, D583–D588 CrossRef CAS.
- J. F. Parker, C. N. Chervin, E. S. Nelson, D. R. Rolison and J. W. Long, Energy Environ. Sci., 2014, 7, 1117–1124 RSC.
- S. Higashi, S. W. Lee, J. S. Lee, K. Takechi and Y. Cui, Nat. Commun., 2016, 7, 11801 CrossRef PubMed.
- W. Li, K. Wang, M. Zhou, H. Zhan, S. Cheng and K. Jiang, ACS Appl. Mater. Interfaces, 2018, 10, 22059–22066 CrossRef CAS PubMed.
- A. Xia, X. Pu, Y. Tao, H. Liu and Y. Wang, Appl. Surf. Sci., 2019, 481, 852–859 CrossRef CAS.
- M. Liu, L. Yang, H. Liu, A. Amine, Q. Zhao, Y. Song, J. Yang, K. Wang and F. Pan, ACS Appl. Mater. Interfaces, 2019, 11, 32046–32051 CrossRef CAS PubMed.
- Y. Tian, Y. An, C. Wei, B. Xi, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2019, 13, 11676–11685 CrossRef CAS PubMed.
- Y. Zeng, X. Zhang, R. Qin, X. Liu, P. Fang, D. Zheng, Y. Tong and X. Lu, Adv. Mater., 2019, 31, 1903675 CrossRef PubMed.
- Z. Wang, J. Huang, Z. Guo, X. Dong, Y. Liu, Y. Wang and Y. Xia, Joule, 2019, 3, 1289–1300 CrossRef CAS.
- S. Akbar, I. I. Naqvi, M. Muhammad and E. Zahir, Asian J. Chem., 2009, 21, 4190–4198 CAS.
- Q. Yang, G. Liang, Y. Guo, Z. Liu, B. Yan, D. Wang, Z. Huang, X. Li, J. Fan and C. Zhi, Adv. Mater., 2019, 31, 1903778 CrossRef CAS PubMed.
- R. E. Doe, R. Han, J. Hwang, A. J. Gmitter, I. Shterenberg, H. D. Yoo, N. Pour and D. Aurbach, Chem. Commun., 2014, 50, 243–245 RSC.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 RSC.
- J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941–5950 RSC.
- N. Wu, Z. Z. Yang, H. R. Yao, Y. X. Yin, L. Gu and Y. G. Guo, Angew. Chem., Int. Ed., 2015, 54, 5757–5761 CrossRef CAS PubMed.
- G. G. Amatucci, F. Badway, A. Singhal, B. Beaudoin, G. Skandan, T. Bowmer, I. Plitza, N. Pereira, T. Chapman and R. Jaworski, J. Electrochem. Soc., 2001, 148, A940–A950 CrossRef CAS.
- E. Levi, M. D. Levi, O. Chasid and D. Aurbach, J. Electroceram., 2009, 22, 13–19 CrossRef CAS.
- D. Aurbach, I. Weissman, Y. Gofer and E. Levi, Chem. Rec., 2003, 3, 61–73 CrossRef CAS PubMed.
- E. Levi, Y. Gofer and D. Aurbach, Chem. Mater., 2010, 22, 860–868 CrossRef CAS.
- S. K. Das, S. Mahapatra and H. Lahan, J. Mater. Chem. A, 2017, 5, 6347–6367 RSC.
- H. Tian, T. Gao, X. Li, X. Wang, C. Luo, X. Fan, C. Yang, L. Suo, Z. Ma, W. Han and C. Wang, Nat. Commun., 2017, 8, 14083 CrossRef CAS PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724 CrossRef CAS PubMed.
- D. Aurbach, G. S. Suresh, E. Levi, A. Mitelman, O. Mizrahi, O. Chusid and M. Brunelli, Adv. Mater., 2007, 19, 4260–4267 CrossRef CAS.
- Y. Liang, R. Feng, S. Yang, H. Ma, J. Liang and J. Chen, Adv. Mater., 2011, 23, 640–643 CrossRef CAS PubMed.
- B. Liu, T. Luo, G. Mu, X. Wang, D. Chen and G. Shen, ACS Nano, 2013, 7, 8051–8058 CrossRef CAS PubMed.
- Y. Liang, H. D. Yoo, Y. Li, J. Shuai, H. A. Calderon, F. C. Robles Hernandez, L. C. Grabow and Y. Yao, Nano Lett., 2015, 15, 2194–2202 CrossRef CAS PubMed.
- M. Okoshi, Y. Yamada, A. Yamada and H. Nakai, J. Electrochem. Soc., 2013, 160, A2160–A2165 CrossRef CAS.
- Y. Mizuno, M. Okubo, E. Hosono, T. Kudo, H. Zhou and K. Oh-ishi, J. Phys. Chem. C, 2013, 117, 10877–10882 CrossRef CAS.
- K. W. Nam, S. Kim, S. Lee, M. Salama, I. Shterenberg, Y. Gofer, J.-S. Kim, E. Yang, C. S. Park, J.-S. Kim, S.-S. Lee, W.-S. Chang, S.-G. Doo, Y. N. Jo, Y. Jung, D. Aurbach and J. W. Choi, Nano Lett., 2015, 15, 4071–4079 CrossRef CAS PubMed.
- Y. Mizuno, M. Okubo, E. Hosono, T. Kudo, K. Oh-ishi, A. Okazawa, N. Kojima, R. Kurono, S.-i. Nishimura and A. Yamada, J. Mater. Chem. A, 2013, 1, 13055–13059 RSC.
- M. Cabello, R. Alcántara, F. Nacimiento, G. Ortiz, P. Lavela and J. L. Tirado, CrystEngComm, 2015, 17, 8728–8735 RSC.
- G. Liu, Q. Chi, Y. Zhang, Q. Chen, C. Zhang, K. Zhu and D. Cao, Chem. Commun., 2018, 54, 9474–9477 RSC.
- C. K. King’ondu, N. Opembe, C.-h. Chen, K. Ngala, H. Huang, A. Iyer, H. F. Garcés and S. L. Suib, Adv. Funct. Mater., 2011, 21, 312–323 CrossRef.
- D. Liu, B. B. Garcia, Q. Zhang, Q. Guo, Y. Zhang, S. Sepehri and G. Cao, Adv. Funct. Mater., 2009, 19, 1015–1023 CrossRef CAS.
- V. V. Krishnan and S. L. Suib, J. Catal., 1999, 184, 305–315 CrossRef CAS.
- S. L. Suib, Acc. Chem. Res., 2008, 41, 479–487 CrossRef CAS PubMed.
- H. Zhang, K. Ye, R. Cang, K. Zhu, J. Yan, K. Cheng, G. Wang and D. Cao, J. Electroanal. Chem., 2017, 807, 37–44 CrossRef CAS.
- H. Zhang, K. Ye, K. Zhu, R. Cang, X. Wang, G. Wang and D. Cao, ACS Sustainable Chem. Eng., 2017, 5, 6727–6735 CrossRef CAS.
- X. Cao, L. Wang, J. Chen and J. Zheng, ChemElectroChem, 2018, 5, 2789–2794 CrossRef CAS.
- Z. Jia, J. Hao, L. Liu, Y. Wang and T. Qi, Ionics, 2018, 24, 3483–3491 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS PubMed.
- L. Li, X. Zhao, Y. Fu and A. Manthiram, Phys. Chem. Chem. Phys., 2012, 14, 12737–12740 RSC.
- X. Wang, Y. Hou, Y. Zhu, Y. Wu and R. Holze, Sci. Rep., 2013, 3, 1401 CrossRef PubMed.
- K. A. See, K. W. Chapman, L. Zhu, K. M. Wiaderek, O. J. Borkiewicz, C. J. Barile, P. J. Chupas and A. A. Gewirth, J. Am. Chem. Soc., 2015, 138, 328–337 CrossRef PubMed.
- X. Sun, V. Duffort, B. L. Mehdi, N. D. Browning and L. F. Nazar, Chem. Mater., 2016, 28, 534–542 CrossRef CAS.
- P. Shan, Y. Gu, L. Yang, T. Liu, J. Zheng and F. Pan, Inorg. Chem., 2017, 56, 13411–13416 CrossRef CAS PubMed.
- H. Zhang, K. Ye, X. Huang, X. Wang, K. Cheng, X. Xiao, G. Wang and D. Cao, J. Power Sources, 2017, 338, 136–144 CrossRef CAS.
- H. Zhang, K. Ye, S. Shao, X. Wang, K. Cheng, X. Xiao, G. Wang and D. Cao, Electrochim. Acta, 2017, 229, 371–379 CrossRef CAS.
- C. Drosos, C. Jia, S. Mathew, R. G. Palgrave, B. Moss, A. Kafizas and D. Vernardou, J. Power Sources, 2018, 384, 355–359 CrossRef CAS.
- Z. Jia, J. Hao, L. Liu, Y. Wang and T. Qi, Ionics, 2018, 24, 3483–3491 CrossRef CAS.
- V. Soundharrajan, B. Sambandam, S. Kim, V. Mathew, J. Jo, S. Kim, J. Lee, S. Islam, K. Kim, Y.-K. Sun and J. Kim, ACS Energy Lett., 2018, 3, 1998–2004 CrossRef CAS.
- J. Zhijun, H. Jiawei, L. Lujing, W. Yi and Q. Tao, Ionics, 2018, 24, 3483–3491 CrossRef.
- L. Chen, J. Bao, X. Dong, D. Truhlar, Y. Wang, C. Wang and Y. Xia, ACS Energy Lett., 2017, 2, 1115–1121 CrossRef CAS.
- F. Wang, Y. Lin, L. Suo, X. Fan, T. Gao, C. Yang, F. Han, Y. Qi, K. Xu and C. Wang, Energy Environ. Sci., 2016, 9, 3666–3673 RSC.
- H. Zhang, K. Ye, K. Zhu, R. Cang, J. Yan, K. Cheng, G. Wang and D. Cao, Chem. – Eur. J., 2017, 23, 17118 CrossRef CAS PubMed.
- H. Zhang, K. Ye, K. Zhu, R. Cang, J. Yan, K. Cheng, G. Wang and D. Cao, Electrochim. Acta, 2017, 256, 357–364 CrossRef CAS.
- L. Chen, J. L. Bao, X. Dong, D. G. Truhlar, Y. Wang, C. Wang and Y. Xia, ACS Energy Lett., 2017, 2, 1115–1121 CrossRef CAS.
- R. Y. Wang, B. Shyam, K. H. Stone, J. N. Weker, M. Pasta, H. W. Lee, M. F. Toney and Y. Cui, Adv. Energy Mater., 2015, 5, 1401869 CrossRef.
- N. A. Hewish, G. W. Neilson and J. E. Enderby, Nature, 1982, 297, 138–139 CrossRef CAS.
- M. Smeu, M. S. Hossain, Z. Wang, V. Timoshevskii, K. H. Bevan and K. Zaghib, J. Power Sources, 2016, 306, 431–436 CrossRef CAS.
- P. Padigi, G. Goncher, D. Evans and R. Solanki, J. Power Sources, 2015, 273, 460–464 CrossRef CAS.
- C. Lee and S. K. Jeong, Chem. Lett., 2016, 45, 1447–1449 CrossRef CAS.
- C. Lee and S. K. Jeong, Electrochim. Acta, 2018, 265, 430–436 CrossRef CAS.
- S. Gheytani, Y. L. Liang, F. L. Wu, Y. Jing, H. Dong, K. K. Rao, X. W. Chi, F. Fang and Y. Yao, Adv. Sci., 2017, 4, 1700465 CrossRef PubMed.
- Z. P. Song, H. Zhan and Y. H. Zhou, Angew. Chem., Int. Ed., 2010, 49, 8444–8448 CrossRef CAS PubMed.
- M. Adil, P. K. Dutta and S. Mitra, ChemistrySelect, 2018, 3, 3687–3690 CrossRef CAS.
- X.-P. Gao and H.-X. Yang, Energy Environ. Sci., 2010, 3, 174–189 RSC.
- C.-X. Zu and H. Li, Energy Environ. Sci., 2011, 4, 2614–2624 RSC.
- N. Jayaprakash, S. Das and L. Archer, Chem. Commun., 2011, 47, 12610–12612 RSC.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- G. A. Elia, K. Marquardt, K. Hoeppner, S. Fantini, R. Lin, E. Knipping, W. Peters, J. F. Drillet, S. Passerini and R. Hahn, Adv. Mater., 2016, 28, 7564–7579 CrossRef CAS PubMed.
- W. I. Al Sadat and L. A. Archer, Sci. Adv., 2016, 2, e1600968 CrossRef PubMed.
- S. Gu, H. Wang, C. Wu, Y. Bai, H. Li and F. Wu, Energy Storage Mater., 2017, 6, 9–17 CrossRef.
- G. A. Elia, K. Marquardt, K. Hoeppner, S. Fantini, R. Lin, E. Knipping, W. Peters, J.-F. Drillet, S. Passerini and R. Hahn, Adv. Mater., 2016, 28, 7564–7579 CrossRef CAS PubMed.
- Y. Hu, D. Sun, B. Luo and L. Wang, Energy Technol., 2019, 7, 86–106 Search PubMed.
- S. Liu, S. Ye, C. Li, G. Pan and X. Gao, J. Electrochem. Soc., 2011, 158, A1490–A1497 CrossRef CAS.
- S. Liu, J. J. Hu, N. F. Yan, G. L. Pan, G. R. Li and X. P. Gao, Energy Environ. Sci., 2012, 5, 9743–9746 RSC.
- K. Fic, G. Lota, M. Meller and E. Frackowiak, Energy Environ. Sci., 2012, 5, 5842–5850 RSC.
- Y. J. He, J. F. Peng, W. Chu, Y. Z. Li and D. G. Tong, J. Mater. Chem. A, 2014, 2, 1721–1731 RSC.
- M. Kazazi, Z. A. Zafar, M. Delshad, J. Cervenka and C. Chen, Solid State Ionics, 2018, 320, 64–69 CrossRef CAS.
- H. Lahan and S. K. Das, Ionics, 2018, 24, 1855–1860 CrossRef CAS.
- H. Lahan, R. Boruah, A. Hazarika and S. K. Das, J. Phys. Chem. C, 2017, 121, 26241–26249 CrossRef.
- Y. Wang, H. M. Shang, T. Chou and G. Z. Cao, J. Phys. Chem. B, 2005, 109, 11361–11366 CrossRef CAS PubMed.
- J. R. González, F. Nacimiento, M. Cabello, R. Alcántara, P. Lavela and J. L. Tirado, RSC Adv., 2016, 6, 62157–62164 RSC.
- F. Nacimiento, M. Cabello, R. Alcántara, P. Lavela and J. L. Tirado, Electrochim. Acta, 2018, 260, 798–804 CrossRef CAS.
- S. Liu, G. L. Pan, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2015, 3, 959–962 RSC.
- A. Holland, R. D. McKerracher, A. Cruden and R. G. A. Wills, J. Appl. Electrochem., 2018, 48, 243–250 CrossRef CAS.
- F. Wang, F. Yu, X. Wang, Z. Chang, L. Fu, Y. Zhu, Z. Wen, Y. Wu and W. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 9022–9029 CrossRef CAS PubMed.
- C. Wu, S. Gu, Q. Zhang, Y. Bai, M. Li, Y. Yuan, H. Wang, X. Liu, Y. Yuan, N. Zhu, F. Wu, H. Li, L. Gu and J. Lu, Nat. Commun., 2019, 10, 73 CrossRef PubMed.
- Y. Liu, S. Sang, Q. Wu, Z. Lu, K. Liu and H. Liu, Electrochim. Acta, 2014, 143, 340–346 CrossRef CAS.
- H. Lahan and S. K. Das, J. Power Sources, 2019, 413, 134–138 CrossRef CAS.
- M. D. Levi, Y. Shilina, G. Salitra, D. Aurbach, E. Guyot, S. Seghir, J. M. Lecuire and C. Boulanger, J. Solid State Electrochem., 2012, 16, 3443–3448 CrossRef CAS.
- C. P. Grey and N. Dupre, Chem. Rev., 2004, 104, 4493–4512 CrossRef CAS PubMed.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef CAS PubMed.
- P. R. Kumar, Y. H. Jung, C. H. Lim and D. K. Kim, J. Mater. Chem. A, 2015, 3, 6271–6275 RSC.
- L. Xue, Q. Zhang, X. Zhu, L. Gu, J. Yue, Q. Xia, T. Xing, T. Chen, Y. Yao and H. Xia, Nano Energy, 2019, 56, 463–472 CrossRef CAS.
- S. Liu, S. H. Ye, C. Z. Li, G. L. Pan and X. P. Gao, J. Electrochem. Soc., 2011, 158, A1490–A1497 CrossRef CAS.
- X. Shan, D. S. Charles, Y. Lei, R. Qiao, G. Wang, W. Yang, M. Feygenson, D. Su and X. Teng, Nat. Commun., 2016, 7, 13370 CrossRef CAS PubMed.
- D. B. Ravnsbaek, K. Xiang, W. Xing, O. J. Borkiewicz, K. M. Wiaderek, P. Gionet, K. W. Chapman, P. J. Chupas and Y. M. Chiang, Nano Lett., 2014, 14, 1484–1491 CrossRef CAS PubMed.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- A. Yamada, Y. Kudo and K. Y. Liu, J. Electrochem. Soc., 2001, 148, A747–A754 CrossRef CAS.
- S. K. Martha, J. Grinblat, O. Haik, E. Zinigrad, T. Drezen, J. H. Miners, I. Exnar, A. Kay, B. Markovsky and D. Aurbach, Angew. Chem., Int. Ed., 2009, 48, 8559–8563 CrossRef CAS PubMed.
- M. S. Zhao, G. L. Huang, B. Zhang, F. Wang and X. P. Song, J. Power Sources, 2012, 211, 202–207 CrossRef CAS.
- J. W. Zhao, Y. Q. Li, X. Peng, S. M. Dong, J. Ma, G. L. Cui and L. Q. Chen, Electrochem. Commun., 2016, 69, 6–10 CrossRef CAS.
- P. Jiang, H. Z. Shao, L. Chen, J. W. Feng and Z. P. Liu, J. Mater. Chem. A, 2017, 5, 16740–16747 RSC.
- C. Liu, X. Wang, W. Deng, C. Li, J. Chen, M. Xue, R. Li and F. Pan, Angew. Chem., Int. Ed., 2018, 57, 7046–7050 CrossRef CAS PubMed.
- S. Liu, G. L. Pan, N. F. Yan and X. P. Gao, Energy Environ. Sci., 2010, 3, 1732–1735 RSC.
- H. Dou, P. Nie and D. R. MacFarlane, J. Mater. Chem. A, 2014, 2, 19536–19541 RSC.
- H. Li, G. Weng, C. Y. V. Li and K.-Y. Chan, Electrochim. Acta, 2011, 56, 9420–9425 CrossRef CAS.
- L. Chen, Z. Guo, Y. Xia and Y. Wang, Chem. Commun., 2013, 49, 2204–2206 RSC.
- P. Ratajczak and F. Béguin, ChemElectroChem, 2018, 5, 2518–2521 CrossRef CAS.
- S. Sur, A. R. Kottaichamy, Z. Manzoor Bhat, M. C. Devendrachari, R. Thimmappa and M. O. Thotiyl, Chem. Phys. Lett., 2018, 712, 160–164 CrossRef CAS.
- C. Li, W. Wu, P. Wang, W. Zhou, J. Wang, Y. Chen, L. Fu, Y. Zhu, Y. Wu and W. Huang, Adv. Sci., 2019, 6, 1801665 CrossRef PubMed.
- H. Li, Y. Wang, H. Na, H. Liu and H. Zhou, J. Am. Chem. Soc., 2009, 131, 15098–15099 CrossRef CAS PubMed.
- M. Zhang, Z. Huang, Z. Shen, Y. Gong, B. Chi, J. Pu and J. Li, Adv. Energy Mater., 2017, 7, 1700155 CrossRef.
- X. Wang, Y. Hou, Y. Zhu, Y. Wu and R. Holze, Sci. Rep., 2013, 3, 1401 CrossRef PubMed.
- X. Wang, Q. Qu, Y. Hou, F. Wang and Y. Wu, Chem. Commun., 2013, 49, 6179–6181 RSC.
- H. T. T. Le, D. T. Ngo, Y.-J. Kim, C.-N. Park and C.-J. Park, Electrochim. Acta, 2017, 248, 232–242 CrossRef CAS.
- H. T. T. Le, R. S. Kalubarme, D. T. Ngo, H. S. Jadhav and C.-J. Park, J. Mater. Chem. A, 2015, 3, 22421–22431 RSC.
- S. Makino, R. Yamamoto, S. Sugimoto and W. Sugimoto, J. Power Sources, 2016, 326, 711–716 CrossRef CAS.
- Q. Zhao, M. J. Zachman, W. I. Al Sadat, J. X. Zheng, L. F. Kourkoutis and L. Archer, Sci. Adv., 2018, 4, eaau8131 CrossRef CAS PubMed.
- J. Zhi, A. Z. Yazdi, G. Valappil, J. Haime and P. Chen, Sci. Adv., 2017, 3, e1701010 CrossRef PubMed.
- M. Ahmed, A. Z. Yazdi, A. Mitha and P. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 30348–30356 CrossRef CAS PubMed.
- K. Nakamoto, R. Sakamoto, Y. Sawada, M. Ito and S. Okada, Small Methods, 2019, 3, 1800220 CrossRef CAS.
- L. Suo, D. Oh, Y. Lin, Z. Zhuo, O. Borodin, T. Gao, F. Wang, A. Kushima, Z. Wang, H. C. Kim, Y. Qi, W. Yang, F. Pan, J. Li, K. Xu and C. Wang, J. Am. Chem. Soc., 2017, 139, 18670–18680 CrossRef CAS PubMed.
- H. G. Jung, M. W. Jang, J. Hassoun, Y. K. Sun and B. Scrosati, Nat. Commun., 2011, 2, 516 CrossRef PubMed.
- F. Wang, L. Suo, Y. Liang, C. Yang, F. Han, T. Gao, W. Sun and C. Wang, Adv. Energy Mater., 2017, 7, 1600922 CrossRef.
- C. Yang, X. Ji, X. Fan, T. Gao, L. Suo, F. Wang, W. Sun, J. Chen, L. Chen, F. Han, L. Miao, K. Xu, K. Gerasopoulos and C. Wang, Adv. Mater., 2017, 29, 1701972 CrossRef PubMed.
- L. Suo, O. Borodin, Y. Wang, X. Rong, W. Sun, X. Fan, S. Xu, M. A. Schroeder, A. V. Cresce, F. Wang, C. Yang, Y.-S. Hu, K. Xu and C. Wang, Adv. Energy Mater., 2017, 7, 1701189 CrossRef.
- J. Han, H. Zhang, A. Varzi and S. Passerini, ChemSusChem, 2018, 11, 3704–3707 CrossRef CAS PubMed.
- S. Liu, L. Wang, J. Liu, M. Zhou, Q. Nian, Y. Feng, Z. Tao and L. Shao, J. Mater. Chem. A, 2019, 7, 248–256 RSC.
- J. Zhao, Y. Li, X. Peng, S. Dong, J. Ma, G. Cui and L. Chen, Electrochem. Commun., 2016, 69, 6–10 CrossRef CAS.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406–A2423 CrossRef CAS.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- S. Wu, Y. Qiao, S. Yang, J. Tang, P. He and H. Zhou, ACS Catal., 2018, 8, 1082–1089 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- H. Ota, K. Shima, M. Ue and J.-i. Yamaki, Electrochim. Acta, 2004, 49, 565–572 CrossRef CAS.
- L. El Ouatani, R. Dedryvere, C. Siret, P. Biensan, S. Reynaud, P. Iratcabal and D. Gonbeau, J. Electrochem. Soc., 2009, 156, A103–A113 CrossRef CAS.
- H. Ota, Y. Sakata, Y. Otake, K. Shima, M. Ue and J. Yamaki, J. Electrochem. Soc., 2004, 151, A1778–A1788 CrossRef CAS.
- C. Yang, J. Chen, T. Qing, X. Fan, W. Sun, A. von Cresce, M. S. Ding, O. Borodin, J. Vatamanu, M. A. Schroeder, N. Eidson, C. Wang and K. Xu, Joule, 2017, 1, 122–132 CrossRef CAS.
- F. Wang, O. Borodin, M. S. Ding, M. Gobet, J. Vatamanu, X. Fan, T. Gao, N. Eidson, Y. Liang, W. Sun, S. Greenbaum, K. Xu and C. Wang, Joule, 2018, 2, 927–937 CrossRef CAS.
- C. Yang, J. Chen, X. Ji, T. P. Pollard, X. Lu, C. J. Sun, S. Hou, Q. Liu, C. Liu, T. Qing, Y. Wang, O. Borodin, Y. Ren, K. Xu and C. Wang, Nature, 2019, 569, 245–250 CrossRef CAS PubMed.
- Z. Chen and J. R. Dahn, Electrochim. Acta, 2004, 49, 1079–1090 CrossRef CAS.
- A. Ramanujapuram, D. Gordon, A. Magasinski, B. Ward, N. Nitta, C. Huang and G. Yushin, Energy Environ. Sci., 2016, 9, 1841–1848 RSC.
- F. Wang, Y. Lin, L. Suo, X. Fan, T. Gao, C. Yang, F. Han, Y. Qi, K. Xu and C. Wang, Energy Environ. Sci., 2016, 9, 3666–3673 RSC.
- N. W. Li, Y. X. Yin, C. P. Yang and Y. G. Guo, Adv. Mater., 2016, 28, 1853–1858 CrossRef CAS PubMed.
- L. Ma, M. S. Kim and L. A. Archer, Chem. Mater., 2017, 29, 4181–4189 CrossRef CAS.
- M. S. Kim, J.-H. Ryu, Deepika, Y. R. Lim, I. W. Nah, K.-R. Lee, L. A. Archer and W. Il Cho, Nat. Energy, 2018, 3, 889–898 CrossRef CAS.
- Z. Liu, G. Pulletikurthi and F. Endres, ACS Appl. Mater. Interfaces, 2016, 8, 12158–12164 CrossRef CAS PubMed.
- A. Tatlisu, Z. Huang and R. Chen, ChemSusChem, 2018, 11, 3899–3904 CrossRef CAS PubMed.
- Y. Zhang, R. Ye, D. Henkensmeier, R. Hempelmann and R. Chen, Electrochim. Acta, 2018, 263, 47–52 CrossRef CAS.
- J. Wei, J. Zhou, S. Su, J. Jiang, J. Feng and Q. Wang, ChemSusChem, 2018, 11, 3410–3415 CrossRef CAS PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- R. Benedek, M. M. Thackeray, J. Low and T. Bučko, J. Phys. Chem. C, 2012, 116, 4050–4059 CrossRef CAS.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS PubMed.
- L. Coustan, G. Shul and D. Bélanger, Electrochem. Commun., 2017, 77, 89–92 CrossRef CAS.
- H. Lahan and S. K. Das, Ionics, 2018, 24, 2175–2180 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem., 1972, 39, 163–184 CrossRef CAS.
- V. Verma, S. Kumar, W. Manalastas, R. Satish and M. Srinivasan, Adv. Sustainable Syst., 2019, 3, 1800111 CrossRef.
- J. K. Norskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and J. K. Norskov, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef CAS.
- B. C. Church, D. T. Kaminski and J. Jiang, J. Mater. Sci., 2014, 49, 3234–3241 CrossRef CAS.
- M. Pourbaix, Corros. Sci., 1974, 14, 25–82 CrossRef CAS.
- S. Gheytani, Y. Liang, Y. Jing, J. Q. Xu and Y. Yao, J. Mater. Chem. A, 2016, 4, 395–399 RSC.
- S. Liu, J. J. Hu, N. F. Yan, G. L. Pan, G. R. Li and X. P. Gao, Energy Environ. Sci., 2012, 5, 9743–9746 RSC.
- S. Hu, Y. Li, J. Yin, H. Wang, X. Yuan and Q. Li, Chem. Eng. J., 2014, 237, 497–502 CrossRef CAS.
- S. Maazi, A. H. Navarchian, M. Khosravi and P. Chen, Solid State Ionics, 2018, 320, 84–91 CrossRef CAS.
- P. Arora and Z. M. Zhang, Chem. Rev., 2004, 104, 4419–4462 CrossRef CAS PubMed.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- F. Zheng, M. Kotobuki, S. Song, M. O. Lai and L. Lu, J. Power Sources, 2018, 389, 198–213 CrossRef CAS.
- Y. Huang, J. Liu, J. Wang, M. Hu, F. Mo, G. Liang and C. Zhi, Angew. Chem., Int. Ed., 2018, 57, 9810–9813 CrossRef CAS PubMed.
- D. Wang, L. Wang, G. Liang, H. Li, Z. Liu, Z. Tang, J. Liang and C. Zhi, ACS Nano, 2019, 13, 10643–10652 CrossRef CAS PubMed.
- J. Xie, Z. Wang, Z. J. Xu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1703509 CrossRef.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- Z. L. Wang, D. Xu, J. J. Xu and X. B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- J.-S. Lee, S. Tai Kim, R. Cao, N.-S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 RSC.
- W. Liu, E. Hu, H. Jiang, Y. Xiang, Z. Weng, M. Li, Q. Fan, X. Yu, E. I. Altman and H. Wang, Nat. Commun., 2016, 7, 10771 CrossRef CAS PubMed.
- Q. Ding, B. Song, P. Xu and S. Jin, Chem, 2016, 1, 699–726 CAS.
- M. Macka, P. Andersson and P. R. Haddad, Anal. Chem., 1998, 70, 743–749 CrossRef CAS.
- W. Sheng, Z. Zhuang, M. Gao, J. Zheng, J. G. Chen and Y. Yan, Nat. Commun., 2015, 6, 5848 CrossRef CAS PubMed.
- L. Suo, F. Han, X. Fan, H. Liu, K. Xu and C. Wang, J. Mater. Chem. A, 2016, 4, 6639–6644 RSC.
- C. Yang, L. Suo, O. Borodin, F. Wang, W. Sun, T. Gao, X. Fan, S. Hou, Z. Ma, K. Amine, K. Xu and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6197–6202 CrossRef CAS PubMed.
- J. Liu, C. Xu, Z. Chen, S. Ni and Z. X. Shen, Green Energy Environ., 2018, 3, 20–41 CrossRef.
- S. Zhao, B. Han, D. Zhang, Q. Huang, L. Xiao, L. Chen, D. G. Ivey, Y. Deng and W. Wei, J. Mater. Chem. A, 2018, 6, 5733–5739 RSC.
- Y. Liu, X. Fang, A. Zhang, C. Shen, Q. Liu, H. A. Enaya and C. Zhou, Nano Energy, 2016, 27, 27–34 CrossRef CAS.
- J. Xu, E. Hu, D. Nordlund, A. Mehta, S. N. Ehrlich, X. Q. Yang and W. Tong, ACS Appl. Mater. Interfaces, 2016, 8, 31677–31683 CrossRef CAS PubMed.
- P. K. Nayak, J. Grinblat, M. Levi, E. Levi, S. Kim, J. W. Choi and D. Aurbach, Adv. Energy Mater., 2016, 6, 1502398 CrossRef.
- Q. Lin, W. Guan, J. Meng, W. Huang, X. Wei, Y. Zeng, J. Li and Z. Zhang, Nano Energy, 2018, 54, 313–321 CrossRef CAS.
- S. Sharifi-Asl, J. Lu, K. Amine and R. Shahbazian-Yassar, Adv. Energy Mater., 2019, 9, 1900551 CrossRef.
- R. Jung, P. Strobl, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A2869–A2879 CrossRef CAS.
- W. Zhang, F. H. Richter, S. P. Culver, T. Leichtweiss, J. G. Lozano, C. Dietrich, P. G. Bruce, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2018, 10, 22226–22236 CrossRef CAS PubMed.
- J. Cabana, B. J. Kwon and L. Hu, Acc. Chem. Res., 2018, 51, 299–308 CrossRef CAS PubMed.
- K.-J. Park, J.-Y. Hwang, H.-H. Ryu, F. Maglia, S.-J. Kim, P. Lamp, C. S. Yoon and Y.-K. Sun, ACS Energy Lett., 2019, 4, 1394–1400 CrossRef CAS.
- X. Shi, J. Du, T. G. J. Jones, X. Wang and H. P. Liang, ACS Appl. Mater. Interfaces, 2018, 10, 29667–29674 CrossRef CAS PubMed.
- L. David, R. E. Ruther, D. Mohanty, H. M. Meyer, Y. Sheng, S. Kalnaus, C. Daniel and D. L. Wood, Appl. Energy, 2018, 231, 446–455 CrossRef CAS.
- K. Hoang, Phys. Rev. Mater., 2017, 1, 075403 CrossRef.
- W. Cho, S. Myeong, N. Kim, S. Lee, Y. Kim, M. Kim, S. J. Kang, N. Park, P. Oh and J. Cho, Adv. Mater., 2017, 29, 1605578 CrossRef PubMed.
- R. Yu, X. Wang, Y. Fu, L. Wang, S. Cai, M. Liu, B. Lu, G. Wang, D. Wang, Q. Ren and X. Yang, J. Mater. Chem. A, 2016, 4, 4941–4951 RSC.
- E. Hu, S.-M. Bak, Y. Liu, J. Liu, X. Yu, Y.-N. Zhou, J. Zhou, P. Khalifah, K. Ariyoshi, K.-W. Nam and X.-Q. Yang, Adv. Energy Mater., 2016, 6, 1501662 CrossRef.
- S. Kalluri, M. Yoon, M. Jo, H. K. Liu, S. X. Dou, J. Cho and Z. Guo, Adv. Mater., 2017, 29, 1605807 CrossRef PubMed.
- B. Qiu, M. Zhang, L. Wu, J. Wang, Y. Xia, D. Qian, H. Liu, S. Hy, Y. Chen, K. An, Y. Zhu, Z. Liu and Y. S. Meng, Nat. Commun., 2016, 7, 12108 CrossRef CAS PubMed.
- Y. Cho, S. Lee, Y. Lee, T. Hong and J. Cho, Adv. Energy Mater., 2011, 1, 821–828 CrossRef CAS.
- X. D. Zhang, J. L. Shi, J. Y. Liang, Y. X. Yin, J. N. Zhang, X. Q. Yu and Y. G. Guo, Adv. Mater., 2018, 30, 1801751 CrossRef PubMed.
- L. Hu, P. Brüner, T. Grehl, H. H. Brongersma and J. Cabana, Chem. Mater., 2017, 29, 5896–5905 CrossRef CAS.
- J. Ahn, J. H. Kim, B. W. Cho, K. Y. Chung, S. Kim, J. W. Choi and S. H. Oh, Nano Lett., 2017, 17, 7869–7877 CrossRef CAS PubMed.
- L. L. Wang, K. W. Huang, J. T. Chen and J. R. Zheng, Sci. Adv., 2019, 5, eaax4279 CrossRef.
- G. Venugopal, J. Power Sources, 2001, 101, 231–237 CrossRef CAS.
- J. H. Tian, T. Jiang, M. Wang, Z. Hu, X. Zhu, L. Zhang, T. Qian and C. Yan, Small Methods, 2019, 1900467, DOI:10.1002/smtd.201900467.
- D. Liu, Z. Shadike, R. Lin, K. Qian, H. Li, K. Li, S. Wang, Q. Yu, M. Liu, S. Ganapathy, X. Qin, Q. H. Yang, M. Wagemaker, F. Kang, X. Q. Yang and B. Li, Adv. Mater., 2019, 31, 1806620 CrossRef PubMed.
- L. Zhang, T. Qian, X. Zhu, Z. Hu, M. Wang, L. Zhang, T. Jiang, J. H. Tian and C. Yan, Chem. Soc. Rev., 2019, 48, 5432–5453 RSC.
- A. J. Ilott, M. Mohammadi, H. J. Chang, C. P. Grey and A. Jerschow, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10779–10784 CrossRef CAS PubMed.
- A. R. Armstrong, C. Arrouvel, V. Gentili, S. C. Parker, M. S. Islam and P. G. Bruce, Chem. Mater., 2010, 22, 6426–6432 CrossRef CAS.
- C. J. Jafta, X. G. Sun, G. M. Veith, G. V. Jensen, S. M. Mahurin, M. P. Paranthaman, S. Dai and C. A. Bridges, Energy Environ. Sci., 2019, 12, 1866–1877 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |