
Advances in ultrahigh-throughput screening for directed enzyme evolution
Ulrich
Markel
 a,
Khalil D.
Essani
a,
Volkan
Besirlioglu
a,
Johannes
Schiffels
a,
Wolfgang R.
Streit
b and
Ulrich
Schwaneberg
*ac
a,
Khalil D.
Essani
a,
Volkan
Besirlioglu
a,
Johannes
Schiffels
a,
Wolfgang R.
Streit
b and
Ulrich
Schwaneberg
*ac
aInstitute of Biotechnology, RWTH Aachen University, Worringer Weg 3, 52074 Aachen, Germany. E-mail: u.schwaneberg@biotec.rwth-aachen.de
bDepartment of Microbiology and Biotechnology, Universität Hamburg, Ohnhorststr 18, 22609 Hamburg, Germany
cDWI – Leibniz Institute for Interactive Materials, Forckenbeckstraße 50, 52074, Germany
First published on 9th December 2019
Abstract
Enzymes are versatile catalysts and their synthetic potential has been recognized for a long time. In order to exploit their full potential, enzymes often need to be re-engineered or optimized for a given application. (Semi-) rational design has emerged as a powerful means to engineer proteins, but requires detailed knowledge about structure function relationships. In turn, directed evolution methodologies, which consist of iterative rounds of diversity generation and screening, can improve an enzyme's properties with virtually no structural knowledge. Current diversity generation methods grant us access to a vast sequence space (libraries of >1012 enzyme variants) that may hide yet unexplored catalytic activities and selectivity. However, the time investment for conventional agar plate or microtiter plate-based screening assays represents a major bottleneck in directed evolution and limits the improvements that are obtainable in reasonable time. Ultrahigh-throughput screening (uHTS) methods dramatically increase the number of screening events per time, which is crucial to speed up biocatalyst design, and to widen our knowledge about sequence function relationships. In this review, we summarize recent advances in uHTS for directed enzyme evolution. We shed light on the importance of compartmentalization to preserve the essential link between genotype and phenotype and discuss how cells and biomimetic compartments can be applied to serve this function. Finally, we discuss how uHTS can inspire novel functional metagenomics approaches to identify natural biocatalysts for novel chemical transformations.
 Ulrich Markel | Ulrich Markel received his bachelor's and master's degree in Molecular and Applied Biotechnology from RWTH Aachen University after conducting research with Prof. Anett Schallmey in Aachen and with Prof. Frances H. Arnold at the California Institute of Technology. In 2017, he joined the group of Prof. Ulrich Schwaneberg at RWTH Aachen University to pursue his doctoral studies on the field of uHTS for the directed evolution of natural and artificial enzymes. |
 Khalil D. Essani | Khalil Essani received his Bachelor's and Master's degree in Biotechnology from the Management Center Innsbruck. He completed his undergraduate research in the group of Prof. Dorothee von Lear and the team of Prof. Anton Glieder. Currently, he is a PhD fellow in the group of Prof. Ulrich Schwaneberg at the RWTH Aachen University. His research focus is on the development of uHTS systems for protein and cell engineering. |
 Volkan Besirlioglu | Volkan Besirlioglu pursued his bachelor studies in Bio- and Nanotechnologies at South Westphalia University of Applied Sciences. After he received his Master degree in Molecular and Applied Biotechnology at RWTH Aachen University, he joined the group of Prof. Ulrich Schwaneberg at RWTH Aachen University. His current research focuses on uHTS systems based on biomimetic compartments for cell-free protein engineering as well as for cell-free metagenomics. |
 Johannes Schiffels | Johannes Schiffels earned his doctoral degree with distinction (summa cum laude) at the Philipps-University Marburg in 2013. Experimental studies were carried out at Aachen University of Applied Sciences, where he subsequently remained for his postdoctoral studies before joining the Schwaneberg group in 2016. Since 2017, he is head of the division “Next generation biocatalysis”, which aims to design novel whole-cell catalysts, artificial metalloenzymes, and uHTS platforms for applications in directed enzyme evolution. |
 Wolfgang R. Streit | Wolfgang Streit studied biology at the Philipps-Universität Marburg. After working as a postdoctoral fellow at the Philipps-University and the University of California, Davis (USA) he started up his own research group with a strong focus on functional metagenomics. Since 2006, Wolfgang Streit is a full Professor of Microbiology and Biotechnology at the University of Hamburg in the Department of Biology. |
 Ulrich Schwaneberg | Ulrich Schwaneberg earned his doctoral degree in chemistry in 1999 at the Institute of Technical Biochemistry, University of Stuttgart, Germany. He then joined Caltech (California Institute of Technology) for two years to pursue his postdoctoral studies. In 2002, he was appointed professor of biochemical engineering at the International University Bremen. In 2009, he moved to RWTH Aachen University as chair of the Institute of Biotechnology. In 2010, he was co-appointed at the DWI (Leibniz Institute for Interactive Materials). |
1. Introduction
In the past two decades, our ability to redesign enzymes has been boosted by directed enzyme evolution methodologies as recently recognized by the 2018 Nobel Prize in Chemistry awarded to Frances H. Arnold (directed evolution of enzymes), George P. Smith, and Sir Gregory P. Winter (phage display of peptides and antibodies).1–5 Today, we can tailor enzymes specifically to the needs of industrial processes and take advantage of their enantio- and regioselectivity as well as their ability to function as green catalysts under mild reaction conditions in aqueous media.First conceptual thoughts of how Darwinian evolution could be used in vitro date back more than 50 years to studies by Spiegelman et al.6,7 With the emergence of polymerase chain reaction (PCR) in the late 1980s8,9 and advancements of PCR technology toward the generation of mutant libraries (e.g., error-prone PCR (epPCR)10–12 and DNA shuffling),13 the field of directed evolution entered a new era: in iterative cycles, genetic variants of an enzyme could be conveniently generated and screened for improved properties.13–16
Today, gene synthesis and sequencing as well as bioinformatic tools are readily available and facilitate the use of directed evolution in many laboratories around the world. Computationally-assisted (semi-) rational design gains increasing importance to establish starting points for the evolution of natural enzymes17–22 or even enables the de novo design of catalytically active proteins.23–31 In cases where screening large numbers of enzyme variants is challenging, smart libraries32–39 with reduced amino acid alphabets40 and recently also machine learning algorithms41–43 have proven to be useful as a trade-off between screening throughput and time investment. In the meantime, it is possible to engineer enzymes to catalyze non-natural reactions44–58 or even to design and engineer artificial enzymes (e.g., containing non-natural cofactors59–66 or non-canonical amino acids).67–77 Notably, directed evolution has recently unlocked novel enzymatic reactivities including the formation of carbon–silicon and carbon–boron bonds.78–80
Directed evolution employs random mutagenesis, gene shuffling and semi-rational site-saturation mutagenesis – all of which rely on some sort of screening technology to identify the most promising enzyme variant among the variants within a library.1 Whereas libraries of >1012 variants can be prepared,81 screening of such large libraries is still challenging. Microtiter plates (MTP) are the most widely used screening format. They allow the analysis of up to 104 variants per day.82 Agar plate-based assays typically can cope with library sizes of up to 105.83,84 Importantly, albeit representing only a very small fraction of the available sequence space, screening of 1000 variants often identifies some beneficial amino acid substitutions.54,85 At the same time, it is obvious that the majority of available sequence space along with potential improvements remains unexplored. For instance, sometimes two substitutions only exhibit beneficial effects when they appear simultaneously86,87 and combinatorial problems can arise88 (e.g., simultaneous saturation at six positions already corresponds to a diversity of 206 = 6.4 × 107 possible variants). Thus, screening methods with much higher throughput are indispensable to explore the generated diversity in reasonable experimental time (hours or days).89 Only then, we can delve into sequence space to identify such synergistic effects by extensive random mutagenesis or combinatorial saturation mutagenesis. Flow cytometric and chip-based microfluidic screening approaches offer unmatched throughput of >106 h−1,90–92 currently making them the benchmark ultrahigh-throughput screening (uHTS) technology to explore such comprehensive libraries.
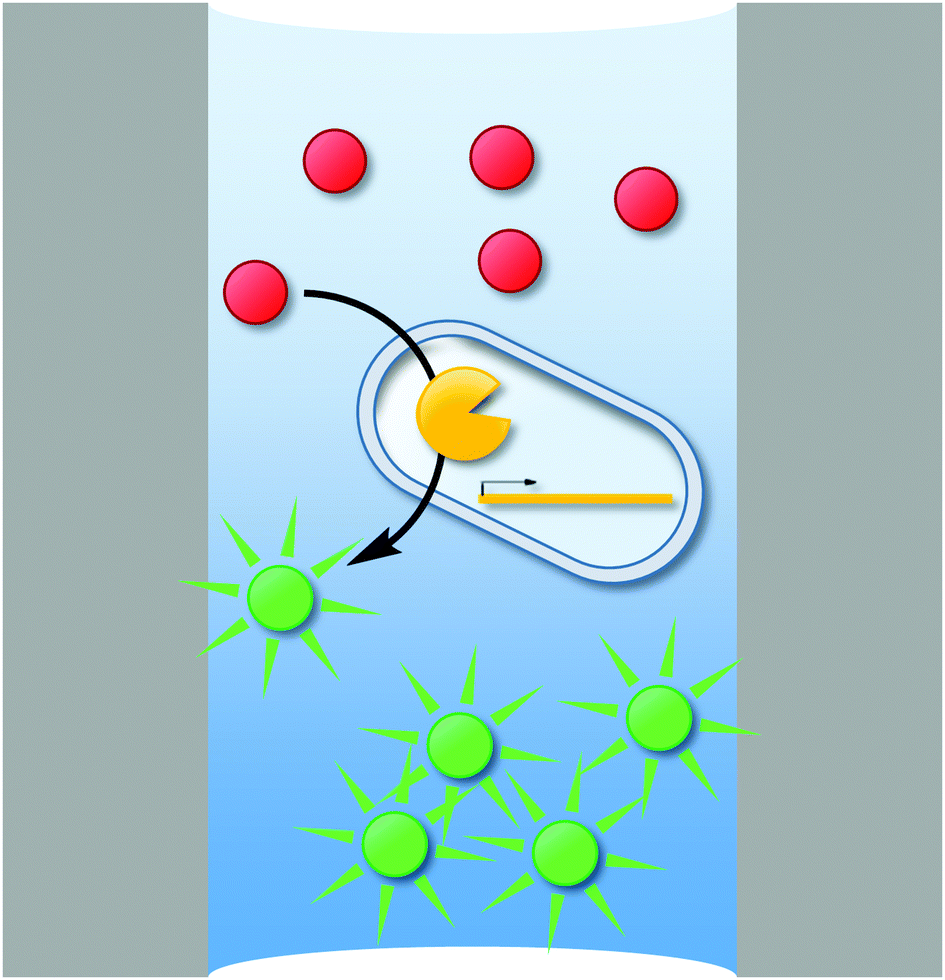
Like MTP or agar plate screens, uHTS requires a signal that corresponds to the activity of the enzyme variant investigated. uHTS typically utilizes fluorescence to provide a signal sufficient for single cell analysis. Thus, the production of fluorescent molecules (typically proteins or small molecules) is a common concept of most uHTS campaigns in directed enzyme evolution. Establishing a link between genotype and this fluorescent signal (phenotype) is the second essential requirement for every successful uHTS platform. Only when the signal is retraced to the enzyme variant that is responsible for the signal (and its encoding gene – genotype), the corresponding variant can be isolated and used for further rounds of evolution.93 Various compartmentalization techniques enable this compulsory genotype–phenotype linkage (Fig. 1).
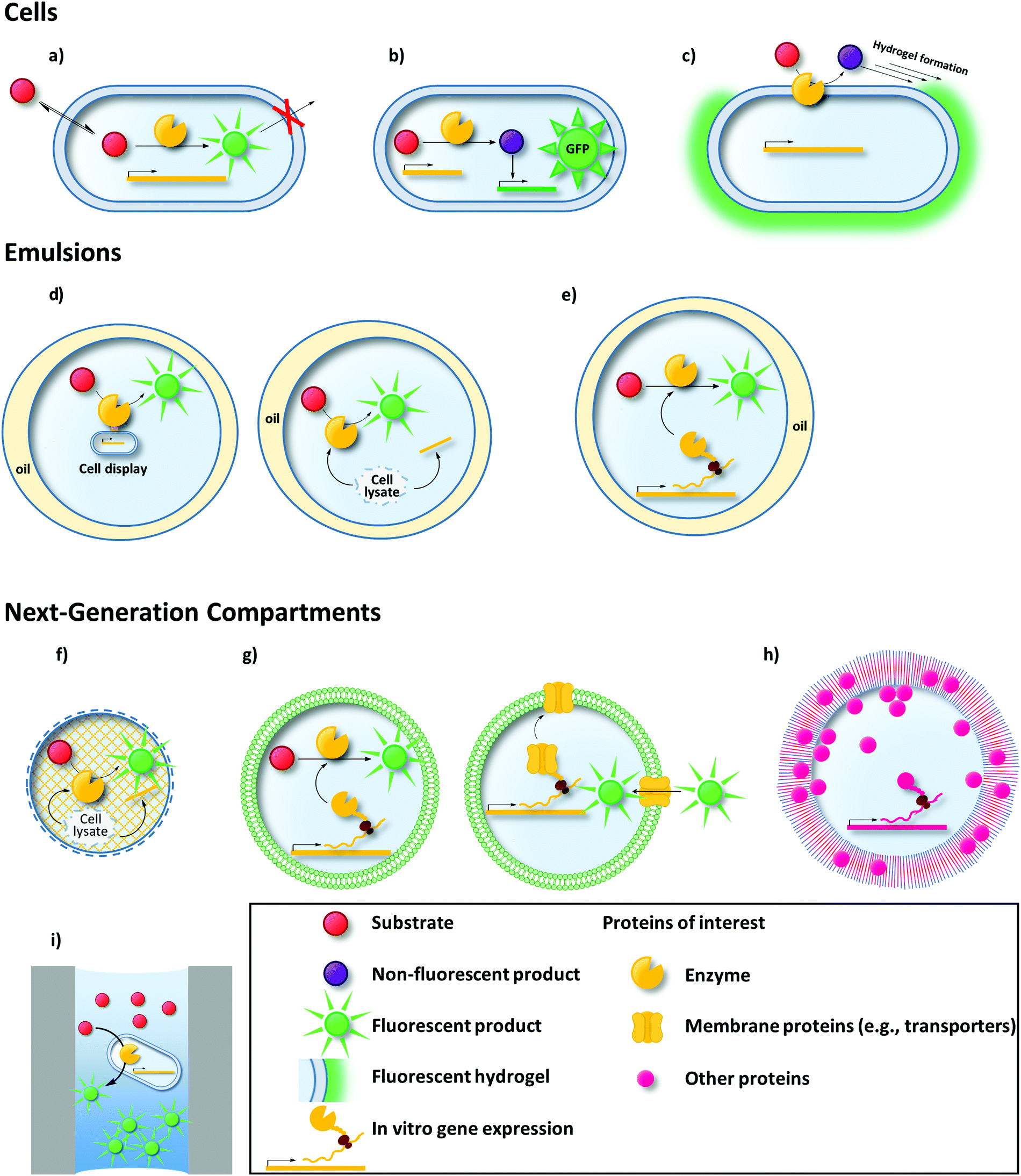 | ||
| Fig. 1 Compartments commonly used in uHTS to ensure genotype–phenotype-linkage: the fluorescent product of a given enzymatic reaction can be trapped inside a cell (a), biosensors (e.g., green fluorescent protein – GFP or variants thereof) can be used (b) or hydrogels (c) can be formed around the cell. Single or double emulsions are biomimetic compartments used to encapsulate cells/cell lysates (d) or in vitro transcription/translation machineries (e). Emerging compartmentalization strategies such as hydrogel beads (f), liposomes (g), polymersomes (h), or microwell/microcapillary arrays (i) are promising new alternatives for uHTS in directed enzyme evolution. In the following, these strategies will be discussed in detail. | ||
Cells are the most natural compartment to ensure a genotype–phenotype-linkage. The cellular membrane can be a powerful, selective separator to entrap fluorescent reporters.94–96 Alternatively, single or double emulsions served as biomimetic compartments to confine substrate, product, and single cells or an in vitro transcription/translation (IVTT) machinery producing the enzyme of interest.97,98
The aim of this review is to familiarize readers with these state-of-the-art uHTS techniques and emerging concepts to facilitate the design of directed enzyme evolution campaigns. First, we will discuss advantages and current limitations of different compartmentalization strategies ranging from cells as compartments (Section 2) to emulsions as biomimetic compartments (Section 3). In addition, we will discuss emerging compartmentalization and detection principles aiming to overcome current challenges of cell- or emulsion-based approaches (Section 4). Finally, we will elaborate on how uHTS methods that were developed in the context of directed enzyme evolution could be applied for the activity-based screening for novel biocatalysts from metagenomic libraries (Section 5). Engineering of peptide or protein binding properties (e.g., antibodies) by uHTS methods will not be described here, as this was covered elsewhere.81,99,100 Furthermore, we will not focus on directed evolution campaigns employing MTP or agar plate-based screening formats, which typically deal with library sizes of <106. For more information on directed evolution at such lower throughputs, we kindly refer the reader to corresponding excellent reviews.47,82,88,101–111
2. Cells – Nature's example for compartmentalization
With the emergence of versatile plasmid-based expression systems, the handling of cells as compartments for enzyme production and screening or selection has in many cases become a straightforward approach and a link between genotype and phenotype is naturally provided. In the following, we show how entrapment of a fluorescent product or dye within the cell (Section 2.1), formation of a fluorescent hydrogel shell around the cell (Section 2.2), or transcription factor and enzyme-mediated regulation of a fluorescent reporter (biosensor; Section 2.3) were utilized for directed enzyme evolution using uHTS methods. Moreover, strategies to display the enzyme of interest on the cell surface are available112 and it will be discussed how in these cases fluorescent reporters can be captured on the cell surface to link genotype and phenotype (Section 2.4).2.1 Entrapment of a fluorescent product or dye within the cell
To ensure genotype–phenotype linkage, fluorescent products can be entrapped within the cell (Fig. 1a). To that end, when cytoplasmic enzymes are confronted with externally supplemented small fluorescent reporter molecules, the fluorogenic substrate must be able to pass through the membrane, whereas the fluorescent product must remain trapped inside the cell.Making use of selective transporters of the cell membrane, Aharoni et al. exemplified how membrane selectivity can be used to entrap a fluorescent product. They described a flow cytometry-based uHTS for the directed evolution of a sialyltransferase (CstII from Campylobacter jejuni) to increase its catalytic efficiency towards various fluorescently labeled acceptor sugars.94 The selectivity of the Escherichia coli strain JM107 NanA− was used, which efficiently transported fluorescently labeled acceptor substrates (lactose or galactose carrying a fluorescein, coumarin or BODIPY moiety) and N-acetylneuraminic acid (Neu5Ac; a precursor of the CMP-Neu5Ac donor substrate) to the cytoplasm. In addition, this strain lacks β-galactosidase (ΔlacZ) and Neu5Ac aldolase (ΔnanA) rendering it unable to catabolize the substrates of the screening reaction. Upon sialylation of the acceptor substrates, the fluorescent product was trapped inside the cell, because the extension by one Neu5Ac monomer impeded transport across the membrane (Fig. 2) due to its negative charge and the increase in size. Unexpectedly, screening for fluorescent cells identified a CstII variant exhibiting a >150-fold improved transfer activity towards the BODIPY-tagged substrate. This indicates an adaptation of the enzyme's selectivity towards the fluorescent dye instead of the acceptor sugar moiety. Now having established a BODIPY binding site in CstII increased the catalytic efficiency towards substrates tagged with hydrophobic moieties. Transfer activity onto a non-natural tagged acceptor substrate increased more than 400-fold compared to the untagged 3-SH-lactose. Notably, none of these acceptor substrates were converted by the wild-type enzyme. In a following study, this uHTS method was refined to reduce bias towards the evolution of dye binding sites by simultaneously using two different acceptor substrates consisting of the same sugar moiety but different dyes.113 Moreover, this modified protocol enabled the directed evolution of β-1,3-galactosyltransferases transferring neutral galactose moieties. In this latter case, genotype–phenotype linkage was provided by a permease that was unable to export the galactose-linked fluorescent product. Glycosynthases (a form of reengineered glycosidases) are an interesting alternative to glycosyltransferases in the context of saccharide synthesis.114,115 Emerging strategies for the release116–118 or the uncaging of fluorophores119 hold great promise for the development of novel uHTS formats for the directed evolution of glycosynthases as recently outlined by Danby and Withers.115
 | ||
| Fig. 2 Transporter-mediated selective entrapment of a fluorescent reporter molecule. The dye-tagged acceptor substrate and Neu5Ac (precursor of the donor substrate) were taken up by selective transporters and converted by sialyltransferase CstII. The sialylated fluorescent product was not accepted by any of the substrate transporters and remained trapped inside the cell.94,113 | ||
Another strategy to entrap a fluorescent reporter makes use of its charge. While hydrophobic molecules can readily pass cellular membranes, charged molecules often do not. This simple but effective principle can be utilized for uHTS. With the aim of screening large libraries of P450 monooxygenases (P450) by flow cytometry, Ruff et al. utilized the uncharged 7-benzoxy-3-carboxycoumarin ethyl ester (BCCE) as a substrate (1a).95 Intracellular cleavage of the ethyl ester moiety by endogenous hydrolases of the E. coli host left a negative charge on the molecule preventing the fluorescent product from escaping through the cellular membrane (Fig. 3a). Upon P450-catalyzed O-dealkylation, a fluorescent coumarin derivative (1d) was formed, which was detectable in a flow cytometer. After one round of directed evolution, a P450 variant with seven-fold increased activity was identified. This illustrates the potential of intracellular cleavage of ester moieties to entrap uHTS compatible substrates by charge. Similarly, fluorescein diacetate derivatives (e.g., carboxy-2′,7′-dichloro-dihydrofluorescein diacetate, 2a) have been used as reporters in the directed evolution of the hydrogen peroxide producing enzyme monoamine oxidase (MAO).96 After 2a enters the cell, endogenous esterases deacetylate the dye trapping the now charged molecule (2b) intracellularly (Fig. 3b). Intracellular peroxidases utilize the hydrogen peroxide produced by MAO to form the fluorescent dye (2c).
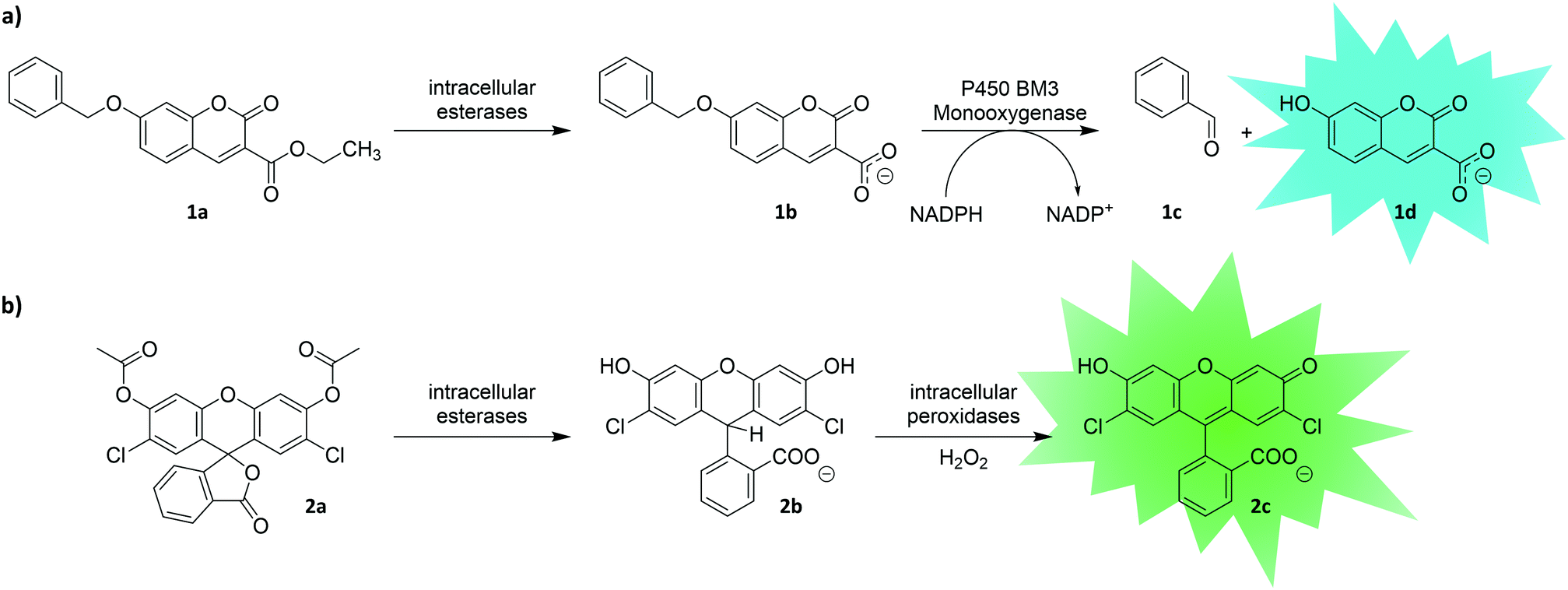 | ||
| Fig. 3 Entrapment of a fluorescent reporter molecule by means of charge. Upon cleavage of the pre-substrates (1a and 2a) by intracellular esterases, the substrates (1b and 2b) become trapped inside the cell due to negative charge of their respective carboxyl groups. (a) P450 BM3 monooxygenase hydroxylates substrate 1b, which leads to decomposition to benzaldehyde (1c) and coumarin derivative 1d.95 (b) ‘Dihydro’ dyes such as 2b can be oxidized by intracellular peroxidases using hydrogen peroxide to yield the respective fluorescein derivative 2c. Thus, they can serve as reporters of enzymes delivering hydrogen peroxide as by-product.96 | ||
Similarly, surrogate substrates have been designed for covalent intracellular entrapment. Inspired by the field of activity-based protein profiling,120 quenched activity-based probes (qABP) were developed by Kalidasan and coworkers. qABPs are substrates consisting of a substrate-mimicking moiety, a fluorescence reporter, and a quencher moiety (Fig. 4). Upon enzyme-mediated attack at the substrate-mimicking moiety, the quencher is released resulting in a fluorescence signal. The group took advantage of an α-N-acetylgalactoseamine-qABP (3a) to evolve an α-N-acetylgalactose aminidase (NAG) to efficiently hydrolyze the terminal sugar subunit of the blood group A antigen.121 Enzymatic cleavage of the sugar moiety released a dabcyl quencher module resulting in a fluorescent and reactive quinone methide intermediate (3b), which was trapped inside the cell by attack of intracellular nucleophiles (Fig. 4). After two rounds of flow cytometric sorting of the top 0.2% of a NAG mutant library, a NAG variant with a more than two-fold improved catalytic efficiency was obtained.
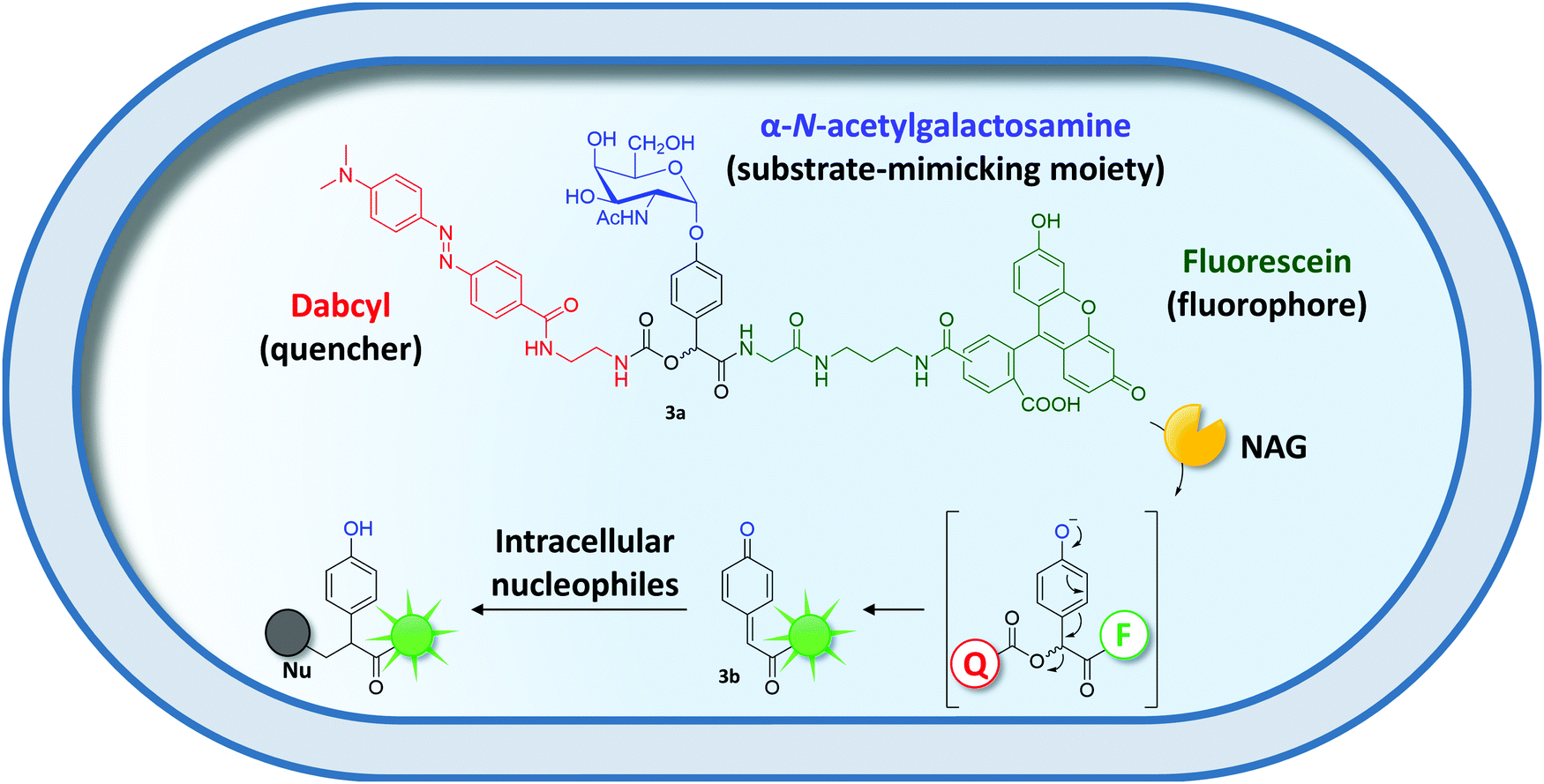 | ||
| Fig. 4 Covalent entrapment of a quenched activity-based probe (qABP) (3a).121 NAG-catalyzed cleavage of the substrate-mimicking moiety initiates cleavage of the quencher moiety (Q) ultimately yielding a reactive, fluorescent quinone methide intermediate (3b). The latter is captured inside the cell by attack of intracellular nucleophiles (Nu). | ||
Recently, Kwok et al. applied strain-promoted azide–alkyne cycloaddition (SPAAC) for the covalent entrapment of fluorescent probes and used it for the directed evolution of p-cyano-L-phenylalanyl aminoacyl-tRNA synthetase (pCNFRS) via uHTS methods.122pCNFRS was evolved to preferentially incorporate the non-canonical amino acid p-azido-L-phenylalanine (pAzF) from mixtures of pAzF and p-cyano-L-phenylalanine (pCNF) into a reporter protein. The addition of a dibenzocyclooctyl-derivatized Cy5 dye followed by intracellular SPAAC probed successful pAzF incorporation and enabled flow cytometric sorting of desired enzyme variants.
Altogether, these studies exemplify the different principles of substrate design to trap a fluorophore within a cell based on its (i) retention by selective cellular transporters, (ii) charge or (iii) covalent immobilization.
Besides surrogate fluorogenic substrates, DNA-binding fluorescent probes can be used for directed evolution. Whereas the former are converted to a fluorescent product, the latter do not interact directly with the enzyme of interest. Instead, DNA-binding fluorescent probes interact with DNA as an indirect consequence of the enzyme's activity and thus label active cells. For instance, this labelling approach was used to evolve Pseudomonas fluorescens aryl esterase (PFE) variants with altered enantioselectivity.123 A pseudo-racemic mixture of two substrates was applied: 3-(R)-phenyl butyric acid glycerol ester (the target substrate) liberated glycerol upon enzyme-mediated hydrolysis, whereas hydrolysis of 3-(S)-phenyl butyric acid 2,3-dibromopropyl ester (the “pseudo-enantiomer”) released a toxic compound. After incubation with the substrate mixture, cells were stained with two DNA-interacting dyes. While the green fluorescent Syto9 dye stained all cells irrespective of their viability, the red fluorescent propidium iodide only stained cells with reduced viability. After flow cytometric sorting for viable clones of a mutant library, which was generated by site-directed mutagenesis, two variants were identified exhibiting increased enantioselectivity towards the non-toxic (R)-enantiomer (E values >80).
2.2 Formation of a fluorescent hydrogel around the cell for flow cytometry
Another way to link genotype and phenotype is to incorporate fluorescent dyes into a hydrogel that surrounds cells producing active enzyme variants. This so-called “Fur-Shell” technology initially was designed to evolve phytases124 and was later expanded to other hydrolases.125E. coli cells producing active Yersinia mollaretii phytase (YmPh) variants converted glucose-6-phosphate (4a) to yield glucose (4b), which in turn was transformed to glucono-δ-lactone and hydrogen peroxide by externally added glucose oxidase (GOx) (Fig. 5a). In the presence of ferrous iron, Fenton's reaction utilized the nascent hydrogen peroxide to form hydroxyl radicals (Fig. 5a), which initiated the polymerization of a monomer (N-vinyl-2-pyrilidone, 5), a crosslinker (poly(ethyleneglycol)-diacrylate, PEG-DA, 6) and a fluorescent co-monomer (Polyfluor 570, 7) on the cell surface (Fig. 5b). Depending on the activity of the phytase variant, cells would exhibit different degrees of fluorescent encapsulation. A random mutagenesis library of YmPh was subjected to Fur-Shell uHTS and cells with a strong fluorescent hydrogel shell were sorted by flow cytometry and rescreened in MTP format. The best phytase variant contained five amino acid substitutions and 97 U mg−1 increased catalytic activity.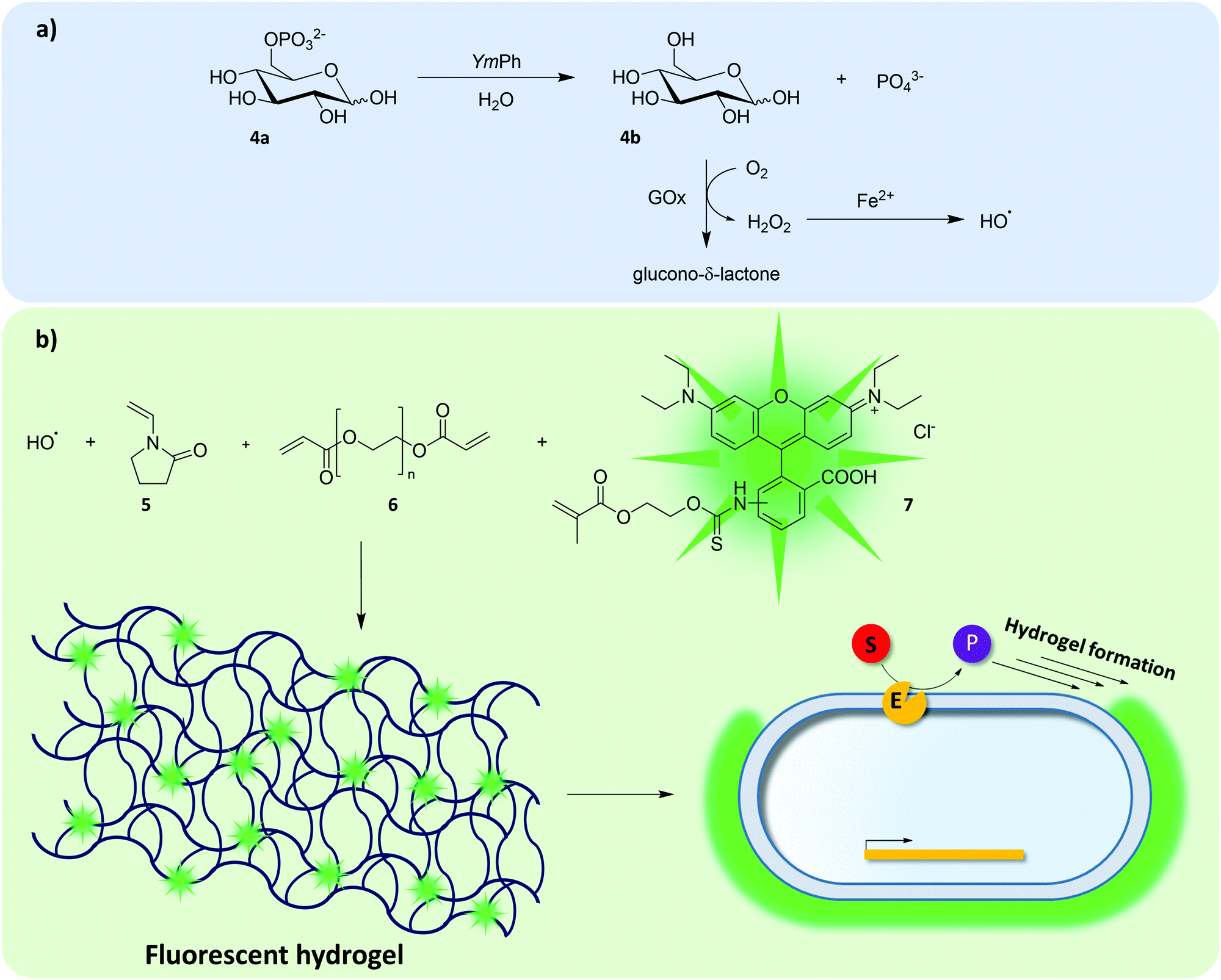 | ||
| Fig. 5 Formation of a fluorescent hydrogel shell. The directed evolution of YmPh was enabled by coupling glucose-6-phosphate hydrolysis with the formation of a fluorescent hydrogel shell (“Fur-Shell”).124,125 Externally added GOx would utilize glucose and produce hydrogen peroxide as by-product, which yields hydroxyl radicals in Fenton's reaction (a). Hydroxyl radicals then initiate hydrogel formation and incorporation of the fluorescent co-monomer Polyfluor 570 (7) enables flow cytometric identification of cells harboring active YmPh variants (b). | ||
Modified Fur-Shell protocols were successfully used to evolve other hydrolytic enzymes including Bacillus licheniformis p-nitrobenzyl esterase, Bacillus subtilis lipase A and a cellulase isolated from a metagenomic library.125,126 After one round of sorting and MTP-based re-screening of enriched populations, variants exhibiting up to 7-fold improved activity were identified. The Fur-Shell strategy is potentially generalizable to any enzymatic (cascade-)reaction producing hydrogen peroxide. Along these lines, a modified alginate-based fluorescent hydrogel has recently been reported for the directed evolution of GOx via uHTS methods.127 Still, encapsulating cells in fluorescent hydrogels currently remains an unexploited but an up-and-coming uHTS approach.
2.3 Transcription factor and enzyme-mediated regulation of fluorescent reporters
Transcription factor-based biosensors are useful tools to link genotype and phenotype. Briefly, substrates and products of enzymatic reactions can lead to up- or down-regulation of gene expression culminating in the formation of a fluorescent signal, thus linking genotype and phenotype (Fig. 6). While being established tools for flow cytometry-based strain engineering,128–131 they remain largely unexploited for directed enzyme evolution. As of late, biosensors based on fluorescent proteins (e.g., enhanced green fluorescent protein – eGFP),132 become more and more relevant in the field of directed enzyme evolution, as well. | ||
| Fig. 6 Overview on transcription factor-mediated regulation of fluorescent reporters. Enzymatic activity can be linked to the formation of a fluorescent reporter protein (e.g., GFP). Regulatory elements of gene expression (so-called transcription factors: activators or repressors) must be identified that change their behavior (up- or down-regulation of gene expression) in the presence of the substrate or product of a given enzymatic reaction. For instance, product formation can be linked to the up-regulation of gene expression (a). Likewise, down-regulators can be used to monitor substrate consumption (b). Typical genetic circuits are of a complexity beyond the simplistic examples shown in this figure. They may involve many regulatory events connected in series (i.e., regulatory cascades) or even several levels of regulation (e.g., regulation on the DNA, RNA and protein level),134–136 which were omitted for clarity. | ||
An example of such a fluorescent biosensor – the “ligand-mediated eGFP expression system” (LiMEX) – was recently introduced.133 With the aim of tuning arginine deiminase (ADI) for application under physiological conditions, a competitive screening strategy was pursued. Inspired by the arginine biosynthetic regulatory system in E. coli, an arginine biosensor was designed. Arginine binds to the cognate repressor ArgR and represses together with ArgR the transcription at the argG promoter. In this study, arginine's co-repressor function was utilized to suppress the expression of the eGFP-encoding gene under the control of the argG promoter. Owing to the depletion of intracellular arginine by highly active ADI variants, eGFP repression was reduced, resulting in a fluorescence signal. After three iterative rounds of random mutagenesis and flow cytometric screening, a variant (M31) was identified exhibiting 970-fold increased catalytic activity compared to the wild type. Notably, as a result of the competition of ADI and ArgR for arginine, the Km value of the M31 variant was reduced from 1.23 mM (parent variant) to 0.17 mM, enabling activity under physiological conditions.
As an alternative to the up-regulation of fluorescent proteins, release of fluorescent dyes can be triggered by transcription factor-mediated systems. Such a biosensor was recently established to evolve nitrogenases for improved production of molecular hydrogen.137 Nitrogenase is an oxygen-sensitive multienzyme complex, which produces H2 as a by-product during nitrogen fixation. A uHTS method to screen for nitrogenase variants with improved H2 formation was designed, utilizing the natural H2 sensing system of Rhodobacter capsulatus. In the presence of molecular hydrogen, transcription of the hup gene cluster (HupUV, HupT, HupR) was upregulated ultimately inducing β-galactosidase expression. The latter released fluorescein from the fluorogenic surrogate substrate fluorescein di-β-D-galactopyranoside and enabled discrimination of H2-producing nitrogenase variants with different activities. After one round of random mutagenesis and very stringent gating (top 0.024% of population), a variant with a 10-fold increased hydrogen gas production was obtained.
Siedler et al. reported an NADPH-biosensor based on the transcriptional regulator SoxR.138 At high intracellular levels of NADPH SoxR remains in its reduced state. At low levels of NADPH, SoxR is present in its oxidized form and promotes transcription at the soxS promoter. This led the authors to assess the SoxR system to sense NADPH levels as indirect measure for the activity of the NADPH consuming Lactobacillus brevis alcohol dehydrogenase (LbADH). To this end, enhanced yellow fluorescent protein (eYFP) was placed under the control of the soxS promoter for flow cytometric detection. The authors showed that cells with high LbADH activity in conversions of methyl acetoacetate to (R)-methyl 3-hydroxybutyrate exhibited higher eYFP fluorescence. Finally, an LbADH mutant library was screened for activity with the less preferred substrate 4-methyl-2-pentanone. Already after one round of flow cytometric screening a variant with 36% increased activity was identified.
Aside from metabolite-regulated pathways, damage response mechanisms provide a valuable tool for directed enzyme evolution. Recently, a GFP reporter was used to evolve nitroreductases toward the activation of DNA-damaging prodrugs.139 A GFP-encoding reporter gene was controlled by an SOS promoter, regulated in response to DNA damage. Upon enzymatic conversion of an inactive nitroaromatic prodrug into a DNA-damaging molecule, GFP-based fluorescence indicated nitroreductase activity and facilitated uHTS. Directed evolution of E. coli nitroreductase NfsA led to the isolation of variants with more than 25-fold reduced Km values towards the prodrug PR-104A compared to wild type.140
Genetic circuits provide the means to regulate fluorescent outputs in response to an enzyme's activity. But they do not remain the only level of regulation that can be taken advantage of for uHTS. Posttranslational enzyme-mediated regulation can be a useful handle, too. For instance, the ssrA peptide sequence, which directs proteins to the cytoplasmic protease ClpXP, was utilized to build a short-lived GFP reporter facilitating the engineering of tobacco etch virus (TEV) protease.141 Kostallas and Samuelson designed a GFP reporter substrate, which was C-terminally fused to a TEV protease recognition site (the TEV substrate), followed by an ssrA degradation tag. ssrA mediated the degradation of GFP unless it was rescued by the TEV protease cleaving off the ssrA sequence. Flow cytometry was used to distinguish active from inactive and soluble from aggregation-prone TEV protease variants based on the GFP signal. Moreover, substrate libraries containing efficiently processed and suboptimal TEV substrates could be separated.
In general, screening conditions should resemble the desired application conditions of the evolved enzyme as closely as possible. Thus, embedding the target enzyme into regulatory networks with physiological substrate concentrations is explicitly useful when it is supposed to function under such physiological conditions (e.g., as a whole cell biocatalyst).
2.4 Cell surface display-mediated fluorescence tethering
Cell surface display (CSD) can be used to present proteins on the surface of microbial cells. Usually, this is achieved by genetically fusing the protein to outer membrane anchoring motifs. Working on the cell surface bypasses any diffusion limitation posed by cellular membranes, but maintaining the mandatory link between genotype and phenotype can become a challenge. Small reporter molecules thus have to be either co-compartmentalized with the cell (see Section 2.1) or must be physically linked to it.Proteases have been a popular target of such display-mediated fluorescence tethering strategies as their substrate – a protein or peptide – can be actively displayed by the cell. That is, no sophisticated chemical anchoring is required. The “yeast endoplasmic reticulum sequestration screening” (YESS) is one example how CSD can facilitate uHTS (Fig. 7).142 The engineering of proteases towards novel substrate specificities often results in decreased selectivity instead of a true change in specificity.143 Thus, a counter selection strategy was developed to evolve TEV protease by co-producing the protease and its polypeptide substrate in the endoplasmic reticulum. The polypeptide substrate was flanked by two different antibody epitopes and additionally contained a counterselection protease recognition sequence (“counter selection substrate”) and the Aga2 protein. Aga2 mediates CSD by covalent linkage to the Aga1 protein via disulfide bridges. After TEV protease-mediated cleavage in the endoplasmic reticulum, the substrate fusion construct was displayed on the cell surface. Depending on the cleavage pattern of the protease (substrate vs. counterselection substrate), different antibody epitopes were accessible for fluorescence labeling, ultimately enabling flow cytometric sorting of TEV protease variants with desired cleavage properties. By screening a TEV protease library against a substrate library, which contained the native protease cleavage site as counter selection sequence, TEV protease variants were identified preferring glutamic acid or histidine over glutamine in the TEV protease recognition sequence. Upon further characterization, selectivity changes in the range between 1100 and 5000-fold were found for the evolved protease variants.142
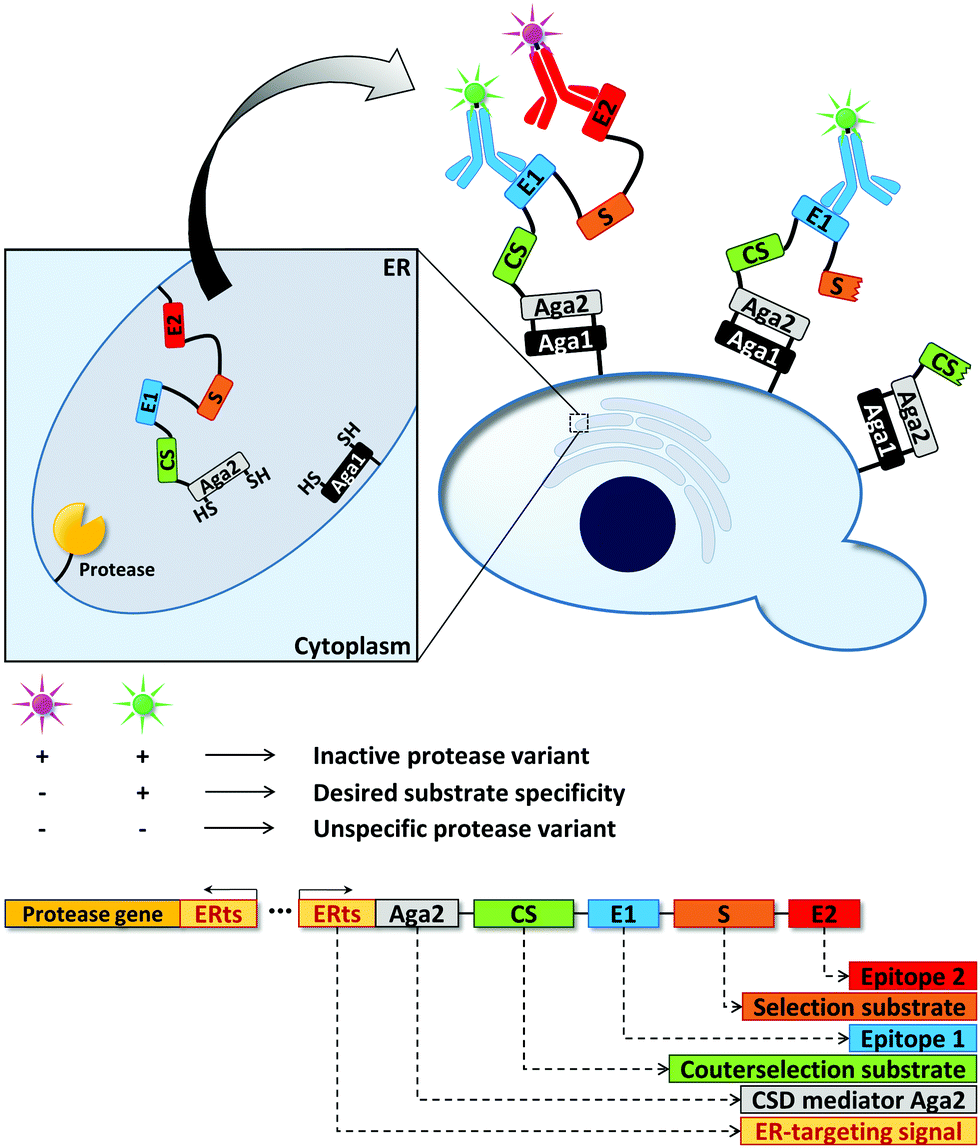 | ||
| Fig. 7 Yeast endoplasmic reticulum sequestration screening (YESS) for the directed evolution of proteases.142 Directing the protease and its peptide substrate into the endoplasmic reticulum (ER) facilitates substrate cleavage and subsequent Aga2-mediated cell surface display (CSD) of the cleaved substrate. The peptide substrate consists of a selection substrate sequence (desired cleavage site), a counterselection substrate sequence (undesired cleavage site) and antibody epitopes. Detection of the latter by fluorescently labeled antibodies enables the fluorimetric read-out of the protease cleavage pattern in a flow cytometer. | ||
Similar counterselection strategies have been applied on bacterial systems. Using CSD strategies, the E. coli outer membrane protease OmpT was subjected to directed evolution.144 As OmpT is naturally located on the cell surface, protease cleavage products had to be captured to the cell surface to prevent loss of the genotype–phenotype connection. To this end, the substrate was equipped with a poly-arginine tag that bound to the negatively charged cell surface by virtue of electrostatic interactions. Finally, cells decorated with desired cleavage products were detected with externally added fluorescent probes.
Cell surface-mediated uHTS protocols have not only been used for the directed evolution of bond-cleaving enzymes (vide supra). As bond-forming enzymes provide the possibility to modify cell surfaces, they are also prone to this kind of screening platform. For instance, the peptide bond-forming enzyme sortase A (SrtA) from Staphylococcus aureus mediates the conjugation between LPXTG- and oligoglycine motifs.145,146 SrtA was evolved to catalyze coupling reactions of LPETG-tagged proteins with 140-fold increased activity compared to wild type using a yeast display-based screening system (Fig. 8).147 The enzyme was genetically fused to the Aga2 domain, which in turn is linked to the cell surface by covalent linkage to the Aga1 protein (vide supra). Aga1 was chemoenzymatically modified with one of the two SrtA substrates (an LPETG peptide or oligoglycine) and incubated with a biotinylated version of the remaining second substrate. Upon successful bond formation, biotinylated cells could be stained using a streptavidin–fluorophore conjugate, whereas cells expressing inactive SrtA variants remained unfunctionalized. Due to the proximity of enzyme and substrate, undesired functionalization of neighboring cells carrying inactive SrtA variants was less favorable. SrtA was subjected to random mutagenesis and improved variants were identified after iterative rounds of screening, reducing the concentration of the biotinylated substrate from round to round.
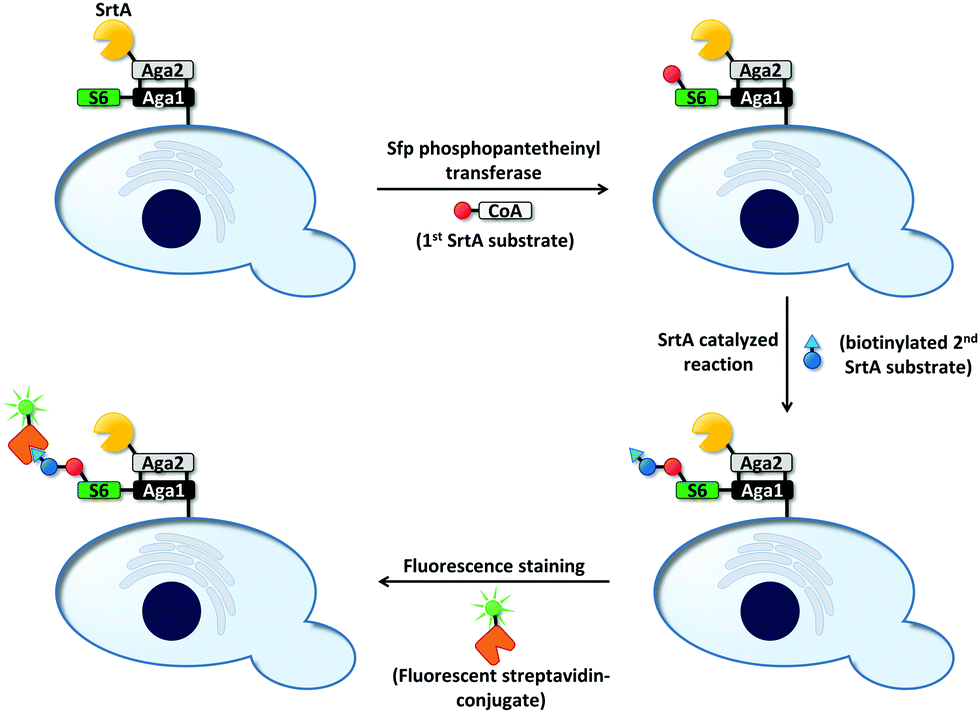 | ||
| Fig. 8 Overview on screening for SrtA-mediated bond formation between an LPETG peptide and an oligoglycine.147 The screening was applicable to link LPETG to oligoglycine and vice versa. First, the S6-peptide was genetically fused to the N-terminus of the Aga1 subunit. Sfp phosphopantetheinyl transferase covalently linked one of the two SrtA substrates (red ball; LPETG or oligoglycine) to the S6-peptide. Subsequently, cells were incubated with a biotinylated derivative of the remaining substrate (blue ball: second SrtA substrate; triangle: biotinylation) to allow SrtA-mediated bond formation. A streptavidin–fluorophore conjugate was used to identify events of successful bond formation. Inactive SrtA variants fail to link the two substrates. | ||
Deweid et al. recently took advantage of yeast surface display using the Aga1–Aga2 system to evolve Streptomyces mobaraensis transglutaminase (mTG).148 Transglutaminases catalyze the bond formation of the glutamine side chain carboxamide group with primary amines and are of interest in the production of antibody–drug conjugates. The authors displayed a variant of mTG on yeast cells. During the uHTS assay, the activated mTG was incubated with a biotin-tagged glutamine donor peptide. Lysine residues on the cell surface served as acceptor substrates. Successfully biotinylated cells were fluorescently labeled using a streptavidin R-phycoerythrin (a fluorescent protein) conjugate and sorted by flow cytometry. After five rounds of screening epPCR libraries mTG variants with only minor improved activity were identified. However, the best engineered mTG variant performed better in antibody-labeling experiments compared to wild-type mTG illustrating its use for the development of antibody–drug conjugates.
Another example of the Aga1–Aga2 surface display was recently reported by Han et al. to engineer a split version of a soybean ascorbate peroxidase termed APEX2, that is, a variant of the protein genetically split in two inactive subunits that regain activity upon mixing.149 The authors investigated 24 potential split sites and subsequently fine-tuned the most promising split APEX2 variant via epPCR and uHTS on the cell surface. Reconstitution of the final enzyme variant was shown in different environments (on RNA motifs, in the mammalian cytosol and at contact sites of mitochondria and the ER) and the authors envisage its application as a tool for proximity labeling.
In contrast to the mostly yeast-based systems reported above, Kwok et al. recently demonstrated the use of the E. coli Lpp-OmpA CSD-scaffold for the directed evolution of Nε-acetyl-lysyl aminoacyl-tRNA synthetase (AcKRS).122 The Lpp-OmpA scaffold consists of 29 amino acids of the E. coli lipoprotein (i.e., Lpp signal peptide + the first nine amino acids of Lpp) and amino acids 46–155 of E. coli outer membrane protein A (OmpA).150 Kwok et al. fused a reporter peptide to the C-terminus of the Lpp-OmpA scaffold. They included an amber stop codon (TAG) into the reporter peptide's sequence to test the incorporation of the non-canonical amino acid m-iodo-L-phenylalanine (mIF) versus the incorporation of Nε-acetyl-lysine (AcK) by variants of AcKRS. Using specific antibodies, the authors were able to fluorescently label the displayed reporter peptide in dependence of incorporated mIF or AcK. They identified AcKRS variants that preferentially recognize mIF in the presence of other non-canonical amino acids.
Today, CSD scaffolds are readily available for yeast (mostly the Aga1–Aga2 system) and E. coli (Lpp-OmpA and others),150,151 which are the organisms mostly used in directed enzyme evolution. The studies highlighted above clearly show that CSD represents an asset for uHTS – especially for the directed evolution of bond-forming/bond-cleaving enzymes.
3. Emulsions – a biomimetic compartment in uHTS
During the last two decades, emulsions have become a notable alternative to cells as compartments in uHTS. They offer high stability over a broad temperature, pH and salt concentration range152 and are easy to prepare. Dispersing an aqueous phase in an oil phase in the presence of stabilizing surfactants yields a water in oil (w/o) emulsion. Stirring, vortexing, extrusion, homogenization, or filtration have successfully been used to prepare such emulsions for directed evolution campaigns.92,97,153–162 However, these methods typically produce polydisperse compartments, which potentially hampers quantitative analysis as already small volumetric deviations can affect the concentration of an enzyme-derived product.163 On that account, sophisticated microfluidic devices have been developed to produce highly monodisperse w/o droplets.157,164–167 Sorting these biomimetic compartments either is accomplished in a custom-made chip device or – after emulsifying the w/o emulsion in an additional water phase (w/o/w emulsion) – in a standard flow cytometer. Using a Poisson distribution, a maximum of one genetic variant is encapsulated, thereby ensuring a monoclonal nature of each compartment and preventing co-sorting of beneficial variants with inactive variants. Thus, either single cells or single copies of a gene are encapsulated allowing in vivo or in vitro approaches of directed evolution in emulsions, respectively. Sometimes, the former technique misleadingly is referred to as “in vitro compartmentalization” (IVC) even when the enzyme is produced by a living cell.158,159,161,168–170 In these cases, however, “in vitro” refers to the compartment (as opposed to living cells; see Section 1) and does not specify whether the source of protein production is an IVTT system (i.e., a truly IVC approach in every sense of the term) or cells. Herein, we chose to deal with both approaches separately, as they rely on different principles accompanied by individually different challenges to overcome.3.1 In vivo applications using emulsions
Cells are the most straightforward way of protein production, yet they often lack the ability to take up the desired screening substrate or to retain the nascent product. They can be co-encapsulated with all compounds required for the screening reaction (substrates, cell lysis agents, buffer etc.) in an emulsion droplet and the enzyme of interest can be released from the cell in situ. This is an elegant approach, as it to combines the advantages of cells and emulsion compartments: that is, the cells produces the enzyme; the emulsion retains the genotype–phenotype connection. Although sorting rates of >106 h−1 are feasible with emulsions in a conventional flow cytometer or in custom-made chip devices,92,171 the restriction of compartmentalizing a maximum of one cell per emulsion droplet by Poisson's distribution causes most of the droplets to be empty. Therefore, the effective throughput is reduced by ∼5-fold.90 Nevertheless, from a sustainability point of view, microcompartments are superior to conventional MTP assays, since only pico- or even femtoliters of reagents are used per sample as previously discussed by Agresti et al.166 Moreover, the compartmentalization of a single cell in such a small reaction volume typically results in high local enzyme concentrations during screening, which benefits the signal-to-noise ratio.152,172The directed evolution of Taq DNA polymerase was a pioneer study using cells in ill-defined emulsions generated by stirring.173 By a selection approach termed “compartmentalized self-replication” (CSR), Ghadessy et al. received a Taq DNA polymerase variant with over 130-fold increased resistance to the inhibitor heparin and a variant with increased thermostability (11-fold). The group expanded their technology towards the directed evolution of a nucleoside diphosphate kinase providing the deoxyribonucleoside triphosphates (dNTPs) for its own replication173 and the directed evolution of a DNA polymerase with a broadened substrate scope.174
The Hollfelder group capitalized on custom-made microfluidic chips for both the generation of monodisperse w/o emulsions and fluorescence-based sorting.98E. coli cells producing Pseudomonas aeruginosa arylsulfatase were co-compartmentalized with the substrate and a lysis agent (Fig. 9). After sorting, DNA was directly recovered from fluorescence-emitting droplets without an additional amplification step and transformed for further rounds of screening.
 | ||
| Fig. 9 Outline of a microfluidic set-up for cell lysis in emulsion droplets. Co-compartmentalization of cells, substrate and cell lysis agents releases the enzyme of interest from the cell and enables conversion of the substrate to a fluorescent product inside an emulsion droplet. Statistically incorporating a maximum of a single cell per droplet ensures genotype–phenotype linkage, i.e., every droplet contains a different gene variant and the respective enzyme of interest. After surrounding the w/o emulsion with an additional water phase, the resulting w/o/w double emulsion can be analyzed in a conventional flow cytometer (not shown). | ||
Using a microfluidic set-up that integrated droplet generation, incubation, and sorting on a single chip,175 the group of Hilvert beautifully demonstrated the advantages of uHTS over conventional MTP-based screening. In only one round of directed evolution and screening of a combinatorial site-saturation mutagenesis (SSM) library comprising 1 × 106 retro-aldolase variants, the group identified a variant (termed RA95.1A-1)176 with comparable catalytic efficiency and stereoselectivity to a variant termed RA95.5. RA95.5 was previously obtained from five rounds of iterative SSM and conventional screening in MTPs. Albeit starting from the same de novo designed parent variant RA95.0177,178 both variants differ in six amino acid substitutions and share only one substitution (T83K). T83K was previously determined to play a key role as a nucleophilic amine, boosting the catalytic activity of the retro-aldolase and inverting the designed (S)-enantioselectivity of the enzyme.
Recently, the same group reported on the directed evolution of a cyclohexylamine oxidase (CHAO) for the deracemization of 1-phenyl-1,2,3,4-tetrahydroisoquinoline to obtain the (S)-enantiomer,87 which is a precursor of the drug solifenacin. Amine oxidases produce hydrogen peroxide as a by-product179 and the authors used horseradish peroxidase (HRP) in a reaction cascade to utilize hydrogen peroxide for the conversion of the fluorogenic dye Amplex UltraRed. Eight amino acid positions were chosen for simultaneous mutation (seven positions in proximity to the native substrate binding site and one in the substrate channel). These positions were randomized by reduced amino acid alphabets using the DYT codon (encoding amino acids A, S, T, V, I, and F) or the BYT codon (encoding A, S, P, V, L, and F), respectively. This corresponds to a theoretical library size of 1.7 × 106. To ensure sufficient oversampling >107 variants were screened using the previously described microfluidic setting. The 0.1% most active droplets were sorted, their DNA was isolated, recovered by PCR, and retransformed into E. coli. The cells were subjected to two more rounds of droplet sorting. Finally, the best variants of the enriched population were validated in a colorimetric screening in MTP format. Notably, according to the authors, the whole procedure from library generation to validation and sequencing of the most promising hits was performed in less than two weeks. This impressively illustrates how uHTS can speed-up directed enzyme evolution. Screening a comparable library in MTP format would consume enormous resources (time and consumables). After one round of evolution, the best CHAO variant contained five amino acid exchanges and exhibited a dramatic change in substrate specificity. While the native substrate (cyclohexylamine) was barely converted, the improved enzyme variant rapidly converted (R)-1-phenyl-1,2,3,4-tetrahydroisoquinoline (960-fold increased catalytic efficiency compared to wild-type). Combining this enantioselective CHAO variant and a non-selective reducing agent enabled a deracemization process in which the (S)-enantiomer could be isolated in high purity (99% ee).
Enantioselectivity is a particularly difficult property to screen for due to the inherent physicochemical similarities of enantiomers. While screening assays distinguishing between two enantiomers can typically be established at lower throughput (e.g., using chiral high-performance liquid chromatography (HPLC)), uHTS of enantioselective enzymes in the presence of both enantiomers is challenging. Recently, Ma et al. developed a dual-channel uHTS to screen for enantioselective variants of Archaeoglobus fulgidus esterase (AFEST) to produce (S)-profens. They established a microfluidic screening assay capable of sorting droplets based on two independent fluorescence signals simultaneously and used the “in-droplet cell lysis strategy” described above (Fig. 9). The evolutionary campaign proceeded in two phases. In the first phase, libraries were simultaneously screened for conversion of two substrates each consisting of an (S)-ibuprofen moiety esterified with one of two different fluorogenic dyes (surrogate substrates (S)-8a and (S,S)-9a; Fig. 10a and b). Only AFEST variants capable of hydrolyzing both substrates were selected to prevent evolutionary bias towards the dye moiety. In the second phase, a mixture of the desired (S)-enantiomer coupled with one fluorogenic dye (surrogate substrate (S)-8a) and the (R)-enantiomer coupled with another fluorogenic dye (surrogate substrate (R,R)-9a) was applied to positively select variants targeting the (S)-enantiomer and negatively select variants targeting the (R)-enantiomer (Fig. 10a and c). After five rounds of evolution (approximately 5 million variants in total), the authors identified an AFEST variant with 700-fold higher enantioselectivity compared to wild-type.170 Similarly, Heemstra and coworkers recently reported on the use of DNA biosensors labeled with two different fluorescent dyes for the FACS-based detection of small molecule enantiomers.180,181
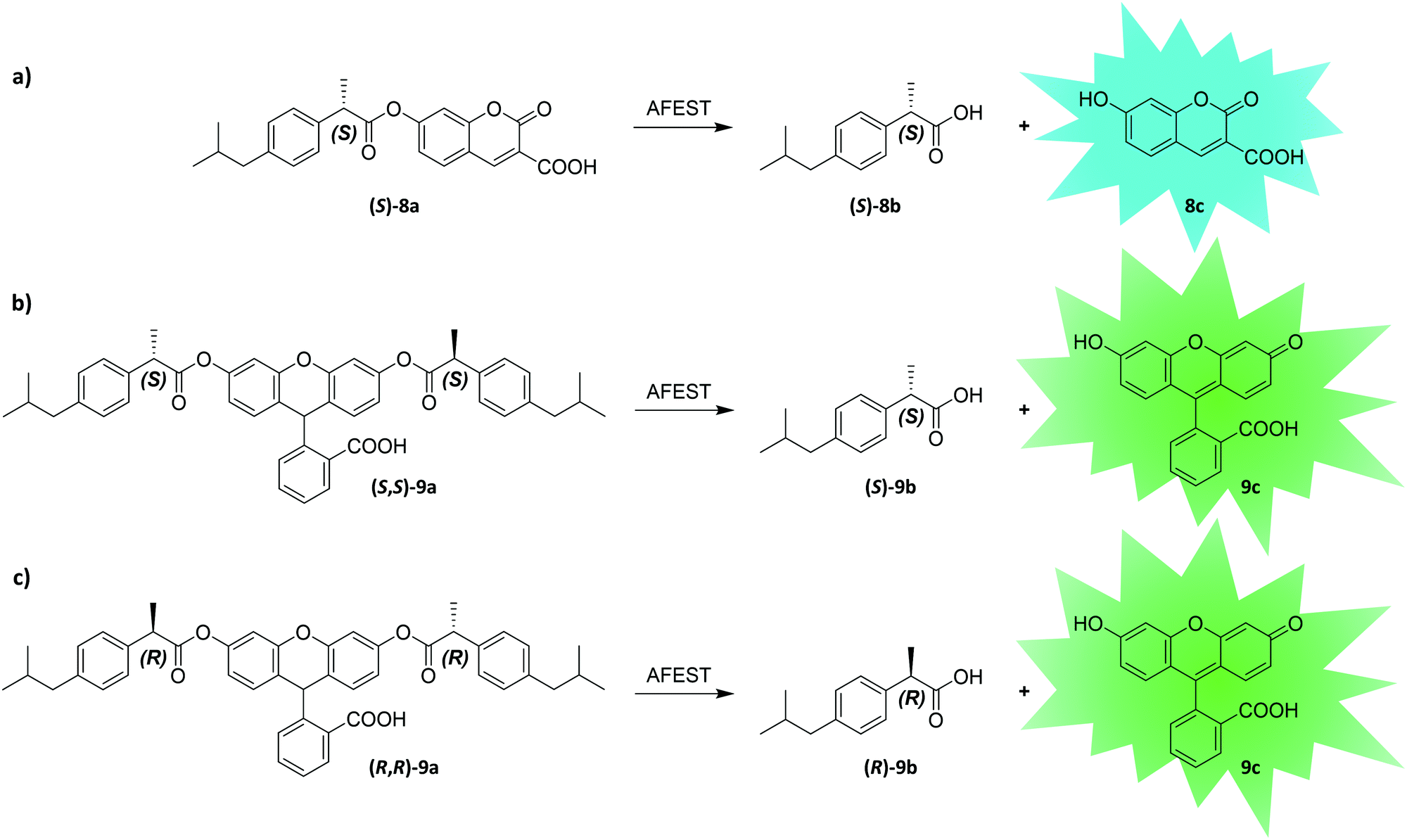 | ||
| Fig. 10 Substrate design and reactions used by Ma et al. to establish an enantioselective uHTS. By combining enantiomers with different fluorescent dyes, the authors screened for enantioselective variants of Archaeoglobus fulgidus esterase (AFEST).170 In a first round of screening, (S)-selective AFEST variants were enriched by screening them for reaction (a and b). Using both surrogate substrate (S)-8a and (S,S)-9a in this step avoids the evolution of AFEST variants with a bias towards the dye moiety of the screening molecule. In a second round AFEST variants were selected more stringently, for increased activity towards the (S)-enantiomer ester and decreased activity towards the (R)-enantiomer by screening them for reaction (a) versus (c). | ||
Romero et al. combined in-droplet cell lysis (Fig. 9) with next-generation DNA sequencing to study the sequence space of a model glycosidase system.182 They investigated amino acid sites throughout the protein for their mutational tolerance. In addition to sites that were known as conserved residues, uHTS identified previously unknown sites that were highly intolerant to amino acid substitutions. Including a heat incubation step into the enzyme activity assay allowed the screening for thermostable enzyme variants. Thus, the authors were able to map amino acid substitutions with beneficial effect on thermostability. Such studies of enzyme function landscapes require processing of enormous numbers of enzyme variants and thus benefit tremendously from uHTS approaches.
All examples highlighted so far depended on fluorescence. Recently, Gielen et al. coupled the NAD+ consumption of phenylalanine dehydrogenase (PheDH) to the formation of a formazan dye (10b) by using 1-methoxy-5-methylphenazinium methyl sulfate (mPMS) as redox mediator (Fig. 11). Ultimately, this led to a massive amplification of the absorbance signal (extinction coefficient of formazan dye 10b: ε455nm = 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 660 M−1 cm−1 in glycine–KOH buffer at pH 10; extinction coefficient of NADH: ε340nm = 6220 M−1 cm−1 in water) allowing for absorbance-based detection in droplets at micromolar concentrations at rates of ∼100 Hz.183 Absorbance-activated droplet sorting (AADS) is a seminal approach towards a more general applicability of uHTS, since the dependency on fluorescent signals often requires the use of industrially irrelevant substrates to liberate a fluorescent product.
660 M−1 cm−1 in glycine–KOH buffer at pH 10; extinction coefficient of NADH: ε340nm = 6220 M−1 cm−1 in water) allowing for absorbance-based detection in droplets at micromolar concentrations at rates of ∼100 Hz.183 Absorbance-activated droplet sorting (AADS) is a seminal approach towards a more general applicability of uHTS, since the dependency on fluorescent signals often requires the use of industrially irrelevant substrates to liberate a fluorescent product.
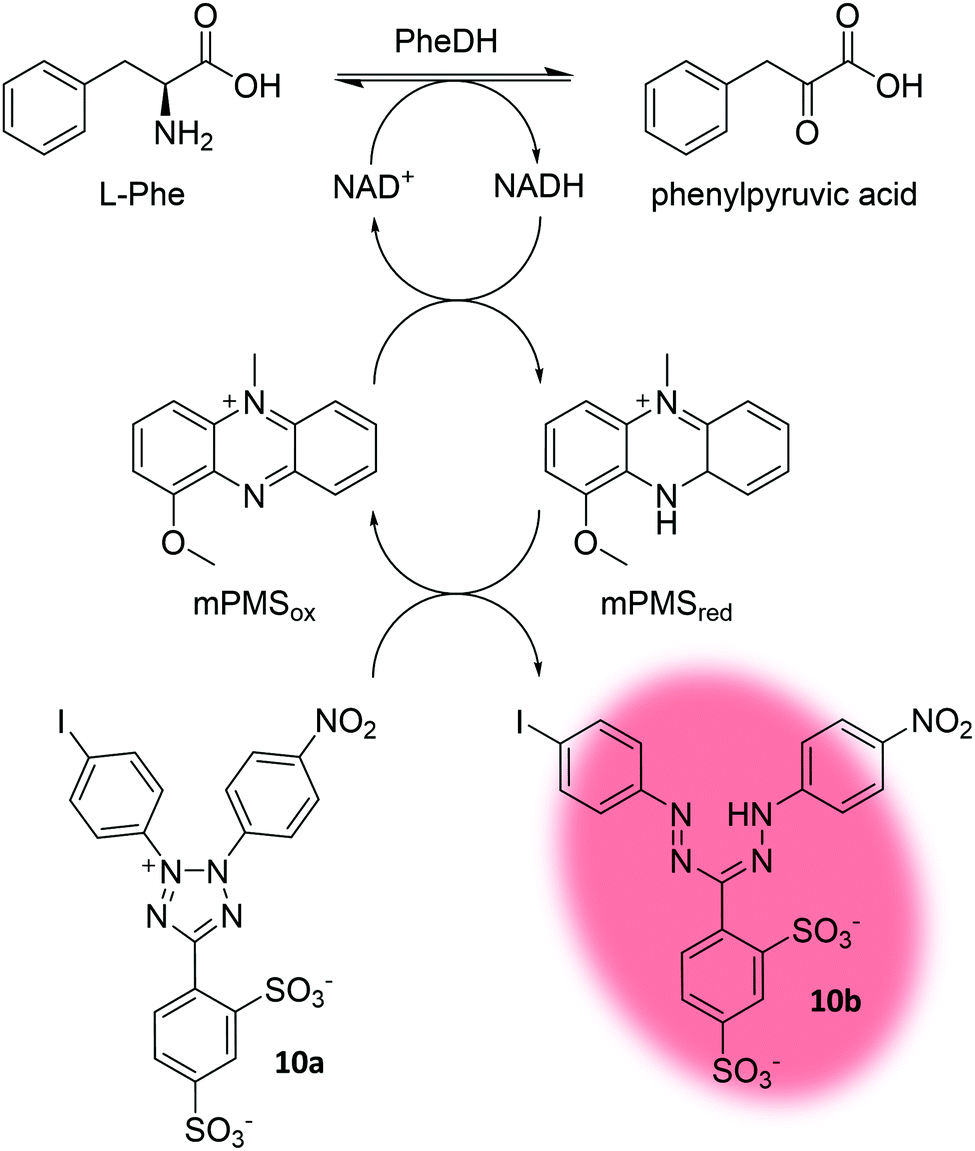 | ||
| Fig. 11 Concept of absorbance-activated droplet sorting (AADS). Phenylalanine dehydrogenase (PheDH) consumes NAD+. The electron mediator 1-methoxy-5-methylphenazinium methyl sulfate (mPMS) then facilitates conversion of the water soluble tetrazolium salt (10a) to the absorbing formazan dye 10b.183 | ||
The studies outlined above illustrate how uHTS not only can identify variants with high levels of activity but also enables researchers to explore different evolutionary trajectories at once. Recently, the in-droplet lysis strategy (Fig. 9) was applied on double emulsions enabling sorting on commercial flow cytometers,164 which expands this method's accessibility beyond laboratories specialized in producing sophisticated microfluidic sorting chips.
As an alternative to the in-droplet cell lysis strategy, CSD and emulsions can be combined. For a survey on how display techniques can be used to engineer biocatalysts, we point the reader to the review of Smith et al.112 Recent examples of display-based uHTS in emulsions mainly include yeast display technologies. Prokaryotic display technologies have proven to be a valuable alternative, too,91,158 albeit they are seemingly superseded by the increasing popularity of in-droplet cell lysis strategies (vide supra).
Agresti et al. used yeast (Saccharomyces cerevisiae) display to evolve horseradish peroxidase (HRP) variants with >10-fold catalytic rates.166 They co-compartmentalized single yeast cells displaying HRP variants on their surface with a non-fluorescent substrate (Amplex UltraRed) in w/o emulsions. After enzymatic conversion to a fluorescent product, compartments exhibiting fluorescence were sorted at rates of 2 kHz.
Enzymatic cascade reactions and yeast surface display enabled the directed evolution of glucose oxidase.184–186 In a w/o emulsion, cell surface-displayed glucose oxidase produced hydrogen peroxide as a by-product. HRP utilized the nascent hydrogen peroxide to form fluorescein tyramide radicals, which in turn undergo phenolic oxidative coupling to tyrosine residues of proteins on the yeast surface (Fig. 12). Compartmentalization of single yeast cells in w/o emulsion reduced the crosstalk between cells carrying different glucose oxidase variants. After breaking the emulsion, cells encoding for highly active glucose oxidase variants were separated from less active variants in a flow cytometer. In a later study, the sensitivity of the assay was increased: the hydrogen peroxide by-product was detected by vanadium bromoperoxidase-coupled formation of a fluorescent coumarin derivative. Coupling of hexose oxidase and a second enzyme (HRP or vanadium bromoperoxidase) further expanded the platform for the uHTS of cellulase variants.187,188
 | ||
| Fig. 12 Directed evolution of glucose oxidase (GOx) on yeast cells.184,185 Externally added HRP uses the GOx reaction by-product hydrogen peroxide to couple fluorescein tyramide (11) to tyrosine residues on the cell surface. Compartmentalizing the reaction in w/o emulsions (omitted for clarity) reduced cross talk between different cells and thus helped preserving the genotype–phenotype linkage. | ||
Enzyme secretion strategies can make enzymes accessible for screening in emulsions as well. Most notably, this has been demonstrated for the screening of hydrolytic enzymes using the yeast Yarrowia lipolytica as secreting host.189 Yet, the latter example represents an exception as most studies still rely on E. coli or S. cerevisiae as enzyme production hosts.
3.2 In vitro applications using emulsions
Cells offer the ease of protein production at the expense of a potentially assay-interfering cellular background and diversity limitations by transformation efficiency (<1010 per μg of DNA for E. coli,81,190 much less for other host organisms).191 As opposed to cells, IVTT has been leveraged for protein expression in artificial emulsion compartments. Each droplet is loaded with a maximum of one molecule of DNA encoding for the gene of interest. After gene expression, the target enzyme is trapped within the compartment.160 Avoiding any transformation step, library sizes exceeding 1012 variants are obtainable,81 which is comparable to other IVTT-based technologies commonly used to evolve protein–ligand interactions (e.g., ribosome or mRNA display).192–194 Moreover, the in vitro approach enables researchers to screen enzyme variants under otherwise unfavorable or even toxic conditions (e.g., the expression of toxic proteins and screening at high substrate concentrations) or to incorporate non-natural cofactors44,45,60,61 or non-canonical amino acids.68–70,195–198Tawfik and Griffiths were the first who capitalized on in vitro compartmentalization for the selection of the DNA methyltransferase HaeIII (M.HaeIII) encoding gene from a mixed library containing the target gene and an excess of up to 107 of a “dummy” gene (encoding dihydrofolate reductase; DHFR).153 Briefly, they attached the M.HaeIII methylation/restriction site – a sequence that is methylated by M.HaeIII and digested by HaeIII endonuclease if not methylated – to the M.HaeIII and the DHFR encoding genes, respectively. Following encapsulation of single copies of the genes, IVTT and enzymatic methylation of the HaeIII site, emulsions were broken and subjected to HaeIII endonuclease digestion. Only successfully methylated HaeIII sites were resistant to digestion and served as template for a subsequent PCR. The authors illustrated that in one round of selection a model library containing 0.1% of the M.HaeIII-encoding gene could be enriched to ∼50% M.HaeIII. In a later study, this platform was used to alter the sequence specificity of M.HaeIII.199 DNA modifying enzymes are best suited as targets for this technology, as the action of the enzyme variant (phenotype) directly influences its encoding gene (genotype) (for more examples see Lee et al.200 and Doi et al.).201 Directed evolution of other enzymes has successfully been demonstrated by IVTT in emulsion compartments, mainly including hydrolases92,97,155,202,203 and oxidoreductases (vide infra).204,205
Combining microbeads and emulsions has been vital in expanding IVTT-based uHTS-approaches towards enzyme classes other than DNA-modifying enzymes. To this end, single copies of a gene and multiple copies of the encoded enzymes are immobilized on microbeads inside an emulsion compartment. This principle was demonstrated on the example of a phosphotriesterase (PTE):97 in a first w/o emulsion, one copy of a biotin-tagged PTE gene was linked to a streptavidin-coated microbead (diameter: 1 μm) (Fig. 13). The same streptavidin–biotin interaction was used to pre-coat the microbeads with antibodies specific for a peptide tag, which was fused to the enzyme encoded by the immobilized gene. After IVTT, the antibodies captured the enzyme fusion construct and the emulsion was broken. Despite the absence of a compartment, the mandatory genotype–phenotype linkage remained intact due to the physical connection of the enzyme and its encoding gene through the microbead. Subsequently, the complex of microbead, immobilized gene, and enzyme was re-encapsulated in emulsions together with a substrate linked to a caged biotin moiety (12a, Fig. 13b). After conversion of the substrate in compartments containing active enzyme variants, biotin was uncaged by photoirradiation to enable product (12b) binding to the microbeads. Finally, the emulsion was broken again and microbeads carrying active enzyme variants (and their respective genes) could be recovered by flow cytometry using a fluorescent anti-product antibody. As IVTT conditions are not necessarily compatible with conditions required for enzymatic catalysis (and vice versa), this example beautifully illustrates how microbeads can serve as physical linkers between genotype and phenotype when emulsion compartments are temporarily absent.
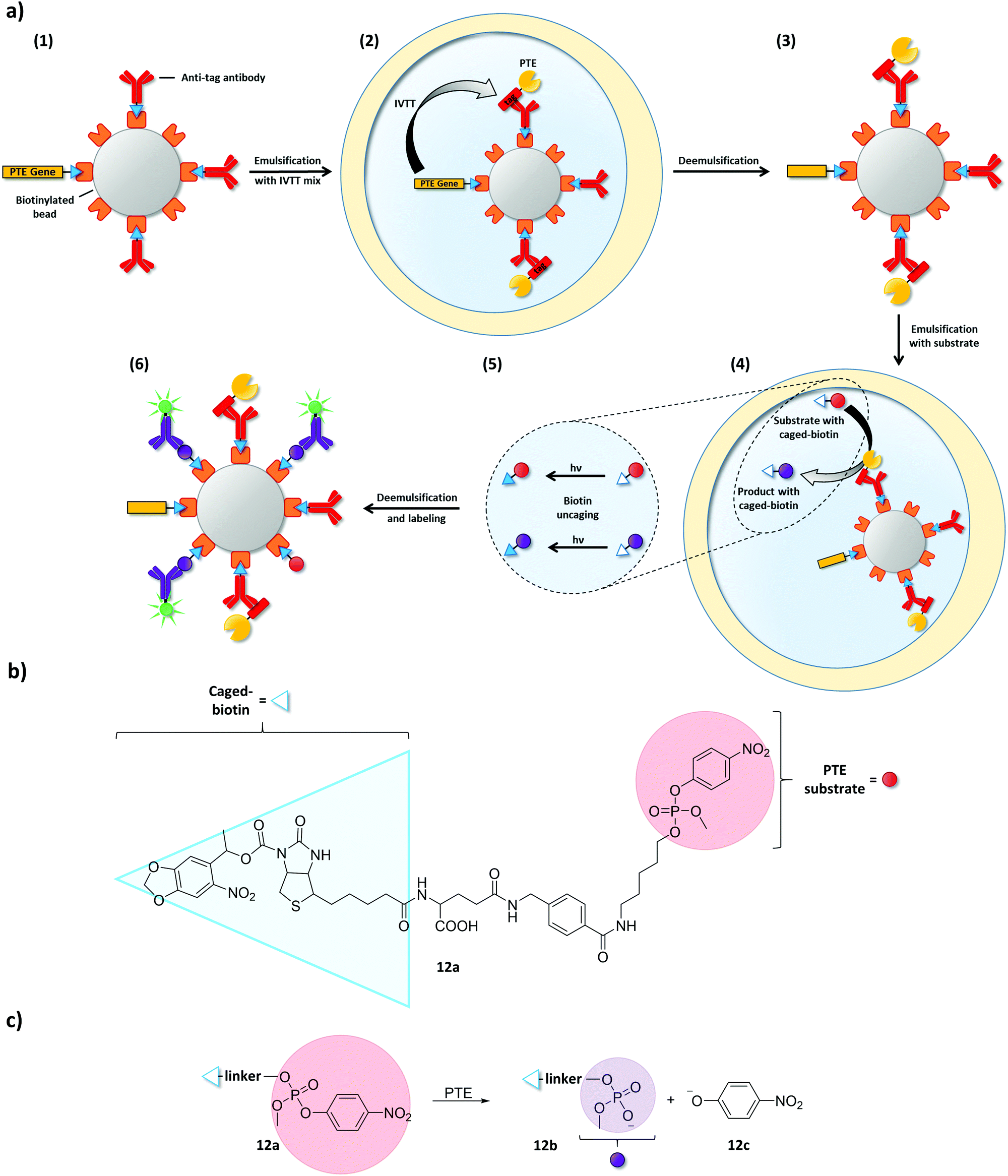 | ||
| Fig. 13 Microbeads maintain the essential genotype–phenotype linkage when emulsions are temporarily broken.97 (a) Streptavidin-coated microbeads were used to immobilize a single copy of a biotinylated phosphotriesterase (PTE) gene and biotinylated antibodies specific for a peptide tag that is genetically fused to the PTE (1). After co-encapsulating the microbeads with an IVTT mixture, PTE expression starts and tagged PTE is captured by the antibodies (2). Thus, PTE gene and PTE remain physically connected via the microbead even when the emulsion is broken to change conditions (3). Microbeads were then co-encapsulated together with PTE substrate equipped with a caged-biotin moiety (4). Product and remaining substrate were uncaged by photoirradiation (5) and captured on the streptavidin-coated microbeads. Product-specific, fluorescently labeled antibodies were used to identify microbeads carrying active PTE variants (6). (b) Structure of the PTE substrate (12a) with a photo-uncagable biotin moiety. (c) PTE catalyzes the cleavage of 12a to product 12b and p-nitrophenolate (12c). | ||
Since this study, the past decade revealed several more examples of uHTS strategies based on microbeads.202,204,205 One striking development of the recent past in microbead-based workflows was the addition of an emulsion PCR step prior to IVTT. Biotinylated primers were linked to microbeads to amplify and immobilize the target gene on the microbead surface. For instance, the Swartz lab followed this method to obtain an oxygen tolerant variant of [FeFe] hydrogenase CpI.205 Their workflow contained three distinct unit operations ((1) emulsion PCR, (2) IVTT and (3) a florescence-generating activity assay), each of which was performed under individual conditions (i.e., requiring the emulsion to be broken and formed again under different conditions). After flow cytometric sorting, genes encoding active CpI variants were recovered in a second PCR amplification step. Recently, a comparable approach was used to evolve HRP.204
In the meantime, in vitro uHTS has also been expanded beyond non-DNA-modifying enzymes without elaborate microbead techniques. For instance, Fallah-Araghi et al. performed emulsion PCR first and subsequently fused the w/o droplets in a microfluidic device with droplets containing an IVTT mixture. Thus, the IVTT mix does not have to endure PCR conditions and emulsion rupture and reformation can be circumvented (i.e., the use of microbeads is avoided).203 Likewise, IVTT and screening reactions have been performed in w/o/w double emulsions for the flow cytometry-based uHTS of hydrolases without the necessity of breaking emulsions during the workflow.92,155 However, this is only possible when IVTT and the screening reaction do not negatively impact each other.
All in all, compartmentalization techniques in w/o or w/o/w emulsions led to the development of cell-independent strategies for directed evolution. These strategies not only allow us to overcome library size limitations posed by transformation steps but eventually enable us to screen these large libraries in dramatically reduced time. Still, most of these techniques cannot qualify yet as being universally applicable. Obviously, well-trained personnel and suitable uHTS technology (e.g., a flow cytometer, encapsulation techniques etc.) are required. Besides these general requirements, special care must be taken when choosing the oil and surfactant for droplet formation. The surfactant could denature the target enzyme and parts of the IVTT machinery. Furthermore, adsorption of entrapped constituents to the oil/water interface might take place.156,206 Based on our experience, we further emphasize that substrate and/or product crosstalk from emulsion compartments is a commonly underestimated problem. Due to publication bias (only successful stories are published), the reader often is confronted with a final (working) system and kept in ignorance of previously failed attempts. In fact, extensive screening for suitable combinations of substrate, oil phase and surfactant can precede the actual enzyme evolution campaign.155,159,181,207–209
4. Emerging concepts
Taking all previous examples together, two major trends can be summarized: (i) emulsions (w/o or w/o/w) have become the major alternative compartment in cases where cells are unsuitable for a given application and (ii) fluorescence-based read-outs are still the gold standard of uHTS assays. In the following chapters, we highlight the few examples where researchers have used alternative methods of (i) compartmentalization or (ii) detection of enzymatic activity in directed evolution campaigns using uHTS. Moreover, an overview is given about technologies that have not yet been used to promote directed evolution but carry the potential to do so in the future.4.1 Beyond emulsions – novel compartments in uHTS
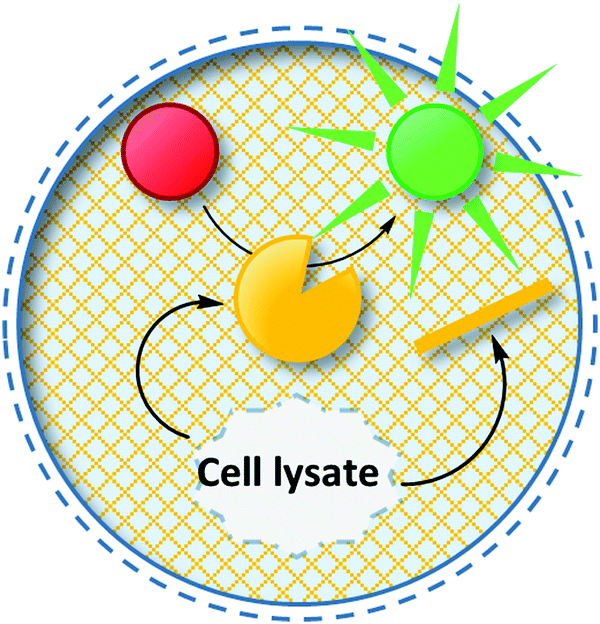 | ||
| Fig. 14 Hydrogel beads as compartments in uHTS. Upon cell lysis inside these hydrogel beads, the enzyme variant and its encoding gene are co-compartmentalized and the genotype–phenotype link is preserved. | ||
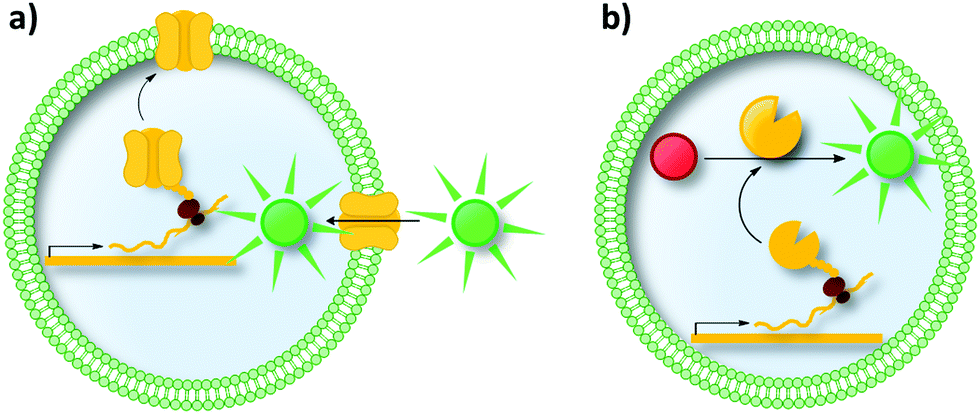 | ||
| Fig. 15 Liposomes as compartments in uHTS. Liposomes are a mimic of the cell's natural membrane as both are formed by a lipid bilayer. (a) Liposomes are promising compartments to study proteins incorporated in/associated with the membrane (e.g., pore-forming proteins, transporters and receptors). (b) Further applications involve enzymatic reactions inside liposomes and the directed evolution of enzymes. IVTT in liposomes has been demonstrated for membrane proteins as well as for soluble enzymes.214–217 | ||
Moreover, liposomes can be used for the directed evolution of enzymes (Fig. 15b). Uyeda et al. reported the directed evolution of LysZ-RS, which is an aminoacyl-tRNA-synthetase variant able to load its cognate tRNA with the non-canonical amino acid N-benzyloxycarbonyl-L-lysine (LysZ).217 The authors followed a strategy reminiscent of GFP-based biosensor systems in cells (compare Section 2.3). They identified LysZ-RS variants with increased LysZ incorporation activity by placing an amber stop codon (encoding LysZ) after the first methionine codon of the GFP gene. Co-encapsulating a single copy of a LysZ-RS variant with copies of the modified GFP gene followed by IVTT of both genes resulted in fluorescence intensity depending on the incorporation efficiency of LysZ. These successes in evolving transporters and enzymes could inspire future endeavors to use liposomes, for instance, for the directed evolution of membrane associated enzymes.
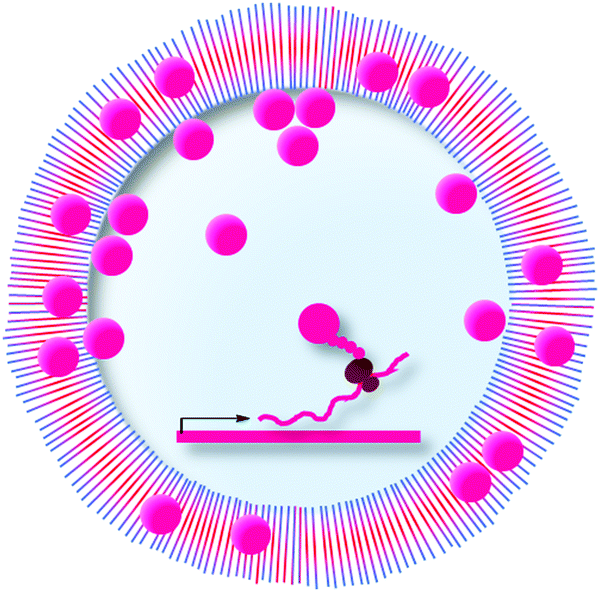 000 microcapillaries per second. However, like in flow cytometric applications, μSCALE relies on fluorescent surrogate substrates.
000 microcapillaries per second. However, like in flow cytometric applications, μSCALE relies on fluorescent surrogate substrates.
 | ||
| Fig. 17 Microcapillary arrays as compartments in uHTS. Microcapillary arrays can serve as compartments for the directed evolution of enzymes. Chemical transformations, fluorescence-based analytics (also kinetics), and the recovery by laser-induced extraction can be performed on each capillary of the array in parallelized fashion. | ||
4.2 Alternative detection principles
With its extraordinary sensitivity and the short time required for a single measurement, fluorescence is an essential detection principle in many uHTS applications. While some enzymatic reactions of interest yield or convert fluorescent substances, the majority does not and thus relies on fluorescent surrogates. In the latter case, screening must be performed with special care to avoid selecting enzyme variants with a bias towards the surrogate substrate, that is, enzyme variants that do not reflect improvements with the actual (industrially relevant) substrate of interest.240 Moreover, in some cases, suitable surrogate substrates do not exist (yet). Hence, there is a desire for alternative screening principles in the field of uHTS. The absorbance-activated droplet sorting approach reported by Gielen et al. was a pleasant exception (compare Section 3.1). It added absorbance – a signal routinely used in assays of lower throughput (primarily MTPs) – to the arsenal of uHTS in cases where NAD(P)H is involved as a cofactor.183 But which alternatives to fluorescence and absorbance are available?Mass spectrometry (MS) is a technique enabling the simultaneous, label-free detection of various analytes with high sensitivity and even the capacity for chemical structure elucidation.241 Recently, MS-based screening platforms are on the rise. Yan et al. utilized DESI-MS (desorption electrospray ionization-mass spectrometry) to screen biocatalytic reactions directly from living colonies on agar plates.242 Ion mobility (IM) spectrometry was installed as intermediate step after DESI and prior to MS (termed DESI-IMMS) to reduce biological background. Cells were grown on a nylon membrane placed on agar plates and gene expression was induced by transferring the membrane to inducing agar plates. The membrane was then soaked with substrate and after incubation analyzed by DESI-IMMS. This direct measurement of bacterial colonies was termed Direct BioTransformation IMMS (DiBT-IMMS). The authors tested their set-up by detecting phenylalanine ammonia lyase (PAL) activity in the asymmetric addition of ammonia to cinnamic acid and its derivatives over time (i.e., repeated DESI-IMMS analysis of the same nylon membrane for several hours). A model library containing PAL (positive control) and empty vector cells (negative control) was subjected to DESI-IMMS-based activity screening. Sequencing results of DNA recovered from positive colonies were in good agreement with DESI-IMMS. In the same study, the authors demonstrated that this set-up can be used to detect hydroxylation of diclofenac by whole cells expressing a cytochrome P450 monooxygenase. Similar high-throughput phenotyping studies of bacterial colonies on agar plates have recently been carried out by Si et al. using MALDI-MS.243 Directed evolution was used to engineer rhamnolipid production via a heterologous two-step pathway in E. coli. In two rounds of evolution a total of ∼6700 colonies were screened for differences in the relative abundance of different rhamnolipids. These examples demonstrate how MS analysis of bacterial colonies on agar plates provides an exciting tool to identify active enzyme variants in a label-free manner.
Keasling et al. recently reported on a process termed PECAN (probing enzymes with click-assisted NIMS).244 NIMS (nanostructure-initiator mass spectrometry) uses laser desorption of perfluoroalkylated analytes on fluorophilic surfaces.245 Substrates can be tagged with a perfluorinated alkyne prior to246,247 or after enzymatic conversion using CuI-catalyzed azide–alkyne cycloaddition (“click chemistry”).244In situ clean-up removes impurities (e.g., from cell lysates) while tagged substrates and products remain associated with the NIMS surface. The authors demonstrated the HTS of a cytochrome P450 BM3 mutant library (combinatorial SSM at two positions close to the active site) for the two step oxidation (hydroxylation and subsequent oxidation toward the ketone) of the sesquiterpene valencene. They used an azide-functionalized derivative of valencene for tagging and subsequent NIMS-based screening. Promising enzyme variants were validated by GC-MS (gas chromatography-mass spectrometry) using untagged valencene. After screening 1208 lysates, several new, highly active enzyme variants were identified, two of which were previously found in a rationally designed minimal library. Automation might help to increase throughput of such MS studies on solid supports in the future.
Droplet microfluidics platforms have proven to be compatible with Matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS)248 and Electrospray ionization-mass spectrometry (ESI-MS),249–252 yet requiring on-chip de-emulsification. Smith et al. successfully sprayed droplets directly into a mass spectrometer. They detected high resolution mass spectra at rates of more than 150 droplets per minute.253 Moreover, high-throughput droplet MS has been used to identify cathepsin B inhibitors from a library of 1280 compounds in a label-free manner.254,255 Samples were re-formatted from eight 384-well MTPs into droplets at rates of 2 samples per s (120 samples per min). In a recent study, carryover between droplets was reduced by using a Teflon needle.256 This modified setting was validated by comparing it to LC-MS (liquid chromatography-mass spectrometry) screening of two transaminase libraries and the authors obtained good correlation (r2 > 0.95). In addition, transaminase activity following IVTT was successfully detected by droplet-MS. Analysis time of a 96-well MTP was reduced from 6 h on the LC-MS device to 120 s (each sample measured in quadruplicates) using the droplet-MS approach. Samples were analyzed at a frequency of 3 Hz. A current limitation of droplet MS is the use of surfactants to stabilize droplets, since surfactants frequently contaminate MS spectra and reduce the ionization efficiency.241
Since more than a decade, capillary electrophoresis (CE) is available for droplet applications, as well.257 Microchip electrophoresis operates at impressive speed (separations in the sub-second range)258 stimulating the interests in this powerful technique for uHTS applications.259 In the last decade, the Kennedy lab made successive progress in connecting the power of CE with droplet microfluidics. Droplet contents were extracted on-chip into a continuous aqueous phase in order to eliminate analysis-interfering influences of the oil phase.260,261 Similar to their droplet MS platform (vide supra), they re-formatted samples located in MTPs into droplets separated by a continuous oil phase.262 In a recent study, they analyzed 1408 samples in 46 min (∼0.5 Hz).263 It is imaginable that further parallelization264 could increase throughput of the platform even more. Unfortunately, the given examples of capillary electrophoresis rely on fluorescent detection, too, and thus require product labeling. Hence, there is a need for label-free detection mechanisms for the capillary electrophoresis of droplets to broaden its scope.
In search for non-invasive techniques, Raman spectroscopy is an interesting measurement principle. As small cross sections impede conventional Raman readout (e.g., in droplets), decoration of gold or silver nanoparticles with Raman-active molecules is necessary and enables surface-enhanced Raman spectroscopy (SERS). For instance, SERS has successfully been used as a detection principle for flow cytometry.265 Instead of conventional fluorescent labeling agents, SERS-active tags were used. Paving the way for uHTS in biological assays, surface-enhanced resonance Raman spectroscopy (SERRS) was used for the characterization of droplets with sub-millisecond resolution.266 Also, cells and their products have been studied by Raman applications in droplets.267,268
Yet another non-invasive method was used by Han et al. They applied electrochemical detection to follow the Michaelis–Menten kinetics of catalase in droplets at time resolutions of 0.05 s.269 Droplets were contacted and measured amperometrically upon flowing past microelectrodes.
Finally, the combination of different detection principles could be used to maximize the information derived from one sample. Chen et al. split droplets and analyzed the daughter droplets off-chip by various principles (fluorescence correlation spectroscopy, MALDI-MS, and fluorescence microscopy). All results where than recombined into one global analysis.270 In a similar fashion, Townsend et al. combined electrochemical and chemiluminescence methods to detect norepinephrine and adenosine triphosphate in parallel.271
Progress in combining alternative detection principles with droplet microfluidics or flow cytometry in general will give uHTS a broader scope of applications. Albeit usually not operating at detection speeds comparable to flow cytometry, these alternative detection principles give us access to information otherwise unobtainable (e.g., mass information or Raman-fingerprints). Many of the groundwork-studies highlighted above have proven to be faster than conventional sample analysis at larger (analytical) scale. Parallelization and automation will gradually increase throughput and thus make these methods attractive for protein engineering laboratories. Ouimet et al. hypothesized that the current dominance of optical plate reader formats has its origin in the strong expertise of assay-developing researchers in chemical biology (in particular the engineering of fluorescence/absorbance changes).259 Commercializing microfluidic devices interfaced with alternative analytical options could make them accessible to a broader non-expert audience.
5. Exploiting uncultivated biodiversity – expanding the scope of uHTS
When the field of metagenomics was introduced by researchers from the US and Europe in the late 1990s, it became quickly clear that this new technology would give access to novel and hitherto unseen microbial biodiversity.272–274 Sequence-based searches were predicted to rapidly find novel genes. However, since a large fraction of metagenome DNA codes for genes and enzymes that have no assigned function and whose sequences are not related to any known protein, mining these sequences (i.e., the metagenomics “dark matter”) using function-based searches can be particularly challenging. In that regard, first publications were focused on technology advancements and the identification of single biocatalysts with superior traits to those known at that time.275–279 Notably, the initial phase of gene discovery from metagenomes was far away from uHTS technologies. It had rather to cope with very basic problems of DNA extraction to establish libraries (depending on the insert size in the form of plasmids, fosmids, cosmids, or bacterial artificial chromosomes – BACs) with sufficient clone numbers to interrogate the available biodiversity. Fosmid and cosmid vectors are commonly used to construct metagenome libraries and they can harbor DNA fragments with sizes up to 40 kb. BAC libraries, in turn, can accommodate inserts up to 200 kb in size, while classical cloning vectors are usually restricted to fragment sizes of 3–5 kb (maximum 10 kb). In parallel, researchers established different types of functionality screens often based on a detection of halo formation on agar plates, color change in cuvettes, TLC based assays, and at the best in 96-well MTPs.280–282Within this framework, successful and remarkable metagenome screening systems have been published very recently. Macdonald et al. used an MTP assay based on the formation of molybdenum blue for the functional screening of a fosmid library for glycoside phosphorylases.283 The colorimetric assay is based on the release of inorganic phosphate upon linking a sugar 1-phosphate (donor) to an acceptor glycan. Phosphate release was then coupled to the production of molybdenum blue.284 The release of phosphate is a common pattern of glycoside phosphorylases and thus can be used to screen for enzymes with different donor/acceptor substrate specificities. In total, 23000 fosmid clones (corresponding to ∼920000 open reading frames) were screened and eight novel glycoside phosphorylases were identified.
New fluorogenic substrates have been designed by Nasseri et al. to screen for glycosidases exhibiting unconventional reaction mechanisms.118 Only enzymes capable of cleaving the S-glycosidic bond present in these substrates would release the fluorophore. These substrates were validated in a test screen of one 96-well MTP (∼200 open reading frames) and are currently used for the functional screening of metagenomic libraries.
In a hallmark study by Coscolín et al., the authors used a functions-based approach that was modified in a way that it allowed the screening of truly large numbers of metagenome clones. Within their study, Coscolín et al. screened a pool of over 500000 fosmid clones containing over 18 Gb of environmental DNA. In their metagenome screen, they applied agar plate-based screening methods using the two amine donors 2-(4-nitrophenyl)ethane-1-amine and o-xylylenediamine hydrochloride. Thereby, they successfully isolated 10 novel transaminases.285 While this study is certainly of high value, it also nicely shows the difficulties these studies usually face, that is, high efforts invested into the screening and rather low numbers of active clones identified.
One strategy to cope with the low hit rate that is associated with metagenome screens employs genetic selection systems that are in fact very sophisticated uHTS systems per se. These strategies target a genetic trait to sort through hundreds of thousands of clones in a very short time. In most cases, a microbial selection strain is used that is either auxotrophic (i) or that has been engineered to produce a toxic protein causing cell death in absence of a functional target within the metagenome library (ii).
(i) The first type of selection aims for a gain of function: the host strain that carries the metagenome clone lacks an essential function and can only grow if the respective metagenome clone provides that function. For instance, this strategy was applied in the search for novel β-galactosidases that were not predictable from metagenome gene sequences286 and for genes involved in the synthesis of polyhydroxyalkanoate.287 Others included the identification of vitamin biosynthesis genes288 located in small operons. Moreover, larger biosynthetic gene clusters linked to the synthesis of natural products have also been isolated using this strategy: metagenome libraries were searched in the background of E. coli and Streptomyces sp. hosts harboring a 4′-phosphopantetheinyl transferase.289,290 Clearly, this approach is highly sophisticated as it quickly delivers a relatively large number of positive clones.
(ii) The second selection strategy aims at a loss of function: in this case, the cell can only grow when a lethal gene product or related system is inactivated as it would otherwise be toxic. In two independent studies by Weiland-Bräuer et al. and by Rasmussen et al., a gene encoding a toxic protein was expressed by using so-called bacterial autoinducer systems.291,292 Bacterial autoinducers are small molecules produced by bacteria to sense cell density and they are relevant for infection. This process is called quorum sensing (QS). The QS molecules and their synthesis are targets for the development of novel drugs. In the study by Weiland-Bräuer et al., the ccdB gene was employed, which encodes a protein that is toxic for E. coli. This gene is part of the toxin-anti-toxin locus ccdA–ccdB. In the absence of CcdA, CcdB inactivates the host DNA gyrase which results in cell death.293 The authors fused the ccdB gene to QS-controlled promoters of Vibrio fischeri (luxI) or E. coli (lsrA) to identify metagenome clones carrying genes coding for quorum quenching (QQ) activities. Based on these reporter systems, the authors identified in a follow-up study 142 out of 46400 metagenomic clones interfering with acyl-homoserine lactones (AHLs), and 13 clones interfering with autoinducer-2 (AI-2)-like molecules.294
Rasmussen et al. and colleagues developed two distinct systems: in the first system, a phospholipase encoding gene (phlA) from Serratia marcescens was cloned into a suitable vector. PhlA is also part of a toxin–anti-toxin system and, if produced in absence of its anti-toxin (PhlB), is lethal to E. coli cells.295 In turn, if the respective antidote protein (PhlB) is present, the cell survives. Thus, this system offered a smart way to select clones of interest from a large pool of metagenome clones with a very low rate of false-positives. In the same publication, the second system employed the levansucrase (sacB) gene under control of a promoter that was activated in the presence of autoinducer molecules. Similar to the above-mentioned strategy, cells could only survive if a QQ-active metagenome fragment was coexpressed in the same cell. Since E. coli does not produce the respective autoinducer molecules, interferences from the host, and thus false-positive clones, were excluded. Altogether, these studies demonstrated that this technique is a powerful tool to identify novel enzymes and gene products from hitherto uncultivated microorganisms. While these are perhaps all examples of early selection-based HTS that do not meet the very strict definition of uHTS in senso stricto, they were superior to classical MTP – or even single colony-based screening systems, as very low numbers of false positives were observed and truly large numbers of clones could be tested.
With respect to the development of uHTS systems for metagenome screening, a Japanese research team has reported on a more advanced system entitled the substrate-induced gene-expression screening (SIGEX).296–298 SIGEX is based on the identification of fluorescent reporter genes on single-cell level. The metagenome fragments are ligated into an operon trap vector that allows the substrate-inducible expression of a fluorescent reporter gene, ultimately affording cell sorting using flow cytometry. The technology has successfully been applied for the search of metagenome-derived DNA fragments comprising genes involved in aromatic compound degradation.299,300 SIGEX, however, was also one of the first reported systems that faced a general problem with fluorescence-based screening in metagenomics. Since fluorescence-based detection is relatively sensitive compared to genetic selection or classical colorimetric screens, very often false positives were obtained. This is in part linked to the presence of corresponding endogenous genes in the host strain used for screening. Moreover, this challenge is also owed to the promiscuity of enzymes acting on different substrates and, thereby, producing weak signals that ultimately could be sorted and identified as candidate enzymes.301 This often makes it necessary to implement a second and third round of time-consuming re-screening.
Interestingly, only a handful of studies have described the use of droplet and flow cytometry-based sorting of metagenome libraries. Hosokawa et al. have reported one of the first studies in which a droplet-based metagenome screening was established. In their study, the authors used gel microdroplets dispersed in oil (forming a picoliter-volume reaction space) and searched in soil-derived metagenome libraries for the presence of lipolytic enzymes. They were able to sort ∼67000 clones, each containing large inserts (>36 kb), within 24 hours. As substrate, fluorescein dicaprylate was used. By cleaving the ester bond, a fluorescein dye was released, enabling the detection of active droplets. In total 69 putatively positive droplets were identified and in part verified by sequencing and massive parallel screening on agar plates. Notably, the assay was based on living and dividing cells within the droplets.302 While the study leaves open some questions with respect to background activities, it is still a groundbreaking study in the field of uHTS of metagenomes. It is notable, that the number of metagenome clones screened in this study is rather low compared to those postulated for evolutionary screenings using the same technique. In such screens, >106 h−1 clones can be screened (see Section 1).90–92,166 While the abovementioned study by Hosokawa et al. was published in 2015, Ferrer et al. and Schaerli et al. had outlined the blueprints of it in earlier reviews from 2009.303,304
In a similar study published in 2015, Colin et al. established a metagenome library comprising 1.2 Mio clones that were derived from different origins (e.g., soil, degraded plant material, and cow rumen).305 They employed fluorogenic substrates as sulfate monoesters or phosphate triesters for screening and sorted the lysed cells (i.e., in picoliter droplets) in two rounds of flow cytometric sorting. They were able to sort 20 Mio. droplets covering the library approximately 15-fold. In total 500 and 300 putatively positive droplets were identified carrying arylsulfatase and phosphotriesterase activity, respectively. In a subsequent plate screening, these activities were verified in 10% of the sorted droplets, and sequencing of the obtained clones finally verified six unique sulfatase and eight unique phosphotriesterase sequences. This relatively low hit rate emphasizes the challenges associated with high background signal to noise ratio, while also indicating redundancy within the libraries. The latter is a major problem that occurs when E. coli clone libraries are subjected to liquid cultures as opposed to agar plates. Apparently, under these conditions certain clones overgrow others and thus the overall diversity is rapidly lost. Notably, despite the larger number of clones screened in this study, the overall amount of analyzed coding information was similar to the study by Hosokawa et al.302
Despite the obvious technical challenges that these early uHTS studies faced, the studies by Colin and Hosokawa were both important landmark studies, as they were the first to report the use of uHTS screening in the field of metagenomics. In this regard, both studies have convincingly demonstrated that uHTS screenings will deliver access to yet unknown biodiversity. Given the overall relatively low number of novel sequences detected, they are currently not superior to classical metagenome screenings and, in particular, genetic selection systems. However, given the rapid technology development in this field, it is likely that uHTS will overcome these bottlenecks soon and deliver novel functions and biodiversity.
With respect to screening of metagenome libraries using droplet-based technologies, an interesting concept study was recently published aiming for the identification of antimicrobials.306 The authors showed that the technology was suitable to identify gene clusters encoding the biosynthetic pathways of small compounds such as violacein in metagenome libraries. While this certainly represents a very interesting approach, the difficulties may lie in the identification and expression of larger secondary metabolite gene clusters using the droplet-based approach. Moreover, the authors had to deal with drawbacks associated with the use of mammalian cells to assay the effects of the natural compounds. Ultimately, the droplets had to be isolated again from the mammalian cell cultures.
In summary, the main challenge that all the above-mentioned approaches faced were high numbers of false positives due to high background activities and promiscuous enzyme activities interfering with the assay. As long as whole cells are screened, problems associated with different cell growth and expression will persist. Further, depending on the insert size and the type of sequence, the host may not recognize the authentic promoters of heterologous genes or might lack the specific chaperones and cofactors required to correctly fold the target proteins. Moreover, we can expect that different codon usage will hamper the expression at a very early level. For more detailed information on these challenges, we point the reader to references in recent reviews on metagenome screenings.280
Cell-free systems (compare IVTT in Section 3.2) might be the key to master the above-mentioned challenges (Fig. 18). These systems must contain chaperones and extra tRNAs to overcome challenges associated with gene expression, codon usage and protein folding. In addition, cell-free systems must address the hurdle of poor promoter recognition. First attempts to develop cell-free screening systems for metagenomics have been published recently.307 In this study, a complex RNA polymerase from Geobacillus sp. was used for the generation of metagenome-derived mRNA and subsequently translated using an in vitro translation cocktail based on E. coli cell extract. Yet, it remains to be shown that in the context of metagenome libraries in vitro transcribed mRNA and ultrahigh-throughput translation provides sufficient amounts of enzyme to generate the required signal intensities for uHTS.
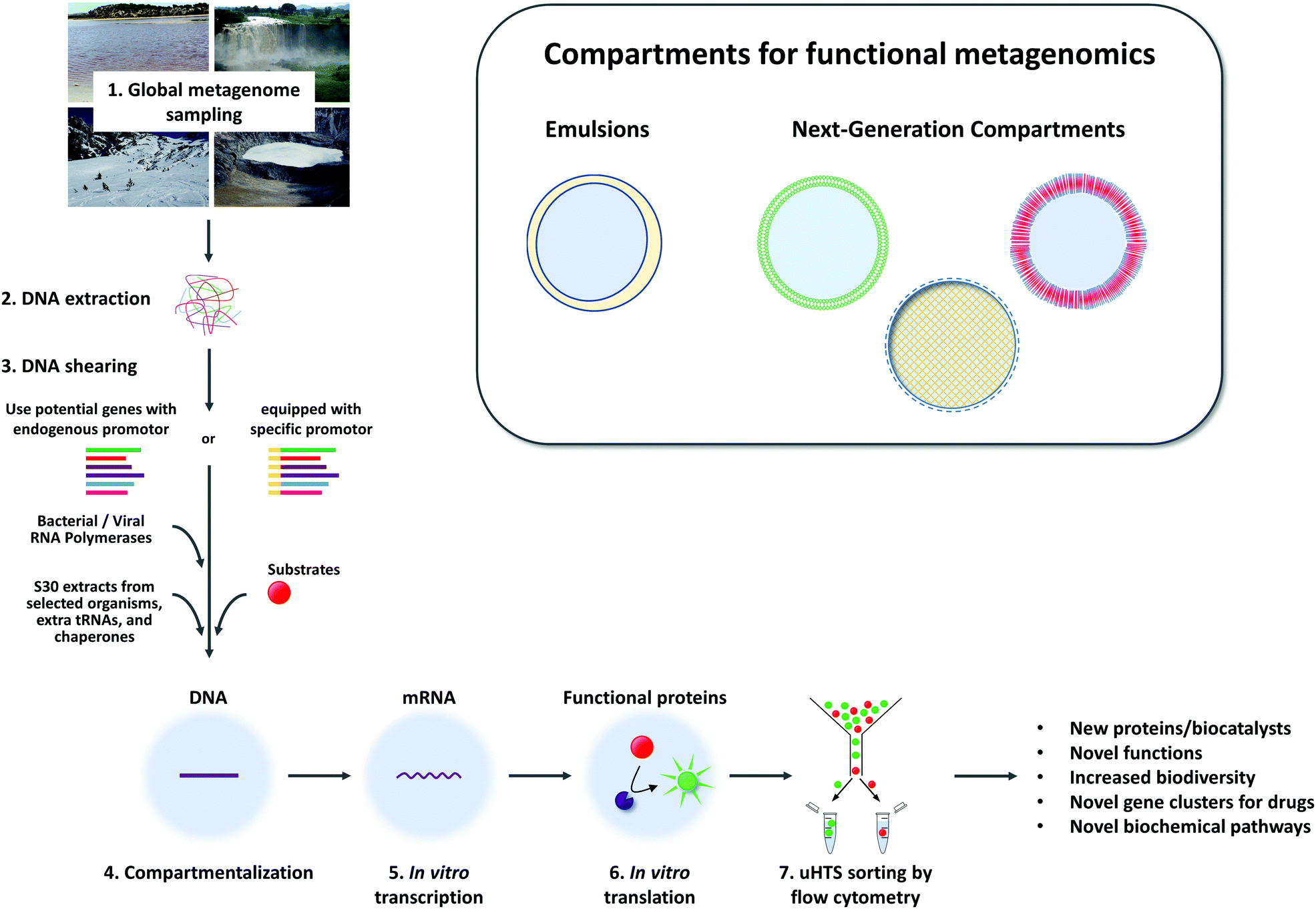 | ||
| Fig. 18 Outline of next-generation uHTS functional screening in metagenomes. Seven major steps are necessary: (1) environmental samples are collected and (2) DNA is extracted. (3) DNA is ligated to generic promoter/primer sequences. (4) All necessary compounds for the following steps are co-compartmentalized (here: only DNA is shown for clarity). (5) Nucleic acids are transcribed in vitro to mRNA from the ligated generic promoters or from native promoters. (6) mRNAs are translated using S30 ribosomal extracts and the substrate is converted to a fluorescent reporter molecule depending on the activity of the translated sample. (7) Compartments are sorted on a flow cytometer to enrich the active population and identify their respective genes. | ||
Yu et al. recently outlined another promising strategy. They developed an approach designated “mini metagenomics” using droplet-based cell sorting. The approach employs droplet microfluidics to separate an environmental sample into many small sub-samples containing 5–10 cells. Thereby, the complexity was reduced and deep sequencing of the obtained mini metagenomes allowed rapid assembly and novel lineages.308
A similar approach was recently reported for single-cell screening.309 In this study, single cells were packed in droplets, sorted, and then analyzed by next-generation sequencing and LC-MS. This resulted in the identification of valuable information on the phenotypes at high frequency and enabled the detection of bacteria producing antimicrobials to suppress growth of the major pathogen Staphylococcus aureus.
6. Conclusion
Random mutagenesis still is the diversity generation method of choice when insufficient knowledge about the enzyme is available. Moreover, even if detailed structural knowledge is at hand and in silico studies readily predict target positions for mutagenesis, it has in many cases proven highly beneficial to perform simultaneous multiple site-saturations at grouped positions to take advantage of cooperative effects brought about by proximal amino acid substitutions. The number of possible variants in such libraries can readily exceed the sampling sizes achievable by MTP- or agar plate screens, that is, the diversity we are nowadays able to generate is not the limiting factor any more. Certainly, speeding up directed evolution (e.g., from 1–1.5 years to few weeks or months) would be highly desirable from an industrial point of view, too. Hence, we need the methodologies to explore the vastness of the given sequence space.uHTS assays can deal with sample sizes at least two orders of magnitude beyond MTP- or agar plate-based assays owing to their superior speed of analysis. Moreover, sample volumes and thus reagent costs per sample are reduced. However, as we have demonstrated in the previous sections, uHTS assay development differs significantly from traditional MTP-based assays as issues like compartmentalization to maintain a genotype–phenotype linkage emerge with the employment of sorting devices instead of MTPs. These issues have inspired a plethora of creative solutions, which hold great potential for adaptation to similar enzymes and challenges, but still cannot qualify as being universally applicable.
The cell, nature's compartment for evolution, still is the most employed compartment in uHTS, but requires inspired solutions for the entrapment or surface tethering of detectable products, the formation of a fluorescent hydrogel, or the utilization of regulatory networks within the cell to elicit a biosensor signal. Limitations like transformation efficiencies, cellular backgrounds interfering with the assay, maintenance of the genotype–phenotype linkage or cross-talk between cells were circumvented in several cases by using w/o or w/o/w emulsions as surrounding compartments.
By adding the emulsion interface as an additional compartment barrier, we can overcome dependencies on the cell as a compartmentalization system but still employ the advantages offered by it (e.g., ease of protein production). Microfluidics provides us with the means to generate monodisperse emulsions and has become vital in making them a true alternative and in many cases a superior, more flexible compartment compared to cells. Undoubtedly, however, they are not universally applicable and cannot offer solutions to all challenges in uHTS attempts. Limitations like instabilities of enzymes within emulsion environments and substrate/product leakage or their adsorption at oil/water interphases have to be addressed. In this context, next-generation compartments like hydrogel beads or polymersomes have emerged as valuable alternatives. Yet, the application of artificial compartments still is highly customized and has not yet spread beyond the laboratories that introduced them. Expert groups mainly utilize highly sophisticated and customized microfluidic devices, which are mostly inaccessible to researchers lacking experience in the field. Double emulsions can be sorted by common flow cytometers, but the generation of monodisperse droplets is often a prerequisite for uHTS and cannot be achieved without some sort of microfluidic device. Therefore, cells still are the preferred compartment in most research groups and uHTS assay design still is adapted to the available equipment within individual laboratories. Inspiring, pioneer technological developments like microarrays can even be used to follow product formation over time (e.g., μSCALE), but are afflicted with the same limitations as microfluidics in terms of customization and unavailability for most researchers outside the inventor's lab. Commercialization of microfluidic unit operations (e.g., monodisperse droplet formation, droplet fusion and mixing) and microcapillary arrays will help us to move beyond the current state, where our laboratory (un-)experience and equipment predetermines the molecular principles and strategies we can apply.
Quo vadis, protein engineering? uHTS can be a powerful adjunct to directed enzyme evolution campaigns not only affording automation and time reduction, but also to explore different evolutionary trajectories at once. Yet, the almost strict dependency on surrogate substrates to achieve fluorescent readouts narrows its current application areas significantly and makes it unattractive for industrial implementation. Thus, the first uHTS assay employing absorbance-activated droplet sorting183 can be considered as a seminal achievement towards more generally applicable uHTS platforms. Attempts to implement methodologies such as mass spectrometry in uHTS are eagerly awaited to lose the dependencies on surrogate substrates. uHTS assays employing surrogate substrates must always be accompanied by a rescreening assay (e.g., in MTP format), in which the preselected population is subjected to the actual substrate of interest. In that case, uHTS assays serve as high-tech prescreening methods to enrich the fractions of active enzymes within a population rather than the one and only screening method. Effects like cell-to-cell variation within a population or the challenge to quantify activity among droplets beyond simple yes/no-type answers further corroborate this notion. Nevertheless, exceptions to this rule exist but are mainly reserved for cases in which the conversion of native substrates can be directly coupled to a fluorescence readout, or where the starting activity of a target biocatalyst is low. Given that most engineered enzymes are ultimately designed for industrial application, future research on the topic of uHTS will have to go the stony road towards universal applicability to eventually transition from academic case studies to “custom engineering” as an affordable endeavor.
In metagenomics, functions-based uHTS represents an enabling technology that will help to overcome the low hit rate associated with metagenome libraries (in some cases <10 ppm).305 However, challenges associated with a high rate of false-positives due to endogenous enzyme activities or substrate promiscuity still make function-based screens (in particular, fluorescence-based ones as required in flow cytometry) a challenge in metagenomics research. The use of whole cells for metagenome library screening introduces the same challenges as in the case of directed enzyme evolution regarding cell-to-cell-variation. Moreover, the host might not recognize authentic promoters from the metagenome. These challenges could be overcome by establishing cell-free metagenomics, which is, however, still in the conceptual phase.
Overcoming these challenges will achieve two main advancements. Firstly, the more frequent use of uHTS will give faster access to the majority of the non-cultivable biodiversity for biotechnological and medical applications. Thereby, it will not only enlarge our portfolio of biomolecules and their diversity, but also unravel novel types of reactions. Secondly, by enlarging the known functional biodiversity, uHTS will boost the assignment of functions to many of the metagenomics “dark matter” (i.e., the proteins whose structure and function cannot be predicted based on encoding sequences). Furthermore, speeding up the discovery of biomolecules from metagenomes will be an important factor with respect to the development of novel products. All in all, the advancement of uHTS will help us to grasp the vastness of enzyme function landscapes to ultimately guide knowledge-driven enzyme discovery and engineering.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG) through the International Research Training Group “Selectivity in Chemo- and Biocatalysis” (SeleCa), the Bundesministerium für Bildung und Forschung (BMBF) (FKZ 031B0297, FKZ 031A571A, FKZ 031A565A, and FKZ 031B0562B), and the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No. 722287.Notes and references
- F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 14420–14426 CrossRef CAS
.
- G. P. Smith, Angew. Chem., Int. Ed., 2019, 58, 14428–14437 CrossRef CAS PubMed
- G. Winter, Angew. Chem., Int. Ed., 2019, 58, 14438–14445 CrossRef CAS PubMed
- Angew. Chem., Int. Ed., 2018, 57, 14683 Search PubMed.
- R. Fasan, S. B. Jennifer Kan and H. Zhao, ACS Catal., 2019, 9, 9775–9788 CrossRef CAS
- D. R. Mills, R. L. Peterson and S. Spiegelman, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 217–224 CrossRef CAS PubMed
- G. F. Joyce, Angew. Chem., Int. Ed., 2007, 46, 6420–6436 CrossRef CAS PubMed
- R. Saiki, D. Gelfand, S. Stoffel, S. Scharf, R. Higuchi, G. Horn, K. Mullis and H. Erlich, Science, 1988, 239, 487–491 CrossRef CAS PubMed
- R. Saiki, S. Scharf, F. Faloona, K. Mullis, G. Horn, H. Erlich and N. Arnheim, Science, 1985, 230, 1350–1354 CrossRef CAS PubMed
- K. R. Tindall and T. A. Kunkel, Biochemistry, 1988, 27, 6008–6013 CrossRef CAS PubMed
- R. C. Cadwell and G. F. Joyce, Genome Res., 1994, 3, S136–S140 CrossRef CAS PubMed
- A. Beaudry and G. Joyce, Science, 1992, 257, 635–641 CrossRef CAS PubMed
- W. P. C. Stemmer, Nature, 1994, 370, 389–391 CrossRef CAS PubMed
- K. Chen and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5618–5622 CrossRef CAS PubMed
- H. Zhao, L. Giver, Z. Shao, J. A. Affholter and F. H. Arnold, Nat. Biotechnol., 1998, 16, 258–261 CrossRef CAS PubMed
- W. P. C. Stemmer, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 10747–10751 CrossRef CAS PubMed
- I. V. Pavlidis, M. S. Weiß, M. Genz, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. T. Bornscheuer, Nat. Chem., 2016, 8, 1076–1082 CrossRef CAS PubMed
- M. Voss, D. Das, M. Genz, A. Kumar, N. Kulkarni, J. Kustosz, P. Kumar, U. T. Bornscheuer and M. Höhne, ACS Catal., 2018, 8, 11524–11533 CrossRef CAS
- M. H. Barley, N. J. Turner and R. Goodacre, ChemBioChem, 2017, 18, 1087–1097 CrossRef CAS PubMed
- I. Mateljak, E. Monza, M. F. Lucas, V. Guallar, O. Aleksejeva, R. Ludwig, D. Leech, S. Shleev and M. Alcalde, ACS Catal., 2019, 9, 4561–4572 CrossRef CAS
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863–10869 CrossRef CAS PubMed
- Z. Sun, Q. Liu, G. Qu, Y. Feng and M. T. Reetz, Chem. Rev., 2019, 119, 1626–1665 CrossRef CAS PubMed
- B. Seelig and J. W. Szostak, Nature, 2007, 448, 828–831 CrossRef CAS PubMed
- L. Jiang, E. A. Althoff, F. R. Clemente, L. Doyle, D. Röthlisberger, A. Zanghellini, J. L. Gallaher, J. L. Betker, F. Tanaka, C. F. Barbas, D. Hilvert, K. N. Houk, B. L. Stoddard and D. Baker, Science, 2008, 319, 1387–1391 CrossRef CAS PubMed
- D. Röthlisberger, O. Khersonsky, A. M. Wollacott, L. Jiang, J. DeChancie, J. Betker, J. L. Gallaher, E. A. Althoff, A. Zanghellini, O. Dym, S. Albeck, K. N. Houk, D. S. Tawfik and D. Baker, Nature, 2008, 453, 190–195 CrossRef PubMed
- H. Kries, R. Blomberg and D. Hilvert, Curr. Opin. Chem. Biol., 2013, 17, 221–228 CrossRef CAS PubMed
- G. Kiss, N. Çelebi-Ölçüm, R. Moretti, D. Baker and K. N. Houk, Angew. Chem., Int. Ed., 2013, 52, 5700–5725 CrossRef CAS PubMed
- P.-S. Huang, S. E. Boyken and D. Baker, Nature, 2016, 537, 320–327 CrossRef CAS PubMed
- S. Studer, D. A. Hansen, Z. L. Pianowski, P. R. E. Mittl, A. Debon, S. L. Guffy, B. S. Der, B. Kuhlman and D. Hilvert, Science, 2018, 362, 1285–1288 CrossRef CAS PubMed
- H. A. Bunzel, H. Kries, L. Marchetti, C. Zeymer, P. R. E. Mittl, A. J. Mulholland and D. Hilvert, J. Am. Chem. Soc., 2019, 141, 11745–11748 CrossRef CAS PubMed
- D. Baker, Protein Sci., 2019, 28, 678–683 CrossRef CAS PubMed
- M. T. Reetz, M. Bocola, J. D. Carballeira, D. Zha and A. Vogel, Angew. Chem., Int. Ed., 2005, 44, 4192–4196 CrossRef CAS PubMed
- M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed
- A. Nobili, M. G. Gall, I. V. Pavlidis, M. L. Thompson, M. Schmidt and U. T. Bornscheuer, FEBS J., 2013, 280, 3084–3093 CrossRef CAS PubMed
- J. Bendl, J. Stourac, E. Sebestova, O. Vavra, M. Musil, J. Brezovsky and J. Damborsky, Nucleic Acids Res., 2016, 44, W479–W487 CrossRef CAS PubMed
- G. Li, M. Garcia-Borràs, M. J. L. J. Fürst, A. Ilie, M. W. Fraaije, K. N. Houk and M. T. Reetz, J. Am. Chem. Soc., 2018, 140, 10464–10472 CrossRef CAS PubMed
- J. B. Rannes, A. Ioannou, S. C. Willies, G. Grogan, C. Behrens, S. L. Flitsch and N. J. Turner, J. Am. Chem. Soc., 2011, 133, 8436–8439 CrossRef CAS PubMed
- A. Currin, J. Kwok, J. C. Sadler, E. L. Bell, N. Swainston, M. Ababi, P. Day, N. J. Turner and D. B. Kell, ACS Synth. Biol., 2019, 8, 1371–1378 CrossRef CAS PubMed
- S. Kille, F. E. Zilly, J. P. Acevedo and M. T. Reetz, Nat. Chem., 2011, 3, 738–743 CrossRef CAS PubMed
- A. Li, G. Qu, Z. Sun and M. T. Reetz, ACS Catal., 2019, 9, 7769–7778 CrossRef CAS
- Z. Wu, S. B. J. Kan, R. D. Lewis, B. J. Wittmann and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8852–8858 CrossRef CAS PubMed
- G. Li, Y. Dong and M. T. Reetz, Adv. Synth. Catal., 2019, 361, 2377–2386 CAS
- K. K. Yang, Z. Wu and F. H. Arnold, Nat. Methods, 2019, 16, 687–694 CrossRef CAS PubMed
- P. Dydio, H. M. Key, A. Nazarenko, J. Y.-E. Rha, V. Seyedkazemi, D. S. Clark and J. F. Hartwig, Science, 2016, 354, 102–106 CrossRef CAS PubMed
- P. Dydio, H. M. Key, H. Hayashi, D. S. Clark and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 1750–1753 CrossRef CAS PubMed
- O. F. Brandenberg, R. Fasan and F. H. Arnold, Curr. Opin. Biotechnol., 2017, 47, 102–111 CrossRef CAS PubMed
- F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed
- K. Chen, X. Huang, S. B. J. Kan, R. K. Zhang and F. H. Arnold, Science, 2018, 360, 71–75 CrossRef CAS PubMed
- C. Zeymer and D. Hilvert, Annu. Rev. Biochem., 2018, 87, 131–157 CrossRef CAS PubMed
- Y. Wei, E. L. Ang and H. Zhao, Curr. Opin. Chem. Biol., 2018, 43, 1–7 CrossRef CAS PubMed
- L. Villarino, K. E. Splan, E. Reddem, L. Alonso-Cotchico, C. Gutiérrez de Souza, A. Lledós, J.-D. Maréchal, A.-M. W. H. Thunnissen and G. Roelfes, Angew. Chem., Int. Ed., 2018, 57, 7785–7789 CrossRef PubMed
- R. K. Zhang, X. Huang and F. H. Arnold, Curr. Opin. Chem. Biol., 2019, 49, 67–75 CrossRef CAS PubMed
- R. K. Zhang, K. Chen, X. Huang, L. Wohlschlager, H. Renata and F. H. Arnold, Nature, 2019, 565, 67–72 CrossRef CAS PubMed
- I. Cho, Z.-J. Jia and F. H. Arnold, Science, 2019, 364, 575–578 CrossRef CAS PubMed
- D. A. Vargas, R. L. Khade, Y. Zhang and R. Fasan, Angew. Chem., Int. Ed., 2019, 58, 10148–10152 CrossRef CAS PubMed
- A. L. Chandgude, X. Ren and R. Fasan, J. Am. Chem. Soc., 2019, 141, 9145–9150 CrossRef PubMed
- S. N. Natoli and J. F. Hartwig, Acc. Chem. Res., 2019, 52, 326–335 CrossRef CAS PubMed
- Y. Gu, S. N. Natoli, Z. Liu, D. S. Clark and J. F. Hartwig, Angew. Chem., Int. Ed., 2019, 58, 13954–13960 CrossRef CAS PubMed
- J. C. Lewis, ACS Catal., 2013, 3, 2954–2975 CrossRef CAS
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed
- U. Markel, D. F. Sauer, J. Schiffels, J. Okuda and U. Schwaneberg, Angew. Chem., Int. Ed., 2019, 58, 4454–4464 CrossRef CAS PubMed
- Y. Cen, W. Singh, M. Arkin, T. S. Moody, M. Huang, J. Zhou, Q. Wu and M. T. Reetz, Nat. Commun., 2019, 10, 3198 CrossRef PubMed
- J. C. Lewis, Acc. Chem. Res., 2019, 52, 576–584 CrossRef CAS PubMed
- K. Oohora, A. Onoda and T. Hayashi, Acc. Chem. Res., 2019, 52, 945–954 CrossRef CAS PubMed
- M. T. Reetz, Acc. Chem. Res., 2019, 52, 336–344 CrossRef CAS PubMed
- A. D. Liang, J. Serrano-Plana, R. L. Peterson and T. R. Ward, Acc. Chem. Res., 2019, 52, 585–595 CrossRef CAS PubMed
- I. Drienovska, L. Alonso-Cotchico, P. Vidossich, A. Lledos, J.-D. Marechal and G. Roelfes, Chem. Sci., 2017, 8, 7228–7235 RSC
- I. Drienovská, C. Mayer, C. Dulson and G. Roelfes, Nat. Chem., 2018, 10, 946–952 CrossRef PubMed
- T. Hayashi, M. Tinzl, T. Mori, U. Krengel, J. Proppe, J. Soetbeer, D. Klose, G. Jeschke, M. Reiher and D. Hilvert, Nat. Catal., 2018, 1, 578–584 CrossRef CAS
- M. Pott, T. Hayashi, T. Mori, P. R. E. Mittl, A. P. Green and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 1535–1543 CrossRef CAS PubMed
- T. Hayashi, D. Hilvert and A. P. Green, Chem. – Eur. J., 2018, 24, 11821–11830 CrossRef CAS PubMed
- G. Roelfes, Acc. Chem. Res., 2019, 52, 545–556 CrossRef CAS PubMed
- Y. Yu, X. Liu and J. Wang, Acc. Chem. Res., 2019, 52, 557–565 CrossRef CAS PubMed
- A. J. Burke, S. L. Lovelock, A. Frese, R. Crawshaw, M. Ortmayer, M. Dunstan, C. Levy and A. P. Green, Nature, 2019, 570, 219–223 CrossRef CAS PubMed
- C. Mayer, C. Dulson, E. Reddem, A.-M. W. H. Thunnissen and G. Roelfes, Angew. Chem., Int. Ed., 2019, 58, 2083–2087 CrossRef CAS PubMed
- K. J. Koebke and V. L. Pecoraro, Acc. Chem. Res., 2019, 52, 1160–1167 CAS
- E. N. Mirts, A. Bhagi-Damodaran and Y. Lu, Acc. Chem. Res., 2019, 52, 935–944 CrossRef CAS PubMed
- S. B. J. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048–1051 CrossRef CAS PubMed
- R. L. Khade, A. L. Chandgude, R. Fasan and Y. Zhang, ChemCatChem, 2019, 11, 3101–3108 CrossRef CAS PubMed
- S. B. J. Kan, X. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132 CrossRef CAS PubMed
- A. Galán, L. Comor, A. Horvatic, J. Kules, N. Guillemin, V. Mrljak and M. Bhide, Mol. BioSyst., 2016, 12, 2342–2358 RSC
- H. Xiao, Z. Bao and H. Zhao, Ind. Eng. Chem. Res., 2015, 54, 4011–4020 CrossRef CAS PubMed
- K. L. Tee and T. S. Wong, Biotechnol. Adv., 2013, 31, 1707–1721 CrossRef CAS PubMed
- H. Leemhuis, R. M. Kelly and L. Dijkhuizen, IUBMB Life, 2009, 61, 222–228 CrossRef CAS PubMed
- T. K. Hyster and F. H. Arnold, Isr. J. Chem., 2015, 55, 14–20 CrossRef CAS
- D. M. Weinreich, N. F. Delaney, M. A. DePristo and D. L. Hartl, Science, 2006, 312, 111–114 CrossRef CAS PubMed
- A. Debon, M. Pott, R. Obexer, A. P. Green, L. Friedrich, A. D. Griffiths and D. Hilvert, Nat. Catal., 2019, 2, 740–747 CrossRef CAS
- N. J. Turner, Nat. Chem. Biol., 2009, 5, 567–573 CrossRef CAS PubMed
- M. D. Truppo, ACS Med. Chem. Lett., 2017, 8, 476–480 CrossRef CAS PubMed
- P. Mair, F. Gielen and F. Hollfelder, Curr. Opin. Chem. Biol., 2017, 37, 137–144 CrossRef CAS PubMed
- S. Becker, H.-U. Schmoldt, T. M. Adams, S. Wilhelm and H. Kolmar, Curr. Opin. Biotechnol., 2004, 15, 323–329 CrossRef CAS PubMed
- G. Körfer, C. Pitzler, L. Vojcic, R. Martinez and U. Schwaneberg, Sci. Rep., 2016, 6, 26128 CrossRef PubMed
- H. Leemhuis, V. Stein, A. D. Griffiths and F. Hollfelder, Curr. Opin. Struct. Biol., 2005, 15, 472–478 CrossRef CAS PubMed
- A. Aharoni, K. Thieme, C. P. C. Chiu, S. Buchini, L. L. Lairson, H. Chen, N. C. J. Strynadka, W. W. Wakarchuk and S. G. Withers, Nat. Methods, 2006, 3, 609–614 CrossRef CAS PubMed
- A. J. Ruff, A. Dennig, G. Wirtz, M. Blanusa and U. Schwaneberg, ACS Catal., 2012, 2, 2724–2728 CrossRef CAS
- J. C. Sadler, A. Currin and D. B. Kell, Analyst, 2018, 143, 4747–4755 RSC
- A. D. Griffiths and D. S. Tawfik, EMBO J., 2003, 22, 24–35 CrossRef CAS PubMed
- B. Kintses, C. Hein, M. F. Mohamed, M. Fischlechner, F. Courtois, C. Lainé and F. Hollfelder, Chem. Biol., 2012, 19, 1001–1009 CrossRef CAS PubMed
- H. R. Hoogenboom, Nat. Biotechnol., 2005, 23, 1105–1116 CrossRef CAS PubMed
- A. K. Brödel, M. Isalan and A. Jaramillo, Curr. Opin. Biotechnol., 2018, 51, 32–38 CrossRef PubMed
- R. J. Kazlauskas and U. T. Bornscheuer, Nat. Chem. Biol., 2009, 5, 526–529 CrossRef CAS PubMed
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed
- N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed
- T. Davids, M. Schmidt, D. Böttcher and U. T. Bornscheuer, Curr. Opin. Chem. Biol., 2013, 17, 215–220 CrossRef CAS PubMed
- M. S. Packer and D. R. Liu, Nat. Rev. Genet., 2015, 16, 379–394 CrossRef CAS PubMed
- C. A. Denard, H. Ren and H. Zhao, Curr. Opin. Chem. Biol., 2015, 25, 55–64 CrossRef CAS PubMed
- P. Molina-Espeja, J. Viña-Gonzalez, B. J. Gomez-Fernandez, J. Martin-Diaz, E. Garcia-Ruiz and M. Alcalde, Biotechnol. Adv., 2016, 34, 754–767 CrossRef PubMed
- I. Slabu, J. L. Galman, R. C. Lloyd and N. J. Turner, ACS Catal., 2017, 7, 8263–8284 CrossRef CAS
- C. P. S. Badenhorst and U. T. Bornscheuer, Trends Biochem. Sci., 2018, 43, 180–198 CrossRef CAS
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef
- G. Qu, A. Li, Z. Sun, C. G. Acevedo-Rocha and M. T. Reetz, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201901491
- M. R. Smith, E. Khera and F. Wen, Ind. Eng. Chem. Res., 2015, 54, 4021–4032 CrossRef CAS PubMed
- G. Yang, J. R. Rich, M. Gilbert, W. W. Wakarchuk, Y. Feng and S. G. Withers, J. Am. Chem. Soc., 2010, 132, 10570–10577 CrossRef CAS
- L. F. Mackenzie, Q. Wang, R. A. J. Warren and S. G. Withers, J. Am. Chem. Soc., 1998, 120, 5583–5584 CrossRef CAS
- P. M. Danby and S. G. Withers, ACS Chem. Biol., 2016, 11, 1784–1794 CrossRef CAS PubMed
- Z. Armstrong, F. Liu, H.-M. Chen, S. J. Hallam and S. G. Withers, ACS Catal., 2019, 9, 3219–3227 CrossRef CAS
- H.-M. Chen and S. G. Withers, Carbohydr. Res., 2018, 467, 33–44 CrossRef CAS PubMed
- S. A. Nasseri, L. Betschart, D. Opaleva, P. Rahfeld and S. G. Withers, Angew. Chem., Int. Ed., 2018, 57, 11359–11364 CrossRef CAS PubMed
- E. Andrés, H. Aragunde and A. Planas, Biochem. J., 2014, 458, 355–363 CrossRef PubMed
- K. T. Barglow and B. F. Cravatt, Nat. Methods, 2007, 4, 822–827 CrossRef CAS PubMed
- K. Kalidasan, Y. Su, X. Wu, S. Q. Yao and M. Uttamchandani, Chem. Commun., 2013, 49, 7237–7239 RSC
- H. S. Kwok, O. Vargas-Rodriguez, S. V. Melnikov and D. Söll, ACS Chem. Biol., 2019, 14, 603–612 CrossRef CAS PubMed
- E. Fernández-Álvaro, R. Snajdrova, H. Jochens, T. Davids, D. Böttcher and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2011, 50, 8584–8587 CrossRef
- C. Pitzler, G. Wirtz, L. Vojcic, S. Hiltl, A. Böker, R. Martinez and U. Schwaneberg, Chem. Biol., 2014, 21, 1733–1742 CrossRef CAS PubMed
- N. Lülsdorf, C. Pitzler, M. Biggel, R. Martinez, L. Vojcic and U. Schwaneberg, Chem. Commun., 2015, 51, 8679–8682 RSC
- N. Ilmberger, D. Meske, J. Juergensen, M. Schulte, P. Barthen, U. Rabausch, A. Angelov, M. Mientus, W. Liebl, R. A. Schmitz and W. R. Streit, Appl. Microbiol. Biotechnol., 2012, 95, 135–146 CrossRef CAS PubMed
- R. Vanella, D. T. Ta and M. A. Nash, Biotechnol. Bioeng., 2019, 116, 1878–1886 CrossRef CAS PubMed
- R. Mahr and J. Frunzke, Appl. Microbiol. Biotechnol., 2016, 100, 79–90 CrossRef CAS
- M. Kortmann, C. Mack, M. Baumgart and M. Bott, ACS Synth. Biol., 2019, 8, 274–281 CrossRef CAS PubMed
- M. Schallmey, J. Frunzke, L. Eggeling and J. Marienhagen, Curr. Opin. Biotechnol., 2014, 26, 148–154 CrossRef CAS PubMed
- G. Schendzielorz, M. Dippong, A. Grünberger, D. Kohlheyer, A. Yoshida, S. Binder, C. Nishiyama, M. Nishiyama, M. Bott and L. Eggeling, ACS Synth. Biol., 2014, 3, 21–29 CrossRef CAS PubMed
- N. C. Shaner, P. A. Steinbach and R. Y. Tsien, Nat. Methods, 2005, 2, 905–909 CrossRef CAS PubMed
- F. Cheng, T. Kardashliev, C. Pitzler, A. Shehzad, H. Lue, J. Bernhagen, L. Zhu and U. Schwaneberg, ACS Synth. Biol., 2015, 4, 768–775 CrossRef CAS PubMed
- T. K. Lu, A. S. Khalil and J. J. Collins, Nat. Biotechnol., 2009, 27, 1139–1150 CrossRef CAS PubMed
- A. Serganov and E. Nudler, Cell, 2013, 152, 17–24 CrossRef CAS
- R. W. Bradley, M. Buck and B. Wang, J. Mol. Biol., 2016, 428, 862–888 CrossRef CAS
- E. Barahona, E. Jiménez-Vicente and L. M. Rubio, Sci. Rep., 2016, 6, 38291 CrossRef CAS
- S. Siedler, G. Schendzielorz, S. Binder, L. Eggeling, S. Bringer and M. Bott, ACS Synth. Biol., 2013, 3, 41–47 CrossRef PubMed
- J. N. Copp, E. M. Williams, M. H. Rich, A. V. Patterson, J. B. Smaill and D. F. Ackerley, Protein Eng., Des. Sel., 2014, 27, 399–403 CrossRef CAS PubMed
- J. N. Copp, A. M. Mowday, E. M. Williams, C. P. Guise, A. Ashoorzadeh, A. V. Sharrock, J. U. Flanagan, J. B. Smaill, A. V. Patterson and D. F. Ackerley, Cell Chem. Biol., 2017, 24, 391–403 CrossRef CAS PubMed
- G. Kostallas and P. Samuelson, Appl. Environ. Microbiol., 2010, 76, 7500–7508 CrossRef CAS PubMed
- L. Yi, M. C. Gebhard, Q. Li, J. M. Taft, G. Georgiou and B. L. Iverson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7229–7234 CrossRef CAS PubMed
- O. Khersonsky and D. S. Tawfik, Annu. Rev. Biochem., 2010, 79, 471–505 CrossRef CAS
- T. H. Yoo, M. Pogson, B. L. Iverson and G. Georgiou, ChemBioChem, 2012, 13, 649–653 CrossRef CAS
- Y. Zong, T. W. Bice, H. Ton-That, O. Schneewind and S. V. L. Narayana, J. Biol. Chem., 2004, 279, 31383–31389 CrossRef CAS
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760–763 CrossRef CAS PubMed
- I. Chen, B. M. Dorr and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11399–11404 CrossRef CAS PubMed
- L. Deweid, L. Neureiter, S. Englert, H. Schneider, J. Deweid, D. Yanakieva, J. Sturm, S. Bitsch, A. Christmann, O. Avrutina, H.-L. Fuchsbauer and H. Kolmar, Chem. – Eur. J., 2018, 24, 15195–15200 CrossRef CAS PubMed
- Y. Han, T. C. Branon, J. D. Martell, D. Boassa, D. Shechner, M. H. Ellisman and A. Ting, ACS Chem. Biol., 2019, 14, 619–635 CrossRef CAS PubMed
- J. A. Francisco, C. F. Earhart and G. Georgiou, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2713–2717 CrossRef CAS
- S. Becker, S. Theile, N. Heppeler, A. Michalczyk, A. Wentzel, S. Wilhelm, K.-E. Jaeger and H. Kolmar, FEBS Lett., 2005, 579, 1177–1182 CrossRef CAS PubMed
- A. D. Griffiths and D. S. Tawfik, Trends Biotechnol., 2006, 24, 395–402 CrossRef CAS PubMed
- D. S. Tawfik and A. D. Griffiths, Nat. Biotechnol., 1998, 16, 652–656 CrossRef CAS PubMed
- G. Houlihan, P. Gatti-Lafranconi, M. Kaltenbach, D. Lowe and F. Hollfelder, J. Immunol. Methods, 2014, 405, 47–56 CrossRef CAS PubMed
- E. Mastrobattista, V. Taly, E. Chanudet, P. Treacy, B. T. Kelly and A. D. Griffiths, Chem. Biol., 2005, 12, 1291–1300 CrossRef CAS PubMed
- M. Kaltenbach, S. R. A. Devenish and F. Hollfelder, Lab Chip, 2012, 12, 4185–4192 RSC
- S. W. Lim and A. R. Abate, Lab Chip, 2013, 13, 4563–4572 RSC
- F. Ma, Y. Xie, C. Huang, Y. Feng and G.-Y. Yang, PLoS One, 2014, 9, e89785 CrossRef PubMed
- F. Ma, M. Fischer, Y. Han, S. G. Withers, Y. Feng and G.-Y. Yang, Anal. Chem., 2016, 88, 8587–8595 CrossRef CAS
- O. J. Miller, K. Bernath, J. J. Agresti, G. Amitai, B. T. Kelly, E. Mastrobattista, V. Taly, S. Magdassi, D. S. Tawfik and A. D. Griffiths, Nat. Methods, 2006, 3, 561–570 CrossRef CAS
- A. Aharoni, G. Amitai, K. Bernath, S. Magdassi and D. S. Tawfik, Chem. Biol., 2005, 12, 1281–1289 CrossRef CAS
- R. D. Gupta, M. Goldsmith, Y. Ashani, Y. Simo, G. Mullokandov, H. Bar, M. Ben-David, H. Leader, R. Margalit, I. Silman, J. L. Sussman and D. S. Tawfik, Nat. Chem. Biol., 2011, 7, 120–125 CrossRef CAS PubMed
- P.-Y. Colin, A. Zinchenko and F. Hollfelder, Curr. Opin. Struct. Biol., 2015, 33, 42–51 CrossRef CAS PubMed
- A. Zinchenko, S. R. A. Devenish, B. Kintses, P.-Y. Colin, M. Fischlechner and F. Hollfelder, Anal. Chem., 2014, 86, 2526–2533 CrossRef CAS
- L.-Y. Chu, A. S. Utada, R. K. Shah, J.-W. Kim and D. A. Weitz, Angew. Chem., Int. Ed., 2007, 46, 8970–8974 CrossRef CAS
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 CrossRef CAS
- L. Mazutis, J. Gilbert, W. L. Ung, D. A. Weitz, A. D. Griffiths and J. A. Heyman, Nat. Protoc., 2013, 8, 870–891 CrossRef CAS PubMed
- E. Hardiman, M. Gibbs, R. Reeves and P. Bergquist, Appl. Biochem. Biotechnol., 2010, 161, 301–312 CrossRef CAS
- B.-Y. Hwang, Biotechnol. Bioprocess Eng., 2012, 17, 500–505 CrossRef CAS
- F. Ma, M. T. Chung, Y. Yao, R. Nidetz, L. M. Lee, A. P. Liu, Y. Feng, K. Kurabayashi and G.-Y. Yang, Nat. Commun., 2018, 9, 1030 CrossRef
- A. Sciambi and A. R. Abate, Lab Chip, 2015, 15, 47–51 RSC
- L. Shang, Y. Cheng and Y. Zhao, Chem. Rev., 2017, 117, 7964–8040 CrossRef CAS PubMed
- F. J. Ghadessy, J. L. Ong and P. Holliger, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4552–4557 CrossRef CAS PubMed
- F. J. Ghadessy, N. Ramsay, F. Boudsocq, D. Loakes, A. Brown, S. Iwai, A. Vaisman, R. Woodgate and P. Holliger, Nat. Biotechnol., 2004, 22, 755–759 CrossRef CAS
- R. Obexer, A. Godina, X. Garrabou, P. R. E. Mittl, D. Baker, A. D. Griffiths and D. Hilvert, Nat. Chem., 2016, 9, 50–56 CrossRef PubMed
- R. Obexer, M. Pott, C. Zeymer, A. D. Griffiths and D. Hilvert, Protein Eng., Des. Sel., 2017, 30, 531 CrossRef PubMed
- L. Giger, S. Caner, R. Obexer, P. Kast, D. Baker, N. Ban and D. Hilvert, Nat. Chem. Biol., 2013, 9, 494–498 CrossRef CAS PubMed
- E. A. Althoff, L. Wang, L. Jiang, L. Giger, J. K. Lassila, Z. Wang, M. Smith, S. Hari, P. Kast, D. Herschlag, D. Hilvert and D. Baker, Protein Sci., 2012, 21, 717–726 CrossRef CAS PubMed
-
S. C. Cosgrove, A. Brzezniak, S. P. France, J. I. Ramsden, J. Mangas-Sanchez, S. L. Montgomery, R. S. Heath and N. J. Turner, in Methods in Enzymology, ed. N. Scrutton, Academic Press, 2018, vol. 608, pp. 131–149 Search PubMed
- T. A. Feagin, D. P. V. Olsen, Z. C. Headman and J. M. Heemstra, J. Am. Chem. Soc., 2015, 137, 4198–4206 CrossRef CAS PubMed
- Z. Tan and J. M. Heemstra, ChemBioChem, 2018, 19, 1853–1857 CrossRef CAS PubMed
- P. A. Romero, T. M. Tran and A. R. Abate, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7159–7164 CAS
- F. Gielen, R. Hours, S. Emond, M. Fischlechner, U. Schell and F. Hollfelder, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7383–E7389 CrossRef CAS PubMed
- R. Prodanovic, R. Ostafe, A. Scacioc and U. Schwaneberg, Comb. Chem. High Throughput Screening, 2011, 14, 55–60 CAS
- R. Ostafe, R. Prodanovic, J. Nazor and R. Fischer, Chem. Biol., 2014, 21, 414–421 CAS
- G. Kovačević, R. Ostafe, A. M. Balaž, R. Fischer and R. Prodanović, J. Biosci. Bioeng., 2019, 127, 30–37 Search PubMed
- R. Ostafe, R. Prodanovic, U. Commandeur and R. Fischer, Anal. Biochem., 2013, 435, 93–98 CAS
- R. Ostafe, R. Prodanovic, W. L. Ung, D. A. Weitz and R. Fischer, Biomicrofluidics, 2014, 8, 041102 CrossRef PubMed
- T. Beneyton, S. Thomas, A. D. Griffiths, J.-M. Nicaud, A. Drevelle and T. Rossignol, Microb. Cell Fact., 2017, 16, 18 CrossRef PubMed
- H. Inoue, H. Nojima and H. Okayama, Gene, 1990, 96, 23–28 CrossRef CAS PubMed
-
D. Hanahan, J. Jessee and F. R. Bloom, in Methods in Enzymology, ed. J. H. Miller, Academic Press, 1991, vol. 204, pp. 63–113 Search PubMed
- A. M. Davis, A. T. Plowright and E. Valeur, Nat. Rev. Drug Discovery, 2017, 16, 681–698 CrossRef CAS PubMed
- J. Hanes and A. Plückthun, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4937–4942 CrossRef CAS PubMed
- D. Lipovsek and A. Plückthun, J. Immunol. Methods, 2004, 290, 51–67 CAS
- Q. Wang, A. R. Parrish and L. Wang, Chem. Biol., 2009, 16, 323–336 CrossRef CAS PubMed
- H. Xiao and P. G. Schultz, Cold Spring Harbor Perspect. Biol., 2016, 8, a023945 CrossRef
- J. Xie, W. Liu and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 9239–9242 CrossRef CAS PubMed
- H. Yang, A. M. Swartz, H. J. Park, P. Srivastava, K. Ellis-Guardiola, D. M. Upp, G. Lee, K. Belsare, Y. Gu, C. Zhang, R. E. Moellering and J. C. Lewis, Nat. Chem., 2018, 10, 318–324 CrossRef CAS PubMed
- H. M. Cohen, D. S. Tawfik and A. D. Griffiths, Protein Eng., Des. Sel., 2004, 17, 3–11 CrossRef CAS PubMed
- Y. F. Lee, D. S. Tawfik and A. D. Griffiths, Nucleic Acids Res., 2002, 30, 4937–4944 CrossRef CAS PubMed
- N. Doi, S. Kumadaki, Y. Oishi, N. Matsumura and H. Yanagawa, Nucleic Acids Res., 2004, 32, e95 CrossRef PubMed
- P. Gianella, E. L. Snapp and M. Levy, Biotechnol. Bioeng., 2016, 113, 1647–1657 CAS
- A. Fallah-Araghi, J.-C. Baret, M. Ryckelynck and A. D. Griffiths, Lab Chip, 2012, 12, 882–891 RSC
- B. Zhu, T. Mizoguchi, T. Kojima and H. Nakano, PLoS One, 2015, 10, e0127479 CrossRef PubMed
- J. A. Stapleton and J. R. Swartz, PLoS One, 2010, 5, e15275 CrossRef CAS
- L. S. Roach, H. Song and R. F. Ismagilov, Anal. Chem., 2005, 77, 785–796 CrossRef CAS PubMed
- N. Wu, F. Courtois, Y. Zhu, J. Oakeshott, C. Easton and C. Abell, Electrophoresis, 2010, 31, 3121–3128 CAS
- F. Courtois, L. F. Olguin, G. Whyte, A. B. Theberge, W. T. S. Huck, F. Hollfelder and C. Abell, Anal. Chem., 2009, 81, 3008–3016 CrossRef CAS PubMed
- G. Woronoff, A. El Harrak, E. Mayot, O. Schicke, O. J. Miller, P. Soumillion, A. D. Griffiths and M. Ryckelynck, Anal. Chem., 2011, 83, 2852–2857 CAS
- M. Fischlechner, Y. Schaerli, M. F. Mohamed, S. Patil, C. Abell and F. Hollfelder, Nat. Chem., 2014, 6, 791–796 CrossRef CAS PubMed
- J. M. Duarte, I. Barbier and Y. Schaerli, ACS Synth. Biol., 2017, 6, 1988–1995 CrossRef CAS PubMed
- M. Li, M. van Zee, C. T. Riche, B. Tofig, S. D. Gallaher, S. S. Merchant, R. Damoiseaux, K. Goda and D. Di Carlo, Small, 2018, 14, 1803315 CrossRef PubMed
- S. Pautot, B. J. Frisken and D. A. Weitz, Langmuir, 2003, 19, 2870–2879 CrossRef CAS
- S. Fujii, T. Matsuura, T. Sunami, Y. Kazuta and T. Yomo, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16796–16801 CrossRef CAS PubMed
- A. Uyeda, S. Nakayama, Y. Kato, H. Watanabe and T. Matsuura, Anal. Chem., 2016, 88, 12028–12035 CrossRef CAS PubMed
- S. Fujii, T. Matsuura, T. Sunami, T. Nishikawa, Y. Kazuta and T. Yomo, Nat. Protoc., 2014, 9, 1578–1591 CrossRef CAS PubMed
- A. Uyeda, T. Watanabe, Y. Kato, H. Watanabe, T. Yomo, T. Hohsaka and T. Matsuura, ChemBioChem, 2015, 16, 1797–1802 CrossRef CAS PubMed
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed
- D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. C. M. Christianen, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2003, 42, 772–776 CrossRef CAS PubMed
- A. Graff, M. Sauer, P. Van Gelder and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5064–5068 CrossRef CAS PubMed
- D. M. Vriezema, P. M. L. Garcia, N. Sancho Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378–7382 CrossRef CAS PubMed
- M. Nallani, R. Woestenenk, H. P. M. de Hoog, S. F. M. van Dongen, J. Boezeman, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Small, 2009, 5, 1138–1143 CrossRef CAS PubMed
- C. Martino, S.-H. Kim, L. Horsfall, A. Abbaspourrad, S. J. Rosser, J. Cooper and D. A. Weitz, Angew. Chem., Int. Ed., 2012, 51, 6416–6420 CrossRef CAS PubMed
- R. B. Liebherr, M. Renner and H. H. Gorris, J. Am. Chem. Soc., 2014, 136, 5949–5955 CrossRef CAS PubMed
- D. M. Rissin, H. H. Gorris and D. R. Walt, J. Am. Chem. Soc., 2008, 130, 5349–5353 CrossRef CAS PubMed
- D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang, A. J. Rivnak, E. P. Ferrell, J. D. Randall, G. K. Provuncher, D. R. Walt and D. C. Duffy, Nat. Biotechnol., 2010, 28, 595–599 CrossRef CAS PubMed
- D. M. Rissin and D. R. Walt, J. Am. Chem. Soc., 2006, 128, 6286–6287 CrossRef CAS PubMed
- H. H. Gorris and D. R. Walt, J. Am. Chem. Soc., 2009, 131, 6277–6282 CrossRef CAS PubMed
- V. Fitzgerald, B. Manning, B. O’Donnell, B. O’Reilly, D. O’Sullivan, R. O’Kennedy and P. Leonard, Anal. Chem., 2015, 87, 997–1003 CrossRef CAS PubMed
- C. J. Ingham, A. Sprenkels, J. Bomer, D. Molenaar, A. van den Berg, J. E. T. van Hylckama Vlieg and W. M. de Vos, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18217–18222 CrossRef CAS PubMed
- S. Lindström, M. Eriksson, T. Vazin, J. Sandberg, J. Lundeberg, J. Frisén and H. Andersson-Svahn, PLoS One, 2009, 4, e6997 CrossRef PubMed
- H.-C. Moeller, M. K. Mian, S. Shrivastava, B. G. Chung and A. Khademhosseini, Biomaterials, 2008, 29, 752–763 CrossRef CAS PubMed
- S. Gobaa, S. Hoehnel, M. Roccio, A. Negro, S. Kobel and M. P. Lutolf, Nat. Methods, 2011, 8, 949–955 CrossRef CAS PubMed
- H. Park, H. Kim and J. Doh, Bioconjugate Chem., 2018, 29, 672–679 CrossRef CAS PubMed
- T. Konry, S. Sarkar, P. Sabhachandani and N. Cohen, Annu. Rev. Biomed. Eng., 2016, 18, 259–284 CrossRef CAS PubMed
- D. R. Walt, Lab Chip, 2014, 14, 3195–3200 RSC
- C. K. Longwell, L. Labanieh and J. R. Cochran, Curr. Opin. Biotechnol., 2017, 48, 196–202 CrossRef CAS PubMed
- B. Chen, S. Lim, A. Kannan, S. C. Alford, F. Sunden, D. Herschlag, I. K. Dimov, T. M. Baer and J. R. Cochran, Nat. Chem. Biol., 2015, 12, 76–81 CrossRef PubMed
- S. Lim, B. Chen, M. S. Kariolis, I. K. Dimov, T. M. Baer and J. R. Cochran, ACS Chem. Biol., 2017, 12, 336–341 CrossRef CAS PubMed
- F. H. Arnold, Acc. Chem. Res., 1998, 31, 125–131 CrossRef CAS
- Y. Zhu and Q. Fang, Anal. Chim. Acta, 2013, 787, 24–35 CrossRef CAS PubMed
- C. Yan, F. Parmeggiani, E. A. Jones, E. Claude, S. A. Hussain, N. J. Turner, S. L. Flitsch and P. E. Barran, J. Am. Chem. Soc., 2017, 139, 1408–1411 CrossRef CAS PubMed
- T. Si, B. Li, T. J. Comi, Y. Wu, P. Hu, Y. Wu, Y. Min, D. A. Mitchell, H. Zhao and J. V. Sweedler, J. Am. Chem. Soc., 2017, 139, 12466–12473 CrossRef CAS PubMed
- T. de Rond, J. Gao, A. Zargar, M. de Raad, J. Cunha, T. R. Northen and J. D. Keasling, Angew. Chem., Int. Ed., 2019, 58, 10114–10119 CrossRef CAS PubMed
- T. R. Northen, O. Yanes, M. T. Northen, D. Marrinucci, W. Uritboonthai, J. Apon, S. L. Golledge, A. Nordström and G. Siuzdak, Nature, 2007, 449, 1033–1036 CrossRef CAS PubMed
- T. R. Northen, J.-C. Lee, L. Hoang, J. Raymond, D.-R. Hwang, S. M. Yannone, C.-H. Wong and G. Siuzdak, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3678–3683 CrossRef CAS PubMed
- T. de Rond, P. Peralta-Yahya, X. Cheng, T. R. Northen and J. D. Keasling, Anal. Bioanal. Chem., 2013, 405, 4969–4973 CrossRef CAS PubMed
- T. Hatakeyama, D. L. Chen and R. F. Ismagilov, J. Am. Chem. Soc., 2006, 128, 2518–2519 CrossRef CAS PubMed
- L. M. Fidalgo, G. Whyte, D. Bratton, C. F. Kaminski, C. Abell and W. T. S. Huck, Angew. Chem., Int. Ed., 2008, 47, 2042–2045 CrossRef CAS PubMed
- L. M. Fidalgo, G. Whyte, B. T. Ruotolo, J. L. P. Benesch, F. Stengel, C. Abell, C. V. Robinson and W. T. S. Huck, Angew. Chem., Int. Ed., 2009, 48, 3665–3668 CrossRef CAS PubMed
- R. T. Kelly, J. S. Page, I. Marginean, K. Tang and R. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 6832–6835 CrossRef CAS PubMed
- Y. Zhu and Q. Fang, Anal. Chem., 2010, 82, 8361–8366 CrossRef CAS PubMed
- C. A. Smith, X. Li, T. H. Mize, T. D. Sharpe, E. I. Graziani, C. Abell and W. T. S. Huck, Anal. Chem., 2013, 85, 3812–3816 CrossRef CAS PubMed
- S. Sun and R. T. Kennedy, Anal. Chem., 2014, 86, 9309–9314 CrossRef CAS PubMed
- S. Sun, T. R. Slaney and R. T. Kennedy, Anal. Chem., 2012, 84, 5794–5800 CrossRef CAS PubMed
- X. W. Diefenbach, I. Farasat, E. D. Guetschow, C. J. Welch, R. T. Kennedy, S. Sun and J. C. Moore, ACS Omega, 2018, 3, 1498–1508 CrossRef CAS PubMed
- J. S. Edgar, C. P. Pabbati, R. M. Lorenz, M. He, G. S. Fiorini and D. T. Chiu, Anal. Chem., 2006, 78, 6948–6954 CrossRef CAS PubMed
- S. C. Jacobson, R. Hergenroder, L. B. Koutny and J. M. Ramsey, Anal. Chem., 1994, 66, 1114–1118 CrossRef CAS
- C. M. Ouimet, C. I. D’amico and R. T. Kennedy, Expert Opin. Drug Discovery, 2017, 12, 213–224 CrossRef CAS PubMed
- G. T. Roman, M. Wang, K. N. Shultz, C. Jennings and R. T. Kennedy, Anal. Chem., 2008, 80, 8231–8238 CrossRef CAS PubMed
- M. Wang, G. T. Roman, M. L. Perry and R. T. Kennedy, Anal. Chem., 2009, 81, 9072–9078 CrossRef CAS PubMed
- E. D. Guetschow, D. J. Steyer and R. T. Kennedy, Anal. Chem., 2014, 86, 10373–10379 CrossRef CAS PubMed
- E. D. Guetschow, S. Kumar, D. B. Lombard and R. T. Kennedy, Anal. Bioanal. Chem., 2016, 408, 721–731 CrossRef CAS PubMed
- J. Pei, J. Nie and R. T. Kennedy, Anal. Chem., 2010, 82, 9261–9267 CrossRef CAS PubMed
- G. Goddard, L. O. Brown, R. Habbersett, C. I. Brady, J. C. Martin, S. W. Graves, J. P. Freyer and S. K. Doorn, J. Am. Chem. Soc., 2010, 132, 6081–6090 CrossRef CAS PubMed
- M. P. Cecchini, J. Hong, C. Lim, J. Choo, T. Albrecht, A. J. deMello and J. B. Edel, Anal. Chem., 2011, 83, 3076–3081 CrossRef CAS PubMed
- A. Walter, A. Marz, W. Schumacher, P. Rosch and J. Popp, Lab Chip, 2011, 11, 1013–1021 RSC
- H. S. Kim, S. C. Waqued, D. T. Nodurft, T. P. Devarenne, V. V. Yakovlev and A. Han, Analyst, 2017, 142, 1054–1060 RSC
- Z. Han, W. Li, Y. Huang and B. Zheng, Anal. Chem., 2009, 81, 5840–5845 CrossRef CAS PubMed
- D. Chen, W. Du, Y. Liu, W. Liu, A. Kuznetsov, F. E. Mendez, L. H. Philipson and R. F. Ismagilov, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16843–16848 CrossRef CAS PubMed
- A. D. Townsend, G. H. Wilken, K. K. Mitchell, R. S. Martin and H. Macarthur, J. Neurosci. Methods, 2016, 266, 68–77 CrossRef CAS PubMed
- J. Handelsman, Microbiol. Mol. Biol. Rev., 2004, 68, 669–685 CrossRef CAS PubMed
- W. R. Streit and R. A. Schmitz, Curr. Opin. Microbiol., 2004, 7, 492–498 CrossRef CAS PubMed
- L. A. Hug, B. J. Baker, K. Anantharaman, C. T. Brown, A. J. Probst, C. J. Castelle, C. N. Butterfield, A. W. Hernsdorf, Y. Amano, K. Ise, Y. Suzuki, N. Dudek, D. A. Relman, K. M. Finstad, R. Amundson, B. C. Thomas and J. F. Banfield, Nat. Microbiol., 2016, 1, 16048 CrossRef CAS PubMed
- F. G. Healy, R. M. Ray, H. C. Aldrich, A. C. Wilkie, L. O. Ingram and K. T. Shanmugam, Appl. Microbiol. Biotechnol., 1995, 43, 667–674 CrossRef CAS PubMed
- A. Henne, R. Daniel, R. A. Schmitz and G. Gottschalk, Appl. Environ. Microbiol., 1999, 65, 3901–3907 CAS
- A. Henne, R. A. Schmitz, M. Bömeke, G. Gottschalk and R. Daniel, Appl. Environ. Microbiol., 2000, 66, 3113–3116 CrossRef CAS PubMed
- M. R. Rondon, P. R. August, A. D. Bettermann, S. F. Brady, T. H. Grossman, M. R. Liles, K. A. Loiacono, B. A. Lynch, I. A. MacNeil, C. Minor, C. L. Tiong, M. Gilman, M. S. Osburne, J. Clardy, J. Handelsman and R. M. Goodman, Appl. Environ. Microbiol., 2000, 66, 2541–2547 CrossRef CAS PubMed
- D. E. Gillespie, S. F. Brady, A. D. Bettermann, N. P. Cianciotto, M. R. Liles, M. R. Rondon, J. Clardy, R. M. Goodman and J. Handelsman, Appl. Environ. Microbiol., 2002, 68, 4301–4306 CrossRef CAS PubMed
- W. Liebl, A. Angelov, J. Juergensen, J. Chow, A. Loeschcke, T. Drepper, T. Classen, J. Pietruszka, A. Ehrenreich, W. R. Streit and K. E. Jaeger, Appl. Microbiol. Biotechnol., 2014, 98, 8099–8109 CrossRef CAS PubMed
- K. N. Lam, J. Cheng, K. Engel, J. D. Neufeld and T. C. Charles, Front. Microbiol., 2015, 6, 1196 Search PubMed
- M. Ferrer, A. Beloqui, K. N. Timmis and P. N. Golyshin, J. Mol. Microbiol. Biotechnol., 2009, 16, 109–123 CrossRef CAS PubMed
- S. S. Macdonald, Z. Armstrong, C. Morgan-Lang, M. Osowiecka, K. Robinson, S. J. Hallam and S. G. Withers, Cell Chem. Biol., 2019, 26, 1001–1012 CrossRef CAS PubMed
- M. R. M. De Groeve, G. H. Tran, A. Van Hoorebeke, J. Stout, T. Desmet, S. N. Savvides and W. Soetaert, Anal. Biochem., 2010, 401, 162–167 CrossRef CAS PubMed
- C. Coscolín, N. Katzke, A. García-Moyano, J. Navarro-Fernández, D. Almendral, M. Martínez-Martínez, A. Bollinger, R. Bargiela, C. Gertler, T. N. Chernikova, D. Rojo, C. Barbas, H. Tran, O. V. Golyshina, R. Koch, M. M. Yakimov, G. E. K. Bjerga, P. N. Golyshin, K.-E. Jaeger and M. Ferrer, Appl. Environ. Microbiol., 2019, 85, e02404 Search PubMed
- J. Cheng, T. Romantsov, K. Engel, A. C. Doxey, D. R. Rose, J. D. Neufeld and T. C. Charles, PLoS One, 2017, 12, e0172545 CrossRef PubMed
- M. Schallmey, A. Ly, C. Wang, G. Meglei, S. Voget, W. R. Streit, B. T. Driscoll and T. C. Charles, FEMS Microbiol. Lett., 2011, 321, 150–156 CrossRef CAS PubMed
- P. Entcheva, W. Liebl, A. Johann, T. Hartsch and W. R. Streit, Appl. Environ. Microbiol., 2001, 67, 89–99 CrossRef CAS PubMed
- J. K. Bitok, C. Lemetre, M. A. Ternei and S. F. Brady, FEMS Microbiol. Lett., 2017, 364, fnx155 CrossRef PubMed
- J. G. Owen, K. J. Robins, N. S. Parachin and D. F. Ackerley, Environ. Microbiol., 2012, 14, 1198–1209 CrossRef CAS PubMed
- T. B. Rasmussen, T. Bjarnsholt, M. E. Skindersoe, M. Hentzer, P. Kristoffersen, M. Köte, J. Nielsen, L. Eberl and M. Givskov, J. Bacteriol., 2005, 187, 1799–1814 CrossRef CAS PubMed
- N. Weiland-Bräuer, N. Pinnow and R. A. Schmitz, Appl. Environ. Microbiol., 2015, 81, 1477–1489 CrossRef PubMed
- H. Afif, N. Allali, M. Couturier and L. Van Melderen, Mol. Microbiol., 2001, 41, 73–82 CrossRef CAS PubMed
- N. Weiland-Bräuer, M. J. Kisch, N. Pinnow, A. Liese and R. A. Schmitz, Front. Microbiol., 2016, 7, 1098 Search PubMed
- K. Shimuta, M. Ohnishi, S. Iyoda, N. Gotoh, N. Koizumi and H. Watanabe, BMC Microbiol., 2009, 9, 261 CrossRef PubMed
- T. Uchiyama, T. Abe, T. Ikemura and K. Watanabe, Nat. Biotechnol., 2005, 23, 88–93 CrossRef CAS PubMed
-
T. Uchiyama and K. Miyazaki, in Methods in Molecular Biology, ed. W. Streit and R. Daniel, Humana Press, Totowa, NJ, 2010, vol. 668, pp. 153–168 Search PubMed
- T. Uchiyama and K. Watanabe, Nat. Protoc., 2008, 3, 1202–1212 CrossRef CAS PubMed
- M. J. Meier, E. S. Paterson and I. B. Lambert, Appl. Environ. Microbiol., 2016, 82, 897–909 CrossRef CAS PubMed
- T. Uchiyama and K. Miyazaki, PLoS One, 2013, 8, e75795 CrossRef CAS PubMed
- M. Martinez-Martinez, C. Coscolin, G. Santiago, J. Chow, P. J. Stogios, R. Bargiela, C. Gertler, J. Navarro-Fernandez, A. Bollinger, S. Thies, C. Mendez-Garcia, A. Popovic, G. Brown, T. N. Chernikova, A. Garcia-Moyano, G. E. K. Bjerga, P. Perez-Garcia, T. Hai, M. V. Del Pozo, R. Stokke, I. H. Steen, H. Cui, X. Xu, B. P. Nocek, M. Alcaide, M. Distaso, V. Mesa, A. I. Pelaez, J. Sanchez, P. C. F. Buchholz, J. Pleiss, A. Fernandez-Guerra, F. O. Glockner, O. V. Golyshina, M. M. Yakimov, A. Savchenko, K. E. Jaeger, A. F. Yakunin, W. R. Streit, P. N. Golyshin, V. Guallar, M. Ferrer and The Inmare Consortium, ACS Chem. Biol., 2018, 13, 225–234 CrossRef CAS PubMed
- M. Hosokawa, Y. Hoshino, Y. Nishikawa, T. Hirose, D. H. Yoon, T. Mori, T. Sekiguchi, S. Shoji and H. Takeyama, Biosens. Bioelectron., 2015, 67, 379–385 CrossRef CAS PubMed
- Y. Schaerli and F. Hollfelder, Mol. BioSyst., 2009, 5, 1392–1404 RSC
- M. Ferrer, A. Beloqui, J. M. Vieites, M. E. Guazzaroni, I. Berger and A. Aharoni, Microb. Biotechnol., 2009, 2, 31–39 CrossRef CAS PubMed
- P.-Y. Colin, B. Kintses, F. Gielen, C. M. Miton, G. Fischer, M. F. Mohamed, M. Hyvönen, D. P. Morgavi, D. B. Janssen and F. Hollfelder, Nat. Commun., 2015, 6, 10008 CrossRef CAS PubMed
- E. Theodorou, R. Scanga, M. Twardowski, M. P. Snyder and E. Brouzes, Micromachines, 2017, 8, 230 CrossRef PubMed
- B. M. Kinfu, M. Jahnke, M. Janus, V. Besirlioglu, M. Roggenbuck, R. Meurer, L. Vojcic, M. Borchert, U. Schwaneberg, J. Chow and W. R. Streit, Biotechnol. Bioeng., 2017, 114, 2739–2752 CrossRef CAS PubMed
- F. B. Yu, P. C. Blainey, F. Schulz, T. Woyke, M. A. Horowitz and S. R. Quake, eLife, 2017, 6, e26580 CrossRef PubMed
- S. S. Terekhov, I. V. Smirnov, A. V. Stepanova, T. V. Bobik, Y. A. Mokrushina, N. A. Ponomarenko, A. A. Belogurov, M. P. Rubtsova, O. V. Kartseva, M. O. Gomzikova, A. A. Moskovtsev, A. S. Bukatin, M. V. Dubina, E. S. Kostryukova, V. V. Babenko, M. T. Vakhitova, A. I. Manolov, M. V. Malakhova, M. A. Kornienko, A. V. Tyakht, A. A. Vanyushkina, E. N. Ilina, P. Masson, A. G. Gabibov and S. Altman, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2550–2555 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2020 |