
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-based imaging agents: progress towards interrogating neurodegenerative disease
Adam C.
Sedgwick
 a,
James T.
Brewster
II
a,
Peter
Harvey
*bc,
Diana A.
Iovan
d,
Graham
Smith
e,
Xiao-Peng
He
*f,
He
Tian
*f,
Jonathan L.
Sessler
*a and
Tony D.
James
*g
a,
James T.
Brewster
II
a,
Peter
Harvey
*bc,
Diana A.
Iovan
d,
Graham
Smith
e,
Xiao-Peng
He
*f,
He
Tian
*f,
Jonathan L.
Sessler
*a and
Tony D.
James
*g
aDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712-1224, USA. E-mail: sessler@cm.utexas.edu
bDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
cSir Peter Mansfield Imaging Centre, Division of Clinical Neuroscience, School of Medicine, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: peter.harvey@nottingham.ac.uk
dDepartment of Chemistry, University of California, Berkeley, California, USA
eDivision of Radiotherapy & Imaging, Institute of Cancer Research, 123 Old Brompton Road, London, SW7 3RP, UK
fKey Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Rd, Shanghai 200237, China. E-mail: xphe@ecust.edu.cn; tianhe@ecust.edu.cn
gDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: t.d.james@bath.ac.uk
First published on 30th March 2020
Abstract
Central nervous system (CNS) neurodegeneration is defined by a complex series of pathological processes that ultimately lead to death. The precise etiology of these disorders remains unknown. Recent efforts show that a mechanistic understanding of the malfunctions underpinning disease progression will prove requisite in developing new treatments and cures. Transition metals and lanthanide ions display unique characteristics (i.e., magnetism, radioactivity, and luminescence), often with biological relevance, allowing for direct application in CNS focused imaging modalities. These techniques include positron emission tomography (PET), single-photon emission computed tomography (SPECT), magnetic resonance imaging (MRI), and luminescent-based imaging (LumI). In this Tutorial Review, we have aimed to highlight the various metal-based imaging techniques developed in the effort to understand the pathophysiological processes associated with neurodegeneration. Each section has been divided so as to include an introduction to the particular imaging technique in question. This is then followed by a summary of key demonstrations that have enabled visualization of a specific neuropathological biomarker. These strategies have either exploited the high binding affinity of a receptor for its corresponding biomarker or a specific molecular transformation caused by a target species, all of which produce a concomitant change in diagnostic signal. Advantages and disadvantages of each method with perspectives on the utility of molecular imaging agents for understanding the complexities of neurodegenerative disease are discussed.
 From left to right: Adam C. Sedgwick and Tony D. James | Adam C. Sedgwick received his MChem (2014) and PhD (2018) with Tony D. James and Steven D. Bull at the University of Bath. He is now a postdoctoral research fellow working under the supervision of Prof. Jonathan L. Sessler at The University of Texas in Austin. His research interests are in the realms of stimuli-responsive materials, molecular imaging agents, and theranostic agents. Tony D. James is a Professor at the University of Bath and Fellow of the Royal Society of Chemistry. He received his BSc from the University of East Anglia (1986), PhD from the University of Victoria (1991), was Postdoctoral Research with Seiji Shinkai (1991–1995) and Royal Society University Research Fellow at the University of Birmingham (1995–2000). He was awarded the Daiwa-Adrian Prize (2013), Inaugural CASE Prize (2015), MSMLG Czarnik Award (2018) and currently holds a prestigious Royal Society Wolfson Research Merit Award. (2017–2022). |
 James T. Brewster | James T. Brewster II grew up in Valley Center, California and obtained his BS with honours in chemistry from the University of California, Irvine with mentorship from Professor Kelvin W. Gee, Dr Derk J. Hogenkamp, and Dr Timothy Johnstone. James recently finished his PhD with Professor Jonathan L. Sessler at The University of Texas. James is currently conducting his postdoctoral work with Professor Andrew G. Myers at Harvard University. |
 Peter Harvey | Pete Harvey received his MChem (2009) and PhD (2013) with Prof David Parker FRS in the Chemistry Department at Durham University. He was then awarded an ESPRC Doctoral Prize Fellowship to work at The University of Manchester. In 2015, he was awarded a Wellcome Trust-MIT Postdoctoral Fellowship in the Department of Biological Engineering at MIT. He recently began his independent career in molecular MRI as a Nottingham Research Fellow. |
 Diana A. Iovan | Diana A. Iovan received her BS in chemistry in 2012 from the University of Richmond. She then moved to Harvard University for her graduate studies with Prof. Ted Betley. After earning her PhD in inorganic chemistry in 2017, Diana joined Prof. Chris Chang's laboratory at UC Berkeley as a Howard Hughes Medical Institute Fellow of the Life Sciences Research Foundation. |
 Graham Smith | Graham Smith obtained his PhD from the University of Surrey and was a PDRA at Newcastle University and Imperial College. In 2011 he became a Lecturer in Molecular Imaging at the University of Hull where he helped establish a new PET research facility. In 2013 he moved to the Institute of Cancer Research to lead radiochemistry development within the Division of Radiotherapy and Imaging. |
 Xiao-Peng He | Xiao-Peng He is professor at Feringa Nobel Prize Scientists Research Center, School of Chemistry and Molecular Engineering, ECUST. He obtained his BSc (2006) and PhD (2011) from ECUST. He completed a co-tutored doctoral program at ENS Cachan (France) (2008 to 2009) and postdoctoral research with Kaixian Chen (SIMM, CAS) from 2011 to 2013 at ECUST. |
 He Tian | He Tian was born in China in 1962. In 1999, he was appointed Cheung Kong Distinguished Professor by the Education Ministry of China. In 2011, he was selected as a member of the Chinese Academy of Science. He has been listed by Thomson Reuters as a Highly Cited Researcher in Chemistry (2014) and Materials Science (2014–2019). |
 Jonathan L. Sessler | Jonathan L. Sessler received a BSc degree in Chemistry in 1977 from the University of California, Berkeley. He obtained his PhD from Stanford University in 1982. After postdoctoral stays in Strasbourg and Kyoto, he accepted a position as an Assistant Professor of Chemistry at the University of Texas at Austin, where he is currently the Doherty-Welch Chair. |
Key learning points(1) The importance of molecular imaging for the understanding of neurodegenerative disease(2) To understand the importance of a specific metal in each imaging modality (3) The strategies to design an effective metal-based molecular imaging agent (4) Current limitations of each imaging modality (5) Future perspectives |
Introduction
Central Nervous System (CNS) degeneration involves a series of neurological conditions that affect millions of people worldwide. These diseases stem from the progressive dysfunction and death of neuronal cells, which lead to loss of motor function and memory. A common theme amongst neurodegenerative processes is the progressive misfolding, aggregation, and accumulation of protein and peptide aggregates resulting in cellular dysfunction, loss of synaptic connections, and brain damage. The most common disorders, Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), Lewy Body dementia, prion diseases, and amyotrophic lateral sclerosis (ALS), also share phenotypic similarities and all result in death.1 Current treatment protocols offer short-term palliative benefits with no long-term improvement for patients or relief for caregivers. Not surprisingly, the pharmaceutical industry and academic institutions alike are seeking to develop new therapies, as well as working to elucidate the biochemical processes responsible for these disorders. High attrition rates in clinical trials have led to the belief that understanding the precise etiology of CNS disease is critical to developing an effective treatment or cure. As discussed below, metal-based imaging modalities may have a critical role to play in this search. The goal of this tutorial review is to summarize recent progress in the area.To date, a variety of metal-based approaches have proved useful in illuminating the biochemical malfunctions that lead to neurodegeneration. These methods include: (1) positron emission tomography (PET), (2) single-photon emission computed tomography (SPECT), (3) magnetic resonance imaging (MRI), and (4) luminescent imaging. This tutorial review will highlight metal-based imaging techniques used to understand aberrant biochemical processes involved in neurodegenerative diseases. Particular focus will be placed on a general overview of each imaging technique, the purpose of each method, and its implementation by highlighting recent demonstrations in the literature.
Positron emission tomography (PET) & single photon emission computed tomography (SPECT)
Major advances in scanner technology over the last 20 years have facilitated adoption of PET and SPECT in the biomedical-imaging field. PET/SPECT-based imaging requires the use of a radiopharmaceutical or radiotracer. Typically, this is a molecule containing a radioisotope within its structure. For PET, the radiotracer contains a positron-emitting isotope. The emitted positron (β+) or “anti-electron” collides with nearby electrons resulting in an annihilation process that produces two γ-rays at exactly 511 keV emitted 180° apart. The specific energy of these γ-rays is dissimilar to other modes of decay possible for most radionuclides. The PET derived γ-rays (511 keV) are detected by specialized instrumentation and used to form the image for diagnosis. SPECT utilizes a similar approach wherein the radiotracer contains a γ-ray emitting short-lived isotope (ideal γ-ray energies: 100–250 keV), which is detected to provide the image for diagnosis. The required dosage for PET/SPECT is commonly within picomolar (pM; 10−12 mol L−1) to nanomolar (nM; 10−9 mol L−1) concentrations reducing any potential toxicology risk arising from radioactive isotope and non-radioactive “carrier” material.The requirement for highly specialized and expensive instruments had previously limited the widespread adoption of PET. Due to early adoption and cheaper instrumentation, more hospitals are equipped with SPECT scanners. As a result, SPECT is more widespread, both in terms of clinical use and current imaging research. However, modern PET setups permit remarkable sensitivity compared to the analogous magnetic resonance imaging (MRI) and X-ray computer tomography (CT) approach, and arguably provides a slight advantage over SPECT-based methods. This has resulted in a shift towards PET-based technology in recent years.
For a PET-based imaging agent to achieve the spatial resolution needed for use in neurological or small animal imaging, a radioisotope with low energy positron emissions is required. The use of such isotopes enables the production of higher resolution images, i.e., lower positron energies correlate to a shorter positron distance giving a more specific location. Tables 1 and 2 highlight the current radioisotopes that have been used towards the development of radiotracers for PET/SPECT imaging in neurodegenerative diseases with corresponding decay energies, half-lives, and production methods. It is important to note, each radioisotope may be subject to one or more decay modes; this can require additional consideration when designing a PET/SPECT radiotracer. The reader is directed to an excellent review by Blower for a more extensive list of radioisotopes used in medical imaging and therapy.2
| Radioisotope | t 1/2 | Production methods | Energy (β+) – MeV | Energy (γ) – MeV | Decay type |
|---|---|---|---|---|---|
| 11C | 20 min | Cyclotron | 1.0 | — | β+ |
| 18F | 110 min | Cyclotron | 0.6 | — | β+ |
| 62Cu | 9.7 min | 62Zn/62Cu generator | 2.9 | 0.12 | β+/γ |
| 64Cu | 12.7 h | Cyclotron | 0.7 | 0.13 | β+, β−, γ, EC |
| 68Ga | 1.1 h | 68Ge/68Ga generator | 1.9 | 0.11 | β+, γ, EC |
| 89Zr | 78.5 h | Cyclotron | 0.9 | — | β+, EC |
| Radioisotope | t 1/2 (h) | Production methods | γ-ray – keV | Decay type |
|---|---|---|---|---|
| 99mTc | 6 | 99mTc/99Mo generator | 141 | γ |
| 123I | 13 | Cyclotron | 159 | γ, EC |
| 111In | 67.9 | Cyclotron | 245 | γ, EC |
The PET/SPECT-based imaging section will be divided according to each metal-based radioisotope with attendant consideration of the neurodegenerative disease-related biomarker target. For use of other radioisotopes and non-metal-based systems for PET/SPECT-based imaging and therapy the reader is directed to a number of excellent reviews.2,4
The development of metal-based radiotracers requires knowledge of metal coordination chemistry and appropriate ligand design. Moreover, the radiotracer itself must:
• Have a high selectivity and binding affinity for the desired target with rapid binding kinetics.
• Target a specific receptor or biochemical process occurring in sufficient concentration so as to provide a good signal-to-noise ratio.
• Permit rapid insertion of the metal radioisotope and produce a complex that does not result in the loss of binding affinity to the biological target. Protocols that allow for metal complexation in aqueous conditions are particularly desired since they may allow for the facile development of “kit-based technology” for use in a clinical setting.
• The imaging agent or administration technique must allow for sufficient localization within the brain, i.e., penetrate the blood brain barrier (BBB). This remains a challenge when designing CNS agents.
Copper (60Cu, 61Cu, 62Cu, 64Cu) – PET
Copper (Cu) has two key biologically relevant oxidation states, Cu+ and Cu2+. Several decades of research from the inorganic chemistry community have resulted in a number of effective chelators able to form stable complexes with both Cu oxidation states. The imaging community has largely relied on Cu2+ systems containing mixed donors (e.g., N and S) with coordination numbers (CNs) of 4–6. To date, a number of Cu positron-emitting isotopes have found utility in PET-based imaging. These include: 60Cu (t1/2 = 20 min), 62Cu (t1/2 = 9.7 min), 61Cu (t1/2 = 3.4 h), and 64Cu (t1/2 = 12.7 h). Copper-62 is an attractive option despite having a relatively short half-life (t1/2 = 9.7 min) as it can be readily produced via an on-site generator. In contrast, the other Cu isotopes are currently produced from solid targets at cyclotron facilities. Copper-64 (t1/2 = 12.7 h) has a longer half-life and has been used extensively to radiolabel a range of small molecules, antibodies, proteins, and nanoparticles.5 The low energy positron emission of 64Cu also enables the production of high-quality images comparable to the gold standard 18F PET (Table 1). To date, [18F]FDG (2-[18F]fluoro-2-deoxy-D-glucose) is the only PET imaging agent that has seen widespread clinical use. It has been extensively used as an imaging agent for studying glucose metabolism in the brain and identifying potential neurological disorders through reduced glucose usage.4 In the following section, we have highlighted select strategies implementing the Cu2+ radioisotopes for the imaging of neurodegenerative disease-related biomarkers. The reader is also directed to an excellent review by Cui and co-workers for more examples of Cu2+-based radiotracers explored in the context of neurodegenerative disease.5To be viable as clinical candidates, metal-based imaging agents must have the appropriate physicochemical properties (e.g., log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) so as to balance the need for cell permeability and water solubility. Optimizing this features often requires planned synthetic efforts and an ability to derivatize easily the core metal-chelating scaffold. Early efforts to develop Cu-based PET agents focused on square planar metal complexes, particularly Cu(II)(diacetyl-bis(N4-methylthiosemicarbazone)) [Cu2+(atsm)] and Cu(II)(glyoxal-bis(N4-methyl-3-thiosemicarbazonato)) [Cu2+(gtsm)] (Fig. 1).
P) so as to balance the need for cell permeability and water solubility. Optimizing this features often requires planned synthetic efforts and an ability to derivatize easily the core metal-chelating scaffold. Early efforts to develop Cu-based PET agents focused on square planar metal complexes, particularly Cu(II)(diacetyl-bis(N4-methylthiosemicarbazone)) [Cu2+(atsm)] and Cu(II)(glyoxal-bis(N4-methyl-3-thiosemicarbazonato)) [Cu2+(gtsm)] (Fig. 1).
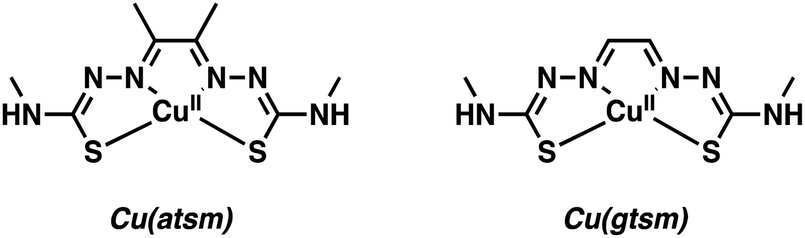 | ||
| Fig. 1 Cu-Based PET agents – Cu(II)(diacetyl-bis(N4-methylthiosemicarbazone)) [Cu(atsm)] and Cu(II)(glyoxal-bis(N4-methyl-3-thiosemicarbazonato)) [Cu(gtsm)]. | ||
Seminal efforts by Yokoyama and co-workers demonstrated that Cu(II)(diacetyl-bis(N4-methylthiosemicarbazone)) Cu(atsm) can be utilized as a PET radiotracer for the imaging of hypoxia in brain and neck cancers.6 Cu(atsm) displayed good stability and blood brain barrier (BBB) permeability with attendant accumulation in regions of the brain suffering from oxidative stress. Under these latter conditions, Cu(atsm) was found to undergo a superoxide oxidase-type conversion wherein superoxide (O2˙−) is oxidized to molecular oxygen (O2) and the Cu2+ center is reduced to Cu+.7 Taking advantage of this redox chemistry, Yoneda and co-workers applied 62Cu(atsm) to image the striatum brain region (i.e., the motor and reward system) in patients with Parkinson's disease (PD).6 High accumulation of 62Cu(atsm) was seen in the striatum of the PD patients compared to the cerebral cortex. This finding was considered consistent with a localized enhancement of oxidative stress due to an increased concentration of superoxide. These efforts by Yoneda and associates are credited with inspiring further imaging research targeting the aberrant oxidative and nitrative processes associated with neurodegeneration.
Continued interest in Cu2+ systems has provided an impetus to develop Cu(atsm) analogues with stimuli-specific properties. Blower and co-workers recently developed an elegant method utilizing Cu(gtsm), the more easily reduced variant of Cu(atsm).8 The use of a glyoxal backbone, as found in gtsm, instead of the diacetyl in atsm, increased the reduction potential of the coordinated Cu2+ making it more susceptible towards intracellular reduction; E1/2 = −0.44 mV versus Ag/AgCl for Cu(gtsm) compared to E1/2 = −0.60 mV versus Ag/AgCl for Cu(atsm).9 In these studies, it was proposed that the Cu(gtsm) complex passes first through the cell membrane via a combination of passive diffusion and protein-carrier mechanisms. Subsequently, the Cu(gtsm) complex disassociates to yield free Cu+ (Scheme 1). In strictly operational terms, these efforts allowed for the PET-based imaging of free Cu+ ions in a transgenic AD mouse model and an initial assessment of the potential of Cu(gtsm) to act as a useful diagnostic biomarker.9
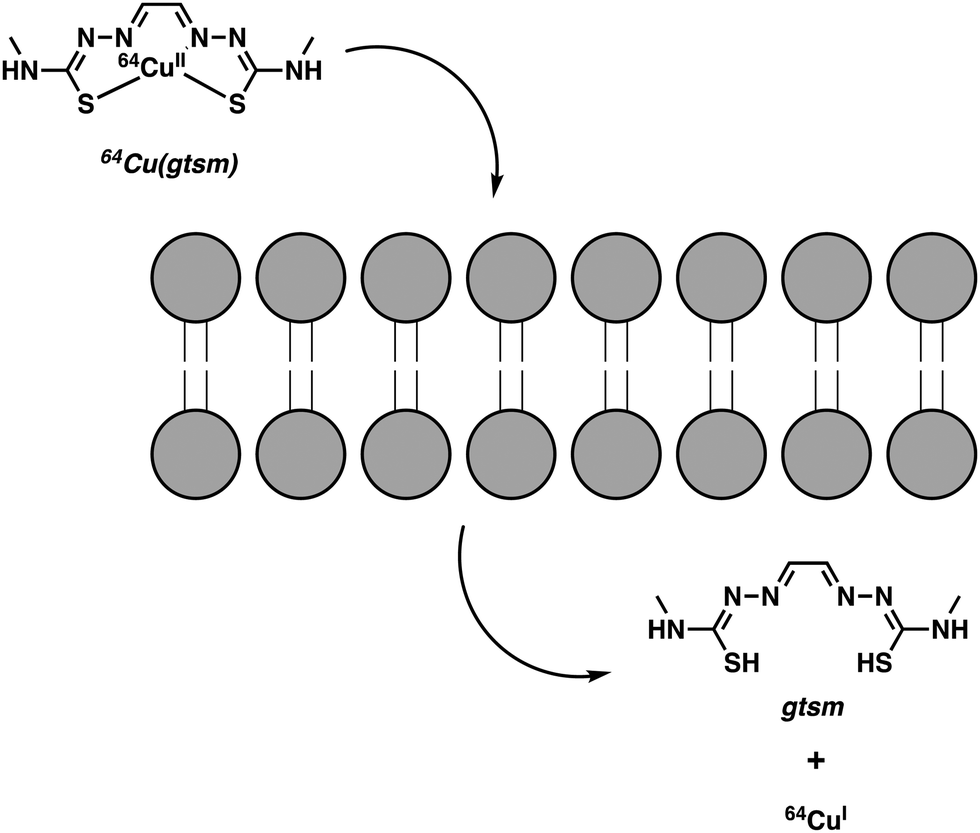 | ||
| Scheme 1 Intracellular reduction of Cu2+ (glyoxal-bis(N4-methyl-3-thiosemicarbazonato)) [Cu(gtsm)], which leads to the release of free Cu+ ions. | ||
Notably, 64Cu(gtsm) was able to penetrate the BBB and increase 64Cu concentrations in the brain. PET imaging revealed statistically significant differences in 64Cu global brain uptake and clearance, as well as in the brain regional 64Cu concentration between AD transgenic mice and age-matched controls.8 These findings were among the first to show that 64Cu could be used as an imaging tool to track changes in the trafficking of free Cu ions in the context of neurodegenerative disease studies.
Free metal ions, such as Cu2+, Fe2+/3+, Mg2+, Mn2+, and Zn2+, are known to induce aggregation of amyloid-beta (Aβ) peptide and support formation of Aβ plaques, a key pathological biomarker of AD.10 Imaging tools enabling the detection of Aβ aggregates are thus of great interest for understanding AD progression. Non-metal based PET imaging of Aβ plaques is possible through the use of radiolabelled benzothiazole and stilbene derivatives, [18F]florbetapir, [18F]flutemetamol, and [18F]florbetaben (Fig. 2).5,11 However, 18F radiotracers (as well as those based on 11C) have a number of disadvantages, such as relatively short half-lives ([18F] t1/2 = 110 min; [11C] t1/2 = 20 min) that require late stage synthetic incorporation of the radiolabel, which is typically produced on site in highly specialized facilities. The development of metal-based strategies that rely on longer lived radionuclides and the insertion of a metal radioisotope into a ligand-conjugate structure with a predetermined high target affinity and selectivity are thus attractive.
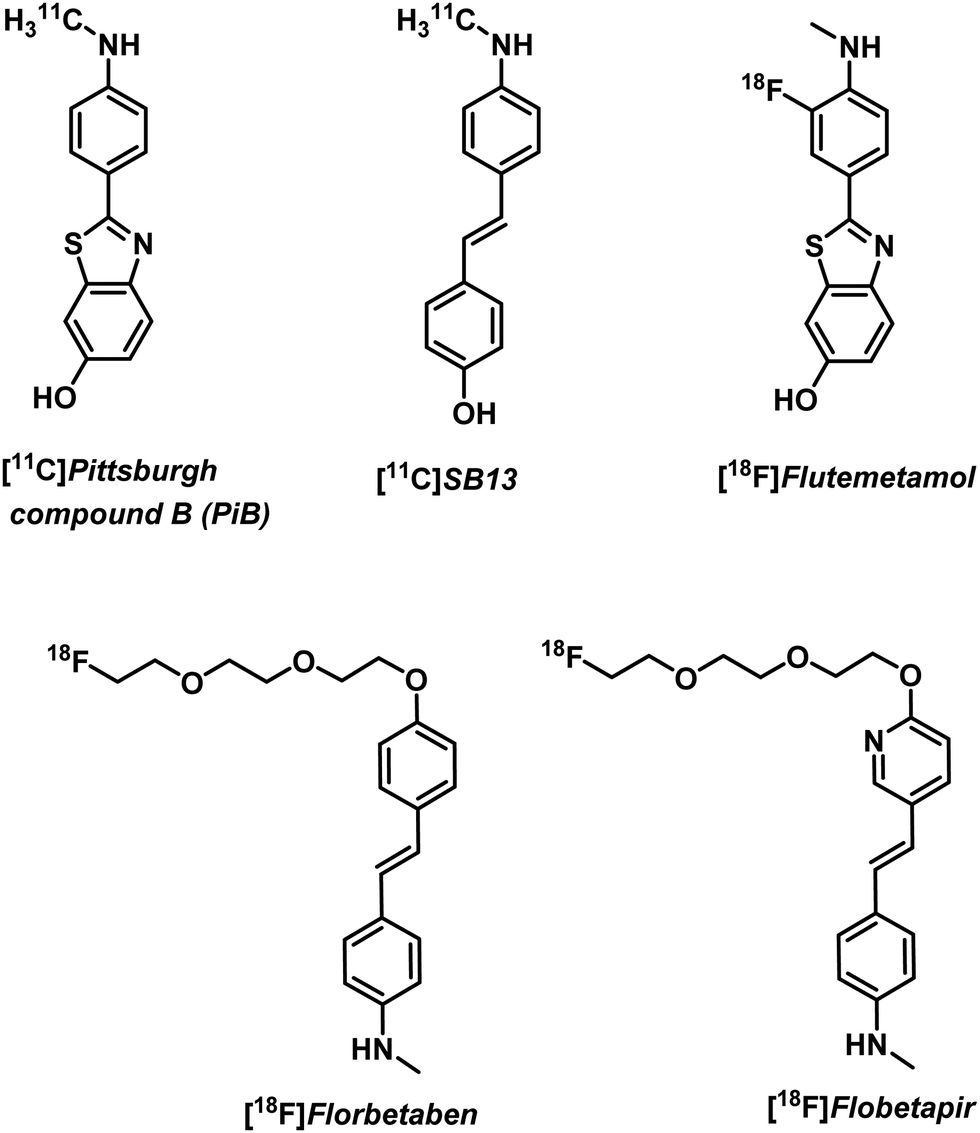 | ||
| Fig. 2 Non-metal-based PET imaging agents for the visualisation of Aβ plaques in vivo. | ||
Donnelly and co-workers have recently reported an elegant demonstration of 64Cu-based PET imaging of Aβ plaques. They did this by coupling the Aβ targeting motifs, benzothiazole and dimethylaminostyrenyl, to Cu(atsm) to generate complexes CuL1–CuL3 (Fig. 3).11 The resultant complexes proved stable in aqueous solutions over a range of pH and avoided the unwanted release of Cu. The authors noted that CuL1 displayed poor aqueous solubility. The majority of their studies thus focused on CuL2 and CuL3. Co-localization experiments with immunostained Aβ plaques provided evidence that CuL2 and CuL3 would bind effectively to Aβ plaques in post-mortem human brain tissue (Fig. 4).
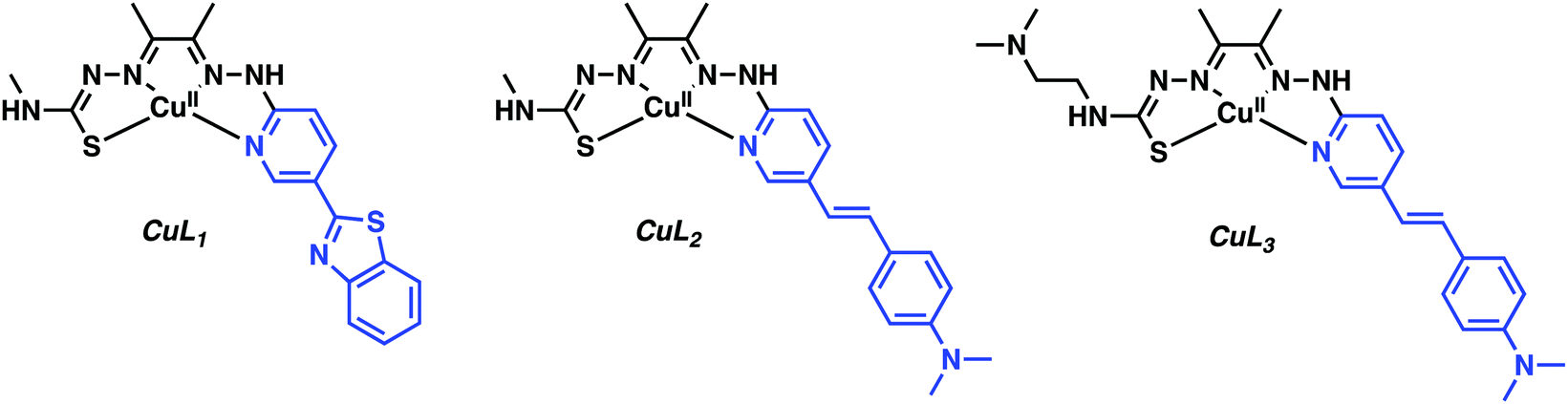 | ||
| Fig. 3 Cu(atsm) hybrids functionalised with the benzothiazole and dimethylaminostyrenyl targeting Aβ ligands highlighted in blue. | ||
 | ||
| Fig. 4 AD human brain sections with 1E8 antibody stained Aβ plaques and fluorescence microscopy of CuL2 (left) and CuL3 (right) binding to Aβ plaques. Reproduced with permission from (J. Am. Chem. Soc., 2013, 135, 16120–16132). Copyright (2019) American Chemical Society. | ||
Subsequent radiolabelling experiments demonstrated marked differences between the aqueous soluble 64CuL2 and –L3, in which 64CuL2 produced only a weak PET signal in the brain after administration, a finding interpreted in terms of poor brain uptake. However, with CuL3, the complex incorporating an appended N,N-dimethylaminoethylene unit, a marked increase in BBB penetration was inferred. Biodistribution experiments in normal mice showed CuL3 had excellent brain uptake: 1.11% ID g−1 at 2 min post injection, and excellent washout with limited accumulation, 0.38% ID g−1 at 30 min. This work highlights some of the challenges associated with developing imaging agents that can effectively penetrate the BBB.
More recently, Mirica and co-workers reported a series of 64Cu-PET agents (TACNL1 and PPL2–5) for the imaging of Aβ plaques.12 These imaging agents consisted of a 1,4,7-triazacylononane (TACN) or 2,11-diaza[3.3](2,6)-pyridinophane (PP) ligands conjugated to 2-phenyl-benzothiazole, which resembles the well-known Aβ-binding dye Thioflavin T (ThT) (Fig. 5). The corresponding Cu complexes were shown to exhibit low nanomolar affinity for Aβ aggregates in vitro and ex vivo with the best binding system proving to be with PPL2. Radiolabelling experiments were then carried out under aqueous conditions (45 °C, 20 minutes, 0.1 M NH4OAc, pH 7). Radiochemical yields of greater than 95% were obtained for L1–L5. The resultant complexes were then used for imaging studies without additional purification. Biodistribution studies demonstrated promising brain uptake in wild-type mice followed by rapid clearance. In vivo PET imaging experiments in AD (Tg2576) transgenic mice revealed radiotracer accumulation in the neck and head for 64Cu-PPL2, 64Cu-PPL4, and 64Cu-PPL5. In contrast, no appreciable brain uptake was seen in the case of 64Cu-TACNL1. The highest brain uptake and tracer accumulation was seen in the case of 64Cu-PPL2, a finding that mirrored what was seen in the in vitro Aβ experiments.
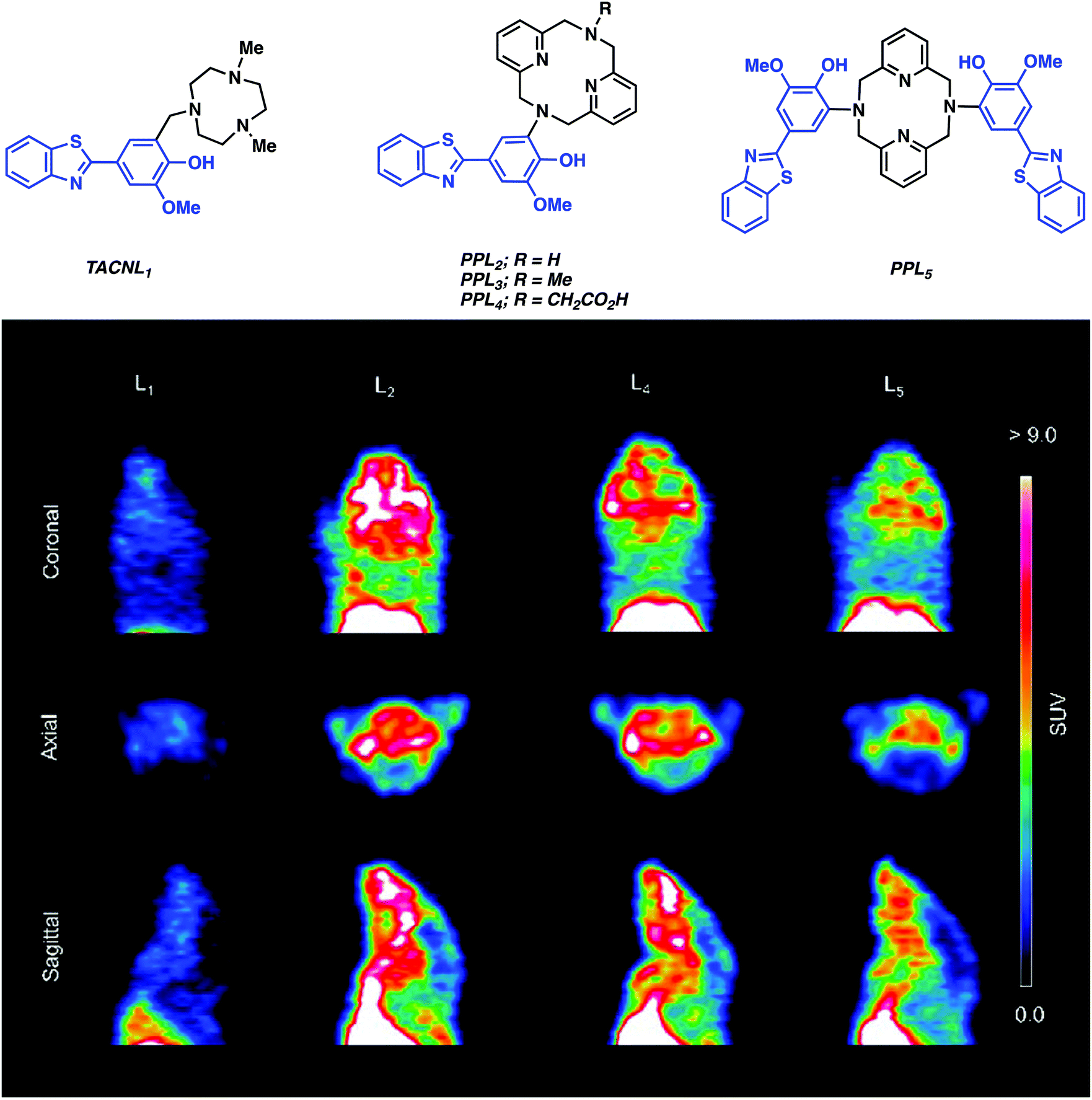 | ||
| Fig. 5 (top) Structures of Cu chelators (TACNL1 and PPL2-5) that are conjugated to a series of Aβ ligands, targeting moieties (highlighted in blue) and (bottom) in vivo PET imaging experiments in AD (Tg2576) transgenic mice carried out using 64Cu-TACNL1, 64Cu-PPL2, 64Cu-PPL4, and 64Cu-PPL5. The best uptake was seen for 64Cu-PPL2 with little or no brain uptake being seen in the case of 64Cu-TACNL1. Adapted with permission from (J. Am. Chem. Soc., 2017, 139, 36, 12550–12558, https://pubs.acs.org/doi/10.1021/jacs.7b05937). Copyright (2019) American Chemical Society. | ||
Efforts with Cu-based PET agents have provided insight into the design criteria that need to be addressed to obtain an effective in vivo imaging agent. In the case of the Donnelly and Mirica studies summarised above, synthetic modification of the chelating motif and judicious choice of targeting unit were crucial in endowing the most effective system with the appropriate properties, particularly BBB penetration. However, continued efforts will be required to evaluate further the potential of these promising candidates as markers for AD. We also believe that additional effort focused on creating next generation Cu-based PET imaging will allow the scope of utility to be expanded to include other neurodegenerative diseases (e.g., Huntington's, ALS, etc.). That researchers will be able to capitalize upon the plethora of Cu ligands found within the bioinorganic and inorganic communities is expected to facilitate these efforts and codify Cu-based PET as a tool to understand better and diagnose more effectively CNS neurodegeneration.
Gallium (68Ga) – PET
Gallium-68 (68Ga, t1/2 = 1.13 h)-based PET radiotracers are becoming attractive as imaging agents owing to the ready availability of gallium-68 from a 68Ge/68Ga generator and an ideal half-life, which complements the biodistribution times typically seen for small molecules. However, widespread adoption of 68Ga-based PET radiotracers has been slow due to concerns associated with the high energy positron emission characteristic of gallium-68 and the resulting high levels of radioactivity (Table 1). However, these concerns are balanced by the commercial availability of 68Ge/68Ga generators, leading us to predict that gallium-68 radiotracers will continue to find utility in the clinic.The most prevalent oxidation state of gallium is the charge dense Ga3+ ion, which forms complexes of CN 4–6. Typical Ga3+ complexes are based on the use of hexadentate ligands containing multiple hard anionic donors, such as 1,4,7,10-tetra-azacyclododecane-1,4,7,10-tetraacetic acid (DOTA), 1,4,7-triazacyclononane-1,4,7-triaceticacid (NOTA), and diethylenetriaminepentaacetic acid (DTPA) (Fig. 6).5 The metallation of these ligands often requires relatively harsh conditions (elevated temperatures, prolonged reaction times) to achieve Ga3+ chelation. Alternative chelators have thus been developed, allowing for improved radiolabelling conditions.2 In this section, we highlight select examples using gallium-68 for the development of PET-based imaging agents that are designed to target neurodegenerative disease.
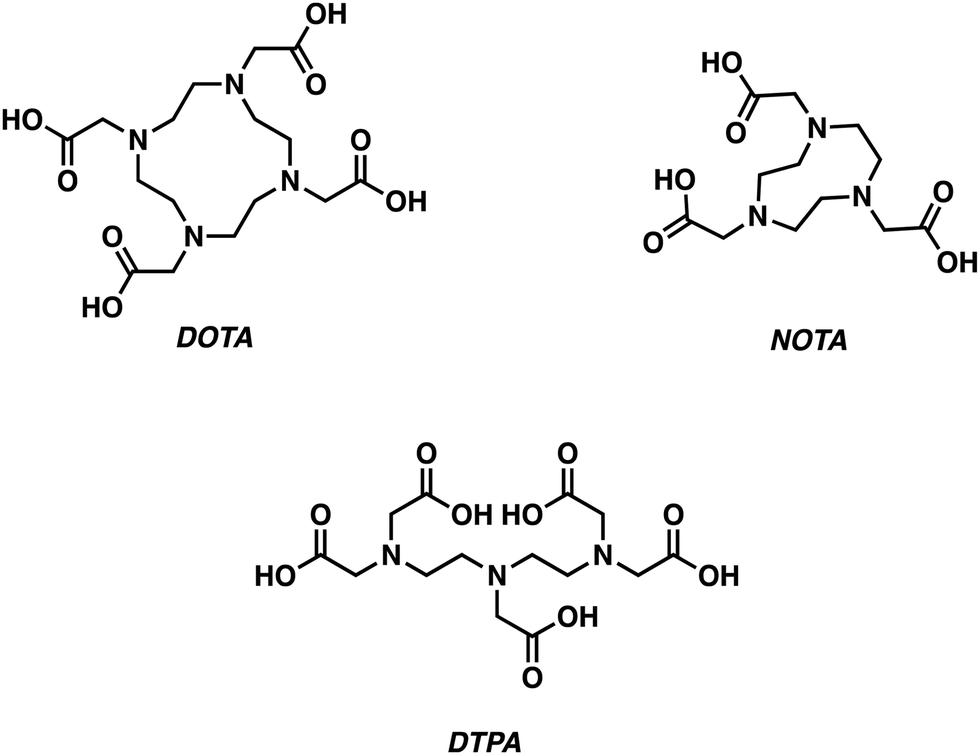 | ||
| Fig. 6 Chemical structures of ligands commonly used to prepare complexes of Ga3+. | ||
Seeking to take advantage of the properties of gallium-68 for use in PET imaging and drawing inspiration from previous small molecule Aβ aggregate probe structure–activity relationship studies (Fig. 2), Saji et al. developed a benzofuran–Ga–DOTA conjugate displaying a high affinity towards Aβ aggregates in vitro and in vivo (Fig. 7).13 Here, the radiolabelled benzofuran–Ga–DOTA conjugate was isolated in 26% yield (>99% purity) by heating the ligand and Ga(NO3)3 in an ammonium acetate buffer (pH 5.8) at 100 °C for 10 min. In vitro binding assays provided support for the conclusion that appending a 68Ga–DOTA metal complex to the benzofuran targeting moiety had little effect on the binding affinity towards Aβ42 aggregates (Ki = 10.8 nM). Subsequent in vitro staining of brain tissue revealed effective binding to Aβ plaques in a tissue section derived from an AD mouse model (Tg2576). Unfortunately, biodistribution experiments in a normal mouse displayed low initial uptake (0.45% ID g−1) in the brain at 2 min post-injection. Currently, it is an open question as to whether additional synthetic modifications will allow for brain uptake. Nevertheless, these results serve to illustrate a simple strategy for developing 68Ga complexes that bind to Aβ aggregates in vitro.
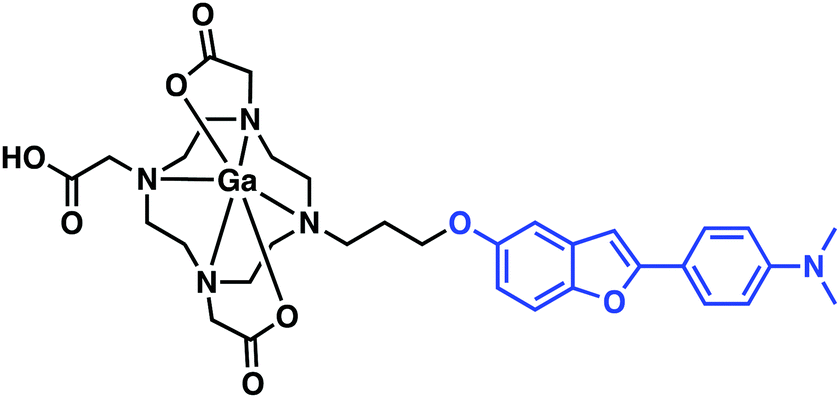 | ||
| Fig. 7 Benzofuran–Ga–DOTA conjugate reported by Saji and co-workers. The benzofuran Aβ targeting ligand is highlighted in blue. | ||
Mishra and co-workers reported a 68Ga chelator-targeting system obtained by conjugating the Aβ binding dimethylamino chalcone motif to DTPA (Fig. 8).14 In this case, rapid 68Ga3+ insertion was achieved in acidic sodium acetate buffer within 10 min (65–70 °C, pH 4.5–5.0). In vitro binding assays demonstrated a high binding affinity towards Aβ aggregates (Ki = 4.18 nM), as monitored by the formation of the fluorescent chalcone-bound Aβ aggregates. In particular, upon presumed binding, a blue shift and a 5.5-fold increase in fluorescence emission intensity ascribed to the Ga–DTPAchalcone complex was observed. This study demonstrated the advantages of a dual-modal PET and fluorescence imaging system as a more refined technique in disease imaging. Animal studies and biodistribution analysis of Ga–DTPAchalcone revealed BBB penetration with brain uptake of 1.24% ID g−1 at 2 minutes post injection and rapid washout with negligible activity (0.36% ID g−1) after 30 minutes. No further biological experiments were performed. Thus, it is difficult at present to assess fully the potential of this PET imaging agent. However, the work as reported does serve to underscore a potentially exciting new approach to developing PET imaging probes for AD.
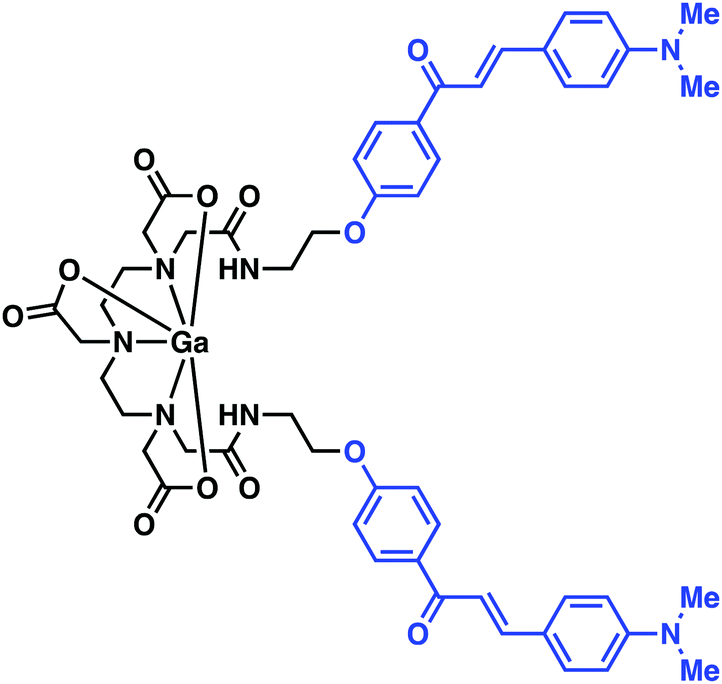 | ||
| Fig. 8 A multi-component complex, Ga–DTPAchalcone, reported by Mishra and co-workers. The chalcone Aβ targeting ligand is highlighted in blue. | ||
Asti and co-workers reported a 68Ga-labeled curcumin complex for the imaging of Aβ plaques (Fig. 9). Curcumin is a phytocompound commonly used in traditional Eastern medicines to which salutary antioxidant, anti-inflammatory, antimicrobial, and anticancer activities have been ascribed.15 The relative planarity of curcumin endows this natural product with an inherent affinity towards Aβ plaques. In the Asti study, curcumin and two synthesised curcumin derivatives, diacetyl-curcumin (DAC) and bis(dihydroxy)curcumin (bDHC), were used to form 2:1 complexes with 68Ga3+ in good-to-excellent yields with a short reaction time when heated at 100 °C in a sodium acetate buffer (pH 5). In vitro studies revealed moderate to high affinities for Aβ plaques. The inherent fluorescence properties of these complexes allowed their cellular uptake to be monitored readily in the A549 human lung cancer cell line. No further biological experiments were carried out; however, this research underscores the potential that 68Ga-curcumin-based systems may hold as potential diagnostic tools in the area of neurodegenerative disease imaging.
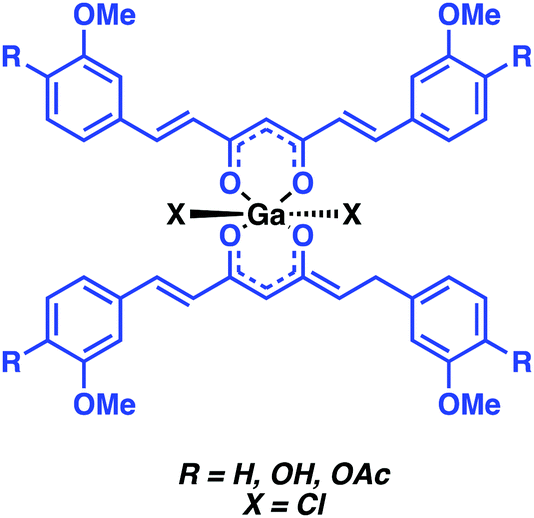 | ||
| Fig. 9 Ga-curcumin complex for the imaging of Aβ plaques reported by Asti and co-workers. The curcumin Aβ targeting ligand is highlighted in blue. | ||
Zirconium (89Zr) – PET
Zirconium-89 (89Zr) has become one of the most popular PET radioisotopes for use with biological targeting/therapeutic agents due to its long half-life (t1/2 = 78.4 h). This has enabled the use of 89Zr monoclonal antibodies (mAb) conjugates (typical biological half-lives = 7 days) for the development of “immuno-PET” radiotracers.16 The use of immuno-PET radiotracers has allowed for more precise targeting of disease-related biomarkers, such as specific neurotoxic protein aggregate conformations. This specificity is expected to provide new insights into neurodegenerative disease and guide the development of treatment protocols that could be individualised from patient to patient.5The high charge and small radius of Zr4+ (CN 4–9) forces a preference toward polyanionic hard donor chelators, such as ethylenediaminetetraacetic acid (EDTA) and diethylenetriaminepentaacetic acid (DTPA). However, the well-known iron (Fe) chelator desferrioxamine B (DFO) has also found utility in in vivo zirconium-89 PET-based applications (Fig. 10).
 | ||
| Fig. 10 Chemical structures of commonly used 89Zr chelators, ethylenediaminetetraacetic acid (EDTA) and desferrioxamine B (Df). | ||
Fissers et al. have recently reported studies detailing the development of a 89Zr immuno-PET strategy for the targeted imaging of Aβ plaques in AD (Fig. 11).17 This work relies on the use of the bifunctional chelator p-isothiocyanatobenzyl desferrioxamine (Df-Bz-NCS), a system routinely used for the rapid and efficient preparation of Zr-89-labeled mAbs. Taking advantage of the isothiocyanate moiety, Df-Bz-NCS was coupled to the amino residue of the antibody JRF/AβN/25, an N-terminal end-specific mAb targeting the full-length human Aβ epitope. Complexation of the resulting construct, Df-Bz-JRF/AβN/25, with 89Zr was successfully achieved under aqueous conditions (HEPES buffer) at room temperature to afford [89Zr]Df-Bz-JRF/AβN/25 in an average yield of 48.39 ± 12.33%. An in vitro binding assay provided evidence of a high binding affinity towards Aβ40 (Kd = 0.85 nM) with no non-specific binding being observed. This high binding affinity was further validated by in vitro autoradiography studies on brain sections from AD Tg mice models. No obvious binding was observed in brain sections of normal mice. [89Zr]Df-Bz-JRF/AβN/25 demonstrated good in vivo stability (i.e., up to 48 hours post injection). [89Zr]Df-Bz-JRF/AβN/25 demonstrated the ability to penetrate the BBB resulting in modest brain uptake in normal mice, thus showing promise as an 89Zr immuno-PET strategy. Unfortunately, no further biological experiments were carried out. Moreover, in vitro studies revealed instability in plasma.
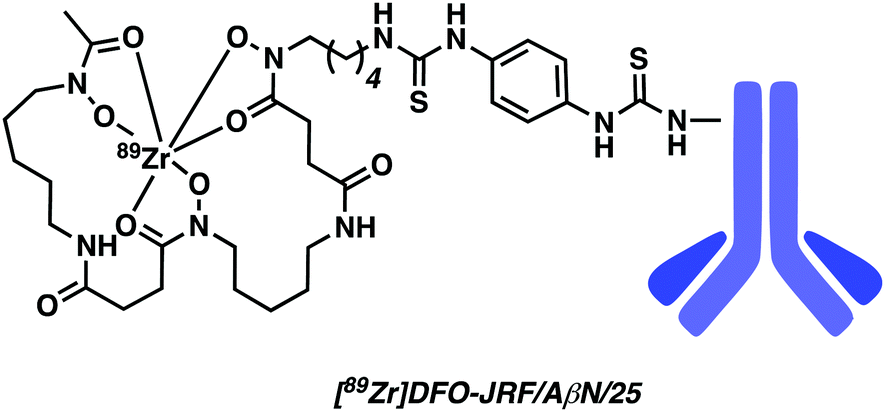 | ||
| Fig. 11 DFO-based 89Zr immuno-PET imaging agent developed by Fissers et al. with the goal of achieving enhanced Aβ plaque imaging. The monoclonal antibody (mAb) Aβ plaque targeting unit is highlighted in blue. | ||
Further studies within the context of developing Zr-based imaging systems should focus on functionalising the best extant systems to improve brain uptake, as well as the synthesis and study of new Zr chelators with improved physicochemical properties. Different mice models should also be screened to allow the utility of these systems across a range of neurodegenerative disease indications.
Technetium (99mTc) – SPECT
The invention of 99mTc/99Mo generators revolutionised nuclear medicine, facilitating the production of the technitium-99m radioisotope and lead to its daily availability in major hospitals. Not surprisingly, this led to a surge in the development of 99mTc-based radiotracers and use of 99mTc as a popular choice for the imaging of a range of diseases. One of the key attributes of 99mTc-based radiotracers, which has led to the predominance of this isotope in the field of nuclear medicine, is the ability to synthesise a range of technitium-99m complexes via a one-pot, one-step “kit” process. This simplicity meant that many sterile 99mTc-based radiotracers could be produced daily with minimal technologist input or without costly equipment.None of the isotopes of Tc are stable enough for routine bench chemistry. However, the similar ionic radius of rhenium (Re) to that of Tc has led to its use as a cold-laboratory (non-radioactive) surrogate for Tc-based complexes. On this basis, various core Tc ion complexes, such as: 99mTc(V)O3+, CpTc(IV)(CO)33+, Tc(IV)N2+, Tc(III)(CO)33+, and Tc(I)(CNR)6+, have been characterised to date.5 One of the most commonly used ligands is the N2S2 donor, bis-aminoethanethiol (BAT); it forms a complex with 99mTc(VI)O3+ that is stable in vivo. For more in-depth information on the history and various biological targets of 99mTc SPECT imaging agents, the reader is directed to the excellent review by Blower.2 Analogous to the metal complex-targeting conjugate systems used in PET, motifs capable of detecting specific biomarkers in neurodegeneration have been developed utilising [99mTc]-SPECT conjugates. In this section, we highlight selected examples of technetium-99m-based systems being explored as SPECT imaging agents for neurodegenerative disease.
An important application of SPECT-based imaging agents has been towards understanding the role of the dopamine transporter (DAT) in a number of neurological processes. Catecholamine neurotransmitters are crucial for maintaining homeostasis throughout the autonomic nervous system. Altered DAT activity has been shown in a number of neurological disorders, with a reduction in DAT being observed in post-mortem examinations of patients with PD and AD. Patients with PD have also been shown to have a selective loss of dopamine neurons in the basal ganglia and substantia nigra.18
DAT antagonists that display a high binding affinity, such as cocaine, are generally constructed around a tropane scaffold (Fig. 12). Influential efforts by Kung and co-workers utilized the tropane motif to design [99mTc]TRODAT-1, a BAT-tropane conjugate (Fig. 12). [99mTc]TRODAT-1 demonstrated nanomolar binding affinity towards DAT in rat striatal homogenate (Ki = 8.7 nM). Ex vivo autoradiography results further confirmed high uptake and retention in the striatal region. These promising results led to the study of [99mTc]TRODAT-1 in normal healthy human subjects. The radiotracer was found to localize in the basal ganglia, a finding consistent with selective DAT binding. Dosimetry studies confirmed that [99mTc]TRODAT-1 was safe for administration to humans and that it could be used in imaging DAT for the diagnosis of PD.19 However, [99mTc]TRODAT-1 has now been largely supplanted by the alternative iodine-123 imaging agent, [123I]FP-β-CIT (DaTSCAN), due to a world-wide shortage of the parent isotope, 99Mo, needed to produce 99mTc.4
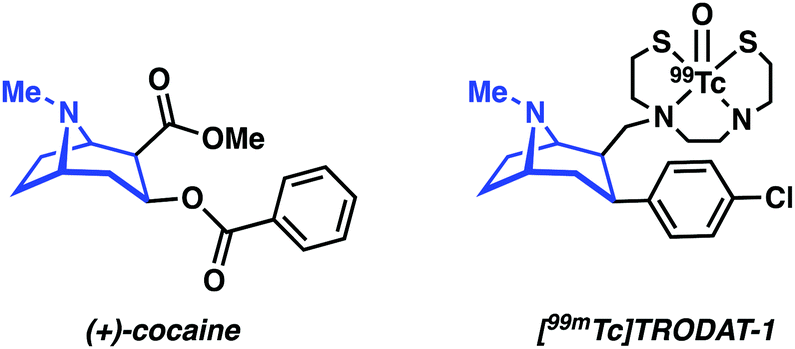 | ||
| Fig. 12 Chemical structure of cocaine, a well-known DAT antagonist, and TRODAT-1, developed as a potential DAT SPECT imaging agent. The tropane core targeting unit is highlighted in blue. | ||
In continuation of their efforts involving the use of labelled benzofuran compounds for Aβ detection, Saji and co-workers reported three [99mTc]-pyridyl benzofuran complexes (Fig. 13).20 The 99mTc and corresponding Re complexes were synthesized using BAT as the chelating ligand. In vitro assays provided evidence of strong affinity towards Aβ42 aggregates (Ki = 13.6–149.6 nM) for the Re complexes. Biodistribution analyses of [99mTc]BAT-Bp-2 in normal mice (ddy mice) revealed brain uptake post injection, 1.80% ID g−1 at 2 minutes, with moderate washout, 0.79% ID g−1 in the brain at 60 minutes, that was deemed sufficient to encourage further study. Furthermore, ex vivo autoradiography with [99mTc]BAT-Bp-2 revealed substantial labelling of Aβ plaques in sections of brain tissue from an AD transgenic (Tg2576) mouse versus age-matched controls (Fig. 13).
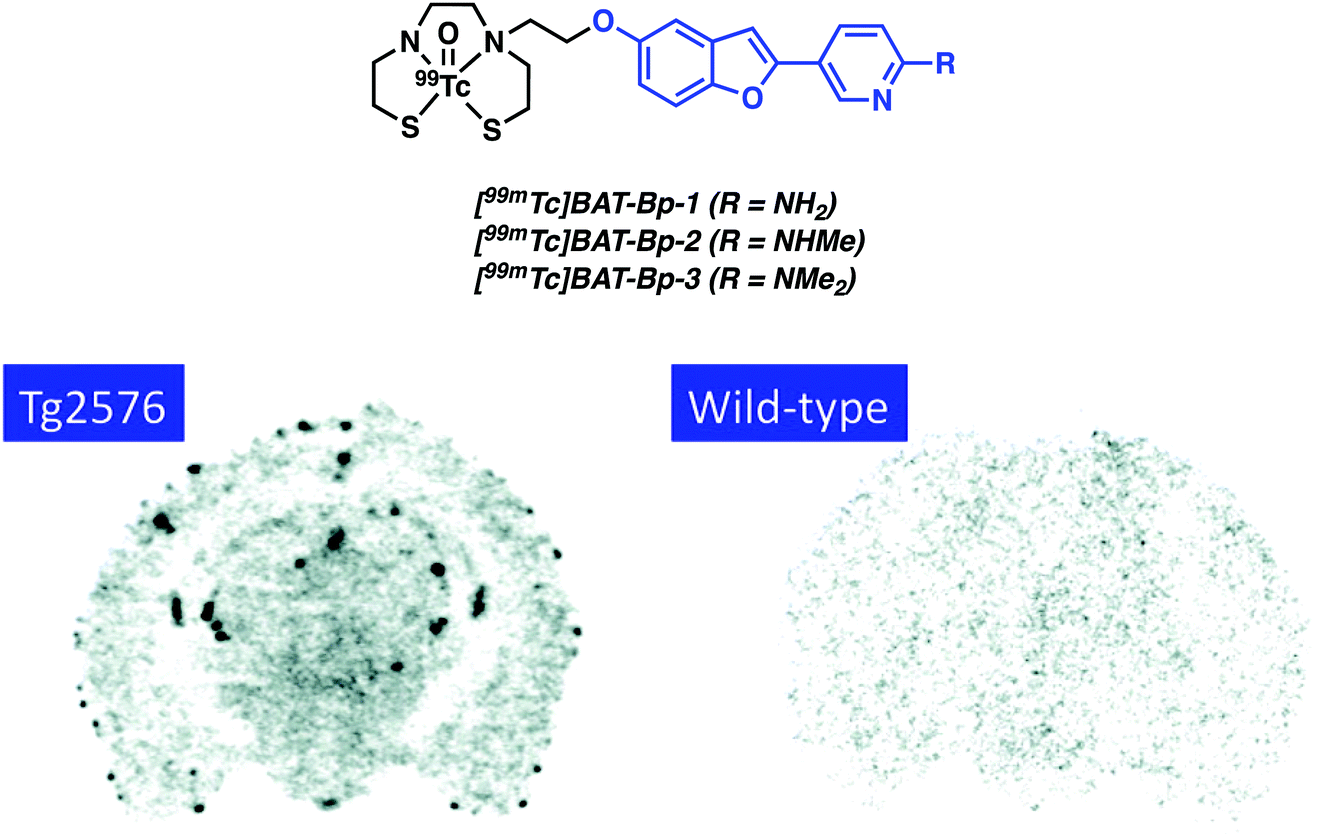 | ||
| Fig. 13 The [99mTc]-pyridyl benzofuran complexes ([99mTc]BAT-Bp-n, where n = 1–3) developed by Saji and co-workers. The 2-pyridylbenzofuran- Aβ targeting ligands are highlighted in blue. Ex vivo autoradiography with [99mTc]BAT-Bp-2 illustrating labelling of Aβ plaques in sections of brain tissue from an AD transgenic (Tg2576) mouse versus age-matched controls (wild-type). Reprinted (adapted) with permission from (J. Med. Chem., 2012, 55, 2279–2286). Copyright (2019) American Chemical Society. | ||
Zhang and co-workers later reported a structural variant of the BAT conjugate containing a 2-arylbenzothiazole moiety.21 A competitive binding assay identified seven Re complexes with potent binding affinity toward Aβ42 aggregates (Ki < 50 nM). Radiolabelling with 99mTc yielded no significant differences in the binding of the [99mTc]BAT conjugate probes with Aβ42 plaques. [99mTc]BAT-benzothiazole conjugate 1 displayed ideal pharmacokinetics with a brain uptake of 2.11% ID g−1 at 2 min and good washout of 0.62% ID g−1 at 60 min in normal mice. Subsequent ex vivo autoradiography experiments revealed extensive labelling of Aβ plaques in brain tissue from AD Tg mice versus age-matched healthy controls. SPECT imaging studies in non-human primates (rhesus monkeys) were also performed. The semiquantitative data resulting from these studies provided support for the notion that [99mTc]BAT-benzothiazole (1) penetrates the BBB of healthy rhesus monkeys (Fig. 14).
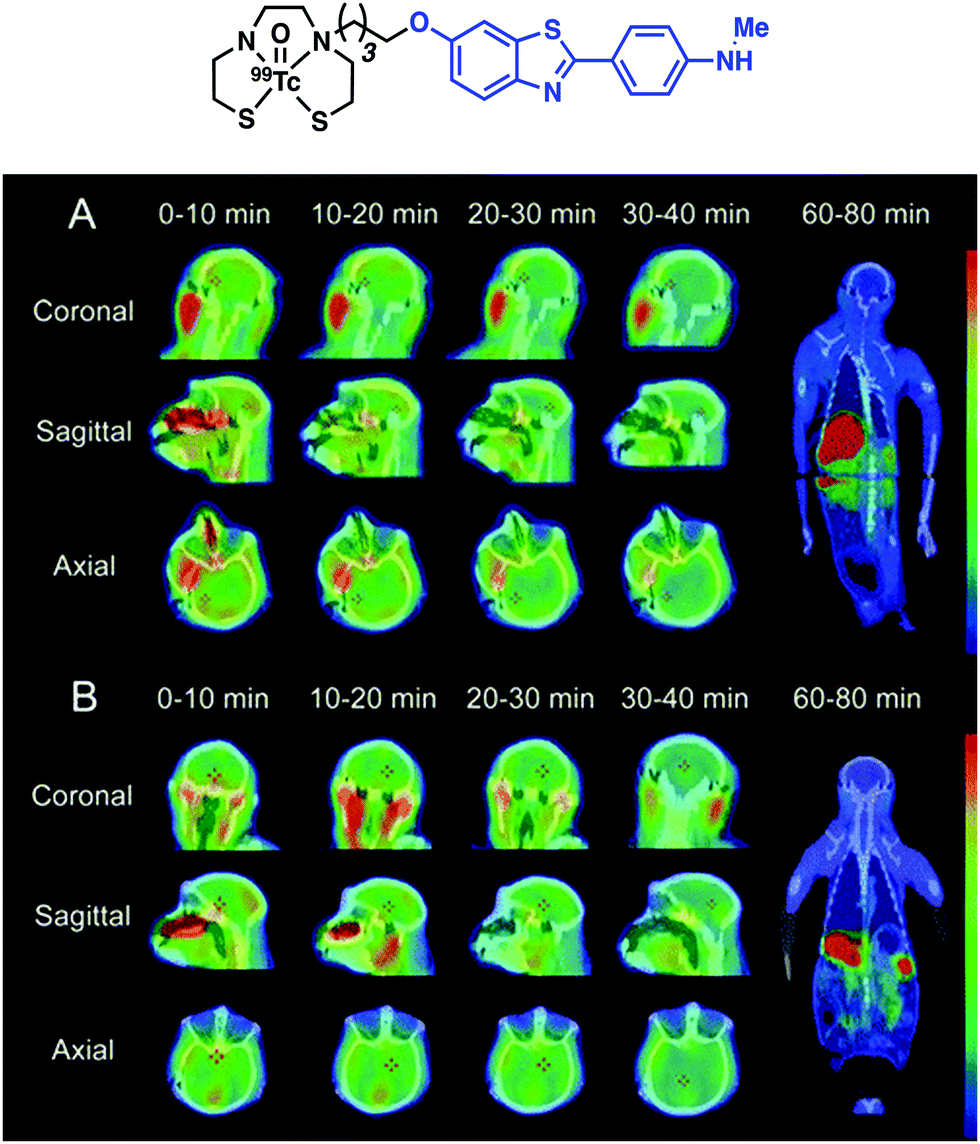 | ||
| Fig. 14 Chemical structure of [99mTc]BAT-benzothiazole conjugate 1 reported by Zhang and co-workers with the benzofuran targeting moiety highlighted in blue. SPECT images of [99mTc]1 in rhesus monkeys (A: 4 years old, male; B: 27 years old, female); SPECT images are overlaid on individual co-registered CT images. Reprinted (adapted) with permission from (Bioconjugate Chem., 2016, 10, 2493–2504). Copyright (2019) American Chemical Society. | ||
Despite metal-based SPECT imaging agents having several advantages, [99mTc]TRODAT-1 is among a select few agents that have actually been used for the clinical diagnosis of neurodegenerative disease. However, as summarised above, a number of promising agents have been reported that underscore how [99mTc]-based systems may have an increased future role in creating improved SPECT imaging agents for CNS disorders. However, balanced against promise are the current supply problems associated with the required parent isotope, 99Mo.
Indium (In) – SPECT
Indium-111 is an isotope characterized by a long half-life (t1/2 = 67.3 h, γ-rays emission – 245 keV). It is thus attractive for conjugation to targeting biomolecules with slow pharmacokinetics, such as mAb. Indium-111 has been the radioisotope of choice for this purpose since the 1980s and was the first radioisotope to be commercially and clinically approved for bioconjugation. Similar to Ga3+, the predominant oxidation state of indium under most aqueous conditions is In3+. Polyanionic, hard donor chelators, such as EDTA, DTPA, and DOTA, have been the ligands of choice for forming In-complexes with good aqueous stability (Fig. 6 and 10).The use of 111In-based SPECT to study neurodegenerative disease has focused on attempts to address issues with BBB penetration and lack of in vivo binding seen with other small-molecule and mAb conjugate systems. For instance, Pardridge et al. reported an Aβ-peptide based radiotracer, which was designed to deposit 111In at pre-existing Aβ plaques. The Pardridge system was constructed by conjugating biotin at the N-terminus of Aβ peptide and attaching DTPA chelator subunits to key lysine residues (Lys16 or Lys28) (Fig. 15).22Ex vivo autoradiography studies provided support for the expectation that significant binding to Aβ plaques from brain sections of patients with AD could be achieved. However, additional quantitative binding studies and in vivo kinetic analysis were not reported. Despite indium-111 being well established in the field of immuno-based imaging, the rise of 89Zr as a more readily available and longer-lived surrogate is seeing 111In-based SPECT agents being increasingly replaced by their 89Zr-based immuno-PET counterparts.
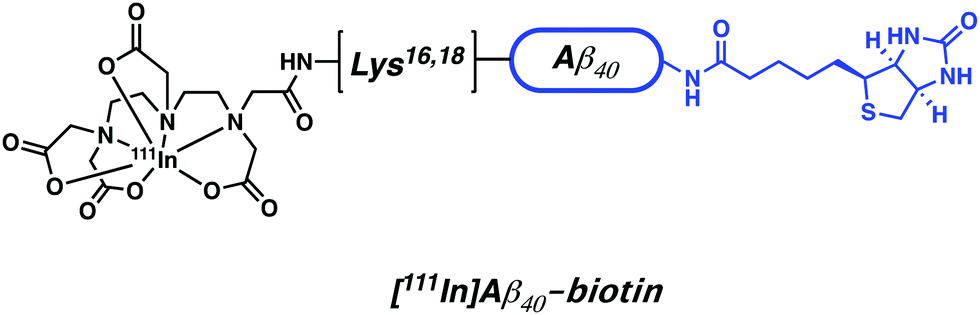 | ||
| Fig. 15 Aβ-peptide based 111In radiotracer developed by Pardridge et al. The biotin targeting unit is highlighted in blue. | ||
Magnetic resonance imaging (MRI) in neurodegenerative disease
Optimization of imaging instruments and the availability of a number of small molecule probes, known as contrast agents (CAs) has made magnetic resonance imaging (MRI) a powerful technique within the imaging paradigm. In comparison to PET and SPECT, MRI is generally cheaper, more widely accessible, and provides higher spatial and temporal resolution (>0.01–1 mm and >0.01–1 s) with enhanced (total) depth penetration through biological tissue. These properties make MRI an excellent tool for in vivo studies and clinical use. A major advantage of MRI over radiotracer (PET/SPECT) approaches is that MRI CA signals can be modulated by environmental changes or via exposure to specific analytes, allowing for real-time functional imaging (i.e., fMRI) in living subjects. However, as a general rule MRI lacks the sensitivity seen for PET and SPECT. Recent efforts within the MR imaging community have focused on the development of tools that enable the imaging of neurodegenerative diseases.23 For extensive coverage of the theory and development of MRI contrast agents, current directions, and challenges for the field, the reader is directed to two excellent reviews by Meade23 and Caravan.24The most common contrast agents for MRI utilize a polydentate ligand with a coordinated paramagnetic metal ion (e.g., Gd3+, Fe2+/3+, and Mn2+/3+). The operational mechanism for MRI involves administration of the contrast agent followed by the application of a short radiofrequency (RF) pulse to a sample (e.g., human, mouse, or test-tube) placed within an external magnetic field (i.e., the scanner). This RF pulse results in the excitation of the nuclear spins of surrounding water protons, which then relax to the equilibrium of the external field. The rates of the relaxation, based upon the longitudinal (T1) and transverse (T2) time constants, are dependent upon the specific environment of the water molecules. It is the variation of these rates that forms the basis of the signal intensity in typical anatomical and contrast-enhanced MRI.
Paramagnetic metal ions, such as Gd3+, Fe2+/3+, and Mn2+/3+, contain unpaired electrons. These ions generate magnetic moments that interact with nearby water molecules to increase proton nuclear relaxation rates. The extent to which a contrast agent can change the T1 or T2 of water is generally defined by relaxivity, r1 or r2, respectively. Relaxivity is the change in bulk relaxation rate per unit of contrast agent, normally in units of mM−1 s−1, and is defined by eqn (1). Briefly, (1/T0i) is the relaxation rate in the absence of the contrast agent, (1/Ti) is the relaxation rate in the presence of contrast agent, and [CA] is the concentration of the MRI contrast agent.
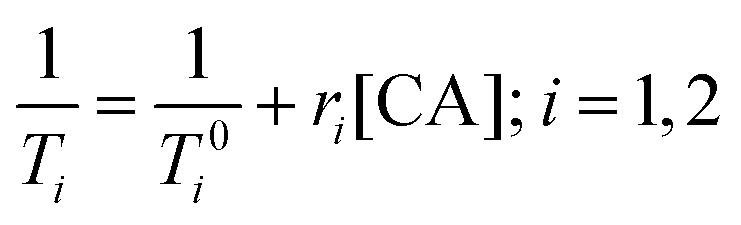 | (1) |
As a general rule, the greater the relaxivity (ri) of a contrast agent, the better the CA increases the rate of relaxation of nearby protons. This enhancement generally manifests as an improved resolution contrast image. Additional changes to the contrast image can also occur depending upon the local environment. Indeed, the relaxation times of endogenous water molecules vary upon interacting with macromolecules (e.g., proteins and DNA) generating a contrast difference in T1- and T2-weighted images compared to standard conditions.
Due to the physical processes underlying relaxation rates, T2 is always shorter than T1. The T2 is thus more difficult to modulate via the addition of contrast agents. In the case of standard Gd3+ contrast agents with relatively similar r1 and r2 values, the slower T1 will display a larger percent modulation than the faster T2. Traditionally, small molecule CAs have been used to increase the signal change in the T1-weighted image (Fig. 16a). For transverse relaxation of these small molecule probes, the observed T2 or T2* is shorter than the true T2 due to field inhomogeneities and other factors. Efforts to generate meaningful changes in T2 have focused on the use of nanoparticle systems. The resulting systems are referred to as T2, or T2* MRI contrast agents; they generally lead to a decrease in the signal of T2/T2*-weighted images.
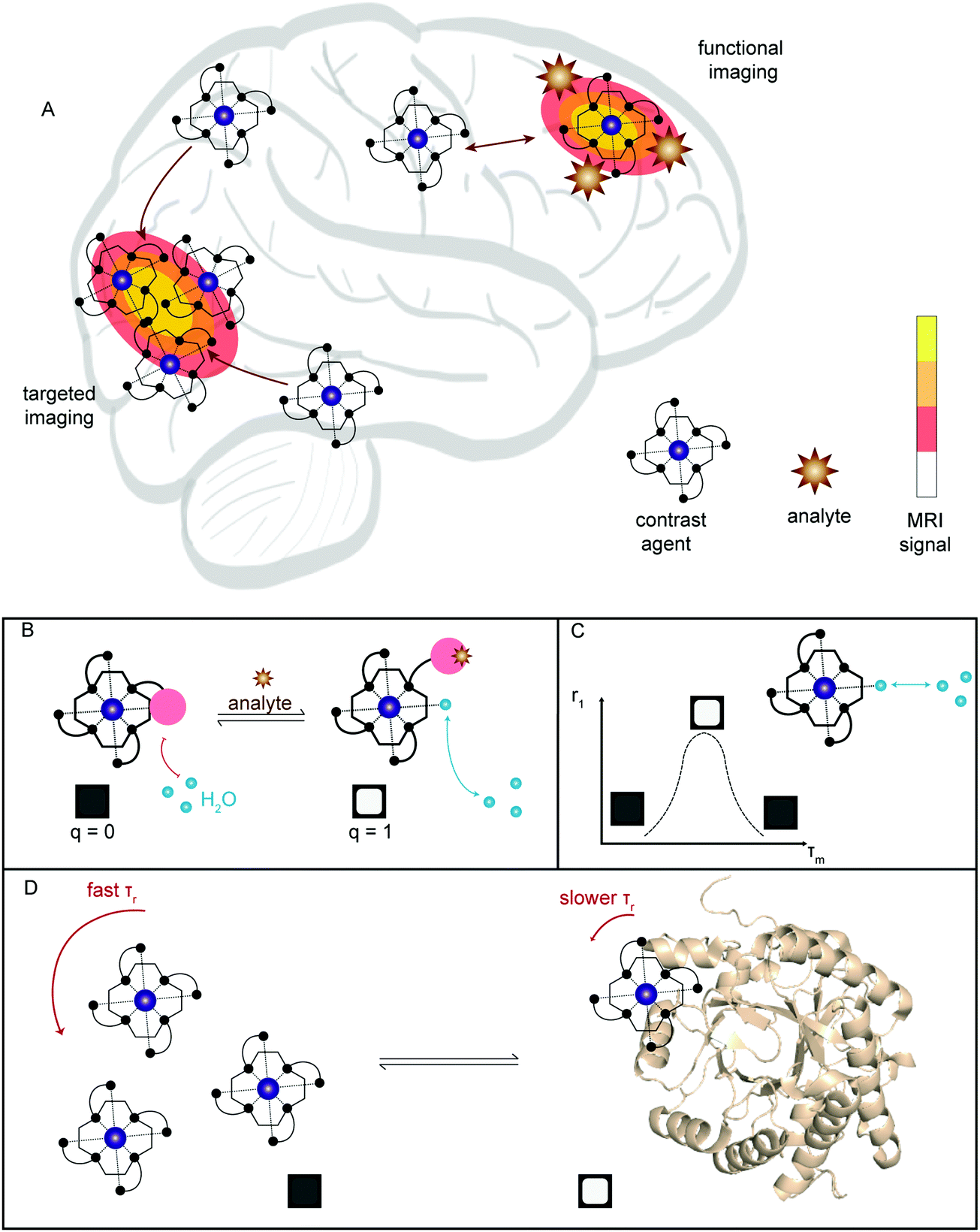 | ||
| Fig. 16 (A) Simplified illustration showing the two main ways in which MRI has been applied towards neurodegenerative imaging; targeted imaging by selective accumulation of contrast agent and functional imaging, where the signal is modulated by presence of an analyte. (B–D) Illustration of the common methods for developing functional T1 contrast agents: (B) modulation of bound water, q; (C) optimisation of water exchange rate, τm; (D) binding to a higher molecular weight conjugate to alter rotational correlation time, τr. | ||
For T1-weighted contrast agents, three main factors determine the relaxivity of the agent:
• q – average number of water molecules bound directly to the paramagnetic metal centre.
• τm – mean residence lifetime of the bound water molecule, i.e., the inverse of the water exchange rate (kex).
• τr – rotational correlation time, i.e., how rapidly the molecule tumbles (rotates) in solution.
In the case of parameter q, water molecules in the first coordination sphere of the metal complex experience the greatest T1 enhancement. Thus, the larger the q-value the higher the r1 value (Fig. 16b). For τm, an exchange rate optimised to the timescale of relaxation will generate the highest relaxation enhancement. To date, τm remains difficult to predict, measure, and implement in CA design (Fig. 16c). However, manipulation of τr is a commonly employed strategy and achieved by increasing the molecular weight of the MRI contrast agent (e.g., by binding to a protein, as larger molecules tend to tumble and rotate much slower) (Fig. 16d). For T1 contrast agents, changes in τr have minimal effects at higher field strengths (>3 T), limiting use of such systems in high-field preclinical and ultra-modern clinical scanners. For T2-weighted agents, changes in tumbling rates are still an effective way to modulate contrast at any field strength. Aggregation remains the primary method for functional imaging with T2-probes. Alternative methods, such as chemical exchange saturation transfer (CEST) and paramagnetically-enhanced chemical shift imaging, are available, but due to the limited applications in neurodegenerative disease imaging they will not be covered in this review. For further details on applications of these techniques, we refer the reader to the aforementioned reviews.23,24
The MRI imaging section will be divided by each paramagnetic metal ions Gd3+, Fe2+/3+, and Mn2+/3+ with focus on the precise method and biomarker relevant to neurodegenerative diseases. These examples will demonstrate both targeted and functional approaches.
Gadolinium (Gd3+) – MRI
Gd3+ has the largest number of unpaired electrons for an aqueous stable ion (S = 7/2), a high available coordination number (CN 7–10), and close-to-ideal electronic relaxation rate that optimises its ability to increase the relaxation rate of water protons through simple water–metal interactions.24 However, free Gd3+ metal ions display off-target toxicity, requiring the use of a tight binding chelator. Polydentate and hard anionic donors, such as those found in diethylenetriaminepentaacetic acid (DTPA) and 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), remain the ligands-of-choice for Gd-based MRI contrast agents in academic research and clinical use. Due to its higher thermal stability and worries of systemic toxicity resulting from leaching of Gd3+ from linear chelators (e.g., DTPA), the macrocyclic DOTA is now preferred for new applications, as illustrated throughout this MRI section.23MRI-based imaging of CNS disorders has focused on methods of targeted (MRI) or functional imaging (fMRI). Recent efforts towards targeted imaging have utilized metal complex-targeting conjugates analogous to PET/SPECT. An exemplary demonstration of such efforts was recently reported by DeBeof and co-workers using arecoline- and tropane-based Gd–DOTA metal complexes for the selective MRI imaging of DAT.25 DeBeof and associates recognized that small molecule binding within a macromolecular biomarker would yield an enhancement in T1via modulation of the molecular rotation (τr). The tropane-derived CA-1 (Fig. 17) produced a 38% increase in the r1 value from 6.32 mM−1 s−1 to 8.71 mM−1 s−1 in the presence of DAT-transfected HEK293 cells at a 7 T magnetic field. Binding studies with tropane-derived CA-2 revealed a high binding affinity to DAT, similar to that of the DAT antagonist cocaine (303 nM). Unfortunately, due to the relative insensitivity of MRI, which typically requires >10 μM detectable concentration of contrast agent, and DAT only being present at nM concentrations in vivo, these probes were not pursued further in the context of potential biological applications. However, this work provided an elegant demonstration of combining targeted molecules with manipulation of τr towards the detection of neurodegenerative biomarkers.
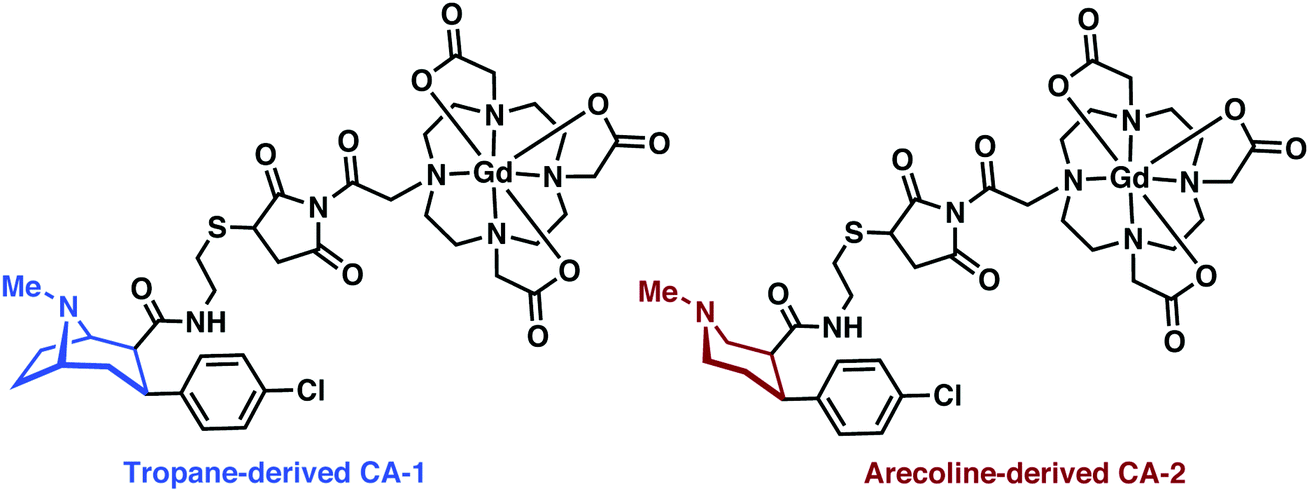 | ||
| Fig. 17 Chemical structures of tropane and arecoline MRI-analogues, CA-1 and CA-2. | ||
A similar Gd–DOTA-targeting moiety conjugate strategy was recently used by Wang and co-workers to effect the MR imaging of myelin.26 Myelination is a fundamental biological process within the nervous system, which involves the formation of myelin sheaths along neuronal axons for rapid and efficient conduction of nerve impulses. Abnormalities or destruction of myelin results in the neurodegenerative disorder Multiple Sclerosis (MS). The ability to visualise and discriminate between healthy and abnormal myelin in vivo could provide insight into this disease and mechanisms behind its progression.
In an attempt to visualize myelination with MRI, Wang and co-workers developed a first-generation conjugate that was prepared by appending a known myelin selective coumarin-based fluorescent probe to Gd–DOTA to form the MRI contrast agent, myelin dye-1 (cf.Fig. 18). Ex vivo imaging of partially demyelinated mouse and rat brain tissue stained with myelin dye-1 revealed discrimination between regions that were high and low in myelin. Subsequent MRI T1 mapping of brain tissue complemented the fluorescence data, and provided support for the suggestion that retention of myelin dye-1 is proportional to the level of myelination in the brain. Due to the polar nature of myelin dye-1, intra-cerebroventricular infusion was required to bypass the BBB and enable in vivo imaging of demyelinated lesions in L-α-lysophosphatidylcholine (LPC) treated rats. MRI mapping yielded relatively long T1 values in demyelinated lesions, a result ascribed to little or no binding of myelin dye-1 in these latter samples. These efforts highlight the potential of MRI contrast agents that can selectively bind to myelin in vivo and underscores the promise they may have in terms of facilitating early diagnoses of MS and monitoring treatments aimed at myelin repair.
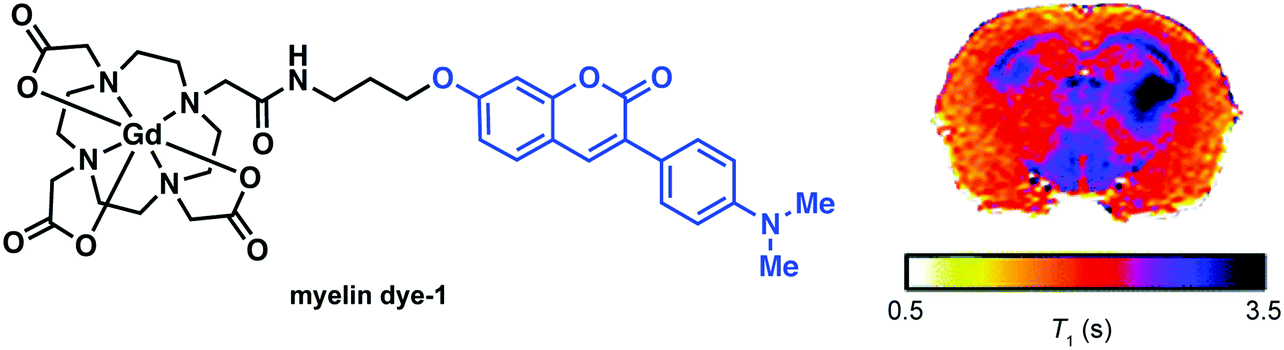 | ||
| Fig. 18 (left) Chemical structure of myelin dye-1 with the coumarin targeting moiety highlighted in blue and (right) T1 mapping of a Sprague–Dawley rat brain proximal after 5 h post injection. The dark regions (short T1) are indicative of increased binding in areas of myelination. Reprinted (adapted) with permission from (J. Med. Chem., 2012, 55, 2279–2286). Copyright (2019) American Chemical Society. | ||
Analogous to the PET and SPECT conjugate literature, Ferroud and co-workers designed a series of Gd3+-based MRI conjugated contrast agents comprising of an Aβ aggregate targeting moiety, a linker, and a Gd3+ chelator (Fig. 19).27 Ferroud et al. carried out a structure activity relationship (SAR) study on 16 different contrast agents, varying the Gd3+ chelator from DOTA to PCTA (3,6,9,15-tetraazabicyclo[9.3.1]-pentadeca-1(15),11,13-triene-3,6,9-triacetic acid), the Aβ targeting ligand between 2-arylbenzothiazole, 2-arylbenzoxazole, and various stilbene derivatives, and the charge of the linker from neutral to overall positive or negative while likewise adjusting its length. The resultant complexes showed good water solubility with nanomolar binding affinities to Aβ aggregates. However, they failed to pass through an intact (healthy) BBB. This shortcoming again highlights the significant challenges associated with developing effective imaging agents that have both high affinity for a neurodegenerative biomarker and the ability to penetrate the BBB.
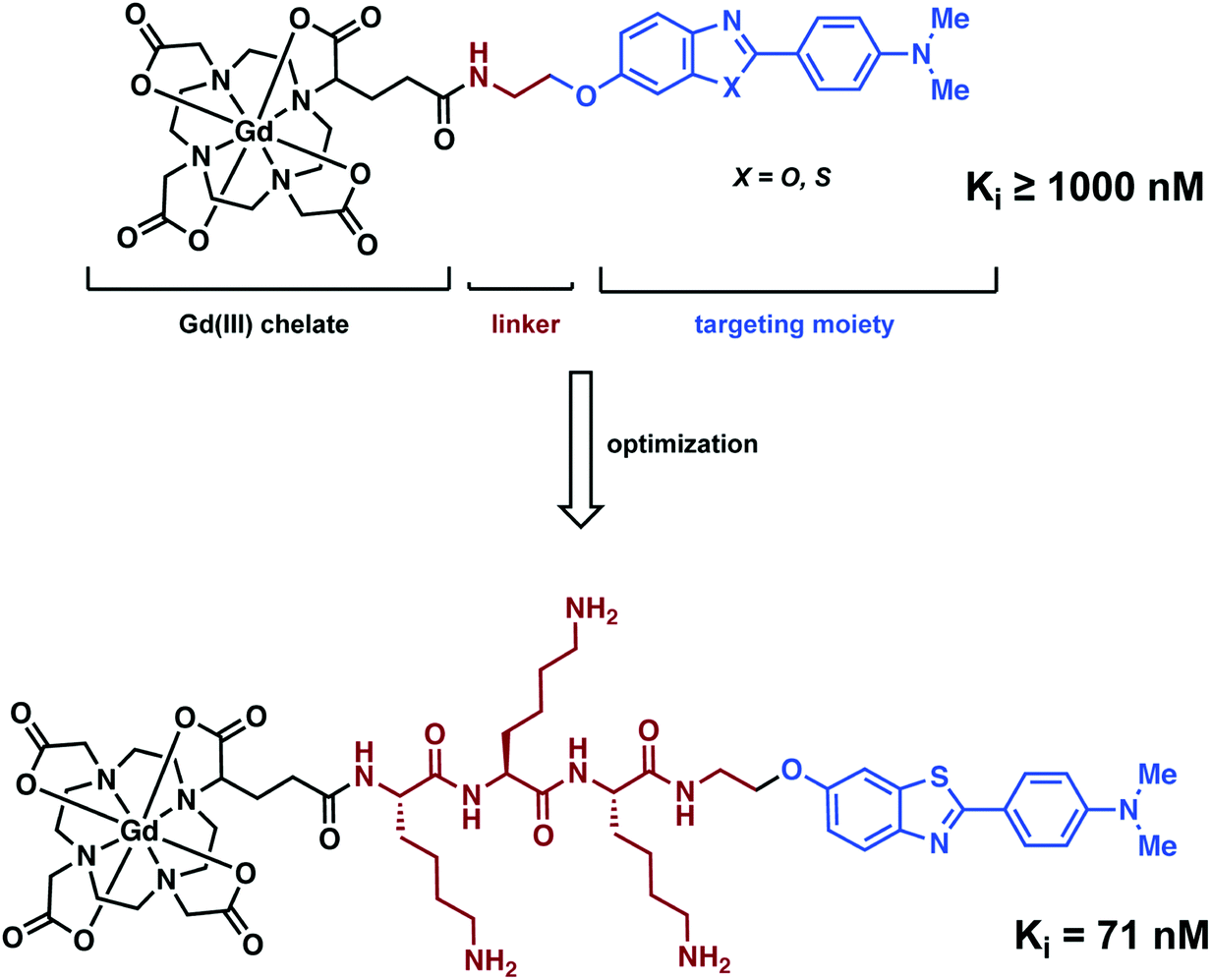 | ||
| Fig. 19 Structural optimisation of a Gd–DOTA complex for the imaging of Aβ plaques. | ||
DOTA-type macrocycles ligate to eight of the nine available Gd3+ coordination sites, leaving one site available for an inner sphere water molecule.23 The Meade group has exploited this free binding site by pioneering the development of q-modulated MRI contrast agents. Such systems are designed to only allow inner-sphere access of water upon binding to the target analyte (biomarker). A seminal contribution by Meade and co-workers was the development of a calcium ion (Ca2+) responsive MRI agent, in which the relaxivity was controlled by the reversible binding of Ca2+ ions.28
Ca2+ ions play a vital role in maintaining proper cell function with deviations in neuronal Ca2+ signalling being correlated with a number of neurodegenerative diseases. The Ca2+-responsive MRI contrast agent developed by Meade and co-workers incorporated a seven-coordinate DOTA analogue, 1,4,7-tris-(carboxymethyl)-1,4,7,10-tetraazacyclododecane (DOPTA), wherein q can vary from 0 to 2 and was linked to the 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) Ca2+ binding subunit via a propyl chain (Scheme 2). In the absence of Ca2+, the aromatic iminoacetates of the BAPTA moiety interact with the Gd3+ centre. The presence of Ca2+ modulated the binding of the aromatic iminoacetates such that they interacted with the Ca2+ ions, rather than the Gd3+ centre. The resulting exposure of the Gd3+ centre to water molecules caused a change in relaxivity. Thus a stimulus-responsive (Gd–DOPTA)2–Ca2+ sensor was produced that was found to have high sensitivity toward Ca2+ (detection range: 0.1 to 10 μM). This MR-based sensor was found to display a 1:1 stoichiometry for the Ca2+ ion triggers and produce an 80% overall increase in relaxivity upon Ca2+ complexation. This work stands as an excellent initial example of how to manipulate various parameters (e.g., q) to enact an effective change in T1 signal. Unfortunately, application of these agents in the context of neurodegenerative disease has been limited due to difficulties in tuning the calcium affinity and MRI sensitivity to match the relative intracellular (0–100 μM) and extracellular (0.1–2 mM) calcium fluctuations typically seen in the brain. Applications may also prove limited by an apparent lack of cell permeability.
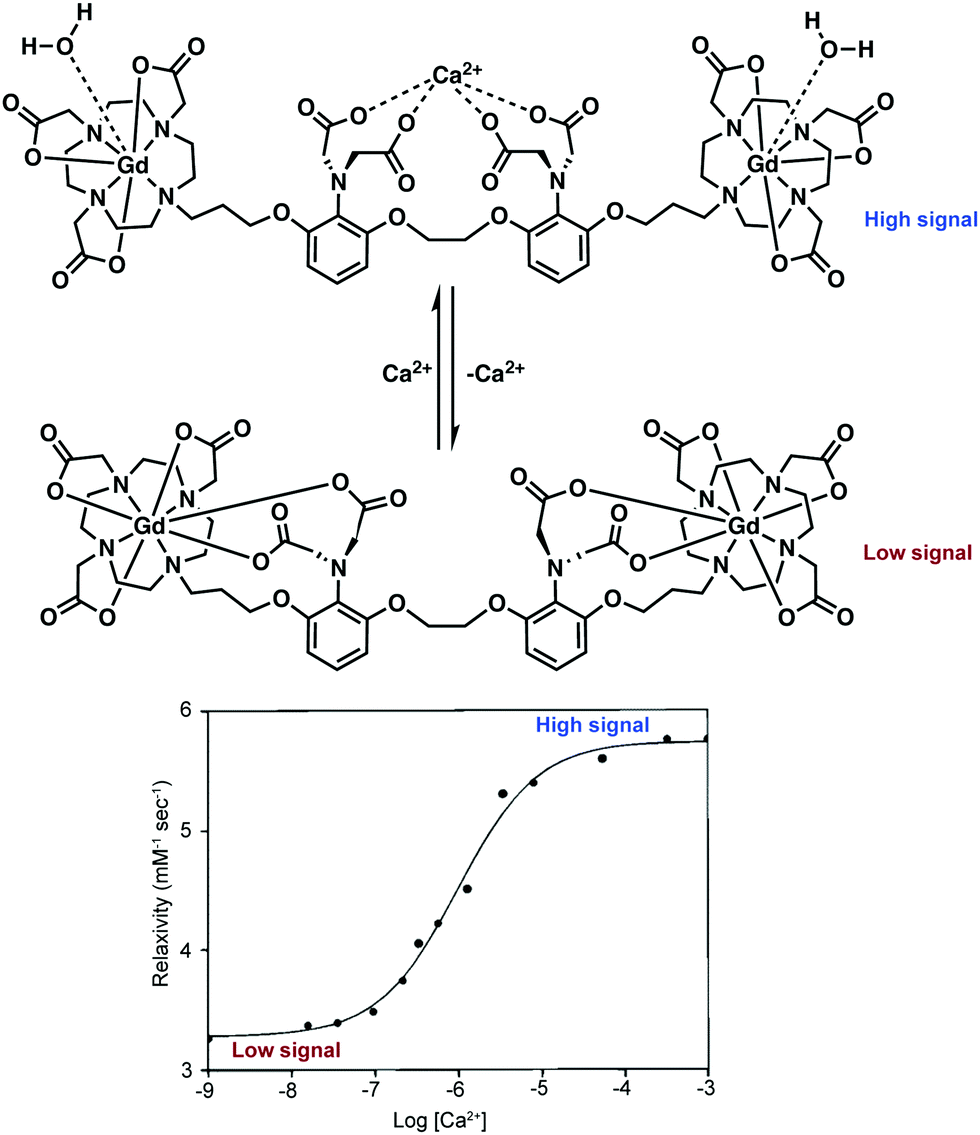 | ||
| Scheme 2 q-Modulating Gd–DOPTA–BAPTA contrast reported by Meade and coworkers that permits the MR-monitored reversible detection of Ca2+ ions. Upon binding of Ca2+, water access to the Gd3+ ion is increased, raising the relaxivity and MRI signal of the contrast agent. The relationship between relaxivity and Ca2+ concentration is displayed in the lower plot. Reprinted (adapted) with permission from (J. Am. Chem. Soc., 1999, 121, 1413–1414). Copyright (2019) American Chemical Society. | ||
Chang and co-workers developed a class of Cu+ responsive q-modulated MRI contrast agents as potential tools for studying copper dysregulation in neurodegenerative diseases.29 The general scaffold was a Gd–DOTA analogue featuring a thioether-rich conjugate (Scheme 3). In the absence of Cu+ ions, the nitrogen on the pyridine ring unit coordinates the Gd3+ centre, restricting inner sphere water coordination. Upon addition of Cu+, the pyridine nitrogen and thioether moiety undergoes a conformational change, a chemical switching event that serves to “turn off” the Gd–pyridine binding interaction. The increased water access to the Gd3+ centre leads to a marked enhancement in the relaxivity (r1), with the largest overall change in r1 (360%; from 1.5 to 6.9 mM−1 s−1) being seen in the case of compound CG2.
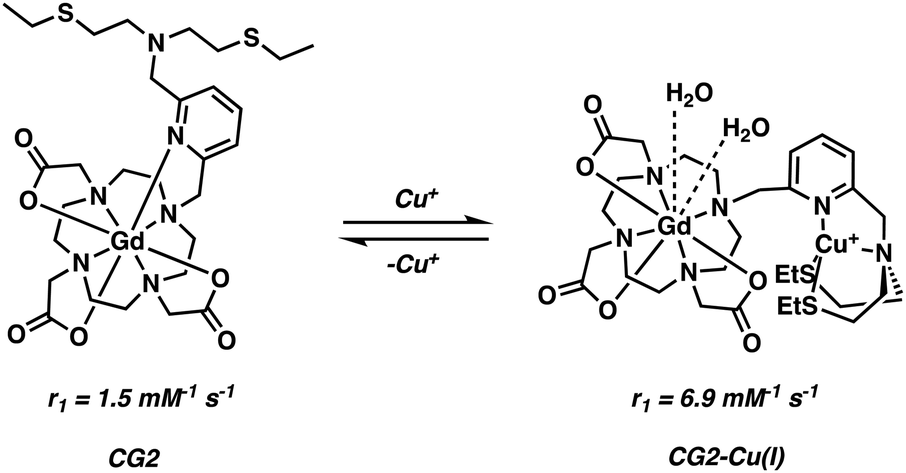 | ||
| Scheme 3 Chang's q-modulating Gd3+-DO3A contrast agent that permits the reversible detection of Cu2+ ions via MRI. | ||
In order to improve cellular uptake, Chang and coworkers included an octa-arginine unit to yield Arg8CG2 (Fig. 20).29 This system was applied towards imaging a Menkes disease model fibroblast cell line and control fibroblasts. Menkes disease is a disorder characterized by a progressive deterioration of the nervous system. In comparison to the untagged CG2, the arginine tagged complex showed a remarkable 9-fold greater accumulation in both cell lines. The overall relaxivity change of Arg8CG2 was slightly lower than with CG2, but gave a higher absolute r1 for both the ‘off’ and ‘on’ states (3.9 to 12.5 mM−1 s−1). However, it should be noted that the buffer system was different for these two studies. While a higher absolute signal will lower the detection threshold, a larger relative change between the bound and unbound conditions should lead to greater sensitivity in determining analyte binding. Phantom MR images of the two cell lines revealed a higher contrast when Arg8CG2 was present than in its absence. Treatment of the cells with a copper chelator allowed monitoring of the depletion of intracellular copper through MRI monitoring, indicating the potential of such an approach for in vivo biological MRI studies.
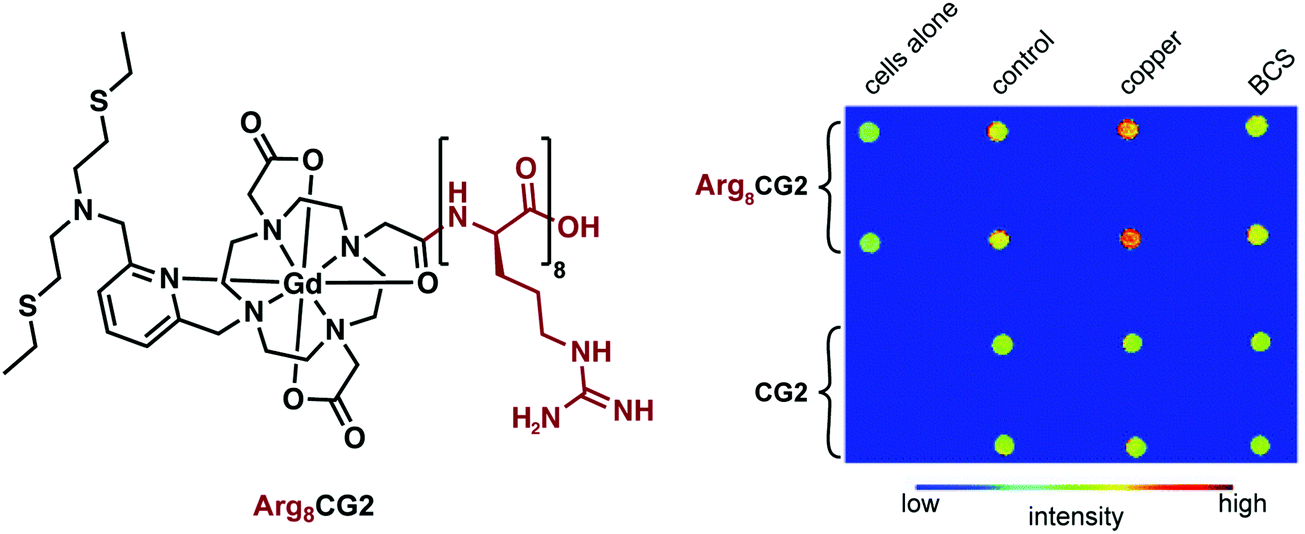 | ||
| Fig. 20 Chemical structure of Arg8CG2 and phantom images of both Arg8CG2 and CG2 in two different cell lines, revealing an increase in signal intensity in the presence of copper. Reproduced from (Chem. Sci., 2012, 3, 1829–1834) with permission from The Royal Society of Chemistry. | ||
Modification of Gd3+-based contrast agents has also yielded functional systems for monitoring enzymatic function (e.g., glutamate decarboxylase (GAD) activity). GAD is an essential enzyme converting L-glutamate (L-Glu), an excitatory neurotransmitter, to γ-aminobutyric acid (GABA), a major inhibitory neurotransmitter. Abnormal variations in GABA-mediated signalling have been linked to a number of neuropathological conditions, such as PD, epilepsy, and various psychiatric disorders. Traditional methods for GAD identification and quantification are based on post-mortem examinations. The development of non-invasive agents that would allow GAD activity to be monitored in vivo would thus be highly desirable; ultimately, they might make it possible to correlate precise deviations in activity with specific neurodegenerative phenotypes.
Hoehn and co-workers recently reported a q-modulated MRI contrast agent activated by GAD.30 The system was constructed from Gd-DO3A conjugated to flexible linkers with pendant glutamate residues (Fig. 21). GAD-mediated decarboxylation of the pendant glutamates unveiled the Gd3+ centre allowing inner sphere water coordination. This increase in the hydration state of the Gd3+ ion (q = 0 to 2) resulted in enhanced relaxivity with attendant complexation to endogenous macromolecules via increased electrostatic interactions. The Gd-DO3A-glutamate conjugate-1 was used to achieve the in vivo imaging of implanted GABAergic neurons that were differentiated from embryonic stem cells. Faster relaxation was observed for the differentiated neurons relative to undifferentiated cells in vitro and in vivo, a result considered consistent with GAD-specific activation of the contrast agent in those neurons.
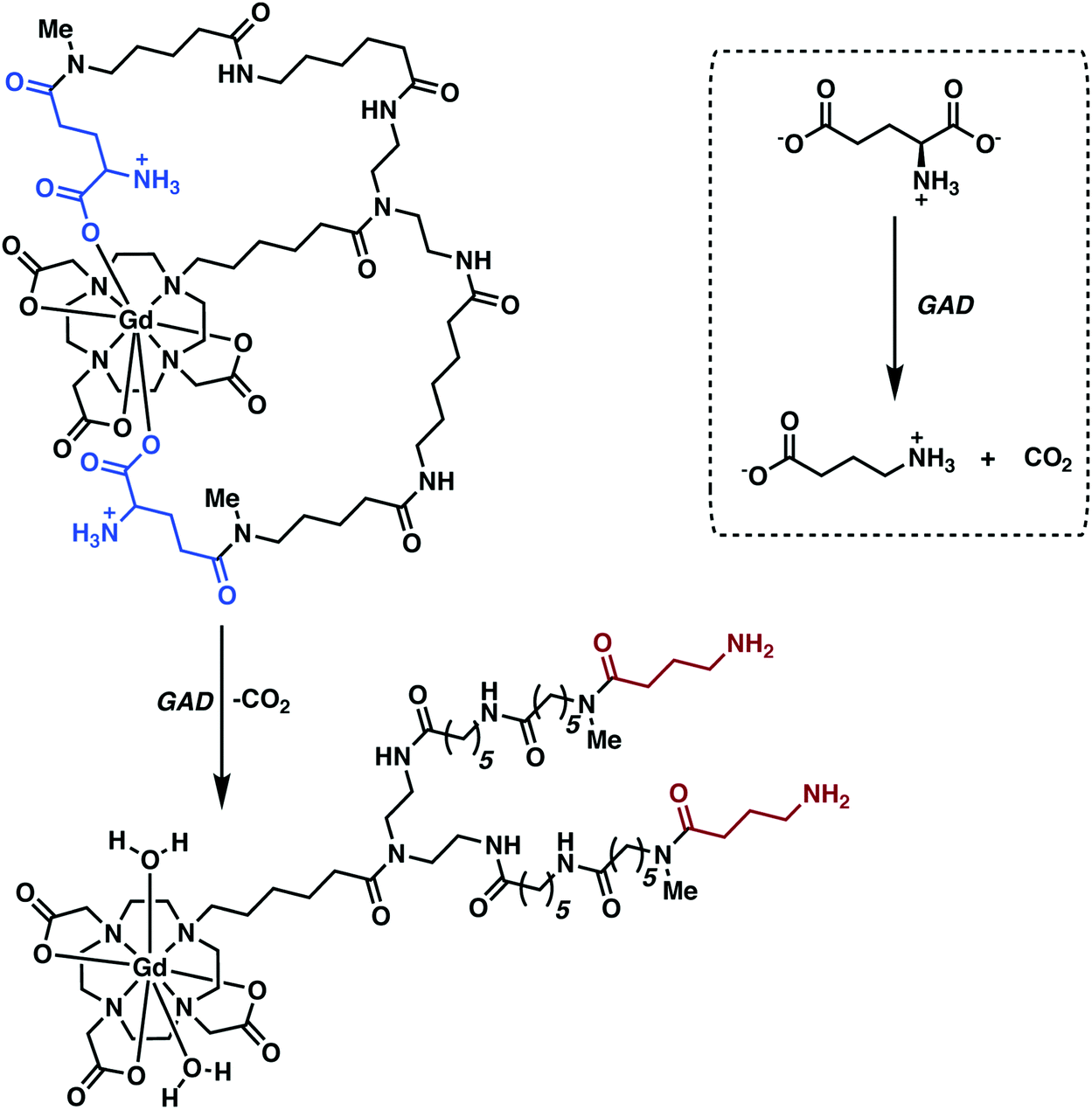 | ||
| Fig. 21 q-Modulated MRI contrast agent reported by Hoehn and co-workers. This system proved responsive towards glutamate decarboxylase (GAD), a finding rationalized in terms of the enzyme serving to cleave the terminal acid group and increase the relaxivity of the contrast agent as the result of an increase in the metal-bound water number, q. | ||
Manganese (Mn2+ and Mn3+) – MRI
Health concerns regarding the use of Gd-based contrast agents have become a focal point in MRI research. Researchers are thus actively searching for non-Gd-based alternatives.24 Manganese (Mn2+/3+) has shown promise as a viable alternative with relatively low inherent toxicity. Manganese is an essential metal ion in biological systems. Indeed, various inherent biochemical mechanisms enable the uptake and cell internalisation of Mn within the brain.31 Even the simple MnCl2 salt has found use in the manganese-enhanced MRI (MEMRI) visualisation of brain architectures, neuronal tracts, and brain activity through T1 and T2 imaging (Fig. 22). However, the relaxivity of Mn2+ is low compared to Gd3+. As a result, significant amounts of Mn2+ are required to produce an effective MRI signal. This can become a limitation since the Mn2+ concentrations in question can produce toxic effects, at least in the case of the free cations.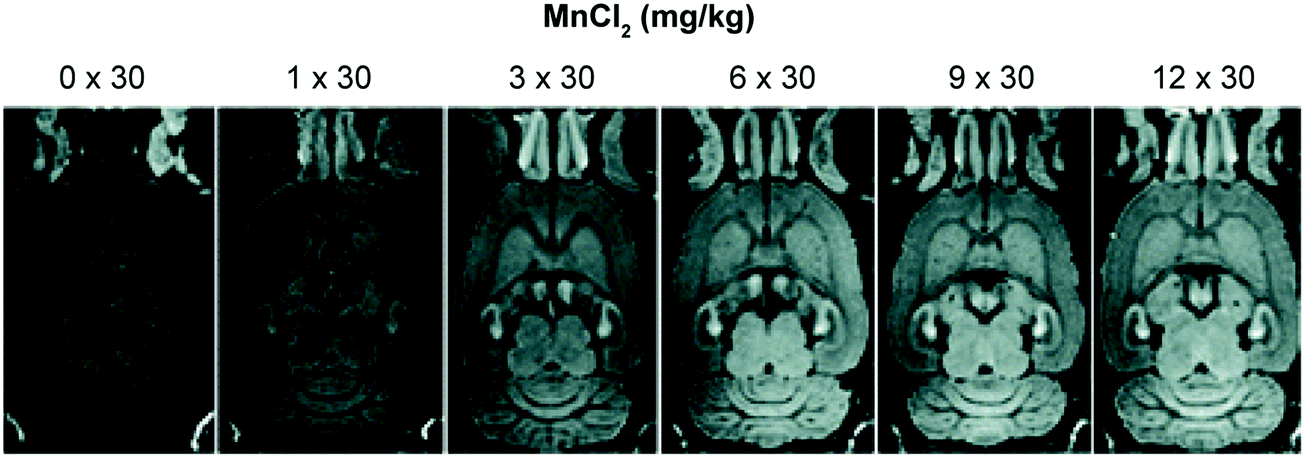 | ||
| Fig. 22 Example of manganese-enhanced MRI (MEMRI), showing an increased signal intensity throughout the brain with increasing concentration of MnCl2. Reprinted (adapted) with permission from (Schizophr. Bull., 2008, 34, 595–604). Copyright (2019) Oxford University Press. | ||
In an attempt to address the toxicity of free Mn2+ at high concentrations, Flesch and co-workers developed a first-generation dendritic DTPA-based Mn2+ complex. The polymeric network acted as a time-release vehicle for the progressive delivery of Mn2+ in the brain (Fig. 23).32 This dendrimeric Mn2+ complex displayed excellent aqueous solubility (0.1 M) with minimal in vitro neuronal toxicity. For instance, a 100% rat survival rate was observed when concentrations as high as 20 mM were given via intraperitoneal (i.p.) administration; presumably, these favourable findings reflect the slow diffusion of the released Mn2+ cation in vivo. The obtained in vivo images in the context of this study were also consistent with what was seen when MnCl2 was injected intravenously. These efforts serve to outline what might emerge as an effective strategy to overcome the unwanted toxicity of free Mn2+ at the concentrations needed to effect the MEMRI-based visualisation of brain structures.
 | ||
| Fig. 23 A dendritic DTPA-based complex reported by Flesch and coworkers that was designed to improved delivery of Mn2+ while decreasing toxic side effects. | ||
To explore imaging beyond MEMRI and compete with current Gd3+-based contrast agents, stable small molecule Mn contrast agents must be developed.24 Jasanoff and co-workers recently reported the preparation of three in vivo stable, cell-permeable phenylenediamido (PDA)-based Mn3+ complexes and showed that they could be used as T1 MRI contrast agents for selective cell labelling (Fig. 24).33 These Mn3+ complexes displayed a higher stability compared to Mn2+ complexes due to favourable chelation between the hard donor, anionic PDA ligand, and the high-spin 3d4 Mn3+ cation. The open coordination sphere of the PDA-based Mn(III) complexes also facilitated exchange with inner-sphere water molecules. Subsequent studies focused on modification of the ligand scaffold with a BAPTA Ca2+ chelating unit. These efforts afforded an effective T1 MRI contrast agent that allowed for the monitoring of intracellular Ca2+ concentrations (Mn-L3-BAPTA). Facile modification of the BAPTA scaffold allowed this system to be tuned to produce an analogue, ManICS1-AM, that displayed good cell permeability. Upon cellular uptake in HEK293 cells, ManICS1-AM undergoes esterase-mediated cleavage to reveal the BAPTA and enable monitoring of intracellular Ca2+ concentrations. In vivo studies provided support for the design expectation that ManICS1-AM could be used to monitor changes in Ca2+ concentrations induced via stimulation with potassium ions (K+). This latter stimulation induces neuronal cell depolarization and gives rise to changes in the Ca2+ concentrations in brain tissue. ManICS1-AM apparently constitutes the first example wherein calcium fluctuations in a living rat brain could be monitored directly at the intracellular level via fMRI.
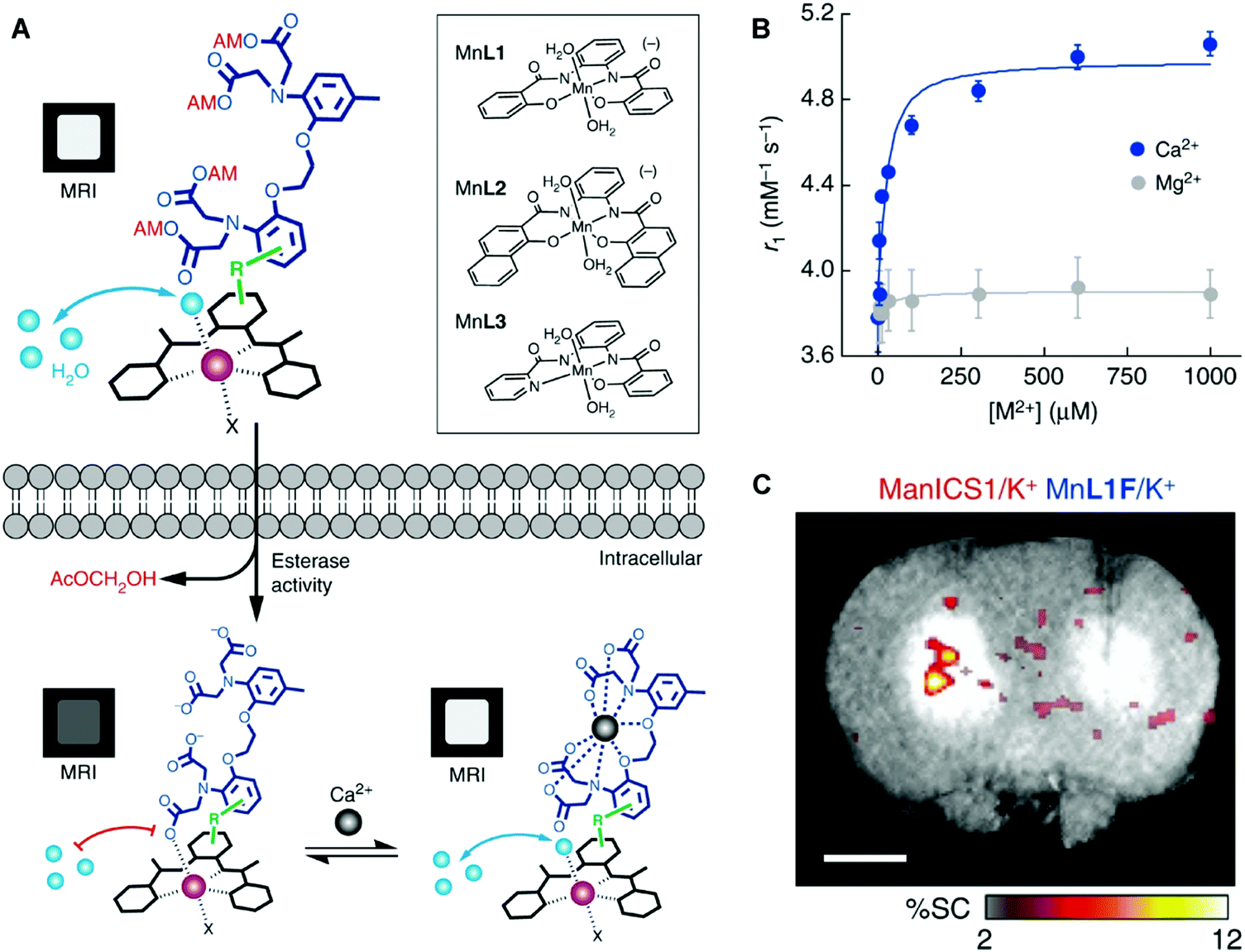 | ||
| Fig. 24 Phenylenediamido (PDA)-based Mn3+ complexes reported by Jasanoff and co-workers. These complexes act as T1 MRI contrast agents and permit the imaging of intracellular and in vivo Ca2+ concentrations. (A) When the cell permeable AM-ester protected complex (ManICS1) is internalised in cells, esterases cleave the AM-ester groups to release the active complex, which is cell permeable and trapped. Binding of Ca2+ causes an increase in signal intensity due to increased water access to the Mn3+ centre. Early iterations of cell permeable Mn3+ complexes are shown in the inset. (B) The relaxivity response of ManICS1 to increasing Ca2+ concentration, in comparison to the similar divalent cation, Mg2+. (C) In vivo signal change in a rat model upon stimulation with potassium, which results in increased neural activity and a spike in intracellular calcium levels. A calcium-insensitive analogue (MnL1F) give rise to no detectable response under otherwise identical experimental conditions. Reproduced from (Nat. Commun., 2019, 10, DOI: http://10.1038/s41467-019-08558-7) – Open Access. | ||
Iron (Fe2+/3+) – MRI
Iron, able to form in vivo stable metal complexes in both its divalent and trivalent oxidation states, is also being explored actively as a relaxation agent for use in MRI.24 In fact, one of the most widespread uses of Fe in MRI involves the use of endogenous haemoglobin (Hb). The difference between paramagnetic deoxygenated Hb and diamagnetic oxygenated Hb can be observed in T2*-weighted images and permits a functional readout through blood oxygenation level-dependent (BOLD) fMRI (Fig. 25). This method provides real-time information on blood oxygenation, blood flow, and blood volume changes. Data gathered through BOLD fMRI can be used to obtain indirect insights into the underlying neural activity, as activation of neurons requires an increased usage of oxygen and blood flow to the firing cells. This technique has shown promise as a biomarker for Alzheimer's disease and other neurodegenerative disorders.34 However, BOLD fMRI remains limited by the indirect coupling between neural activity and vascular response.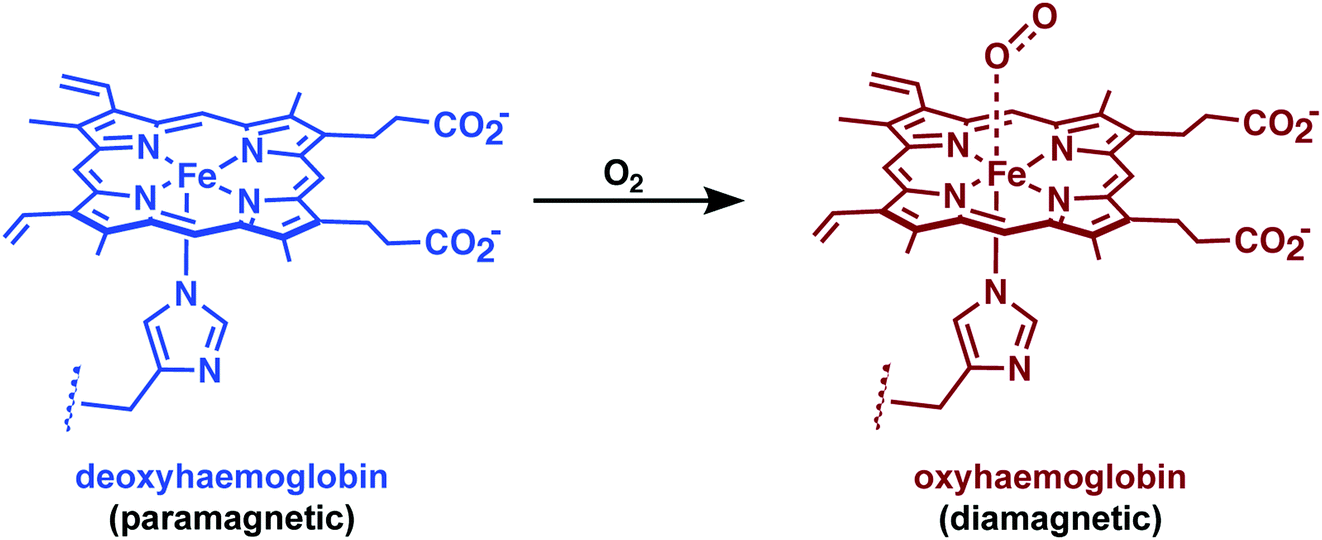 | ||
| Fig. 25 The chemistry underlying blood oxygenation level-dependent (BOLD) functional MRI (fMRI). Oxygenation of haemoglobin causes an decrease in paramagnetism. Neural activity leads to changes in blood flow/oxygenation levels in nearby vessels, which leads to signal changes in MRI. | ||
Currently, synthetic iron-based contrast agents are being explored as an alternative to BOLD. In an attempt to gain direct information on neuronal activity, Jasanoff and co-workers recently reported an Fe3+-based MRI strategy that relies on a directed evolution approach utilising bacterial cytochrome P450-BM3 to afford a series of protein-based MRI contrast agents that permitted dopamine imaging. Dopamine is a monoamine neurotransmitter vital for proper neurological function.35 Therefore, real time imaging of dopamine activity in vivo is particularly desirable as malfunction of the dopaminergic system has been associated with several neurodegenerative diseases. Dopamine is a substrate (endogenous ligand) for the bacterial cytochrome P450-BM3. However, in the absence of this substrate, a water molecule can fill the remaining coordination site of the Fe3+ atom. Subsequent binding of dopamine results in an attenuated MRI signal (0.83 ± 0.01 mM−1 s−1 → 0.10 ± 0.00 mM−1 s−1). This change in signal could be used to achieve the spatiotemporal imaging of dopamine release from PC12 cells and in the ventral striatum of brains in live rats following reward-related lateral hypothalamic brain stimulation.35 This seminal work by Jasanoff and co-workers established a new paradigm in fMRI and demonstrated directly how it could be applied to the problem of mapping neuronal function.
For MRI contrast applications, iron is commonly used in the form of T2 superparamagnetic iron oxide nanoparticles (SPIONs). Wisniewski and co-workers employed bis-functionalised ultra-small SPIO (USPIO) nanoparticles with sufficient BBB permeability for the targeted imaging of Aβ plaques.36 These USPIO nanoparticles were constructed via conjugation to the Aβ42 peptide through a polyethylene glycol (PEG) linker. In vivo and ex vivo imaging of the resulting USPIO-PEG-Aβ42 nanoparticles revealed an ability to image Aβ plaques in vivo by means of T2*-weighted MRI on the μm scale. These imaging-based conclusions were supported by histological analyses (Fig. 26).
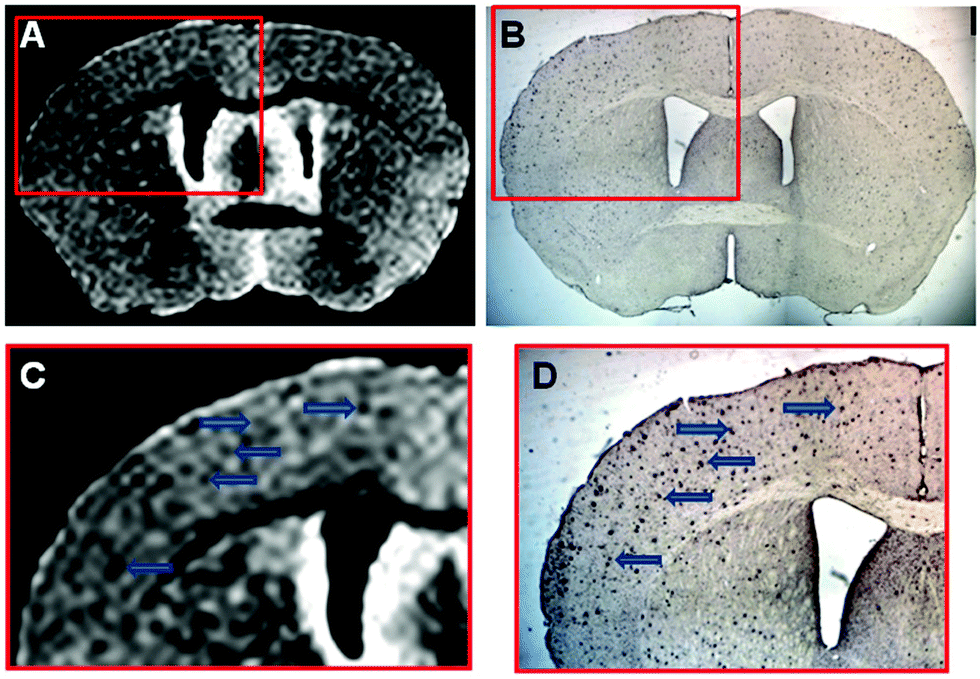 | ||
| Fig. 26 Ex vivo μMRI, at the level of the area cinguli of the cerebral cortex, of a 14 month-old APP/PS1 Tg injected with USPIO-PEG-Aβ1–42, showing numerous dark spots indicating location of Aβ plaques. Reproduced from (PLoS One, 2013, 8, DOI: http://10.1371/journal.pone.0057097) – Open Access. | ||
Jasanoff and co-workers have recently reported an elegant SPION-based imaging strategy for the detection of extracellular Ca2+ fluctuations that exploits the Ca2+ dependent interactions between the calcium-binding protein moiety C2AB and a lipid-coated iron oxide nanoparticle (LCIO).37 In the presence of Ca2+, the magnetic calcium-responsive nanoparticles (MaCaReNa) undergo clustering which serves to enhance the protein–LCIO interactions. Structural changes occur (i.e., aggregation) that lead to an increase in the hydrodynamic diameter from 35 ± 1 nm in the low calcium regime to 262 ± 14 nm in the presence of high calcium concentrations. An almost 2-fold change in r2 accompanies these changes in the Ca2+ concentrations and considered most relevant to extracellular studies (i.e., 0.1 to 1 mM). The possibility of studying brain activation in response to chemical (K+) and electrical stimuli in vivo was then explored using rats. Observed MRI response times were on the order of seconds. This work by Jasanoff and co-workers is arguably the first example of molecular fMRI using an MRI calcium sensor in a living brain model.
Luminescence-based imaging agents for the detection of neurodegenerative disease related biomarkers
Advances in the chemistry of luminescent small molecules and coordination complexes have provided systems that may be used for luminescence-based imaging, either alone or in conjunction with PET, SPECT, or MRI techniques. Luminescence is defined as the emission of light from a substance (luminophore) in its electronically excited state. Upon exciting the luminescent optical imaging agent, the corresponding emission signal can be used to ascertain the location and concentration of the luminophore within a specific biological sample. The resultant emission can be characterised by its wavelength, intensity, and the type of emission – Fig. 26A (i.e., length of the luminescence lifetime; fluorescence < 10 ns versus phosphorescence ∼ μs to ms). A number of optical imaging agents have been designed that may be used to selectively detect an analyte of interest, either through a binding event or by means of a chemical reaction. In comparison to MRI and PET/SPECT, these luminescent optical agents display unrivalled sensitivity, specificity, and temporal and spatial resolution. Optical imaging agents have thus begun to find use in the direct quantification of specific biomarkers involved in physiological or pathological processes in real time at the cellular and subcellular levels. However, due to the limited depth of light penetration through tissue, luminescence-based imaging is currently restricted to small live animals for research purposes only.To achieve optical-based bioimaging, researchers have utilised luminescent imaging agents in the form of organic compounds, transition metal complexes, lanthanide chelates, quantum dots, nanoparticles, and fluorescent proteins.38 In the context of reviewing efforts to image and understand better CNS neurodegeneration, it is convenient to divide the general class of metal-based luminescent imaging agents into those based on transition metal complexes and those that rely on lanthanide cations.
Transition metal-based imaging agents
The photophysical properties of transition metal coordination complexes with luminescent properties have been extensively detailed within the inorganic and material science literature. These complexes often display large Stokes shifts, high photochemical stability, and tuneable chromatic properties achievable via ligand modification. Transition-metal complexes applied to luminescence-based imaging include: Re+, Ru2+, Os2+, Ir3+, Rh3+, Pt2+, Au+, and Cu+. These systems all exhibit spin-forbidden triplet-state phosphorescence involving electronic transitions from the metal ion and ligand (i.e., metal-to-ligand charge-transfer (MLCT) and ligand-to-metal charge-transfer (LMCT)). Intra-ligand (IL) and ligand-to-ligand charge-transfer (LLCT) can also occur and are the result of intra- and inter-ligand transitions, respectively. Many complexes are subject to metal–metal interactions or ligand–ligand stacking that favour triplet-state phosphorescence, dominated by metal–metal-to-ligand charge-transfer (MMLCT), ligand-to-metal–metal charge-transfer (LMMCT), and metal-to-ligand-ligand charge-transfer (MLLCT). As a result of the spin-forbidden nature of the associated relaxation from the triplet back to the ground state long-lived luminescence emission lifetimes are observed for many of these complexes. Researchers have developed various photoluminescence time-resolved techniques, such as time-gated luminescence and photoluminescence lifetime imaging microscopy (TGL and PLIM), that permit discrimination relative to other interfering fluorescent signals with similar wavelengths (Fig. 27).38 This represents a significant advantage for metal-based luminescent species compared to, for instance, traditional organic fluorophores. On the other hand, as a result of the numerous electronic transitions involving both the metal and the ligand, the excited states of transition metal complexes are often sensitive to changes in the surrounding environment and the nature of the coordinating ligand. These combined set of attributes makes transition metal-based luminescence-based imaging agents attractive for the selective detection of analytes, both in vitro and in vivo.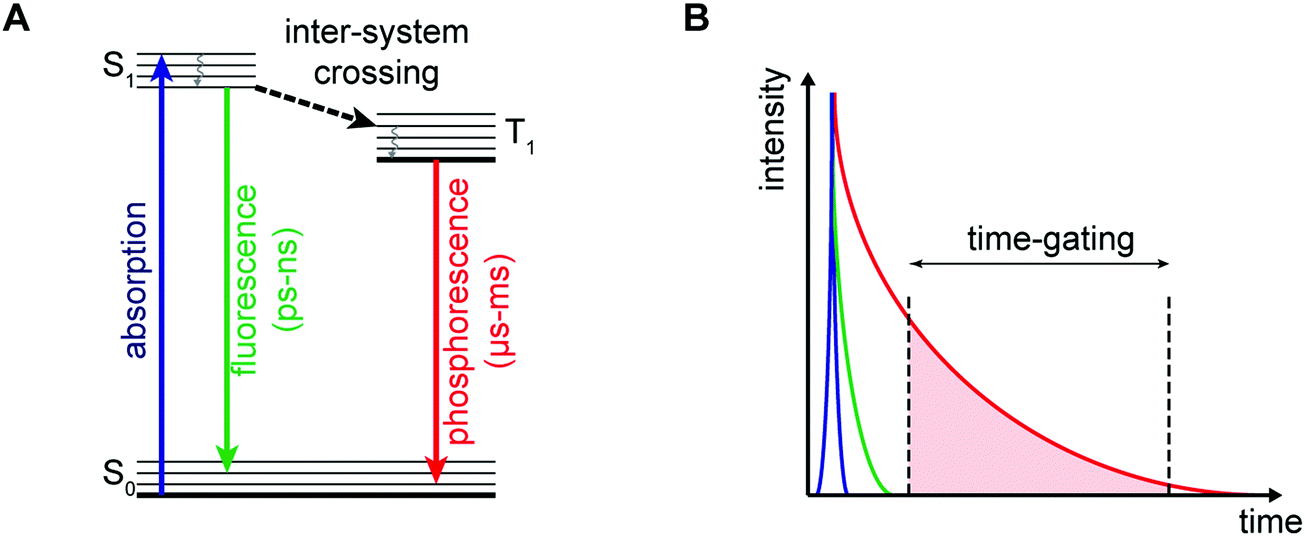 | ||
| Fig. 27 (A) Simplified Jablonksi diagram illustrating the difference between fluorescence and phosphorescence, with representative emission lifetimes. (B) Illustration of the principle behind time-gated imaging and spectroscopy. By applying a time-gate, the excitation pulse (blue) and shorter-lived fluorescence (green) can be removed from the signal to allow selective detection of long-lived phosphorescence (red). | ||
For an extensive coverage of time-resolved photoluminescence techniques and the development of transition-metal complexes for bioimaging applications, the reader is directed to two significant reviews by Huang and Li.38,39 In this section, we present selected examples of transition metal-based luminescent imaging agents (Ir3+, Ru2+, and Re+) that have been used to detect biomarkers implicated in the development of neurodegenerative disease.
Iridium (Ir3+)
Octahedral cyclometallated iridium (Ir3+) complexes remain a current gold-standard in the area of luminescence. They are typically characterized by high photoluminescence efficiencies, good aqueous stability, and excellent colour tunability through synthetic modification of the coordinating ligand. To date, a large number of cyclometallated Ir3+-based complexes have been developed for the imaging of disease-specific biomarkers in vitro and in vivo.38As underscored by the imaging agent development efforts summarised in the earlier portions of this review, the so-called amyloid hypothesis, focused on the role of Aβ plaques in AD progression, remains a focal point in AD research. Within this overall paradigm, researchers have set out recently to develop dual modal tools, so-called theranostics, that are able to detect Aβ peptide and inhibit Aβ plaque formation. Ma and co-workers recently reported a series of Ir3+ and Rh3+ metal complexes capable of inhibiting amyloid fibrillogenesis while permitting the attendant luminescence detection of Aβ fibrils.40 These metal complexes are comprised of two cyclometallated (2-phenyl pyridine, C^N) ligands with two bound solvent molecules (i.e., H2O) (Fig. 28). The labile solvent molecules allow for coordination to the histidine residues (H6, H13, or H14) present within the Aβ fibril framework. A brief structure activity relationship analysis led to the conclusion that Ir3+ and Rh3+ metal complexes bearing C^N phenyl-pyridine (ppy) ligands (i.e., 2a and 2b) provided the greatest inhibition of Aβ aggregation.
 | ||
| Fig. 28 Octahedral cyclometallated iridium (Ir3+) complexes developed by Ma and co-workers for the simultaneous detection of Aβ peptide and inhibition of Aβ plaque formation. | ||
In comparison to 2a, the Rh3+-complex 2b was found to be more active with near complete inhibition of Aβ peptide aggregation at 5 μM, demonstrating its potential as an Aβ peptide inhibitor for the treatment of AD. Unfortunately, 2b proved weakly emissive. On the other hand, the Ir3+-complexes 2a, 3, and 4 displayed luminescence turn-on in the presence of Aβ peptide. This enabled the use of these latter complexes as tools to distinguish between the different states of aggregation, with a higher luminescence emission being observed for Aβ fibril versus Aβ monomers.
In follow up work, Ma and co-workers showed that appropriately designed Ir3+ complexes could function as dual Aβ fibrillogenesis inhibitors and luminescent imaging agents.41 Toward this end, Ma et al. prepared twelve different luminescent Ir3+ complexes and evaluated their ability to bind to the Aβ peptide. It was found that complex 5, comprised of a 2-phenyl-1H-imidazo[4,5-f][1,10]phenanthroline (phenyl-imidazo-phen) (N^N) ligand, was effective in distinguishing between Aβ fibrils over Aβ monomers. Continued optimization yielded complexes 6 and 7, with 6 demonstrating partial inhibition and 7 demonstrating complete inhibition of Aβ fibrillogenesis (Fig. 29). Complex 6 provided for a 6- and 12-fold enhancement in luminescence emission intensity when contacted with Aβ monomers and fibrils, respectively. Complex 7 proved even more effective, providing an 11- and 18-fold luminescence emission enhancement upon treatment with Aβ monomers and fibrils, respectively. In vitro analysis of complex 7 revealed a neuroprotective effect against Aβ-induced cytotoxicity in SH-SY5Y cells and mouse primary cortical cells. The complexes generated by Ma and coworkers thus provide an early demonstration that it is possible to create theranostic systems, namely agents able to inhibit (therapeutic) and monitor (diagnostic) Aβ peptide formation via luminescence.
 | ||
| Fig. 29 Optimised octahedral cyclometallated iridium (Ir3+) phenyl-imidazo-phen-based complexes (5–7) developed by Ma and co-workers for the simultaneous detection of Aβ peptide and inhibition of Aβ plaque formation. | ||
Ruthenium (Ru2+)
As true for Ir-systems, ruthenium (Ru2+) complexes have found extensive use in bioimaging applications. Recent efforts have been devoted to exploring cellular imaging and sensing applications wherein octahedral dipyridophenazine (dppz) Ru2+ complexes are used as molecular “light-switches”. For instance, Marti and co-workers reported a Ru2+-based complex [Ru(bpy)2(dppz)]2+ (bpy = bipyridyl) that undergoes a photoswitching process in the presence of Aβ aggregates (Fig. 30).42 Specifically, upon binding to Aβ fibrils, [Ru(bpy)2(dppz)]2+ displayed a 50-fold increase in luminescence intensity (“turn-on”) as compared to the free Ru complex in aqueous solution. In the presence of monomeric Aβ only a minimal increase in luminescence intensity was observed. By exploiting the long emission lifetimes of [Ru(bpy)2(dppz)]2+, time-resolved experiments enabled the monitoring of Aβ binding within a fluorescent environment. This work demonstrates the potential to overcome a major limitation associated with most current ThT-Aβ inhibition screening assays, since it shows that it is possible to use an appropriately chosen metal complex to screen potential Aβ inhibitors with inherent fluorescent properties. | ||
| Fig. 30 (A) Ru2+-Based dipyridophenazine (dppz) complex reported by Marti and co-workers. (B) [Ru(bpy)2(dppz)]2+ time-gated assay for the monitoring of Aβ fibril formation in a fluorescent medium (rhodamine B). Reprinted (adapted) with permission from (J. Am. Chem. Soc., 2011, 133, 11121–11123). Copyright (2019) American Chemical Society. | ||
PD is often characterized by the abnormal accumulation of Lewy body protein aggregates comprised primarily of the alpha-synuclein (αS) peptide. The role of αS in the development of PD has been studied with data showing a strong correlation between αS misfolding and aggregation and the progression of PD pathogenesis. However, the direct cause of αS misfolding and aggregation as well as the role of αS accumulation in the development of PD has yet to be elucidated. The development of an effective imaging agent that allows αS misfolding and aggregation progression to be monitored readily is thus viewed as being highly desirable.
Building on their earlier work summarised above, Marti and co-workers recently reported a 1,10-phenathroline (phen) supported Ru complex, [Ru(phen)2dppz]2+ and showed it could be used to monitor αS fibrillization.43 Upon binding to αS fibrils, an 18-fold increase in luminescence emission intensity was observed with a remarkable selectivity. [Ru(phen)2dppz]2+ also displayed good cell permeability with low toxicity in human neuroglioma H4 cells, as well as an ability to distinguish between healthy cells and cells overexpressing αS (H4-MG-132) (Fig. 31).
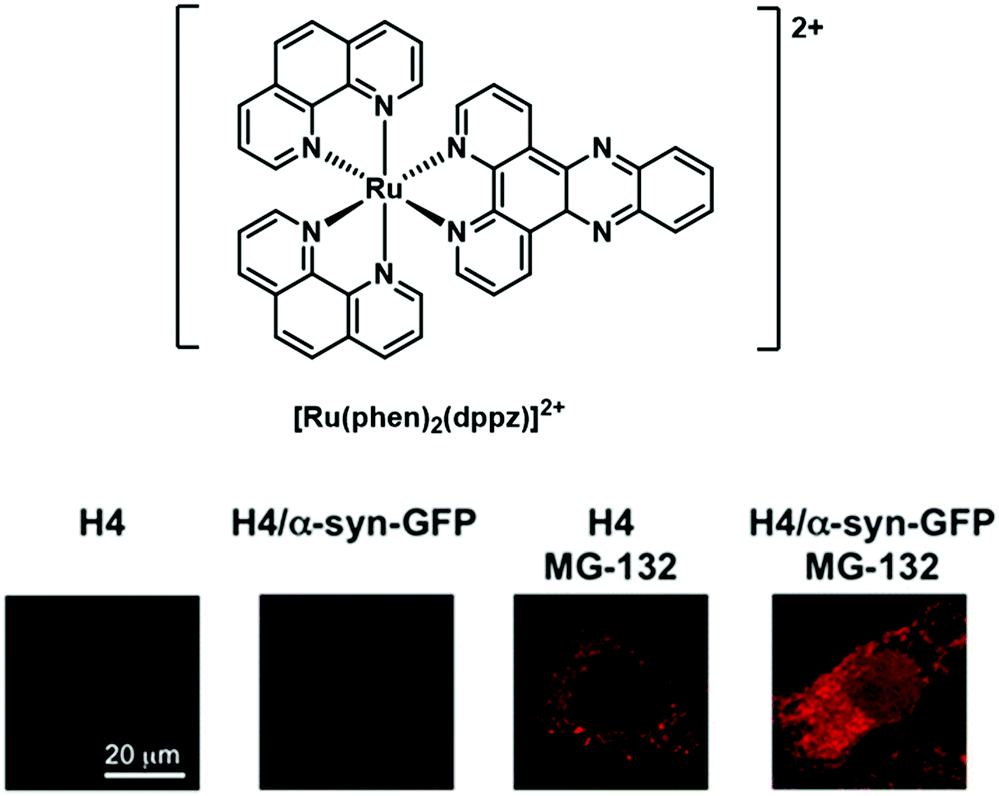 | ||
| Fig. 31 [Ru(phen)2dppz]2+ complex used by Marti and co-workers to monitor in real-time αS fibrillization in human neuroglioma cells (H4). Reprinted (adapted) with permission from (J. Am. Chem. Soc., 2012, 134, 20776–20782). Copyright (2019) American Chemical Society. | ||
The Ru2+ complexes prepared by Marti and associates provide an excellent demonstration of an optical imaging agent. They retain the benefits of the ThT displacement assay currently used in AD research in that they are amenable to use in the high-throughput screening of potential therapeutics targeting αS fibrillization in PD. However, they may be employed under conditions complicated by the presence of a fluorescent background. In addition, these luminescent-based imaging systems may allow the general monitoring of protein aggregation in vitro and do so with high spatial and temporal resolution.
Rhenium (Re+)
Rhenium (Re+) tricarbonyl bipyridine [Re(N^N)(CO)3X]n+ [X = anionic (n = 0) or neutral (n = 1)] complexes have found extensive use in bioimaging applications.39 As true for the [Ru(bpy)2(dppz)]2+ complexes discussed immediately above, [Re(dppz)(CO)3]+ also displays light-switching properties and is capable of detecting biologically important species. In continuation of their efforts on luminescent transition metal probes, Marti and co-workers reported that [Re(CO)3(dppz)(Py)]+ could be used to effect the luminescent-based detection of Aβ aggregates (Fig. 32).44 [Re(CO)3(dppz)(Py)]+ was shown to be non-emissive in aqueous buffer. However, upon exposure to fibrillar Aβ, an instantaneous 18-fold increase in luminescence emission intensity was observed. Upon light irradiation for 6000 s in the presence of oxygen, the Aβ peptide underwent oxidation. The resultant modifications of the Aβ peptide induced an unprecedented second light-switching event with an attendant 105-fold increase in luminescence emission intensity. As used by Marti and coworkers, [Re(CO)3(dppz)(Py)]+ provides an elegant demonstration of a luminescent-based imaging system that may be used to monitor Aβ plaque formation and to obtain insights into the local environment within the Aβ plaque that results from oxidation. | ||
| Fig. 32 [Re(CO)3(dppz)(Py)]+ complex Marti and co-workers that allows for the luminescence detection of Aβ aggregates. As noted in the main text, this system undergoes an unprecedented second light-switching event upon light irradiation. | ||
More recently, Pigge and co-workers reported a series of oligothiophene benzothiazole-based tetraarylethylene (TAE) Re(CO)3 complexes (8–13) and showed that these systems were capable of monitoring Aβ aggregate formation in real time.45 The benzothiazole TAE chelating ligands used in this study contain a different number of thiophene units, a variation that serves to modulate the photophysical properties and the binding affinities toward Aβ fibrils. In aqueous solution, complexes 8–10 displayed minimal luminescence emission intensity (Fig. 33). However, in the presence of Aβ aggregates, a significant increase in the luminescence emission intensity was observed. Complex 8 displayed the smallest response in the presence of aggregated Aβ fibrils (∼13-fold), complex 9 exhibited the largest response with a 34-fold increase in luminescence intensity, and complex 10 demonstrated the largest Stokes shift (105 nm) with a red-shifted emission in the NIR region (λem = 641 nm). Complex 9 was also evaluated against other bio-macromolecules and displayed excellent selectivity towards the Aβ peptide.
 | ||
| Fig. 33 Oligothiophene benzothiazole-based tetraarylethylene (TAE) Re(CO)3 complexes (8–10) reported by Pigge and co-workers. | ||
To confirm further the structural importance of both thiophene and benzothiazole functionalities, complexes 11–13 were synthesised (Fig. 34). In the case of complex 11, each (oligo)thiophene moiety was replaced by a phenyl ring. In 12 and 13 the bis(bithiophene) units were retained, but the benzothiazole core was replaced by ones based on bis(2-pyridyl) or bis(2-quinolyl)Re(CO)3 subunits, respectively. As anticipated, complexes 11–13 displayed only a modest increase in emission intensity in the presence of Aβ fibrils. The high binding affinities and desirable photophysical properties of the oligothiophene benzothiazole-based Re complexes 8–10 supports the contention that these systems may have a role to play as fluorescent Aβ probes, perhaps as an alternative to ThT. Additionally, the fact that this class of complexes is easy to modify leads us to suggest that they could inspire the development of new second-generation TAE-based diagnostic tools for investigating protein aggregation in AD and other neurological diseases.
 | ||
| Fig. 34 Benzothiazole-based tetraarylethylene (TAE) Re(CO)3 complexes (11–13). | ||
Lanthanide-based imaging agents
Trivalent lanthanide ions (Ln3+) display unique intrinsic luminescence properties that arise from the f–f electron transitions. However, as transitions occurring within the same orbitals (4f–4f) are Laporte forbidden, Ln3+ ions display low absorption coefficients. This complicates the efficient harnessing of their luminescence properties. Free Ln3+ ions are also toxic, which makes them unsuitable for bioimaging applications per se. To circumvent toxicity and their poor absorptivity, Ln3+ ions are typically chelated by polyanionic macrocycles, such as DOTA and DTPA, and functionalised with various organic chromophores for use as light excitation “antennas”. These “antennas” enable efficient Ln-based luminescence proceeding via excitation of the organic antennas to their respective S1 excited states, followed by intersystem crossing to their T1 states. Subsequent intramolecular charge transfer populates the excited state of the Ln3+ ion; relaxation of the ion to the ground state then produces a unique fingerprint emission characteristic of the Ln3+ ion in question (Fig. 35).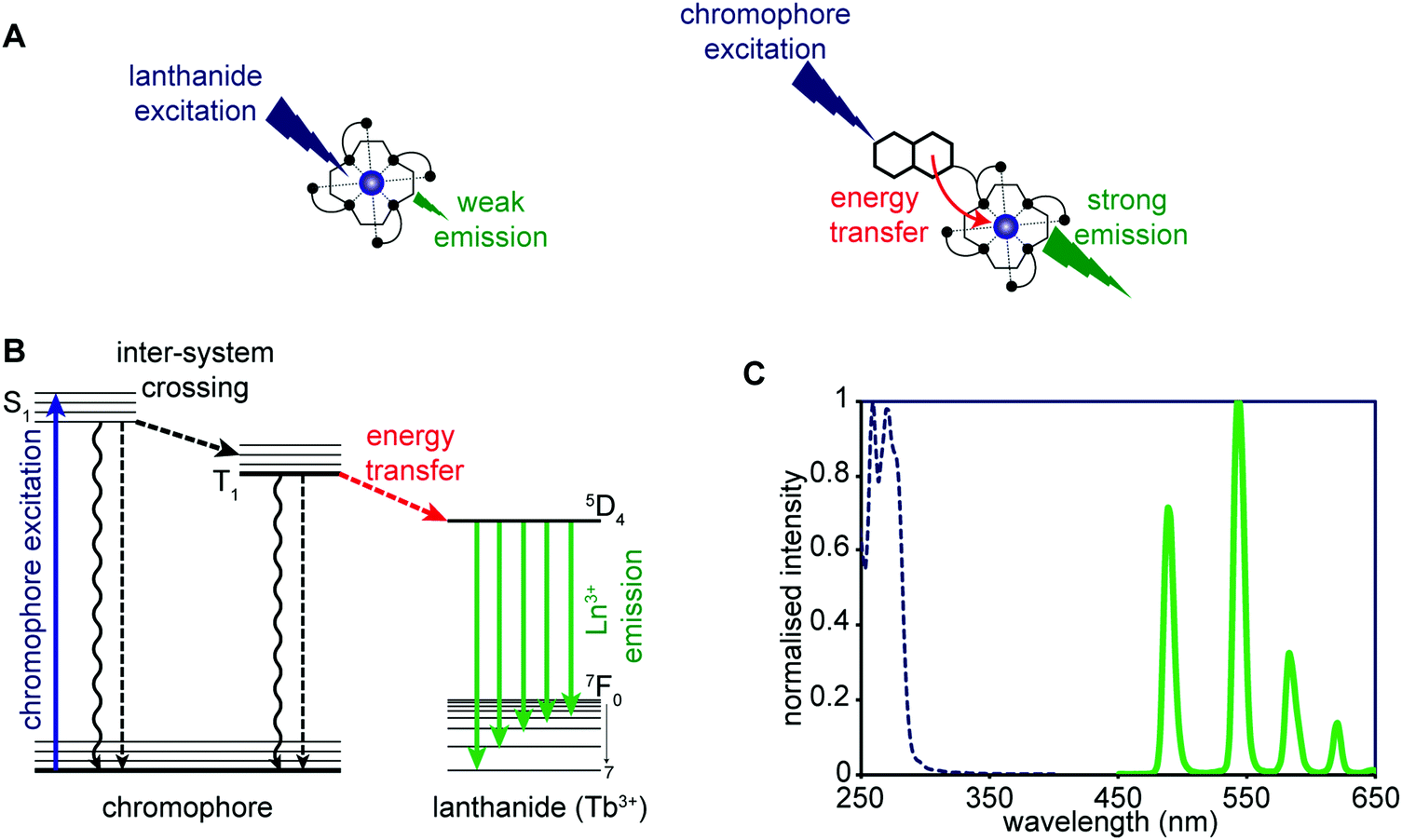 | ||
| Fig. 35 (A) Sensitisation through a chromophore generally leads to more intense emission in comparison to direct excitation of the Ln3+ ion. (B) Simplified Jablonski diagram illustrating the sensitisation of Ln3+ ions, in this case Tb3+. (C) A typical chromophore-sensitised Tb3+ emission spectrum (solid green trace), showing the large Stokes shift relative to the chromophore excitation spectra (dashed blue line) that is typically observed. | ||
Unlike transition metal complexes, lanthanide-based emissions are not significantly affected by the surrounding environment or ligands. Indeed, even under widely different conditions, almost no variation is observed in the emission/excitation wavelengths of the Ln3+ ions. The lack of such interactions is attributed to the valence 4f orbitals being core-like with shielding from chemical bonding by the populated 5s and 5p orbitals. This feature, coupled with the spin and Laporte forbidden nature of these electronic transitions, yields long luminescence lifetimes in the micro- to millisecond range. Luminescent-based lanthanide imaging agents have thus found utility in time-gated or time-resolved live-cell and in vivo imaging. A significant advantage of these lanthanide based optical imaging agents is that varying the Ln3+ centre yields only minor changes in the chemical properties enabling multimodal and multiplexed applications. However, since each Ln3+ ion gives rise to unique luminescent features, in principle it is possible to generate several similar imaging agents based on the same organic framework.
While the wavelengths of Ln3+ transitions are typically fixed, it is possible to modulate the emission intensity and, for some of the ions, the ratio between different emission bands. To date, three main determinants have been used to modulate the luminescence signal of lanthanide-based optical imaging agents for the detection of disease related biomarkers; these are:
• Varying q
• Adjusting the antenna–Ln3+ ion distance
• Modulating the antenna excited state
Varying q, number of bound water molecules, influences the emission intensity, luminescence lifetime, and spectral form of otherwise similar Ln complexes. This influence is due to quenching of the excited state(s) caused by the O–H bond vibrations. Design considerations used to adjust the extent of sensitization produced by the antenna stem from an appreciation that the energy transfer from the triplet excited state (T1) of the antenna to the metal ion is both distance and energy dependent. Current systems have focused on altering the Ln-antenna distance through chemical reactions or binding events. Finally, modulation of the antenna excited state has been achieved. Typically, this has been done by exploiting aqueous-based chemoselective reactions with kinetics that are fast even in complex biological environments to achieve chemical modification to the antenna. This latter approach is particularly attractive in the context of designing agents for the selective detection of neurodegenerative specific analytes.
For the development of molecular lanthanide-based optical imaging agents, three major factors need to be taken into consideration: The Ln3+ ion itself, the photophysical and chemical properties of the chelating ligand, and the photophysical features of the organic-based sensitising antenna. Historically, applications in lanthanide-sensing agents have utilized europium (Eu3+) and terbium (Tb3+), owing to their high intrinsic quantum yields, long-luminescence lifetimes, and emission bands that fall in the visible region. Recent efforts have also focused on neodymium (Nd3+) and ytterbium (Yb3+) as near IR-emitting analogues. For additional and extensive overviews on the use of lanthanide-based imaging agents for general bioimaging applications, the reader is directed to two excellent reviews by Meade and Huang.23,38
Sénèque and co-workers recently reported a responsive system that combines functional readout with multiplexing capabilities. In this system, the authors relied on a bio-inspired zinc finger peptide strategy for the selective luminescent detection of Zn2+ ions through the modulation of the antenna–Ln3+ ion distance (Fig. 36).46 They exploited the tendency of ββα zinc finger (ZF) peptides to change conformation and fold in the presence of Zn2+ over other biologically relevant metal ions. Four different ββα zinc finger (ZF) peptides containing a conjugated Ln3+–DOTA complex and a sensitizing antenna were prepared. Chelation of the Zn2+ cation resulted in a conformational change, basically a folding to the system, which served to decrease the distance between the small molecule sensitizer and the reporter Ln3+–DOTA complex leading to an increase in the luminescence intensity.
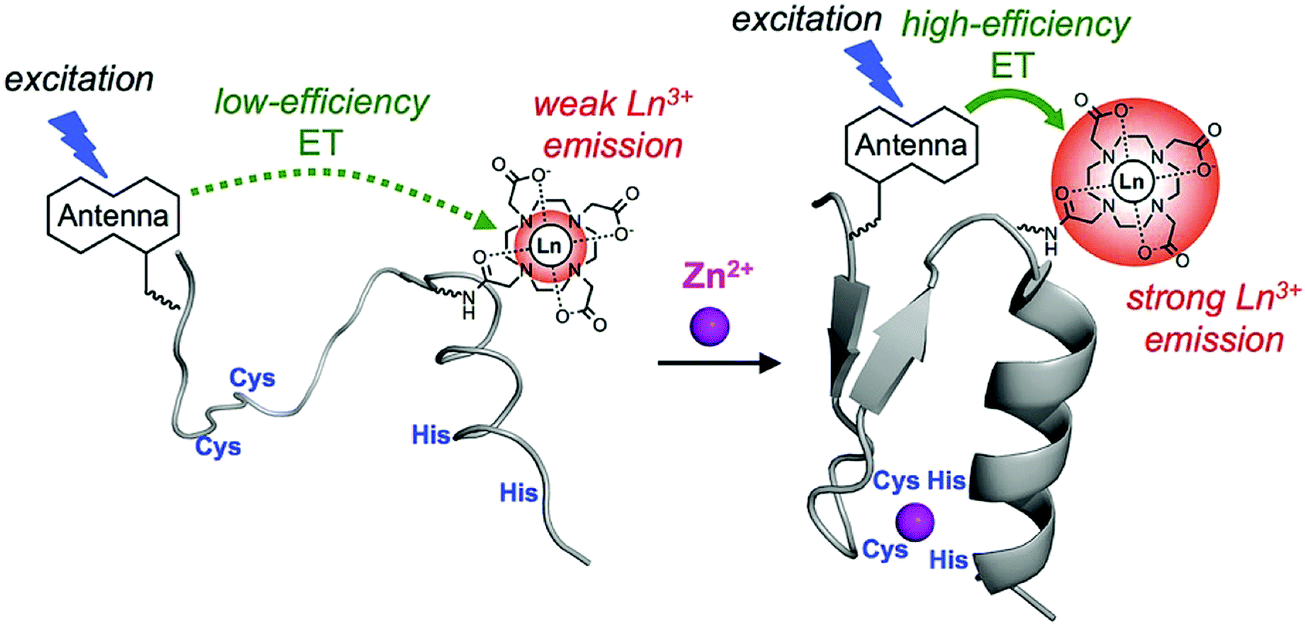 | ||
| Fig. 36 Binding of Zn2+ results in a conformation change of the zinc-finger peptides, which bring the chromophore antenna spatially closer to the Ln3+ ion, resulting in an increase in Ln3+ emission intensity. Reprinted (adapted) with permission from (Chem. – Eur. J., 2017, 23, 10992–10996). Copyright (2019) John Wiley and Sons. | ||
Initial efforts were focused on a Tb3+-based peptide (LZF1Trp|Tb) with a tryptophan antenna. This framework yielded a time-delayed 8.5-fold enhancement in the emission intensity in the presence of Zn2+ (1 equiv.). Luminescence lifetime measurements revealed no change in the number of water molecules within the Tb3+ coordination sphere, providing support for the contention that the observed luminescence enhancement was due to zinc-induced peptide folding. In a similar vein, a neodymium (Nd3+) LZF1NBD|Nd probe was developed that gave rise to a NIR emission upon exposure to the Zn2+ cation in aqueous conditions.
Multimodal systems that combine the emissive Tb3+/Eu3+ ions with the MRI active Gd3+ cation have been studied in an effort to generate imaging agents that permit both luminescence and MRI readouts.47 Tóth and co-workers recently employed this bimodal approach in an effort to image neurotransmitter signalling in neurodegenerative disease models.48 In particular, a Gd-DO3A complex functionalised with a triazacrown ether (Gd-DO3A-3AC) was prepared. This framework enabled the ditopic binding of the excitatory neurotransmitter L-glutamate (L-Glu), comprised of cationic ammonium (–NH3+) and negatively charged carboxylate (–COO−) groups (Fig. 37). The coordinatively unsaturated Gd3+ metal centre (positively charged) provided a binding site for the carboxylate functionality (negatively charged), while the appended triazacrown ether unit enabled binding of the cationic –NH3+ group. The dual coordination of Gd-DO3A-3AC rendered it capable of discriminating zwitterionic neurotransmitters from monoamine neurotransmitter (e.g., serotonin vs. noradrenaline). Upon coordination of the appropriate neurotransmitter, a marked change in relaxivity was observed. This was believed to arise from a decrease in the hydration number (q). Gd-DO3A-3AC was also used to monitor neural activity in ex vivo mouse brain slices by MRI.
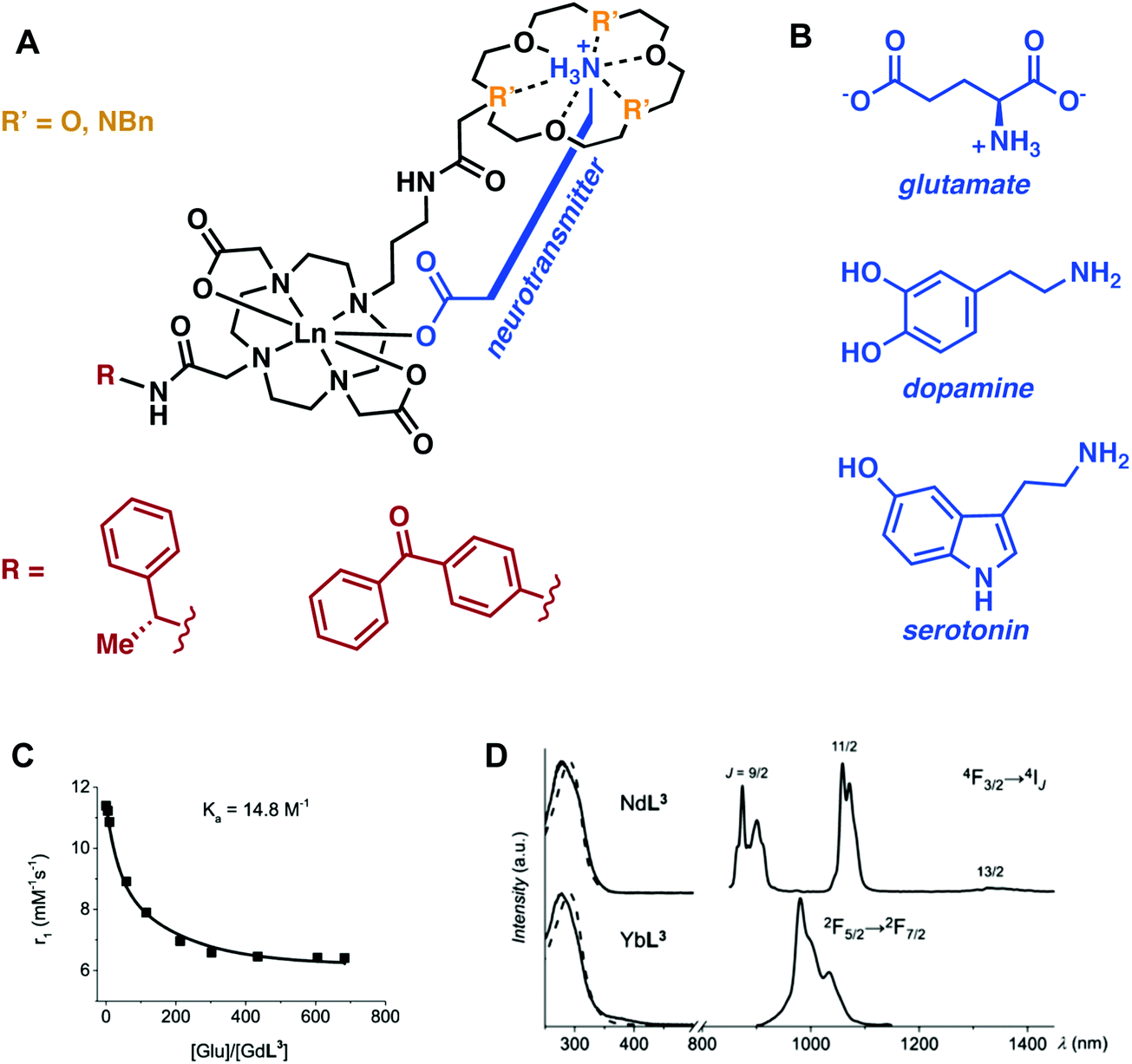 | ||
| Fig. 37 (A) Glutamate-responsive contrast agents. Here Ln = Gd3+, Eu3+, Yb3+, Nd3+. (B) The chemical structure of glutamate is shown along with that of two common neurotransmitters, dopamine and serotonin. (C) Longitudinal relaxivity change seen for the Gd3+ complex as a function of increasing glutamate (Glu) concentration. (D) Excitation and emission spectra of the Nd3+ and Yb3+ analogues. | ||
More recently, Toth and co-workers extended this design strategy by attaching a benzophenone chromophore to the lanthanide ligand site.49 Incorporation of this flat aromatic substituent led to an increased interaction with albumin causing an enhancement in the overall relaxivity. Unfortunately, this came with an overall decrease in the neurotransmitter response. On the other hand, the presence of the benzophenone chromophore allowed for efficient sensitisation of Eu3+, Nd3+, and Yb3+, thus paving the way towards both the visible and near-infrared (NIR) detection of neurotransmitters. These additional studies by Toth and associates provide an illustrative example of the multimodal capabilities possible by creating a system that can function with different Ln3+ ions. It is to be noted, however, that the instability (i.e., photoreactivity) of these first generation systems have precluded their use in more advanced application scenarios.
The efforts with Ln3+ luminescent systems summarised above have provided initial support for the notion that such systems may have utility in the context of elucidating the biochemical mechanisms associated with various disease states. However, this area remains in its infancy in terms of potential applications involving neurodegenerative disorders.
Conclusion and future perspectives
As shown throughout this review, PET/SPECT, MRI and luminescence are non-invasive molecular imaging techniques that show particular promise in terms of visualising various pathological biomarkers that may play a role in the development of neurodegenerative diseases. We have to highlight the fact that each metal-based imaging technique has a set of unique advantages and disadvantages (Table 3). For example, metal complex luminescence-based imaging agents offer a method to visualise specific biomarkers with high sensitivity, specificity, and spatial resolution at the cellular/subcellular level (i.e., mitochondria). On the other hand, they generally suffer from poor tissue penetration leading to limited in vitro and in vivo applications. In contrast, PET/SPECT-based imaging agents have been used extensively to monitor various metabolic/physiological processes and disease related biomarkers with high sensitivity (pM/nM range). However, PET/SPECT provide images with relatively poor resolution and require the use of ionising radiation. On the other hand, small molecule MRI imaging agents have been widely used to effect clinical diagnoses and in the context of research related to neurodegenerative disease. Various small molecule MRI imaging agents have been used to visualise various anatomical architects and pathological biomarkers with high spatial resolution with nearly unlimited depth penetration. However, MRI-based imaging approaches are typically hampered by relatively poor sensitivity (in the μM range); this makes the detection of biologically important species (i.e., DAT) present at nM concentrations in vivo difficult, if not impossible.
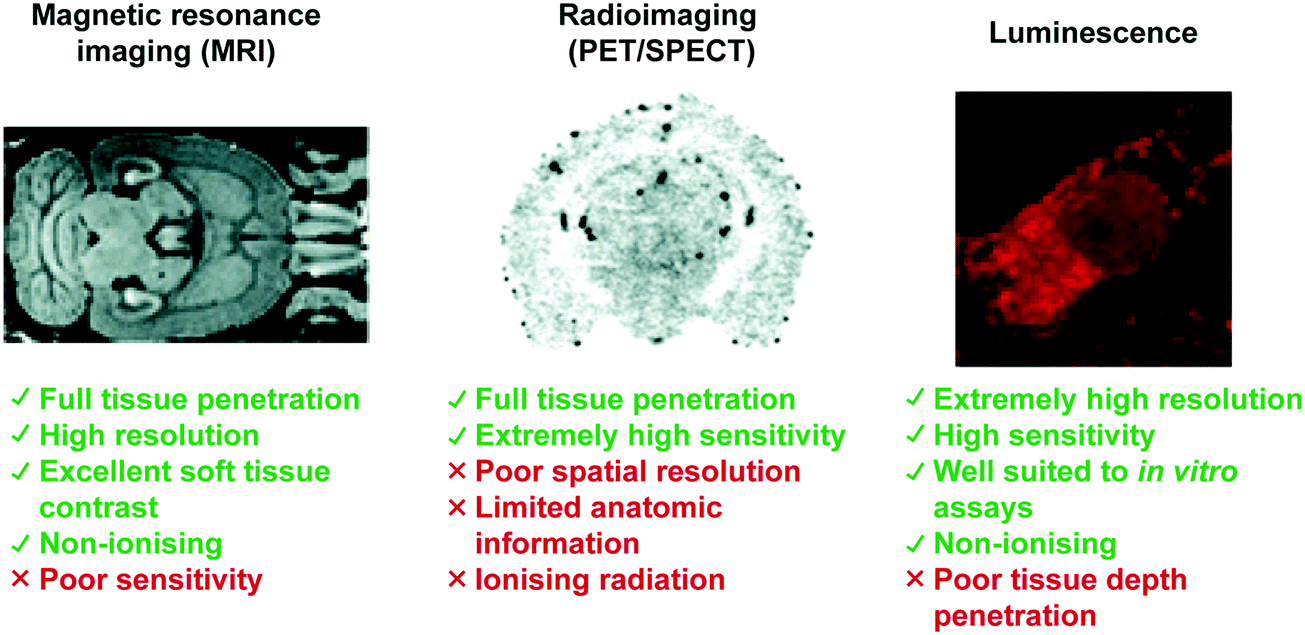
|
Increasingly, researchers in the field are coming to appreciate that no single imaging modality will typically suffice to obtain all the necessary information needed for a clinical diagnosis or for understanding of a specific pathological process. Dedicated efforts in a number of laboratories are thus being made to combine various imaging methods to develop so-called multimodal approaches. For instance, PET/SPECT is being used in conjunction with MRI in an effort to overcome the limitations of each imaging modality so as to unravel more effectively the determinants of various disease states.50 We anticipate a continued growth in metal-based imaging agents, particularly multimodal systems, that will permit a deeper understanding of neurodegenerative disease and its underlying causes. Ultimately, the knowledge gained from the design and study of metal-based imaging agents is expected to translate into new therapeutics and cures for these frightening and debilitating disorders.
Abbreviations
| αS | Alpha-synuclein |
| β+ | Beta positive decay |
| β− | Beta minus decay |
| μM | Micrometre |
| γ | Gamma ray emission |
| 11C | Carbon-11 |
| 61Cu | Copper-61 |
| 62Cu | Copper-62 |
| 64Cu | Copper-64 |
| 18F | Fluorine-18 |
| 68Ga | Gallium-68 |
| 123I | Iodine-123 |
| 99Mo | Molybdenum-99 |
| 89Zr | Zirconium-89 |
| L-Glu | Glutamic acid |
| τ m | Mean residence lifetime |
| τ r | Rotational correlation time |
| Aβ | Amyloid beta |
| AD | Alzheimer's disease |
| Ag | Silver |
| AgCl | Silver chloride |
| Au+ | Gold ion |
| ALS | Amyotrophic lateral sclerosis |
| Atsm | Diacetyl-bis(N4-methylthiosemicarbazone) |
| BBB | Blood brain barrier |
| bDHC | Bis(dihydroxy)curcumin |
| BAPTA | 1,2-Bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid |
| BOLD | Blood-oxygenation level dependent |
| BPy | Bipyridyl |
| Bzq | Benzoquinoline |
| CA | Contrast agent |
| Ca2+ | Calcium ion |
| CEST | Chemical exchange saturation transfer |
| CN | Coordination number |
| CNS | Central nervous system |
| CO | Carbon monoxide |
| Cp | Cyclopentyl |
| Cu+/2+ | Copper ion |
| CT | Computer tomography |
| DAC | Diacetylcurcumin |
| DAT | Dopamine active transporter |
| DFO | Desferrioxamine B |
| DO3A | 1,4,7,10-Tetraazacyclododecane-1,4,7-triacetic acid |
| DOTA | 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid |
| DOPTA | 1,4,7-Tris-(carboxymethyl)-1,4,7,10-tetraazacyclododecane |
| dppz | Dipyridophenazine |
| EC | Electron capture |
| EDTA | Ethylenediaminetetraacetic acid |
| Eu | Europium |
| FDA | Food and drug administration |
| Fe2+/3+ | Iron |
| fMRI | Functional magnetic resonance imaging |
| GABA | γ-Aminobutyric acid |
| GAD | Glutamate decarboxylase |
| Gd3+ | Gadolinium ion |
| Ge | Germanium |
| GFP | Green fluorescent protein |
| gtsm | Glyoxal-bis(N4-methyl-3-thiosemicarbazonato) |
| h | Hours |
| Hb | Haemoglobin |
| HD | Huntington's disease |
| ID g−1 | Injected does per gram tissue |
| IL | Intraligand |
| In3+ | Indium ion |
| Ir3+ | Iridium ion |
| K+ | Potassium ion |
| K d | Dissociation constant |
| keV | Kiloelectron volts |
| K i | Inhibition constant |
| L | Ligand |
| LCIO | Lipid coated iron oxide nanoparticles |
| LLCT | Ligand-to-ligand charge-transfer |
| LMMCT | Ligand-to-metal–metal charge-transfer |
| LMCT | Ligand-to-metal charge-transfer |
| Ln3+ | Lanthanide ion |
| Lys | Lysine |
| mAb | Monoclonal antibody |
| MaCaReNa | Magnetic calcium-responsive nanoparticles |
| MeV | Megaelectron volt |
| MEMRI | Manganese-enhanced magnetic resonance imaging |
| mM | Millimolar |
| MMLCT | Metal–metal-to-ligand charge-transfer |
| MLCT | Metal-to-ligand charge-transfer |
| MLLCT | Metal-to-ligand–ligand charge-transfer |
| Mn2+/3+ | Manganese ion |
| MRI | Magnetic resonance imaging |
| Min | Minute |
| ms | Millisecond |
| mV | MilliVolt |
| Nd3+ | Neodymium ion |
| nM | Nanomolar |
| NOTA | 1,4,7-Triazacyclononane-1,4,7-trisacetic acid |
| ns | Nanosecond |
| O2 | Oxygen |
| O2˙− | Superoxide anion |
| Os2+ | Osmium ion |
| PCTA | 3,6,9,15-Tetraazabicyclo[9.3.1]-pentadeca-1(15),11,13-triene-3,6,9-triacetic acid |
| PD | Parkinson's disease |
| PDA | Phenylenediamido |
| PEG | Polyethylene glycol |
| PET | Positron emission tomography |
| Pt2+ | Platinum ion |
| Phen | 1,10-Phenathroline |
| PiB | Pittsburgh compound B |
| PLIM | Phosphorescence lifetime imaging |
| Phq | 2-Phenylquinoline |
| Ppy | 2-Phenylpyridine |
| Py | Pyridine |
| pM | Picomolar |
| ps | Picoseconds |
| Py | Pyridine |
| q | Average number of water molecules bound directly to the paramagnetic metal centre |
| r 1/2 | Relaxivity |
| Re+ | Rhenium ion |
| Rh3+ | Rhodium ion |
| RF | Radiofrequency |
| Ru2+ | Ruthenium ion |
| SPECT | Single photon emission computed tomography |
| SPION | Superparamagnetic iron oxide nanoparticles |
| USPIO | Ultra-small superparamagnetic iron oxide nanoparticles |
| t 1/2 | Half-life |
| T | Tesla |
| T 1 | Longitudinal time constant |
| T 2 | Transverse time constant |
| TAE | Tetraarylethylene |
| Tb3+ | Terbium ion |
| 99mTc | Technetium-99m |
| Tg | Transgenic |
| TGL | Time-gated luminescence |
| ThT | Thioflavin T |
| Yb3+ | Ytterbium ion |
| Zn | Zinc |
| 89Zr | Zirconium-89 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
DAI thanks the Life Sciences Research Foundation for a postdoctoral fellowship sponsored by the Howard Hughes Medical Institute. PH thanks the University of Nottingham for support through a Nottingham Research Fellowship. XPH would like to thank The National Key Sci-Tech Special Projects of Infectious Diseases of China (2018ZX10732202). XPH and HT thank financial support from Natural Science Foundation of China (No. 21788102, 91853201, 21722801 and 21776078) and the Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX03). A. C. S. and J. T. B. would like to thank Gregory D. Thiabaud (UT Austin) for helpful discussions. JLS would like to thank the National Institutes of Health (grant CA 68682) and the Robert A. Welch Foundation (F-0018) for support. TDJ wishes to thank the Royal Society for a Wolfson Research Merit Award.References
- C. Soto and S. Pritzkow, Nat. Neurosci., 2018, 21, 1332–1340 CrossRef CAS PubMed.
- P. J. Blower, Dalton Trans., 2015, 44, 4819–4844 RSC.
- C. Grupen, Introduction to Radiation Protection, Springer, Berlin, 2010 Search PubMed.
- L. Zhu, K. Ploessl and H. F. Kung, Chem. Soc. Rev., 2014, 43, 6683–6691 RSC.
- K. H. Chen and M. C. Cui, MedChemComm, 2017, 8, 1393–1407 RSC.
- M. Ikawa, H. Okazawa, T. Kudo, M. Kuriyama, Y. Fujibayashi and M. Yoneda, Nucl. Med. Biol., 2011, 38, 945–951 CrossRef CAS PubMed.
- K. Wada, Y. Fujibayashi, N. Tajima and A. Yokoyama, Biol. Pharm. Bull., 1994, 17, 701–704 CrossRef CAS PubMed.
- J. B. Torres, E. M. Andreozzi, J. T. Dunn, M. Siddique, I. Szanda, D. R. Howlett, K. Sunassee and P. J. Blower, J. Nucl. Med., 2016, 57, 109–114 CrossRef CAS PubMed.
- Z. Xiao, P. S. Donnelly, M. Zimmermann and A. G. Wedd, Inorg. Chem., 2008, 47, 4338–4347 CrossRef CAS PubMed.
- C. J. Maynard, A. I. Bush, C. L. Masters, R. Cappai and Q. X. Li, Int. J. Exp. Pathol., 2005, 86, 147–159 CrossRef CAS PubMed.
- J. L. Hickey, S. Lim, D. J. Hayne, B. M. Paterson, J. M. White, V. L. Villemagne, P. Roselt, D. Binns, C. Cullinane, C. M. Jeffery, R. I. Price, K. J. Barnham and P. S. Donnelly, J. Am. Chem. Soc., 2013, 135, 16120–16132 CrossRef CAS PubMed.
- N. Bandara, A. K. Sharma, S. Krieger, J. W. Schultz, B. H. Han, B. E. Rogers and L. M. Mirica, J. Am. Chem. Soc., 2017, 139, 12550–12558 CrossRef CAS PubMed.
- H. Watanabe, M. Ono, S. Iikuni, M. Yoshimura, K. Matsumura, H. Kimura and H. Saji, Bioorg. Med. Chem. Lett., 2014, 24, 4834–4837 CrossRef CAS PubMed.
- K. Chauhan, A. Datta, A. Adhikari, K. Chuttani, A. K. Singh and A. K. Mishra, Org. Biomol. Chem., 2014, 12, 7328–7337 RSC.
- M. Asti, E. Ferrari, S. Croci, G. Atti, S. Rubagotti, M. Iori, P. C. Capponi, A. Zerbini, M. Saladini and A. Versari, Inorg. Chem., 2014, 53, 4922–4933 CrossRef CAS PubMed.
- G. Van Dongen, M. C. Huisman, R. Boellaard, N. H. Hendrikse, A. D. Windhorst, G. W. M. Visser, C. F. M. Molthoff and D. J. Vugts, Q. J. Nucl. Med. Mol. Imaging, 2015, 59, 18–38 CAS.
- J. Fissers, A. M. Waldron, T. De Vijlder, B. Van Broeck, D. J. Pemberton, M. Mercken, P. Van der Veken, J. Joossens, K. Augustyns, S. Dedeurwaerdere, S. Stroobants, S. Staelens and L. wyffels, Mol. Imaging Biol., 2016, 18, 598–605 CrossRef CAS PubMed.
- M. P. Kung, D. A. Stevenson, K. Plossl, S. K. Meegalla, A. Beckwith, W. D. Essman, M. Mu, I. Lucki and H. F. Kung, Eur. J. Nucl. Med., 1997, 24, 372–380 CrossRef CAS.
- P. D. Mozley, J. B. Stubbs, K. Plossl, S. H. Dresel, E. D. Barraclough, A. Alavi, L. I. Araujo and H. F. Kung, J. Nucl. Med., 1998, 39, 2069–2076 CAS.
- Y. Cheng, M. Ono, H. Kimura, M. Ueda and H. Saji, J. Med. Chem., 2012, 55, 2279–2286 CrossRef CAS PubMed.
- X. Y. Zhang, P. R. Yu, Y. P. Yang, Y. Q. Hou, C. Peng, Z. G. Liang, J. Lu, B. A. Chen, J. P. Dai, B. L. Liu and M. C. Cui, Bioconjugate Chem., 2016, 27, 2493–2504 CrossRef CAS PubMed.
- A. Kurihara and W. M. Pardridge, Bioconjugate Chem., 2000, 11, 380–386 CrossRef CAS PubMed.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodriguez-Rodriguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- G. R. Naumiec, G. Lincourt, J. P. Clever, M. A. McGregor, A. Kovoor and B. DeBoef, Org. Biomol. Chem., 2015, 13, 2537–2540 RSC.
- L. Frullano, J. Q. Zhu, C. N. Wang, C. Y. Wu, R. H. Miller and Y. M. Wang, J. Med. Chem., 2012, 55, 94–105 CrossRef CAS PubMed.
- G. Bort, S. Catoen, H. Borderies, A. Kebsi, S. Ballet, G. Louin, M. Port and C. Ferroud, Eur. J. Med. Chem., 2014, 87, 843–861 CrossRef CAS PubMed.
- W. H. Li, S. E. Fraser and T. J. Meade, J. Am. Chem. Soc., 1999, 121, 1413–1414 CrossRef CAS.
- E. L. Que, E. J. New and C. J. Chang, Chem. Sci., 2012, 3, 1829–1834 RSC.
- M. Aswendt, E. Gianolio, G. Pariani, R. Napolitano, F. Fedeli, U. Himmelreich, S. Aime and M. Hoehn, NeuroImage, 2012, 62, 1685–1693 CrossRef CAS PubMed.
- A. C. Silva and N. A. Bock, Schizophr. Bull., 2008, 34, 595–604 CrossRef PubMed.
- A. Bertin, J. Steibel, A. I. Michou-Gallani, J. L. Gallani and D. Felder-Flesch, Bioconjugate Chem., 2009, 20, 760–767 CrossRef CAS PubMed.
- A. Barandov, B. B. Badelle, C. G. Williamson, E. S. Loucks, S. J. Lippard and A. Jasanoff, Nat. Commun., 2019, 10, 897 CrossRef PubMed.
- R. Sperling, Neurobiol. Aging, 2011, 32, S37–S43 CrossRef PubMed.
- T. Lee, L. X. Cai, V. S. Lelyveld, A. Hai and A. Jasanoff, Science, 2014, 344, 533–535 CrossRef CAS PubMed.
- Y. Z. Wadghiri, J. L. Li, J. H. Wang, D. M. Hoang, Y. J. Sun, H. Xu, W. Tsui, Y. S. Li, A. Boutajangout, A. Wang, M. de Leon and T. Wisniewski, PLoS One, 2013, 8 DOI:10.1371/journal.pone.0057097.
- S. Okada, B. B. Bartelle, N. Li, V. Breton-Provencher, J. J. Lee, E. Rodriguez, J. Melican, M. Sur and A. Jasanoff, Nat. Nanotechnol., 2018, 13, 473–477 CrossRef CAS PubMed.
- K. Y. Zhang, Q. Yu, H. J. Wei, S. J. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS PubMed.
- Q. Zhao, C. H. Huang and F. Y. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- B. Y. W. Man, H. M. Chan, C. H. Leung, D. S. H. Chan, L. P. Bai, Z. H. Jiang, H. W. Li and D. L. Ma, Chem. Sci., 2011, 2, 917–921 RSC.
- L. H. Lu, H. J. Zhong, M. D. Wang, S. L. Ho, H. W. Li, C. H. Leung and D. L. Ma, Sci. Rep., 2015, 5, 25774, DOI:10.1038/srep25774.
- N. P. Cook, V. Torres, D. Jain and A. A. Marti, J. Am. Chem. Soc., 2011, 133, 11121–11123 CrossRef CAS PubMed.
- N. P. Cook, K. Kilpatrick, L. Segatori and A. A. Marti, J. Am. Chem. Soc., 2012, 134, 20776–20782 CrossRef CAS PubMed.
- A. Aliyan, B. Kirby, C. Pennington and A. A. Marti, J. Am. Chem. Soc., 2016, 138, 8686–8689 CrossRef CAS PubMed.
- M. T. Gabr and F. C. Pigge, Chem. – Eur. J., 2018, 24, 11729–11737 CrossRef CAS PubMed.
- M. Isaac, L. Raibaut, C. Cepeda, A. Roux, D. Boturyn, S. V. Eliseeva, S. Petoud and O. Seneque, Chem. – Eur. J., 2017, 23, 10992–10996 CrossRef CAS PubMed.
- E. Debroye and T. N. Parac-Vogt, Chem. Soc. Rev., 2014, 43, 8178–8192 RSC.
- F. Oukhatar, S. Meme, W. Meme, F. Szeremeta, N. K. Logothetis, G. Angelovski and E. Toth, ACS Chem. Neurosci., 2015, 6, 219–225 CrossRef CAS PubMed.
- F. Oukhatar, S. V. Eliseeva, C. S. Bonnet, M. Placidi, N. K. Logothetis, S. Petoud, G. Angelovski and É. Tóth, Inorg. Chem., 2019, 58, 13619–13630, DOI:10.1021/acs.inorgchem.9b00887.
- C. T. Yang, K. K. Ghosh, P. Padmanabhan, O. Langer, J. Liu, D. N. C. Eng, C. Halldin and B. Gulyas, Theranostics, 2018, 8, 6210–6232 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |