
Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions
Zi-Lei
Xia
,
Qing-Feng
Xu-Xu
,
Chao
Zheng
 * and
Shu-Li
You
*
* and
Shu-Li
You
*
State Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: zhengchao@sioc.ac.cn; slyou@sioc.ac.cn
First published on 12th December 2019
Abstract
We summarize in this review the recent development of chiral phosphoric acid (CPA)-catalyzed asymmetric dearomatization reactions. A wide array of electron-rich arenes (indoles, phenols, naphthols, benzothiophenes, benzofurans, etc.) and electron-poor arenes (pyridines, quinolines, isoquinolines, etc.) has been proved reactive towards various reaction partners in the presence of a CPA catalyst, enabling asymmetric dearomatization reactions that lead to structurally-diverse polycyclic molecules. The reactions are grouped according to the roles of the arenes in the reactions (as nucleophiles or electrophiles) and the types of reaction partners. This review closes with a personal perspective on the dynamic research area of asymmetric dearomatization reactions by CPAs.
 Zi-Lei Xia | Zi-Lei Xia was born in 1987 in Xuanwei, Yunnan province, China, and received his BS and MS degrees from the Tianjin University of Science and Technology in 2011 and China Pharmaceutical University in 2015, respectively. He joined the Shanghai Institute of Organic Chemistry (SIOC) and obtained his PhD degree under the supervision of Prof. Shu-Li You in 2018. His research was focused on chiral phosphoric acid-catalyzed asymmetric dearomatization reactions. Currently he is a postdoc research fellow at Purdue University with Prof. Arun K. Ghosh. |
 Qing-Feng Xu-Xu | Qing-Feng Xu-Xu was born in 1993 in Wuhan, Hubei province, China. He obtained his BS degree from Central China Normal University in 2015 and joined the Shanghai Institute of Organic Chemistry (SIOC) in the same year under the supervision of Prof. Shu-Li You. His research interests are dearomatization of arenes. |
 Chao Zheng | Chao Zheng was born in Hubei province, China in 1985. He studied chemistry and received his BSc degree at Shanghai Jiao Tong University in 2007. He obtained his PhD degree at the Shanghai Institute of Organic Chemistry (SIOC) under the supervision of Prof. Shu-Li You and Prof. Yu-Xue Li in 2012. Then he joined the group of Prof. Shu-Li You at the SIOC and was promoted to Associate Professor in 2015. His current work is focused on mechanistic understanding of homogeneous organic reactions by employing computational chemistry, and developing new catalytic asymmetric reactions. He has published over 40 journal papers and 4 book chapters. |
 Shu-Li You | Shu-Li You received his BSc in chemistry from Nankai University (1996). He then obtained his PhD from the Shanghai Institute of Organic Chemistry (SIOC) in 2001 under the supervision of Prof. Lixin Dai before doing postdoctoral studies with Prof. Jeffery Kelly at The Scripps Research Institute. From 2004, he worked at the Genomics Institute of the Novartis Research Foundation as a PI before returning to the SIOC as a Professor in 2006. He is currently the director of the State Key Laboratory of Organometallic Chemistry of the SIOC. His research interests mainly focus on asymmetric C–H functionalization and catalytic asymmetric dearomatization (CADA) reactions. He has published over 280 research papers in international peer-reviewed journals and edited two books. |
1. Introduction
With the development of organic chemistry, the demand of novel molecular identities is growing continuously. Relevant research fields including medicinal chemistry, materials sciences, and energy and resources all enjoy the benefits from the exploration of the expanded chemical space. Accordingly, developing efficient synthetic methods that convert readily available starting materials into libraries of molecules with structural complexity and diversity is of great significance. Among the numerous efforts towards this goal, dearomatization reactions have recently emerged as an enabling strategy in this regard.1–3 With cheap aromatic compounds, many of which are derived from bulk chemical feedstocks, as the substrates, the dearomatization reactions provide rapid access to intriguing polycyclic molecules often bearing quaternary stereogenic centers at the ring junctions.4–6 Although they are generally thought of as energetically uphill processes, dearomatization reactions have been proved as, in many cases, thermodynamically feasible, especially when coupled with the formation of quaternary carbon centers or rationally designed intramolecular reactions that harness the entropy compensation. Notably, various chiral transition-metal complexes and organocatalysts have been successfully applied to promote asymmetric dearomatization reactions, delivering valuable enantioenriched polycyclic molecules.7Chiral phosphoric acids (CPAs) have been recognized as powerful organocatalysts8–11 since the seminal contributions by the groups of Akiyama, and Terada in 2004.12,13 Among the many variations of CPAs, the ones derived from privileged skeletons including (H8-)BINOL and SPINOL are the most frequently employed (Fig. 1). In general, CPAs can work as bifunctional catalysts, forming dual hydrogen-bonds with both the electrophile and the nucleophile simultaneously. The structurally well-defined cyclic transition states contribute significantly to the diastereomeric discrimination processes with the crucial influences from the neighboring substituents on the chiral backbone. Besides, the corresponding chiral phosphates have also shown great potential under the scheme of asymmetric counteranion-directed catalysis (ACDC) or asymmetric ion-pairing catalysis.14–17
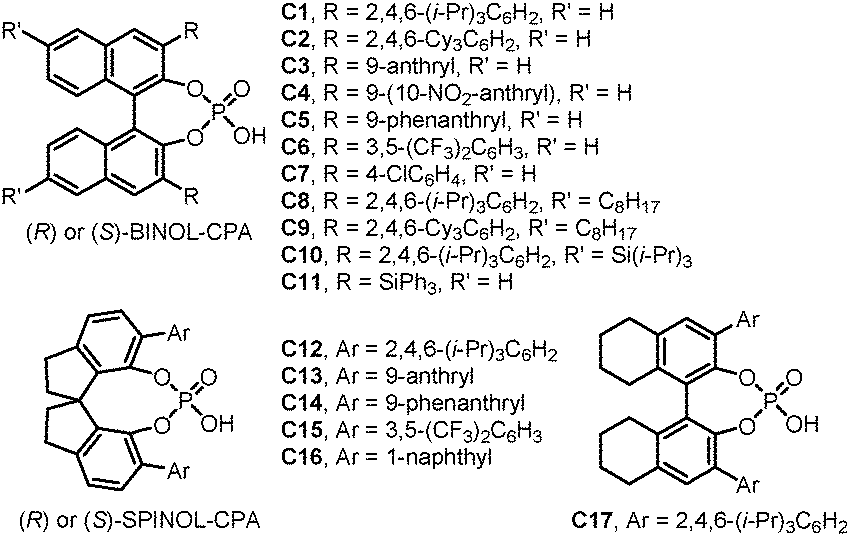 | ||
| Fig. 1 Chiral phosphoric acids discussed in this review. | ||
CPAs have found wide applications in asymmetric Friedel–Crafts reactions, an important class of enabling transformations where aromatic compounds work as the nucleophiles.18 The reactions generally proceed via a dearomatization process, and the subsequent proton abstraction occurs to re-install the aromaticity. If the nucleophilic attack occurs at a substituted position of the aromatic compounds, the subsequent proton abstraction will not be available, thus leading the reaction to dearomatized products. This reaction pattern has been observed for various electron-rich arenes including indoles, pyrroles, naphthols, benzofurans, benzothiophenes, etc. with diverse carbon- or heteroatom-based electrophiles. Notably, thanks to the recently emerging visible-light-catalysis,19–22 novel asymmetric dearomatization of indole derivatives with radical species and electrophiles has appeared. This review aims to provide the readers an overview of the latest progress on the scope and mechanism of CPA-catalyzed asymmetric dearomatization reactions. It should be noted that CPAs are also well known in promoting asymmetric transfer hydrogenation of electron-poor arenes with Hantzsch ester or related hydride sources.23–26 These reactions will not be covered in this review in detail unless they are integrated in a cascade dearomatization reaction.
2. CPA-catalyzed dearomatization reactions of electron-rich arenes
In this section, we will provide detailed discussion on asymmetric dearomatization reactions initiated by CPA-catalyzed nucleophilic attack of electron-rich arenes to various electrophiles. The reactions will be grouped according to the type of electrophiles. The CPA-catalyzed asymmetric dearomatization reactions of indole-derivatives coupled with visible-light-catalysis will also be covered.2.1 Halonium electrophiles
Selectfluor® is among the most applied electrophilic halogenating reagents. The successful applications of Selectfluor® in catalytic asymmetric dearomatization reactions date back to the original contributions from Toste and coworkers. The Toste group developed a chiral anion phase-transfer catalysis (PTC) strategy in which a chiral BINOL-derived phosphate generated in situ underwent anion exchange with halonium reagents to produce a soluble ion pair that participated in the desired catalytic asymmetric reactions. The PTC strategy could effectively eliminate the effects of racemic background reactions and guarantee the high enantioselectivity.In 2011, Toste and coworkers reported the first example of a catalytic asymmetric fluorinated dearomatization reaction using the PTC strategy (Scheme 1).27 With a modified TRIP CPA (R)-C8 [TRIP: 3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate] as the catalyst, the dearomatization of benzothiophene derivatives (1a and 1b) proceeded smoothly under the chiral anion-mediated phase-transfer reaction conditions (Na2CO3, and C6H5F). The desired products (2a and 2b) were delivered in reasonable yields (59% and 69%) with high stereoselectivity (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 89% ee, and >20:1 dr, 90% ee). Notably, only a complex mixture was observed when the reaction was performed under homogeneous conditions (CH3CN).
:1 dr, 89% ee, and >20:1 dr, 90% ee). Notably, only a complex mixture was observed when the reaction was performed under homogeneous conditions (CH3CN).
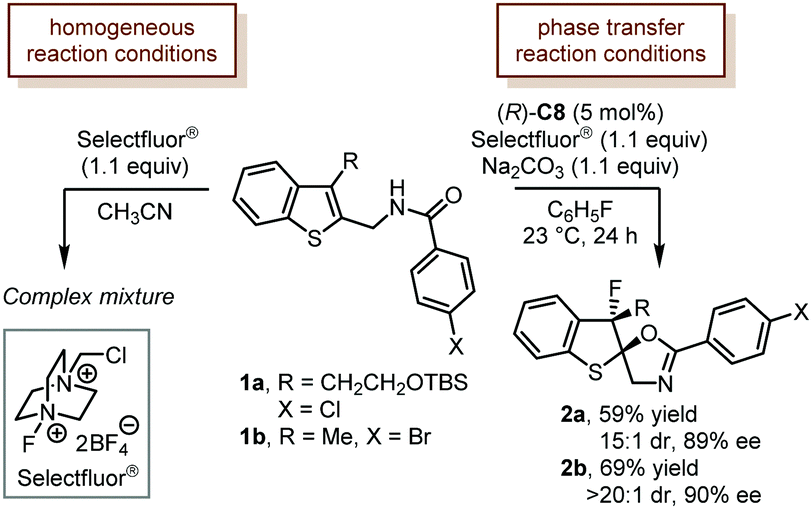 | ||
| Scheme 1 The asymmetric fluorinative dearomatization reaction of benzothiophene derivatives. | ||
Different from conventional hydrogen-bonding interactions in CPA catalysis, the chiral ion pair plays a crucial role in the PTC strategy (Scheme 2). Under the PTC reaction conditions, the in situ generated chiral phosphate I1 undergoes anion exchange with insoluble Selectfluor®, leading to soluble chiral ion pair I2, which consists of two chiral phosphate anions. The observation of a significant nonlinear effect between the ee values of the catalyst and the products supports the existence of chiral ion pair I2 which is believed to be responsible for the asymmetric dearomative fluorination of benzothiophenes.
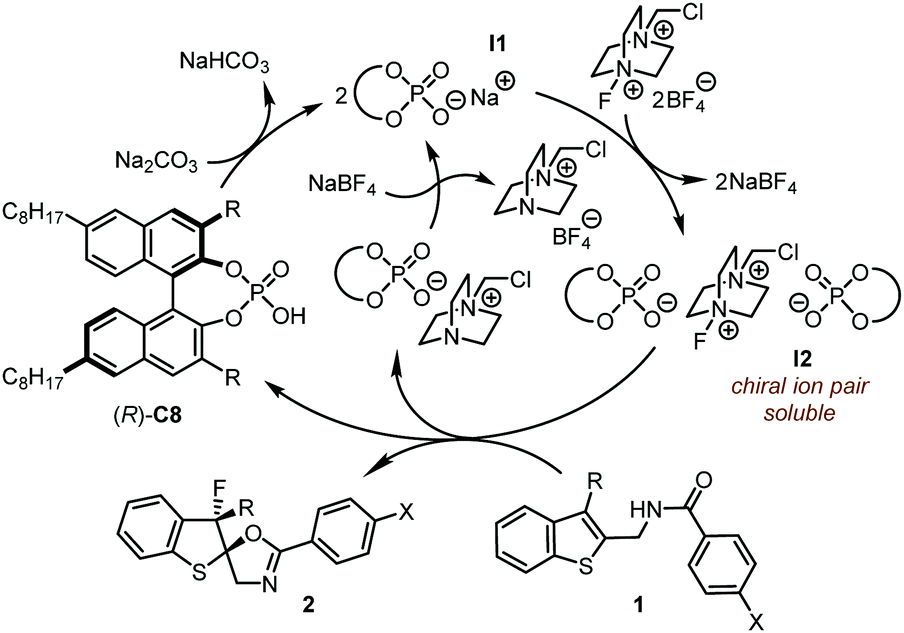 | ||
| Scheme 2 Proposed mechanism of the asymmetric dearomatization reaction under PTC conditions. | ||
Following this seminal report, the Toste group successfully applied the chiral anion PTC strategy in the enantioselective fluorinative dearomatization of phenols in 2013 (Scheme 3).28 The intermolecular dearomatization of phenols is considerably challenging due to the existence of multiple reactive sites which might lead to poor selectivity. By careful optimization of various reaction parameters, the authors discovered that the incorporation of an ortho-substituent in phenols 3 is crucial. With (S)-C2 as the optimal catalyst, the desired fluorinated dearomatized products 4 could be obtained in good yields (up to 75%) and excellent enantioselectivity (up to 96% ee). More interestingly, when phenols without a substituent at the meta position were used, another series of products 5 resulting from fluorinative dearomatization/[4+2] dimerization sequence could be obtained in good yields (up to 96%) with high enantioselectivity (up to 97% ee). This cascade reaction provides valuable building scaffolds with potential interest in synthetic and medicinal chemistry. For instance, an analog of the natural product (−)-grandifloracin, in which the two hydroxyl groups are substituted with fluorine atoms, could be readily synthesized via this dearomatization/dimerization sequence.
 | ||
| Scheme 3 The asymmetric fluorinative dearomatization reaction of phenol derivatives. | ||
In 2017, the You group reported the asymmetric fluorinative dearomatization of tryptamine derivatives 6 using the PTC strategy (Scheme 4).29 Although in previous works polar solvents like acetonitrile were usually detrimental to stereochemical control under the PTC reaction conditions, unexpectedly, a 1:1 mixture of fluorobenzene and acetonitrile was identified as the optimal solvent in this study. The reaction was greatly accelerated but without dramatic decrease of the enantioselectivity. With (S)-C3 as the optimal catalyst, a series of 3-fluoropyrroloindolines 7 could be afforded in moderate to good yields (29–92%) with good enantioselectivity (56–97% ee). Recently, the You group achieved the enantioselective fluorinative dearomatization reactions of more challenging tryptophol derivatives 8 by employing modified PTC reaction conditions (Scheme 4).30 It was found that adding a stoichiometric amount of aryl boronic acid and water was crucial to the reaction outcomes. The possible intermediacy of a tryptophol-derived boronic ester I3 was proposed.
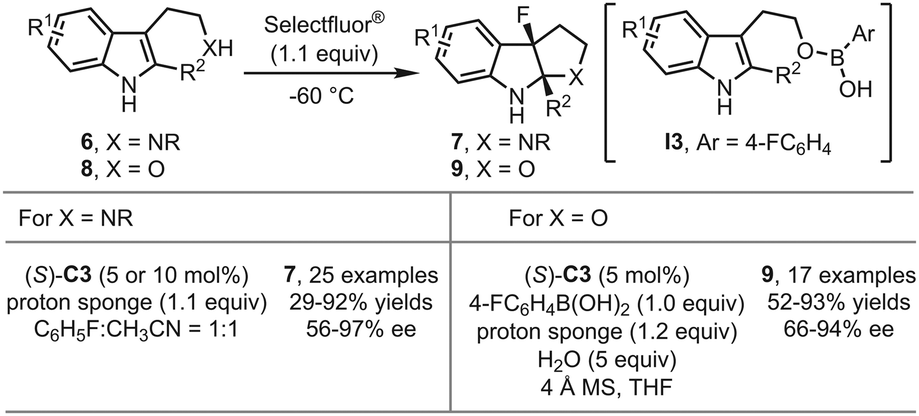 | ||
| Scheme 4 The asymmetric fluorinative dearomatization reaction of tryptamine and tryptophol derivatives. | ||
Besides commercially available Selectfluor®, other electrophilic halogenating reagents were also utilized in asymmetric dearomatization reactions under the chiral phosphate based PTC catalysis. In 2012, the Toste group ingeniously designed a series of electrophilic brominating and iodinating reagents that are dimerized analogs of Selectfluor®.31 Intriguing bromine- or iodine-containing compounds that could be employed in further elaborations were obtained in highly enantioenriched form. Particularly the asymmetric brominative dearomatization of benzothiophene 10 was achieved in the presence of (R)-C10 and B1 affording the corresponding bromocyclized product 11 in 83% yield with 97% ee (Scheme 5).
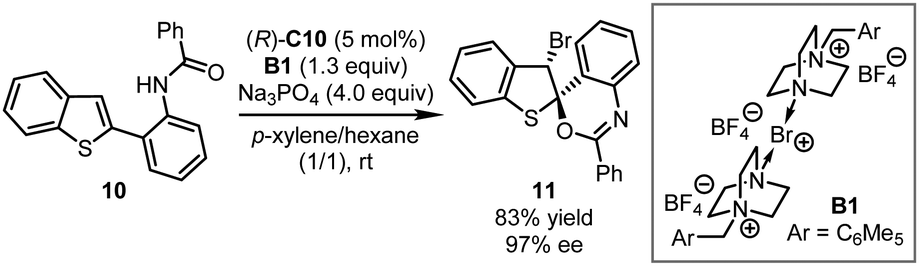 | ||
| Scheme 5 The asymmetric brominative dearomatization reaction of benzothiophene derivatives. | ||
Shortly after, Xie, Lai, Ma and coworkers developed an effective bromocyclization of tryptamine derivatives, leading to brominated pyrroloindoline derivatives (Scheme 6).32 Interestingly, the purity of the brominating reagent significantly influences the reaction outcomes in terms of the stereochemical control. After systematic evaluation, readily available brominating reagent B2 was identified as the optimal one. This bromocyclization protocol could efficiently assemble a series of 3-bromopyrroloindolines 13 in good yields (37%–quant.) with excellent enantioselectivity (79–98% ee). Notably, these bromine-containing compounds could undergo versatile downstream transformations to other functionalized molecules, among which Co-catalyzed stereospecific homodimerization was the most significant one. With this reaction as the key step, an elegant asymmetric total synthesis of the natural product (−)-chimonanthine was achieved. Subsequently, Lai, Xie and coworkers completed the total synthesis of the natural product (−)-conolutinine by applying the bromocyclization of a 2-piperidinone-fused tryptamine derivative 14 with a similar catalytic system (Scheme 6).33 In addition, the authors also accomplished elegant asymmetric bromocyclization of tryptophol derivatives 16 affording a series of 3-bromofuroindolines 17 by utilizing a similar protocol (Scheme 6).34 Interestingly, by adding a catalytic amount of B3, better enantioselectivity and accelerated reaction rate were observed.
 | ||
| Scheme 6 The asymmetric brominative dearomatization reaction of tryptamine and tryptophol derivatives. | ||
2.2 Carbon electrophiles
Various carbon-based electrophiles, including α,β-unsaturated ketones, quinones, imines, allenes, alkynes and their analogs, have been successfully utilized in CPA-catalyzed asymmetric dearomatization reactions. Herein, the most recent progress in this direction will be discussed.In 2012, the Antilla group developed CPA-catalyzed asymmetric dearomatization reactions of tryptamine derivatives with methyl vinyl ketone (MVK) (Scheme 7).35 In the presence of (R)-C1, the asymmetric dearomatization of 18 with three equivalents of MVK proceeded smoothly. Asymmetric C3-Michael addition/cyclization/N-Michael addition sequence led to a series of pyrroloindolines 19 in high yields (85–93%) and enantioselectivity (70–93% ee). The product could be readily transformed to the natural product (−)-debromoflustramine B, highlighting the synthetic utility of this approach. Independently, the You group reported similar results by using aryl vinyl ketone as the electrophile, affording the desired products in good yields (33–97%) with moderate to good enantioselectivity (50–84% ee) (Scheme 7).36 Notably, tryptophol could be tolerated under the optimal conditions in excellent yield (94%) albeit with 55% ee. The same group further achieved the asymmetric dearomatization reactions of indole-tethered anilines 20 with MVK (Scheme 7).37 A series of indolo[2,3-b]quinolines 21 was readily synthesized in moderate to good yields (35–90%) with excellent enantioselectivity (81–98% ee).
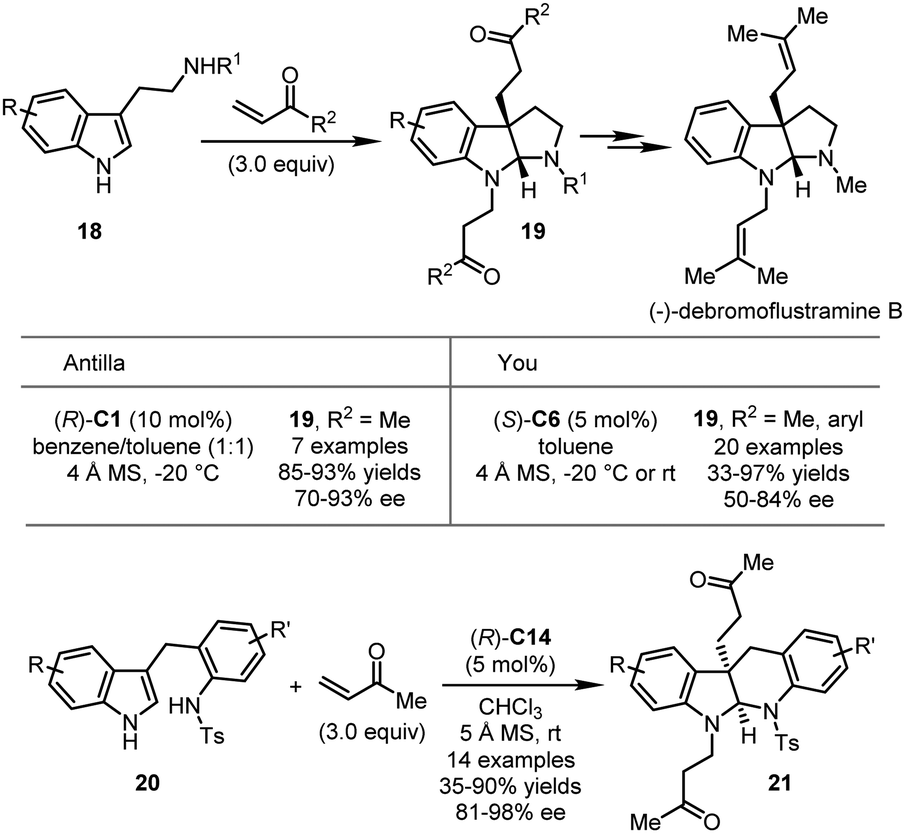 | ||
| Scheme 7 The asymmetric dearomatization reaction of tryptamine derivatives and indole-tethered anilines. | ||
In 2017, the You group developed an intramolecular asymmetric dearomatization reaction of indoles via the CPA-catalyzed Michael addition of the C3 position of the indole ring to a tethered α,β-unsaturated ketone moiety. A series of spiroindolenines 23 was obtained in high yields (56–98%) and enantioselectivity (42–97% ee) (Scheme 8).38 It should be noted that there are two structural requirements of substrates 22, which guarantee this reactivity. First, the aryl substituent at the C2 position of the indole ring is necessary. Friedel–Crafts alkylation at the C2 position will occur predominately if no substituent is attached at the C2 position. Second, a nitrogen atom bearing an electron-withdrawing group or a malonate diester group in the linkage is crucial to stabilize the spiroindolenine product. If an appropriate electron-donating group, like 3,5-(CF3)2C6H3CH2, was incorporated, a skeleton reorganization would be triggered via a retro-Mannich reaction. The iminium intermediate generated in situ will then be trapped by the enol moiety, affording chiral 3,4-disubstituted pyrrolidine products 24 (Scheme 8).39
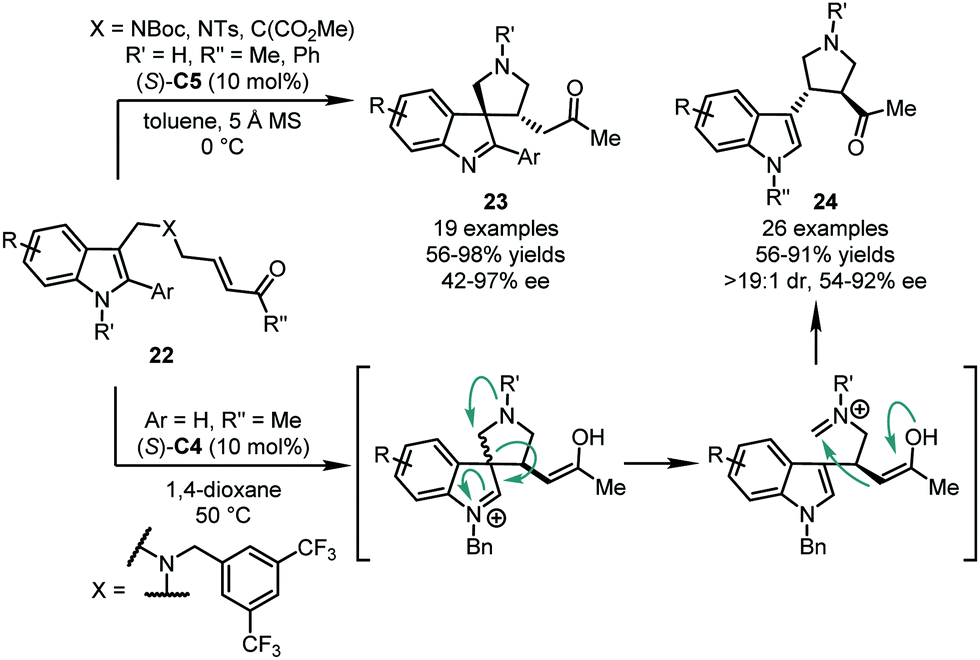 | ||
| Scheme 8 The asymmetric dearomatization reaction of indole-tethered α,β-unsaturated ketones. | ||
Quinones and quinone monoimines (QMIs) are a kind of unique electrophile with inherent oxidability. They could be regarded as activated derivatives of α,β-unsaturated ketones with a special driving force of rearomatization after the nucleophilic attack. Therefore, quinones and QMIs have been widely used as arylative reagents in asymmetric catalysis. In 2014, the Zhang group reported a CPA-catalyzed dearomative [3+2] annulation of 3-substituted indoles 25 with QMI 26, affording a series of benzofuranindolines 27 in high yields (54–98%) with excellent enantioselectivity (80–99% ee) (Scheme 9, eqn 1).40 This reaction showed a wide substrate scope of indoles but only QMI 26 could be well tolerated as the reaction partner. A plausible mechanism for this dearomatization reaction was proposed. (R)-C1 initially acts as a bifunctional catalyst, activating both the indoles and QMI via dual hydrogen-bonding interactions. The nucleophilic addition of indoles to 26 would form indolenine intermediates I4, which undergo rearomatization immediately to afford phenol intermediates I5. Finally, the intramolecular Mannich reaction delivers the target products.
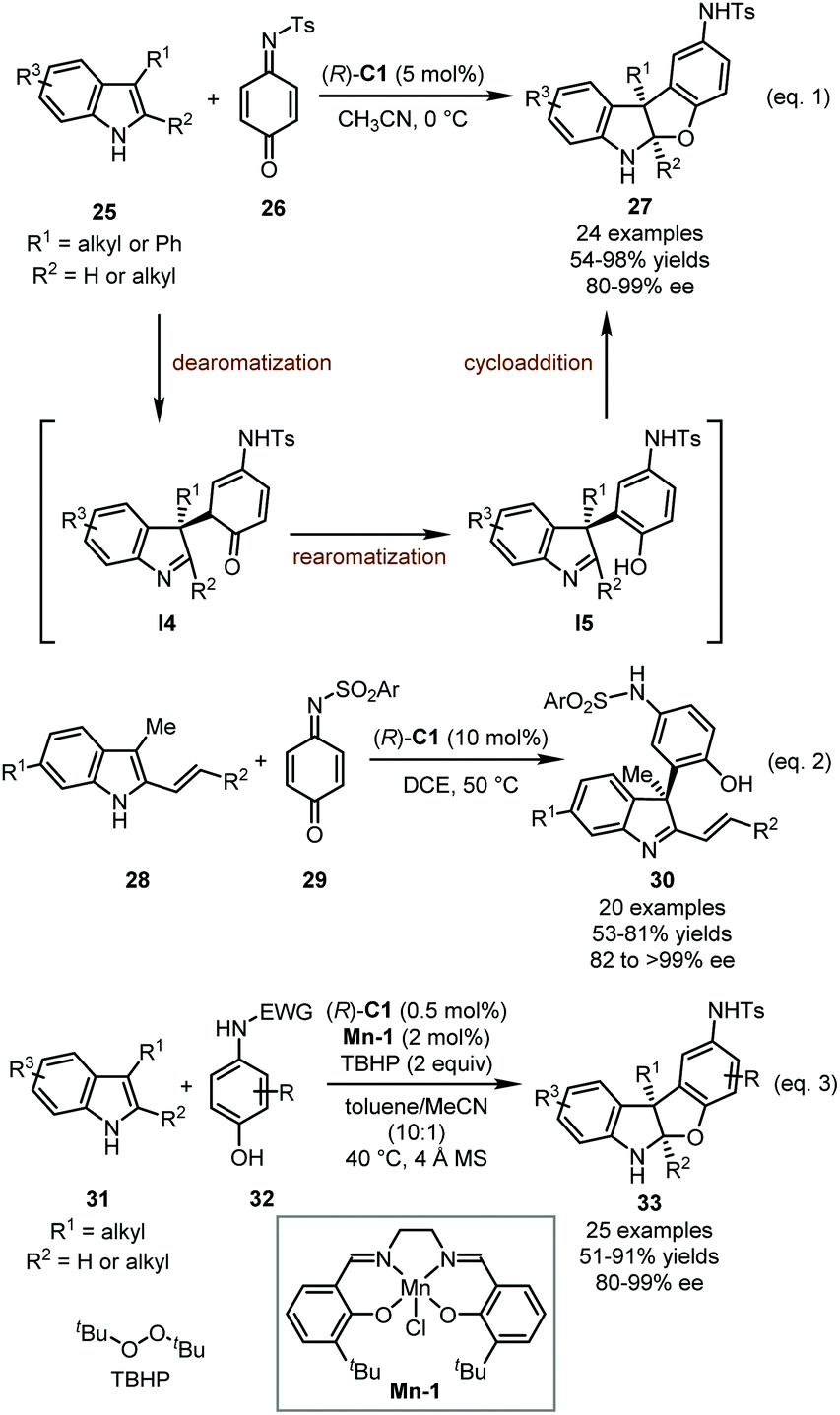 | ||
| Scheme 9 The asymmetric dearomatization reaction of indoles with QMIs. | ||
In 2015, the Shi group designed an elegant reaction in which 3-methyl-2-vinylindoles 28 were used as the coupling partners with QMIs 29.41 Different from 2,3-dialkylsubstituted indoles, introducing a vinyl group on the C2 position would lead to the competition among multiple reaction pathways including [3+2] cycloaddition (vinyl group as a two-carbon unit), dearomative [3+2] annulation, [4+3] annulation (2-vinyl indole as a four-carbon unit), and arylative dearomatization. Notably, thorough screening of the reaction conditions achieved the highly efficient synthesis of arylative dearomatized products 30 in good yields (53–81%) with excellent enantioselectivity (82 to >99% ee) (Scheme 9, eqn 2). The authors also proposed the key role of catalyst (R)-C1 as a dual hydrogen-bond donor/acceptor. When an N-Me indole substrate was employed, the dearomative [3+2] annulation product was obtained with moderate enantiomeric excess.
Very recently, Wu, Zhong, and coworkers reported the dearomative [3+2] annulation between 3-substituted indoles 31 and in situ generated QMIs enabled by a dual catalytic system consisting of a CPA and a (salen)Mn(III) complex.42 With TBHP (tert-butyl hydroperoxide) as the terminal oxidant, para-aminophenols 32 could be oxidized to the corresponding QMIs by a (salen)Mn(III) complex Mn-1. The following dearomative [3+2] annulation between 3-substituted indoles and QMIs occurred smoothly under mild conditions, leading to the desired products 33 in high yields (51–91%) and good enantioselectivity (80–99% ee) (Scheme 9, eqn 3). The catalytic system showed impressive efficiency. The reaction could be performed at gram-scale with the loading of (R)-C1 lowered to 0.05%, and an impressive TON (turnover number) of 4100 was measured with the loading of (R)-C1 at 0.01%.
Quinone imine ketals (QIKs) are another kind of quinone derivative that are less reactive compared with quinones and QMIs. In 2014, the Shi group reported a CPA-catalyzed asymmetric dearomatization of indoles with QIKs (Scheme 10).43 Similar to QMIs, QIKs 34 worked as α,β-unsaturated imines that could be well activated in the presence of (R)-C1. The nucleophilic attack by 3-substituted indoles 35 and the subsequent elimination of one molecule of alcohol via intermediate I6 afforded the corresponding dearomatized indolenines 36 in good yields (54–99%) and high enantioselectivity (82–99% ee). Notably, a one-pot tandem dearomatization/transfer hydrogenation sequence was also developed in which the imine moiety of indolenines was reduced with Hantzsch ester H1 as the hydride donor. Interestingly, moderate kinetic resolutions were observed during the transfer hydrogenation step. A series of indolines 37 was obtained in excellent enantiopurity (>95% ee).
 | ||
| Scheme 10 The asymmetric dearomatization reaction of indoles with QIKs. | ||
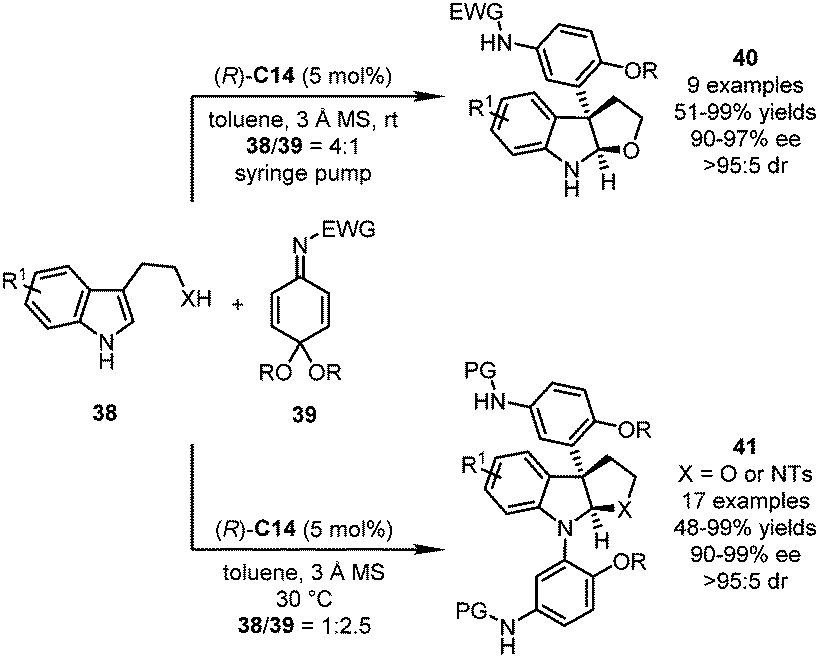
In their follow-up studies, the Shi group further embedded the nucleophilic addition reaction with QIKs into a dearomatization/cyclization sequence (Scheme 11).44 With (R)-C14 as the optimal catalyst, multisubstituted pyrroloindolines and furoindolines were accessed from QIKs 39 and the corresponding tryptamine- and tryptophol-derivatives 38, respectively. It should be noted that the product distribution could be regulated by the molar ratio of both substrates. When a large excess amount of tryptophol-derivatives was employed (38/39 = 4:1, and X = O), an array of 3-aryl furoindolines 40 could be afforded with high efficiency (51–99% yields, and 90–97% ee). On the other hand, if an excess amount of QIKs is present in the reaction system (38/39 = 1:2.5, X = O, and NTs), further aza-Michael addition would occur, leading to pyrroloindoline or furoindoline derivatives 41.
 | ||
| Scheme 11 The asymmetric dearomatization/cyclization sequences with QIKs. | ||
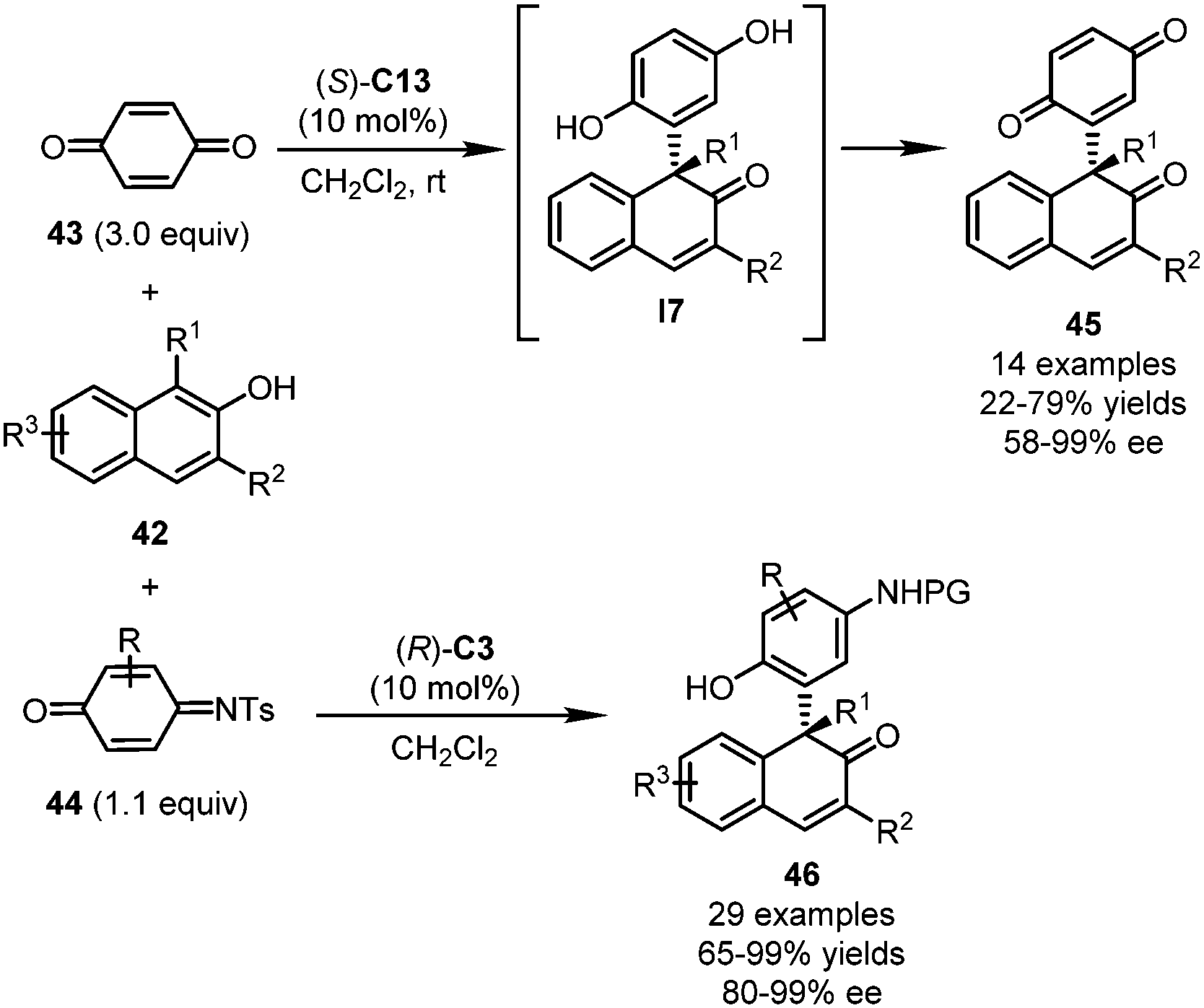
Besides indoles, the dearomatization reactions of naphthols triggered by asymmetric Michael additions with quinones or QMIs have been reported by Sun, Hong, Wang and coworkers,45 and by Chen, Zhou and coworkers,46 independently (Scheme 12). When three equivalents of para-quinone 43 were allowed to react with 1,3-disubstituted 2-naphthols 42 in the presence of (S)-C13, key intermediate I7 was initially formed via Michael addition and then oxidized by the excess amount of 43. Final dearomatized products 45 bearing a substituted para-quinone scaffold were obtained in 22–79% yields with good enantioselectivity (58–99% ee). Notably, lower yields of 45 were observed when decreased equivalents of quinone were employed, which supported the dual roles of para-quinone as both reaction partner and oxidant. Compared with para-quinone, QMIs 44 exhibited attenuated oxidability. In this regard, cyclic conjugated enones 46 bearing a multisubstituted phenyl group incorporated at the α-position was delivered. A much wider substrate scope of 2-naphthols was tolerated. Impressively, the catalyst loading of this reaction could be lowered to 0.5 mol%, without deleterious effects on the yield and enantioselectivity. Notably, substituted para-quinones do not show good reactivity in this reaction.
 | ||
| Scheme 12 The asymmetric dearomatization reaction of 2-naphthols with para-quinone and QMIs. | ||
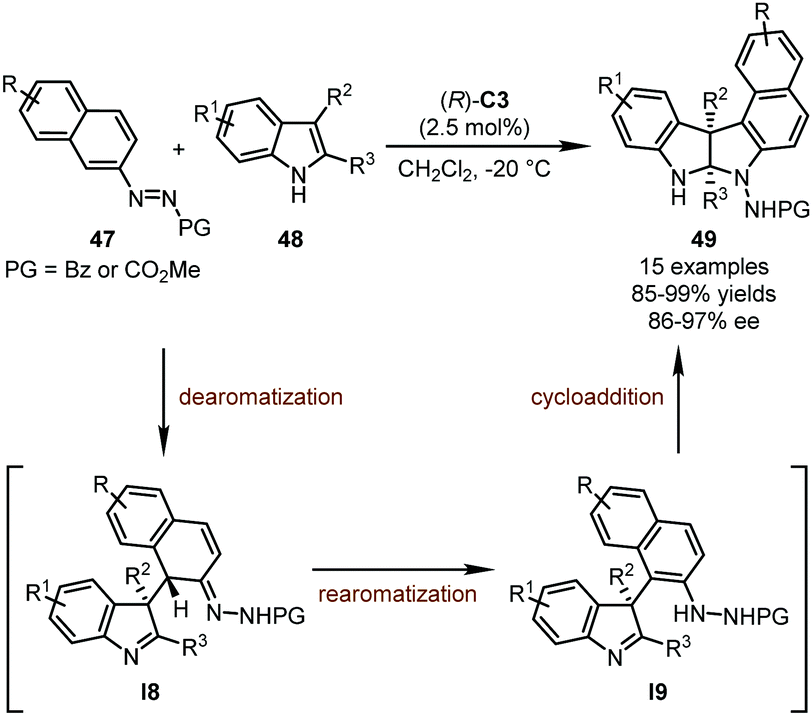
In their systematic work on organocatalytic asymmetric arylation of indoles enabled by azo groups, the Tan group uncovered a novel synthetic strategy towards polycyclic pyrroloindolines initiated by CPA-catalyzed asymmetric Michael addition of 2-azonaphthalenes (Scheme 13).47 It was found that the introduction of an azo group into arenes could notably activate the double bond connected with this group. In the presence of (R)-C3 as the optimal catalyst, the C1 position of 2-azonaphthalenes 47 was readily attacked by 2,3-disubstituted indoles 48, leading to doubly dearomatized intermediates I8. Subsequently, the rearomatization of the 2-azonaphthalene ring resulted in intermediates I9 whose imine functionality was finally trapped by the newly generated hydrazine group, furnishing the desired polycyclic pyrroloindoline derivatives 49 in good yields (85–99%) and enantioselectivity (86–97% ee).
 | ||
| Scheme 13 The asymmetric dearomatization reaction of indoles with 2-azonaphthalenes. | ||
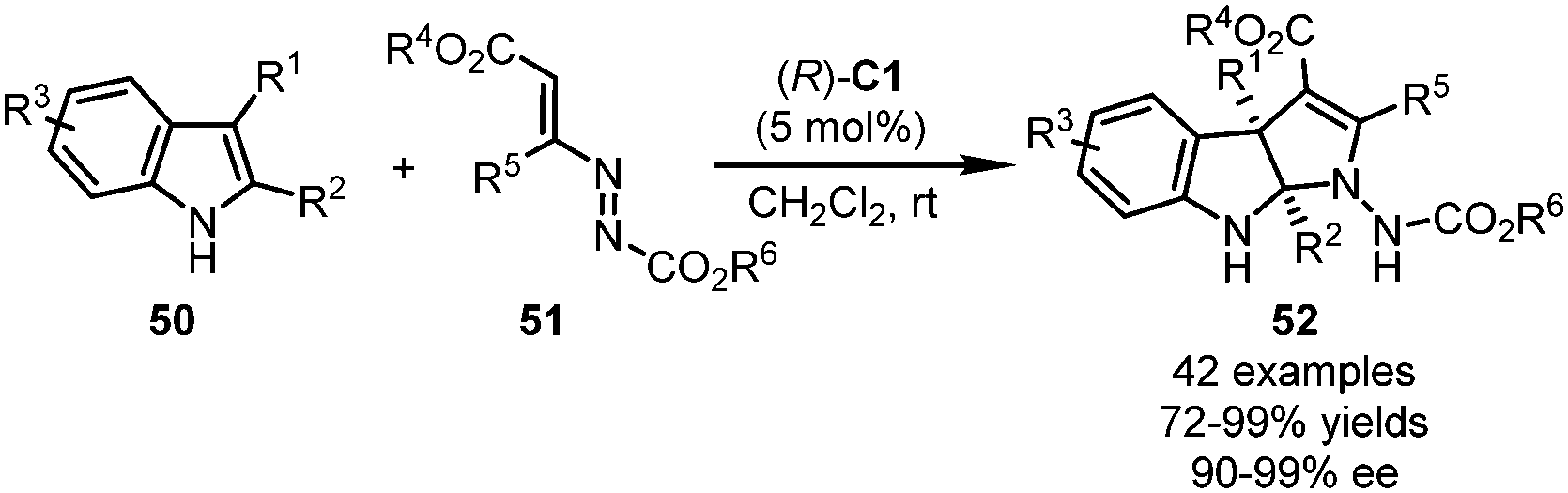
Very recently, Lu and coworkers applied azoalkenes 51 as the precursors of the 1,3-dipolars for the CPA-catalyzed dearomative formal [3+2] cyclization reactions of substituted indoles 50 (Scheme 14).48 This reaction features a wide substrate scope and mild conditions. Structural variation at almost all positions of the pyrroloindoline core of the products 52 could be tolerated. With (R)-C1 as the optimal catalyst, the desired products were delivered in excellent yields (72–99%) with a high level of enantioselective control (90–99% ee).
 | ||
| Scheme 14 The asymmetric dearomatization reaction of indoles with azoalkenes. | ||
In 2016, the Shi group developed an asymmetric dearomatization reaction of tryptamine derivatives 53 with isatin-derived indole-3-yl-methanols 54 (Scheme 15).49 In the presence of CPA (R)-C7, the dehydration of 54 generated key α,β-unsaturated iminium intermediate I10. The following dearomative Michael addition/cyclization sequence between I10 and 53 proceeded smoothly, affording the structurally complex pyrroloindoline-based molecules 55 bearing three contiguous stereogenic centers including two quaternary ones in good to excellent yields (40–99%) and stereoselectivity (70–90% ee, 82:18 to >95:5 dr).
 | ||
| Scheme 15 The asymmetric dearomatization reaction of tryptamines with isatin-derived indol-3-yl-methanols. | ||
In 2017, the Chen group accomplished the catalytic asymmetric total synthesis of natural products (+)- and (−)-paeoveitol.50 The construction of the key tetrahydropyran ring and the establishment of all the three stereogenic centers were realized in the final dearomative hetero-Diels–Alder reaction enabled by a CPA catalyst (Scheme 16). In the presence of a catalytic amount of (R)-C16, a multisubstituted benzofuran derivative paeoveitol D reacted with ortho-quinone methide (o-QM) species I11 that was generated in situ from precursor 56. Therefore, (−)-paeoveitol could be assembled in 92% yield (452 mg) with 90% ee. The ee value could be further enhanced to 96% after a single recrystallization. The enantiomer (+)-paeoveitol could be obtained by employing (S)-C16 as the catalyst.
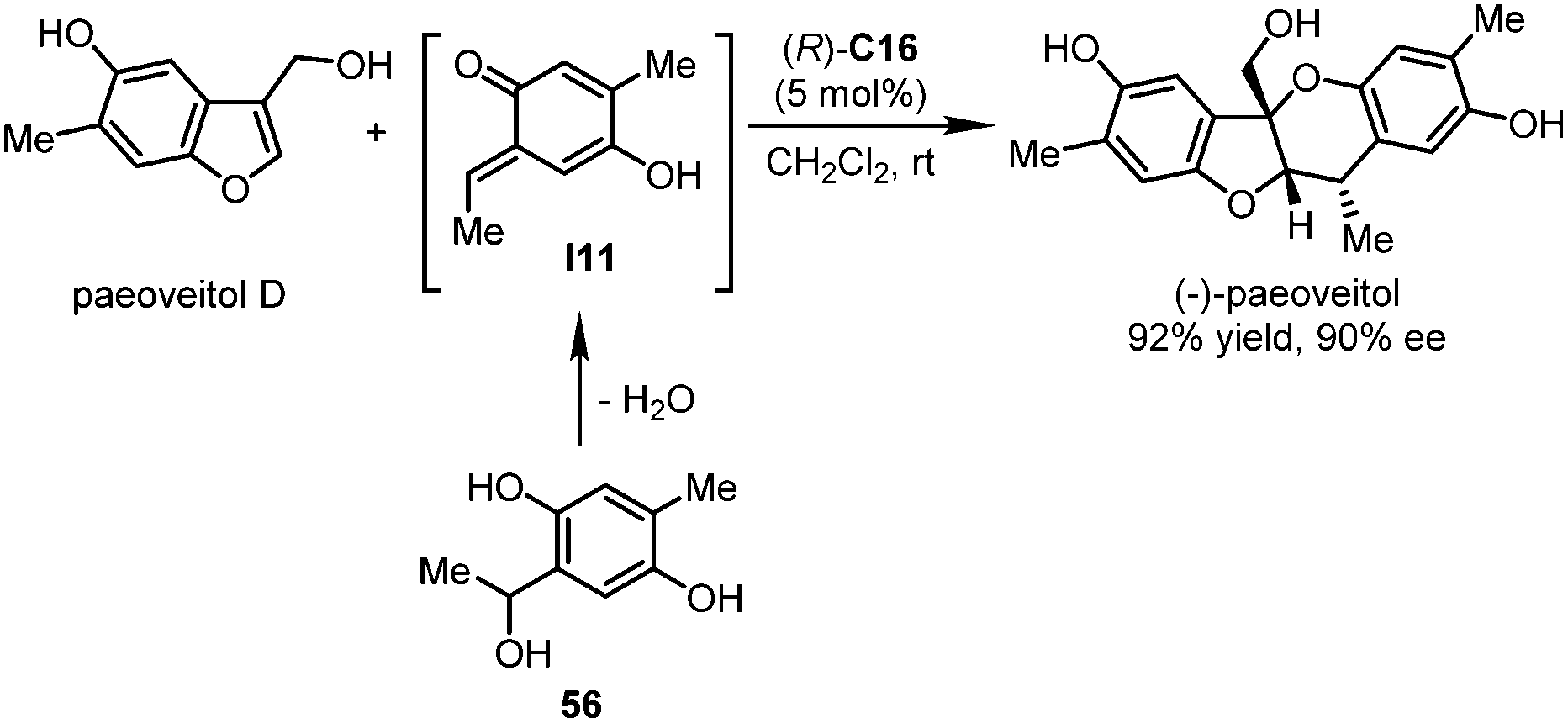 | ||
| Scheme 16 The catalytic asymmetric synthesis of (−)-paeoveitol. | ||
Although imines derived from the condensation between amines with aldehydes or ketones have been widely used as electrophiles in organic syntheses, the application of such electrophiles in the catalytic asymmetric dearomatization reactions by CPAs is relatively underdeveloped. To date, only limited examples in this subject have been reported. In 2013, Zhu, Sun, and coworkers accomplished a remarkable CPA-catalyzed multi-component reaction (Scheme 17),51 in which the imines generated in situ from functionalized benzaldehydes 57 and anilines 58 were involved in a dearomative inverse electron demanding aza-Diels–Alder reaction with indoles 59. A subsequent desymmetrization of an intramolecular oxetane functionality closed the second piperidine ring, delivering the desired products 60 possessing four stereogenic centers in good yields (45–97%) with high stereoselectivity (50–98% ee, up to >99:1 dr). A small positive non-linear effect was observed, indicating the involvement of multiple catalyst components or higher order interactions that were not involved in the catalysis. Notably, some of the products in this study showed inhibitory effects against cell proliferation with certain important cancer cell lines (A549 and HeLa).
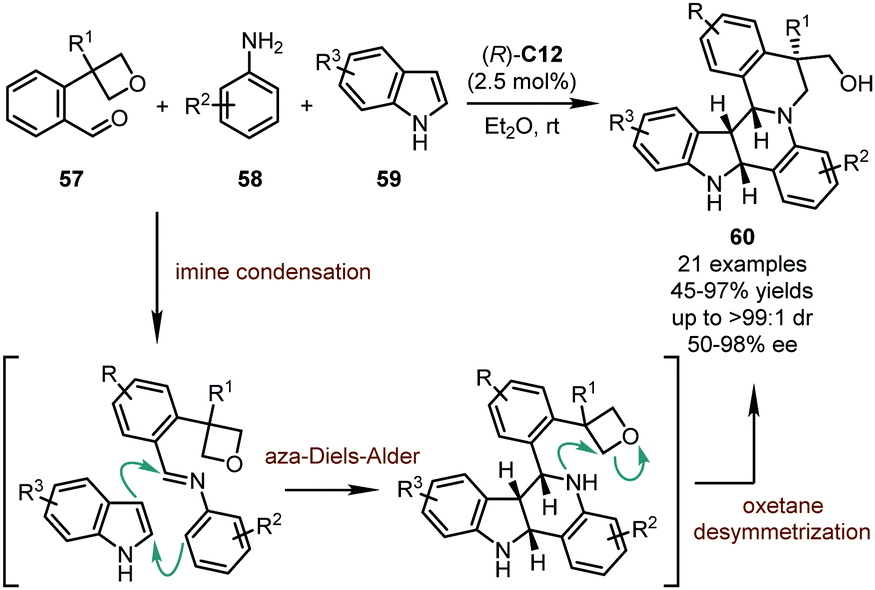 | ||
| Scheme 17 The asymmetric dearomatization reaction of indoles with aza-Diels–Alder reactions. | ||
In 2017, the Shi group reported a CPA-catalyzed cascade dearomatization of tryptophols with isatin-derived imines as the electrophiles.52 The reaction proceeded via nucleophilic addition of the C3 position of the indole ring in tryptophols 61 to imines 62 followed by intramolecular cyclization (Scheme 18). Corresponding furoindoline derivatives 63 bearing three consecutive stereogenic centers were afforded in good yields (49–91%) with high stereoselectivity (82–96% ee, and up to 95:5 dr). Remarkably, the selectivity between the dual nucleophilic sites (C3 vs. O) of tryptophols was well regulated by the chiral catalyst. When (S)-C1 was utilized, double hydrogen-bonding interactions were believed to exist between the hydroxyl group of the catalyst and the imine group of 62, and between the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of the catalyst and the hydroxyl group of tryptophols, which guaranteed the dearomatization cascade. Otherwise, when a bifunctional chiral squaramide-tertiary amine catalyst was employed, the exclusive O-addition of tryptophols to isatin-derived imines was observed (not shown).
O moiety of the catalyst and the hydroxyl group of tryptophols, which guaranteed the dearomatization cascade. Otherwise, when a bifunctional chiral squaramide-tertiary amine catalyst was employed, the exclusive O-addition of tryptophols to isatin-derived imines was observed (not shown).
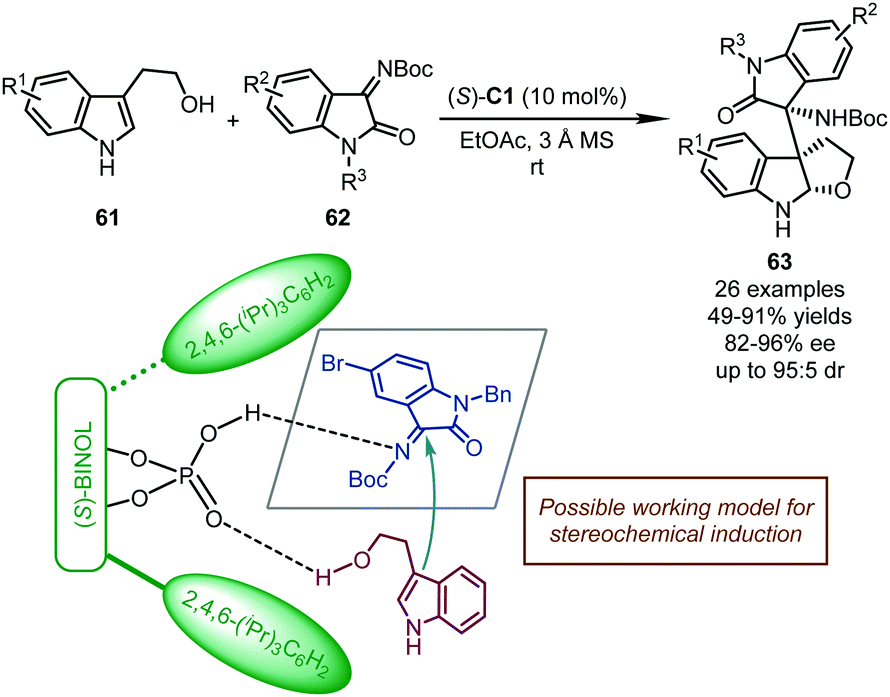 | ||
| Scheme 18 The asymmetric dearomatization reaction of tryptophols with isatin-derived imines. | ||
In 2018, the You group reported a catalytic asymmetric dearomatization reaction of indolyl dihydropyridines 64 with Hantzsch ester H1 (Scheme 19).53 The CPA catalyst contributed to all the three steps integrated in this cascade, namely enamine isomerization, spirocyclization, and transfer hydrogenation. A series of novel polycyclic spiroindolines 65 could be assembled in good yields (51–88%) with excellent stereoselectivity (77–97% ee). Notably, the length of the linker between the indole ring and the dihydropyridine motif has great influence on the reaction. The dearomatization reactions only occur when these two N-heterocycles are connected by three methylene groups, presumably due to the facile spirocyclization of intermediate I12 and the stability of six-membered-ring spiroindolenine intermediate I13 against undesired migration. On the other hand, when tryptamine-derived dihydropyridines were subjected to similar reaction conditions, a Pictet–Spengler-type reaction occurred, leading to polycyclic tetrahydro-β-carboline derivatives.54 DFT calculations were conducted to probe the reaction mechanism and showed that the enamine isomerization and the spirocyclization were reversible, while the transfer hydrogenation was irreversible and the rate-determining step of the reaction. Moreover, the stereochemistry of the reaction was proved to be determined in the transfer hydrogenation process although there was no new stereogenic element established during this step.
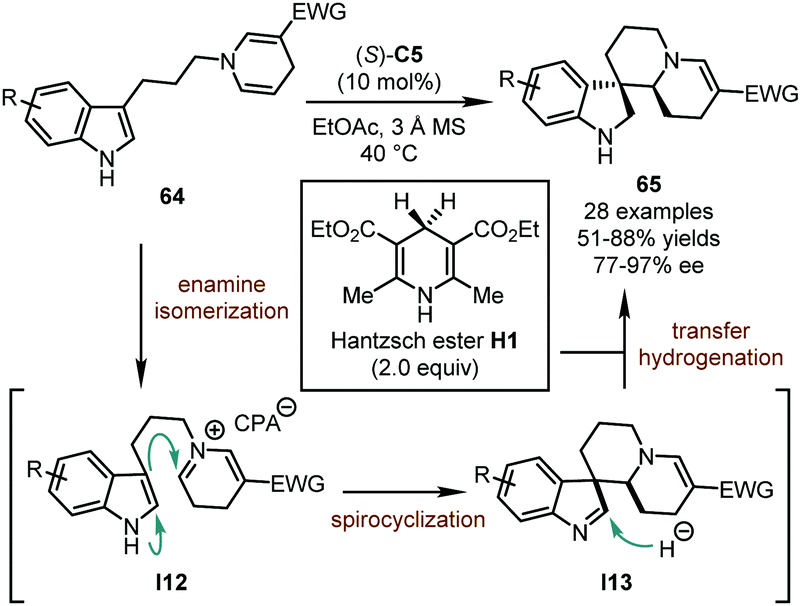 | ||
| Scheme 19 The asymmetric dearomatization reaction of indolyl dihydropyridines with Hantzsch ester. | ||
In 2014, Bandini and coworkers applied allenamides 67 in the CPA-catalyzed dearomatization reaction of 2,3-disubstituted indoles 66 (Scheme 20).55 The allenamides could be activated by a CPA catalyst via hydrogen bonding with the central carbon of the allene moiety. The subsequent nucleophilic attack of the C3 position of 66 afforded the dearomatized products 68 in good to excellent yields (44–98%) with high enantioselectivity (72–94% ee). Besides, a one-pot tandem dearomatization/transfer hydrogenation with Hantzsch ester H2 was also achieved, leading to the corresponding chiral indolines 69 (44–72% yields, up to 99% ee, and up to 19:1 dr).
 | ||
| Scheme 20 The asymmetric dearomatization reaction of indoles with allenamides. | ||
Alkynes could be also utilized as electrophiles after being activated by carbophilic catalysts. Compared with the widespread application of gold catalysts in alkyne activation, the corresponding silver species were relatively less studied due to their relatively lower reactivity. In 2017, the Wang group realized the asymmetric dearomatization reaction of alkyne-tethered tryptamine derivatives 70 (Scheme 21).56 With (S)-C3 as the optimal CPA catalyst and AgBF4 as the silver source, two intramolecular cyclization reactions occurred sequentially, with the terminal alkyne moiety connected to the C3 position of the indole ring, and the protected amine group to the C2 position. Thus, the desired bridged polycyclic indolines 71 were assembled in good to excellent yields (62–99%) with high stereoselectivity (72–96% ee). Interestingly, by exchanging the substituents on the C2 and C3 positions (70a), spirocyclic indoline 72 was obtained in 65% yield with 86% ee. The electron-withdrawing group (p-ClC6H4SO2) on the primary amine group of 70a was finally transferred to the indole nitrogen atom in 72. Extensive mechanistic studies including DFT calculations were carried out to investigate the reaction mechanism. The chiral silver phosphate generated in situ from CPA and AgBF4 enabled the alkyne functionality. Besides, the hydrogen-bonding interactions between the substrate and the chiral phosphate anion accelerated the reaction by promoting proton transfer and played a crucial role in the stereochemical control.
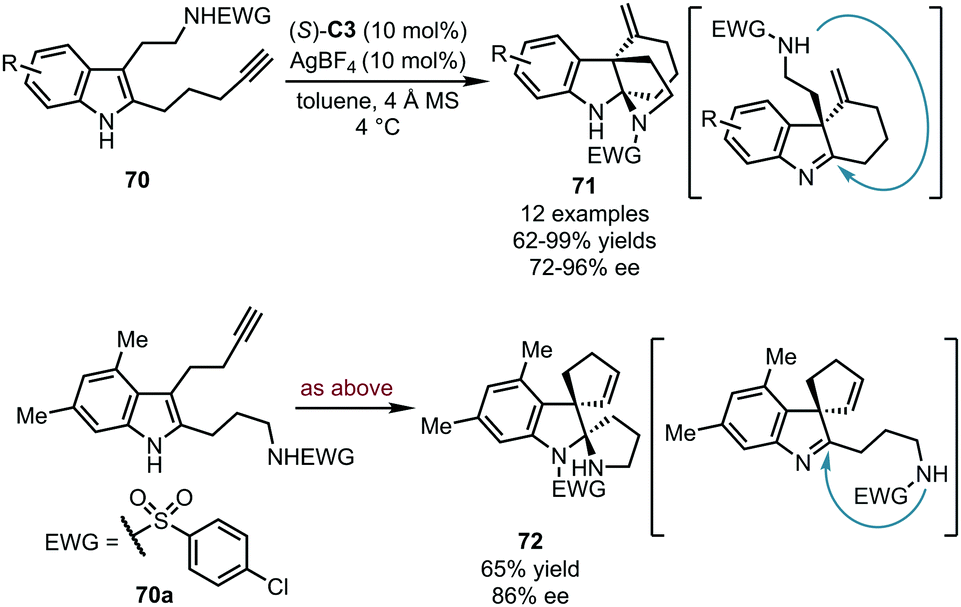 | ||
| Scheme 21 The asymmetric dearomatization reaction of alkyne-tethered tryptamines and analogs. | ||
Sun, Wang and coworkers disclosed a dearomative γ-addition to α-imino-β,γ-alkynyl esters 74 with 2,3-disubstituted indole derivatives 73 by a CPA catalyst (Scheme 22).57 The reaction afforded challenging tetrasubstituted chiral allenes 75 with an adjacent quaternary stereogenic center at the C3 position of the indolenine core. (S)-C13 was identified as the optimal catalyst which provides high yields of 75 (70–99%) with extraordinary stereochemical control (91 to >99% ee, and up to >20:1 dr). Mechanistic studies showed that the substituent at the C3 position of the indole ring of 73 and the ester group of 74 were crucial for the exclusive γ-selectivity, probably due to the steric congestion for the undesired α-attack. A working model for the enantiomeric discrimination process was also provided by DFT calculations.
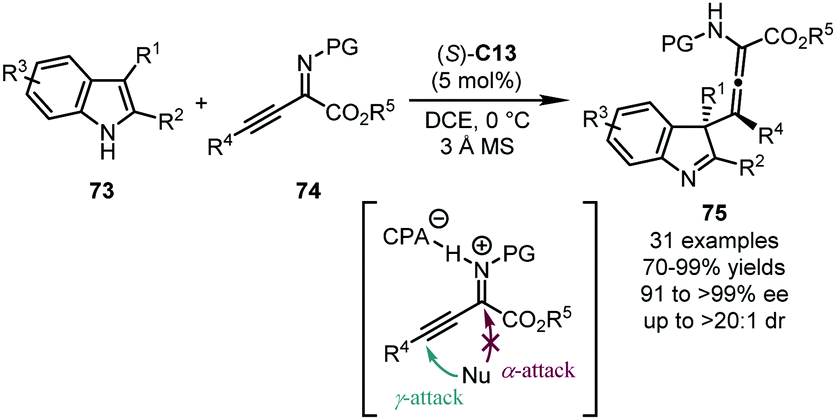 | ||
| Scheme 22 The asymmetric dearomatization reaction of indoles with α-imino-β,γ-alkynyl esters. | ||
2.3 Nitrogen electrophiles
The CPA-catalyzed asymmetric dearomatization reactions coupled with C–N bond-formation have also been well studied. The most frequently utilized nitrogen electrophiles include dialkyl azodicarboxylates and aryldiazonium salts. In 2012, Antilla and coworkers achieved the first aminative dearomatization reaction of tryptamine derivatives 76 with diethyl azodicarboxylate (DEAD) as an electrophile (Scheme 23).35 In the presence of (R)-C1, the desired pyrroloindoline products 77 could be assembled via a dearomatization/cyclization sequence in 56–76% yields with 75–96% ee. | ||
| Scheme 23 The asymmetric dearomatization reaction of tryptamine derivatives with DEAD. | ||
In 2015, the You group applied azodicarboxylates 78 in the CPA-catalyzed asymmetric dearomatization reaction of 2-naphthols (Scheme 24).58 With (R)-C15 as the optimal catalyst, the dearomatization reactions of 1,3-disubstituted 2-naphthols 79 proceeded in CCl4 smoothly, affording dearomatized products 80 in excellent yields (81–99%) with high enantioselectivity (88–96% ee). The reactions also accommodated 2-naphthols without a substituent at the C3 position, a challenging subgroup of substrates in the asymmetric dearomatization reactions of 2-naphthols. Under slightly modified conditions [(S)-C3, and o-xylene], the desired reactions of 1-substituted 2-naphthols 81 generated cyclic α,β-unsaturated ketones 82 efficiently (79–99% yields and 90–96% ee). It should be noted that the catalyst loading of this reaction could be reduced to 0.05 mol% without detectable erosion of the yield and enantioselectivity. Very recently, Zheng, You and coworkers reported cascade aminative dearomatization/Michael additions of 4-substituted 1-naphthols 83 enabled by a CPA catalyst (Scheme 24).59 A series of polycyclic ketones 84 was obtained via a two-step sequence in moderate to excellent yields (up to 93%) with excellent enantioselectivity (up to >99% ee). The 4,4-disbustituted cyclohexenone intermediates I14 that were formed immediately after the dearomatization reaction could be isolated with high enantiopurity. Further mechanistic studies revealed that the stereochemistry of the reaction was established during the aminative dearomatization step while the Michael addition step was rate-limiting.
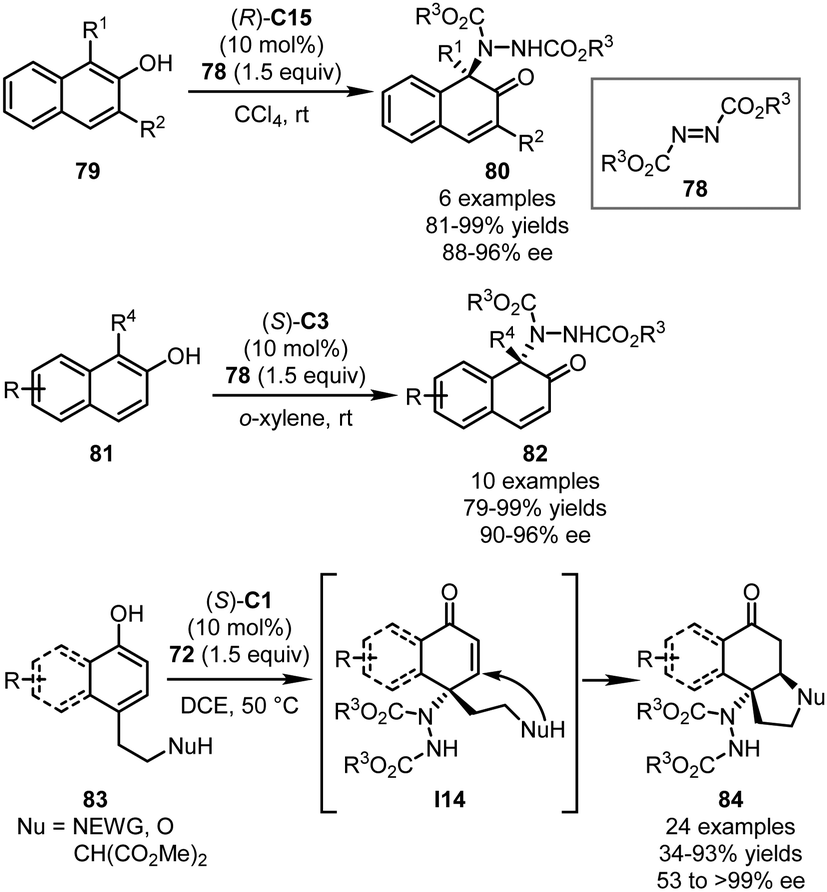 | ||
| Scheme 24 The asymmetric dearomatization reaction of naphthol and phenol derivatives with DEAD. | ||
In 2014, the Toste group applied the PTC strategy in the asymmetric dearomatization of tryptamine derivatives 85 with aryldiazonium salts as the electrophilic nitrogen source (Scheme 25).60 With ArN2BF4 as the reaction partner, a series of 3-diazenated pyrroloindolines 86 could be afforded in good to excellent yields (up to 99%) with high enantioselectivity (69–96% ee) via the aminative dearomatization/cyclization sequence.
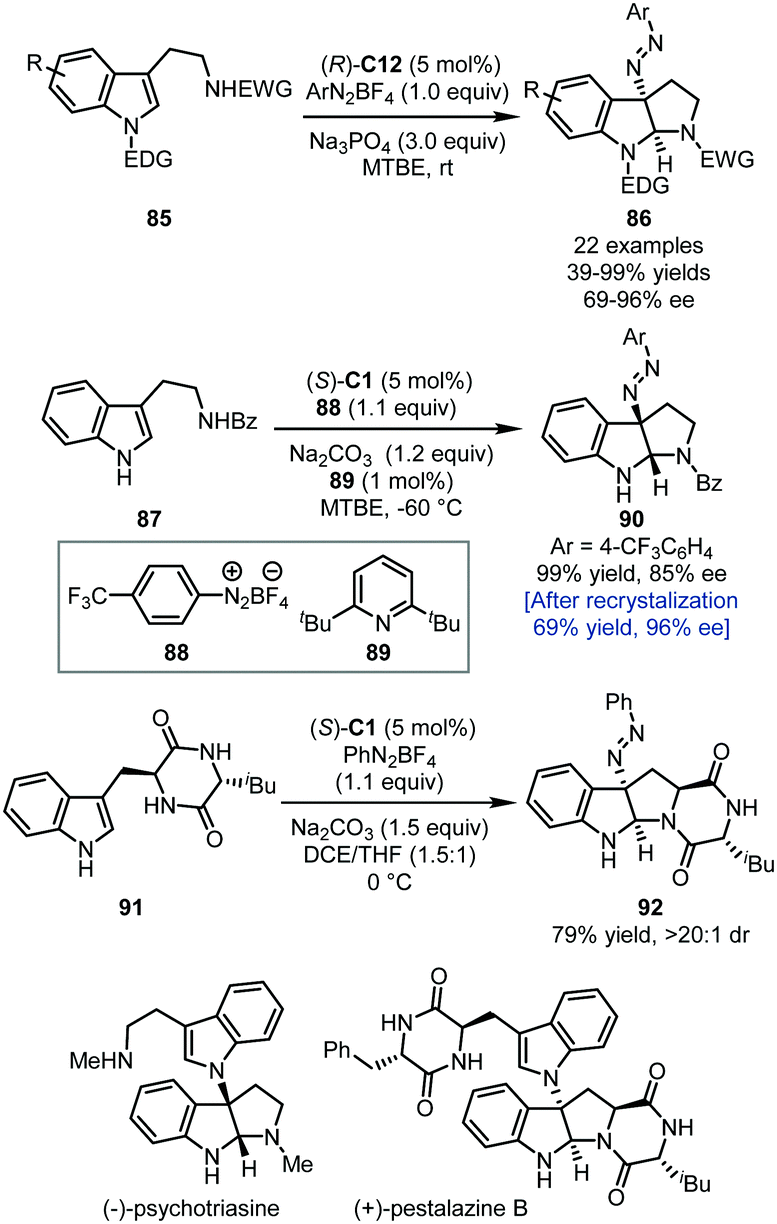 | ||
| Scheme 25 The asymmetric dearomatization reaction of tryptamine derivatives with aryldiazonium salts. | ||
Notably, this method provides an alternative way towards the construction of 3-amino pyrroloindolines since the diazene group can be hydrogenated to afford the corresponding free amino group. By employing a set of slightly different conditions, Deng, Liao, and coworkers also accomplished the construction of 3-diazenated pyrroloindolines initiated by aminative dearomatization reactions of tryptamine derivatives (Scheme 25).61 Notably, in the asymmetric synthesis of 90, it was found that adding a catalytic amount of sterically hindered pyridine 89 could efficiently improve the enantioselectivity (85% ee), presumably by forming a more compact and lipophilic ion pair. The enantiopurity of 90 could be enhanced to 96% ee after a single recrystallization. With this aminative dearomatization/cyclization sequence as a key step (87 → 90 and 91 → 92), the asymmetric total synthesis of (−)-psychotriasine and (+)-pestalazine B was achieved.
2.4 Selenium electrophiles
In 2013, the Gong group reported a CPA-catalyzed dearomatization reaction of tryptamine derivatives 93 utilizing N-phenylselenophthalimides (N-PSPs) 94 as the electrophile (Scheme 26).62 The desired dearomatization/cyclization sequence occurred smoothly, affording the corresponding products 95 in good yields (65–85%) with satisfactory enantioselectivity (71–89% ee). The ee values of the final products could be enhanced to an excellent level by a single recrystallization. | ||
| Scheme 26 The asymmetric dearomatization reaction of tryptamine derivatives with N-PSPs. | ||
2.5 Visible-light-promoted dearomatization reactions of indoles
In all the above sections, the dearomatization of electron-rich arenes was achieved by the reactions with various electrophiles. Remarkably, indole derivatives were found to be transformed into transient radical cationic or cationic species via single-electron oxidation under visible-light irradiation with certain photocatalysts. Thus, the asymmetric dearomatization of the electron-rich arenes with external radicals or nucleophiles becomes possible in the presence of a CPA catalyst (Scheme 27).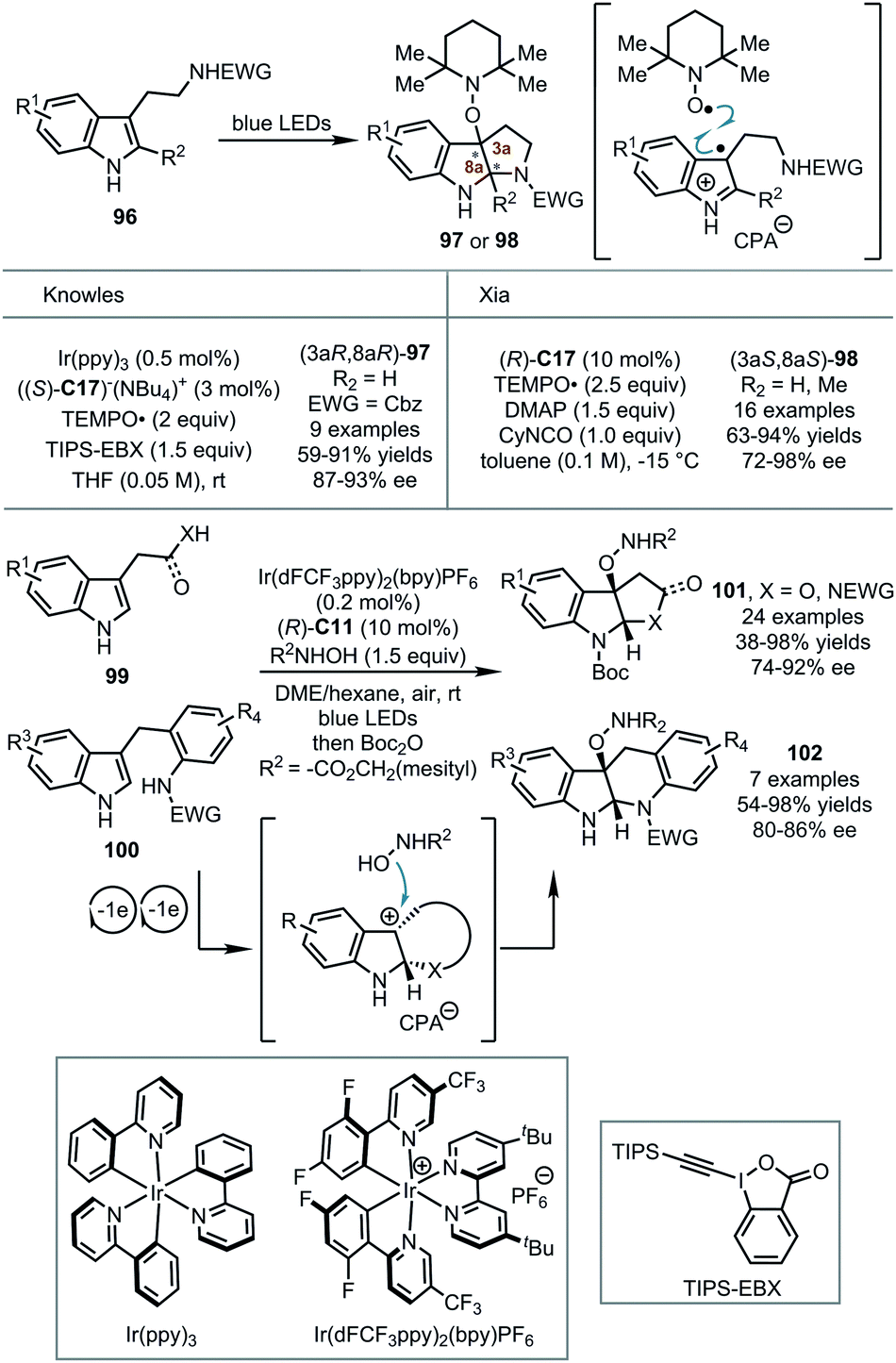 | ||
| Scheme 27 The asymmetric dearomatization reaction of tryptamine derivatives under visible-light irradiation. | ||
In 2018, Knowles and coworkers reported novel enantioselective synthesis of pyrroloindolines from tryptamine derivatives 96 and their applications to the synthesis of alkaloid natural products.63 The reactions were initiated by the visible-light-promoted single-electron-oxidation of chiral phosphate-bonded tryptamine. The subsequent bond-formation with a TEMPO radical and cyclization provided TEMPO-functionalized pyrroloindolines 97 in enantioenriched forms (up to 91% yields and 93% ee). Shortly after, Xia and coworkers reported similar asymmetric dearomative cyclization reactions of tryptamine derivatives with the TEMPO radical.64 An enantioselective capture of a tryptamine radical by the TEMPO radical in the presence of CPA was proposed. However, the utilization of a photocatalyst was not needed.
Very recently, Zhang, You and coworkers reported CPA-catalyzed asymmetric dearomatization of indole derivatives 99 and 100 with N-hydroxycarbamates enabled by photoredox catalysis.65 A series of mechanistic studies showed that in the presence of CPA (R)-C11 and photocatalyst Ir(dFCF3ppy)2(bpy)PF6, and oxygen as the terminal oxidant, tryptophols, tryptamines or indole-tethered anilines underwent two visible-light-induced single-electron-oxidations coupled with cyclization. Thus, configurationally biased tertiary pyrroloindoline-type carbocation intermediates were formed. Finally, these electrophilic species were trapped by N-hydroxycarbamates, furnishing the desired polycyclic indoline products 101 and 102 in up to 98% yield and 92% ee.
3. CPA-catalyzed dearomatization reactions of electron-poor arenes
CPAs are well known in promoting asymmetric transfer hydrogenation (ATH) of electron-poor arenes with Hantzsch ester or related hydride sources, an alternative type of asymmetric dearomatization reactions. Several excellent reviews on this topic are available.22–26 Very recently, Shi, Zhou, and coworkers reported that the ATH reactions of 2-hydroxypyrimidines could be realized by a CPA catalyst (Scheme 28).66 With Hantzsch ester H1 or dihydrophenanthridine (DHPD) as the hydride donor, 4,6-symmetrical or unsymmetrical substituted substrates 103 or 105 were well reduced in the presence of (R)-C1 as the optimal catalyst, leading to their corresponding 3,4-dihydropyrimidin-2(1H)-ones 104 or 106 in good yields with high enantioselectivity. | ||
| Scheme 28 The asymmetric transfer hydrogenation reaction of 2-hydroxypyrimidines. | ||
Notably, the CPA-catalyzed ATH reaction could be coupled with further additions from external nucleophiles, which initiated the asymmetric dearomatization processes of electron-poor arenes.
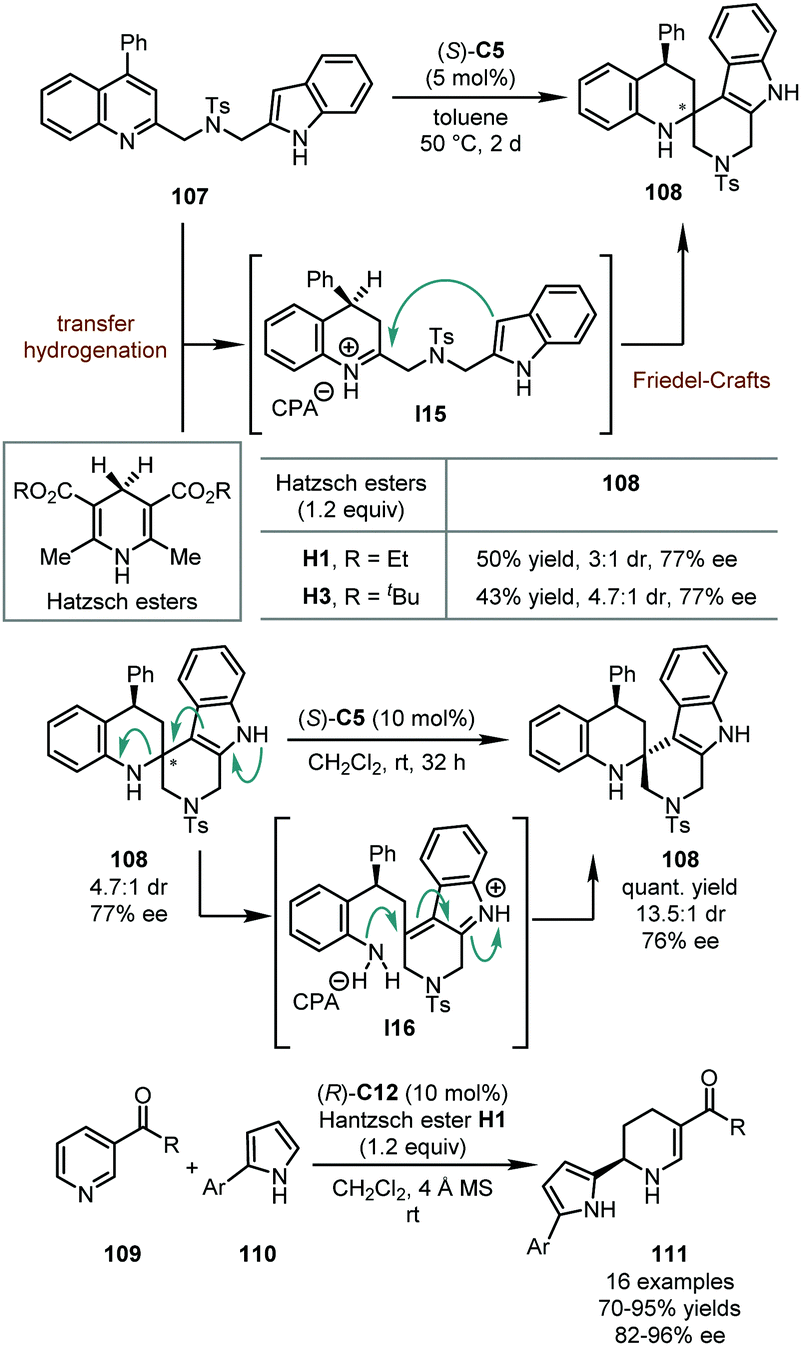
In 2013, the You group developed the hydrogenative dearomatization of indole-tethered quinolines via a cascade transfer hydrogenation/intramolecular Friedel–Crafts alkylation (Scheme 29).67 In the presence of a catalytic amount of (R)-C5, the reaction was triggered by the conjugate addition of quinolines 107 by a Hantzsch ester (H1 or H3). Subsequently, the imine intermediate was trapped by the nucleophilic attack of the indole moiety, affording spirotetrahydroquinoline 108 in moderate yields (50% or 43%) with reasonable stereochemical control (77% ee, and 3:1 or 4.7:1 dr). Notably, the diastereomeric ratio of the isolated 108 could be improved (from 4.7:1 to 13.5:1) when treated with (R)-C5 in DCM, probably via intermediate I16. Following this discovery, the You group further designed an asymmetric intermolecular hydrogenative dearomatization of substituted pyridines 109 (Scheme 29).68 Mechanistically, this reaction shared a similar catalytic cycle as the previous intramolecular reaction, except for the imine intermediate being trapped by 2-arylpyrroles 110 as the external nucleophile. This reaction accommodated a wide substrate scope and a series of chiral tetrahydropyridines 111 was delivered in good to excellent yields (70–95%) with high enantioselectivity (82–96% ee). Of particular note, an electron-withdrawing group at the C3 position of the pyridine ring was crucial to initiate the reaction and stabilize the final products.
 | ||
| Scheme 29 The asymmetric hydrogenative dearomatization reactions with Hantzsch esters. | ||
In 2016, Hong, Li, Wang and coworkers reported a CPA-catalyzed Reissert-type dearomatization reaction of isoquinolines 112 (Scheme 30).69 The isoquinolines could be activated by Boc2O in situ, forming the iminium intermediates I17. The subsequent enantioselective Friedel–Crafts attack from the C3 position of indoles 113 was enabled by (S)-C5, affording the dearomatized products 114 in excellent yields (up to 99%) with good to excellent enantioselectivity (77–97% ee). Interestingly, other electron-rich arenes such as pyrroles and furans did not participate in this reaction. Inspired by this elegant work, the You group recently disclosed the reactions between isoquinolines 115 with 3-substituted indoles 116 under similar conditions (Scheme 30).70 Notably, when tryptamine derivatives were employed, the expected dearomatization of indoles/cyclization sequence did not occur. Instead, the direct N-alkylation of I17 delivered dearomatized products 117 efficiently. The reaction could be performed in gram-scale without a deleterious effect on the enantioselectivity.
 | ||
| Scheme 30 The asymmetric dearomatization reaction of isoquinolines with indoles. | ||
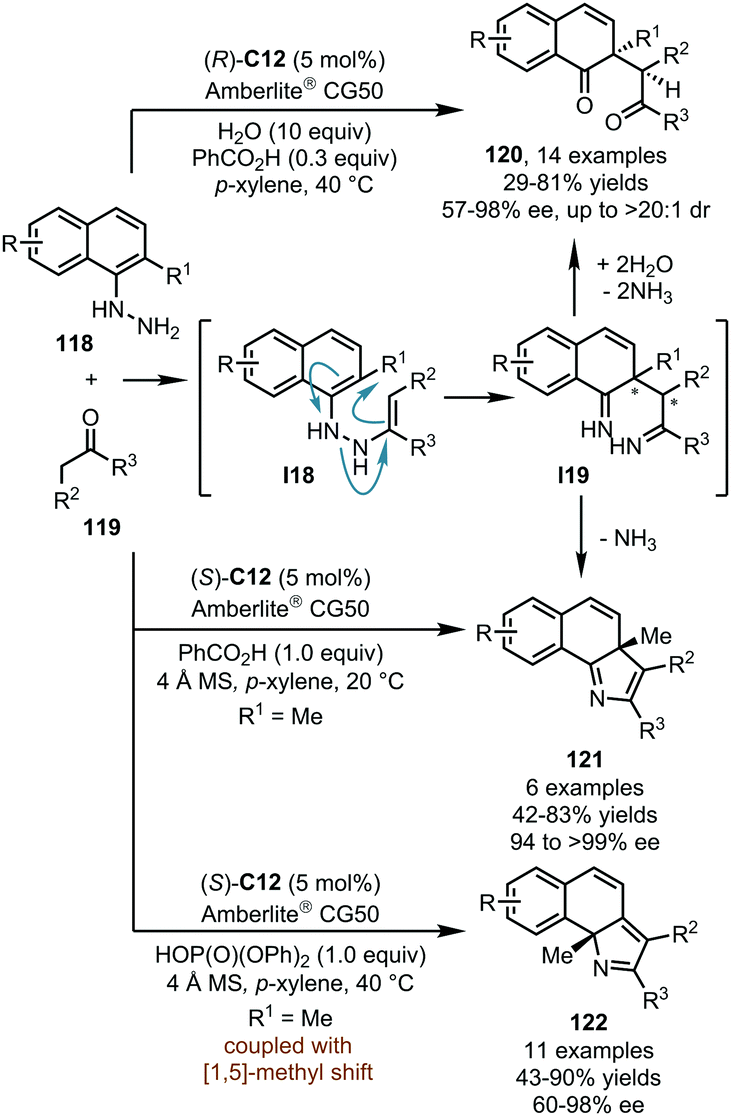
Based on their systematic studies on the interrupted Fischer indole synthesis, List and coworkers reported in 2015 a CPA-catalyzed dearomatization reaction of ortho-substituted α-naphthylhydrazines (Scheme 31).71 In the presence of (R)-C12, the imines generated in situ from α-naphthylhydrazines 118 and ketones 119 can isomerize to enamine intermediate I18. Subsequently, a [3,3]-sigmatropic diaza-Cope rearrangement of I18 delivered the diimine intermediate I19via a boat-like transition state, and the rearomatization process of I19 was prevented due to the ortho-substituent. Finally, the hydrolysis of I19 afforded 1,4-diketones 120 in moderate to high yields (up to 81%) with good to excellent enantioselectivity (57–98% ee, up to >20:1 dr). In this protocol, water is necessary for the hydrolysis of I19, and the weakly acidic Amberlite® CG50 is crucial for the absorption of the released ammonia and the regeneration of the catalyst. It should be noted that benzoic acid as a key additive promotes the target reaction without deleterious effect on the enantioselectivity.
 | ||
| Scheme 31 The asymmetric dearomatization reactions of ortho-substituted naphthylhydrazines. | ||
The List group further achieved the asymmetric construction of 3H-pyrroles or 2H-pyrroles by the dearomatization of ortho-substituted α-naphthylhydrazines 118 (Scheme 31).72 When the reactions were conducted under completely anhydrous conditions, the hydrolysis of I19 was inhibited. Instead, the cyclization would occur to afford chiral 3H-pyrroles 121 in good yields (up to 83%) and excellent enantioselectivity (94 to >99% ee) with the concurrent release of ammonia. Besides, a series of 2H-pyrroles 122 could be obtained via a suprafacial [1,5] sigmatropic alkyl shift of 121 upon the treatment of more acidic reaction conditions.
4. Conclusions
This review has summarized the-state-of-the-art of CPA-catalyzed asymmetric dearomatization reactions. These reactions are not only highly effective in the construction of novel molecules bearing spiro or fused cyclic skeletons with consecutive (quaternary) stereogenic centers, but also feature readily available substrates, mild reaction conditions and excellent stereochemical selectivity.To date, the CPA-catalyzed dearomatization reactions of electron-rich arenes have gained wide research interests and such reactions are initiated by nucleophilic attack from the arenes to electrophiles such as halonium reagents, α,β-unsaturated ketones and their analogues, unconjugated ketones and their derivatives, and other types of electrophiles. On the other hand, the CPA-catalyzed asymmetric dearomatization reactions of electron-poor arenes are relatively underdeveloped, except for sporadic examples of those coupled with transfer hydrogenation or Reissert-type reactions.
The development of catalytic asymmetric dearomatization reactions of simple arenes still remains as a big challenge in this research area. The recent rapid progress on the reactions enabled by visible-light-catalysis and electrochemical catalysis might give promising tools to tackle this problem. It has been proved that CPAs are compatible with the single-electron-transfer processes performed under visible-light irradiation. The CPA-catalyzed asymmetric dearomatization of indole derivatives enabled by photoredox catalysis has been reported very recently, which provided an unprecedented reaction mode featuring the bond-formation between two typical nucleophilic sites.65 Undoubtedly, further exploration in this direction will likely promote future development of CPA-catalyzed asymmetric dearomatization reactions.
The relatively high loading of CPAs (typically 5–10 mol%) is a significant drawback that prevents their scalable industrial application. In the reactions discussed in this review, only limited examples have been known where very high TON was achieved.42 A potential solution to this issue might stem from embedding the CPAs into heterogeneous catalytic systems that might result in recycling and reuse of the catalysts. Preliminary successes on the utilization of metal–organic frameworks (MOFs) with CPAs in the asymmetric dearomatization reactions of 2-naphthols with para-quinone has been reported by Cui and Liu very recently.73 However, additional modification on CPAs is required for the assembly of the MOF structures. The scalable and convenient synthesis of such MOF-based and other types of heterogenized CPAs are the key issues to be addressed in the future.
Despite these challenges, it is quite convincing that CPA-catalyzed asymmetric dearomatization reactions have evolved as powerful methods towards facile synthesis of novel polycyclic molecular identities. We believe that the future development in discovery of new dearomative reaction modes, and the enhanced catalytic efficiency will make this type of reaction more significant both academically and industrially.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Ministry of Science and Technology of China (2016YFA0202900), the National Natural Science Foundation of China (21772219, 21821002, and 91856201), the Science and Technology Commission of Shanghai Municipality (18QA1404900), the Chinese Academy of Sciences (XDB20000000, and QYZDYSSWSLH012) and the Youth Innovation Promotion Association (2017302) of CAS for generous financial support for our works presented in this review.Notes and references
- A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917–2940 CrossRef CAS PubMed.
- S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed.
- S.-L. You, Asymmetric Dearomatization Reactions, Wiley-VCH, Weinheim, 2016 Search PubMed.
- C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef PubMed.
- C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558–2573 CrossRef CAS.
- C. Zheng and S.-L. You, Chem, 2016, 1, 830–857 CAS.
- C. Zheng and S.-L. You, Nat. Prod. Rep., 2019, 36, 1589–1605 RSC.
- T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed.
- M. Terada, Chem. Commun., 2008, 4097–4112 RSC.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- J. Merad, C. Lalli, G. Bernadat, J. Maury and G. Masson, Chem. – Eur. J., 2018, 24, 3925–3943 CrossRef CAS PubMed.
- T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed.
- D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
- R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS PubMed.
- M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed.
- K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed.
- S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS.
- S.-L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190–2201 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- A. A. Festa, L. G. Voskressensky and E. V. Van der Eycken, Chem. Soc. Rev., 2019, 48, 4401–4423 RSC.
- M. Rueping, E. Sugiono and F. R. Schoepke, Synlett, 2010, 852–865 CrossRef CAS.
- C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC.
- A. M. F. Phillips and A. J. L. Pombeiro, Org. Biomol. Chem., 2017, 15, 2307–2340 RSC.
- M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10460–10476 CrossRef CAS PubMed.
- V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681–1684 CrossRef CAS PubMed.
- R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268–1271 CrossRef CAS PubMed.
- X.-W. Liang, C. Liu, W. Zhang and S.-L. You, Chem. Commun., 2017, 53, 5531–5534 RSC.
- X.-W. Liang, Y. Cai and S.-L. You, Chin. J. Chem., 2018, 36, 925–928 CrossRef CAS.
- Y.-M. Wang, J. Wu, C. Hoong, V. Rauniyar and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 12928–12931 CrossRef CAS PubMed.
- W. Xie, G. Jiang, H. Liu, J. Hu, X. Pan, H. Zhang, X. Wan, Y. Lai and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12924–12927 CrossRef CAS PubMed.
- X. Feng, G. Jiang, Z. Xia, J. Hu, X. Wan, J.-M. Gao, Y. Lai and W. Xie, Org. Lett., 2015, 17, 4428–4431 CrossRef CAS PubMed.
- H. Liu, G. Jiang, X. Pan, X. Wan, Y. Lai, D. Ma and W. Xie, Org. Lett., 2014, 16, 1908–1911 CrossRef CAS PubMed.
- Z. Zhang and J. C. Antilla, Angew. Chem., Int. Ed., 2012, 51, 11778–11782 CrossRef CAS PubMed.
- Q. Cai, C. Liu, X.-W. Liang and S.-L. You, Org. Lett., 2012, 14, 4588–4590 CrossRef CAS PubMed.
- D.-H. Duan, Q. Yin, S.-G. Wang and S.-L. You, Acta Chim. Sin., 2014, 72, 1001–1004 CrossRef CAS.
- Y. Zhou, Z.-L. Xia, Q. Gu and S.-L. You, Org. Lett., 2017, 19, 762–765 CrossRef CAS PubMed.
- Z.-L. Xia, C. Zheng, X.-W. Liang, Y. Cai and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 1158–1162 CrossRef CAS PubMed.
- L. Liao, C. Shu, M. Zhang, Y. Liao, X. Hu, Y. Zhang, Z. Wu, W. Yuan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 10471–10475 CrossRef CAS PubMed.
- Y. Wang, M. Sun, L. Yin and F. Shi, Adv. Synth. Catal., 2015, 357, 4031–4040 CrossRef CAS.
- Q. Yu, Y. Fu, J. Huang, J. Qin, H. Zuo, Y. Wu and F. Zhong, ACS Catal., 2019, 9, 7285–7291 CrossRef CAS.
- Y.-C. Zhang, J.-J. Zhao, F. Jiang, S.-B. Sun and F. Shi, Angew. Chem., Int. Ed., 2014, 53, 13912–13915 CrossRef CAS PubMed.
- C. Ma, T. Zhang, J.-Y. Zhou, G.-J. Mei and F. Shi, Chem. Commun., 2017, 53, 12124–12127 RSC.
- G. Zhu, G. Bao, Y. Li, J. Yang, W. Sun, J. Li, L. Hong and R. Wang, Org. Lett., 2016, 18, 5288–5291 CrossRef CAS PubMed.
- X.-Q. Li, H. Yang, J.-J. Wang, B.-B. Gou, J. Chen and L. Zhou, Chem. – Eur. J., 2017, 23, 5381–5385 CrossRef CAS PubMed.
- L.-W. Qi, J.-H. Mao, J. Zhang and B. Tan, Nat. Chem., 2018, 10, 58–64 CrossRef CAS PubMed.
- G.-J. Mei, X. Tang, Y. Tasdan and Y. Lu, Angew. Chem., Int. Ed. DOI:10.1002/anie.201911686.
- F. Jiang, Y.-C. Zhang, X. Yang, Q.-N. Zhu and F. Shi, Synlett, 2016, 575–580 CAS.
- T.-Z. Li, C.-A. Geng, X.-J. Yin, T.-H. Yang, X.-L. Chen, X.-Y. Huang, Y.-B. Ma, X.-M. Zhang and J.-J. Chen, Org. Lett., 2017, 19, 429–431 CrossRef CAS PubMed.
- Z. Chen, B. Wang, Z. Wang, G. Zhu and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 2027–2031 CrossRef CAS PubMed.
- F. Jiang, D. Zhao, X. Yang, F.-R. Yuan, G.-J. Mei and F. Shi, ACS Catal., 2017, 7, 6984–6989 CrossRef CAS.
- Z.-L. Xia, C. Zheng, S.-G. Wang and S.-L. You, Angew. Chem., Int. Ed., 2018, 57, 2653–2656 CrossRef CAS PubMed.
- S.-G. Wang, Z.-L. Xia, R.-Q. Xu, X.-J. Liu, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 7440–7443 CrossRef CAS PubMed.
- C. Romano, M. Jia, M. Monari, E. Manoni and M. Bandini, Angew. Chem., Int. Ed., 2014, 53, 13854–13857 CrossRef CAS PubMed.
- Y. Zhu, W. He, W. Wang, C. E. Pitsch, X. Wang and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 12206–12209 CrossRef CAS.
- J. Yang, Z. Wang, Z. He, G. Li, L. Hong, W. Sun and R. Wang, Angew. Chem., Int. Ed. DOI:10.1002/anie.201911420.
- S.-G. Wang, Q. Yin, C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 647–650 CAS.
- Z.-L. Xia, C. Zheng, R.-Q. Xu and S.-L. You, Nat. Commun., 2019, 10, 3150 CrossRef PubMed.
- H. M. Nelson, S. H. Reisberg, H. P. Shunatona, J. S. Patel and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 5600–5603 CrossRef CAS PubMed.
- Q. Li, T. Xia, L. Yao, H. Deng and X. Liao, Chem. Sci., 2015, 6, 3599–3605 RSC.
- Q. Wei, Y.-Y. Wang, Y.-L. Du and L.-Z. Gong, Beilstein J. Org. Chem., 2013, 9, 1559–1564 CrossRef PubMed.
- E. C. Gentry, L. J. Rono, M. E. Hale, R. Matsuura and R. R. Knowles, J. Am. Chem. Soc., 2018, 140, 3394–3402 CrossRef CAS PubMed.
- K. Liang, X. Tong, T. Li, B. Shi, H. Wang, P. Yan and C. Xia, J. Org. Chem., 2018, 83, 10948–10958 CrossRef CAS PubMed.
- Y.-Z. Cheng, Q.-R. Zhao, X. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 18069–18074 CrossRef CAS PubMed.
- F.-J. Meng, L. Shi, G.-S. Feng, L. Sun and Y.-G. Zhou, J. Org. Chem., 2019, 84, 4435–4442 CrossRef CAS PubMed.
- S.-G. Wang, W. Zhang and S.-L. You, Org. Lett., 2013, 15, 1488–1491 CrossRef CAS PubMed.
- S.-G. Wang and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 2194–2197 CrossRef CAS PubMed.
- M. Zhang, W. Sun, G. Zhu, G. Bao, B. Zhang, L. Hong, M. Li and R. Wang, ACS Catal., 2016, 6, 5290–5294 CrossRef CAS.
- Y. Cai, Q. Gu and S.-L. You, Org. Biomol. Chem., 2018, 16, 6146–6154 RSC.
- S. Huang, L. Kötzner, C. K. De and B. List, J. Am. Chem. Soc., 2015, 137, 3446–3449 CrossRef CAS.
- L. Kötzner, M. Leutzsch, S. Sievers, S. Patil, H. Waldmann, Y. Zheng, W. Thiel and B. List, Angew. Chem., Int. Ed., 2016, 55, 7693–7697 CrossRef.
- W. Gong, X. Chen, H. Jiang, D. Chu, Y. Cui and Y. Liu, J. Am. Chem. Soc., 2019, 141, 7498–7508 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |