
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot mechanochemical ball milling synthesis of the MnOx nanostructures as efficient catalysts for CO2 hydrogenation reactions†
Altantuya
Ochirkhuyag
 a,
András
Sápi
*ab,
Ákos
Szamosvölgyi
a,
Gábor
Kozma
a,
Ákos
Kukovecz
ac and
Zoltán
Kónya
ac
a,
András
Sápi
*ab,
Ákos
Szamosvölgyi
a,
Gábor
Kozma
a,
Ákos
Kukovecz
ac and
Zoltán
Kónya
ac
aUniversity of Szeged, Interdisciplinary Excellence Centre, Department of Applied and Environmental Chemistry, H-6720, Rerrich Béla tér 1, Szeged, Hungary. E-mail: sapia@chem.u-szeged.hu
bInstitute of Environmental and Technological Sciences, University of Szeged, H-6720, Szeged, Hungary
cMTA-SZTE Reaction Kinetics and Surface Chemistry Research Group, University of Szeged, H-6720 Szeged, Rerrich Béla tér 1, Szeged, Hungary
First published on 5th June 2020
Abstract
Here, we report on a one-pot mechanochemical ball milling synthesis of manganese oxide nanostructures synthesized at different milling speeds. The as-synthesized pure oxides and metal (Pt and Cu) doped oxides were tested in the hydrogenation of CO2 in the gas phase. Our study demonstrates the successful synthesis of the manganese oxide nanoparticles via mechano–chemical synthesis. We discovered that the milling speed could tune the crystal structure and the oxidation state of the manganese, which plays an essential role in the CO2 hydrogenation evidenced by ex situ XRD and XPS studies. The pure MnOx milled at 600 rpm showed high catalytic activity (∼20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nmol g−1 s−1) at 823 K, which can be attributed to the presence of Mn(II) besides Mn(III) and Mn(IV) on the surface under the reaction conditions. This study illustrates that the milling method is a cost-effective, simple way for the production of both pure, Pt-doped and Cu-loaded manganese nanocatalysts for heterogeneous catalytic reactions. Thus, we studied the Pt incorporation effect for the catalytic activity of MnOx using different Pt loading methods such as one-pot milling, wet impregnation and size-controlled 5 nm Pt loading via an ultrasonication-assisted method.
000 nmol g−1 s−1) at 823 K, which can be attributed to the presence of Mn(II) besides Mn(III) and Mn(IV) on the surface under the reaction conditions. This study illustrates that the milling method is a cost-effective, simple way for the production of both pure, Pt-doped and Cu-loaded manganese nanocatalysts for heterogeneous catalytic reactions. Thus, we studied the Pt incorporation effect for the catalytic activity of MnOx using different Pt loading methods such as one-pot milling, wet impregnation and size-controlled 5 nm Pt loading via an ultrasonication-assisted method.
1. Introduction
Some of the biggest concerns of our modern world are environmental pollution, global warming, extreme weather changes caused by the emission of CO2 and depleting conventional energy sources such as fossil fuels. CO2 transformation towards valuable fuels (e.g., CH4, CO, methanol, C2+, etc.) is a promising candidate in next-generation energy source production, as well as in the reduction of environmental stress.1,2 New generation plants for CO2 hydrogenation are producing methanol with a high conversion rate, even at a production rate of 5500 tons per year.3 New methods are available to produce C5+ hydrocarbons, however, their industrial production has not yet started.4,5 The reverse water–gas shift reaction (RWGS–CO2 + H2 = CO + H2O) and the subsequent reaction, so-called Sabatier (methanation) reaction (CO2 + 4H2 = CH4 + 2H2O) are also industrial ways to convert CO2 into valuable fuels. In several countries, power-to-gas (PtG) technology is used for the storage of electrical power into chemical energy, where CH4 is the fuel and the product of the CO2 hydrogenation reaction.6In recent years, the most studied CO2 hydrogenation catalysts are nickel-based (Ni/ZrO2, Ni–Ga, and Ni/CeO2);7–10 typically, they have several shortcomings, such as their pyrophoricity, low stability, and fast deactivation.11 Manganese-based materials, on the other hand, are not widely researched; however, they can be cost-effective alternatives for catalyzing the CO2 hydrogenation process.12,13 The fifth most abundant metal in the earth's crust is manganese and it occurs with various types of mineral forms in nature.14,15 The manganese-based compounds are crucial components in supercapacitors, electrochemical cells for rechargeable batteries, the degradation of organic dyes, removal of the heavy metals from the polluted water, water oxidation and soot combustion.16–22 Typically, catalytic activity for CO2 reduction and hydrogenation can be improved by a tiny amount of doping or tuning by promoting noble metals such as platinum.23 Non-noble metal-doped catalysts such as copper24 also exist for CO2 hydrogenation, but they are barely mentioned in the literature.
Different types of manganese oxides can be synthesized at a laboratory scale, but the industrial level production of MnOx, with stable oxidation states and structure, relies on hydrothermal, solvothermal reactions, heating, and electrochemical deposition.25–28 The mechanochemical synthesis is suitable for the large-scale production of a variety of nanomaterials and catalysts,29–33 but a few reports in the literature have described the synthesis of manganese oxides via this process.34–37 The mechanochemical synthesis can be tuned by many parameters such as the milling rotation speed, the milling time, the solvent-template, the weight ratios of the milling mixtures and milling balls, the milling atmosphere, the type of mills, and additional treatments such as sonication or heat treatments as well.38–43 The conventional dry milling method has significant drawbacks such as wider size distribution of materials, material loss, and friction. Nowadays, wet milling methods are more often used for nanomaterial synthesis.44–47 Besides, we can map the milling energy. This is important when the different catalytic activities of each sample is compared as it allows the preparation of a more favourable catalyst.
Here, we report on a simple and cost-effective, mechanochemical, one-pot synthesis method to produce pure as well as Pt-loaded or Cu-loaded manganese oxide nanostructures. Our team is the first to report the adjusted conditions for the synthesis of manganese oxides with this method, such as the particular molar ratio of the precursors, water-assisted grinding (wet grinding), milling time and milling rotation speed. One-pot synthesis can be simplify the additional metal doping process into manganese-oxides as using one-step for the whole synthesis. The MnOx nanostructures were characterized by BET, XRD, Raman, TG, DSC, SEM-EDX, TEM as well as ex situ XPS and XRD to explore the effect of pore structure, specific surface area as well as the oxidation states of the manganese in the catalytic reduction of CO2 with hydrogen. We found that the catalytic activity of the manganese-oxides could be tuned by the milling speed as well as the one-pot addition of active metals where ex situ XPS and XRD showed the crucial role of the oxidation states of the manganese under the reaction conditions.
2 Methods
2.1 Materials
Manganese(II) chloride tetrahydrate (MnCl2·4H2O), potassium permanganate (KMnO4), platinum(IV) chloride (H2PtCl6·H2O), copper(II) chloride (CuCl2), sodium hydroxide (NaOH), platinum(II) acetylacetonate (C10H16O4Pt), polyvinylpyrrolidone (PVP, MW = 40000) and ethylene-glycol (CH2OH)2 were used for the synthesis of the manganese-oxide nanostructures. All chemicals and reagents were of analytical grade, purchased from Sigma Aldrich and used without further purification. Ultrapure water was used for all synthesis and washing.
2.2 The synthetic procedure of the manganese-oxide-based catalysts
All the synthetic procedures are described in Table 1. First, we synthesized pure MnOx (M200, M450, M600) samples by the mechanochemical method with different milling speeds as categorized ball milling (pure samples). Furthermore, we synthesized metal-doped MnOx samples: M200(Cu-milled), M450(Cu-milled), M600(Cu-milled), M200(Pt-milled), M450(Pt-milled), and M600(Pt-milled) using the mechanochemical method as categorized ball milling (metal-doped) (Table 1).| Method | Synthetic procedure | Sample name (code) |
|---|---|---|
| Ball milling (pure samples) | MnCl2·4H2O and KMnO4 with a 1:0.5 molar ratio were mixed with 0.09 M of sodium hydroxide and 5 mL of water. The mixture was filled into a hardened stainless-steel grinding bowl (inner diameter: 7.5 mm – volume: 250 mL) of a Planetary Mono Mill Pulverisette 6 (Fritsch GmbH, Germany) ball-miller equipped with twenty-five hardened stainless-steel grinding balls of 4g weight and 10mm diameter. The mixture was milled at a speed of 200 rpm, 450 rpm and 600 rpm for 4 hours. The products were washed with water and freeze-dried for a night (>8 hours). The resulting products were labelled as M200, M450, and M600 corresponding to manganese-oxide samples prepared with 200 rpm, 450 rpm and 600 rpm milling speeds, respectively. |
M200 |
| M450 | ||
| M600 | ||
| Ball milling (metal-doped samples) | 0.0005 M platinum(IV) chloride or 0.0005 M copper(II) chloride was added into the mixture of the manganese-oxide precursors before the milling process. The same quantity of precursors, milling time and milling speeds (200 rpm, 450 rpm and 600 rpm) were used as in the case of the pure manganese-oxide based catalysts. The final product was obtained after filtration, washing with water followed by freeze-drying overnight. | M200(Cu-milled) |
| M450(Cu-milled) | ||
| M600(Cu-milled) | ||
| M200(Pt-milled) | ||
| M450(Pt-milled) | ||
| M600(Pt-milled) | ||
| Pt loading by wet impregnation method | The required amount of H2PtCl6·xH2O (to reach three wt% of metallic Pt) was dissolved into a determined amount of ethanol. The ethanoluous solution was filtrated into the pores of the M600 manganese oxide support. The supported catalyst was dried overnight. | M600(Pt-impregnated) |
| 5 nm Pt loading by sonication | First, 5 nm Pt nanoparticles with controlled size were produced.48 Here, 0.04 g of platinum(II) acetylacetonate and 0.035 g of polyvinylpyrrolidone (PVP, MW = 40000) were dissolved in 5 mL of ethylene-glycol and ultrasonicated for 30 minutes to obtain a homogenous solution. The solution was poured into a three-necked round bottom flask and was evacuated and purged under atmospheric pressure argon gas for several cycles to get rid of additional oxygen and water. After three purging cycles, the flask was immersed into an oil bath heated up to 473 K under vigorous stirring of the reaction mixture as well as the oil bath. After 10 minutes of the reaction, the flask was cooled down to room temperature. The suspension was precipitated by centrifugation with the co-addition of acetone to the reaction mixture. The nanoparticles were washed by centrifuging with hexane and redispersing in ethanol for at least 2–3 cycles and finally redispersed in ethanol. The concentration of the Pt nanoparticles was measured using an ICP technique. |
M600(5 nm Pt-sonicated) |
| To fabricate manganese-oxide supported Pt nanoparticle catalysts, the ethanoluous suspension of 5 nm Pt nanoparticles and M600 manganese oxide were mixed in ethanol and sonicated in an ultrasonic bath (40 kHz, 80 W) for 3 hours.6 The supported nanoparticles were collected by centrifugation. The products were washed with ethanol three times before they were left for a night (>8 hours) to dry at a temperature of 353 K. |
To study the effect of platinum incorporation, we prepared additionally Pt-loaded M600 samples using two other methods: Pt loading from H2PtCl6 by wet impregnation (M600-Pt-impregnated) and size-controlled 5 nm Pt nanoparticles loading by sonication (M600-5 nm Pt-sonicated) (Table 1).
2.3 Characterization methods
A Rigaku Miniflex II powder X-ray diffractometer using a Cu Kα radiation source (λ = 0.15418 nm) operating at 30 kV and 15 mA at room temperature and a scanning rate of 0.5 degree min−1 in the 10–65° 2θ range was used for crystal structure characterization. A high-resolution Transmission Electron Microscope HR-TEM (FEI TECNAI G2 20 X-TWIN) operated at an accelerating voltage of 200 kV and a Scanning Electron Microscope (Hitachi S-4700 Type II instrument (30 kV accelerating voltage) integrated with EDS) were used to perform morphological and compositional studies.The thermal behaviour of the samples was investigated via Thermogravimetry (TA Instruments Q500 TGA). The instrument worked from RT – 750 °C under both an air and nitrogen atmosphere, where the heating rate was 5 °C min−1. All the samples weighed between 10–20 mg and were placed into high-purity alpha platinum crucibles. Differential scanning calorimetric analysis was performed using Q20 (TA Instruments) at RT – 600 °C under a constant airflow and the heating–cooling rate was 5 °C min−1.
The Raman spectra were collected using a SENTERRA Raman microscope (Bruker Optics, Inc.) at 532 nm with a 1s integration time (with three repetitions) at a resolution of 4 cm−1 and interferometer resolution of 0.5 cm−1. The specific surface area and pore radius were measured using a 3H-2000 BET-A surface area analyzer.
For the analysis of the oxidation states of the manganese in the catalysts, XPS spectra were collected using a SPECS XPS instrument equipped with an XR-50 dual anode X-ray source and a PHOIBOS 150 energy analyzer. All spectra were acquired with a Al Kα radiation source operated at 150 W (14 kV). Survey spectra were collected with a step size of 1 eV and 40 eV pass energy, collecting 1 sweep for each sample. High-resolution spectra of Mn 2p, C 1s, O 1s and Pt 4f regions (latter three not shown) were acquired with 20 eV pass energy and step sizes of 0.1 eV. The Mn 2p regions were fitted with multiplet states taken into consideration, where the method was earlier described by Ilton et al.49 When resolving spectra of mixed oxides of Mn(II), Mn(III) and Mn(IV) data from pure oxides could be used as starting parameters. These parameters include peak positions in binding energies, intensity ratios and FWHM values. Due to the complicated nature of this method, the Mn 2p1/2 regions were not used for the evaluation. A pre-peak at lower binding energies (∼640 eV) was added, which corresponds to lattice defects.
2.4 Catalytic CO2 activation reaction over manganese-oxide catalysts
:H2 molar ratio was 1:4, if not denoted otherwise. The CO2:H2 mixture fed with the help of mass flow controllers (Aalborg), the total flow rate was 50 ml min−1. The reacting gas mixture flow entered and left the reactor through an externally heated tube to avoid condensation. Agilent 6890N gas chromatography analysis of the products and reactants was carried out using an HP-PLOTQ column. The gases were detected simultaneously by thermal conductivity (TC) and flame ionisation (FI) detectors. The CO2 was transformed to methane using a methanizer before being analyzed by FID.
3 Results and discussion
3.1 Mechanochemical synthesis and energy calculation
Fig. 1a represents the high-energy ball milling process. Furthermore, the milling-map of samples milled with different rotation speeds of 200 rpm (M200), 450 rpm (M450), and 600 rpm (M600) are shown in Fig. 1b where the ball-impact energy (Eb)51,52 and cumulative energy (Ecum) were calculated using eqn (1) and (2), respectively.51–55 This model allows the calculation of the impact energy of a single ball hit event as well as the amount of the total energy transferred to the milled material during the process. By comparing the results from a simple statistical model with those of the “Burgio–Rojac model”,52 we were able to demonstrate that the latter provides a more appropriate framework for the interpretation of milling-induced changes in manganese oxide at different oxidation states.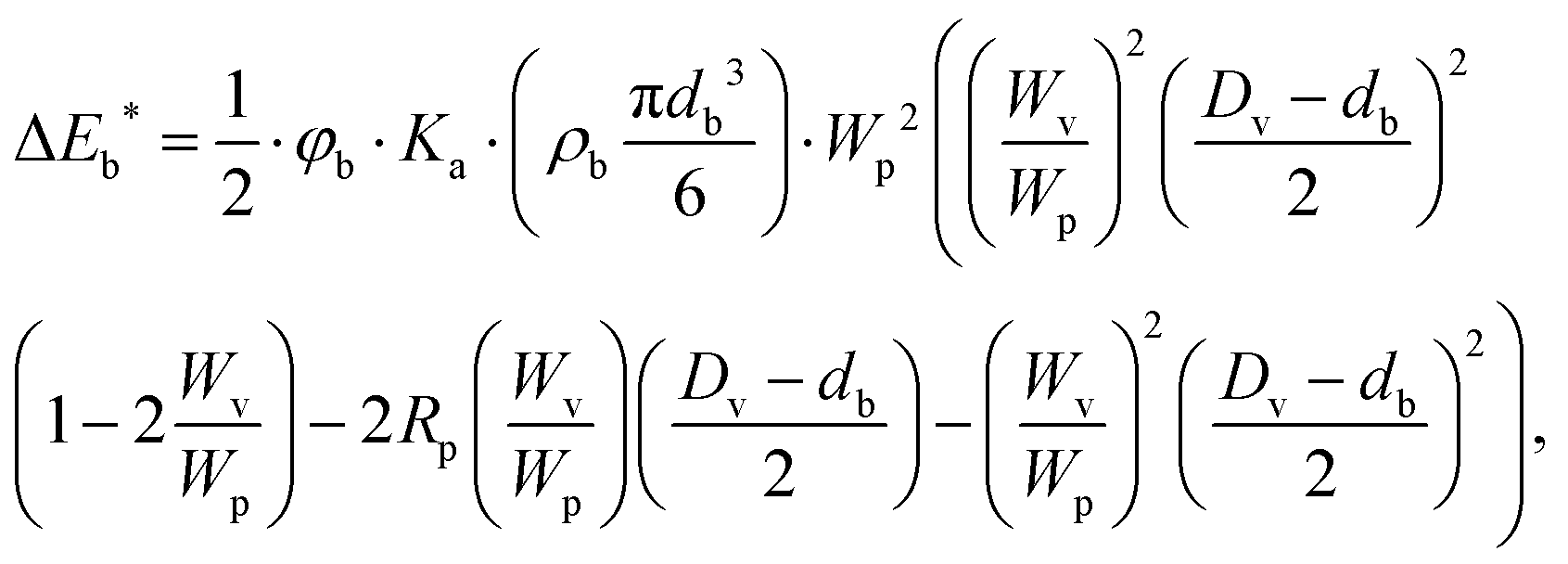 | (1) |
| Ecum = (ΔEb*·vt·t)/mp | (2) |
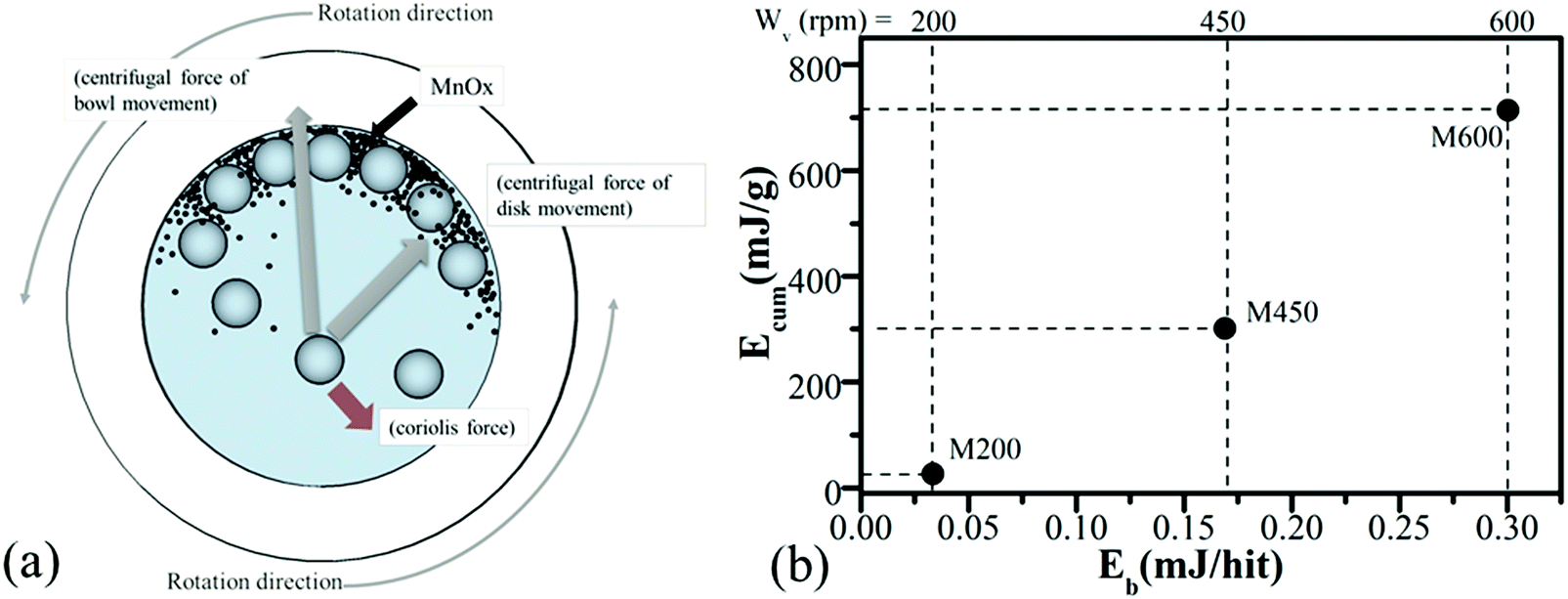 | ||
| Fig. 1 Schematic view of the motion of the ball and powder mixture in the high-energy ball milling process (a) and milling-map of the samples, displaying the grounding process with different ball-impact (Eb) and cumulative energy (Ecum) which is dependent on the rotational speed (Wv) (b). All the samples were milled for 4 hours. | ||
By using the as-calculated Eb and Ecum energies, milling-maps were made, to illustrate the changes in each sample (M200, M450 and M600). As shown in Fig. 1b, as the rotation speed increases, the ball impact energy (Eb) and cumulative energy (Ecum) both increased by a certain amount, which affects the crystal structure and physical–chemical properties of the samples. The following sections (3.2 and 3.3) will describe the crystal structure, chemical and morphological characterization of the samples.
3.2 Structural determination and chemical characterization
Fig. 2a shows the X-ray diffraction patterns of the milled samples. The main reflection peaks of the sample milled at 200 rpm (M200) are at 12.4°, 25.1°, 36.9° and 65.4°, and they correlate with birnessite type σ-MnO2 (JCPDS 421317).56,57 The reflection peaks of the sample milled at 450 rpm (M450) are at 12.4°, 25.3°, 36.5°, 44.6° and 65.4° and they refer to birnessite type σ-MnO2 (JCPDS 421317),58 furthermore, hausmannite-type amorphous Mn3O4 (JCPDS 011127)59 could be detected at 44.6°. The sample milled at 600 rpm (M600) showed only three major reflection peaks at 36.2°, 44.6° and 64.6°, which match with amorphous Mn3O4 (JCPDS 011127).59,60 The crystal structure tuned by birnessite type manganese(IV) oxide (M200) reduces to manganese(III and II) oxides in the case of M450 and M600 samples under alkaline conditions during the milling process. As the rotation speed increases, the energies (Eb and Ecum) increase as well and the temperature rises slightly inside (reference) the milling chamber. Consequently, the interlayer water is partially released, which weakens the layered structure. | ||
| Fig. 2 X-ray diffraction patterns of the pure samples (a) {* birnessite type σ-MnO2, ■ Mn3O4 phase} and the Raman spectra of the samples (b). | ||
Fig. 2b shows the Raman spectra of the samples, where the characteristic Raman shifts of the Mn–O symmetric stretching vibration at 647 cm−1 could be observed for all three manganese oxides. The shift at 575 cm−1 originates from the Mn–O symmetric stretching vibration in the basal plane of [MnO6] sheets of birnessite, and it is dependent on the presence of Mn4+ ions in sample M200 and M450, respectively.61 In the case of M600, which is an amorphous hausmannite-type manganese oxide,62 the Raman shift at 575 cm−1 is absent due to the total transformation of Mn(IV) into Mn3+ and Mn2+ ions in the structure.
X-ray diffraction patterns of the metal (Pt and Cu) doped samples and their Raman shifts are shown in the ESI† (Fig. S1 and S2). No additional peaks were observed in the doped samples for both measurements, which illustrates that loading during the milling process does not alter the original crystal structure of the manganese oxides. One thing to note regarding the Raman spectra: the ratio of the Mn–O liberal stretching and the Mn–O basal plane stretching of the M200 samples are slightly fluctuating due to the Pt or Cu atoms in their structure.
Fig. S3 (ESI†) shows the energy-dispersive X-ray spectra and the elemental analysis results are summarized in Table S1 (ESI†). The EDS spectra reveal that the samples consisted of oxygen, manganese, potassium, sodium, and doped metal elements only, and no impurities from the milling bowl and balls can be found (Fig. S3, ESI†). The sodium and potassium contents were decreased from 0.9 atomic percentage (at%) and 6.51 (at%) to 0.11 (at%) and 2.64 (at%), respectively as the milling speed was increased from 200 rpm to 600 rpm. The presence of a Pt-dopant resulted in a ∼14 times higher sodium and potassium content in the Pt-doped M600 sample compared to the pure M600 sample. The weight percentage of the Pt in M600 (Pt-milled) was ∼3 (wt%) proving that the Pt atoms were fully incorporated into the structures confirming the weight ratio from the precursors (Table S1, ESI†).
3.3 Morphological characterization
Fig. 3a displays the results of the specific surface area measurements. The surface area of the sample was dependent on the milling speed, and it increased from 11 m2 g−1 to 150 m2 g−1 for M200 and both M450, and M600, as presented in Table 2. M200 had mostly micropores, while M450 and M600 were mesoporous with an average pore radius of ∼6 nm (Fig. S4, ESI†). Manganese-oxides milled at a higher milling speed (M450 and M600) presented a higher average specific surface area than the manganese-oxide nanoparticles observed in the literature.63,64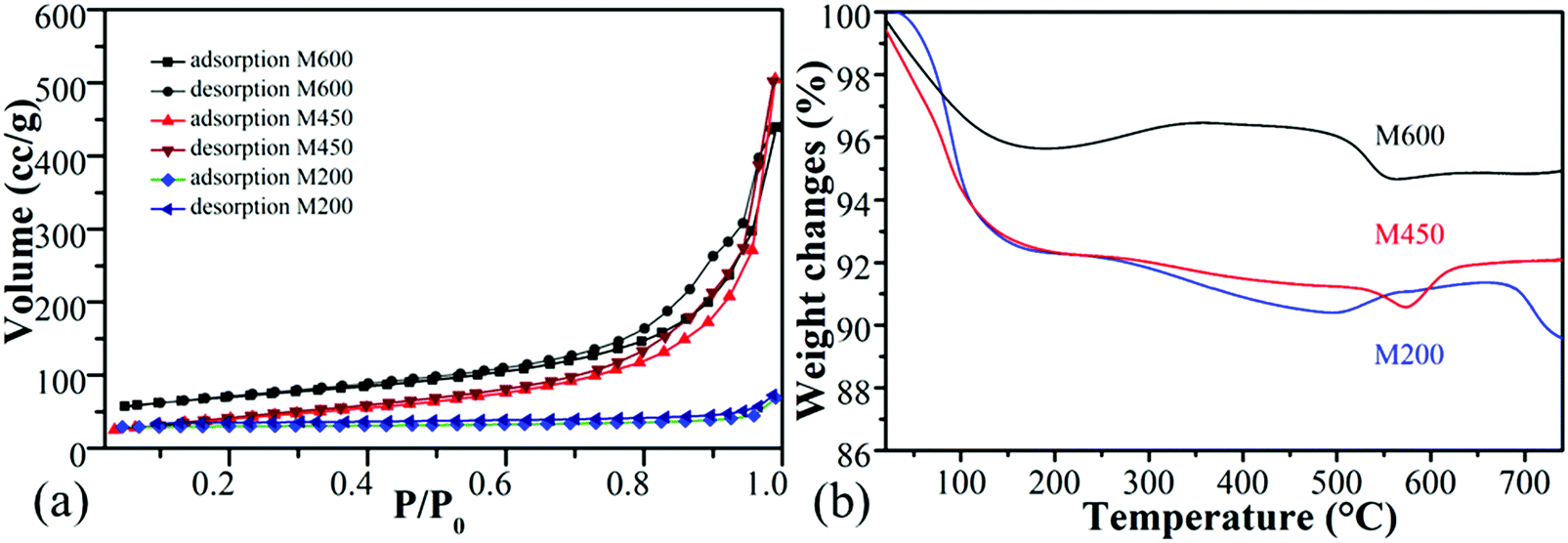 | ||
| Fig. 3 N2 adsorption analysis full isotherm (a) and thermal decomposition of the pure samples at a heating rate of 5 °C min−1 in air (b). | ||
| Samples | Specific surface area, m2 g−1 | Pore radius, nm | Pore volume, cc g−1 |
|---|---|---|---|
| M200 | 11 | <2 | 0.06 |
| M450 | 159 | >5.7 | 0.7 |
| M600 | 159 | >5.8 | 0.6 |
Fig. 3b shows the results of the thermogravimetric analysis of the samples under air. The main thermal changes in the M200 and M450 samples are similar to typical synthetic birnessites. During the first decomposition step at RT – 110 °C a weight loss of ∼6% was caused by the release of physically adsorbed water.65 The second weight loss of ∼2% resulted at 220 °C stemming from the release of physically adsorbed water from the interlayer spacing.66 The third weight loss occurred at 500 °C and 570 °C for M200 and M450, respectively, corresponding to the reduction of Mn(IV) to Mn(III, II) and the formation of Mn2O3.67 The fourth change occurred around 550 °C and 630 °C for M200 and M450, respectively, where a 1% weight gain occurred due to the slow re-oxidation of the manganese Mn3O4 to Mn2O3 and α-MnO2.68,69 It is interesting to note that only in the case of the M200 sample, did a fifth thermal change occur where the rest of the Mn2O3 reduced to Mn3O4 at 680 °C.70
On the other hand, the thermal decomposition behaviour was quite different for the M600 sample due to the presence of Mn(II, III)-oxide phases. The first decomposition occurred at 110 °C with ∼4 wt% weight loss, which corresponds to the release of physically adsorbed water. The second change occurred with ∼1% weight gain at 200 °C possibly stemming from the oxidation of manganese and phase change of Mn3O4 to Mn2O3.68 The third change was a weight loss of ∼2% due to the reduction of Mn2O3 to Mn3O4.70 Oxidation occurred at a low temperature for the M600 sample, which indicates that the amount of Mn(III) is much higher than in the other two samples. It also demonstrates slow crystallization of the amorphous morphology.65 Fig. S5(a–d) (ESI†) discloses the thermogravimetric analysis under a nitrogen flow. No weight loss or gain occurred at 300–550 °C for the M600 sample in a nitrogen flow, helping to prove the results and discussion about the thermal analysis under air.
In Fig. S6a (ESI†), X-ray diffraction patterns of the samples after TG analysis under both air and nitrogen atmospheres showed that α-MnO2 and Mn3O4 oxides were produced by thermal analysis in air, while only Mn3O4 remained for M200. In the case of the M450 sample, α-MnO2 and Mn3O4 phases formed, compared to M600, where only Mn3O4 was created under oxidative conditions. From the thermogravimetric analysis of the samples and the X-ray diffraction patterns of the end products (after TGA), we can derive the same conclusion from the diffraction patterns of the initial samples, such as M200 is mainly consisted of MnO2 (manganese oxidation state: 4+), and M450 consists of MnO2 and a minor amount of Mn3O4 (manganese oxidation state: IV, III, II), while M600 consists of an almost pure phase Mn3O4 (manganese oxidation state: III, II).
Fig. S5e (ESI†) presents the differential scanning calorimetric results of the samples. During the heating process at low temperature (150 °C), endothermic peaks appear for M200 and M450 related to the dehydration of interlayer water and the formation of Mn2O3. Endothermic peaks at higher temperatures illustrate the reduction of Mn(III) to Mn(II) and the possible phase transition to tunnel-structured flakes of Mn3O4.71 In the case of M600, one endothermic peak observed around 550 °C illustrates the phase transition of Mn3O4 from an amorphous state to a nanoflake structure.
Fig. 4(a, c, e) and (b, d, f) show the scanning electron microscopy (SEM) and transmission electron microscopy images, respectively. With the increment of the milling speed, the average particle size also decreased. The M200 sample has larger flakes and aggregated sheets identical to a typical birnessite-structure,35 while at higher milling speeds, the crystal structure was ruined due to the higher mechanical impact as well as the higher temperature resulting in the decreases in the interlayer water. Besides the fact that the average oxidation state of manganese decreased in the case of the M450 and M600 samples compared to the M200 sample, the structure of the M450 sample consisted of a mixture of sheets and flakes and M600 consisted mostly of 10–50 nm small nanoflakes.72
 | ||
| Fig. 4 Scanning electron microscopy images (SEM) and transmission electron microscopy (TEM) images of the pure samples (a and b) M200, (c and d) M450 and (e and f) M600. | ||
4 The catalytic CO2 hydrogenation reaction
Manganese oxides prepared at different milling speeds as well as several Pt- and Cu-doped (loaded with ball milling, wet impregnation method and ultrasonication assisted addition of nanoparticles) manganese oxides tested in CO2 hydrogenation to form carbon-monoxide and methane at 573–823 K in a fixed-bed continuous-flow catalytic reactor at ambient pressure. The catalytic activity was monitored by the consumption rate and selectivity, which is discussed in the following sections. The reaction rates and calculated activation energies for CO2 hydrogenation reactions over all samples are summarized in Table 3.| Sample name | CO2 consumption rate, nmol g−1 s−1 | Activation energy, kcal mol−1 | |
|---|---|---|---|
| 673 K | 873 K | ||
| M200 | — | 10083 |
54.9 |
| M450 | 1038 | 16707 |
23.7 |
| M600 | 1954 | 19523 |
19.3 |
| M200(Cu-milled) | 2811 | 19430 |
19.7 |
| M450(Cu-milled) | 3367 | 19129 |
16.0 |
| M600(Cu-milled) | 6943 | 20722 |
16.7 |
| M200(Pt-milled) | 11186 |
22052 |
13.5 |
| M450(Pt-milled) | 14293 |
22338 |
11.2 |
| M600(Pt-milled) | 12730 |
19996 |
7.6 |
| M600(Pt-impregnated) | 9318 | 20151 |
13.5 |
| M600(5 nm Pt-sonicated) | 9283 | 18895 |
16.7 |
4.1 Effect of milling speed
All the ball-milled manganese oxide prepared at different milling speeds (M200, M450, M600) were active in the CO2 hydrogenation reaction at >600 K and was producing mostly carbon-monoxide besides a small amount of methane (Fig. 5a). In the case of the reaction tested at 873 K, the M600 catalysts showed the highest CO2 consumption rate (∼20000 nmol g−1 s−1) followed by M450 (17500 nmol g−1 s−1). Both M600 and M450 were almost two times more active compared to the catalyst milled at 200 rpm (∼10000 nmol g−1 s−1). The high activity of manganese-oxide milled at higher speeds can be attributed to the high specific surface area73 and porosity as well as the morphological differences. Furthermore, the samples milled at different speeds after the pretreatment in hydrogen showed diversity in the ratio of Mn4+:Mn3+:Mn2+ (Fig. S7a, ESI†), which could also affect the catalytic activity. After the pretreatment, the birnessite type σ-MnO2 ratio decreased, and mostly amorphous or nanoscaled Mn(II, III)3O4 and Mn(II)O presented in the catalyst before the reaction.
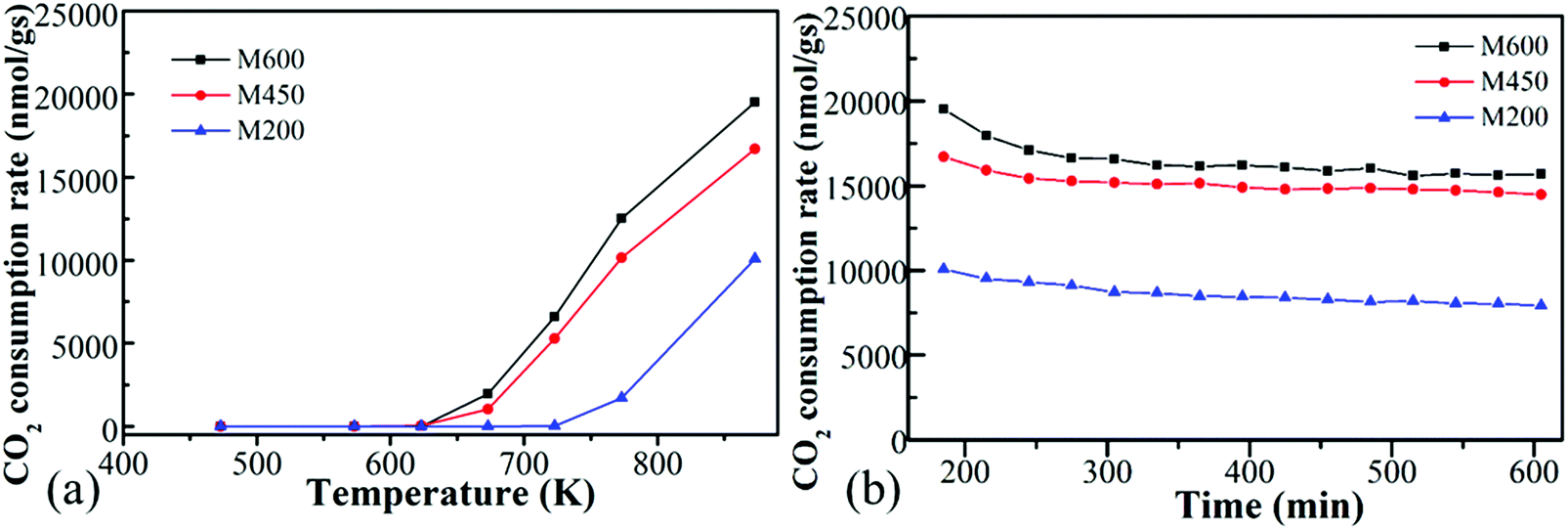 | ||
| Fig. 5 The CO2 consumption rate of manganese oxides synthesized by different milling speed (200 rpm-M200, 450 rpm-M450 and 600 rpm-M600) as a function of temperature (a) and function of time (b). | ||
It is interesting to note that these kinds of noble metal-free oxides have unusual activity compared to mesoporous MnO2 samples. Also, they are competitive with other metal-based (e.g., Ni, Co) oxides in CO2 activation.6 M200, M450 and M600 were stable at 873 K under the reaction conditions for ∼10 hours (Fig. 5b).
As the milling speed was increased, the activation energy of the reaction in the range of 573–873 K was decreased drastically showing the presence of different reaction pathways besides the increasing reaction rate (Table 3). The differences in pathways and reaction rates can be attributed to the different structures of manganese oxides (birnessite type σ-MnO2 for M200, birnessite type σ-MnO2 and hausmannite-type amorphous Mn3O4 for M450 and amorphous Mn3O4 for M600) as evidenced by the XRD results.
4.2 Effect of one-pot loading of Pt- and Cu-doped catalysts
To enhance the catalytic activity, we used a simple one-pot method to dope 3 wt% of Pt and Cu into the manganese oxide nanostructures during the milling process. The catalytic activity was significantly enhanced by adding Pt into the manganese oxide structure at 673 K (Fig. 6a). In the case of the Pt-doped manganese-oxide catalyst milled under different speeds, ∼12–13 times increment in the catalytic activity was observed, showing the presence of the significant role of Pt where the effect of the milling speed was insignificant. Cu-doping resulted in a ∼2.5 times increment in the catalytic activity compared to the pure manganese-oxide catalysts. | ||
| Fig. 6 Temperature effect for the CO2 consumption rate of the samples at (a) 673 K and (b) 873 K. | ||
At the higher temperature (873 K) displayed in Fig. 6b, the catalytic boosting effect of Pt, as well as Cu-doping, was negligible. All the metal-loaded catalysts showed similarly high activity (∼20000 nmol g−1 s−1) as the pure manganese-oxide milled at 600 rpm. This phenomenon shows that the particular mixed Mn(IV, III, II)-oxide role is crucial in the catalytic activity at higher temperatures. These changes illustrate that temperature plays a vital role in the manganese oxide-based catalysts due to crystal structure changes and reducibility of the manganese oxides.
Both copper and platinum doping resulted in the decrease of the activation energy values (Table 3). As the milling speed was increased, the activation energy was decreased. Platinum showed lower values compared to copper-loaded samples.
4.3 Effect of Platinum incorporation (milling vs. impregnation vs. sonication)
Three different types of Pt-loaded M600 prepared by using one-pot synthesis, incipient wetness impregnation as well as loading of pre-synthesized 5 nm Pt nanoparticles to the catalysts by ultrasonication. All Pt-doped catalysts displayed almost the same catalytic activity (Fig. 7a and b). Usually, a wet impregnation method or even surfaces designed by adding size-controlled nanoparticles resulted in specific loading of the metal onto the surface as well as a catalytic activity increment.74 These tests support the idea that the main working surface of these kinds of catalysts is based on the manganese-oxide, regardless of the quality and type of doped metal onto the surface.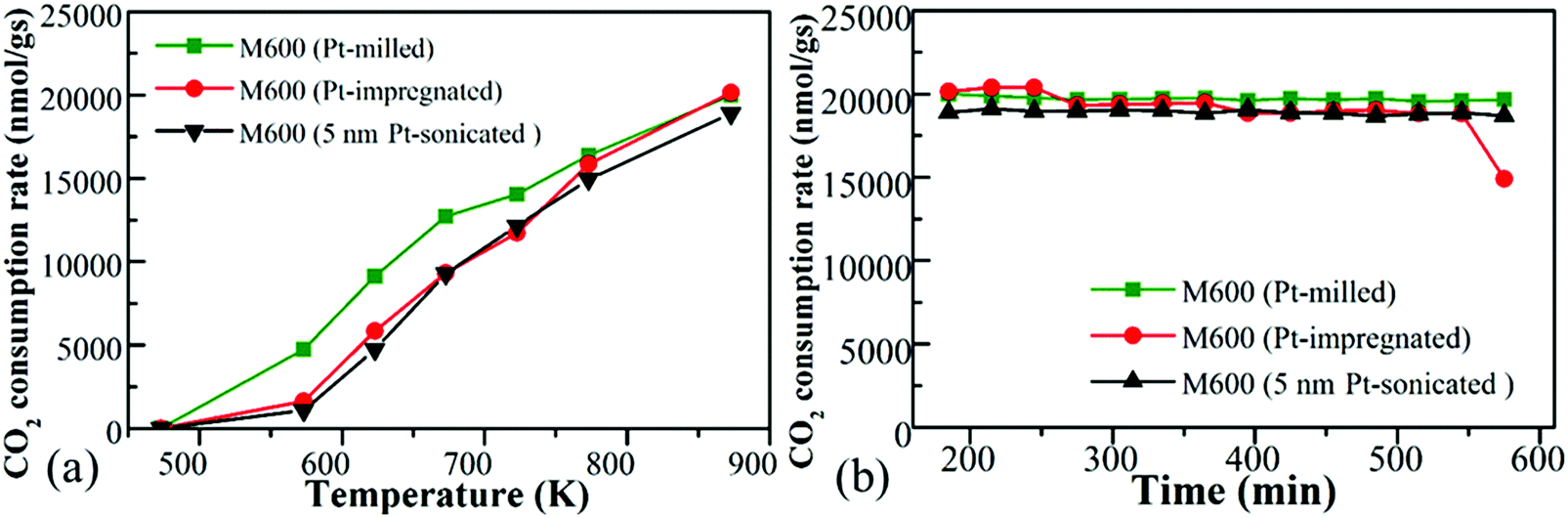 | ||
| Fig. 7 The CO2 consumption rate of the Pt-doped M600 manganese oxide prepared by the one-pot method, incipient wetness impregnation method as well as the designed incorporation of size-controlled 5 nm Pt nanoparticles as a function of temperature (a) and a function of time (b). | ||
It was interesting to note that the activation energies for these catalysts over the CO2 activation showed differences; however, the reaction rate did not show big differences. The samples loaded with Pt prepared by milling showed the lowest activation energies (Table 3).
4.4 Selectivity towards methane
In the case of the selectivity, these catalysts mostly produced carbon monoxide (>95%) and a smaller amount of methane. Most catalysts started to produce methane with a small ratio at an elevated temperature (∼623 K) and after a short increment, shifting of the products into the formation of carbon monoxide was observed (Fig. 8a) as expected from the thermodynamics of the CO2 hydrogenation reaction in the gas phase.6 In the case of the M600(Pt-milled) catalyst produced by the one-pot milling process, methane formation was significant compared to pure and Cu-loaded catalysts prepared by using different milling speeds. However, the results show that the catalytic activity is not profoundly influenced by the metal-loading, however, the Pt/MnOx interphase is crucial in the methane selectivity. The Pt-loaded manganese-oxide prepared via the one-pot synthesis has 1.5–2 times higher selectivity towards methane compared to the Pt-loaded MnOx catalyst synthesized by the wet impregnation and size-controlled Pt sonication, which shows the influence of the formed Pt/MnOx during the reaction (Fig. 8b). | ||
| Fig. 8 The selectivity of methane during the CO2 hydrogenation reaction in the case of (a) pure, Pt-loaded and Cu-loaded catalysts prepared by using different milling speeds and (b) M600 loaded with Pt by different methods. | ||
Table 4 displays the formation rate and selectivity towards CO and CH4 of 3 Pt-incorporated samples: M600(Pt-milled), M600(Pt-impregnated) and M600(5 nm Pt-sonicated) at 673 K. In the presence of Pt in the M600 catalyst prepared by the one-pot ball-milling process, the catalytic activity was ∼17 times higher at 673 K while it was similar at 823 K compared to the Pt-free M600 catalyst. While the main product was CO, the highest amount of CH4 (∼20%) was formed in the case of the Pt-doped M600.
| Catalyst | CO formation rate, nmol g−1 s−1 | CO formation selectivity, % | Methane formation rate, nmol g−1 s−1 | Methane formation selectivity, % |
|---|---|---|---|---|
| M600(Pt-milled) | 12759 |
82 | 2362 | 15 |
| M600(Pt-impregnated) | 9716 | 89 | 992 | 9 |
| M600(5 nm Pt-sonicated) | 9229 | 90 | 822 | 8 |
4.5 Determining the oxidation states (XPS data processing and evaluation) of the catalysts
For a better understanding of the activity differences of the catalysts, the oxidation states of the Mn species were investigated by ex situ XPS for samples prepared with low (M200) and high (M600) milling speeds as well as co-milled with Pt salts at 600 rpm (M600-Pt-milled) after the pretreatment of the samples in O2 followed by H2 at 300 °C as well as after the CO2 hydrogenation reactions. The Mn 2p regions were fitted with multiplet states taken into consideration (Fig. S8, ESI†).49 The ratios of the Mn oxidation states were calculated based on the peak areas of the fittings in Fig. 9. (Data values are summarized in Table S2, ESI.†)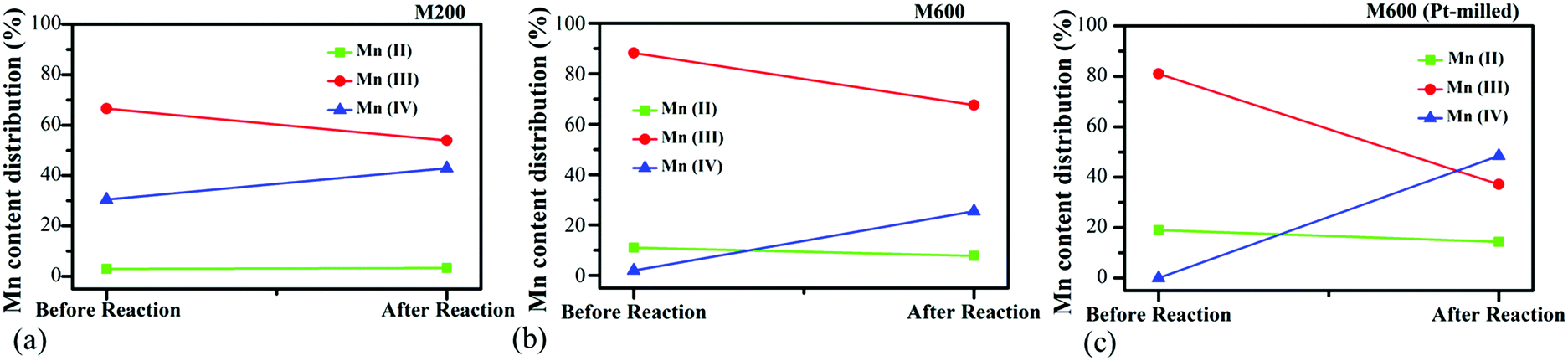 | ||
| Fig. 9 Atomic ratios of Mn with different oxidation states before CO2 hydrogenation (after pretreatment) and after CO2 hydrogenation reactions for (a) M200, (b) M600, and (c) Pt-milled M600. | ||
Before the CO2 hydrogenation (after the pretreatment), in the case of the M200 catalyst, Mn(III) and M(IV) presented with a ratio of Mn(III):Mn(IV) = ∼2. However, after the reaction, this ratio decreased to ∼1 showing the presence of the oxidation of the catalyst during the CO2 hydrogenation. Usually, under the hydrogenation processes, the reduction of the surface is favourable,6,50 but the possibility of CO2 dissociation to CO and *O followed by the oxidation of the metallic or metal–oxide surface as well as the oxidation of the surface by H2O resulting from the Reverse Water Gas Shift Reaction  can be also possible.75,76 In the case of the M600 catalyst, a small amount of Mn(II) (∼10%) was observed next to a higher amount of Mn(III) with the absence of Mn(IV) on the surface before the reaction. After the CO2 hydrogenation reaction, the oxidation of the surface was observed as in the case of the M200 sample. The small amount of the Mn(II) was reduced to ∼3% and Mn(IV) appeared with a ratio of Mn(III):Mn(IV) = ∼3.
can be also possible.75,76 In the case of the M600 catalyst, a small amount of Mn(II) (∼10%) was observed next to a higher amount of Mn(III) with the absence of Mn(IV) on the surface before the reaction. After the CO2 hydrogenation reaction, the oxidation of the surface was observed as in the case of the M200 sample. The small amount of the Mn(II) was reduced to ∼3% and Mn(IV) appeared with a ratio of Mn(III):Mn(IV) = ∼3.
As we could see during the catalytic tests (Section 4.1), the M600 catalysts showed the highest CO2 consumption rate and were almost two times more active compared to M200 at both low and high temperatures. We believe that beside the high specific surface area and porosity as well as the morphological differences, the Mn oxidation states play an important role during the reaction. In the case of the M600 sample, the presence of Mn(II) and the higher concentration of Mn(III) could be the reason for the higher catalytic activity. In manganese-based catalyst driven photosynthesis, the presence of Mn(II)Mn(III)3 is needed for the production of the photo assembly intermediates and Mn(III)Mn(IV)3 was responsible for the oxygen releasing step.77 We also found in our recent studies that Mn(II) helped the reduction of CO2 hydrogenation as a support for metallic Cobalt where the reaction followed the format pathway towards CH4 production.78 We believe that, the presence of Mn(II) as well as the ratio of Mn(III)/Mn(IV) is crucial for this reaction driven by manganese oxide-based catalysts.
Ex situ XPS data show that before the reaction, Mn(II) was observed with the content of ∼20% next to Mn(III). The higher amount of the reduced phase compared to the pure M600 shows the reducing effect of Pt under the H2 pretreatment process. However, after the CO2 activation, a huge amount of Mn(IV) was formed on the surface with a Mn(III)/Mn(IV) ratio of ∼1 beside the slight reduction of the content of Mn(II) to ∼18%. Beside the fact that the presence of Pt usually increases the activity of CO2 reduction,79,80 the perturbation of the Mn oxidation state by Pt can also affect the catalytic activity. At a lower temperature, the Pt-loading resulted in ∼17 times higher catalytic activity, where the CH4 selectivity was drastically increased. This is interesting, as we know that usually, the presence of Pt helps to produce mostly CO in the CO2 hydrogenation reaction.50 On the other hand, at 823 K, the lack of the effect of Pt was observed in the catalytic activity compared to pure M600 catalysts. These phenomena can be attributed to the presence of Mn(II) as well as the different ratios of the oxidation states of the manganese in the oxides. In the future, we will focus on the atomic level understanding of such an easy-to-produce cheap catalyst to reach the highest activity by tuning the oxidation states of the manganese without using precious metals.
Furthermore, we summarized the comparison of the CO2 conversion percentage of our Pt-loaded samples with previously published studies in Table 5. As seen in Table 5, our results prove that efficient manganese oxide catalyst synthesis can be done using a cost-effective one-pot ball milling method, and that even ball milling can be used as a simple method for the loading of metals.
| Catalyst | Synthetic method | Temperature, K | CO2 conversion (%) | Ref. |
|---|---|---|---|---|
| M600(Pt-milled) | Ball milling | 673 | 42 | This study |
| M600(Pt-impregnated) | Ball milling/impregnation | 673 | 28 | This study |
| M600(5 nm Pt-sonicated) | Ball milling/sonication | 673 | 28 | This study |
| Pt/MnO2 | Hard template | 648 | 25 | 6 |
| MnOx–Co3O4 | Sol–gel inverse micelle | 523 | 45 | 73 |
| PtCo/CeO2 | — | 573 | 6 | 81 |
| K–Mn–Fe/Al2O3 | Co-incipient wetness impregnation (IWI) | 836 | 41 | 82 |
| La–Mn–Zn–Cu–O | Sol–gel | 543 | 13 | 83 |
| Mn–Na/Fe | Co-precipitation | 593 | 37 | 84 |
| Pt/TiO2 | — | 673 | 40 | 85 |
| Fe/SiO2-250 | Wet impregnation | 643 | 38 | 86 |
| CuO–ZnO–Al2O3 | Ball milling | 523 | 12 | 87 |
| Ni/SiO2 | Sol–gel | 673 | 35 | 88 |
5 Conclusions
A novel mechanochemical manganese oxide nanostructure synthesis was demonstrated, which can be easily scaled-up for synthetic industries. The shape, porosity, specific surface area, as well as the ratio of the different oxidation states of the Mn-ion in the structure, can be tuned by the milling speed. Pt-loaded and Cu-loaded MnOx structures were prepared with the same one-pot technique and all the samples were tested in a CO2 hydrogenation reaction in the gas phase. Pure MnOx milled at 600 rpm milling speed showed the highest catalytic activity, and the effect of Pt-loading and Cu-loading was insignificant at higher temperatures. It showed that the presence of the different oxidation states of the manganese plays an essential role in the CO2 activation process aside from the advantage of the higher surface area and porosity, which can be tuned by the milling speed of the ball milling process.Also, Pt loading can improve methane selectivity. The one-pot synthesised Pt-loaded MnOx nanostructure showed the highest methane selectivity compared to the Pt-loaded MnOx catalyst synthesised by the wet impregnation and size-controlled Pt sonication, which shows the high impact of the formed Pt/MnOx interface on the selectivity of the catalytic CO2 hydrogenation reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper was supported by the Hungarian Research Development and Innovation Office through grants NKFIH OTKA PD 120877 of AS. ÁK and ZK are grateful for the funds from NKFIH (OTKA) K112531 & NN110676 and K120115, respectively. The financial support of the Hungarian National Research, Development and Innovation Office through the GINOP-2.3.2-15-2016-00013 project “Intelligent materials based on functional surfaces from syntheses to applications” and the Ministry of Human Capacities through the EFOP-3.6.1-16-2016-00014 project and the 20391-3/2018/FEKUSTRAT are acknowledged. Gábor Kozma gratefully acknowledges the support of the Bolyai János Research Fellowship of the Hungarian Academy of Science and the ÚNKP-19-4-SZTE-116 New National Excellence Program of the Ministry for Innovation and Technology.Notes and references
- N. S. Lewis and D. G. Nocera, Powering the Planet: Chemical Challenges in Solar Energy Utilization, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(43), 15729–15735, DOI:10.1073/pnas.0603395103.
- D. Bratt, Catalytic CO2 Hydrogenation – Literature Review: Technology Development since 2014, 2016, https://hdl.handle.net/20.500.11919/779 Search PubMed.
- G. A. Olah, T. Mathew, A. Goeppert and G. K. S. Prakash, Difference and Significance of Regenerative Versus Renewable Carbon Fuels and Products, Top. Catal., 2018, 61(7), 522–529, DOI:10.1007/s11244-018-0964-8.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Directly Converting CO2 into a Gasoline Fuel, Nat. Commun., 2017, 8(May), 15174, DOI:10.1038/ncomms15174.
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Tandem Catalysis for CO2 Hydrogenation to C2–C4 Hydrocarbons, Nano Lett., 2017, 17(6), 3798–3802, DOI:10.1021/acs.nanolett.7b01139.
- A. Sápi, T. Rajkumar, M. Ábel, A. Efremova, A. Grósz, A. Gyuris, K. B. Ábrahámné, I. Szenti, J. Kiss and T. Varga, et al., Noble-Metal-Free and Pt Nanoparticles-Loaded, Mesoporous Oxides as Efficient Catalysts for CO2 Hydrogenation and Dry Reforming with Methane, J. CO2 Util., 2019, 32(January), 106–118, DOI:10.1016/j.jcou.2019.04.004.
- J. Martínez, E. Hernández, S. Alfaro, R. L. Medina, G. V. Aguilar, E. Albiter and M. A. Valenzuela, High Selectivity and Stability of Nickel Catalysts for CO2 Methanation: Support Effects, Catalysts, 2019, 9, 24, DOI:10.3390/catal9010024.
- H. Yang, C. Zhang, P. Gao, H. Wang, X. Li, L. Zhong, W. Wei and Y. Sun, A Review of the Catalytic Hydrogenation of Carbon Dioxide into Value-Added Hydrocarbons, Catal. Sci. Technol., 2017, 7(20), 4580–4598, 10.1039/c7cy01403a.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Discovery of a Ni–Ga Catalyst for Carbon Dioxide Reduction to Methanol, Nat. Chem., 2014, 6(4), 320–324, DOI:10.1038/nchem.1873.
- X. Du, D. Zhang, L. Shi, R. Gao and J. Zhang, Morphology Dependence of Catalytic Properties of Ni/CeO2 Nanostructures for Carbon Dioxide Reforming of Methane, J. Phys. Chem. C, 2012, 116(18), 10009–10016, DOI:10.1021/jp300543r.
- D. Shi, R. Wojcieszak, S. Paul and E. Marceau, Ni Promotion by Fe: What Benefits for Catalytic Hydrogenation?, Catalysts, 2019, 9, 451, DOI:10.3390/catal9050451.
- S. Kar, A. Goeppert, J. Kothandaraman and G. K. S. Prakash, Manganese-Catalyzed Sequential Hydrogenation of CO2 to Methanol via Formamide, ACS Catal., 2017, 7(9), 6347–6351, DOI:10.1021/acscatal.7b02066.
- C.-S. Li, G. Melaet, W. T. Ralston, K. An, C. Brooks, Y. Ye, Y.-S. Liu, J. Zhu, J. Guo and S. Alayoglu, et al., High-Performance Hybrid Oxide Catalyst of Manganese and Cobalt for Low-Pressure Methanol Synthesis, Nat. Commun., 2015, 6, 6538, DOI:10.1038/ncomms7538.
- S. Lee and H. Xu, XRD and Tem Studies on Nanophase Manganese Oxides in Freshwater Ferromanganese Nodules from Green Bay, Lake Michigan, Clays Clay Miner., 2016, 64(5), 523–536, DOI:10.1346/CCMN.2016.064032.
- J. E. Post, Manganese Oxide Minerals: Crystal Structures and Economic And, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(March), 3447–3454 CrossRef CAS PubMed.
- X. Huang, T. Chen, X. Zou, M. Zhu, D. Chen and M. Pan, The Adsorption of Cd(II) on Manganese Oxide Investigated by Batch and Modeling Techniques, Int. J. Environ. Res. Public Health, 2017, 14(10), 1145, DOI:10.3390/ijerph14101145.
- Y. Xin Zhang, X. Long Guo, M. Huang, X. Dong Hao, Y. Yuan and C. Hua, Engineering Birnessite-Type MnO2 nanosheets on Fiberglass for PH-Dependent Degradation of Methylene Blue, J. Phys. Chem. Solids, 2015, 83, 40–46, DOI:10.1016/j.jpcs.2015.03.015.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, In Situ X-Ray Absorption Spectroscopy Investigation of a Bifunctional Manganese Oxide Catalyst with High Activity for Electrochemical Water Oxidation and Oxygen Reduction, J. Am. Chem. Soc., 2013, 135(23), 8525, DOI:10.1021/ja3104632.
- A. C. Thenuwara, S. L. Shumlas, N. H. Attanayake, E. B. Cerkez, I. G. McKendry, L. Frazer, E. Borguet, Q. Kang, M. J. Zdilla and J. Sun, et al., Copper-Intercalated Birnessite as a Water Oxidation Catalyst, Langmuir, 2015, 31(46), 12807–12813, DOI:10.1021/acs.langmuir.5b02936.
- W. Wang, Z. Shao, Y. Liu and G. Wang, Removal of Multi-Heavy Metals Using Biogenic Manganese Oxides Generated by a Deep-Sea Sedimentary Bacterium - Brachybacterium Sp. Strain Mn32, Microbiology, 2009, 155(6), 1989–1996, DOI:10.1099/mic.0.024141-0.
- H. Guan, W. Dang, G. Chen, C. Dong and Y. Wang, RGO/KMn8O16 composite as Supercapacitor Electrode with High Specific Capacitance, Ceram. Int., 2016, 42(4), 5195–5202, DOI:10.1016/j.ceramint.2015.12.043.
- T. B. Atwater and A. J. Salkind, Lithium Potassium Manganese Mixed Metal Oxide Material for Rechargeable Electrochemical Cells, Report of US Army Research Development & Engineering Comd (RDECOM), 2010, pp. 4–8 Search PubMed.
- S. Kattel, B. Yan, J. G. Chen and P. Liu, CO2 Hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of Synergy between Pt and Oxide Support, J. Catal., 2016, 343, 115–126, DOI:10.1016/j.jcat.2015.12.019.
- J. Nakamura, T. Fujitani, S. Kuld, S. Helveg, I. Chorkendorff and J. Sehested, Comment on “Active Sites for CO2 Hydrogenation to Methanol on Cu/ZnO Catalysts”, Science, 2017, 357(6354), 1296–1299, DOI:10.1126/science.aan8074.
- X. Duan, J. Yang, H. Gao, J. Ma, L. Jiao and W. Zheng, Controllable Hydrothermal Synthesis of Manganese Dioxide Nanostructures: Shape Evolution, Growth Mechanism and Electrochemical Properties, CrystEngComm, 2012, 14(12), 4196–4204, 10.1039/c2ce06587h.
- Y. F. Liu, G. H. Yuan, Z. H. Jiang and Z. P. Yao, Solvothermal Synthesis of Mn3O4 Nanoparticle/Graphene Sheet Composites and Their Supercapacitive Properties, J. Nanomater., 2014, 2014, 190529, DOI:10.1155/2014/190529.
- Q. Zhang, J. Luo, E. Vileno and S. L. Suib, Synthesis of Cryptomelane-Type Manganese Oxides by Microwave Heating, Chem. Mater., 1997, 9(10), 2090–2095, DOI:10.1021/cm970129g.
- W. Thanthamrong, R. Songsak, D. Paweena, J. Wirat and W. Winadda, Electrodeposition of Manganese Oxide Nanosheets as Supercapacitor Electrode Materials, Key Eng. Mater., 2016, 675–676, 273–276, DOI:10.4028/www.scientific.net/KEM.675-676.273.
- C. Xu, S. De, A. M. Balu, M. Ojeda and R. Luque, Mechanochemical Synthesis of Advanced Nanomaterials for Catalytic Applications, Chem. Commun., 2015, 51(31), 6698–6713, 10.1039/c4cc09876e.
- R. A. Buyanov, V. V. Molchanov and V. V. Boldyrev, Mechanochemical Activation as a Tool of Increasing Catalytic Activity, Catal. Today, 2009, 144(3–4), 212–218, DOI:10.1016/j.cattod.2009.02.042.
- P. Baláž, Mechanochemistry in Nanoscience and Minerals Engineering, Springer, 2008, pp. 103–132, DOI:10.1007/978-3-540-74855-7.
- S. Ostovar, A. Franco, A. R. Puente-Santiago, M. Pinilla-de Dios, D. Rodríguez-Padrón, H. R. Shaterian and R. Luque, Efficient Mechanochemical Bifunctional Nanocatalysts for the Conversion of Isoeugenol to Vanillin, Front. Chem., 2018, 6(APR), 1–7, DOI:10.3389/fchem.2018.00077.
- V. V. Molchanov and R. A. Buyanov, Mechanochemistry of Catalysts, Usp. Khim, 2000, 69(5), 490–493, DOI:10.1070/rc2000v069n05abeh000555.
- A. Gagrani, J. Zhou and T. Tsuzuki, Solvent Free Mechanochemical Synthesis of MnO2 for the Efficient Degradation of Rhodamine-B, Ceram. Int., 2018, 44(5), 4694–4698, DOI:10.1016/j.ceramint.2017.12.050.
- H. Liu and K. Zhao, Asymmetric Flow Electrochemical Capacitor with High Energy Densities Based on Birnessite-Type Manganese Oxide Nanosheets and Activated Carbon Slurries, J. Mater. Sci., 2016, 51(20), 9306–9313, DOI:10.1007/s10853-016-0177-0.
- A. Iyer, P. Dutta and S. Suib, Water Oxidation Catalysis Using Amorphous Manganese Oxides, Octahedral Molecular Sieves (OMS-2), and Octahedral Layered (OL-1) Manganese Oxide Structures, J. Phys. Chem. C, 2012, 116, 6474–6483 CrossRef CAS.
- S. Anandan, B. Gnana Sundara Raj, G. J. Lee and J. J. Wu, Sonochemical Synthesis of Manganese(II) Hydroxide for Supercapacitor Applications, Mater. Res. Bull., 2013, 48(9), 3357–3361, DOI:10.1016/j.materresbull.2013.05.021.
- T. Prasad Yadav, R. Manohar Yadav and D. Pratap Singh, Mechanical Milling: A Top Down Approach for the Synthesis of Nanomaterials and Nanocomposites, Nanosci. Nanotechnol., 2012, 2(3), 22–48, DOI:10.5923/j.nn.20120203.01.
- T. N. Afonasenko, O. A. Bulavchenko, O. A. Knyazheva, O. N. Baklanova, T. I. Gulyaeva, M. V. Trenikhin, S. V. Tsybulya and P. G. Tsyrul’nikov, Effect of the Mechanical Activation of a Mixture of MnCO3·mMn(OH)2·nH2O and AlOOH as a Stage of the Preparation of a MnOx-Al2O3 Catalyst on Its Phase Composition and Catalytic Activity in CO Oxidation, Kinet. Catal., 2015, 56(3), 359–368, DOI:10.1134/S0023158415030015.
- O. A. Bulavchenko, T. N. Afonasenko, P. G. Tsyrul’nikov, O. A. Knyazheva, O. N. Baklanova and S. V. Tsybulya, MnOx-Al2O3 Catalysts for Deep Oxidation Prepared with the Use of Mechanochemical Activation: The Effect of Synthesis Conditions on the Phase Composition and Catalytic Properties, Kinet. Catal., 2014, 55(5), 639–648, DOI:10.1134/S0023158414050048.
- V. V. Molchanov, V. V. Goidin, R. A. Buyanov, A. V. Tkachev and A. I. Lukashevich, Mechano-Chemical Reactions at High Pressure of the Gas Phase, Chemistry, 2002, 133–140 Search PubMed.
-
Z. Sawlowicz, I. V. Bacherikova and S. M. Shcherbakov, The Effect of Mechanochemical and Ultrasonic Treatments CeO2–MoO3 = 1:1, Nanocomposites, Nanostrcutures and their application, Springer, 2019, ch. 8 Search PubMed.
- R. A. Buyanov, V. V. Molchanov and V. V. Boldyrev, Mechanochemical Activation for Resolving the Problems of Catalysis, KONA Powder Part. J., 2009, 27(27), 38–54, DOI:10.14356/kona.2009007.
- Z. J. Jiang, Z. H. Li, J. B. Yu and W. K. Su, Liquid-Assisted Grinding Accelerating: Suzuki-Miyaura Reaction of Aryl Chlorides under High-Speed Ball-Milling Conditions, J. Org. Chem., 2016, 81(20), 10049–10055, DOI:10.1021/acs.joc.6b01938.
- S. Zhuang, E. S. Lee, L. Lei, B. B. Nunna, L. Kuang and W. Zhang, Synthesis of Nitrogen-Doped Graphene Catalyst by High-Energy Wet Ball Milling for Electrochemical Systems, Int. J. Energy Res., 2016, 40(15), 2136–2149, DOI:10.1002/er.3595.
- V. Šepeĺak, Nanocrystalline Materials Prepared by Homogeneous and Heterogeneous Mechanochemical Reactions, Ann. Chim. Sci. des Mater., 2002, 27(6), 61–76, DOI:10.1016/S0151-9107(02)90015-2.
- V. V. Molchanov and R. A. Buyanov, Scientific Grounds for the Application of Mechanochemistry to Catalyst Preparation, Kinet. Catal., 2001, 42(3), 406–415, DOI:10.1023/A:1010465315877.
- A. Sápi, D. G. Dobó, D. Sebok, G. Halasi, K. L. Juhász, A. Szamosvölgyi, P. Pusztai, E. Varga, I. Kálomista and G. Galbács, et al., Silica Based Catalyst Supports Are Inert, Aren’t They? – Striking Differences in Ethanol Decomposition Reaction Originated from Meso- and Surface Fine Structure Evidenced by Small Angle X-Ray Scattering, J. Phys. Chem. C, 2017, 121(9), 5130–5136, DOI:10.1021/acs.jpcc.7b00034.
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, XPS Determination of Mn Oxidation States in Mn (Hydr)Oxides, Appl. Surf. Sci., 2016, 366, 475–485, DOI:10.1016/j.apsusc.2015.12.159.
- A. Sápi, G. Halasi, J. Kiss, D. G. Dobó, K. L. Juhász, V. J. Kolcsár, Z. Ferencz, G. Vári, V. Matolin and A. Erdöhelyi, et al., In Situ DRIFTS and NAP-XPS Exploration of the Complexity of CO2 Hydrogenation over Size-Controlled Pt Nanoparticles Supported on Mesoporous NiO, J. Phys. Chem. C, 2018, 122(10), 5553–5565, DOI:10.1021/acs.jpcc.8b00061.
- N. Burgio, A. Iasonna, M. Magini, S. Martelli and F. Padella, Mechanical Alloying of the Fe-Zr System. Correlation between Input Energy and End Products, Nuovo Cimento D, 1991, 13(4), 459–476, DOI:10.1007/BF02452130.
- G. Kozma, R. Puskás, I. Z. Papp, P. Bélteky and Z. Kónya, Kukovecz. Experimental Validation of the Burgio-Rojac Model of Planetary Ball Milling by the Length Control of Multiwall Carbon Nanotubes, Carbon, 2016, 105, 615–621, DOI:10.1016/j.carbon.2016.05.005.
- T. Rojac, M. Kosec, B. Malič and J. Holc, The Application of a Milling Map in the Mechanochemical Synthesis of Ceramic Oxides, J. Eur. Ceram. Soc., 2006, 26(16), 3711–3716, DOI:10.1016/j.jeurceramsoc.2005.11.013.
- J. Joardar, S. K. Pabi and B. S. Murty, Milling Criteria for the Synthesis of Nanocrystalline NiAl by Mechanical Alloying, J. Alloys Compd., 2007, 429(1–2), 204–210, DOI:10.1016/j.jallcom.2006.04.045.
- C. X. Wu, S. G. Zhu, J. Ma and M. L. Zhang, Synthesis and Formation Mechanisms of Nanocomposite WC-MgO Powders by High-Energy Reactive Milling, J. Alloys Compd., 2009, 478(1–2), 615–619, DOI:10.1016/j.jallcom.2008.11.100.
- S. Qian, M. A. Cheney, R. Jose, A. Banerjee, P. K. Bhowmik and J. M. Okoh, Synthesis and Characterization of Birnessite and Cryptomelane Nanostructures in Presence of Hoffmeister Anions, J. Nanomater., 2009, 940462, DOI:10.1155/2009/940462.
- J. Zhu and J. He, Facile Synthesis of Graphene-Wrapped Honeycomb MnO2 Nanospheres and Their Application in Supercapacitors, ACS Appl. Mater. Interfaces, 2012, 4(3), 1770–1776, DOI:10.1021/am3000165.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Fast and Reversible Surface Redox Reaction of Graphene-MnO2 Composites as Supercapacitor Electrodes, Carbon, 2010, 48(13), 3825–3833, DOI:10.1016/j.carbon.2010.06.047.
- M. Aghazadeh, A. Bahrami-Samani, D. Gharailou, M. G. Maragheh, M. R. Ganjali and P. Norouzi, Mn3O4 Nanorods with Secondary Plate-like Nanostructures; Preparation, Characterization and Application as High Performance Electrode Material in Supercapacitors, J. Mater. Sci.: Mater. Electron., 2016, 27(11), 11192–11200, DOI:10.1007/s10854-016-5239-1.
- M. Fang, X. Tan, M. Liu, S. Kang, X. Hu and L. Zhang, Low-Temperature Synthesis of Mn3O4 Hollow-Tetrakaidecahedrons and Their Application in Electrochemical Capacitors, CrystEngComm, 2011, 13(15), 4915–4920, 10.1039/c1ce05337j.
- C. Julien, M. Massot, R. Baddour-Hadjean, S. Franger, S. Bach and J. P. Pereira-Ramos, Raman Spectra of Birnessite Manganese Dioxides, Solid State Ionics, 2003, 159(3–4), 345–356, DOI:10.1016/S0167-2738(03)00035-3.
- C. M. Julien, M. Massot and C. Poinsignon, Lattice Vibrations of Manganese Oxides: Part I. Periodic Structures, Spectrochim. Acta, Part A, 2004, 60(3), 689–700, DOI:10.1016/S1386-1425(03)00279-8.
- H. Liu, H. Zhang, L. Shi, X. Hai and J. Ye, Lattice Oxygen Assisted Room-Temperature Catalytic Process: Secondary Alcohol Dehydrogenation over Au/Birnessite Photocatalyst, Appl. Catal., A, 2016, 521, 149–153, DOI:10.1016/j.apcata.2015.10.027.
- S. Chakrabarti, B. K. Dutta and R. Apak, Active Manganese Oxide: A Novel Adsorbent for Treatment of Wastewater Containing Azo Dye, Water Sci. Technol., 2009, 60(12), 3017–3024, DOI:10.2166/wst.2009.758.
- M. A. Cheney, P. K. Bhowmik, S. Qian, S. W. Joo, W. Hou and J. M. Okoh, A New Method of Synthesizing Black Birnessite Nanoparticles: From Brown to Black Birnessite with Nanostructures, J. Nanomater., 2008, 1, 1–8, DOI:10.1155/2008/763706.
- Q. Feng, K. Yanagisawa and N. Yamasaki, Synthesis of Birnessite-Type Potassium Manganese Oxide, J. Mater. Sci. Lett., 1997, 6(3), 5–7, DOI:10.1023/A:1018577523676.
- A. I. Zaitsev, M. A. Zemchenko and B. M. Mogutnov, Thermodynamic Properties of Manganese Silicides, Zh. Fiz. Khim., 1989, 1451–1458 CAS.
- A. M. El-Sawy, C. K. King’ondu, C.-H. Kuo, D. A. Kriz, C. J. Guild, Y. Meng, S. J. Frueh, S. Dharmarathna, S. N. Ehrlich and S. L. Suib, X-Ray Absorption Spectroscopic Study of a Highly Thermally Stable Manganese Oxide Octahedral Molecular Sieve (OMS-2) with High Oxygen Reduction Reaction Activity, Chem. Mater., 2014, 26(19), 5752–5760, DOI:10.1021/cm5028783.
- K. T. Jacob, A. Kumar, G. Rajitha and Y. Waseda, Thermodynamic Data for Mn3O4, Mn2O3 and MnO2, High Temp. Mater. Processes, 2011, 30(4), 459–472, DOI:10.1515/HTMP.2011.069.
- W. Y. Hernández, M. A. Centeno, S. Ivanova, P. Eloy, E. M. Gaigneaux and J. A. Odriozola, Cu-Modified Cryptomelane Oxide as Active Catalyst for CO Oxidation Reactions, Appl. Catal., B, 2012, 123–124, 27–35, DOI:10.1016/j.apcatb.2012.04.024.
- C. González, J. I. Gutiérrez, J. R. González-Velasco, A. Cid, A. Arranz and J. F. Arranz, Application of Differential Scanning Calorimetry to the Reduction of Several Manganese Oxides, J. Therm. Anal. Calorim., 1998, 985–989, DOI:10.1023/A:1010132606114.
- Q. Xue and Q. Zhang, Agar Hydrogel Template Synthesis of Mn3O4 Nanoparticles through an Ion Diffusion Method Controlled by Ion Exchange Membrane and Electrochemical Performance, Nanomaterials, 2019, 9(4), 503, DOI:10.3390/nano9040503.
- K. Stangeland, D. Y. Kalai, Y. Ding and Z. Yu, Mesoporous Manganese-Cobalt Oxide Spinel Catalysts for CO2 Hydrogenation to Methanol, J. CO2 Util., 2019, 32(January), 146–154, DOI:10.1016/j.jcou.2019.04.018.
- A. Sápi, G. Halasi, A. Grósz, J. Kiss, A. Kéri, G. Ballai, G. Galbács, Á. Kukovecz and Z. Kónya, Designed Pt Promoted 3D Mesoporous Co3O4 Catalyst in CO2 Hydrogenation, J. Nanosci. Nanotechnol., 2018, 19(1), 436–441, DOI:10.1166/jnn.2019.15779.
- S. Kattel, P. Liu and J. G. Chen, Tuning Selectivity of CO2 Hydrogenation Reactions at the Metal/Oxide Interface, J. Am. Chem. Soc., 2017, 139(29), 9739–9754, DOI:10.1021/jacs.7b05362.
- Q. Liu, Y. Han, J. Cai, E. J. Crumlin, Y. Li and Z. Liu, CO2 Activation on Cobalt Surface in the Presence of – H2O: An Ambient-Pressure X-Ray Photoelectron Spectroscopy Study, Catal. Lett., 2018, 148(6), 1686–1691, DOI:10.1007/s10562-018-2362-z.
- D. R. J. Kolling, N. Cox, G. M. Ananyev, R. J. Pace and G. C. Dismukes, What Are the Oxidation States of Manganese Required To Catalyze Photosynthetic Water Oxidation?, Biophys. J., 2012, 103, 313–322, DOI:10.1016/j.bpj.2012.05.031.
- G. Varga, A. Sápi, T. Varga, K. Baán, I. Szenti, G. Halasi, R. Mucsi, L. Óvári, J. Kiss and Z. Fogarassy, et al., Ambient Pressure CO2 Hydrogenation over a Cobalt/Manganese-Oxide Nanostructured Interface: A Combined in Situ and Ex Situ Study, J. Catal., 2020, 386, 70–80, DOI:10.1016/j.jcat.2020.03.028.
- J. Gómez-Pérez, D. G. Dobó, K. L. Juhász, A. Sápi, H. Haspel, Á. Kukovecz and Z. Kónya, Photoelectrical Response of Mesoporous Nickel Oxide Decorated with Size Controlled Platinum Nanoparticles under Argon and Oxygen Gas, Catal. Today, 2017, 284, 37–43, DOI:10.1016/j.cattod.2016.10.026.
- H. Wang, K. An, A. Sapi, F. Liu and G. A. Somorjai, Effects of Nanoparticle Size and Metal/Support Interactions in Pt-Catalyzed Methanol Oxidation Reactions in Gas and Liquid Phases, Catal. Lett., 2014, 144(11), 1930–1938, DOI:10.1007/s10562-014-1347-9.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Molybdenum Carbide as Alternative Catalysts to Precious Metals for Highly Selective Reduction of CO2 to CO, Angew. Chem., Int. Ed., 2014, 53(26), 6705–6709, DOI:10.1002/anie.201404109.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. K. Willauer, Mn Doped Iron-Based CO2 Hydrogenation Catalysts: Detection of KAlH4 as Part of the Catalyst's Active Phase, Appl. Catal., A, 2010, 373(1–2), 112–121, DOI:10.1016/j.apcata.2009.11.005.
- H. Zhan, F. Li, C. Xin, N. Zhao, F. Xiao, W. Wei and Y. Sun, Performance of the La–Mn–Zn–Cu–O Based Perovskite Precursors for Methanol Synthesis from CO2 Hydrogenation, Catal. Lett., 2015, 145(5), 1177–1185, DOI:10.1007/s10562-015-1513-8.
- B. Liang, T. Sun, J. Ma, H. Duan, L. Li, X. Yang, Y. Zhang, X. Su, Y. Huang and T. Zhang, Mn Decorated Na/Fe Catalysts for CO2 Hydrogenation to Light Olefins, Catal. Sci. Technol., 2019, 9(2), 456–464, 10.1039/c8cy02275e.
- S. S. Kim, K. H. Park and S. C. Hong, A Study of the Selectivity of the Reverse Water-Gas-Shift Reaction over Pt/TiO2 Catalysts, Fuel Process. Technol., 2013, 108, 47–54, DOI:10.1016/j.fuproc.2012.04.003.
- R. E. Owen, D. Mattia, P. Plucinski and M. D. Jones, Kinetics of CO2 Hydrogenation to Hydrocarbons over Iron–Silica Catalysts, ChemPhysChem, 2017, 18(22), 3211–3218, DOI:10.1002/cphc.201700422.
- Z. J. Liu, X. J. Tang, S. Xu and X. L. Wang, Synthesis and Catalytic Performance of Graphene Modified CuO–ZnO–Al2O3 for CO2 Hydrogenation to Methanol, J. Nanomater., 2014, 690514, DOI:10.1155/2014/690514.
- P. A. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catalytic CO2 Valorization into CH4 on Ni-Based Ceria-Zirconia. Reaction Mechanism by Operando IR Spectroscopy, Catal. Today, 2013, 215, 201–207, DOI:10.1016/j.cattod.2013.02.019.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the characterization data. See DOI: 10.1039/d0cp01855d |
| This journal is © the Owner Societies 2020 |