
Coexistence of three types of sodium motion in double molybdate Na9Sc(MoO4)6: 23Na and 45Sc NMR data and ab initio calculations
Anton L.
Buzlukov
 *a,
Irina Yu.
Arapova
a,
Yana V.
Baklanova
b,
Nadezhda I.
Medvedeva
b,
Tatiana A.
Denisova
b,
Aleksandra A.
Savina
cd,
Bogdan I.
Lazoryak
e,
Elena G.
Khaikina
d and
Michel
Bardet
f
*a,
Irina Yu.
Arapova
a,
Yana V.
Baklanova
b,
Nadezhda I.
Medvedeva
b,
Tatiana A.
Denisova
b,
Aleksandra A.
Savina
cd,
Bogdan I.
Lazoryak
e,
Elena G.
Khaikina
d and
Michel
Bardet
f
aInstitute of Metal Physics, Ural Branch, Russian Academy of Science, S. Kovalevskaya St. 18, Ekaterinburg 620137, Russia. E-mail: buzlukov@imp.uran.ru; buzlukov@mail.ru; Fax: +7-343-3745244; Tel: +7-343-3783839
bInstitute of Solid State Chemistry, Ural Branch, Russian Academy of Science, Pervomayskaya St. 91, Ekaterinburg 620990, Russia
cSkolkovo Institute of Science and Technology, Moscow 121205, Russia
dBaikal Institute of Nature Management, Siberian Branch, Russian Academy of Sciences, Sakh’yanova St. 6, Ulan-Ude 670047, Buryat Republic, Russia
eDepartment of Chemistry, Lomonosov Moscow State University, Leninskie Gory 1, Moscow 119899, Russia
fUniv. Grenoble Alpes, CEA, IRIG-MEM, LRM, 38000, Grenoble, France
First published on 26th November 2019
Abstract
The rechargeable Na-ion batteries attract much attention as an alternative to the widely used but expensive Li-ion batteries. The search for materials with high sodium diffusion is important for the development of solid state electrolytes. We present the results of experimental and ab initio studies of the Na-ion diffusion mechanism in Na9Sc(MoO4)6. The ion conductivity reaches the value of 3.6 × 10−2 S cm−1 at T ∼ 850 K. The 23Na and 45Sc NMR data reveal the coexistence of three different types of Na-ion motion in the temperature range from 300 to 750 K. They are activated at different temperatures and are characterized by substantially different dynamics parameters. These features are confirmed by ab initio calculations of activation barriers for sodium diffusion along various paths.
1. Introduction
The renewal of interest in Na-conducting materials, observed in recent years, is mainly due to the wide availability and low price of sodium. Na-ion batteries can compete with lithium-ion batteries in areas where the mass and energy density are not very critical, for example, for large-scale storage of renewable energy sources such as sun, wind, etc.1–8 Despite the similar electrochemistry for Li and Na, sodium devices have a lower operating voltage and specific capacity than lithium batteries. To compensate for these effects, a viable strategy is to use polymer or inorganic solid electrolytes instead of conventional organic materials. An all-solid battery makes it possible to increase both the battery power and equipment safety due to the thermal stability and the absence of leaks. Besides the well-known sodium-conducting materials based on Na3PS4,9–14 β-Al2O315–18 and the NASICON-related phosphates,19–24 which have extremely high Na-ion transport characteristics, other compounds with an “open” crystal structures are also promising. In particular, a sufficiently high Na-ion mobility has been observed in the double molybdates containing both alkali and di- or trivalent cations. In the Na2MoO4–R2(MoO4)3 systems the NaxRy(MoO4)(x+3y)/2 molybdates are known to exist in a wide range of x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y ratios, 1:1, 5:1, 1:5, 3:1 and 9:125–28 and rich variety of structure types. The ionic conductivity in these compounds, σ > 10−3 S cm−1 at 670–770 K, and activation energy, Ea ∼ 0.6–0.8 eV29–34 are comparable with those for Na2+2xFe2−x(SO4)335 and exceed the corresponding values for isostructural orthophosphates Na2M3(PO4)3 with M3 = GaMn2, GaCd2, InMn2 and FeMnCd (which are characterized by σ ∼ 10−5 S cm−1 at 673 K and Ea = 0.7–0.8 eV).36
:y ratios, 1:1, 5:1, 1:5, 3:1 and 9:125–28 and rich variety of structure types. The ionic conductivity in these compounds, σ > 10−3 S cm−1 at 670–770 K, and activation energy, Ea ∼ 0.6–0.8 eV29–34 are comparable with those for Na2+2xFe2−x(SO4)335 and exceed the corresponding values for isostructural orthophosphates Na2M3(PO4)3 with M3 = GaMn2, GaCd2, InMn2 and FeMnCd (which are characterized by σ ∼ 10−5 S cm−1 at 673 K and Ea = 0.7–0.8 eV).36
Besides practical promise, these compounds are also interesting for fundamental studies of ion dynamics. In particular, our recent researches revealed a non-uniform sodium motion in Na9Al(MoO4)6.37 At T < 490 K it occurs exclusively through ion jumps within the sublattice of sodium sites located far from polyhedral [Al(MoO4)6]9− clusters, forming the structure framework. The activation energy for this type of motion Ea ≈ 0.55 eV. The ions located in the vicinity of [Al(MoO4)6]9− clusters are activated only at T > 490 K with Ea ≈ 0.8 eV. Strong correlation effects in sodium motion are present at low temperatures. They are manifested in a slowdown of ion diffusion and an artificial increase of the Ea value estimated from conductivity data.
A comparison of the electrophysical properties for the NASICON-type compounds19,38,39 shows that the substitution of the transition element can lead to a drastic change of the ionic conductivity (up to several orders of magnitude). In this regard, it was interesting to study the Na diffusion mechanisms in the compound related to Na9Al(MoO4)6: having similar local structure, but another trivalent cation. As such a related compound, Na9Sc(MoO4)6 was chosen. The basic structural units in Na9R(MoO4)6 (R = Al, Sc) are the isolated polyhedral [R(MoO4)6]9− clusters composed of the central RO6 octahedron sharing vertices with six MoO4 tetrahedra to form an open framework, where the Na+ cations are bound to three vertices of the MoO4 tetrahedra (see Fig. 1).
 | ||
| Fig. 1 Crystal structures of Na9Al(MoO4)6 (a and c) and Na9Sc(MoO4)6 (b and d) reproduced with program VESTA40 on structural data from ref. 37 and 41, respectively. Figures a and b represent the [Al/Sc(MoO4)6]9− clusters forming the structure framework and adjacent NaO6 octahedra. Figures c and d represent the Na-sites located in the structure cavities between the [Al/Sc(MoO4)6]9− polyhedra. | ||
The Na9Sc(MoO4)6 is crystallized in a structure with trigonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group.41 Replacing scandium (rVI = 0.74 Å42) with aluminum (rVI = 0.53 Å42) induces the monoclinic distortions (C/2c s.g.) and the “splitting” of sodium sites.37 As a result, there are five nonequivalent structural positions of sodium in Na9Al(MoO4)6, where Na1 and Na2 are located in the vicinity of AlO6 octahedra, and the Na3, Na4 and Na5 positions are placed in the cavities between the [Al(MoO4)6]9− polyhedra. Meanwhile, only three types of sodium positions are present in Na9Sc(MoO4)6: the Na1 ions are located near the ScO6 octahedra, and Na2 and Na3 ones are in between the [Sc(MoO4)6]9− clusters (Fig. 1). It has to be noted however that these structural changes during the Al → Sc substitution can only be detected by a detailed analysis of electronic diffraction, while the powder X-ray diffraction patterns can also be satisfactory fitted in trigonal R and/or R3c space groups.37 So, despite the apparent differences in the crystal structures presented on Fig. 1 they are really quite similar.
space group.41 Replacing scandium (rVI = 0.74 Å42) with aluminum (rVI = 0.53 Å42) induces the monoclinic distortions (C/2c s.g.) and the “splitting” of sodium sites.37 As a result, there are five nonequivalent structural positions of sodium in Na9Al(MoO4)6, where Na1 and Na2 are located in the vicinity of AlO6 octahedra, and the Na3, Na4 and Na5 positions are placed in the cavities between the [Al(MoO4)6]9− polyhedra. Meanwhile, only three types of sodium positions are present in Na9Sc(MoO4)6: the Na1 ions are located near the ScO6 octahedra, and Na2 and Na3 ones are in between the [Sc(MoO4)6]9− clusters (Fig. 1). It has to be noted however that these structural changes during the Al → Sc substitution can only be detected by a detailed analysis of electronic diffraction, while the powder X-ray diffraction patterns can also be satisfactory fitted in trigonal R and/or R3c space groups.37 So, despite the apparent differences in the crystal structures presented on Fig. 1 they are really quite similar.
Here we present the results of detailed studies of the sodium diffusion mechanisms in Na9Sc(MoO4)6 performed at the “atomic level” using the NMR methods and DFT calculations. Differences in the sodium dynamics in comparison with Na9Al(MoO4)6 are discussed.
2. Experimental
2.1. Synthesis
Na9Sc(MoO4)6 was synthesized by annealing of stoichiometric mixtures of Na2MoO4 and Sc2(MoO4)3.41 The sintering temperature was raised from 773 to 873 K in steps of 20°, each temperature being held for 12 h, with intermediate grindings. For grinding the furnace was switched off and cooled down to room temperature. At final step the sample was taken out of the hot furnace. A stoichiometric mixtures of Sc2O3 and MoO3 was used for synthesis of simple molybdate Sc2(MoO4)3 in two stages (573–723 K for 25–40 h and 1023 K for 60 h). Anhydrous Na2MoO4 was obtained by calcination of the corresponding crystalline dihydrate at 823–873 K. The prepared compounds have been checked by using JCPDS PDF-2 Data Base and do not contain reflections of initial or foreign phases.2.2. Characterization
The PXRD patterns were obtained by using a D8 ADVANCE Bruker diffractometer (CuKα radiation, λ = 1.5418 Å, reflection geometry). The PXRD data were collected at RT over the 7°–100° 2θ range with steps of 0.02076°. To determine the lattice parameters, the Le Bail decomposition was carried out using the JANA2006 software. Thermoanalytic studies were carried out with a STA 449 F1 Jupiter NETZSCH thermoanalyser (Pt crucible, heating rate of 10 degree min−1 in Ar stream). Ceramic disks for conductivity measurements were prepared by pressing of powders at 1 kbar and sintering at 863 K for 4 h. The densities of the obtained pellets (measured by the Archimedes method using kerosene as a saturant) were typically 90–95% of theoretical ones. The disks had a diameter of 7–8 mm and a thickness of 1–2 mm, they were electroded by painting of colloidal platinum on their large surfaces with subsequent annealing for one hour at 843 K. Electrical conductivity of the samples was determined by impedance spectroscopy in the temperature range of 343–843 K at heating and cooling rates of 2 K min−1 using two-probe measurements in a NorECs ProboStat cell. The signal was monitored with a Novocontrol Beta-N impedance analyzer at applied voltage of 0.5 V at selected frequencies in the range of 0.3 Hz–1 MHz.2.3. NMR
The static NMR experiments were performed over the temperature range 300–750 K. The experimental data were obtained on an AVANCE III 500WB BRUKER spectrometer in an external magnetic field of 11.74 T. A commercial high-temperature wide-line NMR probe (Bruker Biospin GmbH), including a Pt-wired rf coil and nonmagnetic heater was used to heat a sample in static air atmosphere. The sample was tightly packed inside an open quartz ampule throughout NMR measurements. The static spectra for 23Na (nuclear spin 23I = 3/2, quadrupole moment 23Q = 0.108 barn) were acquired by Fourier transform of both the free induction decay: τp-acq, and, in some cases, the spin echo signals: τp-tdel-2τp-tdel-acq. The pulse duration was in all cases, τp = 2 μs, corresponding to the nuclear magnetization tip angle, θ ∼ 60°. The spin–lattice relaxation times for the 23Na nuclei were measured by the “inversion-recovery” technique: 2τp-tdel-τp-acq. The 45Sc (nuclear spin 45I = 7/2, quadrupole moment 45Q = −0.22 barn) static NMR spectra were acquired by Fourier transform of free induction decay with pulse duration equal to 3 μs.The 23Na MAS NMR spectra were obtained with room temperature bearing gas by using standard Bruker MAS NMR probeheads with 1.3 mm (AVANCE DSX 200 NMR spectrometer, external magnetic field 4.7 T) and 3.2 mm rotors (AVANCE III 500WB spectrometer, 11.7 T), and standard Agilent 4.0 mm MAS Probehead (AGILENT VNMR 400WB spectrometer, 9.4 T).
As for the static regime, the 23Na MAS NMR spectra were acquired by Fourier transform of free induction decay and/or spin echo signals with exciting pulse equal to 2 μs. The spectra deconvolution was performed by using the DMFit program.43
2.4. Computational details
The density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP)44,45 within the projector-augmented wave (PAW) method.46 The exchange–correlation functional was described by the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE).47 The convergence criterion for the total energy was 0.01 meV within a 400 eV cutoff. Integration in the Brillouin zone was done with a mesh of 4 × 4 × 4 irreducible k-points according to the Monkhorst–Pack scheme.48 The lattice parameters were fixed at the experimental values,41 whereas the atomic positions were relaxed via a conjugate gradient algorithm until the forces on all unconstrained atoms were less than 0.01 eV Å−1.The tensors of electric field gradient (EFG) at Na nuclei in different positions were calculated using the method of ref. 49. After diagonalization of the EFG tensor, the principal components Vxx, Vyy and Vzz are chosen as |Vzz| > |Vyy| > |Vxx|. The largest principal component Vzz determines the quadrupole frequency νQ ≡ ωQ/2π = 3eQVzz/2I(2I − 1)h and asymmetry parameter ηQ = (|Vyy| − |Vxx|)/|Vzz|. The values of νQ and η were used for an assignment of the experimental NMR lines to the specific sodium sites.
The nudged elastic band (NEB) method was employed to calculate the energy barriers for Na-ion migration. We considered the diffusion mechanism through migration of single vacancy between two neighbor positions of Na ion. For every path, the total energies were calculated for a few intermediate images. The relaxed maximal energy and coordinates during the sodium migration provided the activation barrier and diffusion pathway between the starting and end states, respectively.
3. Results and discussion
3.1. The characterization of Na9Sc(MoO4)4
The Na9Sc(MoO4)6 sample has a white color and melts at 946 K.The determination of unit cell parameters from PXRD patterns using Le Bail decomposition revealed that all reflections could be indexed in the R space group with a = 15.0047(1) Å, c = 19.1891(1) Å; values of structural R-factors: RP = 3.10% and RwP = 4.37% (Fig. 2). It verifies good phase purity of the powder prepared by solid state synthesis.
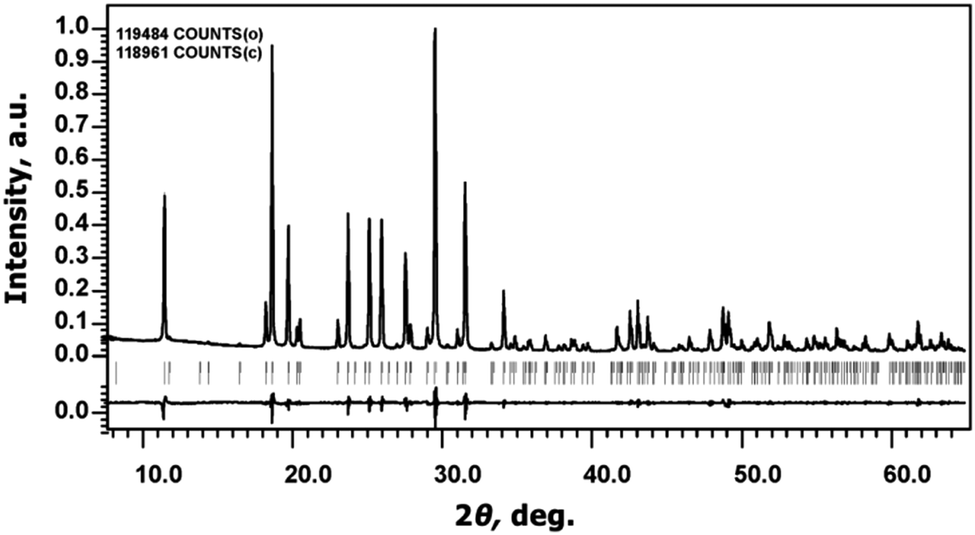 | ||
| Fig. 2 The experimental, calculated, and difference powder X-ray diffraction patterns of Na9Sc(MoO4)6. | ||
3.2. Electric conductivity measurements
The temperature dependence of the conductivity is shown in Fig. 3 in Arrhenius log(σT) − (103/T) coordinates. Conductivity values are given for the impedance frequency 100 kHz since at this frequency the real part of the impedance practically independent on the AC frequencies and corresponds to the bulk resistance of the sample. The temperature dependency of the conductivity can be divided into two approximately linear portions with different slopes. The activation energies for these linear portions are LTEconda ≈ 0.86 eV for the region below 550 K and HTEconda ≈ 0.98 eV for the high-temperature one. Conductivity values for Na9Sc(MoO4)6 are as high as 1.6 × 10−4 S cm−1 at 573 K and 3.63 × 10−2 S cm−1 at 843 K. | ||
| Fig. 3 Temperature dependence of the conductivity measured for Na9Sc(MoO4)6 in the range 343–843 K. The inset shows the Nyquist plot at 573 K. | ||
3.3. 23Na NMR spectra at room temperature: assignment of the observed NMR signals to the distinct Na positions
The Fig. 4 shows the 23Na NMR spectral lines obtained for Na9Sc(MoO4)6 in external magnetic fields 11.7 T (Larmor frequency 23Na, ν0 ≡ ω0/2π = 132.29 MHz) and 4.7 T (ω0/2π = 52.94 MHz). Similar NMR spectra were also recorded in the 9.4 T field (ν0 = 105.82 MHz, not shown here). | ||
| Fig. 4 23Na NMR spectra obtained for Na9Sc(MoO4)6 in magnetic fields 11.7 (a and b) and 4.7 (c and d) T in static and MAS modes. The color lines represent the results of spectra deconvolution (see text). | ||
As can be seen a decrease of the external magnetic field strength leads to a substantial broadening of NMR spectrum. Similar effects can be expected when the main factor determining the shape of NMR spectrum is quadrupole interaction. Nuclei with a spin I > 1/2 have a non-spherical charge distribution in the nucleus. This leads to the appearance of a quadrupole moment that interacts with the Electric Field Gradient (EFG) which in its turn is determined by the features of local environment.50 The influence of quadrupole interaction is usually described as small corrections to the Zeeman energy within the framework of the perturbation theory. In strong magnetic fields or for nuclei with low Q (such as the 7Li), only the first-order effects are usually observed which are manifested in a characteristic splitting of NMR spectrum and appearance (in the case of 23Na with I = 3/2) of three lines: one line corresponding to the central transition, mI = −1/2 ↔ +1/2, and two satellite lines (mI = ±3/2 ↔ ±1/2). For a powder sample, the peaks of satellites are shifted relative to the position of central line at a distance ±1/2νQ(1 − ηQ). In weaker magnetic fields or for nuclei with high Q an important role can be played also by the second-order effects those are manifested in the splitting of central line. The second order splitting is proportional to square quadrupole frequency and inversely proportional to the resonance frequency: ν(2)Q ∼ νQ2/ν0.50,51
Interestingly, three 23Na NMR spectra on Fig. 4: static and MAS spectra acquired at ν0 = 52.94 MHz and MAS spectrum measured at ν0 = 132.29 MHz (panel c, d and b, respectively) can be fitted simultaneously taking into account only the second-order quadrupole effects. This “Quad2nd” model assumes the presence of two spectral components with νQ = 1350 ± 50 and 620 ± 30 kHz; and ηQ = 0.30 ± 0.05 and 0.70 ± 0.05 for Line-1 (red line) and Line-2 (blue one), respectively. Nevertheless, the simulation of static spectrum recorded in 11.7 T yields the result far from ideal with the same fitting parameters (see panel a). It suggests that the situation is more complicated and besides quadrupolar interaction there are another factors affecting the 23Na NMR spectrum in Na9Sc(MoO4)6. This interaction is directly proportional to the magnetic field strength and substantially averaged even at low MAS speeds. The most probable candidate is a chemical shift anisotropy (CSA), reflecting the anisotropic shielding of Na nuclei by surrounding electrons. The parameters of the anisotropy, Δδ, and the asymmetry, ηCS, of chemical shift are determined by the corresponding tensor components: Δδ = δ33 − δiso, ηCS = |δ22 − δ11|/|δ33 − δiso|, where isotropic shift, δiso = (δ11 + δ22 + δ33)/3.52,53 The best fit is achieved with the values of Δδ equal approximately to 20 ppm and −5 ppm for lines 1 and 2, respectively. The parameter ηCS is found to be in the range of 0–0.3 for Line-1. For Line-2 it was difficult to estimate the ηCS value unambiguously since its influence on the line shape is rather weak. The “Quad2nd + CSA” model contains another set of parameters which affect the line shape: the Euler angles φ, χ and ψ (in DMFit43 notations) defining the orientation of principal axes of the EFG and CSA tensors. The best result is achieved at χ = 15 ± 5° for both lines (the changes of φ and ψ do not disturb visibly the line shape).
We assign the detected Line-1 and Line-2 to the 23Na nuclei in the Na1 sites and the Na2 + Na3 sites, respectively. The relative intensities for observed signals ((1)I = 0.30–0.35 and (2)I = 0.65–0.70) agree well with the occupancies of corresponding positions in the crystal structure.41 Moreover this assignment is consistent with the results of ab initio calculations. The calculated quadrupole frequencies νQ and asymmetry parameters ηQ at 23Na nuclei in Na9Sc(MoO4)6 are shown in Table 1.
| Site | ν Q (kHz) | η Q |
|---|---|---|
| Na1 | 1798 | 0.36 |
| Na2 | 661 | 0.61 |
| Na3 | 745 | 0.77 |
As seen, the quadrupole frequencies and asymmetry parameters for Na2 and Na3 sites are very close to each other and we can expect the appearance of two NMR lines with νQ ∼ 1800 and 700 kHz; and ηQ ∼0.4 and 0.7 for Na9Sc(MoO4)6. The relative intensities of these two lines should be (1)I = 0.33, (2)I = 0.67 as follows from the occupancies of different Na sites.41
3.4. Temperature behavior of the 23Na and 45Sc NMR spectra: mechanisms of the long-range sodium diffusion
The Fig. 5 shows the evolution of the 23Na NMR spectra in the Na9Sc(MoO4)6 in temperature range 300–750 K. The Fig. 6 represents the temperature dependencies of characteristic parameters for two spectral components observed in Na9Sc(MoO4)6. The data on central transition linewidth, Δν (a), quadrupole frequency, νQ (b), EFG tensor asymmetry, ηQ (c), chemical shift anisotropy, Δδ (d), isotropic shift, δiso (e) and the relative intensities for two spectral components (f) are present. To reduce the number of fitting parameters the ηCS values as well as the φ and ψ angles were fixed at zero for all temperatures and for both spectral components, χ = 15 ± 5°. | ||
| Fig. 5 23Na NMR spectra for Na9Sc(MoO4)6 acquired in static mode at resonance frequency ω0/2π = 132.29 MHz in temperature range 300–750 K. | ||
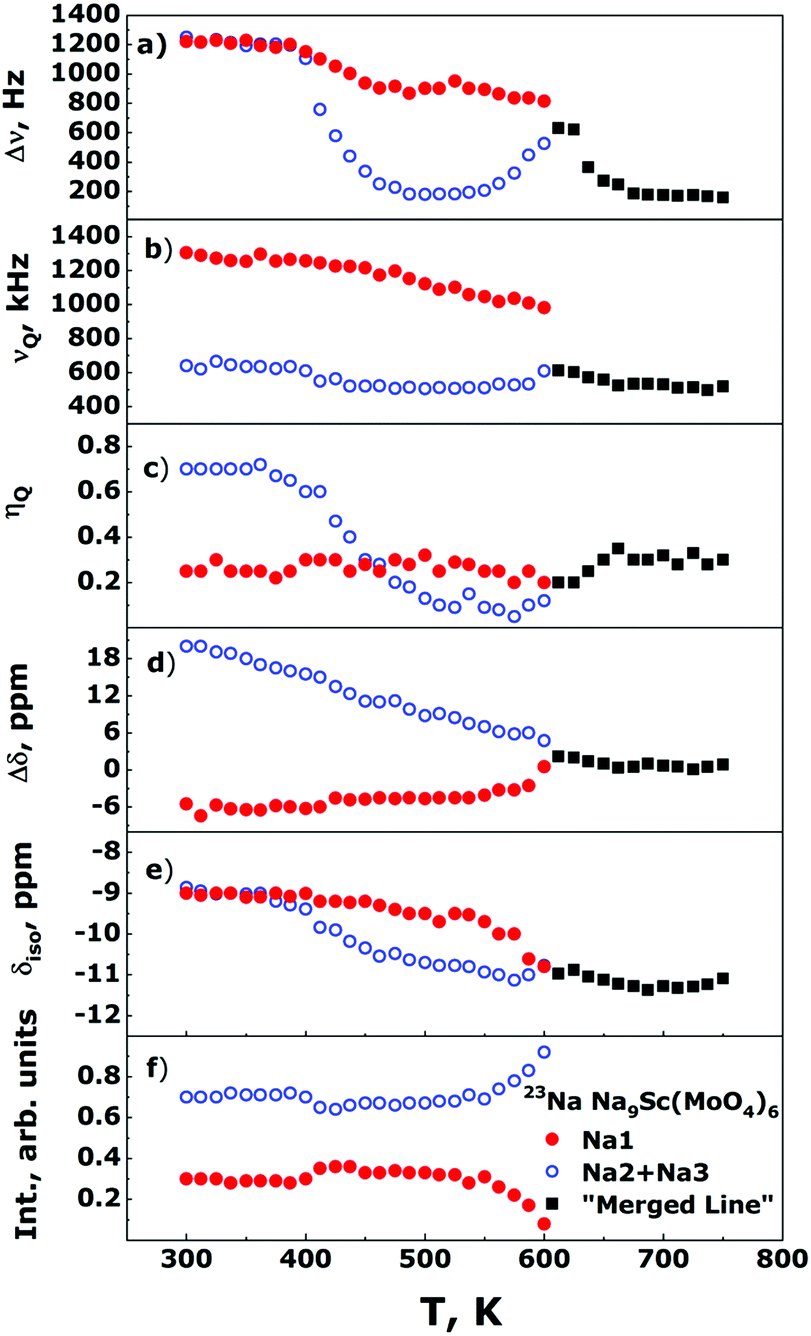 | ||
| Fig. 6 Temperature behavior of the 23Na NMR spectra parameters for Na9Sc(MoO4)6. Filled red and open blue circles correspond to NMR signals, attributed to Na1 and Na2 + Na3 ions, respectively. Black squares represent the corresponding parameters after the “merging” of two NMR components (see text). The data on dipolar central linewidth, Δν (a), quadrupole frequency, νQ (b), EFG tensor asymmetry, ηQ (c), chemical shift anisotropy, Δδ (d), isotropic shift, δiso (e), and the relative intensities for two spectral components (f) are present. | ||
The temperature dependencies of NMR spectra parameters are qualitatively similar to those observed earlier for the related molybdate Na9Al(MoO4)637 and indicate the presence of rather fast Na diffusion in the Na9Sc(MoO4)6. It should be noted however that all “motional” effects in Sc-containing compound are observed at higher temperatures compared to those for Na9Al(MoO4)6 that implies the slower ion diffusion in Na9Sc(MoO4)6. At temperatures above 390 K, the Δν value for a signal corresponding to Na2 + Na3 ions (open blue circles in Fig. 6a) sharply decreases. Such a Δν(T) dependence is typical for the materials with fast ion diffusion.50,54 The Δν value is affected mainly by the internuclear dipolar interaction. For a pair of interacting nuclei, it depends on the distance between nuclei and the orientation of this pair with respect to the external magnetic field. Atomic jumps lead to changes in both the distances and the orientations. As a result, with increasing temperature and growing ion jump frequency, the dipolar interaction is averaged and a sharp decrease of Δν is expected. The weak decrease of Δν for Na1 ions (filled red circles in Fig. 6a) can be explained as follows: for mobile Na2/Na3 ions themselves, the dipolar interaction is fully averaged (excluding some non-averaged part due to inhomogeneity of the external magnetic field etc.). Meanwhile the ions jumps in the Na2/Na3 sublattice affect only weakly the line width of static nuclei in Na1 sites, because for them the Δν is determined mainly by the 23Na–45Sc interaction (indeed, the nearest neighbor for Na1 is the Sc at a distance r ≈ 3.36 Å). At T > 550 K, the Δν for Na2/Na3 ions increases again. Such a Δν(T) dependence is reminiscent of that predicted for “classical” chemical exchange between two non-equivalent sites: with temperature increasing the NMR signals corresponding to different sites broaden at first, then they are merged into one wide line, then a dynamic narrowing of the merged line is expected.50 The parameters of this “merged” line are shown as black squares in Fig. 6. Thus, the experimental data on Δν can be interpreted within the next scenario: at 400 < T < 550 K the Na+ ions motion occurs exclusively within the Na2/Na3 positions which form the continuous paths for long-range ion diffusion, while sodium in Na1 sites remains static (on the NMR frequency scale). At T > 550 K the Na1 ions also start to participate in the diffusion processes through the Na1 ↔ Na2/Na3 jumps. It has to be noted that the latter assumption is in agreement with structure considerations: the Na1 ↔ Na2/Na3 jump it is only one possibility for Na1 because direct jump Na1 ↔ Na1 seems to be quite improbable due to too long jump length (rNa1–Na1 ≈ 5.83 Å).
Temperature behavior of νQ for Na2/Na3 ions is consistent with the proposed picture of ion motion. As is expected,50,55,56 besides the averaging of dipole–dipole interaction the ion jumps in the Na2/Na3 sublattice induce the changes of the EFG tensor components, Vij, for the corresponding NMR signal, those are reflected in the changes of νQ and ηQ values at T > 390 K (see Fig. 6b and c). Meanwhile only monotonous changes of the EFG parameters for spectral component corresponding to Na1 ions are observed at 300 < T < 550 K. Nevertheless, there are some substantial deviations from “classical” behavior expected at two-site exchange. In particular, instead of characteristic coalescence of two spectral components,55,57–60 at T > 550 K (i.e., with the beginning of Na1 ions motion) the corresponding signal sharply decreases. As a result, this line completely disappears at T ≈ 600 K. The changes of line intensity can be explained by a natural assumption that Na1 ions are involved in the motional process gradually with increasing temperature, thus the decreasing signal corresponds to ions still retained in the Na1 sites (whose fraction decreases with temperature). Concerning the absence of spectra coalescence, we have to take into account that the chemical exchange Na1 ↔ Na2/Na3 can not occur directly, but only through intermediate tetrahedral position Naint. For these “two-steps” jumps, Na1 ↔ Naint + Naint ↔ Na2/Na3, the NMR spectrum transformation can significantly differ from that expected at direct Na1 ↔ Na2/Na3 hopping. It has to be noted that some approaches allowing to predict the NMR spectrum shape at two-site exchange have been developed recently (see for example ref. 61–65). However, even for the simplest cases, they require rather complicated numerical calculations. In our case they already can hardly be applied, moreover, if we take into account that the probabilities for the “first step” (Na1 ↔ Naint) jump and for the “second step” (Naint ↔ Na2/Na3) one can be rather different.
Another possible explanation for the disappearance of Line-1 could be the presence of some phase transition at 550–600 K resulting in that the local surrounding of sodium ions in Na1 sites becomes similar to that for Na2/Na3. To trace possible structure transformations we performed also the 45Sc NMR spectra measurements in the temperature range 300–750 K. In contrast to the Na sites (18f in R structure) the point symmetry of the Sc site (6c) contains a 3-fold axis. It applies the strict constraints on the EFG and CSA tensors: they must have axial symmetry (i.e., ηCS = ηQ = 0) and, in addition, their principal axes must be collinear to this 3-fold axis (i.e., φ = χ = ψ = 0°). However, with these constraints we could not find the νQ and Δδ values allowing to fit the experimental spectra. The better result can be achieved either with the parameters φ = χ = ψ = 0° and ηQ = 0, ηCS = 1, or with ηCS = ηQ = 0 and φ = ψ = 0°, χ = 90°. Although the point symmetry plays as it is considered a decisive role, the deviations in both the orientations of principal axes and the axiality of CSA and EFG tensors have been observed in some cases (see, for example, ref. 66 and 67). On the one hand, local symmetry can be distorted due to deviations of the bond lengths/angles from their ideal values. On another hand the electron shielding and especially EFG on the nucleus-probe can be strongly affected by the second and even third coordination spheres. The environment of Sc3+ ions in Na9Sc(MoO4)6 is characterized by the “isotropic” first coordination sphere (containing 3 × O2− ions at a distance r = 2.09 Å and 3 × O2− at r = 2.10 Å), and rather “anisotropic” second one with 3 × Mo6+ (r = 3.49 Å), 3 × Mo6+ (r = 3.66 Å), and 1 × Na1+ (r = 3.37 Å). Thus, as a tentative hypothesis we can infer that the observed features are determined by these peculiarities of local Sc surrounding. Nevertheless we leave a detailed discussion of this issue beyond the scope of present article. It is rather a question of “NMR crystallography” and other studies are required to clarify it.
The Fig. 7a represents the results of 45Sc NMR spectra simulation. For all spectra deconvolutions the parameters of φ = χ = ψ = 0°, ηQ = 0 and ηCSA = 1, the estimates of dipolar line width, Δν, quadrupole frequency, νQ, anisotropy of chemical shift, Δδ, and isotropic shift value, δiso, are presented in Fig. 7b–e.
 | ||
| Fig. 7 45Sc NMR spectra acquired for Na9Sc(MoO4)6 at resonance frequency ω0/2π = 121.49 MHz in temperature range 300–750 K (a), temperature behavior of the central line width, Δν (b), quadrupole frequency, νQ (c), anisotropy of chemical shift, Δδ (d), and isotropic shift value, δiso (e). | ||
As can be seen, the parameters of CSA and EFG on the 45Sc nuclei show only monotonous changes with temperature indicating the absence of any structure transformations in the entire temperature range 300–750 K (excepting, of course, uniform lattice expansion). Meantime, the Δν(T) dependence reveals two characteristic “steps” at T ≈ 365 and 585 K which are most likely induced by the averaging of 23Na–45Sc internuclear dipolar interaction due to the activation at these temperatures of Na2/Na3 and Na1 ions, respectively. Thus, the 45Sc NMR data confirm our conclusions on the mechanisms of sodium diffusion in the Na9Sc(MoO4)6 compound.
The features of local structure allow us to explain why the Na2 and Na3 ions are activated at substantially lower temperature while the Na1 ions remain static up to 550 K (although, considering the jump length as a main criterion we should get the opposite situation because the interatomic distances rNa1–Na3 ≈ 3.65 Å, rNa1–Na2 ≈ 3.69 Å are shorter than rNa2–Na2 ≈ 3.85 Å and rNa3–Na3 ≈ 4.07 Å). We have to take into account the fact that the probability of ion jump is determined not only by the jump length directly but also by other factors such as the type of adjacent cation (that defines the force of coulombic attraction) and the size of the “saddle point” (triangular oxygen window that has to be passed for ion release from a distinct site).38,68–70 In some cases even more tiny features such as the “geometry” of saddle point can play an important role and determine the mechanisms of ion diffusion.60 From this point of view the “immobility” of Na1 ions at T < 550 K seems to be quite natural. Indeed, the jump Na1 → Na3 which is the most expected from interatomic distance is restricted most likely due to a very small square of a triangle face: the corresponding O–O distances for this face are only 3.528, 3.257 and 2.921 Å. The jump Na1 → Na2 seems to be more probable from “geometrical” point of view: the corresponding O–O distances are equal to 3.257, 3.835 and 4.463 Å. However, the adjacent tetrahedral site for this jump is very close to highly charged Mo6+ ion (r < 2 Å).
Besides the clarifying of ion transport mechanisms, the temperature evolution of NMR spectrum allows us to estimate the parameters of ion diffusion, in particular, the characteristic ion jump frequency, τd−1. As it is expected,50 the motional narrowing occurs at a temperature where the characteristic frequency of ion jumps exceeds the “rigid lattice” (i.e., in the absence of ion motion) line width: τd−1 ∼ 2πΔνRL. Taking into account the value of ΔνRL ≈ 1.2 kHz (see Fig. 6a) we can estimate τd−1 ∼ 104 s−1 at T ≈ 400 K for Na-ions jumps within the Na2/Na3 sublattice. Similarly, for the “two-site exchange” it is expected71 that in the point of spectra coalescence the τd−1 ∼ |ωQ1(2)RL − ωQ2(2)RL|. Thus, for Na1 ↔ Na2/Na3 jumps we can estimate the τd−1 ∼ 104 s−1 at T ≈ 600 K. The analysis of Δν(T) allows also to estimate roughly the activation energy for ion motion, Ea. Several approaches have been developed for such an analysis (see for example, ref. 72–74). The simplest one, proposed by Waugh and Fedin74 suggests that:
| Ea (meV) = 1.617T0 (K), | (1) |
3.5. 23Na spin–lattice relaxation rate: localized sodium jumps
The eqn (1) yields usually only rough estimates of Ea.75 The well known NMR technique allowing to evaluate the ion transport parameters more precisely is the spin–lattice relaxation rate, T1−1, measurements. The Fig. 8 represents temperature behavior of the 23Na relaxation rates measured for Na9Sc(MoO4)6 over the temperature range 300–750 K in the magnetic field of 11.7 T. For the entire temperature range, the nuclear magnetization recovery after inverting pulse was characterized by nonexponential behavior and at least two relaxation components were required for the experimental data approximation: Mz(t) = Mz,eq − [Mz,eq − Mz(0)](csexp(−t/T1S) + cfexp(−t/T1F)), where Mz,eq is the equilibrium value of nuclear magnetization, T1F and T1S are the fast and slow spin–lattice relaxation components, respectively, the values of cs and cf determine the fractions of corresponding components (cs + cf = 1). As in the case of Na9Al(MoO4)637 the appearance of two “branches” of spin–lattice relaxation for 23Na nuclei (I = 3/2) is most likely due to dominant contribution of the quadrupole mechanisms in nuclear relaxation (see, for example, ref. 76 and 77). For the entire range of temperatures the value of cs = 0.6 ± 0.1. The final estimates of T1F−1 and T1S−1 were obtained with fixed cs = cf = 0.5, that is expected for the case when only central transition is excited and detected.78
 | ||
| Fig. 8 Semi-logarithmic Arrhenius plot of the 23Na spin–lattice relaxation rate for Na9Sc(MoO4)6. The filled and empty circles correspond to the slow (T1S−1) and fast (T1F−1) relaxation components, respectively. The green line corresponds to fit of experimental data on T1F−1 by eqn (5). Red and blue solid lines are the contributions from high- and low-temperature T1F−1 peaks (see text). The inset shows the values of τd−1 = 109 s−1 estimated from (T1−1)HTmax (red circle) and (T1−1)LTmax (blue circle), as well as τd−1 = 104 s−1 (black square and triangle) which are expected from the 23Na Δν(T) and νQ(T) data for Na2/Na3 and Na1 jumps, respectively. The red and blue solid lines correspond to (τHTd)−1 and (τLTd)−1 values obtained from eqn (5), black dashed line – the estimates of τd−1 for Na1 ↔ Na2/Na3 jumps from eqn (4) (see text). | ||
The main feature of the T1F−1 temperature dependence in Na9Sc(MoO4)6 is the maximum at Tmax ≈ 730 K. The appearance of a maximum on the T1−1(T) dependence is typical for materials with fast ion diffusion. The simplest model describing the dynamic contribution to T1−1 was proposed by Bloembergen, Purcell and Pound.79 Within the framework of BPP model, the T1−1(T) dependence is determined by the expression:
| T1−1(T) ∝ C{J(1)(ω0) + 4J(2)(2ω0)}, | (2) |
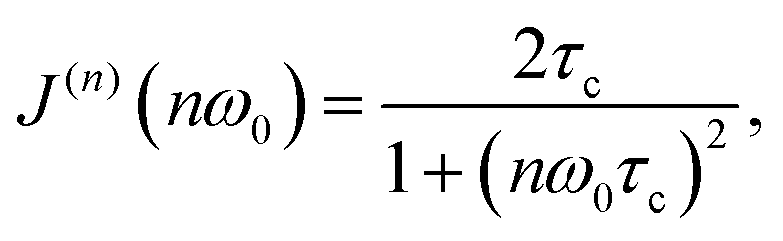 | (3) |
| τd = τd0exp(Ea/kBT), | (4) |
T1−1vs. T−1 dependence. The maximum of relaxation rate is observed at the temperature Tmax where the ion jump frequency becomes comparable with the resonance frequency: τd−1 ∼ ω0 (i.e., reaches ∼109 s−1 in our case). The slopes of the T1−1 peak at high- and low-temperature limits are equal to Ea/kB and −Ea/kB, respectively.
Nevertheless, as can be seen from Fig. 8 the T1−1vs. T−1 dependence for Na9Sc(MoO4)6 significantly deviates from the expected asymptotic behavior due to the presence of an additional low-temperature maximum at Tmax ≈ 500 K. Similar dependences with two peaks of T1−1 were observed earlier for some lithium and proton-conducting materials and have been interpreted in the assumption of a coexistence of different types of atomic motion with highly differing jump frequencies and activation energies.80–82 For this case the experimental data on T1−1 can be fitted by the modified equation:
| T1−1(T) ∝ C1{J(1)HT(ω0) + 4J(2)HT(2ω0)} + C2{J(1)LT(ω0) + 4J(2)LT(2ω0)}, | (5) |
 | (6) |
| τHTd = τHTd0exp(EHTa/kBT), τLTd = τLTd0exp(ELTa/kBT). | (7) |
As can be seen, the high-temperature maximum of T1−1 at Tmax ≈ 730 K and the 23Na NMR line narrowing observed at T ≥ 390 K are induced by one motional process, namely by the long-range Na diffusion within the Na2/Na3 sublattice. The maximum of T1−1 induced by the Na1 ↔ Na2/Na3 jumps should be observed at Tmax ∼ 1300 K. The most interesting feature of the T1−1(T) data is the low-temperature spin–lattice relaxation maximum at Tmax ≈ 500 K. It must be obviously induced by some much faster motional process. The most probable candidate for this fast ion motion is the localized “back and forth” jumps Na3 ↔ Na2. Indeed, the crystal structure of Na9Sc(MoO4)6 implies the formation of adjacent Na3O6 and Na2O6 octahedra (see Fig. 1). These sites have the shortest interatomic distance, rNa2–Na3 = 3.027 Å. Moreover, these Na3O6 and Na2O6 octahedra share faces, not edges, so this jump can occur directly without any intermediate position. Furthermore, this pair of ions is distant from other available sites at rNa2–Na2 = 3.847 Å and/or rNa3–Na3 = 4.074 Å. So, it seems to be quite reasonable to suppose the realization of localized jumps within this Na3–Na2 pair in the temperature range where the heat energy is already enough for these short “back and forth” jumps but not yet enough for the long-range diffusion onset. These considerations are consistent with the results of ab initio calculations.
3.6. Ab initio calculations: activation energies and most probable paths for sodium motion
The activation barriers for sodium diffusion calculated with the NEB method are shown in Table 2. Our results indicate that the Na-ion diffusion extends across a 3D network of migration pathways (Fig. 9) with activation barriers from 0.1 to 0.8 eV. The lowest barrier of 0.12 eV corresponds to the shortest path (3.03 Å) between the Na2 and Na3 sites. This migration is almost not blocked by oxygen atoms all the way and therefore the Na2–Na3 path is linear, as seen in Fig. 9. The linear paths for the Na2–Na2 and Na3–Na3 migrations are impossible due to the very small distances (1.3–1.5 Å) to the nearest oxygen atoms. As a result, these jumps occur through the curved trajectories, which provide the barriers of 0.59 and 0.53 eV, respectively. The Na1–Na3 and Na1–Na2 migrations (path ∼3.7 Å) have the highest energy barriers (0.69 and 0.84 eV) among the considered sodium hoppings.| Path | R Na–Na | E a, eV |
|---|---|---|
| Na2–Na3 | 3.04 | 0.12 |
| Na3–Na3 | 4.05 | 0.53 |
| Na2–Na2 | 3.93 | 0.59 |
| Na1–Na3 | 3.78 | 0.69 |
| Na1–Na2 | 3.84 | 0.84 |
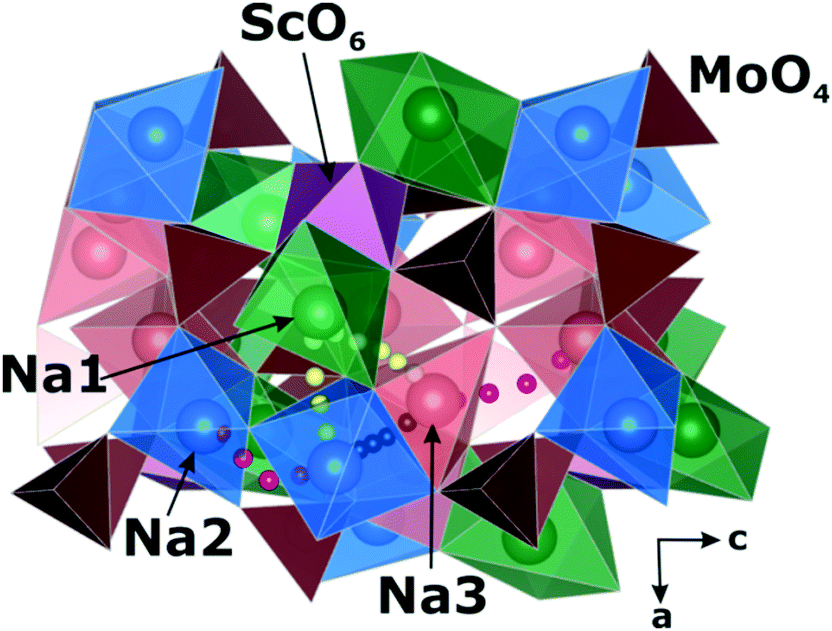 | ||
| Fig. 9 Calculated diffusion paths of sodium jumps in Na9Sc(MoO4)6: Na1 ↔ Na2/Na3 (yellow circles), Na2 ↔ Na2 and Na3 ↔ Na3 (purple circles), Na2 ↔ Na3 (black circles). | ||
4. Discussion
Based on NMR data and the DFT calculations, we propose the following mechanism of sodium diffusion in Na9Sc(MoO4)6. With increasing temperature, the localized Na3 ↔ Na2 jumps are activated first. The characteristic frequency for these jumps reaches τd−1 ∼ 109 s−1 at T ≈ 500 K and activation energy for these local jumps is Ea ≈ 0.45 eV. At temperatures above 390 K, the Na2 ↔ Na2 and Na3 ↔ Na3 jumps are activated, that leads to the long-range sodium diffusion. The characteristic frequency for these jumps is τd−1 ∼ 104 s−1 at T ≈ 400 K and ∼ 109 s−1 at T ≈ 730 K with activation energy Ea ≈ 0.8 eV (remind that from conductivity data we estimated LTEconda = 0.86 eV, NMR data yield Ea = 0.65–0.72 eV, ab initio calculations predict a similar trend for jumps within Na2/Na3 sublattice). Above 550 K, the Na1 ions also start to participate in diffusion processes through the Na1 → Na2/Na3 jumps. The characteristics of these jumps: τd−1 ∼ 104 s−1 at T ≈ 600 K, Ea = 0.95–1 eV. This scenario allows to explain the σ(T) dependence (Fig. 3) with characteristic bend at T ≈ 590 K. Moreover, the measured σ values are consistent with our estimates of τd−1. Indeed, these “macro” and “atomic-scale” data can be roughly compared using the well-known Nernst–Einstein and Einstein–Smoluchowski equations: σ = NqD/kBT and D = l2/6td, where D is the diffusion coefficient, N is the concentration of ions in a cubic centimeter of matter (∼1022), q is the ion charge, and l is the average jump length. Taking into account the characteristic frequencies of ion jumps in the Na2/Na3 sublattice, which are responsible for the main Na diffusion mechanism in Na9Sc(MoO4)6, we can estimate σ ≈ 1.2 × 10−7 and ≈6.6 × 10−3 S cm−1 at T ≈ 400 and 730 K, respectively. These values are close to the experimental data (Fig. 3).The mechanism of the long-range sodium diffusion is similar to that revealed in the related Na9Al(MoO4)6. With increasing temperature, Na-ions located far from the [R(MoO4)6]9− (R = Al, Sc) clusters are activated first, while ions located in the vicinity of the [R(MoO4)6]9− polyhedra start to participate in diffusion processes at significantly higher temperatures. Nevertheless, there are substantial differences in the Na dynamics: first, all motional effects are detected by NMR at lower (on ∼100 degrees) temperatures in Na9Al(MoO4)6 and estimates of Ea for different ion jumps yield lower values (on ∼0.2 eV).37 Second, in contrast to Na9Sc(MoO4)6, the localized sodium jumps in Na9Al(MoO4)6 are not observed in explicit form. Some deviations from the linear behavior of lnT1−1(T1) at T ∼ 360 K (see Fig. 10 in ref. 37) can be interpreted by the presence of additional low-temperature peak of T1−1. Nevertheless, these deviations are rather small, so it is difficult to conclude whether this is the real presence of an additional peak of relaxation rate or just the errors in the T1−1 measurements. Moreover, even if we assume the presence of local Na jumps in Na9Al(MoO4)6, the amplitude of the corresponding T1−1 maximum is very low reflecting low fraction of sodium participating in this localized motion. The features of sodium dynamics correlate with the features of crystal structures for Na9Sc(MoO4)6 and Na9Al(MoO4)6. Indeed, Na9Al(MoO4)6 also contains the pairs of adjacent octahedra with a common face: Na3O6–Na5O6. These sites are even closer to each other (rNa3–Na5 = 2.86 Å) than the corresponding sites in Na9Sc(MoO4)6. However, due to very large distortions of the Na3O6 and Na5O6 octahedra37 the O–O distances at the common face are rather short (4.16, 2.93, and 2.95 Å), so the probability of direct Na3 ↔ Na5 jump should be rather low. The intermediate tetrahedral site must be involved for this jump, which elongates the total path and increases the total value of Ea. Meanwhile, other distances in the sublattice of the Na3–Na5 positions (rNa4–Na4 = 3.29 Å, rNa3–Na4 = 3.56 Å, rNa4–Na5 = 3.88 Å) are significantly shorter than the corresponding values in Sc-containing compound: rNa2–Na2 = 3.847 Å, rNa3–Na3 = 4.074 Å. Thus, it becomes clear that there are no structural prerequisites for the realization of localized ion motion in Na9Al(MoO4)6, meanwhile the overall ion dynamics should be higher than that in Na9Sc(MoO4)6.
Note, that the correlation effects in sodium motion that were found in Na9Al(MoO4)637 were not observed in Na9Sc(MoO4)6. For Na9Al(MoO4)6 we assumed that the correlation effects slowing down the Na motion at T < 575 K are induced by the interaction between the neighboring mobile ions Na3–Na5 and the “static” Na1, Na2 ones.37 Due to similar interatomic distances, we expected to observe similar effects also in Na9Sc(MoO4)6. However, the characteristic features,37,57,83–86 such as the bending on the σ(T) dependence with a higher slope in the low-temperature region and a lower slope at higher temperatures and/or a large difference between the values of ENMRa and Econda was not found in Na9Sc(MoO4)6. Thus, the question on the origin of the correlation effects in sodium motion in Na9M3+(MoO4)6 (R = Al, Sc) remains open so far.
5. Conclusions
Summarizing our results we can make some general conclusions on the mechanisms of ion transport in Na9Sc(MoO4)6. The total conductivity was found to reach 3.6 × 10−2 S cm−1 at 843 K. The mechanisms of ion transport are rather complicated: the coexistence of at least three different types of Na-ions motion has been found in the temperature range 300–750 K. With increasing temperature the localized ion jumps within the pairs of adjacent sites Na2 ↔ Na3 located far from the [Sc(MoO4)6]9− clusters are activated first. These local jumps are characterized by the activation energy Ea ≈ 0.45 eV and jump frequency τd−1 ∼ 109 s−1 already at T ≈ 500 K. At temperatures higher 390 K the jumps between the Na2 ↔ Na2 and Na3 ↔ Na3 sites belonging to different pairs are activated leading to the appearance of long-range sodium diffusion. The characteristic frequency for these jumps is about 104 s−1 at T ≈ 400 K and ∼109 s−1 at T ≈ 730 K with activation energy Ea ≈ 0.8 eV. Finally, at T > 550 K the ions in Na1-sites placed in the vicinity of [Sc(MoO4)6]9− clusters also become involved in diffusion processes through the Na1 → Na2/Na3 jumps. The characteristics of these jumps: τd−1 ∼ 104 s−1 at T ≈ 600 K, Ea = 0.95–1 eV.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was carried out within the state assignment of Ministry of Science and Higher Education of the Russian Federation (Themes No. AAAA-A19-119012990095-0, A19-119031890025-9 and 0339-2016-0007), supported in part by the Russian Foundation for Basic Research (Projects No. 16-03-00164, 17-03-00333).Notes and references
- M. D. Slater, D. Kim, E. Lee and Ch. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Adv. Energy Mater., 2016, 6, 1600943 CrossRef.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 RSC.
- S. P. Ong, V. L. Chevrier, G. Hautier, A. Jain, C. Moore, S. Kim, X. Ma and G. Ceder, Energy Environ. Sci., 2011, 4, 3680–3688 RSC.
- Y. Huang, L. Zhao, L. Li, M. Xie, F. Wu and R. Chen, Adv. Mater., 2019, 31, 1808393 CrossRef PubMed.
- S. Y. Hong, Y. Kim, Y. Park, A. Choi, N. S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6, 2067–2081 RSC.
- C. Zhao, L. Liu, X. Qi, Y. Lu, F. Wu, J. Zhao, Y. Yu, Y.-S. Hu and L. Chen, Adv. Energy Mater., 2018, 8, 1703012 CrossRef.
- J. J. Kim, K. Yoon, I. Park and K. Kang, Small Methods, 2017, 1, 1700219 CrossRef.
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 856 CrossRef PubMed.
- N. Tanibata, K. Noi, A. Hayashi and M. Tatsumisago, RSC Adv., 2014, 4, 17120–17123 RSC.
- R. P. Rao, H. Chen, L. L. Wong and S. Adams, J. Mater. Chem. A, 2017, 5, 3377–3388 RSC.
- H. Wang, Y. Chen, Z. D. Hood, J. K. Keum, A. S. Pandian, M. Chi, K. An, Ch. Liang and M. K. Sunkara, ACS Appl. Energy Mater., 2018, 1, 7028–7034 CrossRef CAS.
- C. Yu, S. Ganapathy, N. J. de Klerk, E. R. van Eck and M. Wagemaker, J. Mater. Chem. A, 2016, 4, 15095–15105 RSC.
- C. K. Moon, H. J. Lee, K. H. Park, H. Kwak, J. W. Heo, K. Choi, H. Yang, M.-S. Kim, S.-T. Hong, J. H. Lee and Y. S. Jung, ACS Energy Lett., 2018, 3, 2504–2512 CrossRef CAS.
- J. Wang, X. P. Jiang, X. L. Wei, H. Yang and X. D. Shen, J. Alloys Compd., 2010, 497, 295–299 CrossRef CAS.
- L. P. Yang, S. J. Shan, X. L. Wei, X. M. Liu, H. Yang and X. D. Shen, Ceram. Int., 2014, 40, 9055–9060 CrossRef CAS.
- K. Zhao, Y. Liu, S. Zhang, S. He, N. Zhang, J. Yang and Z. Zhan, Electrochem. Commun., 2016, 69, 59–63 CrossRef CAS.
- S. J. Shan, L. P. Yang, X. M. Liu, X. L. Wei, H. Yang and X. D. Shen, J. Alloys Compd., 2013, 563, 176–179 CrossRef CAS.
- M. Guin and F. Tietz, J. Power Sources, 2015, 273, 1056–1064 CrossRef CAS.
- M. Guin, F. Tietz and O. Guillon, Solid State Ionics, 2016, 293, 18–26 CrossRef CAS.
- Y. S. Zhu, L. L. Li, C. Y. Li, L. Zhou and Y. P. Wu, Solid State Ionics, 2016, 289, 113–117 CrossRef CAS.
- Y. Noguchi, E. Kobayashi, L. S. Plashnitsa, S. Okada and J. I. Yamaki, Electrochim. Acta, 2013, 101, 59–65 CrossRef CAS.
- S. Lunghammer, D. Prutsch, S. Breuer, D. Rettenwander, I. Hanzu, Q. Ma, F. Tietz and H. M. R. Wilkening, Sci. Rep., 2018, 8, 11970 CrossRef CAS PubMed.
- Y. Deng, C. Eames, L. H. Nguyen, O. Pecher, K. J. Griffith, M. Courty, B. Fleutot, J. N. Chotard, C. P. Grey, M. S. Islam and C. Masquelier, Chem. Mater., 2018, 30, 2618–2630 CrossRef CAS.
- Yu. A. Velikodnyj and V. K. Trunov, Zhurnal Neorganicheskoj Khimii, 1977, 22, 1496–1498 Search PubMed.
- V. A. Efremov, Yu. A. Velikodnyj and V. K. Trunov, Kristallografiya, 1975, 20, 287–292 CAS.
- A. A. Evdokimov, V. A. Efremov, V. K. Trunov, I. A. Kleiman and B. F. Djurinsky, Soedineniya Redkozemelnykh Elementov. Molibdaty. Volframaty [Rare-earth Compounds. Molybdates, Tungstates], Nauka, Moscow, 1991 (in Russian) Search PubMed.
- V. K. Trunov, V. A. Efremov and Yu. A. Velikodnyj, Kristallokhimiya, Svoistva Dvoinykh Molibdatov i Volframatov [Crystal Chemistry and Properties of Double Molybdates and Tungstates], Nauka, Leningrad, 1986 (in Russian) Search PubMed.
- I. Ennajeh, S. Georges, Y. B. Smida, A. Guesmi, M. F. Zid and H. Boughazala, RSC Adv., 2015, 5, 38918–38925 RSC.
- J. Grins and M. Nygren, Solid State Ionics, 1983, 9, 859–862 CrossRef.
- N. I. Sorokin, Phys. Solid State, 2009, 51, 1128–1130 CrossRef CAS.
- A. L. Kruglyashov and E. M. Skou, Solid State Ionics, 1988, 28, 233–236 CrossRef.
- N. I. Medvedeva, A. L. Buzlukov, A. V. Skachkov, A. A. Savina, V. A. Morozov, Y. V. Baklanova, I. E. Animitsa, E. G. Khaikina, T. A. Denisova and S. F. Solodovnikov, J. Phys. Chem. C, 2019, 123, 4729–4738 CrossRef CAS.
- A. A. Savina, S. F. Solodovnikov, O. M. Basovich, Z. A. Solodovnikova, D. A. Belov, K. V. Pokholok, I. A. Gudkova, S. Y. Stefanovich, B. I. Lazoryak and E. G. Khaikina, J. Solid State Chem., 2013, 205, 149–153 CrossRef CAS.
- J. Lu and A. Yamada, ChemElectroChem, 2016, 3, 902–905 CrossRef CAS.
- C. Durio, A. Daidouh, N. Chouaibi, C. Pico and M. L. Veiga, J. Solid State Chem., 2002, 168, 208–216 CrossRef CAS.
- A. A. Savina, V. A. Morozov, A. L. Buzlukov, I. Y. Arapova, S. Y. Stefanovich, Y. V. Baklanova, T. A. Denisova, N. I. Medvedeva, M. Bardet, J. Hadermann, B. I. Lazoryak and E. G. Khaikina, Chem. Mater., 2017, 29, 8901–8913 CrossRef CAS.
- A. Martínez-Juárez, C. Pecharromán, J. E. Iglesias and J. M. Rojo, J. Phys. Chem. B, 1998, 102, 372–375 CrossRef.
- B. Lang, B. Ziebarth and C. Elsasser, Chem. Mater., 2015, 27, 5040–5048 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- A. A. Savina, V. A. Morozov, O. M. Basovich, E. G. Khaikina and B. I. Lazoryak, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1301–1303 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calve, B. Alonso, J.-O. Durand, B. Bujoli, Zh. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- H. M. Petrilli, P. E. Blöchl, P. Blaha and K. Schwarz, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 14690 CrossRef CAS.
- A. Abragam, The Principles of Nuclear Magnetism, Clarendon Press, Oxford, UK, 1961 Search PubMed.
- P. P. Man, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, John Wiley & Sons Ltd, Chichester, 2000, pp. 12224–12265 Search PubMed.
- D. Massiot, R. Conanec, W. Feldmann, R. Marchand and Y. Laurent, Inorg. Chem., 1996, 35, 4957–4960 CrossRef CAS PubMed.
- H. Saitô, I. Ando and A. Ramamoorthy, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 181–228 CrossRef PubMed.
- C. P. Slichter, Principles of Magnetic Resonance, Springer-Verlag, Berlin, 1980 Search PubMed.
- H. W. Spiess, Colloid Polym. Sci., 1983, 261, 193–209 CrossRef CAS.
- M. Wilkening and P. Heitjans, Diffusion Fundamentals, 2007, 6, 40 Search PubMed.
- K. Arbi, M. Tabellout, M. G. Lazarraga, J. M. Rojo and J. Sanz, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 094302 CrossRef.
- B. Bader, P. Heitjans, H.-J. Stockman, H. Ackerman, W. Buttlert, P. Freilander, G. Kiese, C. van der Marel and A. Schirmer, J. Phys.: Condens. Matter, 1992, 4, 4779–4800 CrossRef CAS.
- Ya. V. Baklanova, T. A. Denisova, I. Yu. Arapova, A. L. Buzlukov, A. P. Gerashenko, S. V. Verkhovskii, K. N. Mikhalev, I. R. Shein and L. G. Maksimova, J. Solid State Chem., 2013, 208, 43–49 CrossRef CAS.
- A. L. Buzlukov, I. Yu. Arapova, Y. V. Baklanova, N. I. Medvedeva, T. A. Denisova and S. V. Verkhovskii, J. Phys. Chem. C, 2016, 120, 23911–23921 CrossRef CAS.
- R. W. Schurko, S. Wi and L. Frydman, J. Phys. Chem. A, 2002, 106, 51–62 CrossRef CAS.
- J. H. Kristensen, G. L. Hoatson and R. L. Vold, J. Chem. Phys., 1999, 110, 4533–4553 CrossRef CAS.
- J. H. Kristensen and I. Farnan, J. Chem. Phys., 2001, 114, 9608–9624 CrossRef CAS.
- M. Witschas, H. Eckert, H. Freiheit, A. Putnis, G. Korus and M. Jansen, J. Phys. Chem. A, 2001, 105, 6808–6816 CrossRef CAS.
- R. L. Vold and G. L. Hoatson, J. Magn. Reson., 2009, 198, 57–72 CrossRef CAS PubMed.
- W. P. Power, S. Mooibroek, R. E. Vasylishen and T. S. Cameron, J. Phys. Chem., 1994, 98, 1552–1560 CrossRef CAS.
- S. Ding and C. A. McDowell, Chem. Phys. Lett., 2001, 333, 413–418 CrossRef CAS.
- K. Kang and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 094105 CrossRef.
- J. C. Bachman, S. Muy, A. Grimaud, H. H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- J. Langer, D. L. Smiley, A. D. Bain, G. R. Goward and M. Wilkening, J. Phys. Chem. C, 2016, 120, 3130–3138 CrossRef CAS.
- S. E. Ashbrook, S. Antonijevic, A. J. Berry and S. Wimperis, Chem. Phys. Lett., 2002, 364, 634–642 CrossRef CAS.
- H. S. Gutowsky and G. E. Pake, J. Chem. Phys., 1950, 18, 162–170 CrossRef CAS.
- J. R. Hendrickson and P. J. Bray, J. Magn. Reson., 1973, 9, 341–357 CAS.
- J. S. Waugh and E. I. Fedin, Sov. Phys. Solid State, 1963, 4, 1633–1636 Search PubMed.
- M. Wilkening, D. Bork, S. Indris and P. Heitjans, Phys. Chem. Chem. Phys., 2002, 4, 3246–3251 RSC.
- P. S. Hubbard, J. Chem. Phys., 1970, 53, 985–987 CrossRef CAS.
- J. R. C. Van der Maarel, Concepts Magn. Reson., Part A, 2003, 19A, 97–116 CrossRef CAS.
- L. C. A. Groot, J. R. C. Van der Maarel and J. C. Leyte, J. Phys. Chem., 1994, 98, 2699–2705 CrossRef CAS.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–715 CrossRef CAS.
- A. Kuhn, P. Sreeraj, R. Pottgen, H.-D. Wiemhofer, M. Wilkening and P. Heitjans, J. Am. Chem. Soc., 2011, 133(29), 11018–11021 CrossRef CAS PubMed.
- A. V. Skripov, A. L. Buzlukov, V. N. Kozhanov, T. J. Udovic and Q. Huang, J. Alloys Compd., 2003, 359, 27–34 CrossRef CAS.
- A. L. Buzlukov, A. V. Soloninin and A. V. Skripov, Solid State Commun., 2004, 129, 315–318 CrossRef CAS.
- O. Kanert, J. Steinert, H. Jain and K. L. Ngai, J. Non-Cryst. Solids, 1991, 131–133, 1001–1010 CrossRef CAS.
- K. L. Ngai and O. Kanert, Solid State Ionics, 1992, 53–56, 936–946 CrossRef CAS.
- K. L. Ngai, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13481–13485 CrossRef CAS PubMed.
- K. L. Ngai and A. K. Rizo, Phys. Rev. Lett., 1996, 76, 1296–1299 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2020 |