
The pH dependent mechanisms of non-enzymatic peptide bond cleavage reactions†
Yi
Sun
ab,
Moran
Frenkel-Pinter
 bc,
Charles L.
Liotta
acb and
Martha A.
Grover
*ab
bc,
Charles L.
Liotta
acb and
Martha A.
Grover
*ab
aSchool of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA, USA. E-mail: martha.grover@chbe.gatech.edu
bNSF/NASA Center for Chemical Evolution, USA
cSchool of Chemistry & Biochemistry, Georgia Institute of Technology, Atlanta, GA, USA
First published on 22nd November 2019
Abstract
The non-enzymatic cleavage rates of amide bonds located in peptides in aqueous solution is pH-dependent and involves two distinct mechanisms: direct hydrolysis (herein termed “scission”) and intramolecular aminolysis by the N-terminal amine (herein termed “backbiting”). While amide bond cleavage has been previously characterized using a variety of peptides, no systematic study has yet been reported addressing the effect of the pH on the interplay between the two amide bond cleavage pathways. In this study, the cleavage rates of the glycine dimer (GG), the glycine trimer (GGG), and the cyclic dimer (cGG), as well as the alanine trimer (AAA), were measured at pH 3, 5, 7, and 10 at 95 °C employing quantification based on 1H NMR. The distinct rate constants for scission and backbiting processes were obtained by solving the differential rate equations associated with the proposed kinetic model. Generalizations concerning the relative importance of the various amide bond cleavage pathways at pH 3, 5, 7, and 10 are presented. In particular, scission dominates at pH 10, while backbiting dominates at neutral pH. At the acidic pH of 3, both backbiting and scission are significant. The model of the reaction network, used in this work, enables the quantification of these multiple competing mechanisms and can be applied to longer peptides and to similar types of reaction networks.
Introduction
Fundamental studies of the reaction kinetics and mechanisms associated with non-enzymatic peptide cleavage in aqueous solution are critical in a wide variety of scientific areas, including enzymatic catalysis,1,2 peptide synthesis,3 geochemistry4 and prebiotic chemistry.4,5 On a practical note, these studies also provide comparison for enzymatic peptide hydrolase reactivity studies1,2,6 and storage of peptide-based drugs.7–9 From a prebiotic chemistry point of view, peptides have been shown to form under various conditions simulating environments on the early Earth, such as hydrothermal conditions mimicking deep-sea environments and shallow pools on land.10–17 Investigation of the kinetics and mechanisms associated with the stepwise cleavage of the polypeptides back to the amino acid building blocks and smaller polypeptides are critical to understanding the survival and selection of functional polypeptides related to the origin of life on early Earth.18The literature contains several reports addressing dipeptide hydrolysis within a range of pH and temperature conditions.19,20 Wolfenden and co-workers2 reported the amide bond cleavage rates of diglycine at neutral pH and temperatures ranging from 120 °C to 200 °C. The half-life of diglycine hydrolysis was determined to be approximately 350 years when the data were extrapolated to 25 °C. Yokoyama and co-workers21 showed that both pH and temperature affect the cleavage of diglycine and modeled the reaction kinetics taking into account the different ionization states of the dipeptide as the pH of the aqueous medium changed. Amide cleavage investigations of longer polypeptides have also been reported under various pH conditions. Bada and co-workers22,23 investigated the decomposition of a tripeptide and a hexapeptide at elevated temperatures (130 °C) under neutral pH conditions and showed the formation of diketopiperzines. They proposed that the diketopiperzines were derived from an internal aminolysis (backbiting) mechanism where the N-terminal amine attacked a proximate carbonyl group via a 6-membered ring transition state. Goolcharran and Borchardt employed a simple model peptide, phenylalanine-proline-p-nitroaniline, to investigate the backbiting reaction pathway as a function of pH.23 The overall amide cleavage rates increased with increasing pH. The backbiting pathway was found to dominate within the pH range 3–8 while the direct scission reaction pathway was dominant below pH 3 and above pH 8. Fig. 1 shows the transition state for the acid-catalyzed backbiting process. A free amine group is necessary for this process and the amount of free amine decreases as the pH decreases. At the same time, the equilibrium protonation of the carbonyl oxygen, which increases the electrophilicity of the carbonyl carbon, increases with decreasing pH. In addition, to complete the formation of the diketopiperazine product, protonation of the amine leaving group is necessary. This last step in the overall process is facilitated as the pH decreases. Thus, a delicate pH balance must be achieved in order for substantial backbiting to take place.
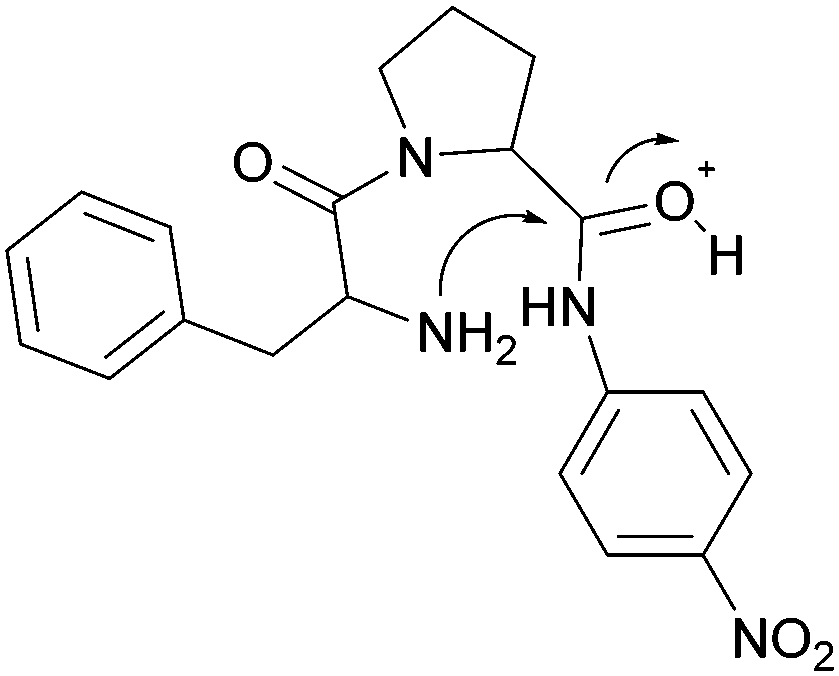 | ||
| Fig. 1 Transition state for the acid-catalyzed backbiting pathway as described in ref. 22. | ||
Recently, Savage et al.24 reported studies related to amide cleavage reactions of tetra-alanine as a function of temperature (170–230 °C) and pH in high-pressure/high-temperature water to mimic hydrothermal vent conditions. They presented a kinetic model to describe their experimental results that contained both backbiting and scission pathways. It is known that the auto-ionization constant of water increases and the dielectric constant of water decreases as the temperature of water increases. These two factors could potentially influence the relative reaction pathways (backbiting vs. scission) associated with amide bond cleavage when compared to similar reactions at lower temperatures.25,26 Moreover, acid catalysis was not included in their model, and could potentially play a crucial role affecting the interplay between the two amide bond cleavage mechanisms. Indeed, as will be shown in this paper, scission is significant at acidic pH.
The objectives of this paper are (a) to examine the effect of pH on diglycine (GG), cyclic glycine dimer (cGG) and triglycine (GGG) bond cleavage, and (b) to quantify the two competing reaction pathways (scission and backbiting) across acidic, neutral and basic pH conditions at 95 °C. Here we report the results of non-enzymatic peptide cleavage, addressing two mechanistic pathways. No previous studies on peptide cleavage have considered atmospheric pressure and temperatures below 100 °C. For comparison with GGG, we also investigated the effect of pH on trialanine (AAA) amide bond cleavage. Using quantitative 1H NMR analysis, the amounts of each species were measured as a function of time and the rate constants for each reaction pathway were then estimated based on the proposed kinetic models. We characterized the cleavage kinetics of amide bonds both within the starting oligomer and in the accompanying products. Specifically, the hydrolysis reactions of GG, cGG, and GGG were conducted at 95 °C and pH values ranging from 3 to 10. Neither the decomposition of the glycine monomer nor polymerization of the glycine monomer nor any of the oligomers were observed under the studied experimental conditions. Only amide cleavage reactions and cyclization reactions to form diketopiperazine were observed. Fig. 2 shows the postulated reaction pathways for GGG and GG amide bond cleavage, the opening of the cyclic dimer (cGG) and the closing of the linear dimer (GG) along with the accompanying rate constants. These are the rate processes which form the basis of the kinetic model reported herein. Two reaction pathways are possible for the hydrolysis of GGG, either forming G and GG through the scission pathway or forming G and cGG through the backbiting pathway. GG can react further to produce two Gs through amide scission or to produce cGG via a reversible cyclization. Reversibility for the ring opening of cGG or the ring closure of GG takes place only under acidic or neutral conditions. Under basic condition the ring opening process is irreversible. When fitting the four rate constants, multiple data sets including all GG, cGG and GGG hydrolysis data at a specific pH are fitted with a shared set of rate constants. It is assumed that the scission rate constant is the same in both the trimer and the dimer. The model does not include the rate constants associated with every ionization state of G, GG, cGG, and GGG. Instead the more compact model is applied as outlined in Fig. 2, having distinct rate constants at each of the four pH levels.
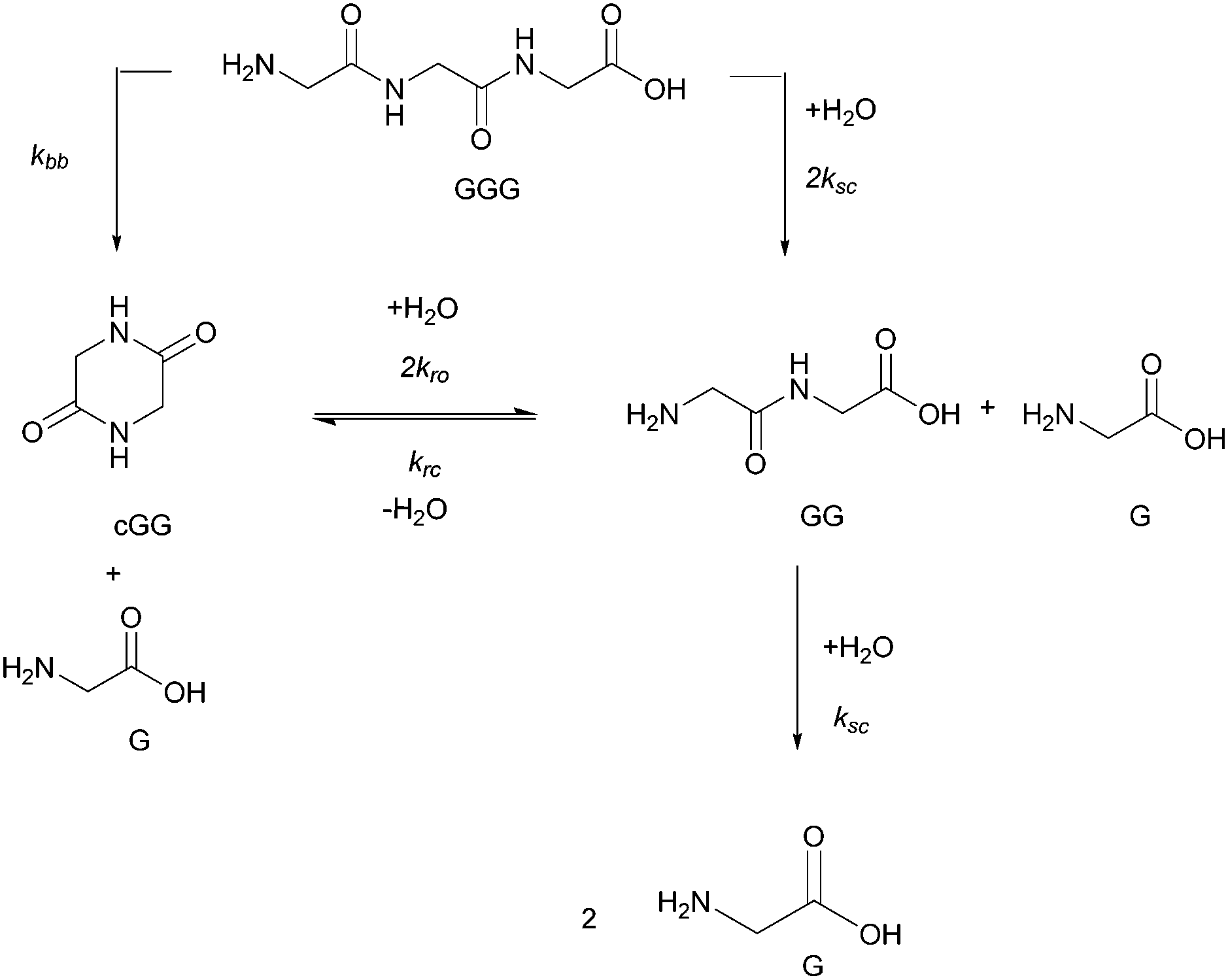 | ||
| Fig. 2 Reaction pathways for the amide cleavage of the glycine trimer (GGG), the glycine dimer (GG), and the cyclic glycine dimer (cGG) and the ring closing reaction of the linear dimer (GG) along with the associated rate constants. ksc is the rate constant corresponding to the hydrolysis of the glycine oligomers through a scission pathway; kbb is the rate constant for the backbiting pathway of GGG, and krc and kro are the rate constants for cGG ring closing and opening, respectively. | ||
Methods
Glycine (Sigma G7126), glycine dimer (Sigma G1002), glycine trimer (Sigma G1377), cyclic glycine dimer (Sigma G7251), L-alanine trimer (Sigma A9627), hydrochloric acid, sodium hydroxide, potassium hydrogen phthalate (Sigma P1088), deuterium oxide (99.9 mol%) and HPLC-grade water were all obtained from Sigma-Aldrich.Glycine dimer and glycine trimer were dissolved in water to a concentration of 200 mM. The initial concentration of cyclic glycine dimer and alanine trimer were 100 mM due to low solubility. The initial pH of the solutions was measured using a FiveEasy Benchtop F20 pH/mV Meter with an InLab Micro pH electrode probe from Mettler-Toledo.
In each experiment, peptide solutions were prepared and the initial pH was adjusted using HCl or NaOH to 3, 5, 7 or 10 at room temperature. Reactions were held in 2 mL glass vials (Supelco 29381-U) with a starting volume of 200 μL. The vials were sealed and heated at 95 °C for up to 120 hours in an oven. At various time points (0 h, 12 h, 24 h, 36 h, 48 h, 72 h, 96 h and 120 h), three replicates were removed from the oven. We noted that less than 10% of water evaporated during the heating process. All the results are expressed in moles instead of concentrations due to the volume changes during the heating process. Changes in pH of 0.2–1.0 pH units were observed during the heating process.
Before analysis, all the solutions were transferred into new tubes and water was removed using a Speedvac for 5 hours at ambient temperature to suppress the water signal during NMR analysis. The dried samples were then rehydrated with 600 μL of D2O, and potassium hydrogen phthalate (25 mM final concentration) was added as an internal standard before 1H NMR analysis was undertaken. The samples were then analyzed using a Bruker Avance IIIHD 700 spectrometer and the concentrations for G, GG, cGG, GGG, and AAA were determined. The data were collected using a 30 degree pulse program with a 15 second relaxation delay to ensure quantitative integration of the resonances. The signals for each species were separated on the 1H NMR spectra and the quantification of each species was based on the integrated signal intensities relative to the internal standard intensity. All the 1H NMR spectra were plotted and analyzed using MestReNova 9.1.
The kinetic model describing the degradation rates of the glycine oligomers is
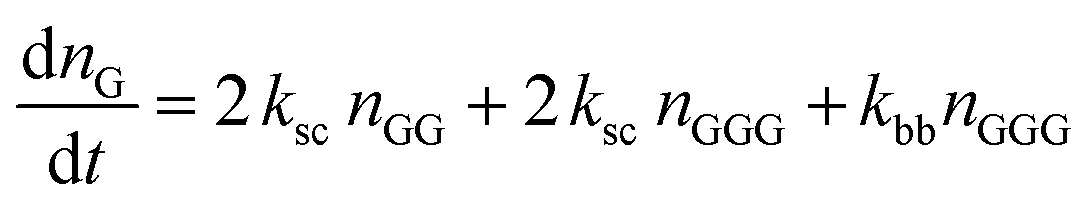 | (1) |
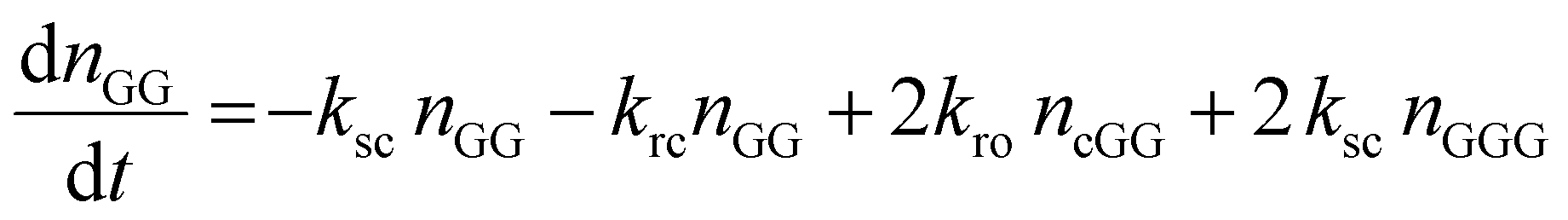 | (2) |
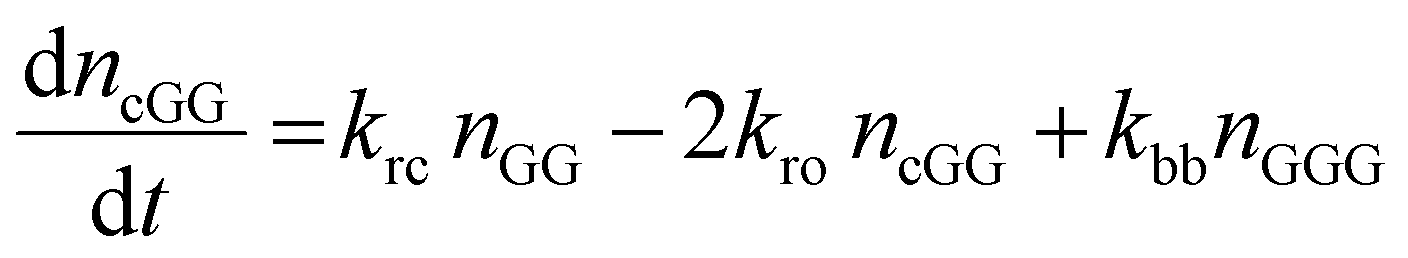 | (3) |
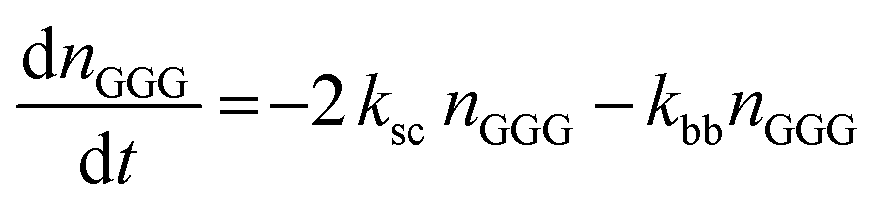 | (4) |
All the reactions are assumed to be pseudo-first order, according to elementary reaction kinetics based on stoichiometry, and assuming a significant excess of water. As shown in Fig. 2, there are seven parameters in the fitting process, including the four rate constants, as well as the initial concentrations of the reactants: GG, cGG and GGG. In this investigation, the initial concentrations are estimated so as not to give inordinate weight to the first measurement compared to the subsequent measurements. The parameter estimates are obtained using MATLAB, using the ode45 function to solve the differential equations, and the patternsearch function to find the parameter values that minimize the overall sum-squared error of the model fit. The model predictions capture the initial rates, as well as the decrease in the rates as the reactions progress. Due to the slow rates of these processes, the equilibrium values of the species are not measured directly, although they can be inferred from the estimated rate constants. The model fits are best fits given the assumed reaction model, although there are some deviations particularly for GG in the experiments beginning with cGG.
Results
1. Glycine dimer reaction kinetics
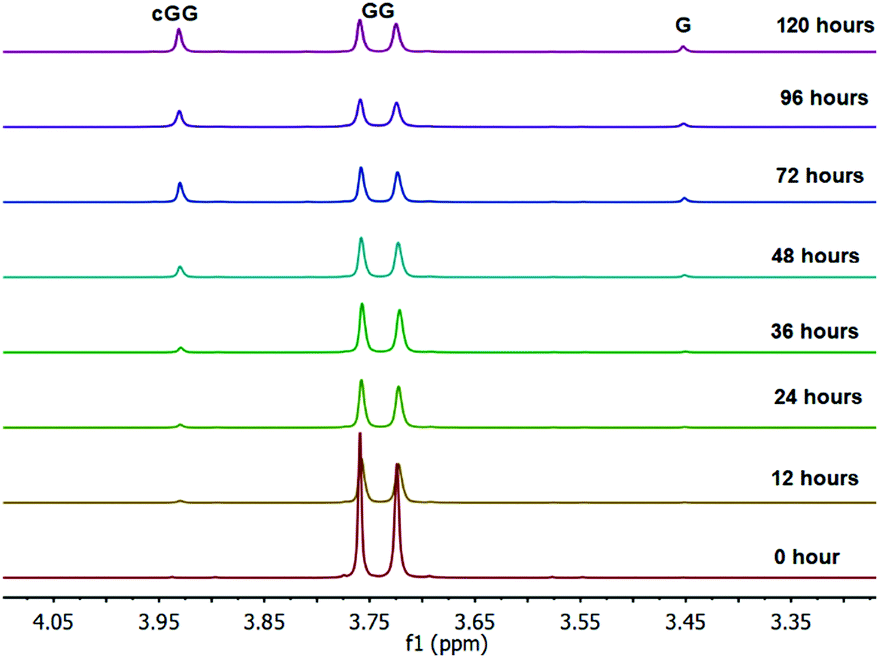
The linear dimer GG can undergo two reactions (Fig. 2): (a) the amide bond in GG can be cleaved via an acid-catalyzed attack by a water molecule at the internal carbonyl carbon or by a direct attack of hydroxide ion yielding two glycine monomers G, and (b) the terminal amino group of GG can react with the terminal carboxyl group to form the six-membered ring cGG by acid-catalyzed cyclization. 1H NMR was used to monitor the reactions of GG at pH 3, 5, 7, and 10. (Note all pH values are those measured at the beginning of the experiment at ambient temperature.) As examples, Fig. 3 and 4 show the stacked 1H NMR spectra for GG degradation at pH 5 and 7 at 95 °C over a period of five days. The signals for G, GG, and cGG are well resolved. The 1H NMR spectra for reactions of linear GG degradation all four pH values are shown in ESI,† Fig. S1–S4. For linear GG degradation, the amount of the reactant (GG) and products (G and cGG) were determined from the integrated NMR signals, shown in ESI,† Tables S1–S4. For cGG degradation, the related stacked 1H NMR spectra for the reactions of GG at pH 3, 5, 7, and 10 are shown in ESI,† Fig. S5–S8, and the amount of the reactant (cGG) and products (G and GG) are shown in ESI,† Tables S5–S8. Fig. 5 shows the experimental (squares) and model fit (solid lines) profiles for GG, cGG, and G at pH 3, 5, 7, and 10 starting with GG and cGG. Overall, the experimental rate of reaction for GG is faster at pH 3 and 10 compared to pH 5 and 7. The mechanistic pathways for the reaction of GG under acidic and basic pH are outlined in Fig. 6 and 7, respectively. In acidic media, the electrophilicity of the carbonyl carbon of the peptide bond is enhanced by the coordination of a proton with the carbonyl oxygen. This increased electrophilicity facilitates reaction with weakly nucleophilic water to form a tetrahedral intermediate which subsequently decomposes into two glycine monomers. In contrast, under basic conditions, hydroxide ion may directly attack the peptide carbonyl carbon to form an analogous tetrahedral intermediate. This intermediate then decomposes to form the glycine products. | ||
| Fig. 3 Stacked 1H NMR spectra for GG reactions at 95 °C at pH 5. | ||
 | ||
| Fig. 4 Stacked 1H NMR spectra for GG reactions at 95 °C at pH 7. | ||
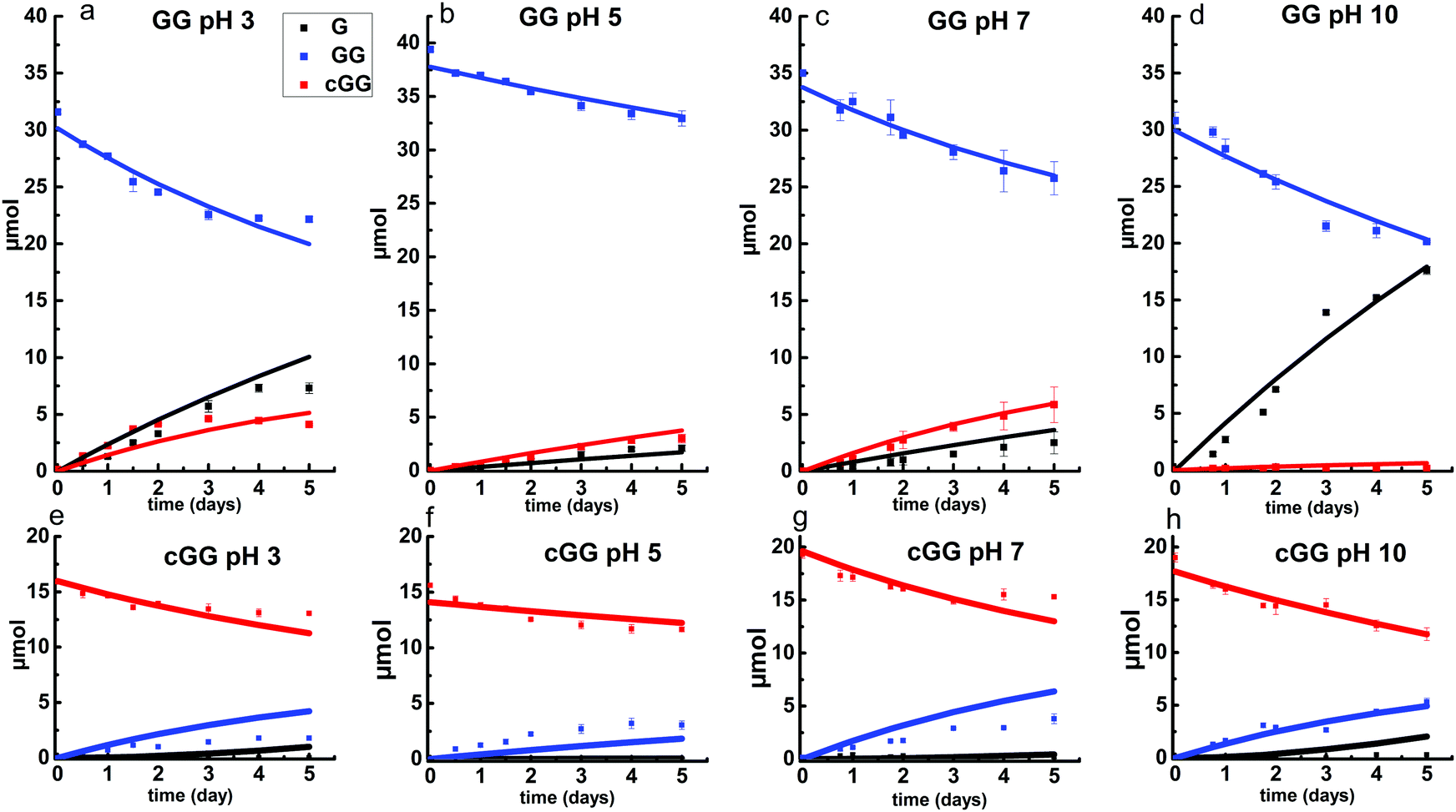 | ||
| Fig. 5 Glycine dimer (GG) (a–d) and cyclic glycine dimer (cGG) (e–h) reaction kinetics at pH 3, 5, 7 and 10 at 95 °C over a period of five days. The squares represent experimental data obtained from 1H NMR measurements, while the solid lines represent the model prediction. The error bars represent standard deviation over three replicates. (GG) = blue; (G) = black; (cGG) = red. | ||
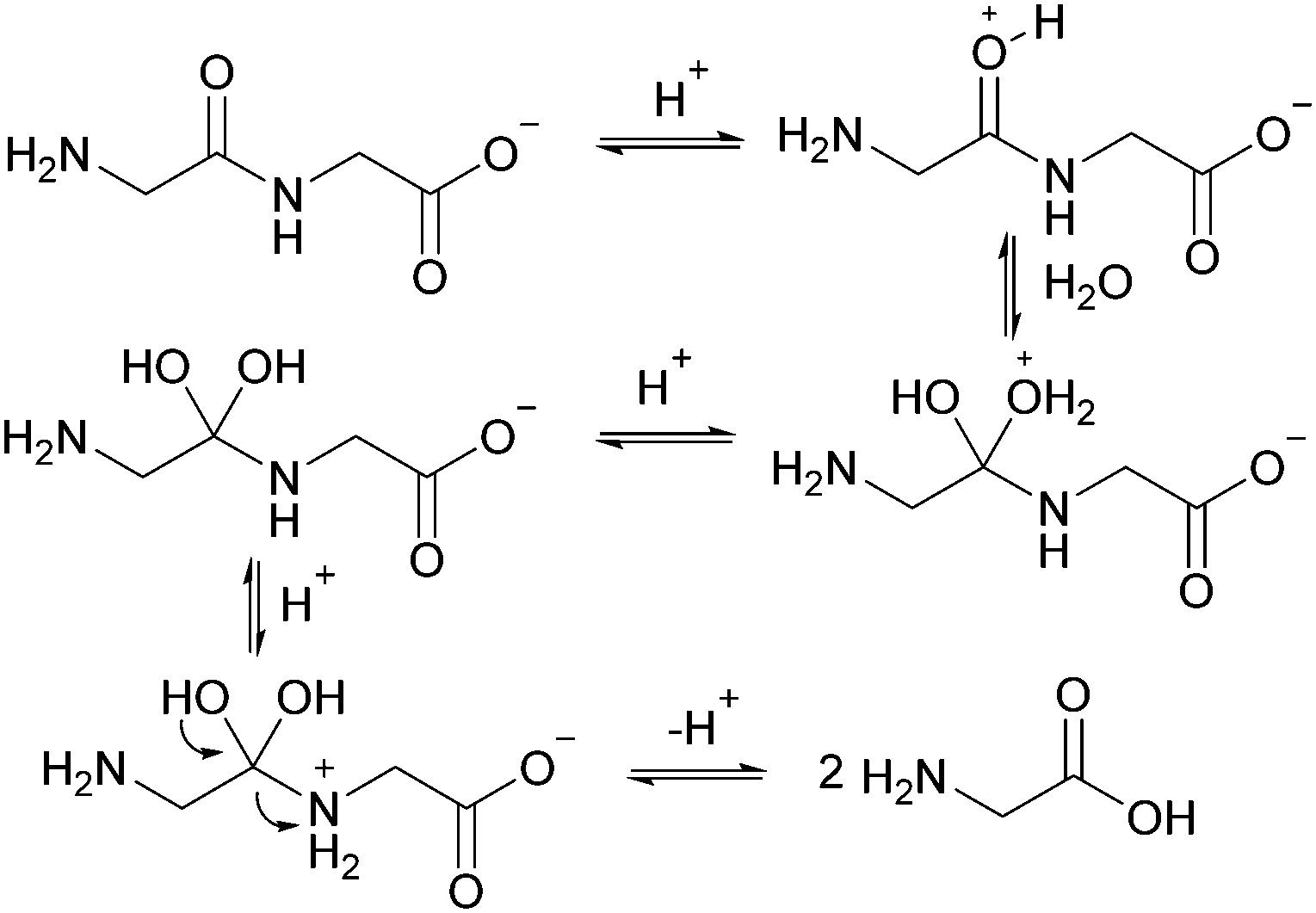 | ||
| Fig. 6 Mechanism for GG hydrolysis at pH 3. | ||
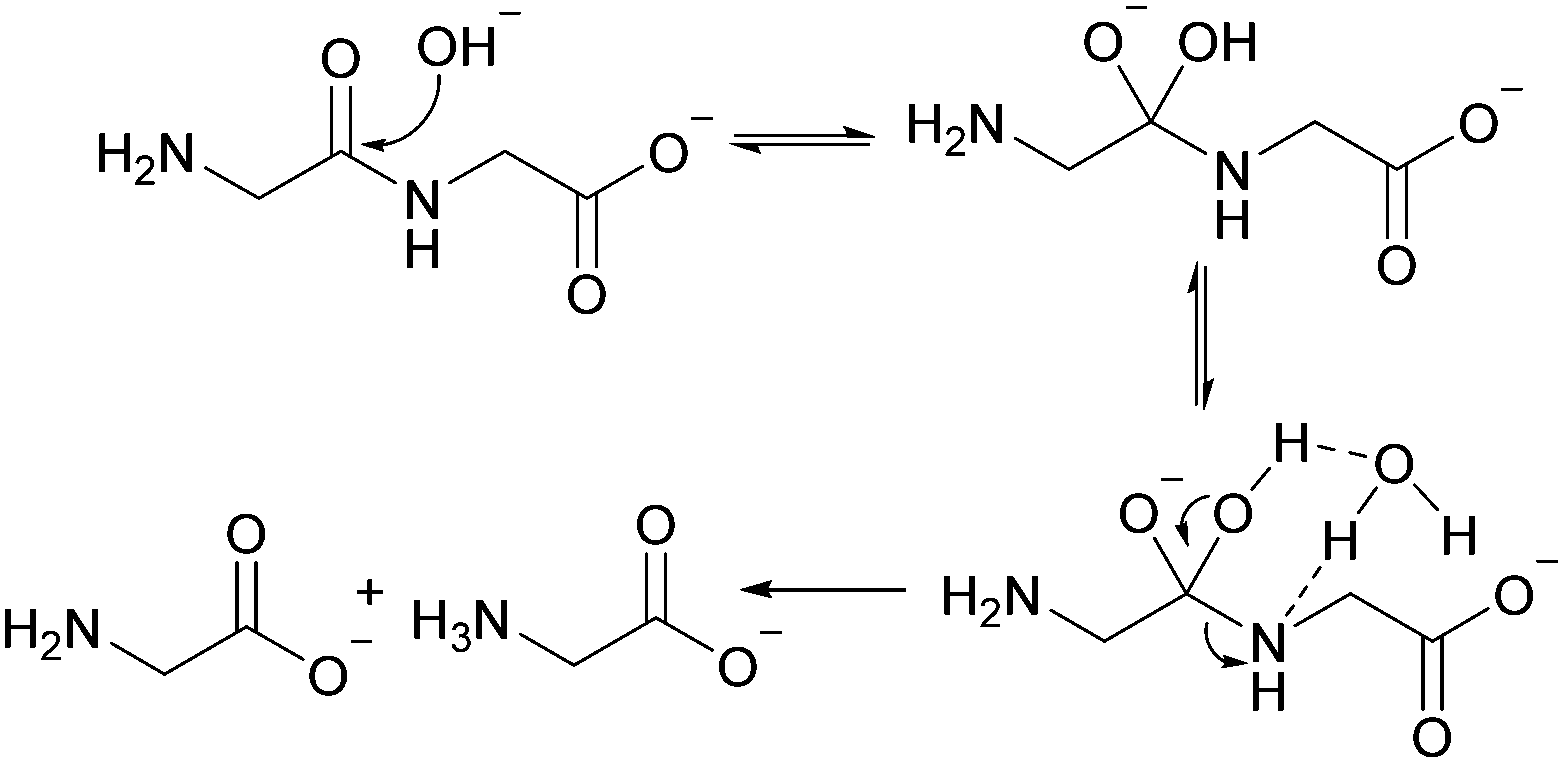 | ||
| Fig. 7 Mechanism pathway for GG hydrolysis at pH 10. | ||
Fig. 5 also demonstrates that the rates of ring opening are relatively slow at 95 °C over the pH range studied. Earlier investigations have considered the interconversion between linear glycine dimer GG and the cyclic dimer cGG.27–30Fig. 8 shows the acid-catalyzed mechanism for the acid-catalyzed ring-opening of cGG; every step is reversible. The principle of microscopic reversibility dictates that the corresponding acid-catalyzed ring-closure is just the reverse of the pathway shown in Fig. 8. In contrast, under basic conditions at pH 10, the product of reaction (the linear glycine dimer GG) has a terminal carboxylate anion which is not susceptible to nucleophilic attack by the proximate amino group to form cGG; thus the final step in the ring opening at pH 10 is essentially irreversible. Indeed, at pH 10, the experimental formation of cGG is essentially zero (Fig. 5d).
 | ||
| Fig. 8 Mechanism for the acid-catalyzed ring-opening of cGG. | ||
2. Glycine trimer (GGG) reaction kinetics
As shown in Fig. 2, there are two principle reaction pathways for GGG: (a) the formation of cGG and G via backbiting and (b) the formation of the linear dimer GG and glycine G via direct scission of one of the peptide linkages. The detailed description of the mechanism for each of these processes depends on the ambient pH. The acid-catalyzed backbiting process is described in Fig. 9 while the mechanism for the competing acid-catalyzed scission to the linear glycine dimer GG and glycine G is essentially the same as that shown in Fig. 6. The corresponding backbiting and peptide cleavage mechanisms in basic media are similar to Fig. 7. The subsequent reactions of the glycine dimer GG were discussed in Section 1. It should be emphasized that the backbiting pathway is an intramolecular process which always begins at the N-terminal amino acid unit and, in order to proceed, the amine group must not be protonated. In contrast, the intermolecular peptide hydrolysis pathway by water or hydroxide can, in principle, take place anywhere along the polypeptide chain.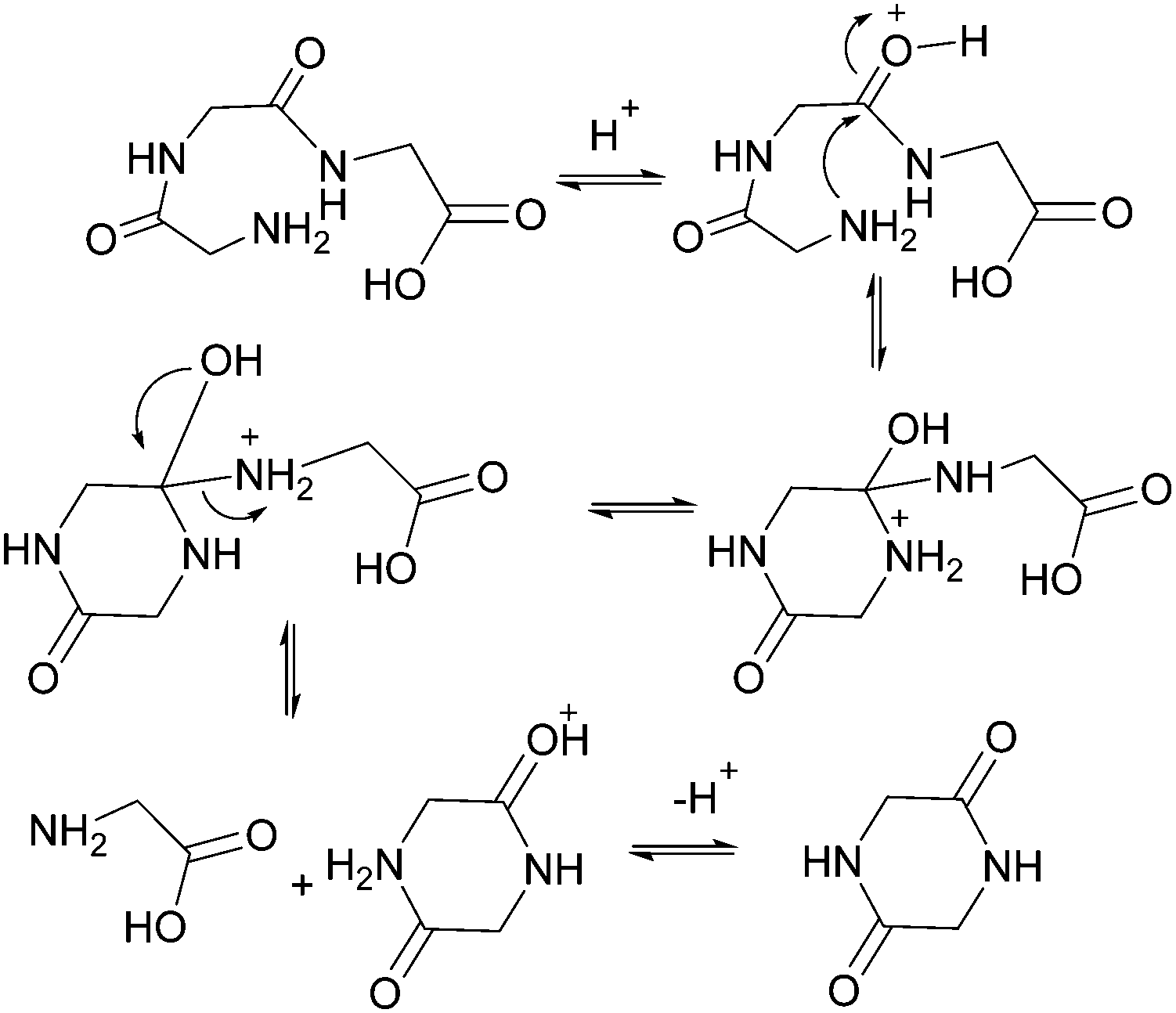 | ||
| Fig. 9 Acid-catalyzed backbiting mechanism for the formation of cGG and glycine. | ||
Fig. 10 shows the stacked 1H NMR spectra for GGG degradation at pH 5. The remaining NMR spectra and tabulated peak integration values for GGG degradation are summarized in ESI,† Fig. S9–S12 and Tables S9–S12. Fig. 11 graphically illustrates the experimental (squares) and model-fit (solid lines) profiles for GGG, GG, and cGG with respect to time at 95 °C at pH 3, 5, 7 and 10 starting with GGG. Interestingly, the degradation rate of GGG is fastest at pH 7 and appears to decrease as the medium becomes more acidic or basic. At pH 10 the rate is substantially slower compared to the other pH conditions studied. These results are in stark contrast to the GG kinetic profiles discussed in Section 1 where the fastest rates occur at pH 3 and 10. This suggests that the dominant reaction pathway for GGG degradation may be different from that of GG.
 | ||
| Fig. 10 Stacked 1H NMR spectra for GGG reactions at 95 °C at pH 5. | ||
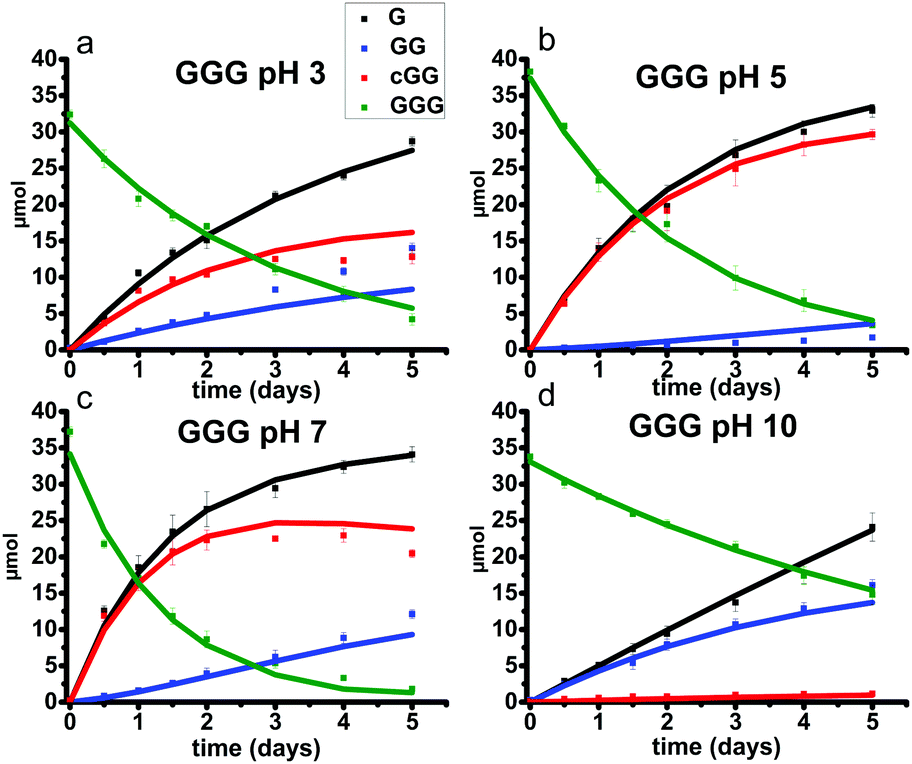 | ||
| Fig. 11 GGG reaction kinetics at pH 3, 5, 7 and 10 at 95 °C over five days (a–d). The symbols denote 1H NMR measured abundance and solid lines are the model predictions. The error bars represent standard deviation over three replicates. (GGG) = green; (GG) = blue; (G) = black; (cGG) = red. | ||
Fig. 11 shows that the initial rates for the reaction of GGG via the backbiting process steadily increase from pH 3 to pH 7. At pH 5 and 7, cGG and G are initially produced in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio which is consistent with the operation of the GGG backbiting pathway. As the reaction progresses, however, a deviation from the 1:1 ratio is observed due to the accompanying ring opening reaction of cGG producing GG which can subsequently form G. These results clearly demonstrate that the backbiting reaction mechanism is favored under neutral pH conditions.
:1 molar ratio which is consistent with the operation of the GGG backbiting pathway. As the reaction progresses, however, a deviation from the 1:1 ratio is observed due to the accompanying ring opening reaction of cGG producing GG which can subsequently form G. These results clearly demonstrate that the backbiting reaction mechanism is favored under neutral pH conditions.
Backbiting at pH 10 appears to be negligible. At pH 10 the initial production of GG and G occurs in approximately a 1:1 molar ratio suggesting that the scission pathway operates. The amount of cGG formed at pH 10 is negligible indicating that the scission mechanism is the favored pathway. At pH 3 both the scission and the backbiting mechanisms contribute to the reaction process. It is concluded that the competitive pathways (backbiting and random scission) for the reaction of GGG is strongly dependent on the pH of the aqueous medium and that backbiting is an important pathway for GGG decomposition in both acidic and neutral media.
3. Rate constants based on proposed kinetics model
Since all three sets of kinetic experiments (GG, cGG and GGG) share similar reaction pathways, the three sets of experimental data were fit with a shared set of rate constants at pH 3, 5, 7, and 10. Fig. 12 compares the pH-dependent rate constants shown in Fig. 2 based on the fit to the kinetic model. The rate constants and the initial concentrations for the starting materials derived from the kinetic model for each of these reactions are also tabulated in ESI,† Tables S13 and S14.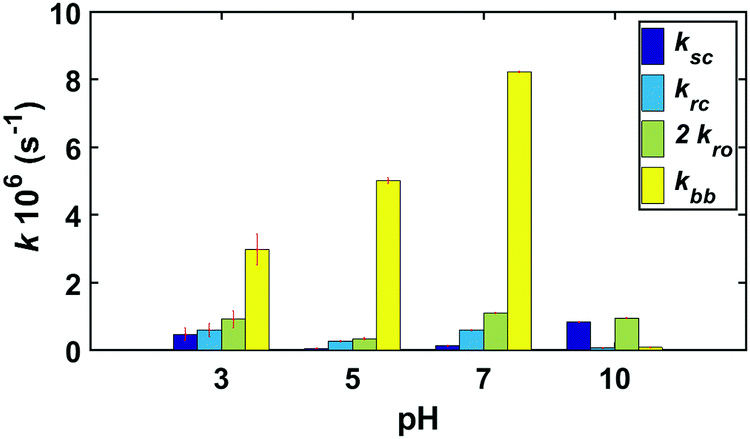 | ||
| Fig. 12 Estimated rate constants for GG, cGG and GGG kinetics at pH 3, 5, 7 and 10 at 95 °C. Confidence intervals were calculated following the chi-squared method (discussed in ESI,†) at the 95% confidence level. | ||
Overall, the fastest rate constants are associated with backbiting, over the range of pH from 3–7, though the rate of backbiting is extremely low at pH = 10. The highest values of the scission rate constant are observed at acidic and basic pH, with lower rates near neutral pH.
4. Alanine trimer (AAA) reaction kinetics
To support the generality of the glycine studies, tri-alanine (AAA) was also investigated. The results are shown in Fig. 13. The stacked 1H NMR spectra and the tabulated peak integration values for AAA are summarized in ESI,† Fig. S13–S16 and Tables S15–S18. Alanine racemization is not significant at 95 °C and was not included in the model.31 The optimized reaction rates for each reaction pathway and the initial starting AAA concentrations at each pH condition are listed in ESI,† Tables S19 and S20.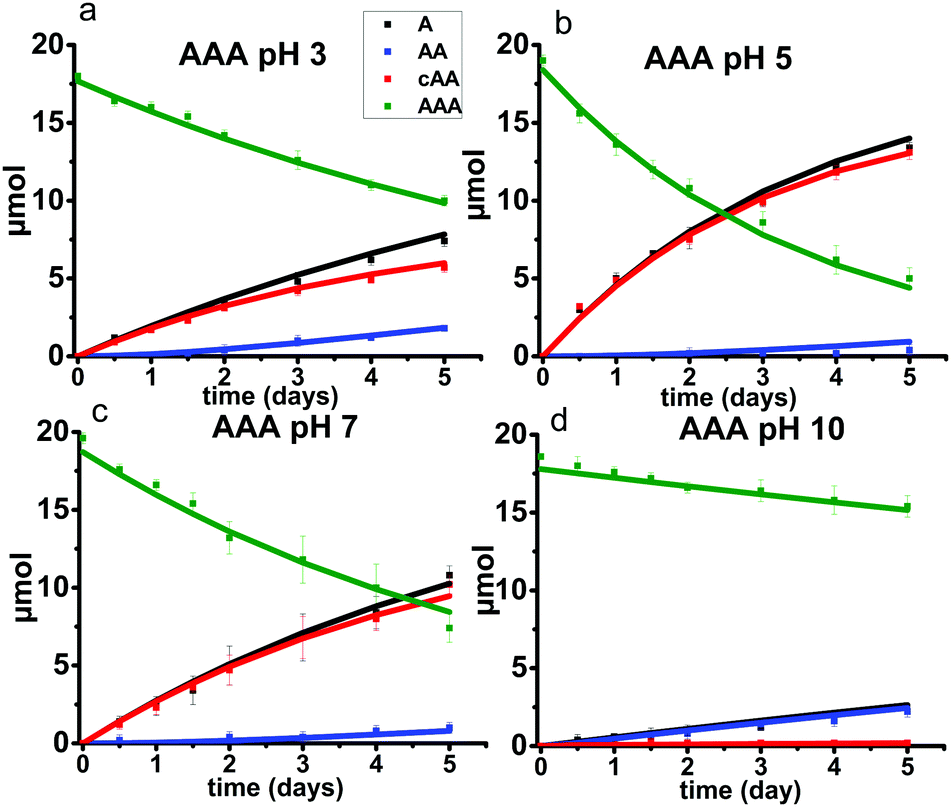 | ||
| Fig. 13 Alanine trimer (AAA) degradation reaction kinetics under pH 3, 5, 7 and 10, at 95 °C, over 5 days (a–d). The symbols denote 1H NMR measured quantifications and solid lines are the model predictions. The error bars represent standard deviation over three replicates. (AAA) = green; (A) = black; (AA) = blue; (cAA) = red. | ||
Overall, the same trends in pH are observed for AAA as for GGG. Specifically, backbiting is the major reaction pathway at pH 3, 5, and 7 where a 1:1 molar ratio of cyclic alanine dimer (cAA) to monomer (A) are produced. This observation is similar to that of the GGG reaction within the same pH range. A limited amount of linear dimer (AA) is detected, suggesting a slower rate of ring opening of cAA compared to cGG (Fig. 5 and 11). In contrast, only the scission pathway is observed at pH 10. The overall reaction rates are slower for AAA compared to GGG, likely due to steric hindrance from the methyl group on the alpha carbon of alanine.
Discussion
In the model presented here, the rate of scission for each peptide bond is equal, independent of peptide length. Thus, peptide trimers will degrade by scission at twice the rate of linear dimers, since the trimers contain two peptide bonds. More generally, the degradation rate of any homopolymeric peptide with the length of n units could be described as the summation of two terms, the backbiting reaction rate and the random internal scission rate:| kobs = kbb + (n − 1)krs | (5) |
The assumption of equal scission rates for all n would be valid when no macromolecular structures are formed; folding of longer peptides could alter the cleavage at particular sites. Thus, even though backbiting appears to be dominant at pH 3–7, scission might also be important for longer peptides, especially at acidic pH. A model of the reaction network, such as the one presented here, enables the quantification of these multiple competing mechanisms.
Previous kinetic modeling studies of peptide cleavage have not used atmospheric pressure and temperatures below 100 °C. Radzicka and Wolfenden2 studied peptide cleavage at neutral pH with higher temperatures. Extrapolation of their degradation rate constant to 95 °C yields ksc = 1.5 x 10−7 s−1, similar to our prediction of ksc = 1.4 × 10−7 s−1. Similarly, extrapolation of the model from Sakata et al.21 at pH = 9.8 yields ksc = 6.6 × 10−7 s−1, compared to our prediction of ksc = 8.4 × 10−7 s−1 at pH = 10. Thus, the results presented here are consistent with previous reports, while providing a comprehensive quantitation of the cleavage reaction network, from acidic to basic pH, and measured at atmospheric pressure.
While scission and backbiting are both significant in this study, the ring opening reaction of diketopiperazines is very slow at all pH values considered. The ease of formation and the stability of the cyclic dimers presents one of the greatest obstacles in our understanding of the prebiotic origin of polypeptides.32,33 Once formed, the cyclic dimer is extremely stable and presents a dead-end for further polymerization under plausible prebiotic conditions. However, as shown here, basic conditions can be used to retard the ring-closure reaction.
Conclusions
Dipeptide cleavage in water at atmospheric pressure occurs by direct scission, and is much faster at acidic and basic pH, compared to neutral pH. In contrast, tripeptide cleavage under the same conditions occurs by both scission and backbiting. The overall observed cleavage in tripeptides is fastest at neutral pH, due to backbiting. At acidic pH, both backbiting and scission are active pathways.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Jeff Bada, Prof. Paul Bracher, Prof. Jay Forsythe, Prof. Sheng-Sheng Yu, Prof. Luke Leman, and Prof. Aikomari Guzman for their discussions. This work was funded by the National Science Foundation and the NASA Astrobiology Program, under the NSF Center for Chemical Evolution (CHE-1504217). Dr. Moran Frenkel-Pinter was supported by the NASA Postdoctoral Program, administered by Universities Space Research Association under contract with NASA.References
- L. Lawrence and W. J. Moore, J. Am. Chem. Soc., 1951, 73, 3973–3977 CrossRef CAS.
- A. Radzicka and R. Wolfenden, J. Am. Chem. Soc., 1996, 118, 6105–6109 CrossRef CAS.
- B. F. Gisin and R. Merrifield, J. Am. Chem. Soc., 1972, 94, 3102–3106 CrossRef CAS PubMed.
- F. S. van Kleef, W. W. de Jong and H. J. Hoenders, Nature, 1975, 258, 264 CrossRef CAS PubMed.
- G. Danger, R. Plasson and R. Pascal, Chem. Soc. Rev., 2012, 41, 5416–5429 RSC.
- K. Marshall-Bowman, S. Ohara, D. A. Sverjensky, R. M. Hazen and H. J. Cleaves, Geochim. Cosmochim. Acta, 2010, 74, 5852–5861 CrossRef CAS.
- J. Battersby, W. Hancock, E. Canova-Davis, J. Oeswein and B. O'onnor, Int. J. Pept. Protein Res., 1994, 44, 215–222 CrossRef CAS PubMed.
- L. Gu and R. G. Strickley, Pharm. Res., 1987, 4, 392–397 CrossRef CAS PubMed.
- A. R. Oyler, R. E. Naldi, J. R. Lloyd, D. A. Graden, C. J. Shaw and M. L. Cotter, J. Pharm. Sci., 1991, 80, 271–275 CrossRef CAS PubMed.
- S.-S. Yu, R. Krishnamurthy, F. M. Fernández, N. V. Hud, F. J. Schork and M. A. Grover, Phys. Chem. Chem. Phys., 2016, 18, 28441–28450 RSC.
- M. Rodriguez-Garcia, A. J. Surman, G. J. Cooper, I. Suárez-Marina, Z. Hosni, M. P. Lee and L. Cronin, Nat. Commun., 2015, 6, 8385 CrossRef CAS PubMed.
- G. Martra, C. Deiana, Y. Sakhno, I. Barberis, M. Fabbiani, M. Pazzi and M. Vincenti, Angew. Chem., Int. Ed., 2014, 53, 4671–4674 CrossRef CAS PubMed.
- L. Leman, L. Orgel and M. R. Ghadiri, Science, 2004, 306, 283–286 CrossRef CAS PubMed.
- E.-I. Imai, H. Honda, K. Hatori, A. Brack and K. Matsuno, Science, 1999, 283, 831–833 CrossRef CAS PubMed.
- J. Greenwald, M. P. Friedmann and R. Riek, Angew. Chem., 2016, 128, 11781–11785 CrossRef.
- J. G. Forsythe, S. S. Yu, I. Mamajanov, M. A. Grover, R. Krishnamurthy, F. M. Fernández and N. V. Hud, Angew. Chem., Int. Ed., 2015, 54, 9871–9875 CrossRef CAS.
- J.-P. Biron and R. Pascal, J. Am. Chem. Soc., 2004, 126, 9198–9199 CrossRef CAS PubMed.
- S. I. Walker, M. A. Grover and N. V. Hud, PLoS One, 2012, 7, e34166 CrossRef CAS PubMed.
- T. D. Campbell, C. A. Hart, R. Febrian, M. L. Cheneler and P. J. Bracher, Tetrahedron Lett., 2018, 59, 2264–2267 CrossRef CAS.
- R. M. Smith and D. E. Hansen, J. Am. Chem. Soc., 1998, 120, 8910–8913 CrossRef CAS.
- K. Sakata, N. Kitadai and T. Yokoyama, Geochim. Cosmochim. Acta, 2010, 74, 6841–6851 CrossRef CAS.
- S. M. Steinberg and J. L. Bada, J. Org. Chem., 1983, 48, 2295–2298 CrossRef CAS.
- C. Goolcharran and R. T. Borchardt, J. Pharm. Sci., 1998, 87, 283–288 CrossRef CAS PubMed.
- J. D. Sheehan, A. Abraham and P. E. Savage, React. Chem. Eng., 2019, 4(7), 1237–1252 RSC.
- N. Akiya and P. E. Savage, Chem. Rev., 2002, 102, 2725–2750 CrossRef CAS PubMed.
- W. Medina-Ramos, M. A. Mojica, E. D. Cope, R. J. Hart, P. Pollet, C. A. Eckert and C. L. Liotta, Green Chem., 2014, 16, 2147–2155 RSC.
- H. Cleaves, A. Aubrey and J. Bada, Origins Life Evol. Biospheres, 2009, 39, 109–126 CrossRef CAS PubMed.
- S. Steinberg and J. L. Bada, Science, 1981, 213, 544–545 CrossRef CAS PubMed.
- D. Long, T. Truscott, J. Cronin and R. Lee, Trans. Faraday Soc., 1971, 67, 1094–1103 RSC.
- Y. Qian, M. H. Engel, S. A. Macko, S. Carpenter and J. W. Deming, Geochim. Cosmochim. Acta, 1993, 57, 3281–3293 CrossRef CAS.
- S. Yamada, C. Hongo, R. Yoshioka and I. Chibata, J. Org. Chem., 1983, 48, 843–846 CrossRef CAS.
- L. E. Orgel, J. Mol. Evol., 1989, 29, 465–474 CrossRef CAS PubMed.
- N. Lahav, D. White and S. Chang, Science, 1978, 201, 67–69 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Mathematical model, the detailed NMR spectra, and the kinetics data of peptide bond degradation under the given pH condition, the. See DOI: 10.1039/c9cp05240b |
| This journal is © the Owner Societies 2020 |