
Theoretical insights into the effect of the overpotential on CO electroreduction mechanisms on Cu(111): regulation and application of electrode potentials from a CO coverage-dependent electrochemical model†
Lihui
Ou
 *a and
Junxiang
Chen
b
*a and
Junxiang
Chen
b
aHunan Province Cooperative Innovation Center for the Construction & Development of Dongting Lake Ecologic Economic Zone, College of Chemistry and Materials Engineering, Hunan University of Arts and Science, Changde, 415000, China. E-mail: oulihui666@126.com
bCAS Key Laboratory of Design and Assembly of Functional Nanostructures, Fujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, 350002, China
First published on 25th November 2019
Abstract
A recently proposed CO coverage-dependent electrochemical model combined with the calculation of electronic structure is applied for the first time to study the effect of the overpotential on Cu-catalyzed CO electroreduction mechanisms by changing the coverage of surface adsorbed CO. The results show that the presently defined CH2O and CHOH pathways may be able to occur parallelly under different overpotentials. However, high overpotentials will facilitate CO electroreduction, thus explaining why a high overpotential is required during CO2 electroreduction in experiments on Cu electrodes. The potential-limiting step may be further CO electroreduction into CHO, which is considered as the origin of the experimentally observed high overpotentials. The analyses of electronic structure show that an adsorbed COδ− species is formed on the Cu electrodes, validating the previous experimental speculations on electron transfer between CO and Cu electrodes. More and more electrons are transferred into the π antibonding orbitals of the adsorbed CO with increasing surface CO coverage, leading to increasing overpotential and weaker and weaker CO bonding with the Cu surface. Thus, the significantly lower barrier of further CO electroreduction at higher overpotential can be correlated with lower CO adsorption energy. Interestingly, it is found that there is greater localization of electrons around the C than the O atom in the adsorbed CO molecule, explaining why the hydrated proton prefers to reach the C atom to form intermediate CHO rather than COH.
1. Introduction
Electrocatalytic reduction technologies of CO2 offer a promising pathway to sustainable fuels and chemicals by using renewable electricity and carbon sources.1,2 Cu with a medium hydrogen overvoltage is the focus of present CO2 electroreduction investigations and has a unique ability among metals to catalyze CO2 into hydrocarbons with significant current density and reasonable current efficiency.3–9 Furthermore, the crystal faces of Cu exhibit high selectivity for the CO2 electroreduction product, in which the (111) facet of Cu favors CO2 conversion into CH4 and more open Cu(100) exhibits greater selectivity toward C2H4 production, as reported experimentally by the group of Prof. Hori.10–15 However, high overpotentials are required on Cu electrodes to achieve significant current efficiency and selectivity to CH4 and C2H4. Also, the formation mechanisms of both of these products remain ambiguous due to the complexities of electrochemical interfaces so far.3–19 In particular, the mechanism for CH4 formation on Cu(111) needs more evidence from both theory and experiment.3–8,10–16,20–22 Identifying the influence of the overpotential on CO2 electroreduction mechanisms on Cu electrodes will accelerate the design of efficient Cu-based alloy electrocatalysts that operate at low overpotentials.In situ infrared spectroscopy has offered some insights into the nature of adsorbed CO2, CO and formate. Moreover, the observed CH2 species is able to lead to the formation of hydrocarbons CH4 and C2H4 as shown by in situ X-ray photoelectron spectroscopy and Auger electron spectroscopy on Cu electrodes.5,23,24 However, experimental understanding of CO2 electroreduction mechanisms has been challenged and hindered by difficulties in performing spectroscopic characterizations of the various possible intermediates that are crucial to the production of the observed electroreduction products at electrochemical interfaces. Most recently, the feeding technique conducted by Schouten et al. has been used to analyze experimentally CO2 electroreduction mechanisms on Cu by feeding postulated intermediates into reaction systems and the subsequent product distribution is taken as evidence that the intermediates may indeed play a crucial role in the electrochemical reduction mechanisms.7 However, this approach has significant limitations since only stable intermediates can be studied. Thus, only rough and incomplete CO2 electroreduction pathways can be speculated based on early experiments. In recent decades, theoretical calculations have become a powerful tool for investigating various key electrocatalytic reactions and numerous theoretical studies have contributed to our understanding of CO2 electroreduction mechanisms on Cu electrodes by using various theoretical models,8,15,20–22,25–29 which can provide mechanistic insights that are not accessible from experiments alone and identify favored intermediates. Taking CO2 electroreduction into the CH4 product on Cu(111) as an example with the application of various theoretical models, Nørskov et al. reported the first full studies of CO2 electroreduction into CH4 on the Cu surface by using the computational hydrogen electrode (CHE) model with a hexagonal H2O overlayer.8 They concluded that CO, CHO, CH2O and CH3O are key intermediates during CH4 formation along which CO electroreduction into CHO is the potential-limiting step. The CHE model could calculate conveniently the reaction free energy changes of the proton-coupled electron transfer (PCET) steps at applied potentials. However, the activation barriers of all PCET steps are neglected by the CHE model and thus accurate kinetics are not available. The kinetic barriers for each PCET step are subsequently considered with incorporation of applied potentials at the electrode/aqueous interfaces. In contrast to the study of the CHE model, Asthagiri et al. reported the complete CO2 electroreduction pathways by calculating the applied potential-dependent barriers on Cu(111) in the aqueous phase based on Butler–Volmer theory,15,20,27 in which an adsorbed H atom is used to represent a proton–electron couple under the equilibrium potential and the COH species is predicted to be the kinetically dominant key intermediate, sequentially resulting in CHx (x = 0–4) species. The difference in electroreduction pathways between both of these models may show the importance of considering kinetic barriers. However, the generality of the assumption that the adsorbed H direct transfer represents the kinetics of a PCET step may be still in question. Moreover, the inclusion of only one or two explicit H2O molecules or a fixed H2O bilayer would be insufficient for the modeling of the aqueous-phase environment. By changing the number of H atoms added to the bilayer H2O structure to tune the electrode potentials, a DFT-based extrapolation scheme was applied to studies of CO2 electroreduction into CH4 on Cu(111) by extrapolating the calculated barriers in model systems of increasing cells.28,29 Skúlason et al. speculated that the COH, CHOH, CH2OH, CH2 and CH3 species are the reduction intermediates after CO formation. The major issue for this scheme is that it is extremely costly computationally due to extrapolation of an increasingly larger size. However, inconsistent conclusions were obtained by Nørskov et al. by using a similar electrochemical model,21 in which CHO formation is predicted to be more favorable than COH on Cu(111) thermodynamically and kinetically, subsequently leading to the formation of CHOH and CHx (x = 2–4), in contradiction to the report from Asthagiri and Skúlason. By using the implicit solvation model, CO2 electroreduction pathways were studied based on a DFT methodology that predicted onset potentials at constant electrochemical potentials by Goddard et al.22 They proposed that forming COH is more favorable than forming CHO although the difference in the barrier is subtle on Cu(111). The subsequent COH electroreduction results in the formation of CHOH and CHx (x = 1–4), which is different from the pathways reported by Asthagiri et al. The drawback of this model may be that the implicit solvation model cannot describe hydrogen bonds and interactions between adsorbates and H2O molecules. Despite numerous theoretical efforts, the mechanism for CH4 formation on Cu(111) is still under debate, and the difference in the CO2 electroreduction pathways may lie in the different electrochemical models. Furthermore, most previous theoretical investigations were done at constant charge, resulting in changes of the work function/electrode potential during a PCET step due to the limited cell size, whereas the experimental PCET steps take place at a constant electrode potential. Thus, the modeling of electrode/aqueous interfaces remains a subject of ongoing discussion to obtain sufficiently accurate computational models for describing electrochemical systems and fill in the lacking intermediate in the uncertain reaction mechanisms.30–32
The previous experimental reports from the groups of Frese, Ito and Hori felt that adsorbed CO was a crucial intermediate during CO2 electroreduction into CH4, with further CO electroreduction as the slowest step in the overall reduction pathways, which can dominate most of the Cu electrode surface.11,33–36 Thus, it can be speculated that the CO2 electroreduction kinetics is dependent on the coverage of CO. Based on this, an improved CO coverage-dependent electrochemical model can be used to predict CO coverage-dependent equilibrium potentials and elucidate CH4 production mechanisms during CO2 electroreduction on Cu(111) by calculating potential-dependent free energy profiles at the predicted equilibrium potentials. The solvent effect is essential in modeling electrode/aqueous interfaces and the previous study from Nørskov et al. validated that even a single H2O bilayer is sufficient to describe the electrical double layer.37 An explicit solvation model containing two relaxed H2O bilayers is included in the present model which allows us to better simulate the interactions between H2O molecules and adsorbates. Furthermore, the interactions between solvent and adsorbents have been elaborated in our most recent work.38,39 It is aim of this paper to offer in depth insight focused on specifically the influence of the overpotential on the CO2 reduction mechanisms on Cu(111) by applying the CO coverage-dependent electrochemical model, which may be able to result in better understanding of the possible electroreduction pathways.
2. Determination of CO coverage-dependent equilibrium potentials
Computational details including the surface and solvation model and computational parameters have been elaborated in the ESI.† The method for determining CO coverage-dependent equilibrium potentials has been reported in our most recent study,40 which was first employed to model the electroreduction pathways of CO2 at low overpotentials. For clarity, the corresponding method is again elaborated in this paper. The CO intermediate is formed during CO2 electroreduction by the following reaction:| CO2 + * + 2(H+ + e−) → CO* + H2O | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(θ/1 − θ) represent the differential adsorption energy of CO, entropy, zero-point energy, and configurational entropy contributions to ΔG(θ), respectively. Here coverage θ = n/N, in which n is the number of adsorbed CO molecules on the surface and N is the total number of surface Cu atoms. Thereby, the CO coverage-dependent equilibrium potential U (vs. RHE) can be determined when the Gibbs free energy ΔG(θ) is zero.
ln(θ/1 − θ) represent the differential adsorption energy of CO, entropy, zero-point energy, and configurational entropy contributions to ΔG(θ), respectively. Here coverage θ = n/N, in which n is the number of adsorbed CO molecules on the surface and N is the total number of surface Cu atoms. Thereby, the CO coverage-dependent equilibrium potential U (vs. RHE) can be determined when the Gibbs free energy ΔG(θ) is zero.| ΔG(θ) = ΔE(θ) + 2eU − TΔS(θ) + ΔZPE + 2kBTln(θ/1 − θ) | (2) |
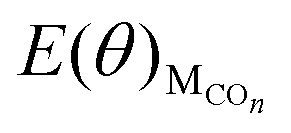 is the total energy of the surface with different CO coverage.
is the total energy of the surface with different CO coverage. | (3) |
Considering that the entropies of the adsorbed molecules are small when compared with the entropies of gases and that the zero-point energy of the adsorbed CO molecules on Cu(111) is little based on our present calculations, the contribution from the changes of entropy and ZPE together to ΔG(θ) has been estimated to be −0.42 eV for standard temperature (298 K) according to the available data from Nørskov et al.8 Thus, eqn (3) can be converted to the following form of eqn (4).
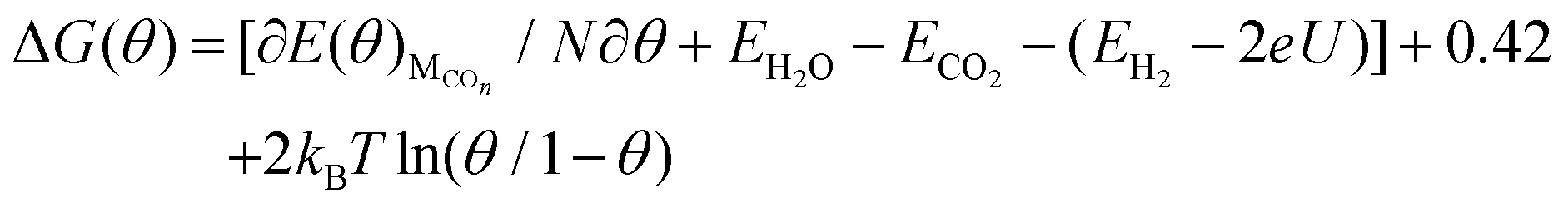 | (4) |
The values of 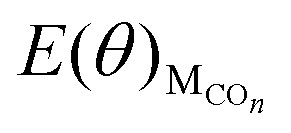 , EH2O, ECO2 and EH2 are directly available via DFT calculations. By changing the coverage of adsorbed CO on Cu(111), a series of values of
, EH2O, ECO2 and EH2 are directly available via DFT calculations. By changing the coverage of adsorbed CO on Cu(111), a series of values of 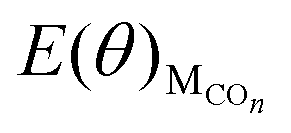 can be obtained. The values of ΔE(θ) at various coverages can be obtained according to eqn (3) by differentiating the plots of
can be obtained. The values of ΔE(θ) at various coverages can be obtained according to eqn (3) by differentiating the plots of 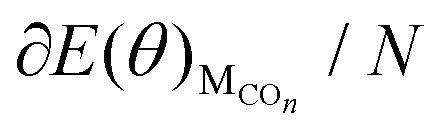 against θ.
against θ.
3. Results and discussion
3.1 Relationships between the CO coverage and equilibrium potentials
Various possible surface CO adsorption sites and the relationship between CO coverage and equilibrium potentials have been also given in our most recent study.40 As shown in Fig. 1(a), the differential adsorption energy of CO, ΔE(θ), exhibits reasonable linear dependence on θCO on Cu(111), implying that CO adsorption on Cu(111) may nearly follow the Termkin adsorption isotherms. Linearly fitting the present ΔE(θ) ∼ θCO data to a straight line enables us calculate ΔE(θ) at any θCO. Thus, the linear relationships between CO coverage (θCO) and the calculated equilibrium potentials (U) can be obtained on Cu(111) based on the above eqn (4), as shown in Fig. 1(b). It can be observed that more CO coverage will lead to a more negative equilibrium potential for CO2 electroreduction, suggesting that the overpotential may mainly originate from the adsorbed CO on the Cu electrodes. The presently calculated equilibrium potential is ca. 0.27 V (vs. RHE) when θCO is close to zero (ca. 0.05), which is comparable with the thermodynamically required equilibrium potential of 0.17 V (vs. RHE) for CO2 electroreduction and confirms the reasonability of the present theoretical model to some degree. Our present study will focus on the conditions of low and high overpotentials, which correspond to CO adsorption with different coverage on Cu(111). Thus, the effect of the overpotential on the electroreduction pathways of CO2 can be obtained.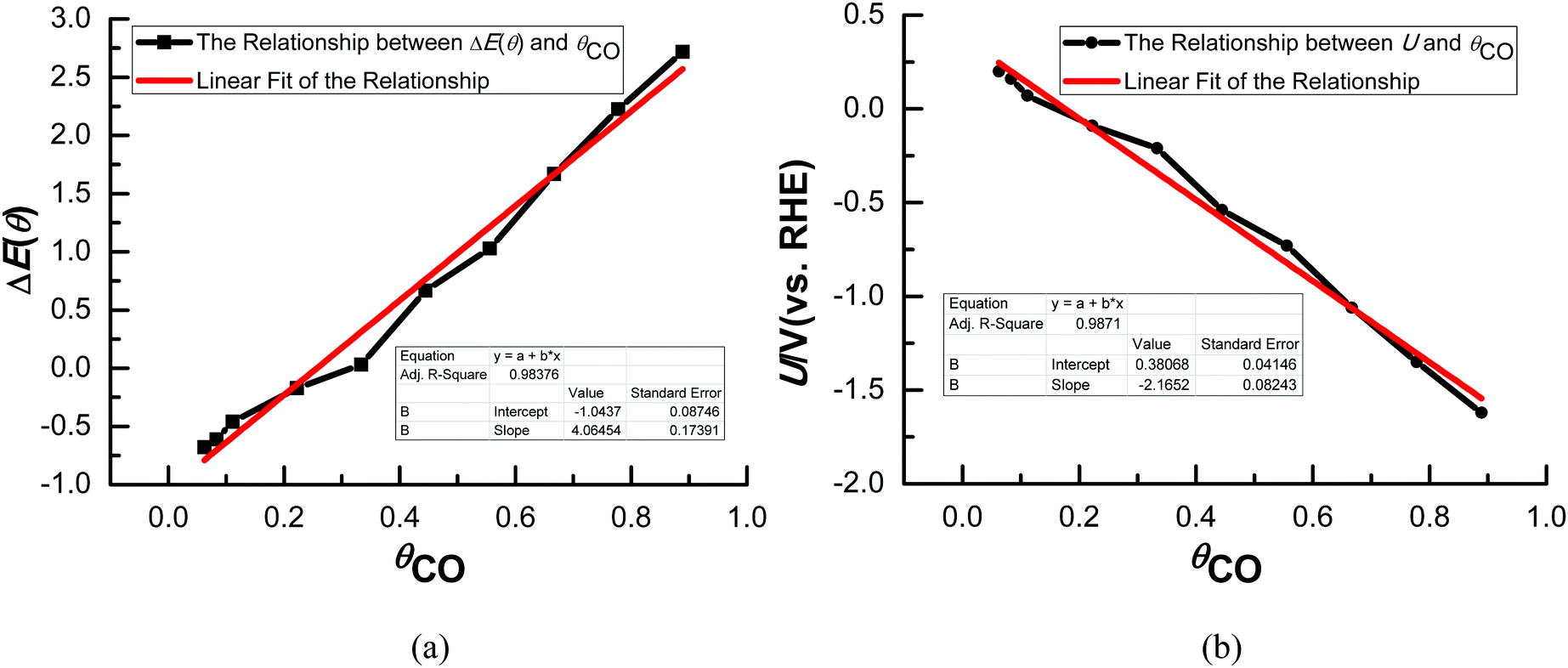 | ||
| Fig. 1 (a) The relationships between CO coverage (θCO) and the differential adsorption energy of CO, ΔE(θ); (b) the relationships between CO coverage (θCO) and the calculated equilibrium potentials (U). | ||
3.2 CO coverage-dependent electroreduction mechanisms
The previous experimental studies clearly showed that the Cu electrode surface is dominated by adsorbed CO during CO2 electroreduction and similar spectra and onset potentials for CO electrochemical reduction in experiments to those observed for that of CO2 on Cu electrodes have established that CO is a crucial and essential intermediate for producing hydrocarbons. Because of the high implied CO coverage during CO2 electroreduction, it was felt that the reduction of adsorbed CO was the key rate-determining step for the overall electroreduction of CO2 into hydrocarbons. Thus, we mainly focus on the CO electroreduction pathways on Cu(111) at the presently simulated equilibrium potentials in this paper, which has the advantage of simplifying the complication found in previous theoretical studies of CO2 electroreduction with the aim of applying the improved CO coverage-dependent model.Subsequently, further CH2O and CHOH electroreduction are considered. Further hydrogenation of intermediate CH2O may be able to form CH2OH and CH3O species on Cu(111). Barriers of 0.32 and 0.23 eV are required for CH2OH and CH3O formation at the overpotential of 0.13 V, respectively, being slightly higher than those of the corresponding processes (0.19 and 0.16 eV) at the relatively higher overpotential of 0.37 V, as shown in Fig. S3 (ESI†). Thus, the results show that further CH2O electroreduction is more favorable at the higher overpotentials. However, both of these intermediates can be easily formed in the range of low overpotentials due to the surmountable barriers at room temperature. Our presently calculated barriers for further CH2O electroreduction into CH2OH and CH3O are significantly lower than those reported by Asthagiri et al. at low overpotentials,20 in which barriers of ca. 0.90 and 0.60 eV need to be overcome at an applied potential of ca. −0.10 V (vs. RHE). The difference in the used solvation models may lead to the inconsistencies, where only a single H2O molecule or fixed H2O bilayer structure was employed in previous theoretical studies. Due to the favorable CHOH formation, the CH2OH species can be also formed by CHOH hydrogenation with almost identical barriers of ca. 0.50 eV at the low overpotentials of 0.13 and 0.37 V (see Fig. S4, ESI†). Simultaneously, we also note that the CH species can be formed by CHOH hydrogenative dissociation along with H2O formation with extremely low barriers of ca. 0.20 eV in the range of low overpotentials, which are significantly lower than those of CHOH hydrogenation into CH2OH, thereby suggesting that CH formation is more favorable during further CHOH electroreduction at low overpotentials. Considering the almost identical and extremely low barriers between further CH2O electroreduction into CH2OH and CH3O species and further CHOH electroreduction into CH species, we can speculate that parallel pathways may occur through the CH2O and CHOH intermediates during CO2 electroreduction in the range of low overpotentials.
Further electroreduction of intermediate CH2OH into CH3OH through direct hydrogenation and into the CH2 species through hydrogenative dissociation along with H2O formation are considered on Cu(111). The calculated barriers for further CH2OH electroreduction into CH3OH and CH2 are ca. 0.36 and 0.07 eV at the overpotential of 0.13 V, which is in good agreement with the previously reported values of ca. 0.29 and 0.05 eV from Goddard et al. for the formation of product CH3OH and intermediate CH2,46 further validating the rationality of our presently employed electrochemical model. However, they are 0.34 and 0.31 eV when the overpotential is increased to 0.37 V, respectively, as shown in Fig. S5 and S6 (ESI†). The results suggest that CH2 formation is more and more difficult, whereas CH3OH formation becomes more and more favorable with increasing overpotential. However, it is observed that the CH3OH and CH2 species may be able to be formed simultaneously due to the surmountable barriers at room temperature in the range of low overpotentials. Although the CH3O species can be easily formed, a significantly higher barrier of ca. 0.70 is required for its further hydrogenation into CH3OH compared to CH2OH electroreduction at the overpotentials of 0.13 and 0.37 V (see Fig. S5 and S6, ESI†). Thus, we can speculate that the CH3O species is only a spectator during CO2 electroreduction at low overpotentials. Additionally, the CH2 intermediate can be also produced with a low barrier of ca. 0.35 eV at low overpotentials by further hydrogenation of the CH species formed by CHOH hydrogenative dissociation. Subsequently, the final CH4 product can be formed by CH2 serial hydrogenation, in which almost identical barriers of ca. 0.32 and 1.30 eV need to be overcome for CH2 hydrogenation into CH3 and then CH4 at the overpotentials of 0.13 and 0.37 V, respectively, as shown in Fig. S7 (ESI†). Almost identical barriers for CH serial hydrogenation into CH4 are also obtained at low overpotentials based on the electrochemical model proposed by Asthagiri et al.20 Thus, the present results suggest that the barriers for CH serial hydrogenation into CH4 will be almost not affected in the range of low overpotentials. The corresponding energies of various possible elementary steps during CO2 electroreduction into CH4 and CH3OH on Cu(111) under conditions of low overpotentials are listed in Tables S1 and S2 (ESI†). Based on the energetics, the presently defined CH2O and CHOH pathways on Cu(111) may be able to be proposed at low overpotentials, as can be seen in Fig. 2 and 3.
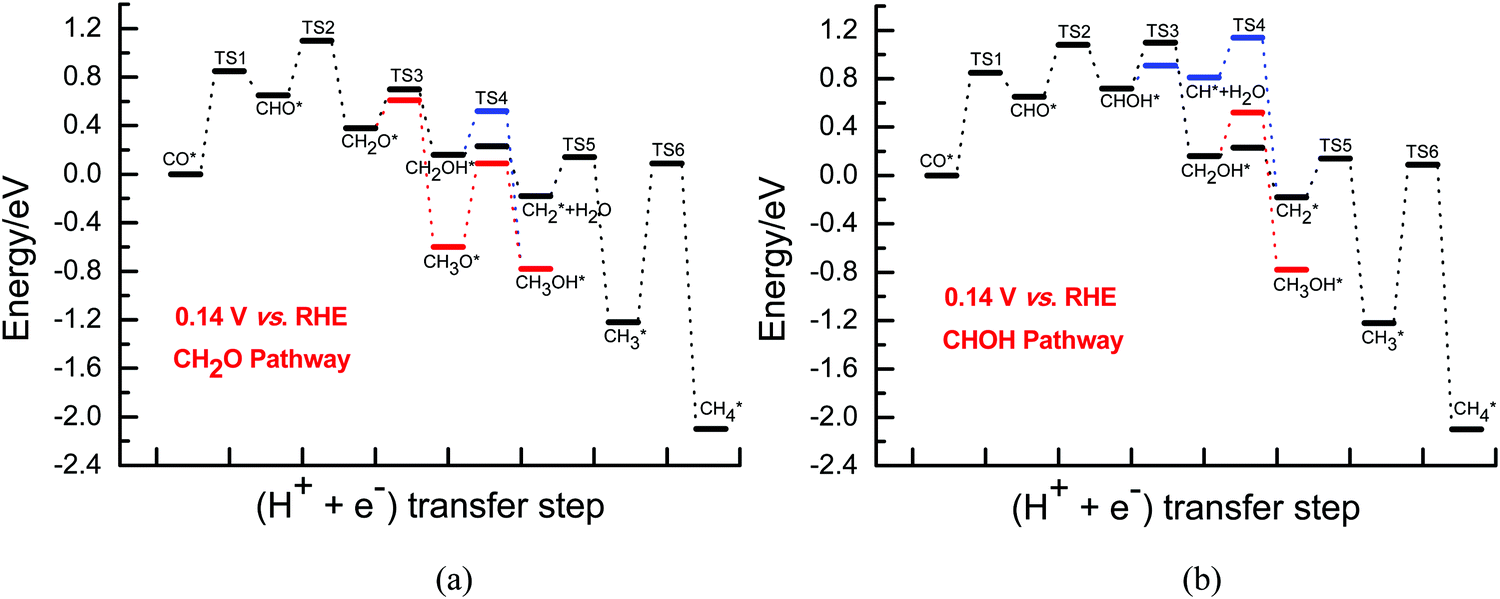 | ||
| Fig. 2 The overall energy diagram of CO electroreduction into CH4 and CH3OH on Cu(111) at the low overpotential of 0.13 V: (a) CH2O pathway; (b) CHOH pathway. | ||
 | ||
| Fig. 3 The overall energy diagram of CO electroreduction into CH4 and CH3OH on Cu(111) at the low overpotential of 0.37 V: (a) CH2O pathway; (b) CHOH pathway. | ||
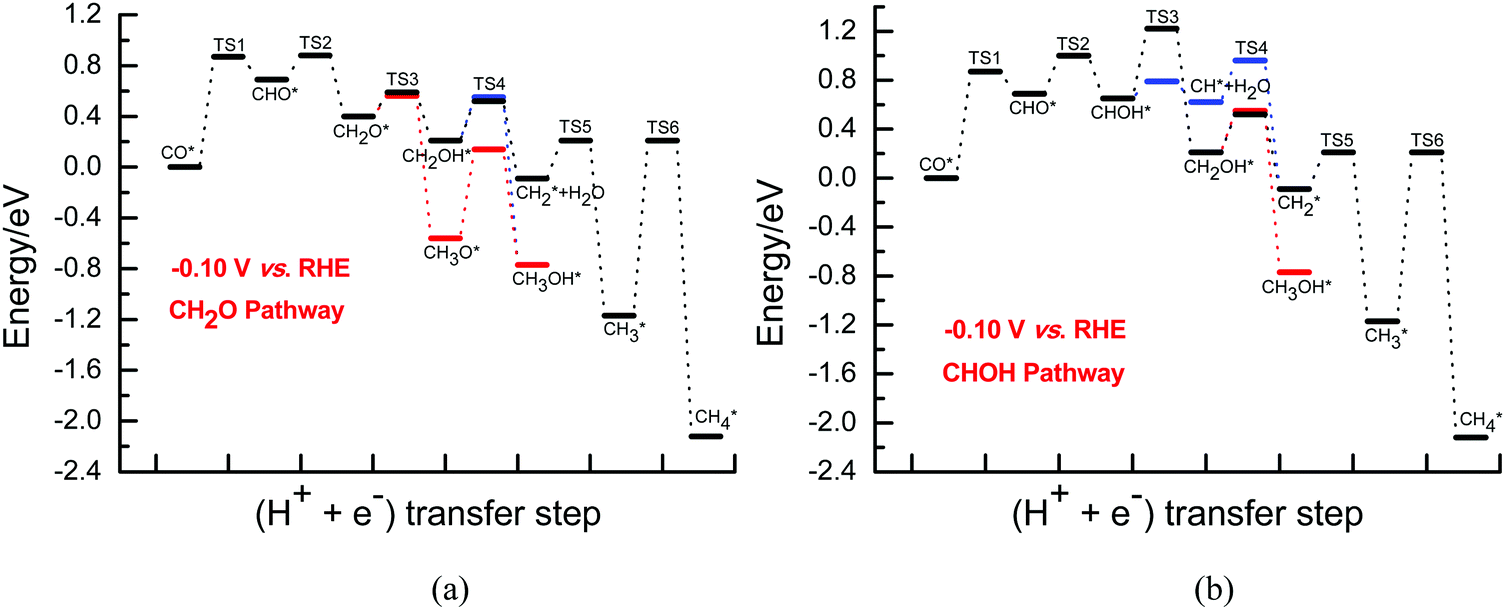
Once CH2O is formed, the further hydrogenation of CH2O into CH3O occurs more easily with a barrier of ca. 0.29 eV compared to that into CH2OH with a barrier of ca. 0.54 eV on Cu(111) under conditions of high overpotentials (see Fig. S10, ESI†). In fact, based on the electrochemical model previously proposed by Asthagiri et al.,20 activation barriers of ca. 0.25 and 0.60 eV for CH3O and CH2OH formation on Cu(111) at an applied potential of ca. −0.80 V (vs. RHE) can be obtained, respectively. Thus, the results calculated by using our present and previous electrochemical models show that CH3O formation is more favorable during further CH2O electroreduction on Cu(111) at high overpotentials. However, CH2OH may be able to be easily formed by further CHOH hydrogenation with a surmountable barrier of ca. 0.24 eV at room temperature at high overpotentials, as shown in Fig. S11 (ESI†). Furthermore, we find that the barrier for further CHOH electroreduction into the CH2OH species is considerably decreased on Cu(111) compared with that at low overpotentials, again indicating that the effect of the overpotential on the barriers is significant. A barrier of ca. 0.29 eV is required for CHOH hydrogenative dissociation into the CH species along with H2O formation at high overpotentials, which is almost identical to CHOH hydrogenation into CH2OH (see Fig. S11, ESI†), suggesting that intermediates CH2OH and CH may be able to be formed simultaneously through further CHOH electroreduction. Due to more favorable formation of CH3O and CH2OH at high overpotentials, CH3OH may be able to be produced by further CH3O and CH2OH hydrogenation with surmountable barriers of ca. 0.01 and 0.40 eV at room temperature, respectively. Moreover, hydrogenative dissociation of CH2OH into CH2 and H2O needs to overcome a higher barrier of ca. 0.56 eV than into CH3OH, as shown in Fig. S12 (ESI†). Compared with those under conditions of low overpotentials, the barriers of CH3OH production are considerably reduced, suggesting that CH3OH formation may be kinetically favorable at high overpotentials. Intermediate CH2 may be also able to be formed by CH direct hydrogenation. The calculated barrier is 0.36 eV under conditions of high overpotentials, which is lower than that of CH2 formation through CH2OH hydrogenative dissociation, showing that the CH2 species is mainly formed by CH direct hydrogenation. Finally, the product CH4 can be formed by CH2 serial hydrogenation, in which barriers of ca. 0.44 and 0.01 eV need to be overcome for CH2 hydrogenation into CH3 and then its further hydrogenation into the CH4 product at high overpotentials (see Fig. S13, ESI†), respectively. We note that the hydrogenation barriers for CH into CH2 and then CH3 will be almost not affected with increasing overpotential. However, compared with that under conditions of low overpotentials, the barrier for CH3 hydrogenation into CH4 is considerably reduced to barrierless at high overpotentials. In fact, based on the electrochemical model from Asthagiri et al.,20 the hydrogenation barrier of CH3 to CH4 on Cu(111) also goes from ca. 0.90 eV at low overpotentials to barrierless at high overpotentials, confirming the rationality of the presently proposed electrode/aqueous interface model. The energies of the various possible elementary steps during CO2 electroreduction into CH4 and CH3OH on Cu(111) under conditions of high overpotentials are listed in Table S3 (ESI†). The corresponding CH2O and CHOH pathways on Cu(111) are given in Fig. 4.
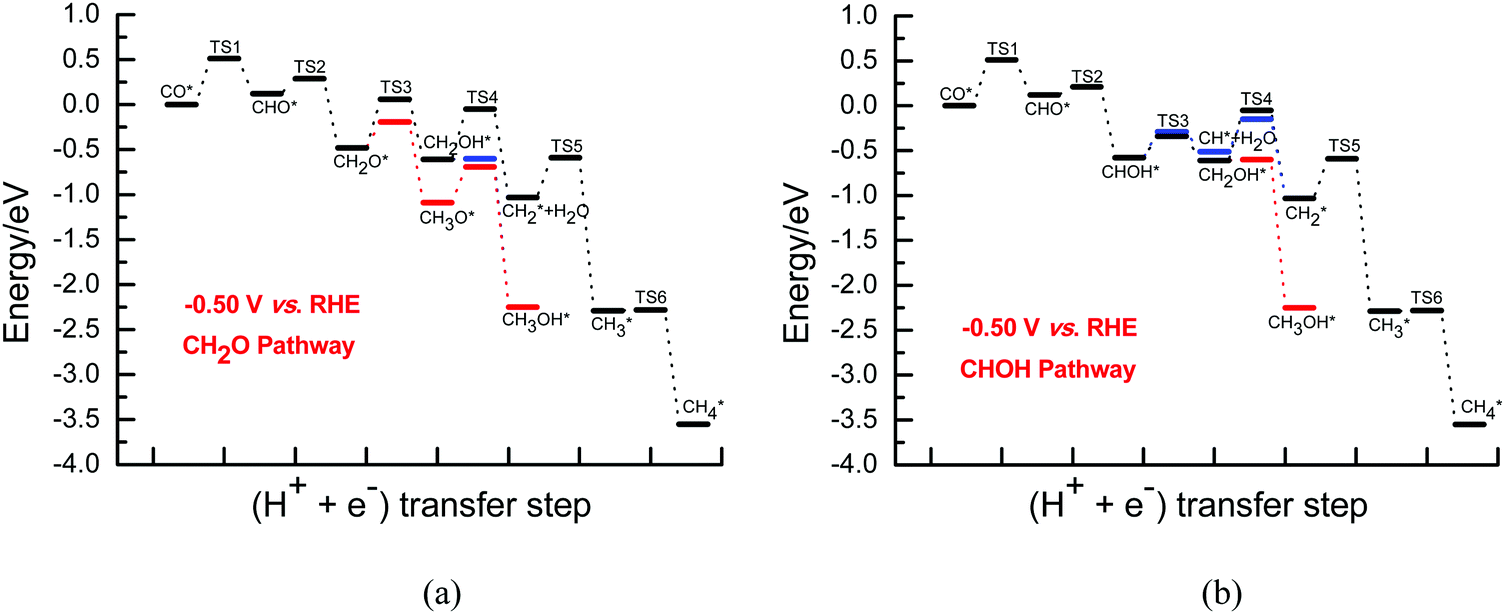 | ||
| Fig. 4 The overall energy diagram of CO electroreduction into CH4 and CH3OH on Cu(111) at the high overpotential of ca. 0.80 V: (a) CH2O pathway; (b) CHOH pathway. | ||
By comprehensive consideration of the overall energy diagrams during CO2 electroreduction into the CH4 product at the overpotentials of ca. 0.13 and 0.37 V (see Fig. 4 and 5), we speculate that the presently defined CH2O pathway can occur through the CHO, CH2O, CH2OH and CHx (x = 2–4) species in the range of low overpotentials with barriers of ca. 1.10 and 0.88 eV, respectively. Considering that there are two possibilities for further CHOH electroreduction into CH4, the overall energy diagrams are scrutinized in the CHOH pathways. It is found that the CHOH pathway can parallelly occur through CHOH electroreduction into the CH and CH2OH species due to almost identical barriers of ca. 1.10 eV at the overpotential of ca. 0.13 V, whereas it may be only able to take place through CHOH electroreduction into the CH species due to the lower required barrier than that of CHOH electroreduction into CH2OH (1.00 eV vs. 1.22 eV) at the relatively higher overpotential of 0.37 V. In the meantime, we note that the presently defined CH2O and CHOH pathways may be able to parallelly occur due to the subtle difference in the barriers in the range of low overpotentials. However, higher overpotentials will facilitate CO2 electroreduction based on the above difference in the barriers. The optimal CO electroreduction pathways on Cu(111) can be proposed under conditions of low overpotential by the above analysis, as can be seen in Fig. 5.
 | ||
| Fig. 5 The optimal CO electroreduction mechanisms on Cu(111) under conditions of low and high overpotentials: (a) CH2O pathway; (b) CHOH pathway through CHOH electroreduction into CH and CH2OH; (c) CHOH pathway through CHOH electroreduction into CH. | ||
To further determine the effect of the overpotential on CO2 electroreduction into the CH4 product, the overall energy diagrams at the higher overpotential of ca. 0.80 V are scrutinized (see Fig. 5). The CH2O pathway can occur through the CHO, CH2O, CH2OH and CHx (x = 2–4) species at high overpotentials with barriers of ca. 0.51 eV. In the CHOH pathways, two possibilities via further electroreduction of the CHOH species into CH along with H2O formation and the CH2OH species may be able to parallelly occur due to the identical barriers of ca. 0.51 eV. Thus, it is also speculated that the CH2O and CHOH pathways can parallelly occur and the optimal CO electroreduction pathways on Cu(111) can be proposed at high overpotentials, as shown in Fig. 5. The present results are found to be in excellent agreement with the available experimental data. For example, Dewulf et al. presented that adsorbed CHO and CH2 species are crucial intermediates for CH4 formation at Cu electrodes using in situ X-ray photoelectron spectroscopy and Auger electron spectroscopy during electroreduction of CO2,5 and the most recent experiment from Koper et al. studied CO2 electroreduction into CH4 on two basal planes, Cu(111) and Cu(100), using online electrochemical mass spectrometry, in which the electrochemical reduction of the various species can be measured online when the electrode potentials are changed by the tip-based sampling technique and the formation and consumption of intermediates during CO2 reduction can be followed, and showed that CHO is possibly the key intermediate toward the formation of the CH2 and CH3 species and CH4 product through the cleavage of the C–O bond,7,14 further validating the rationality of the presently employed methodology. However, the presence of high overpotentials notably reduces the barriers of the overall energy pathway. Simultaneously, it is observed that CH4 formation via further CH3 electroreduction requires a high barrier of ca. 1.40 eV at low overpotentials, whereas it is an almost barrierless process (ca. 0.01 eV) under conditions of high overpotentials. Thus, the present results can conclude that high overpotentials will be in favor of CO2 electroreduction into CH4, again explaining why a high overpotential is required during CO2 electroreduction into hydrocarbons in experiments on Cu electrodes.3–6
Additionally, we observed that an extremely low barrier of ca. 0.20 eV is required for the inverse process of further CO electroreduction into CHO, which is lower than that of further CHO electroreduction under conditions of low overpotentials. Thus, it can be speculated that intermediate CHO may be not stable in the range of low overpotentials, and can easily transform back to CO by C–H bond cleavage, leading to not easy formation of key intermediate CHO and preventing CO2 electroreduction on Cu electrodes. However, the barrier for the inverse process of further CO electroreduction into CHO is increased to ca. 0.39 eV at the high overpotential of ca. 0.80 V, which is notably higher than that of further CHO electroreduction (ca. 0.10 eV). Simultaneously, the barrier for further CO electroreduction into CHO is also significantly lowered at high overpotentials. Thus, we can speculate that the potential-limiting step may be further CO electroreduction into CHO during CO2 electroreduction into the CH4 product, which is considered as the origin of the observed experimentally high overpotentials. In fact, the previous experiments from Hori et al. and Frese et al. also showed that adsorbed CO electroreduction is the key rate-determining step for the overall reduction reaction of CO2 into hydrocarbons.11,35 Thus, the present finding on the potential-limiting step is also in excellent agreement with the available experimental data.
To further determine the origin of the overpotential, partial density of states (PDOS) analyses of an adsorbed CO molecule that can be further electrochemically reduced into hydrocarbons are performed on Cu(111) with different surface CO coverages in the present study since it can describe the number of electronic states near the Fermi energy level that are available to be occupied, as shown in Fig. 6. A high intensity of the PDOS near the Fermi energy level means high localization of electrons. For comparison, the PDOS of free CO is also included. As can be seen in Fig. 6(a) and (c), almost no obvious peaks exist in the PDOS of s orbitals of C and O atoms of adsorbed CO compared with that of the free CO molecule near the Fermi level, suggesting that maybe no significant electron transfer occurs into the s state of adsorbed CO. However, obvious peaks are observed in the p orbitals of C and O atoms near the Fermi level (ca. 0–3 eV), as shown in Fig. 6(b) and (d), indicating that electrons from the surface and electrochemical interfaces are transferred into π antibonding orbitals of the adsorbed CO molecule. Thus, an adsorbed COδ− species is formed on Cu(111), validating the previous experimental speculations on electron transfer between CO and Cu electrodes.5,11 To ascertain the effect of the CO coverage on electron transfer, the PDOS of p orbitals near the Fermi level in Fig. 6 is amplified, as can be seen in Fig. 7. The calculated value of the Fermi level is ca. 3.45, 3.49, 3.72 and 3.79 eV in the present research range of CO coverage, respectively, which gradually increases with increasing CO coverage. Furthermore, it is clearly observed that the intensity of the peaks becomes larger and larger with increasing CO coverage, indicating more and more electron transfer into π antibonding orbitals of the adsorbed CO molecule and higher and higher localization of electrons around C and O atoms. The higher peak intensities of π antibonding orbitals near the Fermi level (ca. 0–3 eV) indicate weaker CO bonding with the Cu surface, as can be confirmed by the calculated less and less negative CO adsorption energies of −2.86, −1.15, −0.76 and −0.59 eV with increasing surface CO coverage at the presently simulated electrochemical interfaces, suggesting that higher overpotentials will reduce the CO adsorption strength. Thus, the abovementioned significantly lower activation barrier of further CO electroreduction into CHO on Cu(111) at higher overpotential can be correlated with lower CO adsorption energy. Simultaneously, higher peak intensities in the PDOS of p orbitals of C atoms are observed compared to those of O atoms (see Fig. 7), suggesting greater localization of electrons around C atoms, which will be further confirmed by the following Löwdin population analysis.
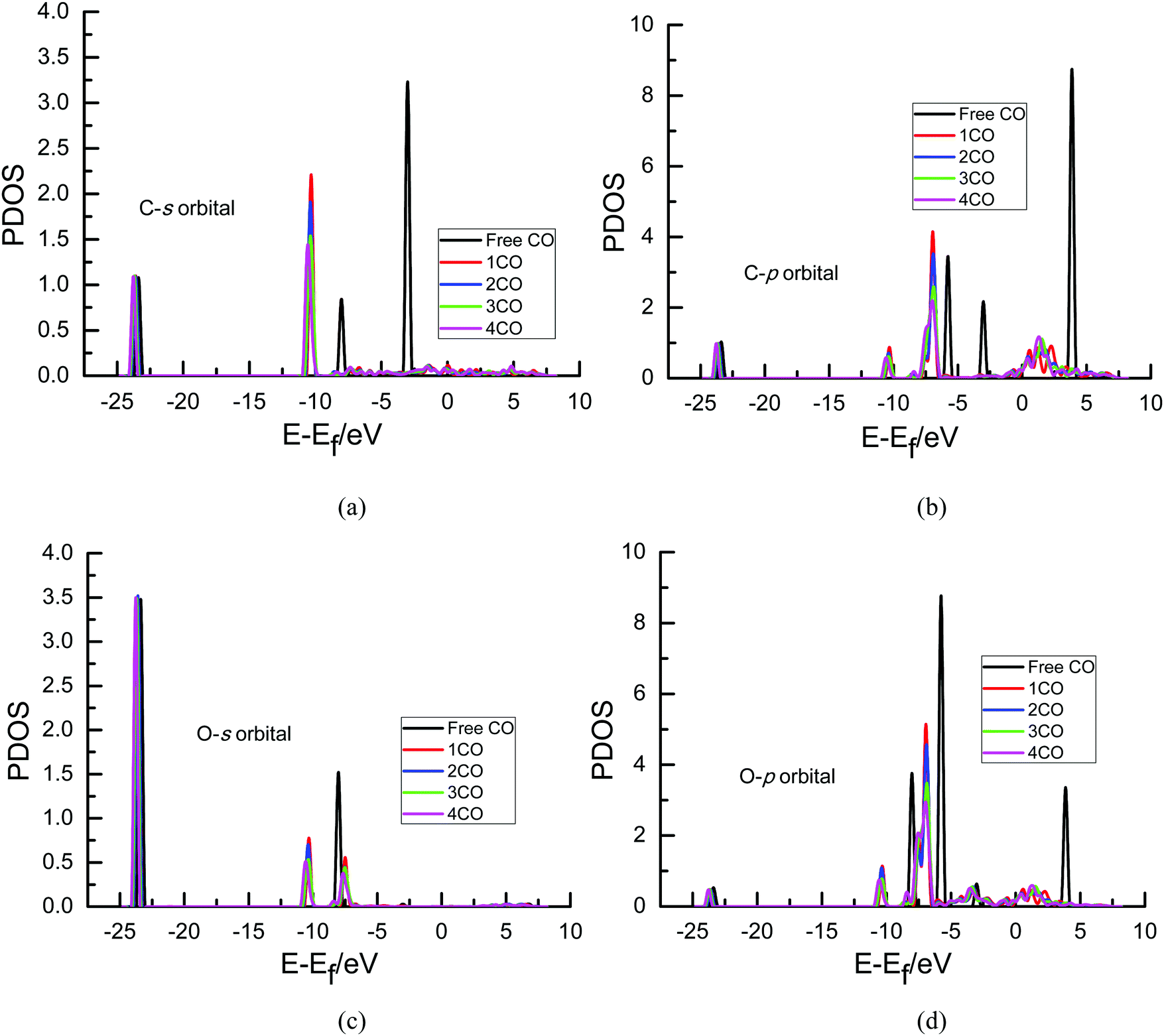 | ||
| Fig. 6 Local density of states of free and adsorbed CO on the Cu(111) surface with different CO coverages: (a) s orbital of C; (b) p orbital of C; (c) s orbital of O; (d) p orbital of O. The Fermi level is zero. | ||
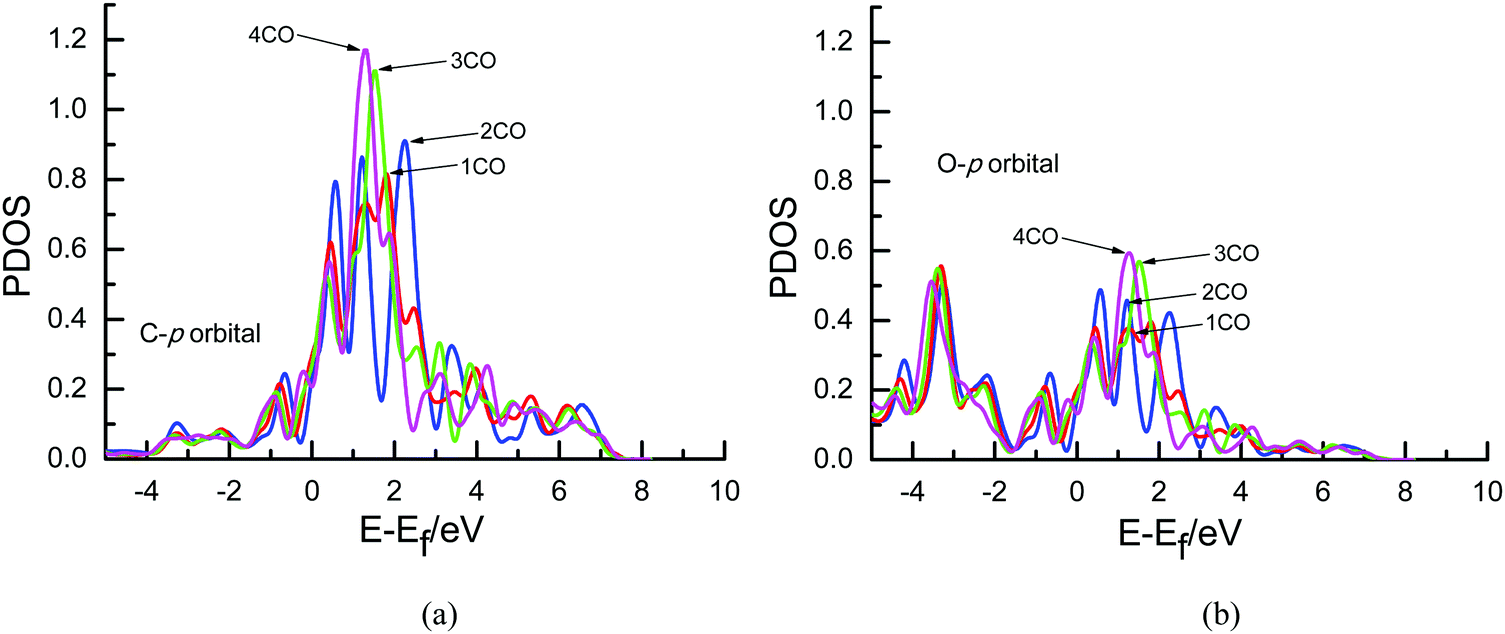 | ||
| Fig. 7 The amplified local density of states of an adsorbed CO molecule that can be further electrochemically reduced into hydrocarbons on Cu(111) with different surface CO coverages: (a) p orbital of C; (b) p orbital of O. The Fermi level is zero. | ||
The quantative analysis of electronic structures will facilitate the further understanding of the origin of the overpotentials and the effect of surface CO coverage on electron transfer during CO2 electroreduction. The Löwdin charge of an adsorbed CO molecule that can be further electrochemically reduced into hydrocarbons on Cu(111) with different surface CO coverages can be obtained from Löwdin population analysis based on the projected electron densities of states. As shown in Table 1, the net electron gains of the p orbitals of the C and O atoms in the adsorbed CO molecule that can be further electrochemically reduced into hydrocarbons gradually increased with increasing surface CO coverage, which are filled into π antibonding orbitals based on the above PDOS analysis. Furthermore, the total net electron gain of adsorbed CO also gradually increased, indicating that the net electron gain of the s orbital is negligible and further conforming the results obtained by the above PDOS analysis. Interestingly, it is noted that the C atom obtains more electrons than the O atom in the adsorbed CO molecule, which quantatively confirms the abovementioned greater localization of electrons around C atoms and explains why the hydrated proton tends to reach the C atom to form intermediate CHO rather than COH as indicated in the above MEP analysis. Simultaneously, the greater and greater electron transfer into the π antibonding orbitals of the adsorbed CO molecule with increasing CO coverage may lead to increasing overpotential and easier and easier occurrence of further CO electroreduction.
| Difference of electron/atom (Δq)a | |||||
|---|---|---|---|---|---|
| Orbital | 1CO | 2CO | 3CO | 4CO | |
| a Positive values of Δq imply an electron gain by the component. | |||||
| C | s | −0.2467 | −0.2363 | −0.2356 | −0.2308 |
| p | +0.7740 | +0.7809 | +0.7833 | +0.8039 | |
| Total | +0.5237 | +0.5446 | +0.5477 | +0.5730 | |
| O | s | +0.0208 | +0.0192 | +0.0169 | +0.0179 |
| p | +0.0452 | +0.0619 | +0.0778 | +0.0920 | |
| Total | +0.0660 | +0.0811 | +0.0947 | +0.1099 | |
4. Conclusions
In this paper, a recently proposed CO coverage-dependent electrochemical model combined with the calculation of electronic structure is used for the first time to study the effect of the overpotential on Cu-catalyzed CO electroreduction mechanisms by changing the coverage of surface adsorbed CO. The results show that the presently defined CH2O and CHOH pathways may be able to parallelly occur at the presently simulated low and high overpotentials based on the analyses of potential-dependent energetics at electrode/aqueous interfaces. However, high overpotentials will be in favor of CO electroreduction into CH4 due to the notably reduced kinetic barrier of the overall energy pathway, thus explaining why a high overpotential is required during CO2 electroreduction into hydrocarbons in experiments on Cu electrodes. The potential-limiting step may be further CO electroreduction into CHO during CO2 electroreduction by analyzing the potential-dependent energetics for the inverse process of CO electroreduction into CHO and further CHO electroreduction, which is considered as the origin of the experimentally observed high overpotentials. The present studies also suggest that CH4 may be more facile to form than CH3OH on Cu(111) in the range of low overpotentials, whereas the CH3OH product may be able to be formed along with the CH4 product at high overpotentials. Based on the analysis of electronic structure, an adsorbed COδ− species is formed on Cu(111), validating the previous experimental speculations on electron transfer between CO and Cu electrodes. Moreover, more and more electrons are transferred into the π antibonding orbitals of the adsorbed CO molecule with increasing surface CO coverage, leading to increasing overpotential and weaker and weaker CO bonding with the Cu surface. Thus, the significant lower activation barrier of further CO electroreduction into CHO on Cu electrodes at higher overpotential can be correlated with lower CO adsorption energy. Interestingly, it is found that there is greater localization of electrons around the C than the O atom in the adsorbed CO molecule, explaining why the hydrated proton prefers to reach the C atom to form intermediate CHO rather than COH. The excellent consistencies between our predicted pathways and the available experimental and partial theoretical data confirm the reasonability of the presently employed electrode/aqueous interface model.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Hunan Province (Grant No. 2018JJ2273); the Outstanding Youth Foundation of Education Department of Hunan Province (Grant No. 16B178) and the National Natural Science Foundation of China (Grant No. 21303048).References
- S. Chu, Y. Cui and N. Liu, The Path towards Sustainable Energy, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- B. Obama, The Irreversible Momentum of Clean Energy, Science, 2017, 355, 126–129 CrossRef CAS PubMed.
- Y. Hori, K. Kikuchi and S. Suzuki, Production of CO and CH4 in Electrochemical Reduction of CO2 at Metal Electrodes in Aqueous Hydrogencarbonate Solution, Chem. Lett., 1985, 1695–1698 CrossRef CAS.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Production of Methane and Ethylene in Electrochemical Reduction of Carbon Dioxide at Copper Electrode in Aqueous Hydrogencarbonate Solution, Chem. Lett., 1986, 897–898 CrossRef CAS.
- D. W. Dewulf, T. Jin and A. J. Bard, Electrochemical and Surface Studies of Carbon Dioxide Reduction to Methane and Ethylene at Copper Electrodes in Aqueous Solutions, J. Electrochem. Soc., 1989, 136, 1686–1691 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, A Review of the Aqueous Electrochemical Reduction of CO2 to Hydrocarbons at Copper, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, A New Mechanism for the Selectivity to C1 and C2 Species in the Electrochemical Reduction of Carbon Dioxide on Copper Electrodes, Chem. Sci., 2011, 2, 1902–1909 RSC.
- A. A. Peterson, F. Abild-Pederson, F. Studt, J. Rossmeisl and J. K. Nørskov, How Copper Catalyzes the Electroduction of Carbon Dioxide into Hydrocarbon Fuels, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- W. J. Durand, A. A. Peterson, F. Studt, F. Abild-Pederson and J. K. Nørskov, Structure Effects on the Energetics of the Electrochemical Reduction of CO2 by Copper Surfaces, Surf. Sci., 2011, 605, 1354–1359 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, Electrochemical Reduction of Carbon Dioxide at Various Series of Copper Single Crystal Electrodes, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- Y. Hori, A. Murata and R. Takahashi, Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- C. Hahn, T. Hatsukade, Y. G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Engineering Cu surfaces for the electrocatalytic conversion of CO2: Controlling selectivity toward oxygenates and hydrocarbons, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5918–5923 CrossRef CAS PubMed.
- K. J. P. Schouten, Z. Qin, E. Perez Gallent and M. T. M. Koper, Two Pathways for the Formation of Ethylene in CO Reduction on Single-Crystal Copper Electrodes, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- W. J. Luo, X. W. Nie, M. J. Janik and A. Asthagiri, Facet Dependence of CO2 Reduction Paths on Cu Electrodes, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- F. Calle-Vallejo and M. T. M. Koper, Theoretical Considerations on the Electroreduction of CO to C2 Species on Cu(100) Electrodes, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, Theoretical Insights into a CO Dimerization Mechanism in CO2 Electroreduction, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, Insights into C-C Coupling in CO2 Electroreduction on Copper Electrodes, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- X. W. Nie, W. J. Luo, M. J. Janik and A. Asthagiri, Reaction Mechanisms of CO2 Electrochemical Reduction on Cu(111) Determined with Density Functional Theory, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- X. Y. Liu, J. P. Xiao, H. J. Peng, X. Hong, K. Chan and J. K. Nørskov, Understanding Trends in Electrochemical Carbon Dioxide Reduction Rates, Nat. Commun., 2017, 8, 15438 CrossRef CAS PubMed.
- H. Xiao, T. Cheng, W. A. Goddard III and R. Sundararaman, Mechanistic Explanation of the pH Dependence and Onset Potentials for Hydrocarbon Products from Electrochemical Reduction of CO on Cu (111), J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, Mechanistic Insights into the Reduction of CO2 on Tin Electrodes using in Situ ATR-IR Spectroscopy, ACS Catal., 2015, 5, 3148–3156 CrossRef CAS.
- M. C. Figueiredo, I. Ledezma-Yanez and M. T. M. Koper, In Situ Spectroscopic Study of CO2 Electroreduction at Copper Electrodes in Acetonitrile, ACS Catal., 2016, 6, 2382–2392 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the Computational Design of Solid Catalysis, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Density Functional Theory in Surface Chemistry and Catalysis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- G. Rostamikia, A. J. Mendoza, M. A. Hickner and M. J. Janik, First-Principles Based Microkinetic Modeling of Borohydride Oxidation on a Au(111) Electrode, J. Power Sources, 2011, 196, 9228–9237 CrossRef CAS.
- J. Hussain, H. Jónsson and E. Skúlason, Calculations of Product Selectivity in Electrochemical CO2 Reduction, ACS Catal., 2018, 8, 5240–5249 CrossRef CAS.
- J. Hussain, E. Skúlason and H. Jónsson, Computational Study of Electrochemical CO2 Reduction at Transition Metal Electrodes, Procedia Comput. Sci., 2015, 51, 1865–1871 CrossRef.
- M. Nielsen, M. E. Björketun, M. H. Hansen and J. Rossmeisl, Towards First Principles Modeling of Electrochemical Electrode-Electrolyte Interfaces, Surf. Sci., 2015, 631, 2–7 CrossRef CAS.
- E. Skúlason, Modeling Electrochemical Reactions at the Solid-Liquid Interface Using Density Functional Calculations, Procedia Comput. Sci., 2015, 51, 1887–1896 CrossRef.
- E. Skúlason and H. Jónsson, Atomic Scale Simulations of Heterogeneous Electrocatalysis: Recent Advances, Adv. Phys.: X, 2017, 2, 481–495 Search PubMed.
- H. Noda, S. Ikeda, Y. Oda and K. Ito, Potential Dependencies of the Products on Electrochemical Reduction of Carbon Dioxide at a Copper Electrode, Chem. Lett., 1989, 289–292 CrossRef CAS.
- Y. Hori, A. Murata and Y. Yoshinami, Adsorption of CO, Intermediately Formed in Electrochemical Reduction of CO2 at a Copper Electrode, J. Chem. Soc., Faraday Trans., 1991, 87, 125–128 RSC.
- J. J. Kim, D. P. Summers and K. W. Frese Jr, Reduction of CO2 and CO to Methane on Cu Foil Electrodes, J. Electroanal. Chem., 1988, 245, 223–244 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, Electroreduction of Carbon Monoxide to Methane and Ethylene at a Copper Electrode in Aqueous Solutions at Ambient Temperature and Pressure, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS.
- E. Skúlason, V. Tripković, M. E. Björketun, S. Gudmundsdóttir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jónsson and J. K. Nørskov, Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the basis of Density Functional Theory Calculations, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef.
- L. H. Ou, J. X. Chen, Y. D. Chen and J. L. Jin, Mechanistic Study of Pt-Catalyzed Electrooxidation of HCOOH in Acid Medium: Kinetic Considerations on the Effect of Solvation, J. Phys. Chem. C, 2018, 122, 24871–24884 CrossRef CAS.
- L. H. Ou, Theoretical Insights into the Effect of Solvation and Sublayer Ru on Pt-Catalytic CH3OH Oxidation Mechanisms in the Aqueous Phase, J. Phys. Chem. C, 2018, 122, 14554–14565 CrossRef CAS.
- L. H. Ou, J. X. Chen, Y. D. Chen and J. L. Jin, Mechanistic study on Cu-catalyzed CO2 Electroreduction into CH4 at Simulated Low Overpotentials Based on An Improved Electrochemical Model, Phys. Chem. Chem. Phys., 2019, 21, 15531–15540 RSC.
- G. S. Karlberg, T. F. Jaramillo, E. Skúlason, J. Rossmeisl, T. Bligaard and J. K. Nørskov, Cyclic Voltammograms for H on Pt(111) and Pt(100) from First Principles, Phys. Rev. Lett., 2007, 99, 126101 CrossRef CAS PubMed.
- E. Skúlason, G. S. Karlberg, J. Rossmeisl, T. Bligaard, J. Greeley, H. Jónsson and J. K. Nørskov, Density Functional Theory Calculations for the Hydrogen Evolution Reaction in an Electrochemical Double Layer on the Pt(111) Electrode, Phys. Chem. Chem. Phys., 2007, 9, 3241–3250 RSC.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- F. Yang, Q. F. Zhang, Y. W. Liu and S. L. Chen, A Theoretical Consideration on the Surface Structure and Nanoparticle Size Effects of Pt in Hydrogen Electrocatalysis, J. Phys. Chem. C, 2011, 115, 19311–19319 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadóttir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- H. Xiao, T. Cheng and W. A. Goddard III, Atomistic Mechanisms Underlying Selectivities in C1 and C2 Products from Electrochemical Reduction of CO on Cu(111), J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS PubMed.
- A. A. Peterson and J. K. Nørskov, Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- K. S. Rawat, A. Mahata and B. Pathak, Thermochemical and Electrochemical CO2 Reduction on Octahedral Cu Nanocluster: Role of Solvent towards Product Selectivity, J. Catal., 2017, 349, 118–127 CrossRef CAS.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, Electrocatalytic Conversion of Carbon Dioxide to Methane and Methanol on Transition Metal Surfaces, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- E. Pérez-Gallent, G. Marcandalli, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Structure- and Potential-Dependent Cation Effects on CO Reduction at Copper Single-Crystal Electrodes, J. Am. Chem. Soc., 2017, 139, 16412–16419 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp05043d |
| This journal is © the Owner Societies 2020 |