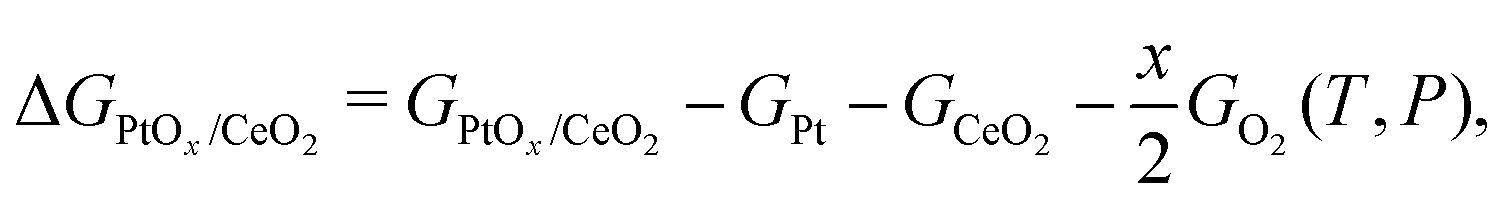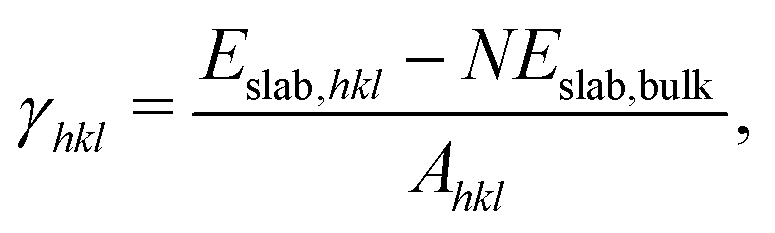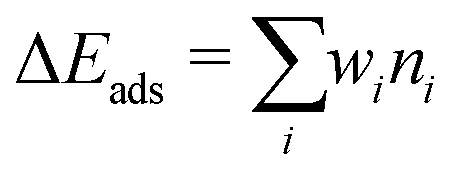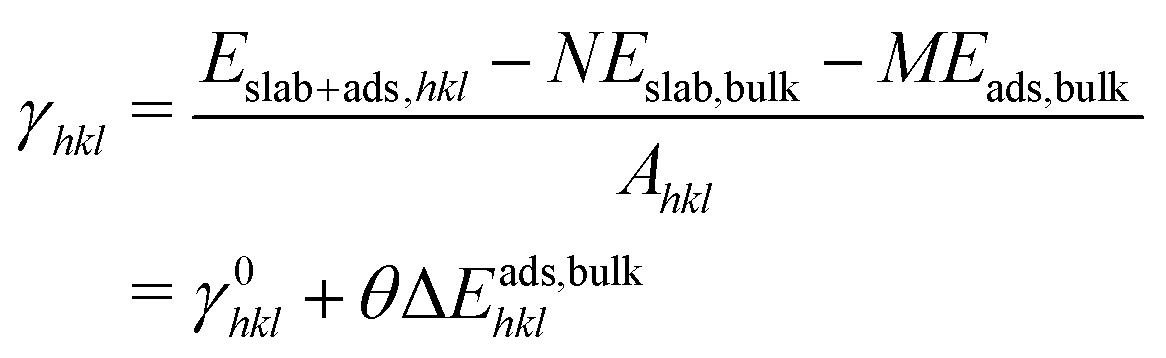Atomically dispersed platinum on low index and stepped ceria surfaces: phase diagrams and stability analysis†
Received
7th September 2019
, Accepted 7th October 2019
First published on 7th October 2019
Abstract
Through the combination of density functional theory calculations and ab initio atomistic thermodynamics modeling, we demonstrate that atomically dispersed platinum species on ceria can adopt a range of local coordination configurations and oxidation states that depend on the surface structure and environmental conditions. Unsaturated oxygen atoms on ceria surfaces play the leading role in stabilization of PtOx species. Any mono-dispersed Pt0 species are thermodynamically unstable compared to bulk platinum, and oxidation of Pt0 to Pt2+ or Pt4+ is necessary to stabilize mono-dispersed platinum atoms. Reduction to Pt0 leads to sintering. Both Pt2+ and Pt4+ prefer to form the square-planar [PtO4] configuration. The two most stable Pt2+ species on the (223) and (112) surfaces are thermodynamically favorable between 300 and 1200 K. The most stable Pt4+ species on the (100) surface tends to desorb from the surface as gas phase above 950 K. The resulting phase diagrams of the atomically dispersed platinum in PtOx clusters on various ceria surfaces under a range of experimentally relevant conditions can be used to predict dynamic restructuring of atomically dispersed platinum catalysts and design new catalysts with engineered properties.
1 Introduction
Atomically dispersed metals on a support attract great attention due to their high surface to volume ratio, thus 100% atomic utilization efficiency, as well as their catalytic activity and selectivity.1–7 Such highest atomic utilization is especially important for precious platinum group metal (PGM) catalysts, due to their low abundance but a wide usage in a broad range of applications, such as vehicle emissions control,8,9 water–gas shift reaction,10–14 methanol reforming,15–17 carbon monoxide oxidation18 and electro-catalysis.19–22 Various oxidic supports, including iron oxide,1,23 alumina,24–26 titania,27,28 ceria,29–32 and copper(I) oxide,33 have been used to anchor platinum atoms with atomic dispersion.
At the same time, atomically dispersed platinum atoms often exhibit highly dynamic behavior, and thus can aggregate into nanoparticles and change configurations under reaction conditions.34–37 As a result, low loading of metal and careful control of conditions are often required to achieve and maintain site isolation. There is also an active debate regarding the catalytic activity of such species as well as their chemical nature.32,38,39 Therefore, stabilization and determination of the structure of the dispersed species are central goals in the development of atomically dispersed catalysts.
Ceria inhibits sintering of platinum by strong metal–support interaction.40,41 In catalysts comprising atomically dispersed platinum on ceria, platinum is claimed to be present as Pt0, Pt+, Pt2+ and Pt4+ species. CeO2(100) can strongly anchor Pt2+ by coordinating to four surface oxygen atoms in a square-planar configuration.29,42 Recent experimental and theoretical data also emphasized that defect sites and step edges on the ceria(111) surface provide stable sites for atomically dispersed platinum.32,43 Dvořák et al.30 demonstrated that by carefully engineering the density of surface steps, high loading of platinum could be stabilized on the support in the form of atomically dispersed platinum ions. Jones et al.31 showed that volatile platinum species can be trapped by ceria step sites to form atomically dispersed platinum species that are stable at temperatures as high as 800 °C.
The structure of the active sites of atomically dispersed platinum on ceria is debated. Pereira-Hernández et al.32 showed that different synthesis methods (conventional wet chemistry synthesis and high temperature vapor phase synthesis) exhibit differences in the carbon monoxide infrared peak position, suggesting that there are different atomically dispersed platinum species on ceria. Moreover, the structure after activation had changed, resulting in different activity. Derevyannikova et al.44 found the main platinum species to be Pt2+ and Pt4+ in the Pt–CeO2 nanocomposites calcined in air at 450 °C to 1000 °C with a platinum loading over a range of 1–30 wt%. The formation of Pt4+ species on thin films of ceria was observed, while Pt2+ species on the ceria surface exhibited high electrocatalytic activity.45 Theoretical studies demonstrated that the excess oxygen atoms at the surface step edges are essential for the stability of Pt2+ ions.43,46,47 Our previous work showed that trapped platinum species have a square-planar [PtO4] structure at the step site.48 It has also been demonstrated that isolated platinum species on titania can adopt a range of local coordination environments and oxidation states that evolve in response to varied environmental conditions.27 Very recently, molecular dynamics simulation revealed that several oxidation and charge states coexist for monodispersed platinum on CeO2(100) surface.49
Due to the existence of various anchor sites on the ceria surface and the complex nature of bonding between platinum and ceria, there is a large number of possible atomically dispersed platinum species. The population of these platinum species depend strongly on the synthesis methods, temperature, activation procedure and the reaction gas environment. In this study, we have carried out systematic density functional theory (DFT) calculations corrected by on-site Coulomb interactions (DFT+U) to investigate the structure and stability of atomically dispersed platinum on a range of ceria surfaces: (100), (110), (111) lower index surfaces as well as (221), (331), (112) and (223) high index stepped surfaces. Effects of temperature and oxygen pressure have also been considered using the ab initio atomistic thermodynamics (aiAT) framework, allowing the construction of phase diagrams of supported platinum species.
2 Computational models and methods
2.1 Electronic structure method
Spin-polarized DFT calculations were performed using the Quickstep module in the CP2K simulation package.50 The generalized gradient approximation (GGA) using the Perdew, Burke and Ernzerhof (PBE) functional was chosen to evaluate the exchange–correlation energy.51 Valence electrons were treated explicitly, whereas interactions with the frozen atom core were described using Goedecker–Teter–Hutter (GTH) pseudo potentials52 with 30, 6, 4, and 18 valence electrons for Ce, O, C and Pt, respectively. A double-zeta valence plus polarization (DZVP) basis set, optimized according to the Mol-Opt method,53 has been used. A cutoff of 1000 Ry has been used for the auxiliary plane wave expansion of the charge density. Brillouin zone integration was performed with a reciprocal space mesh consisting of only the gamma point. The strict convergence criteria of 10−7 Ha was used for the SCF calculations. We carry out spin-polarized calculations assuming singlet and triplet states of every considered structure. All discussed structures correspond to the lowest energy spin state of the given system. The DFT+U method, based on the Mulliken 4f state population analysis, was used to describe the Ce 4f electrons. A U value of 4.5 eV was used in order to get a balanced description of different properties of ceria, in line with the previous works using typical U values in the range of 3.0–5.5 eV.54,55 Discussion of the choice of the U value can be found in the ESI† (Section SI).
In this study, the adsorption energy was calculated as follows:
| | | ΔEads = Eslab+ads − Eads − Eslab | (1) |
where
Eslab+ads,
Eslab, and
Eads are the calculated energies of the surface with adsorbed species, a clean surface, and a free PtO
x species, respectively.
In order to allow a comparison with experimental data, bulk modulus (B) values have been obtained by fitting a Birch–Murnaghan equation of state to the energy–volume data. Bader charge analysis has also been performed using the program developed by the Henkelman group.56
2.2
Ab initio thermodynamics
The effect of the gaseous environment on the relative stability of the considered surface structure was captured by the method of ab initio atomistic thermodynamics.57 The Gibbs free energy of a gas phase at temperature T and partial pressure P is given by:| | G(T,P) = EDFT + EZPE + ΔG(T,P0) + kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(P/P0), ln(P/P0), | (2) |
where EDFT is the energy calculated by DFT at 0 K, EZPE is the zero point energy, P0 is the standard pressure, and ΔG(T,P0) includes the contributions from translational, rotational, vibrational and electronic free energy terms of the species under consideration. The detailed derivation for ΔG(T,P0) can be found elsewhere.58 These were implemented in the Atomic Simulation Environment (ASE) Python package.59 The Gibbs free energy of the solid phase change with T and P is much smaller compared to the gas phase, thus is neglected in this study. The stability of surface structure PtOx/CeO2 compared to the bulk platinum bulk, oxygen molecule and clean ceria surface is given by:| | 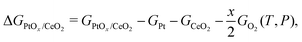 | (3) |
where GPtOx/CeO2, GCeO2, GPt and GO2(T,P) are the free energies of the surface with adsorbed species PtOx, a clean surface, bulk platinum phase and oxygen molecule, respectively.
The equilibrium phase diagram for different surface structures PtOm/CeO2 and PtOn/CeO2 is calculated by solving the following equation:
| | | GPtOm/CeO2(T,P) − GPtOn/CeO2(T,P) − (m − n)μO(T,P) = 0, | (4) |
where
m and
n is the number of oxygen atoms in the surface species and
μO is the chemical potential of oxygen atoms. The equilibrium point (
T,
P) is then plotted.
3 Results and discussion
3.1 Bare slabs
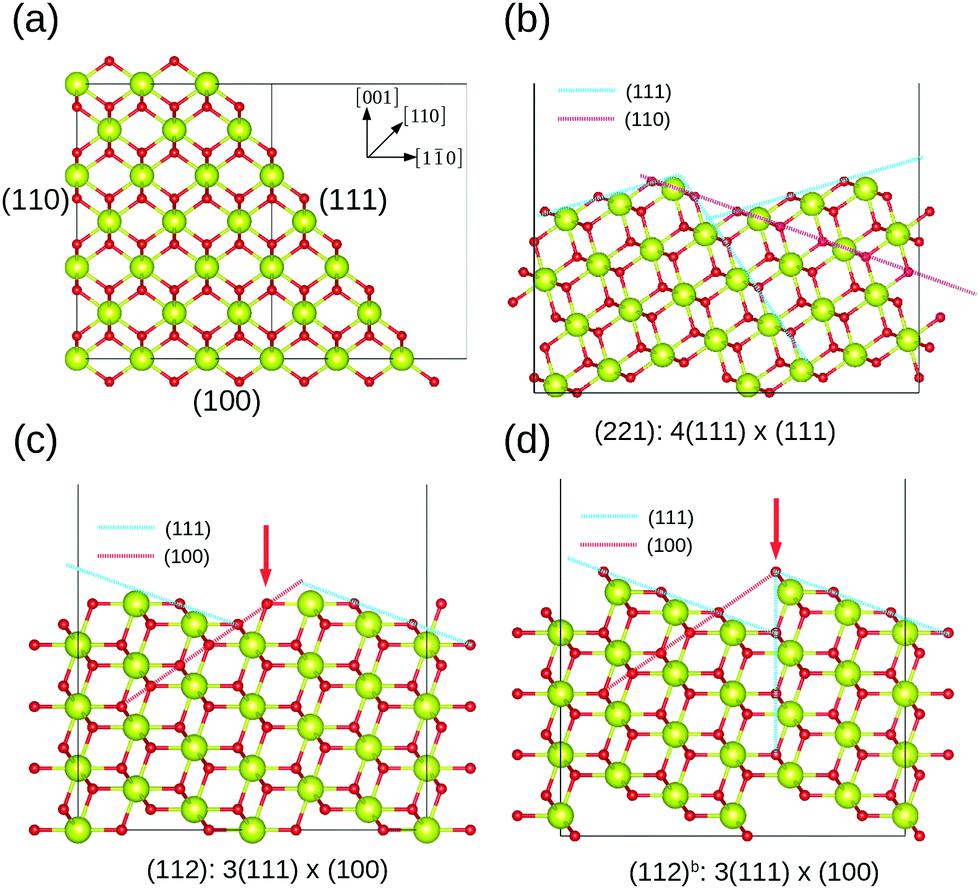
Ceria has a cubic fluorite structure (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). In this structure, all cerium atoms are eight-coordinated, while oxygen atoms are four-coordinated, located at the center of a tetrahedron of cerium atoms. Fig. 1 shows the low index and high index surface models. (110) and (111) are additionally terminated by a threefold-coordinated oxygen atom, and (100) by a twofold-coordinated oxygen atom, as within this crystal structure it is otherwise impossible to define a cerium-terminated slab model without breaking the stoichiometry. The coordination number of surface cerium at the (100) and (110) surfaces is six; the coordination number at the (111) surface is seven. High index surfaces are morphologically characterized by combinations of m rows of terraces with the (htktlt) orientation separated by n layers of steps with the (hsksls) orientation, forming the so-called step notation [n(htktlt) × m(hsksls)].60 The (221) and (331) surfaces are represented as [4(111) × (111)] and [3(111) × (111)] respectively, indicating the same type of the terraces and steps, but a different length of the terrace. This is the same for the (112) and (223) surfaces. The difference between the (112) and the (112)b surfaces is marked by the red arrow in Fig. 1(c) and (d), indicating a reconstruction of the oxygen atoms at the step. After the reconstruction, the step of the (112)b surface becomes a combination of the (100) and (111) step orientations. Table 1 lists the coordination numbers of all surfaces. The surface energies were calculated according to
m). In this structure, all cerium atoms are eight-coordinated, while oxygen atoms are four-coordinated, located at the center of a tetrahedron of cerium atoms. Fig. 1 shows the low index and high index surface models. (110) and (111) are additionally terminated by a threefold-coordinated oxygen atom, and (100) by a twofold-coordinated oxygen atom, as within this crystal structure it is otherwise impossible to define a cerium-terminated slab model without breaking the stoichiometry. The coordination number of surface cerium at the (100) and (110) surfaces is six; the coordination number at the (111) surface is seven. High index surfaces are morphologically characterized by combinations of m rows of terraces with the (htktlt) orientation separated by n layers of steps with the (hsksls) orientation, forming the so-called step notation [n(htktlt) × m(hsksls)].60 The (221) and (331) surfaces are represented as [4(111) × (111)] and [3(111) × (111)] respectively, indicating the same type of the terraces and steps, but a different length of the terrace. This is the same for the (112) and (223) surfaces. The difference between the (112) and the (112)b surfaces is marked by the red arrow in Fig. 1(c) and (d), indicating a reconstruction of the oxygen atoms at the step. After the reconstruction, the step of the (112)b surface becomes a combination of the (100) and (111) step orientations. Table 1 lists the coordination numbers of all surfaces. The surface energies were calculated according to| |  | (5) |
where Eslab,hkl is the energy of the (hkl) supercell, N is the number of ceria bulk units in the supercell, Eslab,bulk is the reference energy of one bulk unit, and Ahkl is the surface area of the supercell. The relative stability of low index surfaces is well known both from experimental61 and theoretical54,62 studies: (111) > (110) > (100). Table 1 shows the calculated surface energies for the low index and high index surfaces. The relative stability of the surfaces follows the order: (111)[0.75 eV] > (221)[0.82 eV] > (331)[0.85 eV] > (223b)[0.86 eV] > (112b)[0.96 eV] > (223)[1.04 eV] > (110)[1.11 eV] > (112)[1.24 eV] > (100)[1.43 eV]. For the high index surfaces that can be represented as the combination of low index surfaces, the more of the (111) component and less of the (100) component the surface comprises, the more stable the surface is. The reconstructions of oxygen atoms on the step site of the (112) and (223) surfaces result in a large surface energy difference (0.28 eV and 0.18 eV, respectively), rendering the (112)b and (223)b surfaces more stable than the (110) one.
 |
| | Fig. 1 Side view of the surface structures. (a) Low index (100), (110) and (111) surfaces; (b) high index stepped (221) surface; (c) and (d) high index stepped (112) surfaces with different reconstruction of the oxygen atoms at the step. Green spheres represent cerium atoms, while red ones represent oxygen atoms. | |
Table 1 Supercell, area (nm2), coordination number of surface oxygen and cerium atoms, surface energy (J m−2) and step notation for the slab models
| Surface |
Cell |
Area |
Coordination |
γ
hkl
|
Step notation |
| (111) |
4 × 4 |
2.03 |
O(3); Ce(7) |
0.75 |
|
| (221) |
2 × 4 |
3.93 |
O(3); Ce(7; 6) |
0.82 |
4(111) × (111) |
| (331) |
1 × 4 |
2.93 |
O(3); Ce(7; 6) |
0.85 |
3(111) × (111) |
| (223)b |
1 × 4 |
2.58 |
O(3; 2); Ce(7; 5) |
0.86 |
4(111) × (100) |
| (112)b |
2 × 4 |
1.54 |
O(3; 2); Ce(7; 5) |
0.96 |
3(111) × (100) |
| (223) |
1 × 4 |
2.58 |
O(3; 2); Ce(7; 5) |
1.04 |
4(111) × (100) |
| (110) |
4 × 3 |
2.49 |
O(3); Ce(6) |
1.11 |
|
| (112) |
2 × 4 |
1.54 |
O(3; 2); Ce(7; 5) |
1.24 |
3(111) × (100) |
| (100) |
3 × 3 |
2.64 |
O(2); Ce(6) |
1.43 |
|
3.2 PtOx adsorption
During the calcination process, due to the interaction of platinum atoms with the oxide support and the oxygen in the environment, mono-dispersed platinum atoms can have a variable number of coordinating oxygen atoms and charge states. Hence, three models of PtOx (x = 0, 1, 2) adsorbed on ceria surfaces are considered:| | | (PtOx)gas + CeO2 → (PtOx)ads/CeO2 | (6) |
The considered PtOx (x = 0, 1, 2) species correspond to the adsorption of free, gas-phase Pt atom, PtO and PtO2 molecules. PtO3 molecules, to the best of our knowledge, has not been reported in the experiment. Besides, platinum in (PtO3)ads would have to have +6 oxidation state, which is not observed in the Pt/CeO2 catalysts. Thus adsorption of PtO3 species is not considered here. The exact number of coordinated oxygen atoms after adsorption depends on the adsorption site and is discussed for every case.
PtOx species are stabilized by the interaction with oxygen and cerium atoms of the ceria surface. Since all computed systems are charged neutrally, we assign formal oxidation states of 0, +2, and +4 to platinum in (Pt)ads, (PtO)ads, and (PtO2)ads, respectively. Bader charges were used to analyze the electronic structure after the adsorption process. Table 2 shows the Bader charge values of the platinum atoms, and Fig. S1 (ESI†) shows the Bader charge difference before and after PtOx adsorption for cerium and oxygen atoms in the surfaces model. As references, we calculated Bader charges for platinum in the bulk platinum(II) and platinum(IV) oxides, yielding +0.88 |e| and +1.51 |e|, respectively. Platinum atoms in (Pt)ads are in a nearly neutral electronic structure (Bader charge between −0.03 and 0.08 |e|), in line with the results for ceria(111) surfaces.63,64 The interaction between these neutral (Pt)ads and ceria support can be described as a polarized covalent bond with little charge transfer from the metal to the oxide. This has been previously discussed by the example of transition-metal atoms in various oxides, such as MgO,65 TiO228 and Ce21O42 nanoparticle.64 Another way to define the charge state of platinum is to count how many electrons are transferred from platinum to the substrate, i.e. to count the number of Ce3+ ions. Fig. S1 and Table S2 (ESI†) show that no Ce3+ is formed on the support surfaces at the most stable configurations. Further analysis shows that platinum atoms oxidized to Pt+ with the reduction of a single Ce4+ to Ce3+ have a slightly higher energy compared to platinum atoms that retain their neutrality (see Fig. S2 and S3, ESI†). Therefore, Pt+ is less stable than Pt0. Platinum atoms in (PtO)ads exhibit Bader charge values (between 0.50 and 0.79 |e|) close to the value in platinum(II) oxide. Platinum atoms in (PtO2)ads exhibit Bader charge values (between 1.07 and 1.21 |e|) close to that in platinum(IV) oxide.64
Table 2 Adsorption energy (ΔEads) (eV), stability of the surface structure PtOx/CeO2 compared to the platinum bulk, oxygen molecule and clean ceria surface (ΔEs) (eV), Bader charge (Q) (|e|) for platinum in PtOx species adsorbed on ceria surfaces. The corresponding structures are displayed in Fig. 2, 3 and 4
| Surface |
Pt |
PtO |
PtO2 |
| ΔEads |
ΔEs |
Q
|
ΔEads |
ΔEs |
Q
|
ΔEads |
ΔEs |
Q
|
| (100) |
−5.02 |
0.61 |
−0.01 |
−4.40 |
−0.60 |
0.60 |
−3.88 |
−2.30 |
1.07 |
| (110) |
−3.86 |
1.77 |
0.01 |
−3.77 |
0.04 |
0.62 |
−2.97 |
−1.39 |
1.18 |
| (111) |
−2.64 |
2.99 |
0.05 |
−2.06 |
1.74 |
0.50 |
−0.84 |
0.74 |
1.08 |
| (221) |
−2.87 |
2.76 |
0.02 |
−3.26 |
0.55 |
0.79 |
−2.30 |
−0.71 |
1.18 |
| (331) |
−2.86 |
2.77 |
0.08 |
−3.41 |
0.39 |
0.67 |
−2.24 |
−0.65 |
1.18 |
| (112) |
−4.69 |
0.94 |
−0.02 |
−6.23 |
−2.42 |
0.72 |
−2.75 |
−1.17 |
1.21 |
| (112)b |
−4.68 |
0.95 |
−0.03 |
−3.60 |
0.20 |
0.65 |
−3.02 |
−1.44 |
1.06 |
| (223) |
−4.76 |
0.87 |
−0.02 |
−6.34 |
−2.53 |
0.74 |
−2.75 |
−1.17 |
1.19 |
| (223)b |
−4.71 |
0.92 |
−0.03 |
−3.84 |
−0.04 |
0.60 |
−3.09 |
−1.50 |
1.07 |
3.2.1 Structure and energy.
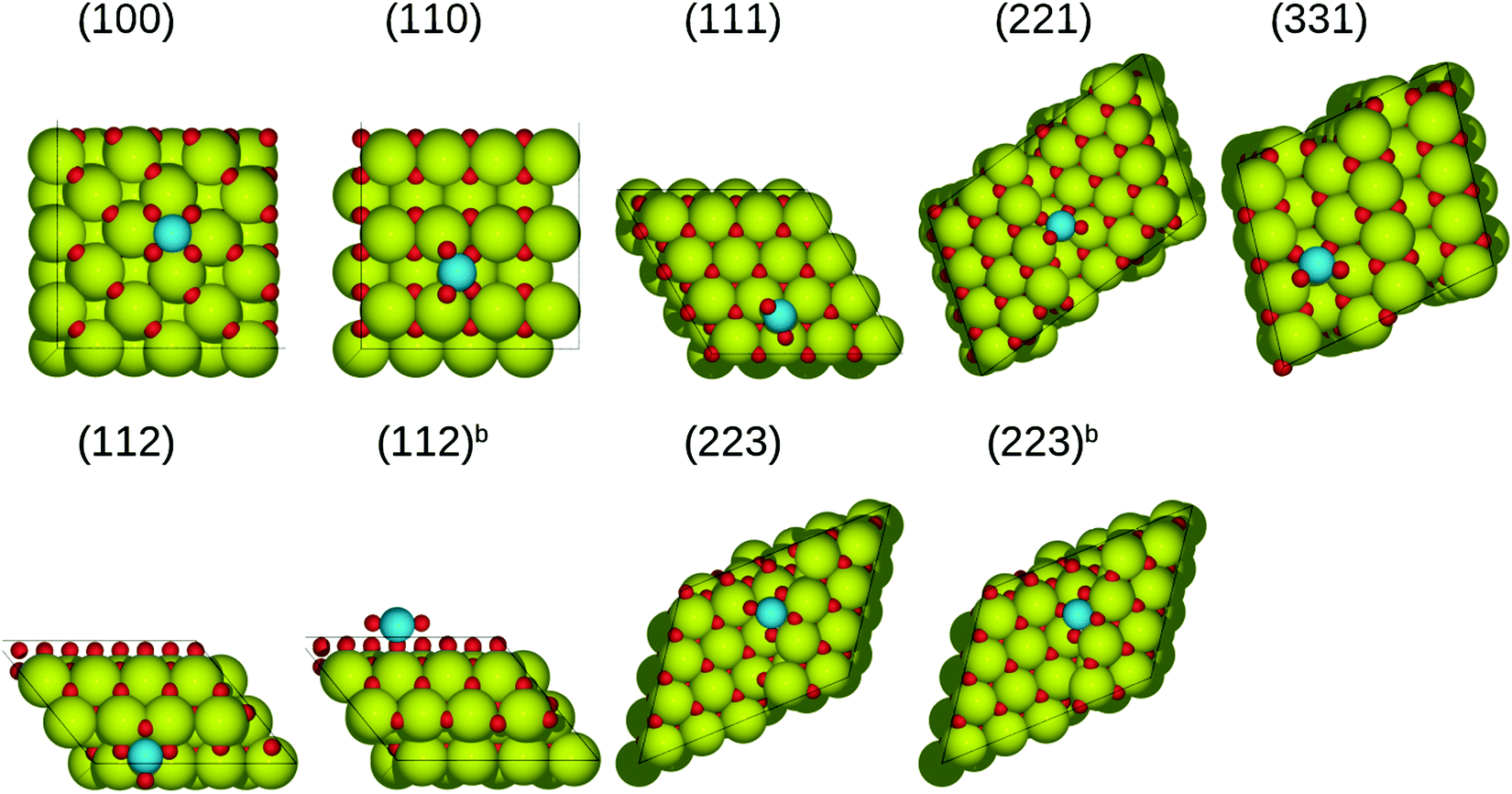
Fig. 2 and Table 2 show the configurations and the adsorption energies of the most stable (Pt)ads on all surfaces. Optimization of (Pt)ads on the ceria(100), (110) and (111) surfaces resulted in the convergence to the Obri, Cetop and Otop sites. (Pt)ads on the ceria(100) surface is most favorable on the bridge site between two surface oxygen atoms (Fig. 2 and Table 2). The platinum-oxygen bond length is 2.03 Å, which is close to that in the platinum(II) oxide (2.05 Å) and platinum(IV) oxide (2.03 Å). The adsorption energy of (Pt)ads on the Obri site on ceria(100) surface is −5.02 eV. Adsorption on the Cetop site results in a platinum atom positioned 2.41 Å above a surface cerium atom and bonded with two oxygen atoms with the bond length of 2.23 Å (Fig. S4, 100-pt-2, ESI†). However, this structure is much less favorable than the Obri site with an adsorption energy of −2.59 eV. Adsorption on the Otop site results in a short bond length (1.90 Å) and adsorption energy of −2.26 eV (Fig. S4, 100-pt-3, ESI†). On the (110) surface, the most favorable adsorption occurs on the Obri site with adsorption energy of −3.86 eV and the platinum atom positioned between two surface oxygen atoms with the platinum-oxygen bond length of 1.98 Å. The Otop site (Fig. S5, 110-pt-2, ESI†) is the next most favorable site for (Pt)ads on the (110) surface, followed by the Cetop site (Fig. S5, 110-pt-3, ESI†). On the (111) surface, the most favorable adsorption occurs on the Obri site with the adsorption energy of −2.64 eV and the platinum atom positioned between two surface oxygen atoms with the Pt–O bond length equal to 2.16 Å. The Otop site (Fig. S6, 111-pt-2, ESI†) is the next most favorable site for (Pt)ads on the (111) surface (−2.36 eV), followed by the Cetop site (−1.28 eV) (Fig. S6, 111-pt-3, ESI†).
 |
| | Fig. 2 The most stable structures of (Pt)ads on ceria surfaces in side and top views. In each case, the Obri, Cebri, Cetop and Otop sites on the terrace and step are considered. The most stable configuration then was chosen for all subsequent analysis. Green spheres represent cerium atoms, blue ones represent platinum atoms, while red ones represent oxygen atoms. | |
All of the high index surfaces have the (111) terrace site. (Pt)ads on these terrace sites show the same trend as on the (111) surface with the adsorption energies (over a range of −2.88 to −2.46 eV) on the Obri site close to that on the (111) surface (−2.64 eV). In the case of the step site on the (221) and (331) surfaces, platinum bonds with three oxygen atoms with two short Pt–O bonds and one long Pt–O, resulting in a similar binding energy compared to the Obri site on the terrace. The (112), (112b), (223) and (223b) surfaces have (100) steps. On these step sites, (Pt)ads adsorbs more strongly than on the terrace sites. On (112b) and (223b), platinum bonds with two oxygen atoms on the step site, while on (112) and (223), platinum bonds with three oxygen atoms with two short Pt–O bonds and one long Pt–O bond on the step site. However, all binding energies on step sites are similar to those on the (100) surface. Overall, the adsorption of a single platinum atom on the ceria surface is most favorable on the (100) surface termination. The relative stability decreases in the following series: (100)[−5.02 eV] > (223)[−4.76 eV] > (223b)[−4.71 eV] > (112)[−4.69 eV] > (112b)[−4.68 eV] > (110)[−3.86 eV] > (221)[−2.87 eV] > (331)[−2.86 eV] > (111)[−2.64 eV]. These results show that the (111) termination exhibits the weakest interaction with (Pt)ads and this interaction is up to 2.3 eV higher on the (100) surfaces. The stability energies (ΔEs in Table 2) of the surface structure PtOx/CeO2 compared to the bulk platinum, oxygen molecule and clean ceria surface are all positive, and thus the corresponding species are unstable with respect to bulk platinum.
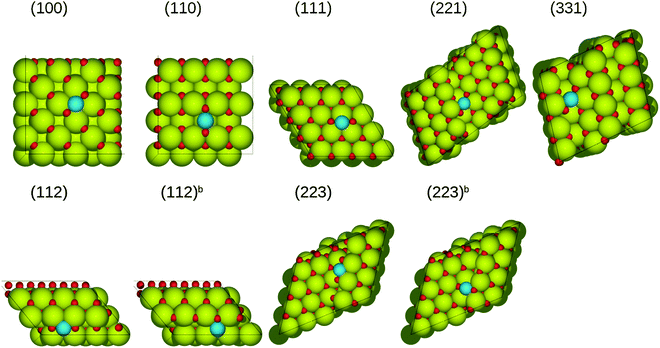
The most exothermic adsorption for both (PtO)ads and (PtO2)ads on each surface occurs on the sites where oxygen atoms interact with cerium atoms, and platinum atoms interact with surface oxygen atoms. Fig. 3 and 4 display the most favorable adsorption structures on each of the ceria surfaces. The relative stability of (PtO)ads decreases in the following series: (223)[−6.34 eV] > (112)[−6.23 eV] > (100)[−4.40 eV] > (223b)[−3.84 eV] > (112b)[−3.60 eV] > (331)[−3.41 eV] > (221)[−3.26 eV] > (111)[−2.06 eV]. On the (223) and (112) surfaces, (PtO)ads together with three surface oxygen atoms form a square-planar unit [PtO4], facilitating a strong interaction with the surface, with binding energy of −6.23 eV and −6.34 eV, respectively, much larger than that on the flat CeO2(111) surface (−2.06 eV). Since sixfold-coordinated cerium, which can bond with the oxygen atoms of the (PtO)ads, exists on the step, adsorption on the step sites yields configurations that are more stable compared to the terrace sites and the (111) surface.
 |
| | Fig. 3 The most stable structures of (PtO)ads on stoichiometric ceria surfaces in side and top views. Green spheres represent cerium atoms, blue ones represent platinum atoms, while red ones represent oxygen atoms. | |
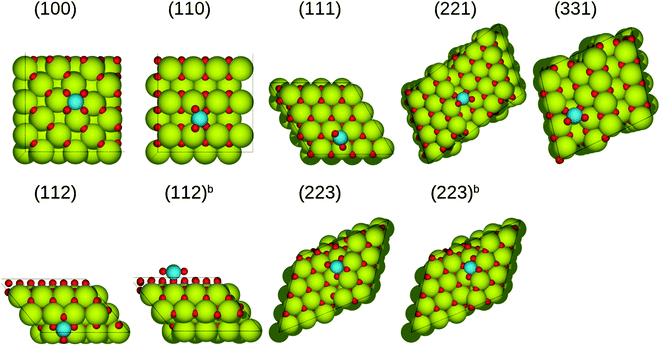
 |
| | Fig. 4 The most stable structures of (PtO2)ads on stoichiometric ceria surfaces in side and top views. Green spheres represent cerium atoms, blue ones represent platinum atoms, while red ones represent oxygen atoms. | |
In the case of (PtO2)ads supported on the ceria(111) surface, platinum bonds with three oxygen atoms, of which one has no direct bond to ceria, resulting in a weak adsorption energy (−0.84 eV). On the rest of the surfaces, all most stable configurations feature the platinum atoms bound to four oxygen atoms. On the (100) surface, (PtO2)ads together with two surface oxygen atoms form a square-planar unit [PtO4], resulting in a strong interaction with the surface, with a binding energy of −3.88 eV, which is much more negative than that on the flat CeO2(111) surface. Adsorption on the (221) and (331) surfaces is relatively weak (−2.30 eV and −2.24 eV), because of the smaller number of sixfold cerium atoms compared to other surfaces. Besides, (PtO2)ads species adsorbed on the (221) and (331) surfaces have distorted structures compared to other cases. The relative stability of (PtO2)ads decreases in the following series: (100)[−3.88 eV] > (223b)[−3.09 eV] > (112b)[−3.02 eV] > (110)[−2.97 eV] > (112)[−2.75 eV], (223)[−2.75 eV] > (221)[−2.30 eV] > (331)[−2.24 eV] > (111)[−0.84 eV]. With the exception of the (111) surface, all the stability energies (ΔEs in Table 2) are negative, and thus the (PtO2)ads is more stable than platinum bulk and oxygen molecule.
Strong correlation between the adsorption energy and the number of oxygen and cerium atoms that bind to the (PtOx)ads suggests the existence of a relation between the adsorption energy of (PtOx)ads and the topological features of the PtOx–CeO2 species, namely the number of Pt–O, O–Ce and Pt–Ce bonds. The bond lengths of the different types are used as descriptors to identify the main topological components contributing to the stability of various species. Bond lengths in the PtOx molecules and bulk phases are used as starting points. In total, four to seven features are used for the binding energies of (Pt)ads or (PtO)ads and (PtO2)ads, respectively. Table 3 provides a full description of the parameters. The linear regression model can be written as:
| |  | (7) |
where
ni is the number of the feature
i in the topology, and the coefficients
wi represent the contribution of the feature
i to the binding energy. The root-mean-squared-error (RMSE) was used to compare the model predicted energies (
Êj) with the DFT calculated energies (
Ej):
| | 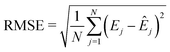 | (8) |
where
N is the number of the data points in the dataset.
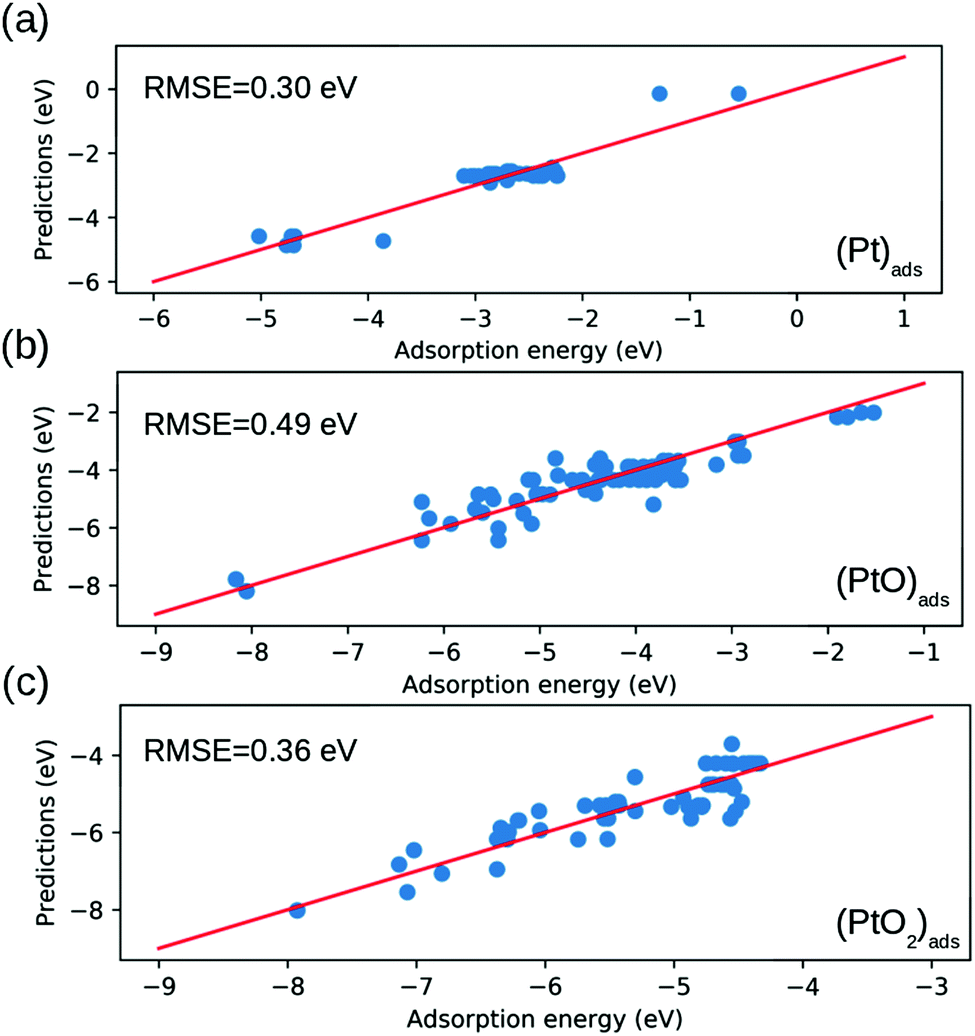
Fig. 5 shows that our model exhibits an average RMSE of 0.30 eV, 0.49 eV and 0.36 eV for Pt, (PtO)
ads and (PtO
2)
ads binding energies, respectively. Given the simple linear relationship, this can be considered sufficient.
Table 3 shows that all
wi are negative for Pt–O and O–Ce features, which indicates that all these interactions contribute to the adsorption and stabilization of the (PtO
x)
ads species on the ceria surfaces. The longer the bond length the smaller the value of
wi, and thus the weaker the bond strength. Generally,
wi for the Pt–O features is larger than that of the O–Ce and Pt–Ce features, thus suggesting that unsaturated oxygen atoms play the leading role in stabilization of (PtO
x)
ads species. In our dataset, the most stable configurations for the (PtO
x)
ads adsorption have Pt–O bonds with the twofold-coordinated oxygen atoms.
wi for the Pt–Ce features are close to zero suggesting that the interactions between platinum and cerium atoms are small and contribute little to the stabilization of PtO
x.
Table 3 Features of the linear regression model. Bond length range corresponding to the gas phase (PtO and PtO2 molecules, 1.75 Å and 1.72 Å, respectively), bulk platinum phase (platinum(II) oxide and platinum(IV) oxide, 2.05 Å and 2.03 Å, respectively), bulk ceria phase (cerium(IV) oxide and cerium(III) oxide, 2.31 Å and 2.37 Å respectively) and bulk platinum-cerium alloy phase (3.03 Å) are labeled accordingly
| Feature i |
Bond |
Bond length |
w
i
|
| Pt |
PtO |
PtO2 |
| 1 |
Pt–O |
1.70–1.80 (gas) |
|
−2.38 |
−2.10 |
| 2 |
Pt–O |
1.80–1.95 |
−2.56 |
−1.51 |
−1.37 |
| 3 |
Pt–O |
1.95–2.05 (bulk) |
−2.30 |
−1.13 |
−1.07 |
| 4 |
Pt–O |
2.05–2.30 |
−1.25 |
−0.72 |
−0.83 |
| 5 |
O–Ce |
2.00–2.40 (bulk) |
|
−1.33 |
−0.78 |
| 6 |
O–Ce |
2.40–3.00 |
|
−0.66 |
−0.54 |
| 7 |
Pt–Ce |
2.70–3.20 (bulk) |
−0.14 |
−0.35 |
0.10 |
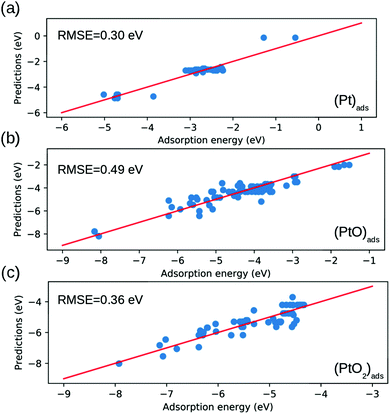 |
| | Fig. 5 Comparison between the binding energy estimated by eqn (7) and that calculated by DFT. The root mean square error (RMSE) are depicted in each panel. (a) (Pt)ads; (b) (PtO)ads; (c) (PtO2)ads. | |
3.2.2 Phase diagrams of (PtOx)ads on ceria: local equilibrium.
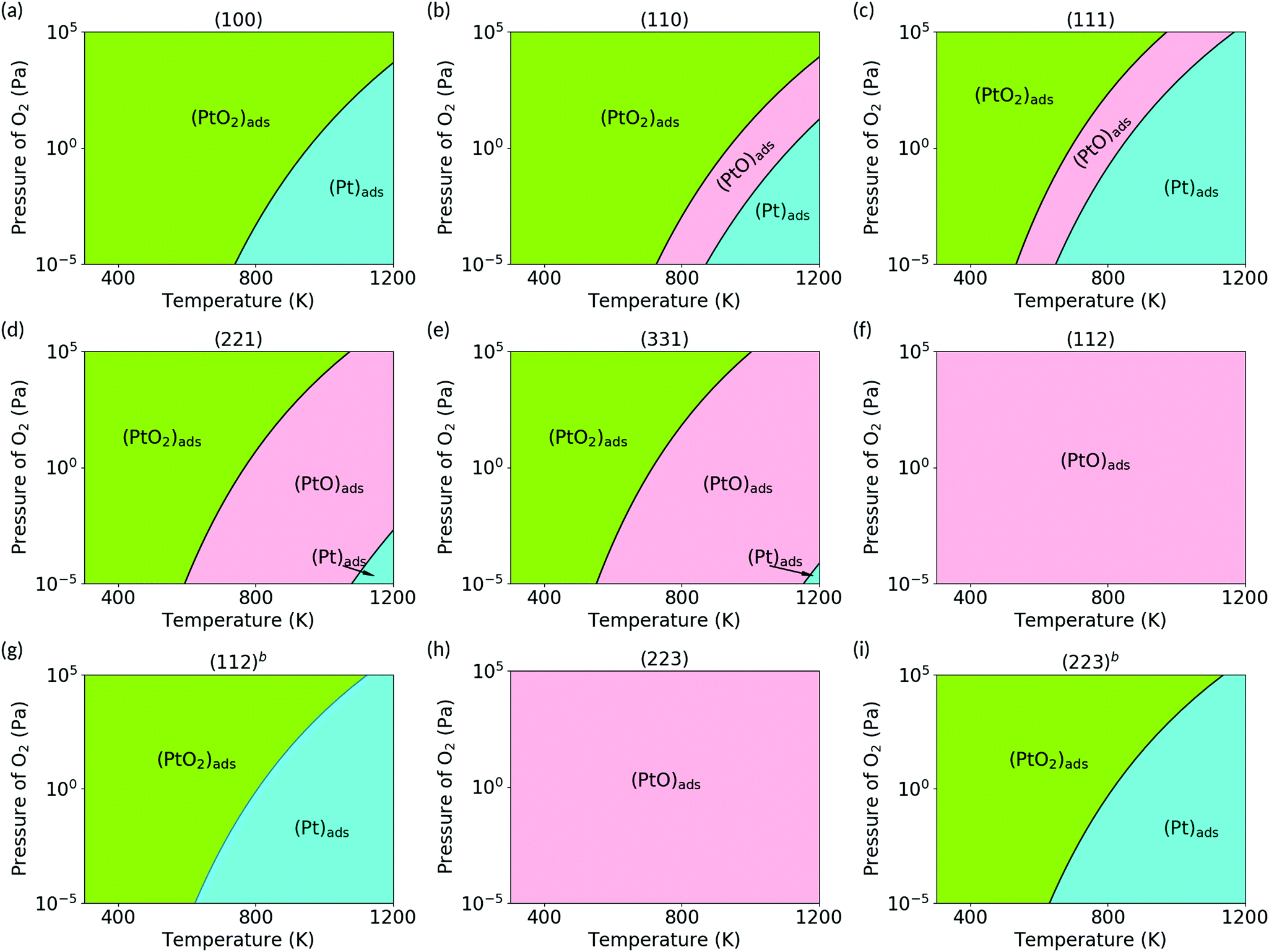
Using ab initio atomistic thermodynamics framework, we calculated the Gibbs free energy of the (PtOx)ads species on the ceria surfaces, ΔGads, as a function of temperature and oxygen pressure. We first assume the local equilibrium of subsystems, considering that only adsorbed (PtOx)ads species are in thermodynamic equilibrium. Platinum bulk phase is removed from the equilibrium, and (PtOx)ads are not allowed to desorb. All other cluster configurations (PtxOy) with more than one Pt atom may be interesting, but are out of the scope of the current work and are not considered. Oxygen vacancy formation energies are calculated as 2.40 eV, 2.74 eV and 3.15 eV for the (100), (110) and (111) surfaces at 0 K and 105 Pa oxygen. The oxygen chemical potential changes by around −1.0 eV from 300 to 1200 K, and by around −0.8 eV from 10−5 Pa to 105 Pa at 600 K for the oxygen molecule. This is smaller than the oxygen vacancy formation energy. Thus the stoichiometric ceria surface is preserved without reduction at these pressure and temperature ranges (300–1200 K and 105 to 10−5 Pa). Fig. 6 shows the phase diagram of (PtOx)ads on the ceria surfaces. The stability range of (PtOx)ads strongly correlates with the number of unsaturated oxygen and ceria atoms, and thus changes dramatically with the structure of the supporting surface. On the (112) and (223) surfaces, (PtO)ads species is stable under all conditions. On other surfaces, all the three (PtOx)ads (x = 0, 1, 2) species have a stability range, which is sensitive to temperature and pressure. At higher temperatures and lower oxygen partial pressures, the (Pt)ads species is energetically favorable. By either lowering the temperature or increasing the oxygen partial pressure, the species transform in the order of (Pt)ads → (PtO)ads → (PtO2)ads.
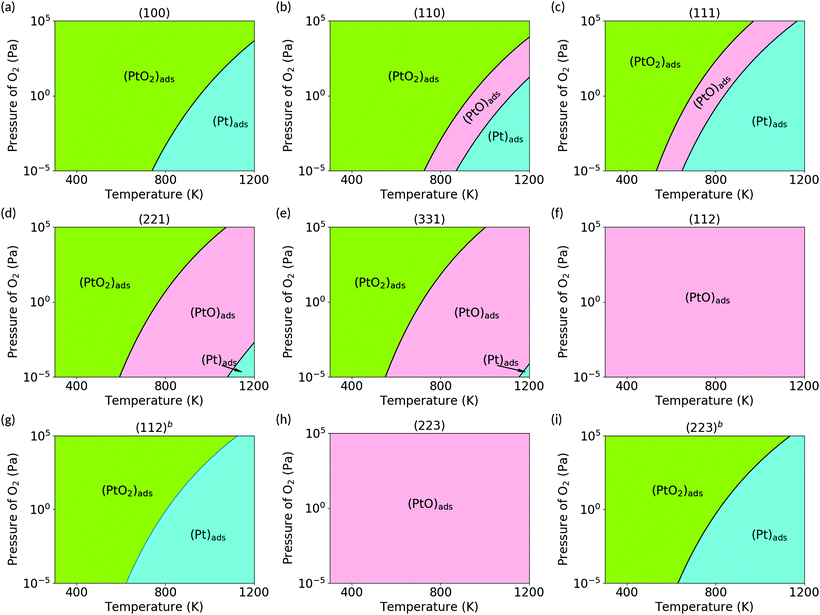 |
| | Fig. 6 Phase diagram obtained for PtOx (x = 0, 1, 2) species on ceria surfaces at the temperature range of 300 to 1200 K, and oxygen pressure range from 10−5 Pa to 105 Pa. Each region shows the most stable species under the corresponding conditions. (a) (100); (b) (110); (c) (111); (d) (221); (e) (331); (f) (112); (g) (112)b; (h) (223); (i) (223)b. | |
3.2.3 Phase diagrams of (PtOx)ads on ceria: global equilibrium.
We now move to the analysis of the global thermodynamic equilibrium, which takes into account the bulk and gas phases of platinum. Transformation of (PtOx)ads species to the bulk phase and the gas phase corresponds to the sintering and evaporation processes, which are the main ways for the deactivation of catalysts. For platinum, evaporation through volatile PtO2 gas species at high temperature and in an oxygen environment is well-known.66–68 The equilibrium pressure of PtO2(g) is proportional to oxygen pressure. According to the study by Alcock and Hooper,66 the partial pressure of (PtO2)ads at 1 bar oxygen and 1000 K is 1.5 × 10−4 Pa. However, the exact partial pressure of (PtO2)ads in the gas phase depends on the ability of the newly formed molecules of (PtO2)ads to escape from the vicinity of the metal surface to the surrounding atmosphere. Therefore, a range of partial pressures of (PtO2)ads between 10−5 and 10−3 Pa was considered in this study. Fig. 7 and Fig. S7 (ESI†) display the relative energy for (PtOx)ads species on each ceria surface compared to the bulk platinum and (PtO2)ads gas phase, as a function of temperature at 1 bar oxygen pressure. The Gibbs free energy of (PtO2)ads becomes lower than that of the bulk platinum at 800–900 K, indicating the start of the evaporation process. The energy plot of (Pt)ads, (PtO)ads and (PtO2)ads have different slopes as they have a different number of oxygen atoms. Temperature affects the stability of the species, and, as indicated by the non-negative slopes for all lines, the increase of the temperature goes along with the destabilization of all adsorbed species. All mono-dispersed Pt0 species are unstable compared to the bulk platinum formation, therefore, oxidation of Pt0 to Pt2+ or Pt4+ is necessary to stabilize mono-dispersed platinum atoms. The two most stable Pt2+ species with the [PtO4] structure on the (223) and (112) surfaces are thermodynamically favorable in the entire temperature range. The most stable Pt4+ species on the (100) surface is more stable than the bulk phase below 1100 K. However, it is less stable compared to the PtO2 gas phase above 950 K, indicating the desorption of (PtO2)ads from the surface. When we consider both terrace and step sites, platinum species can coexist as nanoparticles and single atoms on the high index surfaces, with nanoparticles located on the terrace site and single atoms on the step site.
 |
| | Fig. 7 Relative stability of PtOx (x = 0, 1, 2) phases on ceria surfaces compared to bulk platinum and PtO2 gas phase. Symbols (circle: (Pt)ads; triangle: (PtO)ads; square: (PtO2)ads) on each colored line correspond to the PtOx species adsorbed on a corresponding surface. | |
3.2.4 Effects of coverage.
To achieve atomic dispersion of metal, low loading of metal is often required.27 We, therefore, explored the influence of the loading on the stability of supported platinum species. The coverages (θ) are calculated as the number of adsorbed PtOx divided by the corresponding area of the surfaces models (Table 1). For the low index (100), (110) and (111) slab models with one platinum species, θ = 0.4, 0.4 and 0.5 atoms per nm−2, respectively. Platinum loading of 0.5 atoms per nm−2 on (111) surface corresponds to platinum loading of 0.06 monolayer (1 monolayer = 7.9 atoms per nm−2). For the high index surfaces, where platinum species are only adsorbed on the step site, the coverage of (PtOx)ads species also depend on the density of such one-dimensional step sites. The shorter the terrace, the larger the density of the step sites, the higher the coverage of (PtOx)ads species. Fig. 8 and Fig. S17 (ESI†) display the relative stability of (PtOx)ads at different coverages. Fig. S8–S16 (ESI†) show the corresponding structures. On low index (100), (110) and (111) surfaces, there are many more adsorption sites (maximum coverages for (Pt)ads are 5.7, 3.6 and 4.4 nm−2, respectively) compared to the one-dimensional step sites. (PtOx)ads species on these surfaces are not in contact with each other directly under the coverages considered here, thus the stability changes little with the increasing coverage. On the (221) and (331) surfaces, the stability of (Pt)ads first decreases and then increases. Fig. S11 and S12 (ESI†) show that the stabilization at high coverages is due to the formation of Pt–Pt bonds. Even through the initial structures are set to be mono-dispersed Pt atoms bonded with oxygen atoms, platinum atom trends to bond with another platinum atom after geometry optimization at high coverages, thus facilitating the formation of energetically favorable platinum clusters instead of mono-dispersed platinum species. Therefore, the stability of the system increases at high coverages.
 |
| | Fig. 8 Relative stability of (PtOx)ads (x = 0, 1, 2) phases on ceria surfaces with respect to bulk platinum changes with coverage. | |
In the case of (PtO)ads on the (112) and (223) surfaces, the stability decreases at high coverages, because two [PtO4] species share the same twofold-coordinated oxygen atoms on the step site. On the (112)b and (223)b step sites, at lower coverages, platinum bonds with three oxygen atoms at the step site. Fig. S14(b) and S16(b) (ESI†) show that, at high coverages, one oxygen atom from the (PtO)ads species connects with the neighboring platinum atom, forming a square-planar [PtO4] chain on the step site, becoming more stable at high coverages. On these step sites, mono-dispersed platinum is less stable than platinum in the [–Pt–O–Pt–] chain.
On the (221) surface, the stability increases due to the formation of the Pt–Pt bond. On the (331) surface, which has the same type of step as (221), (PtO)ads binds stronger than on the (221), and no Pt–Pt bond is formed at higher coverages.
For (PtO2)ads, the stability decreases almost linearly with the coverage. In the case of the (100) surface, it decreases from −2.4 eV to −1.6 eV for the coverage of 0.38 to the coverage of 1.52. On the (112)b and (223)b surfaces, the maximum coverage corresponds to two (PtO2)ads species on the step site when using the slab model listed in Table 1. Higher coverage results in the overlap of the oxygen atoms on the step site.
3.3 Surface formation energies
While ceria helps to trap atomically dispersed platinum species, the strong interaction between platinum species and ceria also helps to stabilize the surface of ceria itself. Large surface area and small crystallite size was observed experimentally upon ionic platinum deposition.31,32 The surface energy after adsorption of different species was calculated according to:| | 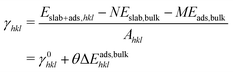 | (9) |
where γ0hkl is the surface energy of the clean surface (hkl), θ is the adsorption coverage with a unit of 1 nm−2, and ΔEads,bulkhkl(θ) is the energy required to disperse bulk species on the (hkl) surface at coverage θ. The maximum platinum coverage reported in experiment31 is around 1.0 platinum atoms per nm2. Platinum loading of 0.5 atoms per nm2 is considered here. ΔEads,bulkhkl(θ) is interpolated from the energies in Fig. 8. Table 4 shows the surface energies calculated by eqn (9) for ceria surfaces with and without (PtOx)ads adsorption at 0 K and 1 atm oxygen pressure. Since (Pt)ads on all surfaces is unstable, the surface energies increase. For (PtO)ads and (PtO2)ads, surface energies decrease for the surfaces with the negative ΔEads,bulkhkl(θ) value, indicating the stabilization of the ceria surface. The relative stability of the high index stepped surfaces is altered due to the interaction with platinum species (e.g. (112) versus (112)b). The relative stability of the low index surface ((111) versus (110) versus (100)) is not altered at this platinum loading. However, energy differences between surfaces are reduced due to the interaction with platinum. For example, the bare (100) surface has a surface energy, which is 0.68 (J m−2) higher than that of the (111) surface; however, this difference is reduced to 0.20 (J m−2) for (PtO2)ads due to the stronger bonding of (PtO2)ads to the CeO2(100) surface.
Table 4 Surface formation energies for ceria surfaces with (PtOx)ads loading of 0.5 atoms per nm2
| Surface |
γ
hkl
(J m−2) |
| (Pt)ads |
(PtO)ads |
(PtO2)ads |
| (111) |
1.23 |
1.03 |
0.87 |
| (221) |
1.26 |
0.96 |
0.71 |
| (331) |
1.30 |
0.92 |
0.75 |
| (223)b |
1.01 |
0.86 |
0.63 |
| (112)b |
1.11 |
0.99 |
0.73 |
| (223) |
1.18 |
0.64 |
0.86 |
| (110) |
1.38 |
1.12 |
0.89 |
| (112) |
1.39 |
0.85 |
1.05 |
| (100) |
1.53 |
1.33 |
1.07 |
4 Conclusions
In summary, density functional theory combined with ab initio atomistic thermodynamics has been used to investigate the structure and stability of atomically dispersed platinum species on CeO2(100), (110), (111) lower index surfaces as well as on (221), (331), (112), (223) high index stepped surfaces. Surface energies of all surfaces were calculated and the order of stability was found to depend on the structure of the step: the more of the (111) component and less of the (100) component the step has, the more stable the surface is. Unsaturated oxygen atoms on the surface play the leading role in stabilization of (PtOx)ads species. All mono-dispersed Pt0 species are thermodynamically unstable compared to the bulk platinum, and oxidation of Pt0 to Pt2+ or Pt4+ is necessary to stabilize adsorbed mono-dispersed platinum atoms. Both Pt2+ and Pt4+ prefer to form the square-planar [PtO4] structure. Phase diagrams for the dispersed (PtOx)ads species supported on a range of CeO2 surfaces were constructed. Temperature, oxygen pressure and surface structure significantly affect the stability of (PtOx)ads species (cf.Fig. 9). The (223) and (112) surfaces stabilize two most stable Pt2+ species that are thermodynamically favorable in the entire considered temperature range (300–1200 K). The (100) surface stabilizes Pt4+ species that are more stable than the bulk phase below 1100 K. However, such species start desorbing as PtO2 gas above 950 K. The stability of ceria surfaces is also increased by the metal–support interaction. The resulting phase diagrams of the atomically dispersed (PtOx)ads species on various ceria surfaces under a range of experimentally relevant conditions can be used to predict dynamic restructuring of atomically dispersed platinum catalysts.
 |
| | Fig. 9 Schematic representation of the structure of platinum species on ceria surfaces depending on external conditions. (PtOx)ads species are depicted in red, while the surface atoms are depicted in black. | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the computational resources provided by the Paul Scherrer Institut (PSI) and the Swiss National Supercomputing Centre (CSCS), as well as financial support from the China Scholarships Council (No. 201506370019).
References
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS.
- J. D. Kistler, N. Chotigkrai, P. Xu, B. Enderle, P. Praserthdam, C.-Y. Chen, N. D. Browning and B. C. Gates, Angew. Chem., Int. Ed., 2014, 53, 8904–8907 CrossRef CAS.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS.
- M. Yang, J. Liu, S. Lee, B. Zugic, J. Huang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2015, 137, 3470–3473 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- R. Lang, W. Xi, J.-C. Liu, Y.-T. Cui, T. Li, A. F. Lee, F. Chen, Y. Chen, L. Li, L. Li, J. Lin, S. Miao, X. Liu, A.-Q. Wang, X. Wang, J. Luo, B. Qiao, J. Li and T. Zhang, Nat. Commun., 2019, 10, 234 CrossRef.
- J. Zhang, M. P. Everson, T. J. Wallington, F. R. Field, R. Roth and R. E. Kirchain, Environ. Sci. Technol., 2016, 50, 7687–7695 CrossRef CAS.
- J. Lee, J. R. Theis and E. A. Kyriakidou, Appl. Catal., B, 2019, 243, 397–414 CrossRef CAS.
- D. M. Meira, R. U. Ribeiro, O. Mathon, S. Pascarelli, J. M. C. Bueno and D. Zanchet, Appl. Catal., B, 2016, 197, 73–85 CrossRef CAS.
- D. Miao, G. Cavusoglu, H. Lichtenberg, J. Yu, H. Xu, J.-D. Grunwaldt and A. Goldbach, Appl. Catal., B, 2017, 202, 587–596 CrossRef CAS.
- A. Kaftan, M. Kusche, M. Laurin, P. Wasserscheid and J. Libuda, Appl. Catal., B, 2017, 201, 169–181 CrossRef CAS.
- S. C. Ammal and A. Heyden, ACS Catal., 2017, 7, 301–309 CrossRef CAS.
- E. A. Walker, D. Mitchell, G. A. Terejanu and A. Heyden, ACS Catal., 2018, 8, 3990–3998 CrossRef CAS.
- D. Liu, Y. Men, J. Wang, G. Kolb, X. Liu, Y. Wang and Q. Sun, Int. J. Hydrogen Energy, 2016, 41, 21990–21999 CrossRef CAS.
- M. Martinelli, G. Jacobs, U. M. Graham and B. H. Davis, Catalysts, 2017, 7, 148 CrossRef.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y.-W. Li, C. Shi, X.-D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS.
- F. Calle-Vallejo, M. D. Pohl and A. S. Bandarenka, ACS Catal., 2017, 7, 4355–4359 CrossRef CAS.
- C. E. Blackmore, N. V. Rees and R. E. Palmer, Phys. Chem. Chem. Phys., 2015, 17, 28005–28009 RSC.
- S. Sakong and A. Groß, ACS Catal., 2016, 6, 5575–5586 CrossRef CAS.
- M. Dunwell, Y. Yan and B. Xu, ACS Catal., 2017, 7, 5410–5419 CrossRef CAS.
- S. Sui, X. Wang, X. Zhou, Y. Su, S. Riffat and C.-J. Liu, J. Mater. Chem. A, 2017, 5, 1808–1825 RSC.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS.
- M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS.
- C. K. Narula, L. F. Allard, G. M. Stocks and M. Moses-DeBusk, Sci. Rep., 2014, 4, 7238 CrossRef CAS.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS.
- L. DeRita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang, A. S. Hoffman, I. Ro, G. W. Graham, S. R. Bare, G. Pacchioni, X. Pan and P. Christopher, Nat. Mater., 2019, 18, 746–751 CrossRef CAS.
- H. V. Thang, G. Pacchioni, L. DeRita and P. Christopher, J. Catal., 2018, 367, 104–114 CrossRef CAS.
- A. Bruix, Y. Lykhach, I. Matolínová, A. Neitzel, T. Skála, N. Tsud, M. Vorokhta, V. Stetsovych, K. Ševčíková, J. Mysliveček, R. Fiala, M. Václavů, K. C. Prince, S. Bruyère, V. Potin, F. Illas, V. Matolín, J. Libuda and K. M. Neyman, Angew. Chem., Int. Ed., 2014, 53, 10525–10530 CrossRef CAS.
- F. Dvořák, M. Farnesi Camellone, A. Tovt, N.-D. Tran, F. R. Negreiros, M. Vorokhta, T. Skála, I. Matolínová, J. Mysliveček, V. Matolín and S. Fabris, Nat. Commun., 2016, 7, 10801 CrossRef.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS.
- X. I. Pereira-Hernández, A. DeLaRiva, V. Muravev, D. Kunwar, H. Xiong, B. Sudduth, M. Engelhard, L. Kovarik, E. J. M. Hensen, Y. Wang and A. K. Datye, Nat. Commun., 2019, 10, 1358 CrossRef.
- A. J. Therrien, A. J. R. Hensley, M. D. Marcinkowski, R. Zhang, F. R. Lucci, B. Coughlin, A. C. Schilling, J.-S. McEwen and E. C. H. Sykes, Nat. Catal., 2018, 1, 192–198 CrossRef CAS.
- Y.-G. Wang, D. Mei, V.-A. Glezakou, J. Li and R. Rousseau, Nat. Commun., 2015, 6, 6511 CrossRef CAS.
- P. L. Gai, K. Yoshida, M. R. Ward, M. Walsh, R. T. Baker, L. van de Water, M. J. Watson and E. D. Boyes, Catal. Sci. Technol., 2016, 6, 2214–2227 RSC.
- M. Mao, H. Lv, Y. Li, Y. Yang, M. Zeng, N. Li and X. Zhao, ACS Catal., 2016, 6, 418–427 CrossRef CAS.
- S. Zhang, Z. Xia, T. Ni, Z. Zhang, Y. Ma and Y. Qu, J. Catal., 2018, 359, 101–111 CrossRef CAS.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS.
- A. Figueroba, G. Kovacs, A. Bruix and K. M. Neyman, Catal. Sci. Technol., 2016, 6806–6813 RSC.
- J. Lee, Y. Ryou, X. Chan, T. J. Kim and D. H. Kim, J. Phys. Chem. C, 2016, 120, 25870–25879 CrossRef CAS.
- M. Hatanaka, N. Takahashi, T. Tanabe, Y. Nagai, K. Dohmae, Y. Aoki, T. Yoshida and H. Shinjoh, Appl. Catal., B, 2010, 99, 336–342 CrossRef CAS.
- D. Kunwar, S. Zhou, A. DeLaRiva, E. J. Peterson, H. Xiong, X. I. Pereira-Hernández, S. C. Purdy, R. ter Veen, H. H. Brongersma, J. T. Miller, H. Hashiguchi, L. Kovarik, S. Lin, H. Guo, Y. Wang and A. K. Datye, ACS Catal., 2019, 9, 3978–3990 CrossRef CAS.
- E. A. Derevyannikova, T. Y. Kardash, A. I. Stadnichenko, O. A. Stonkus, E. M. Slavinskaya, V. A. Svetlichnyi and A. I. Boronin, J. Phys. Chem. C, 2019, 123, 1320–1334 CrossRef CAS.
- R. Fiala, A. Figueroba, A. Bruix, M. Vaclavu, A. Rednyk, I. Khalakhan, M. Vorokhta, J. Lavkova, F. Illas, V. Potin, I. Matolinova, K. M. Neyman and V. Matolin, Appl. Catal., B, 2016, 197, 262–270 CrossRef CAS.
- A. Tovt, L. Bagolini, F. Dvořák, N.-D. Tran, M. Vorokhta, K. Beranová, V. Johánek, M. F. Camellone, T. Skála, I. Matolínová, J. Mysliveček, S. Fabris and V. Matolín, J. Mater. Chem. A, 2019, 7, 13019–13028 RSC.
- Y.-Q. Su, Y. Wang, J.-X. Liu, I. A. Filot, K. Alexopoulos, L. Zhang, V. Muravev, B. Zijlstra, D. G. Vlachos and E. J. Hensen, ACS Catal., 2019, 9, 3289–3297 CrossRef CAS.
- X. Wang, J. A. van Bokhoven and D. Palagin, Phys. Chem. Chem. Phys., 2017, 19, 30513–30519 RSC.
- N. Daelman, M. Capdevila-Cortada and N. López, Nat. Mater., 2019 DOI:10.1038/s41563-019-0444-y.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS.
- G. Lppert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–488 CrossRef.
- M. M. Branda, R. M. Ferullo, M. Causà and F. Illas, J. Phys. Chem. C, 2011, 115, 3716–3721 CrossRef CAS.
- S. M. Kozlov, F. Viñes, N. Nilius, S. Shaikhutdinov and K. M. Neyman, J. Phys. Chem. Lett., 2012, 3, 1956–1961 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- K. Reuter and M. Scheffler, Phys. Rev. Lett., 2003, 90, 046103 CrossRef.
-
K. Reuter, C. Stampf and M. Scheffler, Ab Initio Atomistic Thermodynamics and Statistical Mechanics of Surface Properties and Functions, Springer, Netherlands, Dordrecht, 2005, pp. 149–194 Search PubMed.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef.
- B. Lang, R. W. Joyner and G. A. Somorjai, Surf. Sci., 1972, 30, 440–453 CrossRef CAS.
- Y. Lin, Z. Wu, J. Wen, K. R. Poeppelmeier and L. D. Marks, Nano Lett., 2014, 14, 191–196 CrossRef CAS.
- O. Matz and M. Calatayud, ACS Omega, 2018, 3, 16063–16073 CrossRef CAS.
- A. Bruix, K. M. Neyman and F. Illas, J. Phys. Chem. C, 2010, 114, 14202–14207 CrossRef CAS.
- H. A. Aleksandrov, K. M. Neyman and G. N. Vayssilov, Phys. Chem. Chem. Phys., 2015, 17, 14551–14560 RSC.
- I. Yudanov, G. Pacchioni, K. Neyman and N. Rösch, J. Phys. Chem. B, 1997, 101, 2786–2792 CrossRef CAS.
- C. B. Alcock and G. W. Hooper, Proc. R. Soc. London, Ser. A, 1960, 254, 551–561 CrossRef CAS.
- H. Jehn, J. Less-Common Met., 1984, 100, 321–339 CrossRef CAS.
- T. R. Johns, R. S. Goeke, V. Ashbacher, P. C. Thüne, J. W. Niemantsverdriet, B. Kiefer, C. H. Kim, M. P. Balogh and A. K. Datye, J. Catal., 2015, 328, 151–164 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp04973h |
|
| This journal is © the Owner Societies 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab and
Dennis
Palagin
ab and
Dennis
Palagin