
Light emission colour modulation study of oxyluciferin synthetic analogues via QM and QM/MM approaches†
Madjid
Zemmouche
 a,
Cristina
García-Iriepa
ab and
Isabelle
Navizet
*a
a,
Cristina
García-Iriepa
ab and
Isabelle
Navizet
*a
aUniversité Paris-Est, Laboratoire Modélisation et Simulation Multi Échelle, MSME UMR 8208 CNRS, UPEM, 5 bd Descartes, 77454 Marne-la-Vallée, France. E-mail: isabelle.navizet@u-pem.fr
bDepartamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain
First published on 18th November 2019
Abstract
Firefly oxyluciferin is the chemical product of bioluminescence responsible for light emission. Experiments have already shown that different analogues of natural oxyluciferin, exhibit different emission colours. In particular, the structure of natural oxyluciferin has been modified by atom or group substitutions. However, a rationalization of the origin of the bioluminescence emission colour modulation of these analogues has still not been reported. For these reasons, the aim of this study is to explain the influence of structural modifications within the natural oxyluciferin on the colour modulation of bioluminescence. To do this, natural firefly oxyluciferin and three synthetic analogues whose experimental bioluminescence spectra are red- and blue-shifted compared to the natural one were studied. The absorption and emission transition energies have been calculated at the Time-dependent density functional theory (TD-DFT) level using both quantum mechanics (QM) and quantum mechanics/molecular mechanics (QM/MM) methods. Moreover, the solvent (water using the PCM model) and the protein surrounding effect have also been considered. The predicted emission spectra are in quite good agreement with the available experimental spectra, validating the methodology followed in this study. In particular, it was demonstrated that using the QM/MM approach, and considering explicitly the protein environment, the experimental bioluminescence spectra can be reproduced. Furthermore, this study shows that the substitution within the oxyluciferin structure causes a change of its electronic distribution and energies of the HOMO and LUMO orbitals involved in the vertical transitions, leading to different light emission colours. This work will promote future studies focused on luciferin mutations guided by the prediction of their bioluminescence emission spectra.
Introduction
Bioluminescence, the conversion of chemical energy into light emission by some living organisms such as fireflies, is a spectacular phenomenon of flashing light. In fireflies, the yellow-green light comes from the relaxation of the light emitter, the so-called oxyluciferin, from its excited to its ground state by photon release. In particular, oxyluciferin (described as Natural_Oxy in this paper) is generated in the excited state after oxidation of D-luciferin in the presence of dioxygen (O2), magnesium ions (Mg2+) and adenosine triphosphate (ATP), catalysed by an enzyme (luciferase) (Fig. 1a).1–5 | ||
| Fig. 1 (a) General reaction of light emission in fireflies through D-luciferin oxidation catalysed by luciferase. (b) Structure of the natural firefly light emitter, Natural_Oxy, and the synthetic analogues under study (chemical modification depicted in blue). The values in nanometers (nm) correspond to the experimental bioluminescence taken from ref. 14–16. | ||
Beyond its visual appeal, firefly bioluminescence has inspired scientists and has motivated extensive studies of this natural phenomenon due to its widespread applications. For instance, firefly bioluminescence has been used as a bioanalytical tool for in vivo imaging,6–8 real time monitoring of cell dynamics,9,10 gene expression and regulation,11,12 bacterial contamination, food quality control13 and other applications.
It has been previously reported that different factors can tune the colour of firefly bioluminescence, from blue to red, for instance by modifying the Natural_Oxy structure14–16 (Fig. 1b), by luciferase mutations,17–21 as well as changes of the temperature22 and pH.23 While the vast majority of the studies have been focused on luciferase (enzyme) mutations,17–21 other studies have discussed the chemical modification of the Natural_Oxy structure, presumably hampered by the difficulty in designing and synthesizing bioluminescent analogues.14–16,24,25 The main aim of synthesizing novel oxyluciferin analogues is to enhance biological applications. For this purpose, analogues emitting in the red and near-IR spectral window (>600 nm) are highly desirable due to their ability to penetrate deeper into living tissues.26
With this aim several synthetic analogues have been designed to obtain a red emission. However, the chemical modifications required are not straightforward and, in some cases, blue or even no light emission have been observed. Despite the efforts focused on understanding the factors responsible for colour modulation, many details regarding this system still remain under debate.27–29 According to previous studies, the emission wavelength of oxyluciferin analogues strongly depends on the molecular structure. For instance, Conley et al.25 designed an oxyluciferin analogue with a 40 nm red-shifted emission by replacing the sulfur atom of the thiazolone moiety with a selenium atom and the hydroxyl group of the phenol moiety by an amino group. An even more red-shifted analogue has been reported by Jathoul et al.30 (near-IR emission of 670 nm), extending the π-conjugation by introducing a double bond between the benzothiazole and thiazolone moieties. A remarkable instance of the effect of the sensitivity of the emitted colour on the chemical structure is the case of two hydroxypyridine-like analogues with blue-shifted (30 nm) and red-shifted (10 nm) emission, differing only in the position of an extra nitrogen atom in the benzothiazole ring closed to the phenol group.31 Moreover, not only a spectral shift of the emission can be achieved by structural modifications but also the emission quenching as is the case of the analogues synthesized by McCutcheon et al.32 and Ioka et al.15 for which the sulfur atom of the thiazolone moiety is replaced by nitrogen or oxygen atoms, respectively. Hence, the emission wavelength of oxyluciferin analogues can be blue- or red-shifted or even no light emission can be observed depending on the chemical modifications made in the benzothiazole14,16 and thiazolone15 moieties or in the π-backbone.29
In addition, some theoretical studies were focused on understanding the effect of the substitution on the colour tuning,33–37 showing that the change in the electronic density modified by the substitution of some of the atoms in the emitter structure influences the light colour. Even if the experimental reaction takes place in a protein cavity, most of the previous computational studies, either do not consider the effect of the surroundings or take into account the solvent only with an implicit model [polarizable continuum-model (PCM)].33,37–39 However, the effect of the protein is in some cases crucial to properly describe the system and reproduce the experimental values. Only a few computational studies have been reported in which the emission colour of the synthetic analogues has been calculated within the protein at the quantum mechanics/molecular mechanics (QM/MM) level.36,40
In this work, a computational study of the natural firefly oxyluciferin (Natural_Oxy) and three synthetic analogues was performed to investigate the origin of the modulation of their colour emission (Fig. 1b). The phenolate-keto chemical form of Natural_Oxy has been selected as it has been considered as the most probable light emitter in fireflies.41–44 Regarding the selected synthetic analogues, two of them exhibit a blue-shifted and the other one a red-shifted emission, compared to the natural bioluminescence. Regarding the blue-shifted analogues, in one case the structural modification is located in the benzothiazole moiety (nitrogen atom replaced by a methine, benzothiophene_Oxy in Fig. 1b) and for the other one in the thiazolone moiety (sulfur atom replaced by a methylene, dihydropyrrolone_Oxy in Fig. 1b). For the red-shifted analogue, the modification is placed in the benzothiazole moiety by introduction of an allyl group (allylbenzothiazole_Oxy, Fig. 1b). The study of this last analogue is quite interesting as a red-shift of the emission has been experimentally found despite the fact that the allyl group is not included in the π-conjugated system.16 Moreover, the allyl group is not part of the planar geometry, and so, the arrangement of the protein active site could be different due to the steric effects of the allyl group and its free rotation. The vertical transition energies corresponding to the absorption and emission of the four compounds under study (Fig. 1b) have been simulated to not only consider, in particular, the protein environment but also in the gas phase and in the implicit solvent (water) for comparison. The influence of the substituents and solvent effects on the absorption and emission wavelengths and on the oscillator strength (f) have been analysed and compared with experimental data.
Computational details
QM calculation
Density functional theory (DFT) was employed for the optimization of the ground state (GS) minima. Time-dependent density functional theory (TD-DFT) was used for the optimization in the excited state (ES) and calculation of the vertical transition energies. All calculations were carried out using the B3LYP45,46 functional with the 6-311G(2d,p) basis set. This functional and basis set were selected because they have been reported to be suitable for this chromophore.36,38,39,47 The energies of the S0 → S1 transition at the GS minimum, corresponding to the absorption, and the S1 → S0 transition at the first ES minimum, corresponding to the emission, were calculated in the gas phase and in implicit solvent (water using the polarization continuum model (PCM)48 with a dielectric constant of 78.355) for the four compounds under study (Fig. 1b). When considering the implicit solvent, the non-equilibrium regime within the linear response-PCM approach has been used for the calculation of the absorption and emission energies, whereas the equilibrium regime was used for the geometry optimizations. Moreover, the charge transfer (CT) character of the vertical transitions (both absorption and emission) was evaluated in implicit water by calculating the difference in the thiazolone charge (defined as the sum of the Mulliken charges of the atoms included in this moiety) between the GS and ES. To further investigate the charge transfer character, the natural transition orbitals (NTOs) were computed and their isosurfaces plotted with the Avogadro code,49 considering a contour threshold of 0.02 au. Furthermore, the HOMO and LUMO energies of the compounds and their energy gap (ΔEHOMO–LUMO) were calculated in the gas phase and in solution. All the calculations were carried out using the Gaussian 09 program package.50MD simulation
A classical molecular dynamic (MD) simulation of 20 ns was performed with the Amber14 program51 for each system under study within the protein in order to obtain a statistical number of snapshots to simulate the absorption and emission spectra. The crystallographic structure of firefly engineered luciferase (the North American firefly, Photinus pyralis), characterized by a disulfur bridge (linkage) between ILE108 and TYR447 residues, built to keep the second catalytic conformation of luciferase,52 was downloaded from the Protein Data Bank (PDB code 4G37).52 The choice of using the luciferase 4G37 conformation for this study was related to the fact that, during the bioluminescence process, light emission occurs in this second catalytic luciferase conformation.53 It should be noted that the substrate inside the crystallographic 4G37 structure, 5′-O-[N-dehydroluciferyl)-sulfamoyl]-adenosine (DLSA), has been removed and replaced by the Natural_Oxy structure (or by its synthetic analogues) and adenosine monophosphate (charged negatively −1 and further noted as AMPH). Moreover, the linkage between ILE108 and TYR447 residues has been removed, as no accurate parameters are available to describe this linkage, which is essential to perform the MD simulations. It was checked that the second catalytic conformation of the protein was kept along the classical MD simulations although the linkage had been deleted.Starting from the crystallographic structure, the residues were protonated by using LEaP from the Amber14 program.51 To have a neutral charge for the system, seven histidines (76, 171, 310, 332, 419, 461 and 489) were protonated by computing their pKa with the H++ program.54 The system was solvated within an octahedral box of water molecules, ensuring a solvent shell of at least 10 Å around the chromophore, which corresponded to around 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 water molecules. Then, after the minimization step, the system was heated from 100 K to 300 K in 20 ps. Finally, under isothermal–isobaric ensemble (NPT) conditions (T = 300 K and P = 1 atm), a 20 ns MD simulation with periodic boundary conditions was performed with a 2 fs time step. During these simulations, the pressure and temperature were maintained by using the Berendsen algorithm.55
000 water molecules. Then, after the minimization step, the system was heated from 100 K to 300 K in 20 ps. Finally, under isothermal–isobaric ensemble (NPT) conditions (T = 300 K and P = 1 atm), a 20 ns MD simulation with periodic boundary conditions was performed with a 2 fs time step. During these simulations, the pressure and temperature were maintained by using the Berendsen algorithm.55
The force field, Amberff14SB was used for the protein residues and the TIP3P water model56 was used for the water molecules. The parameters of AMPH were previously designed by our group.5,28,57 Regarding the force field of Natural_Oxy and its analogues, two sets of parameters were obtained for each molecule: one for the GS and another for the ES. First, a preliminary MD simulation was performed with a set of parameters (bonds, angles, torsions and charges) obtained in the gas phase for both the ground or first excited state minima (see section on QM calculation). One snapshot was extracted from each MD simulation and optimized at the QM/MM level. The parameters resulting from this QM/MM optimization were used for the final MD simulation, from which a statistical number of snapshots were extracted for further QM/MM calculations to simulate the absorption and emission spectra. This procedure had already been used in a previous study,58 and showed a good convergence in the shape of both absorption and emission spectra.
QM/MM calculation
One hundred snapshots were randomly extracted along the MD simulation and taken as the starting structures to compute the absorption and emission spectra at the QM/MM level. The Natural_Oxy and its analogues were described at the QM level while the water, AMPH and the protein were treated at the MM level. For each snapshot, water molecules within a radius of 15 Å from the QM part were selected and only the QM part and the surrounding residues within a radius of 9 Å were allowed to move. A coupling scheme59 between Gaussian09 and Tinker60 was used to perform the QM/MM calculation. The QM/MM optimizations of the ground and the first excited states were performed, followed by the calculation of the vertical S0 → S1 and S1 → S0 transition energies, corresponding to the absorption and emission, respectively. The electrostatic potential fitted method (ESPF)59 was used in particular to compute the interaction between the Mulliken charges of the QM part and the external electrostatic potential of the MM part within 9 Å from the QM part. The micro-iterations technique was used to converge the MM part for every QM minimization step. For the absorption spectra simulation, the first three singlet excited states were considered. A Gaussian function convolution of the absorption or emission energies of 100 snapshots was performed, using a half-width at half-maximum (HWHM) of 0.20 eV, to simulate the absorption and emission spectra.Results and discussion
Next, the origin of the bioluminescence colour tuning was investigated using the structural modifications of Natural_Oxy in terms of chemical physical properties: (i) the vertical transition energies (Te) and oscillator strength (f) of both absorption and emission, (ii) the CT character and the NTOs, and (iii) the HOMO and LUMO energies. This section is presented in two parts depending on the environment being considered: (i) compounds in the gas phase and implicit solvent, and (ii) compounds inside the protein active site used to investigate the H-bonds networks.Compounds in the gas phase and implicit solvent
First, the vertical transition (S0 → S1 and S1 → S0) energies were computed for the Natural_Oxy and its synthetic analogues in the gas phase and in implicit water at the QM level (Table 1). By analysing the data (Table 1), it was found that the absorption energies were shifted to lower energies (between 0.07–0.11 eV) and the emission energies were shifted to higher energies (between 0.04–0.10 eV) when considering implicitly the water solvent. In general, the differences in the transition energy values in the gas phase and in PCM may be related to the fact that the interaction with the solvent can sharply stabilize/destabilize the ground or excited states.| Absorption | Emission | Stokes shift (eV) | ||||||
|---|---|---|---|---|---|---|---|---|
| Environments | Compounds | T abse (eV) | λ abs (nm) | f | T emie (eV) | λ emi (nm) | f | T abse − Temie |
| Gas phase | Benzothiophene_Oxy | 2.74 | 452 | 0.63 | 2.40 | 515 | 0.45 | 0.33 |
| Dihydropyrrolone_Oxy | 2.67 | 465 | 0.59 | 2.32 | 534 | 0.39 | 0.35 | |
| Natural_Oxy | 2.60 | 476 | 0.60 | 2.27 | 545 | 0.41 | 0.33 | |
| Allylbenzothiazole_Oxy | 2.56 | 484 | 0.55 | 2.22 | 559 | 0.36 | 0.32 | |
| PCM (water) | Benzothiophene_Oxy | 2.66 | 465 | 0.81 | 2.50 | 495 | 0.7 | 0.16 |
| Dihydropyrrolone_Oxy | 2.59 | 478 | 0.72 | 2.41 | 514 | 0.60 | 0.18 | |
| Natural_Oxy | 2.50 | 496 | 0.73 | 2.32 | 533 | 0.61 | 0.18 | |
| Allylbenzothiazole_Oxy | 2.45 | 506 | 0.68 | 2.26 | 549 | 0.55 | 0.19 | |
| Protein | Benzothiophene_Oxy | 2.73 | 454 | 0.52 | 2.35 [2.37] | 528 [523] | 0.36 | 0.38 |
| Dihydropyrrolone_Oxy | 2.63 | 471 | 0.51 | 2.22 [2.26] | 557 [547] | 0.30 | 0.41 | |
| Natural_Oxy | 2.52 | 491 | 0.51 | 2.18 [2.21] | 568 [560] | 0.31 | 0.34 | |
| Allylbenzothiazole_Oxy | 2.49 | 498 | 0.50 | 2.02 [2.05] | 614 [605] | 0.24 | 0.47 | |
Moreover, the Stokes shifts (difference between absorption and emission energies) in both the gas phase and the implicit water environments (Table 1) were calculated. It was found that the order of magnitude of the Stokes shifts calculated with the implicit solvent (PCM) were half of those calculated in gas phase, and this was a consequence of the absorption red-shift and emission blue-shift when considering the solvent. In addition, the oscillator strengths (f), of both the absorption and emission transitions, increase substantially in the presence of the implicit solvent. To rationalize this finding, the square of the transition dipole moments (μge) between the ground state and excited states for the absorption and the emission, respectively, in the gas phase and in the implicit water (PCM) were calculated (Table 2). It was observed that the square of the transition dipole moment (μge)2 increased as long as the oscillator strength (f) increased (f values in Table 1), as the magnitude of f for an electronic transition is proportional to the square of μge.
| Gas phase | Water (PCM) | |||
|---|---|---|---|---|
| Compounds | μ ge 2 at S0 | μ ge 2 at S1 | μ ge 2 at S0 | μ ge 2 at S1 |
| Benzothiophene_Oxy | 89 | 59 | 155 | 130 |
| Dihydropyrrolone_Oxy | 81 | 48 | 130 | 104 |
| Natural_Oxy | 86 | 55 | 140 | 114 |
| Allylbenzothiazole_Oxy | 78 | 45 | 128 | 99 |
The influence of the structural modifications on the absorption and emission transition was also investigated. The computed emission energies of the Natural_Oxy and the three analogues followed the same trend as the experimentally observed bioluminescence: the benzothiophene_Oxy and dihydropyrrolone_Oxy maximum wavelengths were blue-shifted compared to the ones of Natural_Oxy, whereas those of allylbenzothiazole_Oxy were red-shifted (Table 1). Unfortunately, a direct comparison with the experimental data cannot be made as the absorption and emission experimental spectra of the synthetic analogues in water solution were not available.
The frontier molecular orbital energies (EHOMO and ELUMO) and their energy difference (ΔEHOMO–LUMO) were computed for the absorption and the emission transitions in PCM (water) and in the gas phase (see Table S1, ESI†). We found that the energy differences (ΔEHOMO–LUMO), in both absorption and emission transitions, were affected by the oxyluciferin structural modifications, which was in line with the differences found in the transition energies (Table 1). For instance, the energy difference (ΔEHOMO–LUMO) was smaller for allylbenzothiazole_Oxy than for Natural_Oxy, which explained the red-shift of the maxima absorption and emission wavelengths (Table 1). Whereas, in the benzothiophene_Oxy and dihydropyrrolone_Oxy, the HOMO–LUMO energies difference was larger compared to that of the Natural_Oxy, which was in agreement with the computed blue-shift in the maximum absorption and emission wavelengths (Table 1).
Furthermore, the CT character (see Computational details) of the S0 → S1 and S1 → S0 transitions corresponding to the absorption and emission (Table 3 and Table S2, ESI†) for the compounds under study were analysed. In detail, for both the ground and excited state minima, the sum of the Mulliken charges of the atoms included in the thiazolone moiety in the ground (S0) and in the first excited state (S1) were calculated. The difference between the charge of the thiazolone moiety in S0 and S1 shows the through space CT character of the transition (between the thiazolone and the benzothiazole moieties) (Table 3 for the emission and Table S2 (ESI†) for the absorption). As defined in this work, positive values of CT indicate an electronic CT from the thiazolone to the benzothiazole moiety. By analysing the calculated CT character for the emission transition, it was found that the CT character of the studied compounds were quite similar, being slightly smaller for benzothiophene_Oxy and dihydropyrrolone_Oxy (Table 3). This means that the substitution within the Natural_Oxy structure scarcely influenced the electronic nature of the absorption and emission vertical transitions, that is, the CT character.
 character calculated as the difference of the thiazolone moiety Mulliken charges between the ground
character calculated as the difference of the thiazolone moiety Mulliken charges between the ground 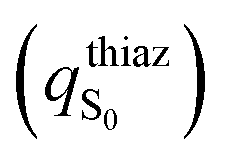 and the first
and the first  excited states for the emission (S1 → S0) in water (PCM), at the excited state minima
excited states for the emission (S1 → S0) in water (PCM), at the excited state minima
| Compounds |
|
|
|
|---|---|---|---|
| Benzothiophene_Oxy | −0.279 | −0.438 | 0.159 |
| Dihydropyrrolone_Oxy | −0.254 | −0.409 | 0.155 |
| Natural_Oxy | −0.294 | −0.456 | 0.162 |
| Allylbenzothiazole_Oxy | −0.296 | −0.468 | 0.172 |

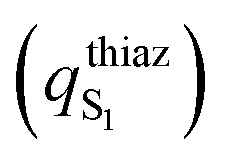

Finally, the NTOs involved in the absorption and emission transitions were computed for the gas phase and for PCM for the four compounds under study (Fig. 2 and Fig. S1, S2, ESI†). First, it should be noted that the electron density of the NTOs computed for the gas phase and the water were quite similar. By analysing the NTOs, it was observed that for all the compounds, the electron density was transferred from the benzothiazole to the thiazolone moiety upon absorption and the other way around for the emission transition (Fig. 2 and Fig. S1, S2, ESI†). If the difference of the electron density distribution in the Natural_Oxy and in its synthetic analogues is analysed in more detail, it was observed that it was sensitive to the structural modifications. Starting with benzothiophene_Oxy, the electron density located in the CH group (which replaces the N atom of the benzothiazole moiety in Natural_Oxy), of the unoccupied NTO is slightly larger than the one computed for Natural_Oxy (dotted circle in Fig. 2).
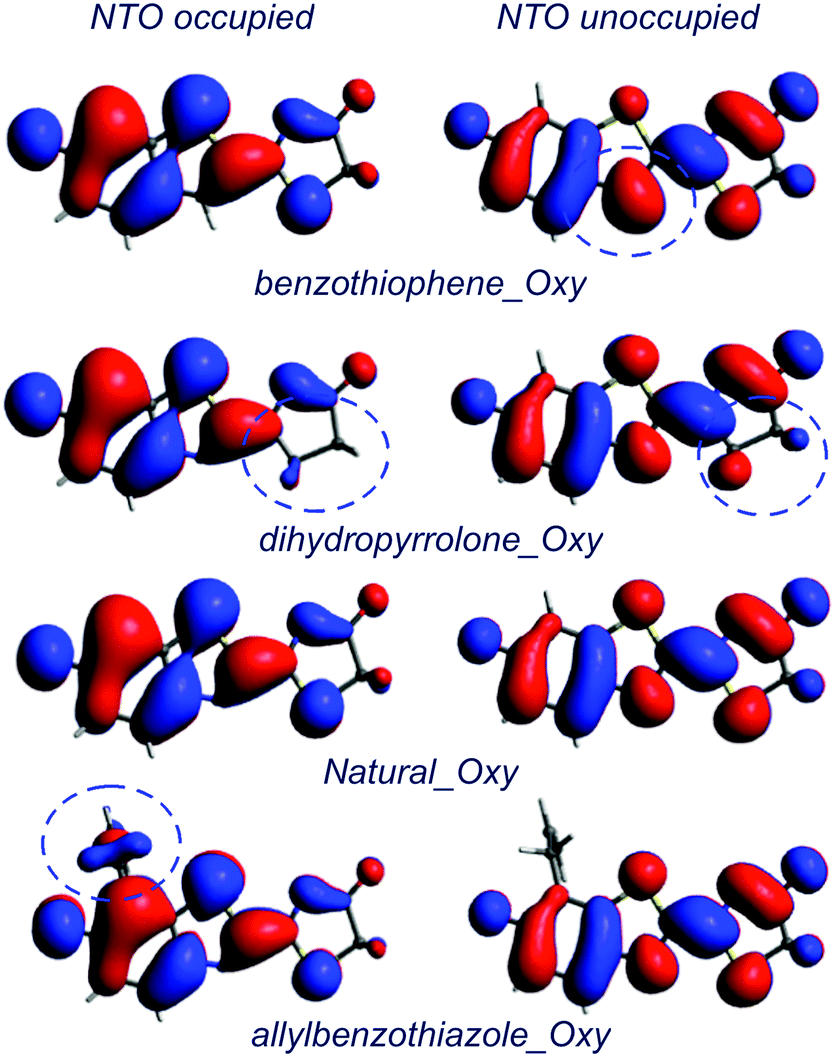 | ||
| Fig. 2 Natural transition orbitals (NTOs) of Natural_Oxy and its synthetic analogues computed at the excited state minimum corresponding to the emission in water (PCM). | ||
Regarding dihydropyrrolone_Oxy (substitution of the S atom of the thiazolone moiety by a CH2 group), its unoccupied and occupied NTOs have a lower electron density located in the CH2 group, compared to the one located on the S atom of the Natural_Oxy (dotted circle in Fig. 2), due to the larger electronegativity of the S atom.
For the allylbenzothiazole_Oxy analogue, although the allyl group is not included in the π backbone (it is not conjugated), the occupied NTO has some electronic density focused in this substituent (dotted circle in Fig. 2). Hence, it can be noted that the structural modifications of Natural_Oxy slightly influence the electron density of the NTO orbitals involved, both in the absorption and emission transitions, which is in line with the slight variations of their CT character.
In conclusion, the difference in the electronic densities and the CT in the four compounds are slight but consistent with the trend of the calculated emission and absorption wavelengths.
Compounds in a protein environment
To investigate in more detail the structural modification effects on the experimental colour tuning of the bioluminescence of the fireflies, the absorption and emission spectra of the four compounds at the QM/MM level considering the protein environment (see Computational details) were simulated. In addition, the computed values considering the protein environment to those in gas phase (Table 1) were compared. It was found that when considering, in particular, the protein environment, both the absorption and emission energies were shifted to lower energies (between 0.01–0.08 eV and 0.05–0.2 eV, respectively), compared to the calculations in gas phase (Table 1). In addition, the Stokes shifts calculated considering the protein environment were larger (between 0.05–0.15 eV) than those calculated in the gas phase. The oscillator strengths (f) in both the gas phase and the protein were of the same order of magnitude, being slightly smaller in the protein than in the gas phase. Comparing the f values of the compounds in all the environments, in general the ones of benzothoiphene_Oxy were slightly larger than the ones of Natural_Oxy, whereas the ones of dihydropyrrolone_Oxy and allylbenzothiazole_Oxy were smaller than the one of the Natural_Oxy. The range of magnitude was small but still consistently independent of the environment. However, this information cannot be exploited yet as no relative experimental intensities are available.Moreover, the NTOs involved in the absorption and emission transitions for the compounds inside the protein active site were similar to the ones obtained in the gas phase (Fig. S1 and S2, ESI†).
With the aim of determining the structural effect on the bioluminescence, the simulated emission spectra of the Natural_Oxy and its synthetic analogues inside the protein were compared with the experimental ones. The theoretical spectral shapes were quite similar to the experimental ones (Fig. 3). Moreover, the difference between the computed and the experimental maximum emission wavelengths of the four compounds in the protein were less than 10 nm. The qualitative trend of the maximum emission wavelengths of the calculated spectra was the same as the one for the experimental spectra14–16 (Fig. 3). For instance, for allylbenzothiazole_Oxy the emission spectrum was red-shifted 46 nm theoretically and 45 nm experimentally, for the Natural_Oxy. Similarly, for benzothiophene_Oxy and dihydropyrrolone_Oxy, quite good agreement was achieved between the simulated and experimental emission spectra. In particular blue-shifts of 40 nm and 11 nm were found theoretically, for benzothiophene_Oxy and dihydropyrrolone_Oxy, respectively, whereas experimentally the shifts were of 37 nm and 11 nm, respectively. The maximum emission wavelength shifts of the analogues were compared to those of the Natural_Oxy, and were therefore smaller in the protein simulated spectra than in the experimental one. It should be noted that the allylbenzothiazole_Oxy simulated spectrum was wider than the other ones (Fig. 3a). The oscillator strengths for the 100 vertical S1 → S0 transitions of the four compounds under study were also computed (Fig. S3 and S4, ESI†). For all the compounds, the optically bright vertical transition energies were located in a narrow range of energy, except for allylbenzothiazole_Oxy, for which they were found in a larger range (Fig. S3 and S4, ESI†). The origin of this could be due to the large dihedral angle fluctuation between 40° and −160° of the allyl group found during the MD simulations (Fig. S8, ESI†). Hence, a Gaussian function convolution of the vertical transition emission energies of these compounds gave a simulated allylbenzothiazole_Oxy emission spectrum which was broader by comparison to the others. The relative order of the calculated wavelength maxima in the gas phase and water solvent (Table 1) are the same as the ones in the protein for both emission and absorption (Fig. S6, ESI†).
 | ||
| Fig. 3 (a) Emission spectra of Natural_Oxy and its analogues simulated with QM/MM methods considering 100 statistical MD snapshots (HWHM of 0.20 eV), and (b) experimental bioluminescence spectra14–16 adapted with permission from (C. C. Woodroofe, P. L. Meisenheimer, D. H. Klaubert, et al., Novel Heterocyclic Analogues of Firefly Luciferin, Biochemistry, 2012, 51, 9807–9813). Copyright (2012) American Chemical Society, and from (S. Ioka, T. Saitoh, S. Iwano, et al., Synthesis of Firefly Luciferin Analogues and Evaluation of the Luminescent Properties, Chem. – Eur. J., 2016, 22, 9330–9337. Copyright (2016) Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Due to the crucial role of the protein and its surrounding on the emission spectra, the interactions (H-bonds) between the compounds under study and the active site environment were investigated, to better understand the influence of the structural modification. In order to determine the effect of substitution within the oxyluciferin structure on the H-bond networks with the protein and solvent, the MD simulations of Natural_Oxy and its analogues were performed using the parameters of the excited state (see Computational details). The statistical average number of H-bond interactions between Natural_Oxy, the protein, water molecules and AMPH are reported in Table S3 (ESI†). Fig. 4 presents a representative snapshot of each compound studied showing the H-bond interactions with the surroundings. Starting with benzothiophene_Oxy, for which the benzothiazole moiety is replaced by a benzothiophene, it was observed that the H-bond interaction for the Natural_Oxy between the N2 atom (see Fig. 4 for atom numbering) of the benzothiazole moiety and the amino acids SER347 and GLY246 no longer existed due to the structural modification (Fig. 4). Moreover, along the simulation of benzothiophene_Oxy, the H-bond interaction between the phenolate (O10) and ARG337 was not observed, contrary to what occurred in the simulation of Natural_Oxy. However, this fact can be explained by the flexibility of the ARG side chain whose position can fluctuate significantly. The structural modification could not be the reason for this finding. This hypothesis was confirmed by a second MD simulation (Fig. S7c, ESI†) of benzothiophene_Oxy for which ARG337 comes closer to benzothiophene_Oxy leading to the H-bond interaction (as already observed in previous work with Natural_Oxy61). In addition, by analysing the MD simulation of benzothiophene_Oxy and Natural_Oxy, it was found that benzothiophene_Oxy gets closer to AMPH, leading to more H-bond interactions (Fig. 4, Fig. S8 and Table S3, ESI†).
 | ||
| Fig. 4 Representative snapshots extracted from the MD simulations performed with the excited state parameters of Natural_Oxy and its analogues. Atom numbering is given. H-bond interactions are depicted as dotted lines. | ||
Furthermore, analysing the MD simulation of the dihydropyrrolone_Oxy (for which the thiazolone moiety has been replaced by a dihydropyrrolone moiety) it was observed that it behaves almost identically to Natural_Oxy inside the protein cavity and it has the same tendency to form H-bonds with either the surrounding water molecules, the amino acids (SER314, ARG218, ARG337) and AMPH (Fig. 4 and Fig. S9, ESI†). In addition, the distance between the atoms O10 or O11 (see the atom numbers in Fig. 4) of both molecules and some of the atoms of either amino acids (SER314, ARG218 and ARG337) or AMPH were also measured (Fig. S7, ESI†). It was found that in both molecules all the amino acids and AMPH were located almost at the same distances during the MD simulations. Moreover, the probabilities of the H-bond formation were quite similar in both dihydropyrrolone_Oxy and Natural_Oxy MD simulations (Table S3, ESI†). Hence, this structural modification does not have significant influence on the interaction with the environment for dihydropyrrolone_Oxy. This means that the origin of the blue-shifted experimental and simulated emission spectra does not lie on different H-bond interactions.
Finally, the MD simulations of allylbenzothiazole_Oxy were analysed and compared with those of Natural_Oxy to check the steric effect of the allyl group on the interactions with the surroundings (Fig. 4 and Fig. S10, ESI†). It was found that in general, the same H-bonds were formed with the closer amino acids (SER347, ARG337, ARG218 and GLY246) as well as with AMPH. However, no H-bond network between SER314 (close to the allyl group) and O10 of allylbenzothiazole_Oxy atom were found (Fig. 4). Measuring the distance between SER314 and the O10 atom of both molecules (Fig. S7, ESI†), it was observed that SER314 was located around 7.5 Å away from the O10 atom of allylbenzothiazole_Oxy, whereas it lies around 5.5 Å away from the O10 atom of the Natural_Oxy. Moreover, the root mean square deviation (RMSD) has been computed from the MD simulations of the compounds under study (Fig. S11, ESI†). It was found that the RMSD was within 1 Å and 3 Å, confirming that there was a stable trajectory except for the one of allylbenzothiazole_Oxy, in which large fluctuations have been observed due to the allyl group rotation during the MD simulation (Fig. S5 (ESI†) shows a large dihedral angle fluctuation between 40° and −160°). In addition, the average number of H-bonds formed between the O10 atom of allylbenzothiazole_Oxy and the surrounding water molecules is half of the number found in the Natural_Oxy simulations (Table S3 and Fig. S12, ESI†). This lack of water molecules may be due to the steric effect of the allyl group as the rotation of this group during the MD simulation prevents the water molecules getting closer to the O10 atom.
Conclusions
In this work, QM and QM/MM methods together with the TD-DFT have been used to simulate the absorption and emission wavelengths of firefly Natural_Oxy (in its phenolate-keto form) and some synthetic analogues that emit different light emission colours, ranging from blue to red. The predicted results in the gas phase and in solution (water) indicate that considering the solvent, the absorption energies are shifted to lower energies and the emission energies are shifted to higher energies (0.07–0.11 eV and 0.04–0.1 eV, respectively), and as well the oscillator strength (f) increased substantially. Moreover, the analysis of the calculated vertical transition energies (Te) in gas phase and in protein indicates that considering the protein environment, shifts to lower energies occur giving absorption and emission energies shifts of 0.01–0.08 eV and 0.05–0.2 eV, respectively. The substitution effects on the emission values can be summarized as follows: substitutions by less electron donor groups in the benzothiazole (N by CH) or thiazolone (S by CH2) moieties induce a blue-shift of the maximum absorption and emission wavelengths, whereas substitutions by more electron donor groups on benzothiazole (H by CH2CHCH2) induce a red-shift of the maximum absorption and emission wavelengths. In addition, from this study, it was found that the substitutions within the Natural_Oxy benzothiazole and thiazolone moieties on the one hand, slightly influence the electronic density distribution, and on the other hand, influence the H-bond interactions with the protein environment, water molecules or AMPH. Furthermore, the steric effects generated by the introduction of an allyl group have an impact on the formation of the H-bond interactions. It should be noted that the computed trend of the maximum emission wavelengths of the four compounds is in quite good agreement with the experimental data from bioluminescence. It has been demonstrated that the experimental bioluminescence data can be reproduced by the QM/MM approach by considering explicitly the protein environment (theoretical maximum emission wavelengths less than 10 nm away from the experimental bioluminescent wavelengths). Further studies on novel synthetic analogues could make use of the present results as it has been shown that reasonable calculations in the gas phase can already give a good qualitative trend (see Table S4 and Fig. S13, ESI†), although QM/MM calculations are needed for quantitative agreement or study of the interactions with the surroundings.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors are grateful to the French Agence Nationale de la Recherche (Grant: ANR-BIOLUM ANR-16-CE29-0013). Cristina García-Iriepa acknowledges the Fundación Ramón Areces for a postdoctoral fellowship.References
- E. H. White, E. Rapaport, H. H. Seliger and T. A. Hopkins, The chemi- and bioluminescence of firefly luciferin: An efficient chemical Z production of electronically excited states, Bioorg. Chem., 1971, 1, 92–122 CrossRef CAS.
- M. Deluca, Firefly luciferase, Adv. Enzymol. Relat. Areas Mol. Biol., 1976, 44, 37–68 CAS.
- V. R. Viviani, F. G. C. Arnoldi, A. J. S. Neto, T. L. Oehlmeyer, E. J. H. Bechara and Y. Ohmiya, The structural origin and biological function of pH-sensitivity in firefly luciferases, Photochem. Photobiol. Sci., 2008, 7, 159–169 RSC.
- P. Naumov, Y. Ozawa, K. Ohkubo and S. Fukuzumi, Structure and Spectroscopy of Oxyluciferin, the Light Emitter of the Firefly Bioluminescence, J. Am. Chem. Soc., 2009, 131, 11590–11605 CrossRef CAS PubMed.
- I. Navizet, Y.-J. Liu, N. Ferré, D. Roca-Sanjuán and R. Lindh, The Chemistry of Bioluminescence: An Analysis of Chemical Functionalities, ChemPhysChem, 2011, 12, 3064–3076 CrossRef CAS PubMed.
- C. C. Woodroofe, P. L. Meisenheimer, D. H. Klaubert, Y. Kovic, J. C. Rosenberg, C. E. Behney, T. L. Southworth and B. R. Branchini, Novel Heterocyclic Analogues of Firefly Luciferin, Biochemistry, 2012, 51(49), 9807–9813 CrossRef CAS PubMed.
- L. F. Greer and A. A. Szalay, Imaging of light emission from the expression of luciferases in living cells and organisms: a review, Luminescence, 2002, 17, 43–74 CrossRef CAS PubMed.
- T. Kuchimaru, et al., A luciferin analogue generating near-infrared bioluminescence achieves highly sensitive deep-tissue imaging, Nat. Commun., 2016, 7, 11856 CrossRef CAS PubMed.
- Q. Shao, T. Jiang, G. Ren, Z. Cheng and B. Xing, Photoactivable bioluminescent probes for imaging luciferase activity, Chem. Commun., 2009, 4028 RSC.
- T. C. Doyle, S. M. Burns and C. H. Contag, In vivo bioluminescence imaging for integrated studies of infection, Cell. Microbiol., 2004, 6, 303–317 CrossRef CAS PubMed.
- L. J. Kricka, Application of bioluminescence and chemiluminescence in biomedical sciences, Methods Enzymol., 2000, 305, 333–345 CAS.
- H. Ohkuma, K. Abe, Y. Kosaka and M. Maeda, Detection of luciferase having two kinds of luminescent colour based on optical filter procedure: application to an enzyme immunoassay, Luminescence, 2000, 15, 21–27 CrossRef CAS PubMed.
- M. W. Griffiths, The role of ATP bioluminescence in the food industry: new light on old problems, Food Technol., 1996, 50, 62–72 CAS.
- C. C. Woodroofe, P. L. Meisenheimer, D. H. Klaubert, Y. Kovic, J. C. Rosenberg, C. E. Behney, T. L. Southworth and B. R. Branchini, Novel Heterocyclic Analogues of Firefly Luciferin, Biochemistry, 2012, 51, 9807–9813 CrossRef CAS PubMed.
- S. Ioka, T. Saitoh, S. Iwano, K. Suzuki, S. A. Maki, A. Miyawaki, M. Imoto and S. Nishiyama, Synthesis of Firefly Luciferin Analogues and Evaluation of the Luminescent Properties, Chem. – Eur. J., 2016, 22, 9330–9337 CrossRef CAS PubMed.
- Y. Ikeda, T. Saitoh, K. Niwa, T. Nakajima, N. Kitada, S. Maki, M. Sato, D. Citterio, S. Nishiyama and K. Suzuki, An allylated firefly luciferin analogue with luciferase specific response in living cells, Chem. Commun., 2018, 54, 1774–1777 RSC.
- K. A. Jones, W. B. Porterfield, C. M. Rathbun, D. C. McCutcheon, M. A. Paley and J. A. Prescher, Orthogonal Luciferase–Luciferin Pairs for Bioluminescence Imaging, J. Am. Chem. Soc., 2017, 139, 2351–2358 CrossRef CAS PubMed.
- D. M. Mofford, G. R. Reddy and S. C. Miller, Aminoluciferins Extend Firefly Luciferase Bioluminescence into the Near-Infrared and Can Be Preferred Substrates over D-Luciferin, J. Am. Chem. Soc., 2014, 136, 13277–13282 CrossRef CAS PubMed.
- Y. Wang, H. Akiyama, K. Terakado and T. Nakatsu, Impact of site-directed mutant luciferase on quantitative green and orange/red emission intensities in firefly bioluminescence, Sci. Rep., 2013, 3, 2490 CrossRef PubMed.
- B. R. Branchini, et al., Red-emitting luciferases for bioluminescence reporter and imaging applications, Anal. Biochem., 2010, 396, 290–297 CrossRef CAS PubMed.
- B. R. Branchini, D. M. Ablamsky and J. C. Rosenberg, Chemically Modified Firefly Luciferase Is an Efficient Source of Near-Infrared Light, Bioconjugate Chem., 2010, 21, 2023–2030 CrossRef CAS PubMed.
- T. Mochizuki, Y. Wang, M. Hiyama and H. Akiyama, Robust red-emission spectra and yields in firefly bioluminescence against temperature changes, Appl. Phys. Lett., 2014, 104, 213704 CrossRef.
- A. P. Jathoul, H. Grounds, J. C. Anderson and M. A. Pule, A dual-color far-red to near-infrared firefly luciferin analogue designed for multiparametric bioluminescence imaging, Angew. Chem., Int. Ed., 2014, 53, 13059–13063 CrossRef CAS PubMed.
- G. R. Reddy, W. C. Thompson and S. C. Miller, Robust Light Emission from Cyclic Alkylaminoluciferin Substrates for Firefly Luciferase, J. Am. Chem. Soc., 2010, 132, 13586–13587 CrossRef CAS PubMed.
- N. R. Conley, A. Dragulescu-Andrasi, J. Rao and W. E. Moerner, A Selenium Analogue of Firefly D-luciferin with Red-Shifted Bioluminescence Emission, Angew. Chem., Int. Ed., 2012, 51, 3350–3353 CrossRef CAS PubMed.
- R. Weissleder, A clearer vision for in vivo imaging, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- S. Hosseinkhani, Molecular enigma of multicolor bioluminescence of firefly luciferase, Cell. Mol. Life Sci., 2011, 68, 1167–1182 CrossRef CAS PubMed.
- I. Navizet, Y.-J. Liu, N. Ferré, H.-Y. Xiao, W.-H. Fang and R. Lindh, Color-Tuning Mechanism of Firefly Investigated by Multi-Configurational Perturbation Method, J. Am. Chem. Soc., 2010, 132, 706–712 CrossRef CAS PubMed.
- S. Iwano, et al., Development of simple firefly luciferin analogs emitting blue, green, red, and near-infrared biological window light, Tetrahedron, 2013, 69, 3847–3856 CrossRef CAS.
- A. P. Jathoul, H. Grounds, J. C. Anderson and M. A. Pule, Designed for Multiparametric Bioluminescence Imaging, 2014 Search PubMed.
- B. S. Zhang, K. A. Jones, D. C. McCutcheon and J. A. Prescher, Pyridone Luciferins and Mutant Luciferases for Bioluminescence Imaging, ChemBioChem, 2018, 19, 470–477 CrossRef CAS PubMed.
- D. C. McCutcheon, M. A. Paley, R. C. Steinhardt and J. A. Prescher, Expedient synthesis of electronically modified luciferins for bioluminescence imaging, J. Am. Chem. Soc., 2012, 134, 7604–7607 CrossRef CAS PubMed.
- Y. Y. Cheng, J. Zhu and Y. J. Liu, Theoretical tuning of the firefly bioluminescence spectra by the modification of oxyluciferin, Chem. Phys. Lett., 2014, 591, 156–160 CrossRef CAS.
- M. Zhang, M. Zhang and G. Zhao, Time-dependent density functional theory (TDDFT) study on the electronic spectroscopic blue-shift phenomenon and photoinduced charge transfer of firefly luciferin anion in aqueous solution: Insight into the excited-state hydrogen bond weakening mechanism, J. Lumin., 2018, 195, 116–119 CrossRef CAS.
- C. G. Min, Y. Leng, X. K. Yang, S. J. Huang and A. M. Ren, Systematic color tuning of a family of firefly oxyluciferin analogues suitable for OLED applications, Dyes Pigm., 2016, 126, 202–208 CrossRef CAS.
- R. Berraud-Pache and I. Navizet, QM/MM calculations on a newly synthesised oxyluciferin substrate: new insights into the conformational effect, Phys. Chem. Chem. Phys., 2016, 18, 27460–27467 RSC.
- L. Pinto Da Silva and J. C. G. Esteves Da Silva, Theoretical fingerprinting of the photophysical properties of four firefly bioluminophores, Photochem. Photobiol. Sci., 2013, 12, 2028–2035 RSC.
- C. G. Min, Y. Leng, Y. Q. Zhu, X. K. Yang, S. J. Huang and A. M. Ren, Modification of firefly cyclic amino oxyluciferin analogues emitting multicolor light for OLED and near-Infrared biological window light for bioluminescence imaging: a theoretical study, J. Photochem. Photobiol., A, 2017, 336, 115–122 CrossRef CAS.
- C. Garcia Iriepa, M. Zemmouche, M. Ponce-Vargas and I. Navizet, The role of solvation models on the computed absorption and emission spectra: the case of fireflies oxyluciferin, Phys. Chem. Chem. Phys., 2019, 21, 4613–4623 RSC.
- Y. Y. Cheng and Y. J. Liu, Theoretical Development of Near-Infrared Bioluminescent Systems, Chem. – Eur. J., 2018, 24(37), 9340–9352 CrossRef CAS PubMed.
- P. Naumov and M. Kochunnoonny, Spectral-Structural Effects of the Keto–Enol–Enolate and Phenol Phenolate Equilibria of Oxyluciferin, J. Am. Chem. Soc., 2010, 132, 11566–11579 CrossRef CAS PubMed.
- B. R. Branchini, M. H. Murtiashaw, R. A. Magyar, N. C. Portier, M. C. Ruggiero and J. G. Stroh, Yellow-green and red firefly bioluminescence from 5,5-dimethyloxyluciferin, J. Am. Chem. Soc., 2002, 124, 2112–2113 CrossRef CAS PubMed.
- M. Rebarz, et al., Deciphering the protonation and tautomeric equilibria of firefly oxyluciferin by molecular engineering and multivariate curve resolution, Chem. Sci., 2013, 4(10), 3803–3809 RSC.
- S. F. Chen, Y. J. Liu, I. Navizet, N. Ferré, W. H. Fang and R. Lindh, Systematic theoretical investigation on the light emitter of firefly, J. Chem. Theory Comput., 2011, 7(3), 798–803 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- C. Min, A. Ren, J. Guo, Z. Li, L. Zou, J. D. Goddard and J. Feng, A Time-Dependent Density Functional Theory Investigation on the Origin of Red Chemiluminescence, ChemPhysChem, 2010, 11, 251–259 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 17 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- D. A. Case, V. Babin, J. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, S. LeGrand, T. Luchko, R. Luo, B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu and P. A. Kollman, Amber 14, 2014 Search PubMed.
- J. A. Sundlov, D. M. Fontaine, T. L. Southworth, B. R. Branchini and A. M. Gulick, Crystal structure of firefly luciferase in a second catalytic conformation supports a domain alternation mechanism, Biochemistry, 2012, 51, 6493–6495 CrossRef CAS PubMed.
- B. R. Branchini, J. C. Rosenberg, D. M. Fontaine, T. L. Southworth, C. E. Behney and L. Uzasci, Bioluminescence is produced from a trapped firefly luciferase conformation predicted by the domain alternation mechanism, J. Am. Chem. Soc., 2011, 133, 11088–11091 CrossRef CAS PubMed.
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations, Nucleic Acids Res., 2012, 40, W537–W541 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- I. Navizet, D. Roca-sanjuán, L. Yue, Y. Liu, N. Ferré and R. Lindh, Are the Bio- and Chemiluminescence States of the Firefly Oxyluciferin the Same as the Fluorescence State?, Photochem. Photobiol., 2013, 319–325 CrossRef CAS PubMed.
- C. García-Iriepa, P. Gosset, R. Berraud-Pache, M. Zemmouche, G. Taupier, K. D. Dorkenoo, P. Didier, J. Léonard, N. Ferré and I. Navizet, Simulation and Analysis of the Spectroscopic Properties of Oxyluciferin and Its Analogues in Water, J. Chem. Theory Comput., 2018, 14, 2117–2126 CrossRef PubMed.
- N. Ferré and J. G. Ángyán, Approximate electrostatic interaction operator for QM/MM calculations, Chem. Phys. Lett., 2002, 356, 331–339 CrossRef.
- J. W. Ponder, TINKER, Software Tools for Molecular Design, version 6.3, Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. Louis, MO, 2004 Search PubMed.
- C. Garcia-iriepa and I. Navizet, Effect of Protein Conformation and AMP Protonation State on Fireflies’ Bioluminescent Emission, Molecules, 2019, 24, 1565 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp04687a |
| This journal is © the Owner Societies 2020 |