
Critical nucleus of Greek-key-like core of α-synuclein protofibril and its disruption by dopamine and norepinephrine†
Yu
Zou
ab,
Zhenyu
Qian
c,
Yehong
Gong
b,
Yiming
Tang
d,
Guanghong
Wei
 *d and
Qingwen
Zhang
*b
*d and
Qingwen
Zhang
*b
aDepartment of Sport and Exercise Science, College of Education, Zhejiang University, 148 Tianmushan Road, Hangzhou 310007, Zhejiang, People's Republic of China
bCollege of Physical Education and Training, Shanghai University of Sport, 399 Changhai Road, Shanghai 200438, People's Republic of China. E-mail: zqw@sus.edu.cn
cKey Laboratory of Exercise and Health Sciences (Ministry of Education) and School of Kinesiology, Shanghai University of Sport, 399 Changhai Road, Shanghai 200438, People's Republic of China
dState Key Laboratory of Surface Physics, Key Laboratory for Computational Physical Science (Ministry of Education), and Department of Physics, Fudan University, 220 Handan Road, Shanghai 200433, People's Republic of China. E-mail: ghwei@fudan.edu.cn
First published on 18th November 2019
Abstract
The formation of amyloid fibrils by α-synuclein (αS) protein inside the Lewy bodies and Lewy neurites is the prominent pathological hallmark of Parkinson's disease (PD). The fibrillation of αS in vitro is described by a nucleation–elongation process involving the formation of a critical nucleus. Finding the critical/smallest nuclei and effective inhibitors of αS aggregation is a crucial step for the development of drugs against PD. Recent experiments reported that dopamine (DA) and norepinephrine (NE), two prominent naturally occurring neurotransmitters, can effectively disrupt the preformed αS fibrils. The level of DA/NE in blood can be markedly increased by exercise. However, the size and structure of the critical nucleus and the disruptive mechanism by DA/NE are largely unknown. In this work, we performed multiple molecular dynamics (MD) simulations to find the critical nucleus size and examine the influences of DA/NE molecules on preformed αS44–96 (Greek-key-like core of full length αS) protofibrils. Our results show that the trimer is the critical nucleus for the αS44–96 fibril formation, and the tetramer is the minimal stable nucleus. When DA/NE molecules bind to the fibril-like trimer and tetramer, they strongly destabilize the αS protofibrils by disrupting the β-sheet structure and inter-chain E46-K80 salt bridges. Two common binding sites are identified for both DA and NE molecules on αS oligomers: residues 57–70 and 81–83. A different binding site is also observed, which is located at the N-terminal region (residues 45–52). The binding of DA/NE molecules to αS oligomers is mostly driven by hydrophobic and electrostatic interactions. We found two disruptive modes, and binding to the turn region of αS oligomers but disrupting the adjacent β-sheet structure is the dominant one. Our work identified the critical nucleus of Greek-key-like core of αS protofibrils and revealed the disruptive mechanism of αS protofibrils by DA/NE molecules, which may be helpful to the design of effective drugs against αS aggregation.
Introduction
Parkinson's disease (PD), one of the major neurodegenerative disorders, affects 1–2% of the population over the age of 65 worldwide currently.1 One prominent pathological hallmark of PD is the formation of fibrils by α-synuclein (αS) protein inside the Lewy bodies and Lewy neurites, making αS a potential therapeutic target for the development of treatments.2,3 The structure of αS fibrils is observed to have an amyloid feature including hydrophobic core residues and parallel, in-register β-sheets.4–7 This structural feature is also shown in fibrils formed by other amyloid proteins such as amyloid-β and tau associated with Alzheimer's disease.8,9 The aggregation of αS is characterized by a nucleation–elongation process, in which once a nucleus is formed, the fibrils grow rapidly by further association of either monomers or oligomers.10,11 Thus, assessing the size of the nucleus is quite important to understand the nucleation processes of αS.αS is a 140-residue intrinsically disordered protein (IDP) that is composed of three distinct polypeptide regions: the amphipathic N-terminal domain (residues 1–60), which is responsible for membrane binding, the central hydrophobic non-amyloid component (NAC) domain (residues 61–95) with a high propensity to aggregate, and the negatively charged C-terminal domain (residues 96–140) containing several sites of post-translational modifications.12–14 Recently, a high-resolution three-dimensional (3D) structure model of full-length αS fibril (residues 1–140, PDB ID: 2N0A) has been determined based on solid-state NMR data, providing a framework for understanding the interactions of αS with other proteins and inhibitors.4 The core of the αS fibril is arranged in parallel, in-register β-sheets with a Greek-key topology, which is stabilized by the E46-K80 salt bridges, the Q79 glutamine ladder, and hydrophobic packing of aromatic residues F94. This full-length αS fibril and its Greek-key-like core (αS44–96) have been investigated in a few computational studies.15–17 Beyond the Greek-key structure, other possible folds of αS have been reported. Jiang et al. determined the cryo-EM structures of two αS fibril polymorphs (“rod” and “twister”) sharing a conserved kernel structure of a bent β-arch motif from the full-length recombinant human αS.7 Stahlberg et al. reported a cryo-EM structure of a truncated αS (residues 1–121) fibril, with a structure similar to the rod polymorph of the full-length protein.5
A number of strategies of molecular intervention to reduce the toxic effects of the αS oligomers have been proposed, including the use of antibodies18,19 and molecular chaperones,20 or the employment of small molecules.21–23 Among the small molecules, dopamine (DA) and norepinephrine (NE) have been identified for their remarkable abilities to inhibit the αS aggregation.23–29 DA and NE are two of the prominent naturally occurring neurotransmitters. In vivo, DA is synthesized in the cytosol of dopaminergic neurons and released in the inter-synaptic space to interact with its physiological receptors.30,31 However, DA synthesis is altered in PD patients because over 80% DA innervations are reduced in the basal ganglia of the patients.32 Central deficiency of NE has been associated with many nonmotor features of PD.33 Interestingly, numerous studies showed that exercise can be considered as a stressor that is able to stimulate the release of DA/NE.34,35 The exercise type, duration and intensity are major factors that influence the DA/NE responses.34
Increasing experimental studies have explored the effects of DA/NE molecules on the aggregation of αS peptide and the preformed αS protofibrils. For example, Conway et al. found that the catecholamines, including DA/NE, can effectively inhibit αS fibrillization by screening a commercially available compound library.24 Using a combined method of thioflavin T assay, light scattering, electron microscopy, and atomic force microscopy, Li et al. reported that DA/NE at micromolar concentration can inhibit the in vitro formation of αS amyloid fibrils and dissolve the pre-existing fibrils.25 Matera et al. reported that both DA and NE have the ability to strongly inhibit the formation of large fibrillar aggregates of αS and disaggregate the existing fibrils.29 By means of microscopic and spectroscopic methods, Maji et al. studied the effect of DA and other three amyloid inhibitors on the oligomerization, fibrillization and preformed fibrils of αS, and they found that high concentrations of DA can slow down the fibrillization kinetics of αS, decrease the cytotoxicity of αS aggregates and reduce the fibrillar length of the preformed αS fibrils.23 Despite these extensive studies, the size and structure of the critical nucleus and the disruptive mechanism by DA/NE have not been well explored.
In this study, we performed extensive atomistic molecular dynamics (MD) simulations on the Greek-key-like core of αS protofibril in the absence and presence of DA/NE molecules. The initial states of each system and the chemical structures of the DA/NE molecules are given in Fig. 1. Our aim is to identify the critical nucleus of αS Greek-key-like core fibril and its disruption by DA/NE molecules. The simulation results show that the αS44–96 protofibrillar trimer is the critical nucleus for the fibril formation, and tetramer is the smallest stable protofibril. By further examining the effect of DA/NE molecules on those αS44–96 trimer and tetramer, we found that DA/NE molecules could disrupt the β-sheet structure and inter-chain E46-K80 salt bridges, which are critical for the stability of αS fibril. DA and NE molecules bind to two common regions: residues 57–70 and 81–83. The interactions of DA/NE molecules with αS44–96 oligomers are mostly driven by hydrophobic and electrostatic interactions. Two disruptive modes are found for DA/NE molecules, and the mode of binding to the turn region of αS oligomers but disrupting the adjacent β-sheet structure is the dominant one.
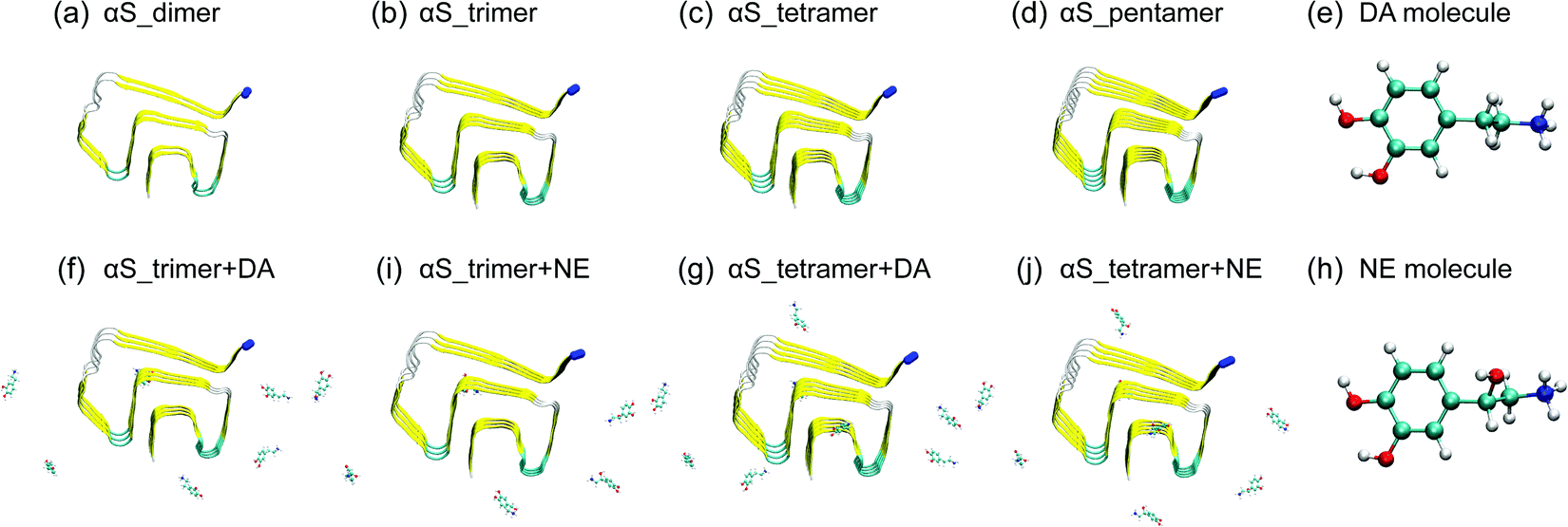 | ||
| Fig. 1 Initial states of the eight simulated systems and the chemical structures of DA/NE molecules. The Cα atom in the N-terminal of each αS chain is represented by the blue bead. | ||
Materials and methods
Simulated systems: αS44–96 protofibrillar oligomers with and without DA/NE molecules
We simulated the protofibrillar oligomers (from dimer to pentamer) consisting of residues 44–96 obtained from αS1–140 fibril structure in the absence and presence of DA/NE molecules. The reasons we chose this region are that it has the β-sheet-rich Greek-key-like structure and is the core of αS1–140 fibril. The initial structures of αS44–96 protofibrillar oligomers were built by deleting the N-terminal residues 1–43 and C-terminal residues 97–140 of the solid-state NMR derived αS1–140 fibril structure (PDB ID: 2N0A).4 The αS44–96 sequence is 44TKEGVVH50GVATVAEKTK60EQVTNVGGAV70VTGVTAVAQK80TVEGAGSIAA90ATGFVK96. We first investigated the stability of different sizes of αS44–96 protofibrillar oligomers. Subsequently, we examined the effect of DA/NE molecules on critical and minimal stable nucleus for αS fibrillization. Eight systems were simulated including αS_dimer, αS_trimer, αS_tetramer, αS_pentamer, αS_trimer + DA, αS_trimer + NE, αS_tetramer + DA, and αS_tetramer + NE. The molar ratio of DA/NE molecules to αS peptide chains is 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which is equal to that used in the experimental study.29 DA/NE molecules are expected in protonated forms (DA+ and NE+) in biological environments, and thus protonated DA/NE are considered in our simulations. The initial structures of the DA/NE molecules were taken from ChemSpider. They were first roughly optimized using Spartan’1036 and then finely energy-minimized by the GAMESS software.37 The atomic partial charges were obtained by the Amber Tools REP package.38 The topologies of DA/NE molecules were generated using the GlycoBioChem PRODRG2 Server.39
:1, which is equal to that used in the experimental study.29 DA/NE molecules are expected in protonated forms (DA+ and NE+) in biological environments, and thus protonated DA/NE are considered in our simulations. The initial structures of the DA/NE molecules were taken from ChemSpider. They were first roughly optimized using Spartan’1036 and then finely energy-minimized by the GAMESS software.37 The atomic partial charges were obtained by the Amber Tools REP package.38 The topologies of DA/NE molecules were generated using the GlycoBioChem PRODRG2 Server.39
MD simulation details
We performed MD simulations in the isothermal–isobaric ensemble using the GROMACS-5.1.4 software package40 with Amber99SB-ILDN force field.41 The αS oligomers were placed in the cubic water box. Six and eight DA/NE molecules were initially placed randomly around the energy-minimized αS trimer and tetramer, respectively. The initial minimum distance between DA/NE molecules and αS oligomers was at least 1.5 nm. Counterions (Cl−) were added to keep the systems neutrally charged. Constrains were applied for bond lengths of peptides using the LINCS algorithm42 and for those of water molecules using the SETTLE algorithm,43 with an integration time step of 2 fs. The protein, water, counterion and molecule groups were separately coupled to an external heat bath at 310 K using a velocity rescaling coupling method,44 with a coupling constant of 0.1 ps. Pressure was maintained under 1 bar by applying Parrinello–Rahman's method with the coupling constant of 1.0 ps.45 Long-range electrostatic interaction were calculated using the Particle Mesh Ewald (PME) method46 with a real space cutoff of 1.0 nm, and van der Waals interactions were cutoff at 1.4 nm. The MD setup details are summarized in Table 1.| Name of the system | Simulation time (ns) × Number of MD | Size of the box (nm3) | Number of atoms |
|---|---|---|---|
| αS_dimer | 300 × 2 | 7.75 × 7.75 × 7.75 | 46417 |
| αS_trimer | 300 × 2 | 7.82 × 7.82 × 7.82 | 47037 |
| αS_trimer + DA | 300 × 3 | 7.82 × 7.82 × 7.82 | 47046 |
| αS_trimer + NE | 300 × 3 | 7.82 × 7.82 × 7.82 | 47043 |
| αS_tetramer | 300 × 2 | 7.93 × 7.93 × 7.93 | 49349 |
| αS_tetramer + DA | 300 × 3 | 7.93 × 7.93 × 7.93 | 49353 |
| αS_tetramer + NE | 300 × 3 | 7.93 × 7.93 × 7.93 | 49361 |
| αS_pentamer | 300 × 2 | 8.08 × 8.08 × 8.08 | 51874 |
Analysis methods
MD trajectories were analyzed using in-house-developed codes and the GROMACS toolkits. Secondary structures were calculated by the DSSP method.47 A hydrogen bond (H-bond) is considered to be formed if the distance between donor D and acceptor A is ≤0.35 nm and the angle of D–H⋯A is ≥150°. An E46-K80 salt bridge is considered to be formed if the minimum distance between the nitrogen atom of the side-chain NH3+ group of K80 and the side-chain COO− group of E46 is ≤0.4 nm. The binding affinity of DA/NE molecules with αS oligomers was estimated using contact probability. An atomic contact between DA/NE molecules and αS oligomers is considered when the heavy atom pairs are ≤0.54 nm.48,49Results and discussion
Protofibrillar trimer is the critical nucleus for the αS fibril formation, and the tetramer is the minimal stable nucleus
The critical nucleus is defined as the least thermodynamically stable species in solution, which is the minimal size oligomer capable of initiating further growth into protofibril.50 It is also defined as the smallest aggregate that is stable enough to grow faster by monomer addition than dissolving.51 Thus, in order to estimate the critical nucleus size, one has to study the dependence of the free energy51,52 and structural stability of fibril for different seed sizes53,54 in studied systems. In this study, to determine the critical nucleus and the smallest stable size for αS fibril formation, we examined the structural stability of different oligomer size by comparing the average Cα-root-mean-square-deviation (Cα-RMSD) and β-sheet probability of each size of αS44–96 (αS for short) oligomers. As seen in Fig. 2(a), the average Cα-RMSD of αS dimer is 0.92 nm, indicative of a highly unstable dimer. When the oligomer size increases to 3, the average Cα-RMSD value sharply decreases to 0.39 nm, implying an enhanced structural stability. However, the N-terminal region is partly in a disordered state. With the further increment in the oligomer size, the average Cα-RMSD values of the tetramer and pentamer decrease slightly to 0.33 and 0.31 nm, respectively. Similar trend is also observed for β-sheet probability. The enhanced structural stability with the increased oligomer size can also be seen from the snapshots in Fig. 2(b). These data indicate that the trimer is the critical nucleus for fibril formation, in which the protofibrillar shape is preserved and part disassociation in the N-terminal region is observed (Fig. 2(b)), and the tetramer is the minimal stable nucleus. Interestingly, a recent replica exchange molecular dynamics (REMD) simulation study suggested that the minimal stable nucleus of αS NACore (residues 68–78) protofibril consist of two 4-stranded β-sheet layers.15 | ||
| Fig. 2 The average Cα-RMSD and β-sheet probability as a function of αS oligomer size (a). The representative structures at t = 300 ns for the αS trimer and tetramer (b). The data were averaged over the last 100 ns of the MD runs. The Cα atom in the N-terminal of each αS chain is represented by the blue bead. Error bars represent standard deviations of two independent MD trajectories. | ||
DA/NE molecules destabilize αS oligomers by disrupting β-sheet structures and inter-chain E46-K80 salt bridges
To monitor the effects of DA/NE molecules on the secondary structure probabilities, we calculated the average probability of each secondary structure (coil, β-sheet, β-bridge, bend and turn) over all residues and the residue-based β-sheet probabilities of αS trimer and tetramer with and without DA/NE molecules. As seen in Fig. 3(a), the β-sheet probability in the αS_trimer system is 39.1%, while it is reduced to 34.1% with the addition of DA molecules. The β-sheet disruptive effect is more prominent in the presence of NE molecules, with a β-sheet probability of 31.1% in the αS_trimer + NE system. The probabilities of coil, bend and β-bridge are all enhanced upon the addition of DA/NE molecules, compared to those in the isolated αS trimer system. The β-sheet probability of αS tetramer also has a remarkable decrease from 46.5% (without DA/NE molecules) to 37.9% (with DA molecules) and 36.4% (with NE molecules), while the coil probability increases separately from 30.9% to 37.9% and 38.5%. These results indicate that DA/NE molecules can serve as a β-sheet breaker to disrupt the β-sheet structure of αS oligomers, and NE displays a higher disruptive effect than DA. Fig. 3(b) and (d) shows the β-sheet probabilities of each amino acid residue of αS trimer and tetramer in all six systems. For the αS trimer, the β-sheet probabilities for residues 46–49, 53–55, 63–66 and 88–90 in the αS_trimer + DA system and residues 45–49, 52–55, 63–66, 74–80 and 88–90 in the αS_trimer + NE system are significantly reduced compared to those in the αS_trimer system. Similar results were observed in the tetramer systems. Besides, the residues 72–75 and 79–82 also have a reduced β-sheet probability (Fig. 3(d)). Previous studies showed that the NAC domain of αS, especially for the fibril-forming NACore (residues 68–78) region, plays a critical role in both aggregation and cytotoxicity of αS.55–57 Moreover, the NACore sequence is the minimal entity necessary to recapitulate all the features of full length αS aggregation.57 The decrease of β-sheet probabilities in the NACore region observed in our MD simulations indicates that DA/NE could effectively disrupt the αS protofibrils, thus preventing the αS fibrillization. | ||
| Fig. 3 Secondary structure probabilities and residue-based β-sheet probability of αS trimer (a and b) and tetramer (c and d) in the absence and presence of DA/NE molecules. Error bars represent standard deviations of different secondary structures. | ||
To examine the effect of DA/NE molecules on the stability of αS oligomers, we calculated the probability distribution function (PDF) of the backbone H-bond number. As shown in Fig. 4(a), the H-bond number in the αS trimer is obviously reduced upon the addition of DA/NE molecules, and the peak value separately decreases from 68 (without DA/NE molecules) to 56 (with DA molecules) and 56 (with NE molecules). For αS tetramer, the presence of DA/NE molecules also leads to a decreased backbone H-bond number, shifting from 106 (without DA/NE molecules) to 96 (with DA molecules) and 96 (with NE molecules). These data demonstrate that the interactions of DA/NE with αS oligomers disrupt the backbone H-bonds. It has been demonstrated that the inter-molecular E46-K80 salt-bridges play a critical role in the stabilization of the αS fibrils.4,58 The distribution of the minimum distance between the nitrogen atom of the K80 side-chain NH3+ group and E46 side-chain COO− group was calculated to examine whether the presence of DA/NE molecules can influence the inter-chain E46-K80 salt bridges. It can be seen from the distance distribution in Fig. 4(c) that almost half of E46-K80 salt bridges in the αS_trimer system are preserved. When the oligomer size increases to four, most of the E46-K80 salt bridges are maintained. These results also indicate that trimer is the critical nucleus and tetramer is the smallest stable nucleus for fibril formation. Upon addition of DA/NE molecules, E46-K80 distance becomes larger and its distribution becomes much broader, indicating that the binding of DA/NE molecules to αS oligomers disrupts the inter-chain E46-K80 salt bridges. Taken together, the results of the β-sheet probability, H-bond number and inter-chain E46-K80 salt bridges demonstrate that DA/NE molecules can effectively disrupt the αS protofibrils, which provide a microscopic mechanism for the experimentally observed fibril disruption by DA/NE molecules.23,25,29
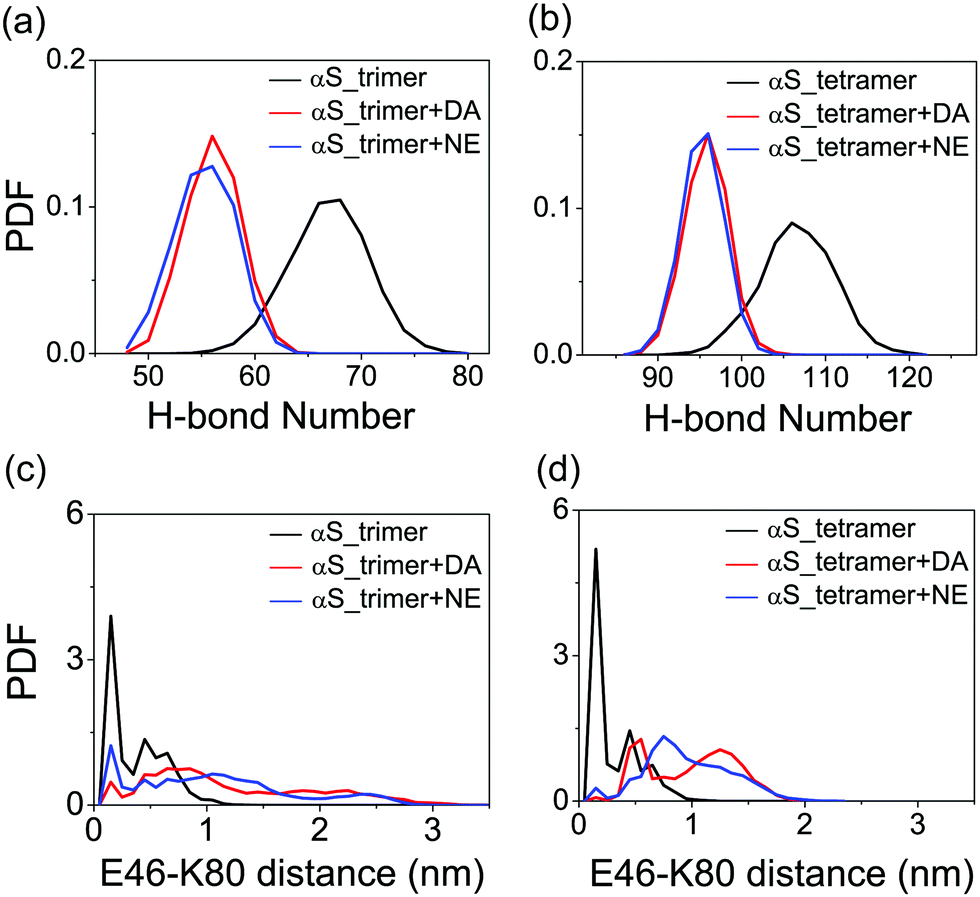 | ||
| Fig. 4 Probability distribution function of backbone H-bond number and inter-chain E46-K80 distance in αS trimer (a and c) and tetramer (b and d) systems with and without DA/NE molecules. | ||
The dominant binding sites and disruptive modes of DA/NE molecules with αS oligomers
To identify the binding sites of DA/NE molecules on αS oligomers, we calculated the residue-based binding probability of DA/NE molecules to αS trimer and tetramer using the last 100 ns data of each MD run. As shown in Fig. 5, both DA and NE molecules have three dominant binding sites on αS trimer and tetramer. When comparing the binding sites of DA on αS oligomers with those of NE, we observed that two binding sites are quite similar: residues 57–70 and 81–83. In these two binding sites, negatively charged residues E57, E61, E83 and hydrophobic residues V63, V66, V70, V82 display a high binding probability with DA/NE molecules, indicating the strong hydrophobic and electrostatic interactions. This finding is consistent with a previous experimental study showing that DA mostly binds to residue E61 on αS fibril.27 Our results also coincide with early combined computational and experimental studies of full-length αS monomer by Carloni et al. suggesting that DA binds to residues V66 and E83 in the NAC region and residues 125–129 in the C-terminal region.26,28In vitro experiment further showed that the replacement of glutamate by alanine at position 83 (E83A) abolishes the ability of DA to inhibit αS fibrillization.26 The different binding site is located at the terminal region of αS Greek-key-like core. DA is more prone to binding to the N-terminal region (residues 45KEGVVHGV52), while NE is not. Most of the residues in this N-terminal region are hydrophilic, while NE has much lower probability to bind than DA. Thus, this observation could be attributed to the different size of DA and NE molecules: DA is smaller than NE, which has a hydroxyl group in the ethyl chain. The smaller size of DA enables more DA molecules to bind to residues 45–52, which are located at the entry of the Greek-key-like core (see Fig. S1, ESI†). This is also consistent with the NMR study by Maji et al. showing that DA strongly interacts with residue G51 and the nearby residues V48, H50, and V52.23 | ||
| Fig. 5 Residue-based binding probability of DA/NE molecules to the αS trimer (a and b) and tetramer (c and d). The number of hydrogen bond formed between the DA/NE molecules and each amino acid residue of the αS trimer (e and f) and tetramer (g and h). The main-chain and side-chain H-bonds are colored by red and black, respectively. The results were averaged over the last 100 ns data of all MD runs. | ||
We then calculated the number of main-chain (MC) and side-chain (SC) H-bonds formed between each amino acid residue of αS44–96 peptide and DA/NE molecules to identify the residues contributing mostly to the H-bond disruption. As shown in Fig. 5(e) and (g), DA molecules form MC H-bonds mostly with residues E57, V71, Q79 and I88 (red font), and form SC H-bonds mostly with the polar residues T59, T64 and N65 (black font). For the NE molecules (Fig. 5(f) and (h)), the dominant MC H-bond contributors are residues E61, Q62, V63 and G67, and the dominant SC H-bond contributors are polar residues T64 and N65. DA/NE molecules form H-bonds mostly with MC atoms, and thus disturb the backbone hydrogen bonds of αS. In addition, NE molecules form more backbone H-bonds (trimer: 1.7, tetramer: 2.2) with αS than DA (trimer: 1.4, tetramer: 1.7). This observation explains the stronger disruptive effect of NE on αS oligomers. We also calculated the potential of mean force (PMF) as a function of the number of H-bonds and the β-sheet content of αS oligomers in the presence of DA/NE molecules (Fig. S2, ESI†). The first four most-populated clusters of each of the four different systems were presented at the side and bottom of the PMF. The free energy landscapes of αS trimer/tetramer with DA (Fig. S2(a) and (c), ESI†) and NE (Fig. S2(b) and (d), ESI†) molecules display different shapes. The locations of the dominant minimum-energy basins for the αS oligomers with DA and NE are also very different. For the αS trimer/tetramer with DA molecules, the energy basins are separately centered at the (H-bond number, β-sheet content) values of (57, 35%)/(93, 40%), while for the αS trimer/tetramer in the presence of NE molecules, they are located at (55, 29%)/(91, 35%). The reduced β-sheet contents and H-bond number in the αS_trimer + NE/αS_tetramer + NE systems compared to those in the αS_trimer + DA/αS_tetramer + DA systems further indicate that NE displays a higher disruptive effect than DA.
The binding probability and fraction of reduced β-sheet as a function of the residue index were calculated to examine the relationship between the binding sites and the β-sheet disruptive sites. As shown in Fig. 6, there are two disruptive modes of DA/NE molecules on αS trimer/tetramer, which can be briefly described as following. Mode I: DA/NE molecules bind to the turn region of αS oligomers but disrupt the adjacent β-sheet structure. This disruptive mode is reminiscent of the allosteric effect. Mode II: they bind to a certain β-sheet region of αS oligomers and disrupt the β-sheet structure of this region. Specifically, for Mode I, residue E57 in the αS_tetramer + NE system displays a high binding probability with NE molecules, while the β-sheet probabilities of the adjacent β-sheet region spanning residues 52–55 have an obvious reduction (Fig. 6(d)). For Mode II, as seen in Fig. 6(c), the region around K80 in the αS_tetramer + DA system has a high binding probability with DA molecules, and it displays a decreased β-sheet probability. Fig. 6(a)–(d) shows most of the peaks of the residue-based binding probability distributions and those of the reduced β-sheet fraction curves do not appear at the same residue region, indicating that Mode I has a higher incidence than Mode II. We also performed cluster analyses, and found that when DA/NE molecules are bound to turn regions of αS oligomers, in most cases, the adjacent β-sheet structures are disrupted. This can be clearly seen from the representative conformations in the first four most-populated clusters (especially for cluster-2 and cluster-4) of αS_tetramer + NE system in Fig. S3 (ESI†). Mode I was not often reported previously. Mode II was reported in numerous computational studies. For example, Sun et al. investigated the effect of a fullerene derivative on the Aβ1–42 dimer by performing explicit-solvent REMD simulation, and found that this fullerene derivative preferentially interacts with the central hydrophobic region (residues 17–21) of the Aβ peptide and greatly disrupts the β-sheet of this region.59 Our previous MD simulations showed that O4 molecules mostly bind to residues 22–26 of hIAPP oligomers and significantly disrupt the β-sheet structure in this amyloid core region.60 The snapshots in Fig. 6(e) show the two disruptive modes of DA/NE molecules on αS trimer and tetramer. It is noteworthy that DA/NE molecules can also stabilize the terminal β-sheet (β1 and β5). Overall, DA/NE molecules predominantly display a disruptive effect on αS oligomers.
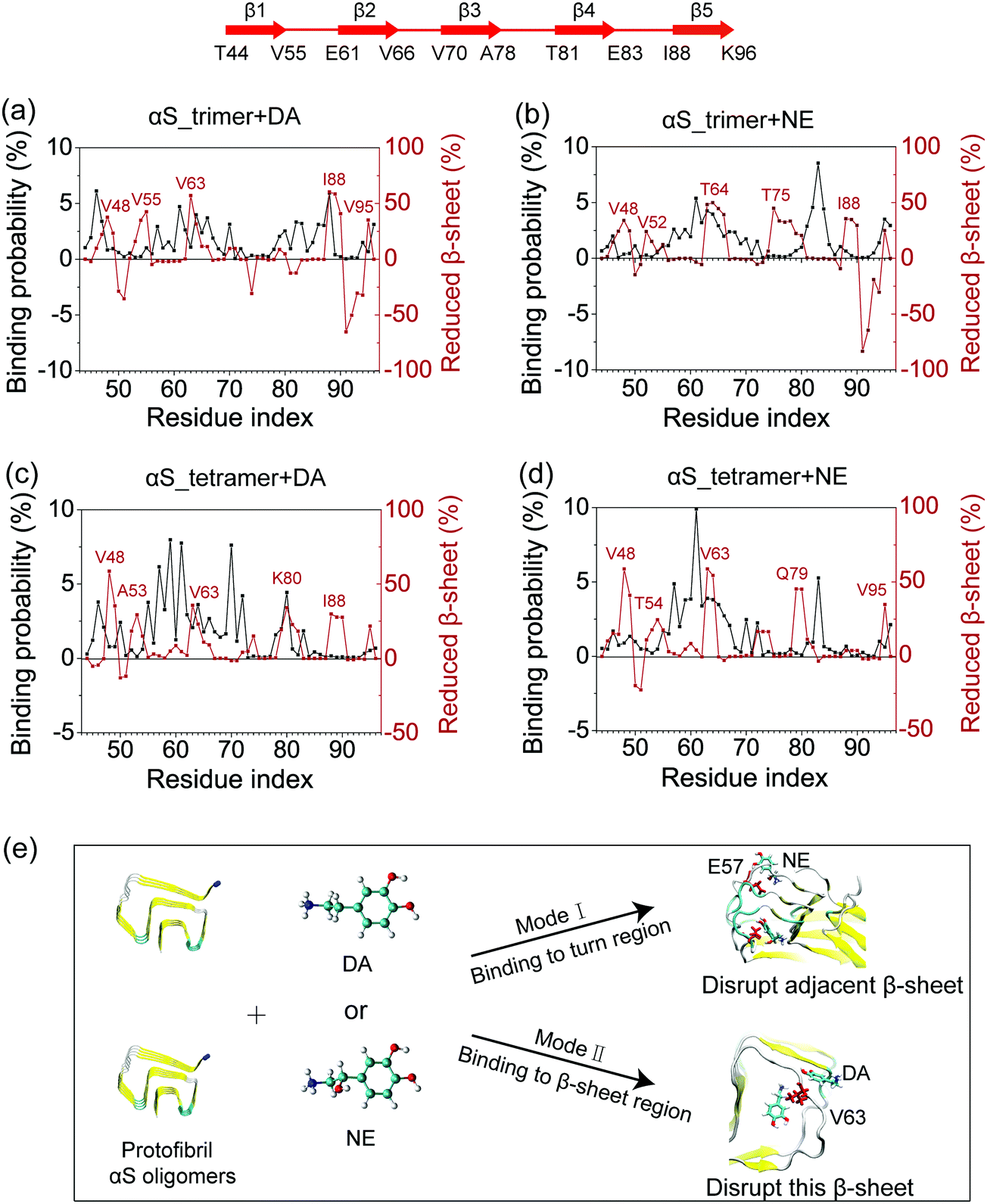 | ||
| Fig. 6 Disruptive mode analyses: the residue-based binding probability and fraction of reduced β-sheet in αS_trimer + DA (a), αS_trimer + NE (b), αS_tetramer + DA (c), and αS_tetramer + NE (d) systems; schematic diagram for two disruptive modes of DA/NE molecules on αS trimer and tetramer (e). Residues E57 and V63 are in CPK representation and colored in red. Reduced β-sheet probabilities were calculated using the residue-based β-sheet probabilities in the isolated αS systems minus those in the αS systems with DA/NE molecules. | ||
Recent MD simulation studies by the Thompson group showed that the flexible C-terminal group of αS plays a critical role in stabilizing folded helical conformations.61–63 This finding indicates that small molecules may bind to the C-terminal residues and disrupt/stabilize αS protofibril. In our study, as the first step to understand how DA/NE molecules disrupt the αS protofibril at atomic level, we chose the β-sheet-rich Greek-key-like structure as a model system. This region is the core of αS1–140 fibril and contributes to the stability of the in-register β-sheet of full-length αS fibril. The influence of small molecules on the full-length Greek-key fold protofibrils will be examined in our future work.
Conclusions
In this study, we identified the critical nucleus of the pre-formed αS Greek-key-like core and investigated the disruptive mechanisms of DA/NE on this region by performing multiple all-atom MD simulations. Our simulation results show that the αS trimer is the critical nucleus size for the fibril formation, and tetramer is the smallest stable protofibril. By analyzing the influence of DA/NE molecules on αS trimer and tetramer, we find that DA/NE can disrupt the β-sheet structure and the inter-chain E46-K80 salt bridges of αS trimer and tetramer, and the disruptive effect of NE is stronger than that of DA. Through binding probability analyses, we identified two common dominant binding sites for both DA and NE molecules on αS oligomers: residues 57–70 and 81–83. We also observed a different binding site located at the terminal region of αS Greek-key-like core. The hydrophobic and electrostatic interactions together contribute to the binding of DA/NE molecules to αS oligomers. Two disruptive modes are found, and the mode of binding to the turn region but disrupting the adjacent β-sheet is the dominant one. Our work determined the critical nucleus of Greek-key-like core of αS protofibrils and revealed the disruptive mechanism of αS protofibrils by DA/NE molecules, which provides new clues for the development of treatment (drug design or exercise therapy) on PD.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the financial support from the Natural Science Foundation of Shanghai (Grant No. 19ZR1453100), National Natural Science Foundation of China (Grant No. 11674065 and 11704256). All simulations were performed using the High-Performance Computing Server at Shanghai University of Sport and National Supercomputer Center in Guangzhou.References
- G. B. D. Disease, I. Injury and C. Prevalence, Lancet, 2016, 388, 1545–1602 CrossRef.
- M. G. Spillantini, M. L. Schmidt, V. M. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef CAS PubMed.
- M. G. Spillantini, R. A. Crowther, R. Jakes, M. Hasegawa and M. Goedert, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6469–6473 CrossRef CAS PubMed.
- M. D. Tuttle, G. Comellas, A. J. Nieuwkoop, D. J. Covell, D. A. Berthold, K. D. Kloepper, J. M. Courtney, J. K. Kim, A. M. Barclay, A. Kendall, W. Wan, G. Stubbs, C. D. Schwieters, V. M. Lee, J. M. George and C. M. Rienstra, Nat. Struct. Mol. Biol., 2016, 23, 409–415 CrossRef CAS PubMed.
- R. Guerrero-Ferreira, N. M. Taylor, D. Mona, P. Ringler, M. E. Lauer, R. Riek, M. Britschgi and H. Stahlberg, eLife, 2018, 7, e36402 CrossRef.
- Y. Li, C. Zhao, F. Luo, Z. Liu, X. Gui, Z. Luo, X. Zhang, D. Li, C. Liu and X. Li, Cell Res., 2018, 28, 897–903 CrossRef CAS PubMed.
- B. Li, P. Ge, K. A. Murray, P. Sheth, M. Zhang, G. Nair, M. R. Sawaya, W. S. Shin, D. R. Boyer, S. Ye, D. S. Eisenberg, Z. H. Zhou and L. Jiang, Nat. Commun., 2018, 9, 3609 CrossRef PubMed.
- L. Gremer, D. Scholzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schroder, Science, 2017, 358, 116–119 CrossRef CAS PubMed.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. W. Scheres, Nature, 2017, 547, 185–190 CrossRef CAS PubMed.
- A. Lomakin, D. S. Chung, G. B. Benedek, D. A. Kirschner and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1125–1129 CrossRef CAS PubMed.
- M. Stefani and C. M. Dobson, J. Mol. Med., 2003, 81, 678–699 CrossRef CAS PubMed.
- M. Hashimoto, T. Takenouchi, M. Mallory, E. Masliah and A. Takeda, Am. J. Pathol., 2000, 156, 734–736 CrossRef CAS.
- D. P. Hong, W. Xiong, J. Y. Chang and C. Jiang, FEBS Lett., 2011, 585, 561–566 CrossRef CAS PubMed.
- N. Lorenzen, L. Lemminger, J. N. Pedersen, S. B. Nielsen and D. E. Otzen, FEBS Lett., 2014, 588, 497–502 CrossRef CAS PubMed.
- T. D. Romo, A. K. Lewis, A. R. Braun, A. Grossfield and J. N. Sachs, ACS Chem. Neurosci., 2017, 8, 1859–1864 CrossRef CAS PubMed.
- L. Xu, B. Ma, R. Nussinov and D. Thompson, ACS Chem. Neurosci., 2017, 8, 837–849 CrossRef CAS.
- X. Liu, S. Zhou, D. Shi, Q. Bai, H. Liu and X. Yao, Chem. Biol. Drug Des., 2018, 91, 162–171 CrossRef CAS PubMed.
- T. Nasstrom, S. Goncalves, C. Sahlin, E. Nordstrom, V. Screpanti Sundquist, L. Lannfelt, J. Bergstrom, T. F. Outeiro and M. Ingelsson, PLoS One, 2011, 6, e27230 CrossRef PubMed.
- M. Iljina, L. Hong, M. H. Horrocks, M. H. Ludtmann, M. L. Choi, C. D. Hughes, F. S. Ruggeri, T. Guilliams, A. K. Buell, J. E. Lee, S. Gandhi, S. F. Lee, C. E. Bryant, M. Vendruscolo, T. P. J. Knowles, C. M. Dobson, E. De Genst and D. Klenerman, BMC Biol., 2017, 15, 57 CrossRef PubMed.
- D. Cox and H. Ecroyd, Cell Stress Chaperones, 2017, 22, 589–600 CrossRef CAS PubMed.
- J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. Wobst, K. Neugebauer and E. E. Wanker, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7710–7715 CrossRef CAS PubMed.
- N. Ahsan, S. Mishra, M. K. Jain, A. Surolia and S. Gupta, Sci. Rep., 2015, 5, 9862 CrossRef CAS PubMed.
- N. N. Jha, R. Kumar, R. Panigrahi, A. Navalkar, D. Ghosh, S. Sahay, M. Mondal, A. Kumar and S. K. Maji, ACS Chem. Neurosci., 2017, 8, 2722–2733 CrossRef CAS PubMed.
- K. A. Conway, J. C. Rochet, R. M. Bieganski and P. T. Lansbury Jr, Science, 2001, 294, 1346–1349 CrossRef CAS PubMed.
- J. Li, M. Zhu, A. B. Manning-Bog, D. A. Di Monte and A. L. Fink, FASEB J., 2004, 18, 962–964 CrossRef CAS PubMed.
- F. E. Herrera, A. Chesi, K. E. Paleologou, A. Schmid, A. Munoz, M. Vendruscolo, S. Gustincich, H. A. Lashuel and P. Carloni, PLoS One, 2008, 3, e3394 CrossRef PubMed.
- D. Latawiec, F. Herrera, A. Bek, V. Losasso, M. Candotti, F. Benetti, E. Carlino, A. Kranjc, M. Lazzarino, S. Gustincich, P. Carloni and G. Legname, PLoS One, 2010, 5, e9234 CrossRef PubMed.
- D. Dibenedetto, G. Rossetti, R. Caliandro and P. Carloni, Biochemistry, 2013, 52, 6672–6683 CrossRef CAS PubMed.
- A. F. Fischer and K. M. Matera, Neurochem. Res., 2015, 40, 1341–1349 CrossRef CAS PubMed.
- L. L. Venda, S. J. Cragg, V. L. Buchman and R. Wade-Martins, Trends Neurosci., 2010, 33, 559–568 CrossRef CAS PubMed.
- M. Bisaglia, R. Filograna, M. Beltramini and L. Bubacco, Ageing Res. Rev., 2014, 13, 107–114 CrossRef CAS PubMed.
- R. Deumens, A. Blokland and J. Prickaerts, Exp. Neurol., 2002, 175, 303–317 CrossRef CAS PubMed.
- H. Kaufmann and D. S. Goldstein, Handb. Clin. Neurol., 2013, 117, 259–278 Search PubMed.
- H. Zouhal, C. Jacob, P. Delamarche and A. Gratas-Delamarche, Sports Med., 2008, 38, 401–423 CrossRef PubMed.
- H. Zouhal, S. Lemoine-Morel, M. E. Mathieu, G. A. Casazza and G. Jabbour, Sports Med., 2013, 43, 591–600 CrossRef PubMed.
- Y. Shao, L. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. Brown, A. Gilbert, L. Slipchenko, S. Levchenko, D. O’Neill and R. A. DiStasio Jr, Phys. Chem. Chem. Phys., 2006, 8, 3172–3191 RSC.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen and S. Su, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- D. François-Yves, P. Adrien, Z. Thomas, S. Corentin, L. Rodolphe, G. Nicolas, L. Dimitri, R. Wilfried and C. Piotr, Phys. Chem. Chem. Phys., 2010, 12, 7821–7839 RSC.
- A. W. Schüttelkopf and D. M. F. Van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- D. S. D. Van, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef PubMed.
- K. Lindorfflarsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CAS.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1998, 98, 10089–10092 CrossRef.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- Z. Qian, Y. Zou, Q. Zhang, P. Chen, B. Ma, G. Wei and R. Nussinov, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1818–1825 CrossRef CAS PubMed.
- Z. Liu, Y. Zou, Q. Zhang, P. Chen, Y. Liu and Z. Qian, Int. J. Mol. Sci., 2019, 20, 2048 CrossRef PubMed.
- A. M. Morris, M. A. Watzky and R. G. Finke, Biochim. Biophys. Acta, 2009, 1794, 375–397 CrossRef CAS PubMed.
- F. Ferrone, Method. Enzymol., 1999, 309, 256–274 CrossRef CAS PubMed.
- R. D. Hills Jr and C. L. Brooks III, J. Mol. Biol., 2007, 368, 894–901 CrossRef PubMed.
- N. L. Fawzi, Y. Okabe, E. H. Yap and T. Head-Gordon, J. Mol. Biol., 2007, 365, 535–550 CrossRef CAS PubMed.
- Y. Zou, Y. Sun, Y. Zhu, B. Ma, R. Nussinov and Q. Zhang, ACS Chem. Neurosci., 2016, 7, 286–296 CrossRef CAS PubMed.
- B. I. Giasson, I. V. Murray, J. Q. Trojanowski and V. M. Lee, J. Biol. Chem., 2001, 276, 2380–2386 CrossRef CAS PubMed.
- M. Periquet, T. Fulga, L. Myllykangas, M. G. Schlossmacher and M. B. Feany, J. Neurosci., 2007, 27, 3338–3346 CrossRef CAS PubMed.
- J. A. Rodriguez, M. I. Ivanova, M. R. Sawaya, D. Cascio, F. E. Reyes, D. Shi, S. Sangwan, E. L. Guenther, L. M. Johnson, M. Zhang, L. Jiang, M. A. Arbing, B. L. Nannenga, J. Hattne, J. Whitelegge, A. S. Brewster, M. Messerschmidt, S. Boutet, N. K. Sauter, T. Gonen and D. S. Eisenberg, Nature, 2015, 525, 486–490 CrossRef CAS PubMed.
- A. Sanjeev and V. S. K. Mattaparthi, Cent. Nerv. Syst. Agents Med. Chem., 2017, 17, 209–218 CAS.
- Y. Sun, Z. Qian and G. Wei, Phys. Chem. Chem. Phys., 2016, 18, 12582–12591 RSC.
- Y. Zou, Z. Qian, Y. Sun, G. Wei and Q. Zhang, J. Phys. Chem. B, 2017, 121, 9203–9212 CrossRef CAS PubMed.
- L. Xu, S. Bhattacharya and D. Thompson, Chem. Commun., 2018, 54, 8080–8083 RSC.
- S. Bhattacharya, L. Xu and D. Thompson, ACS Chem. Neurosci., 2019, 10, 2830–2842 CrossRef CAS PubMed.
- L. Xu, S. Bhattacharya and D. Thompson, Phys. Chem. Chem. Phys., 2019, 21, 12036–12043 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp04610k |
| This journal is © the Owner Societies 2020 |