
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ortho-Substituent effect on the crystal packing and solid state speciation of aromatic C-nitroso compounds†
Sarah J.
Pike
 *ab,
Armelle
Heliot
b and
Colin C.
Seaton
b
*ab,
Armelle
Heliot
b and
Colin C.
Seaton
b
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: s.j.pike@bham.ac.uk
bSchool of Chemistry and Biosciences, Faculty of Life Sciences, University of Bradford, Bradford, West Yorkshire BD7 1DP, UK
First published on 16th July 2020
Abstract
The solid state behaviour of a short series of aromatic C-nitroso compounds bearing a range of ortho-substituents, including amides, alcohols, methyl and fluorine have been investigated using single crystal X-ray diffraction. Solid state analysis has confirmed the contrasting speciation behaviour of aromatic C-nitroso compounds incorporating strongly and weakly electron-donating groups at the ortho-position with the former group adopting monomers or oxo-oxime tautomers whilst the latter group adopts either the cis- or trans-azodioxy dimer depending on the nature of the solvent employed in the crystallisation conditions. The presence of weakly electron-withdrawing fluorine substituents on the nitrosoarene leads to the formation of the trans-azodioxy dimer. Single crystal X-ray diffraction analysis has identified the presence of a diverse array of hydrogen-bonding interactions and π–π stacking interactions that support the adoption of a range of supramolecular aggregates, including chains and tunnels, in the crystal packing of this small library of aromatic C-nitroso compounds.
Introduction
Since their discovery in 1894, aromatic C-nitroso compounds have found widespread applications in synthetic organic chemistry1–3 coordination chemistry,4–6 and as spin traps in biological and synthetic systems.7,8 These compounds may also possess important biological properties with potential antiviral activity.9Aromatic C-nitroso compounds are known to exist as a dynamic equilibrium of monomers, cis-azodioxy or trans-azodioxy dimers (Fig. 1) and the complex speciation behaviour of aromatic C-nitroso compounds has excited much interest.10,11 Understanding the complex behaviour of this important class of compounds, and the factors that influence it, is critical for their effective use and application in synthesis and for optimising their biological activity. These monomer–dimer isomerisations have also been shown to occur in the solid state leading to insights into the mechanisms of thermally controlled solid-state reactivity12 and potential applications as photochromic or thermochromic materials.13 Recently, the polymerisation of monomeric aromatic C-nitroso compounds has been employed to construct well-ordered, monocrystalline, covalent organic networks with potential emerging materials science applications.14
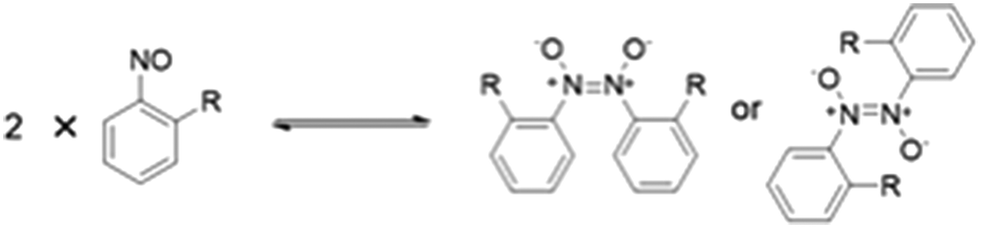 | ||
| Fig. 1 Monomer–dimer equilibrium that exists for aromatic C-nitroso compounds involving the monomer, cis-azodioxy dimer and trans-azodioxy dimer species. Illustrated for an ortho-substituted nitroarene. | ||
Solution-based studies of aromatic C-nitroso compounds have probed the influence of a range of factors including the nature of the substituent on the aromatic ring,15 temperature16 and pressure17 on the monomer–dimer equilibrium. However, the factors that influence the adoption of monomers and azodioxy dimers in the solid-state of aromatic C-nitroso compounds have not been as systematically or widely investigated although some general trends have been established.10,18,19 The position of the monomer–dimer equilibrium is highly dependent on the nature and the position of the substituent on the ring, relative to the electron-withdrawing nitroso group. Generally, ortho-substituted nitrosoarenes have a propensity to form dimers in the solid, with the trans-azodioxy dimer more often favoured over the analogous cis-azodioxy dimer, as the trans-geometry about the central N–N bond typically helps to better alleviate the strain of any sterically demanding ortho-substituents.
The first trans-azodioxy dimers of aromatic C-nitroso compounds were reported, in 1950, for 4-bromonitrosobenzene, 4-chloronitrosobenzene and 2,4,6-tribromonitrosobenzene.20,21 Since these initial reports, there have been a diverse array of C-nitrosoarenes incorporating a range of substituents on the aromatic ring that have been shown to adopt trans-azodioxy dimers in the solid state. These include nitrosobenzene,22ortho-substituted nitrosoarenes bearing alkyl groups (methyl and iso-propyl)23,24 and carboxylic acids,25 as well as para-substituted, 4-nitronitrosobenzene,26 4-iodonitrosobenzene,27 and additional reports of 4-chloronitrosobenzene,28 and 4-bromonitrosobenzene wherein the latter is formed via single-crystal-to-single-crystal transformation from the monomer.29a
In 1970, the first crystal structure of a cis-azodioxy dimer of an aromatic C-nitroso compound was reported when Dieterich et al., identified cis-azodioxybenzene in the solid state.30 However, since this first report there have only been a limited number of reports of crystal structures of cis-azodioxy dimers for other aromatic C-nitroso compounds, these include; 2-methyoxynitrosobenzene,31 nitrosotoluene,32 and pentafluoronitrosobenzene,33 in addition to N-heterocycles; 2-nitrosopyridine34 and 2-nitrosoquinozaline.35
Whilst dimers are more commonly found in the solid state, monomers can be favoured when there are resonance electron-donating groups (e.g. amines or amides) present at ortho (or para) position of the ring, relative to the nitroso substituent.10,36–41 The preferential formation of monomers over azodioxy dimers in these compounds is most likely a consequence of the reduced ability of the azodioxy dimers to stabilise the structure of the C-nitrosoarene through resonance effects.10 The preparation procedure can also play an important role in determining the solid state speciation of certain C-nitrosoarenes which are known to be polymeric.10 In such systems, metastable disordered monomeric forms can be accessed through sublimation of the dimer.10,29b
In this paper, we report a study on the influence of the electronic and steric nature of the ortho-substituent on the arene ring, relative to the nitroso group, on the solid state speciation behaviour of a short series of aromatic C-nitroso compounds. For one compound, nitrosotoluene, we have also probed the significance of the nature of solvent employed in the crystallisation conditions on the preferential adoption of the corresponding cis- and trans-azodioxy dimers. The importance of intermolecular non-covalent interactions, including hydrogen-bonding and π–π stacking interactions, on the crystal packing of each of the studied nitrosoarenes has been established and the adoption of supramolecular aggregates, including chains and tunnels, has been identified.
Results and discussion
Design of aromatic C-nitroso compounds
A small library of five aromatic C-nitroso compounds was designed to investigate the influence of the electronic and steric nature of the ortho-substituent on their speciation and crystal packing behaviour (Fig. 2). Aromatic C-nitroso compounds incorporating the strongly electron-donating acetyl (Ac), tert-butyloxycarbonyl (Boc) and hydroxyl groups, the weakly electron-donating methyl substituent, and weakly electron-withdrawing fluorine ortho-substituents, relative to the nitroso group, were investigated: 2-nitrosoacetanilide 1, tert-butyl-N-(2-nitrosophenyl)carbamate 2, 1-nitroso-2-naphthol 3, nitrosotoluene 4 and 2,6-difluoronitrosobenzene 5.42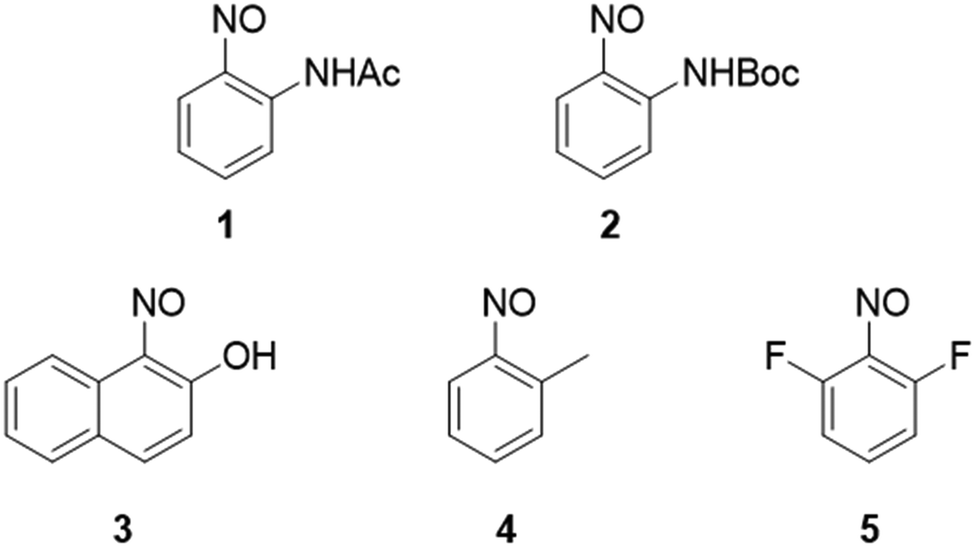 | ||
| Fig. 2 Library of aromatic C-nitroso compounds 1–5 that are employed in this study. | ||
Single crystal X-ray diffraction analysis carried out on 1–5 allowed for determination of the species (monomers, oxo-oxime tautomers, cis- and trans-azodioxy dimers) adopted by each compound in the solid state. X-ray crystallographic analysis also identified the presence of π–π stacking and hydrogen-bonding interactions, including neutral O–H⋯O, C–H⋯O, N–H⋯O, C–H⋯F, C–H⋯π hydrogen bonding interactions and charge assisted C–H⋯O− hydrogen bonding interactions, involving the nitroso group in the azodioxy dimers,43,44 that support the adoption of supramolecular aggregates in the crystal packing of 1–5. Selected crystallographic data and details for 1–5 are presented in Table 1. The nature of the species adopted, selected bond lengths and crystal colour observed in 1–5 are presented in Table 2.
| 1 | 2 | 3 | cis-4 | trans-4 | 5 | |
|---|---|---|---|---|---|---|
| Empirical formula | C8H8N2O2 | C11H14N2O3 | C10H7NO2 | C14H14N2O2 | C14H14N2O2 | C12H6F4N2O2 |
| Formula weight | 164.16 | 222.24 | 173.17 | 242.27 | 242.27 | 286.19 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Monoclinic | Orthorhombic | Triclinic |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | I2/a | Pbca |
P |
| a (Å) | 7.6784(3) | 5.6472(8) | 5.4498(9) | 15.1760(13) | 8.1552(6) | 7.0409(7) |
| b (Å) | 10.7811(4) | 14.1772(19) | 9.1487(16) | 10.4211(6) | 10.3206(8) | 7.1841(8) |
| c (Å) | 9.5685(4) | 14.2690(18) | 15.499(3) | 15.5264(12) | 14.3800(12) | 7.1860(9) |
| α (°) | 90 | 93.043(8) | 90 | 90 | 90 | 61.943(7) |
| β (°) | 97.899(2) | 92.475(8) | 98.193(3) | 90.000(7) | 90 | 65.010(7) |
| γ (°) | 90 | 93.340(8) | 90 | 90 | 90 | 64.427(7) |
| V (Å3) | 784.58(5) | 1137.6(3) | 764.9(2) | 2455.5(3) | 1210.32(16) | 278.14(6) |
| Z | 4 | 4 | 4 | 8 | 4 | 1 |
| Temperature (K) | 169.99 | 100.11 | 150.01 | 100.15 | 104.71 | 149.99 |
| Calc. density (g cm−3) | 1.390 | 1.298 | 1.504 | 1.311 | 1.330 | 1.709 |
| Compound | Speciation | C–Nnitroso (Å) | N–O (Å) | Crystal colour |
|---|---|---|---|---|
| a Disorder about the nitroso group prevents determination of the C–N and N–O nitroso bond lengths in 2. b See ref. 46. c Refers to the N–O bond in the oxime (not nitroso) functionality. d See ref. 47. e Asymmetry in the bond lengths for the nitroso and arene ring is observed in cis-4, both sets of bond lengths are provided. f See ref. 24a. | ||||
| 1 | Monomer | 1.4280(10) | 1.2261(9) | Green |
| 2 | Monomera | — | — | Green |
| 3 | oxo-Oxime tautomer | 1.3099(12)b | 1.3614(12)c | Red |
| 4 | cis-Azodioxy dimere | 1.447(2) | 1.2671(18) | Colourless |
| 1.457(2) | 1.2695(18) | Colourless | ||
| 4 | trans-Azodioxy dimer | 1.4529(17) | 1.2679(16) | Colourless |
| 5 | trans-Azodioxy dimer | 1.444(2) | 1.262(2) | Colourless |
Influence strongly electron-donating ortho-substituents
2-Nitrosoacetanilide 1, tert-butyl-N-(2-nitrosophenyl)carbamate 2 and 1-nitroso-2-naphthol 3 bearing the strongly electron-donating acetyl, Boc and hydroxyl groups respectively were studied using X-ray crystallographic analysis.Single crystals of 1 suitable for X-ray crystallography diffraction were grown through slow evaporation of dichloromethane at ambient temperature from a saturated solution to yield bright green crystals. 1 exists as a monomer which correlates well with the observed green colour of the crystal which is characteristic of C-nitrosoarene monomers (Table 2 and Fig. 3a).10 The crystal structure of 1 displays disorder in the CH3 group of the acetate group (Fig. 3a). In 1, the ω torsion angle (C3–N1–C2–C1) of ±178.12° lies close to the anticipated value for the trans peptide bond. The C–N and N–O nitroso bond lengths of 1.4280(10) Å and 1.2261(9) Å respectively (Table 2). The acetyl group of the amide bond extends below the plane of the nitroso group on the arene ring whilst the NH bond extends above the plane of the nitroso group (Fig. 3b). Consequently, the mesomeric effects observed in 1 are minimised leading to only the slight observed shortening of the C–N bond length in the nitroso group.10
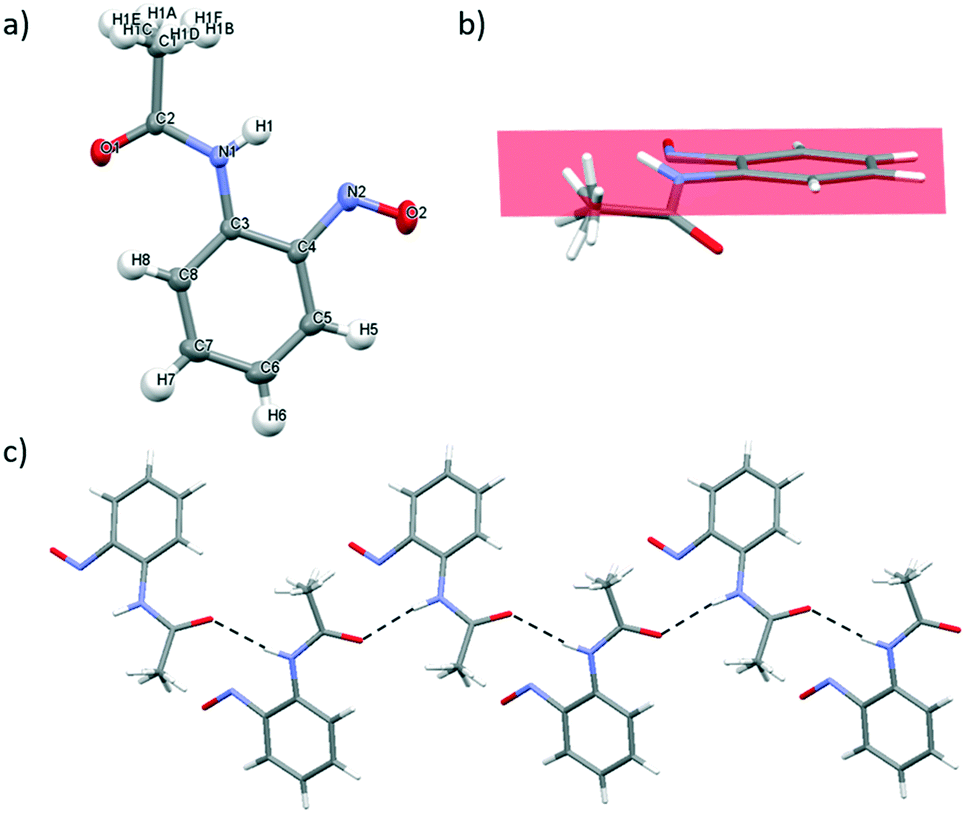 | ||
| Fig. 3 a) Mercury drawing of 1 with thermal ellipsoids drawn at 50% probability level and numbering is also shown. b) Viewed along the plane of the arene ring to highlight the orientation of the acetyl group and the NH bond deviating away from coplanarity with the nitroso group, c) X-ray structure of 1 highlighting the one-dimensional supramolecular chain involving intermolecular N–H⋯O hydrogen-bonding interactions running along the c axis. The hydrogen bonding interactions are shown as dashed black lines. The plane of the arene ring is shown in pink. | ||
In 1, there is one intermolecular hydrogen bonding interaction present between the carbonyl O atom and the NH of the amide functionality (2.124(13) Å) (Fig. 3c) and these N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C interactions form the basis of a one-dimensional, supramolecular hydrogen bonding chain orientated along the c axis. Within the unit cell, the aromatic rings of adjacent molecules are alternatively orientated above and below the plane of the supramolecular hydrogen bonding chain (Fig. 3c). The disorder about the methyl group prevents any information about the parameters (i.e. bond length and angle) of any potential intermolecular hydrogen-bonding interactions between the aryl protons of the methyl group and the O atom of nitroso group from being determined.
C interactions form the basis of a one-dimensional, supramolecular hydrogen bonding chain orientated along the c axis. Within the unit cell, the aromatic rings of adjacent molecules are alternatively orientated above and below the plane of the supramolecular hydrogen bonding chain (Fig. 3c). The disorder about the methyl group prevents any information about the parameters (i.e. bond length and angle) of any potential intermolecular hydrogen-bonding interactions between the aryl protons of the methyl group and the O atom of nitroso group from being determined.
The slow evaporation of chloroform from a saturated solution of 2 at ambient temperature yielded single green crystals that were suitable for X-ray diffraction. As for 1, and in line with the observed green colour of the crystals, 2 exists as a monomer in the solid state (Fig. 4a). There are two molecules in the asymmetric cell and both display disorder about the nitroso bond which prevents the C–N and N–O bonds lengths in 2 from being determined. The ω torsion angles for the amide bond lie close to the anticipated value for the trans peptide bond (173.80–177.70°). The carbamate group at the ortho position of 2 is coplanar with the aromatic ring and the nitroso group, presumably as the bulky tert-butyl group is far enough removed from the amide bond to prevent any significant distortion that could impact on any resonance stabilisation within the structure (see Fig. S10†).
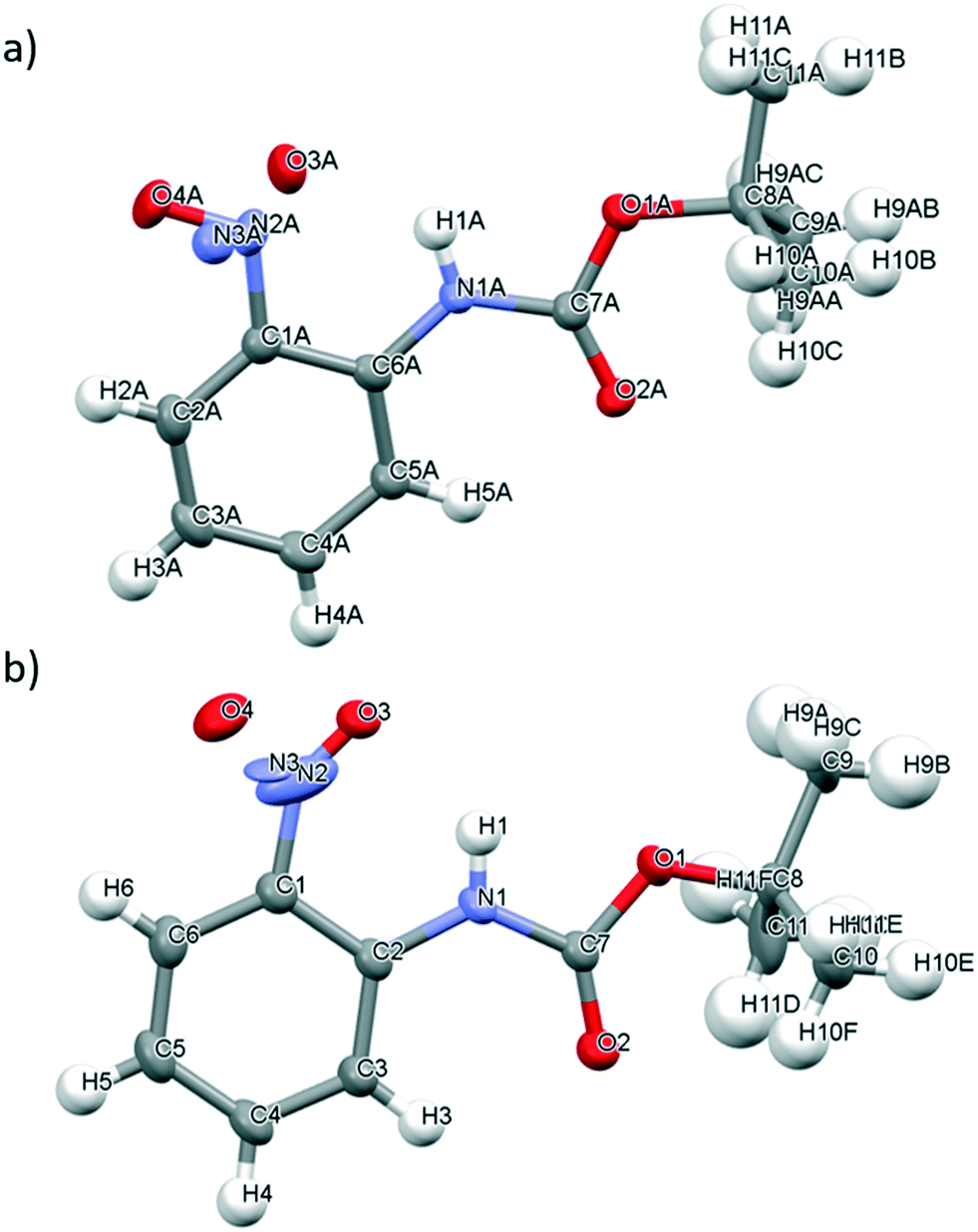 | ||
| Fig. 4 a) Mercury drawing of 2 with thermal ellipsoids drawn at 50% probability level and numbering is also shown. a) Showing one molecule in the asymmetric unit; b) showing the second molecule in the asymmetric unit. | ||
It appears that 2 has the potential to form intramolecular hydrogen bonding interactions involving the O atom of the nitroso group and the H atom of the amide bond (i.e. N(1A)–H(1A)⋯O(3A) and N(1)–H(1)⋯O(3)) but disorder about the nitroso group prevents determination of information about the bond lengths or angles (Fig. 4). Both of the amide containing nitrosoarenes, 1 and 2, have the potential to form two different types of competing hydrogen bonding interactions involving the NH atom of the amide bond and either the O atom of the nitroso group or the O atom of the carbonyl group in the amide bond but the nature of the amide substituents (both the electronic and steric demands) will play an important role in dictating the adoption of a hydrogen bond network in the solid state for these nitroso compounds.
In the crystal packing of 2, alternating asymmetric molecules are arranged perpendicular to each other in stacks running along the b axis (see Fig. S5†). This packing arrangement is supported by a series of three intermolecular C–H⋯O hydrogen bonding interactions.45 These include a set of weak bifurcated C–H⋯O hydrogen bonding interactions involving the O atom on the nitroso group and the aromatic protons of two neighbouring molecules (see Fig. S6†) and intermolecular hydrogen bonding interactions between the O atom of the carbonyl group in the ester moiety and either a proton on the tert-butyl group of an adjacent molecule (for C(11)–H(11E)⋯O(2A), see Fig. S7†) or an aromatic proton on an adjacent molecule (for C(2)–H(2A)⋯O(2), see Fig. S8†).
Slow evaporation of a saturated solution of 3 in methanol at ambient temperature generated single crystals suitable for X-ray diffraction. 3 exists as the 1,2-napthoquinone-1-oxime tautomer in the solid state, wherein the proton has been transferred from the hydroxyl group to the nitroso group to generate the corresponding oxo-oxime species (Fig. 5a).46 The N(1)–O(1) bond length of 1.361 Å observed in 3, is significantly longer than those observed for monomer 1 due to the differing bond order character in these species (Table 2). The C(1)–N(1) and C(2)–O(2) bond lengths of 1.3099(12) Å and 1.2502(12) Å respectively are close to those values expected for double bonds and, thus, are consistent with the presence of the oxime group due to tautomerization (Fig. 5a). The syn geometry of the oxime and carbonyl groups is stabilised by the formation of an intramolecular O–H⋯O(2) hydrogen-bonding interaction (1.646(2) Å). The crystal packing of the oxo-oxime tautomer of 3, when viewed along the a axis, arranges columnar stacks of molecules wherein the molecules are orientated in alternating head-to-tail and tail-to-tail fashion (Fig. 5b). This packing arrangement is supported by reciprocal intermolecular C(3)–H(3)⋯O(2) hydrogen-bonding interactions45 observed between two adjacent molecules (see Fig. S12†) and C–H–π stacking interactions (see Fig. S16†).48 Unlike in 1 and 2, where the presence of the amide groups at the ortho position leads to favourable formation of the monomer species, the incorporation of the hydroxyl group at the ortho position of 1-nitroso-2-napthol induces tautomerization to form the corresponding oxo-oxime in this aromatic C-nitroso compound.10
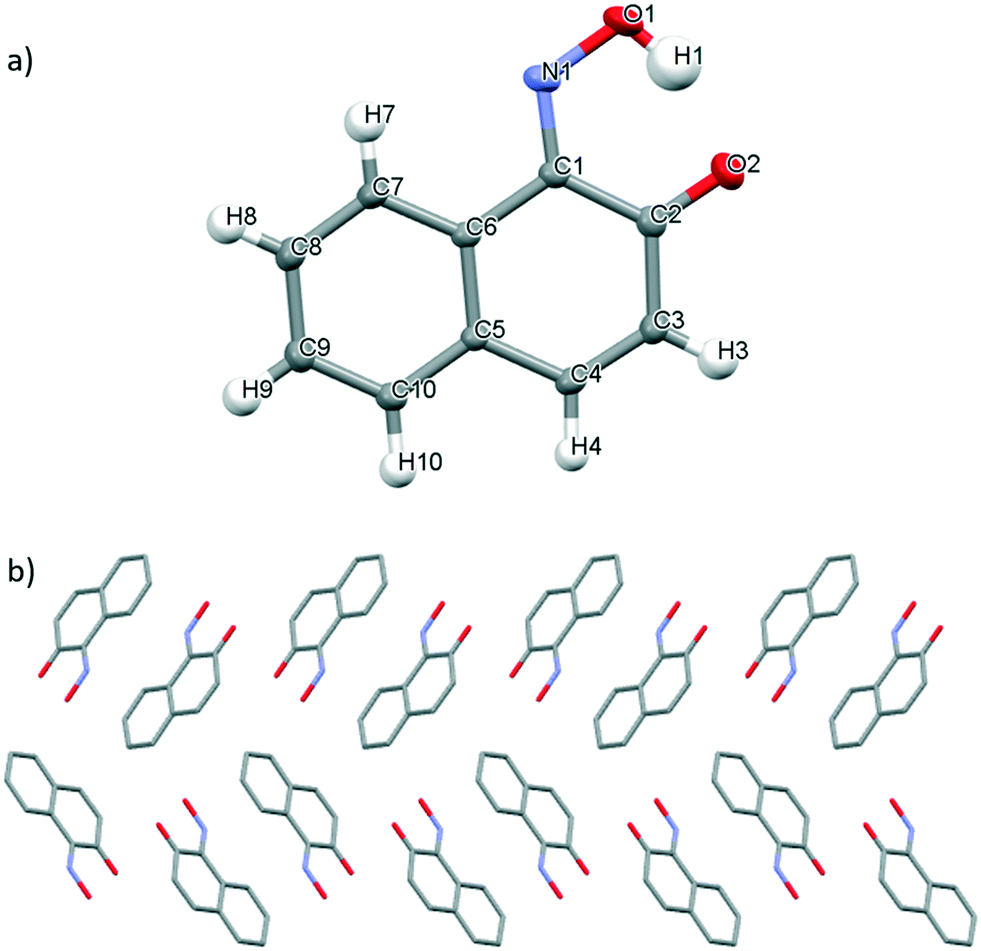 | ||
| Fig. 5 a) Mercury drawing of oxo-oxime tautomer of 3 with thermal ellipsoids drawn at 50% probability level and numbering is also shown. b) Crystal packing of 3 as viewed along the a axis. | ||
Influence of weakly electron-donating ortho-substituents
The solid state behaviour of nitrosotoluene 4, bearing a weakly inductive electron-donating methyl group at the ortho-position of the ring was investigated using X-ray crystallographic analysis. Both the cis- and trans-azodioxy dimers of 4 (cis-4 and trans-4, respectively) could be obtained through variation of the solvent employed in the crystallisation conditions (Fig. 6).49 Crystal structures of organic C-aromatic nitroso compounds that adopt both the cis- and trans-azodioxy dimers in the solid state are rare.24a,47,50 Single colourless block crystals of cis-4, suitable for X-ray crystallography diffraction were grown through the slow evaporation of a 1/1 chloroform/methanol solvent mixture at ambient temperature whilst single colourless block crystals of trans-4 were grown through the slow evaporation of a chloroform solution at ambient temperature (Fig. 6).51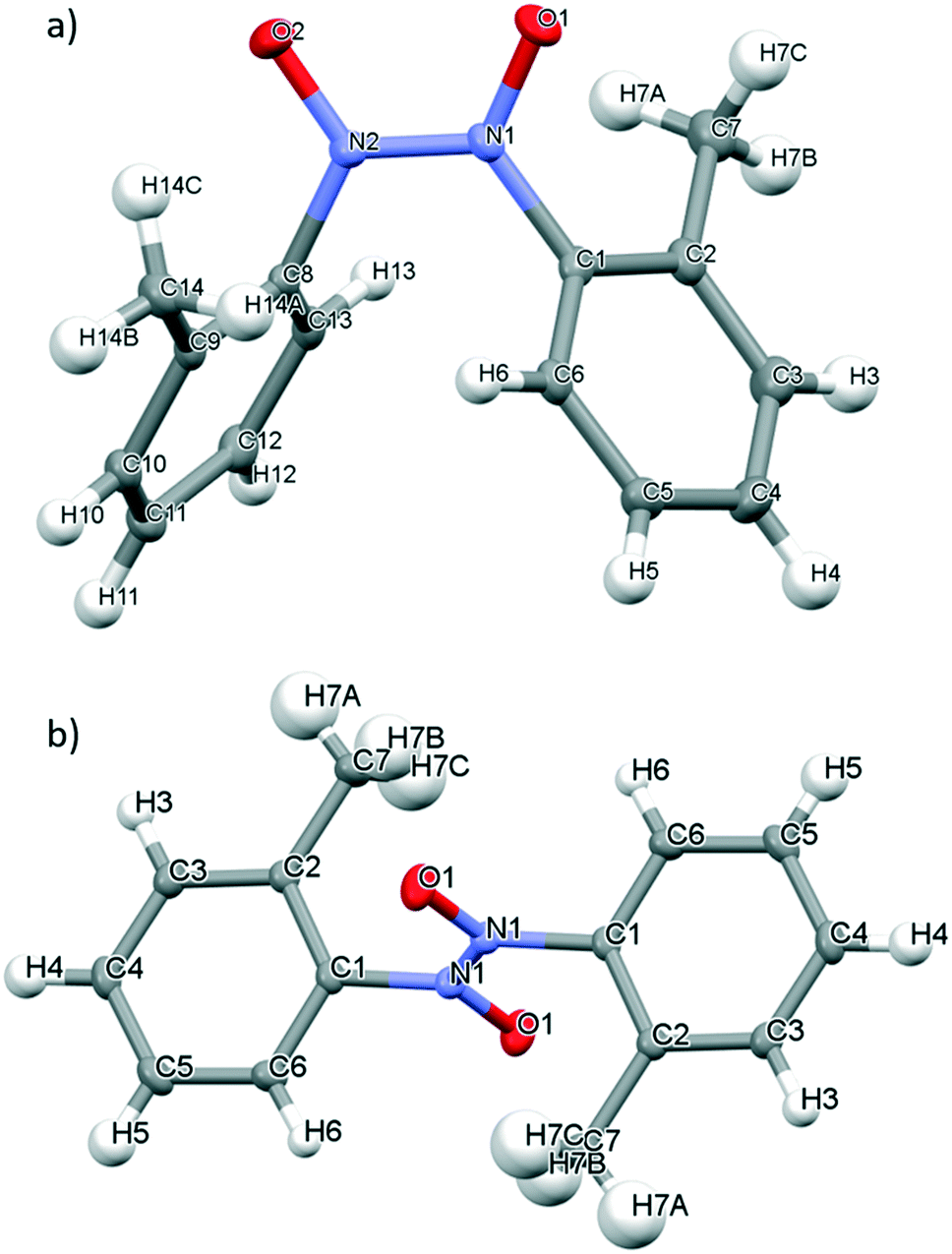 | ||
| Fig. 6 Mercury drawing of a) cis-4 and b) trans-4 with thermal ellipsoids drawn at 50% probability level and numbering is also shown. | ||
In cis-4 and trans-4, the N–N bond lengths are 1.3241(19) Å and 1.319(2) Å respectively, both of which are close to that expected of a double bond.10 In cis-4, the N–O bond lengths are 1.2671(18)–1.2695(18) Å and the C–N bond lengths are 1.447(2)–1.457(2) Å whilst in trans-4 comparable bond lengths of 1.2679(16) Å (N–O) and 1.4529(17) Å (C–N) are observed (Table 2). All the C–N and N–O bond lengths observed in 4 are consistent with those typically expected for azodioxy dimers of C-nitrosoarenes.10
The crystal packing of cis-4 leads to layered stacking of the molecules generating tunnels with central hollow cavities running along the c axis (Fig. 7a).52 The porous packing arrangement observed in cis-4, is built from a series of intermolecular bifurcated C–H⋯O hydrogen bonding interactions between O1 on the nitroso group and H10 and H14B on the aromatic ring and methyl group respectively (see Fig. S17†). The crystal packing arrangement between molecules in adjacent off-set channels (Fig. 7a) is supported by a second set of intermolecular bifurcated C–H⋯O hydrogen-bonding interactions53 between O2 on the nitroso group and H4 and H13 which are aromatic protons on two different adjacent molecules (Fig. S18†).
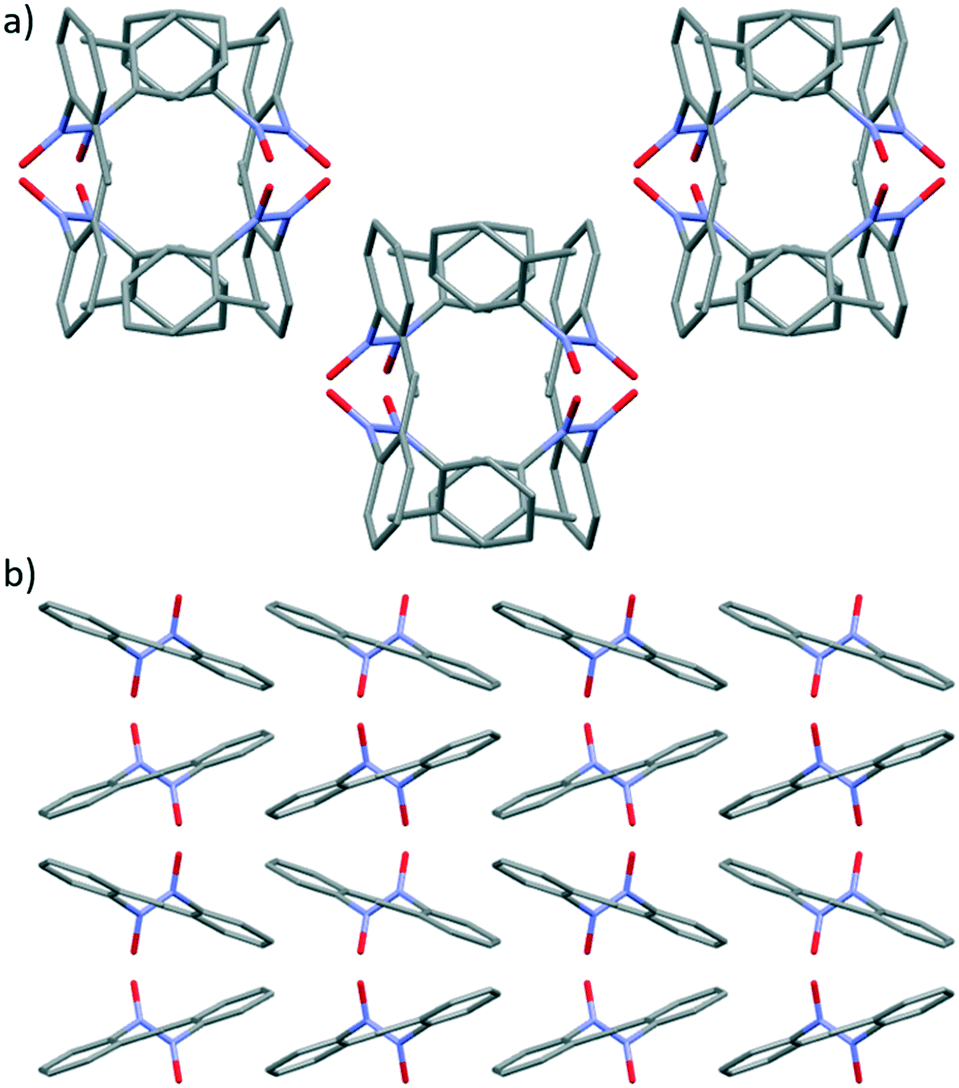 | ||
| Fig. 7 a) Packing arrangement in crystals of cis-4 displaying the channel with a central cavity as viewed along the c axis. b) Packing arrangement in crystals of trans-4 showing the type of zigzag structure observed when viewed along the b axis. H atoms have been removed for clarity. | ||
In trans-4, a zigzag crystal packing arrangement is observed, when the structure is viewed along b axis, due to the off-set alternating packing of the molecules within stacks (see Fig. 7b).24a The crystal packing of trans-4 is supported by two types of non-covalent interactions; firstly, intermolecular C-H⋯π interactions54 between H7A of the methyl group and the aromatic ring of an adjacent molecule (see Fig. S20†) and secondly, an intermolecular charged assisted C–H⋯O− hydrogen-bonding interactions between the O atom of the nitroso group and the proton at the meta position (H5) of the aromatic ring (see Fig. S21†).19 The supramolecular intermolecular interactions that promote the formation of tunnel structure in cis-4 are lacking in trans-4 (Fig. 7).
It can be seen that modification of the ortho-substituent from a resonance electron-donating group, in 1–3, to a weakly inductive electron-donating group as in 4, has a significant effect on the speciation behaviour of the studied nitrosoarenes, in the studied conditions, leading to the preferential formation of azodioxy dimers in the solid state.10 The ready accessibility of both the cis- and trans-azodioxy dimers of 4 indicates that the energetics controlling the adoption of both dimers in the solid state is finely balanced and can be readily tuned through the choice of solvent employed in the crystallisation conditions. The presence of polar protic solvents in the crystallisation conditions favours the cis-azodioxy dimer over the trans isomer. Notably, solution studies into the influence of solvent effects on the monomer–dimer equilibrium of nitroso compounds, have identified that polar protic solvents generally favour the formation of azodioxy dimers over monomers10,15 and that they, typically, lead to preferential adoption of cis-azodioxy dimers over trans-azodioxy isomers.10 Specifically, the influence of solvent on the equilibrium constants for dissociation of the azodioxy dimers of 4, in solution, has been systematically studied using NMR spectroscopy and it was observed that dimers were more likely to persist in solution when polar protic solvents (e.g. methanol-d4) and polar aprotic solvents with high dielectric constants (e.g. DMSO-d6, acetonitrile-d3) were employed.10,15 Conversely, polar aprotic solvents with low dielectric constants (e.g. carbontetrachloride) favoured formation of the monomer in solution studies.10
Generally, it has been suggested that the observed preference for dimerization over adoption of the monomer form is due to the stabilisation of the more polar dimer species over the less polar monomer species by solvent-solute dipole–dipole moments.10,55 Additionally, as cis-azodioxy dimers are typically known to display larger net dipole moments than analogous trans-azodioxy dimers, solvent studies have suggested that the cis-azodioxy dimer is better stabilised by solvent–solute interactions established in polar protic solvents than the analogous trans-isomer.10,56 It is possible that the observed dependence of the adoption of cis- and trans-4 in the solid state, on the nature of the solvent employed in the crystallisation conditions, could in a similar manner be influenced by the differing nature of the solvent–solute interactions present in solution for each isomer.57 However, it should be noted that attempts to derive direct correlation between the solution and solid state behaviour of these systems is challenging and complex due to complications from a number of factors, including the crystal packing forces observed in the solid state, that are often so delicately balanced and governed by a wide range of influences. Further in-depth solvent screening studies of this system are required to ascertain the role of the solvent in the crystallisation conditions on the adoption of the azodioxy dimers.
Influence of weakly electron-withdrawing fluorine ortho-substituents
The solid state behaviour of 2,6-difluoronitrosobenzene 5, incorporating the weakly inductive electron-withdrawing fluorine groups at both ortho-positions of the ring, relative to the nitroso group, was studied using X-ray crystallographic analysis.Slow evaporation of a chloroform solution of 5 at ambient temperature, yielded single colourless crystals suitable for X-ray diffraction. 5 exists as the trans-azodioxy dimer in the solid state (Fig. 8a), which is in line with literature reports on the general trends observed for the solid state speciation of C-nitrosoarene bearing inductive electron-withdrawing substituents on the ring.10,58 The trans-azodioxy dimer in 5 has two symmetrically-equivalent monomer units linked by an N–N bond (1.325(3) Å) which is close to that expected of a bond (Fig. 8a).10 The centre of inversion for 5 is located in the centre of the N–N bond. The N–O bond length is 1.262(2) Å whilst the C(1)–N(1) bond length is 1.444(2) Å and these values are comparable to both dimers of 4 (Table 2).
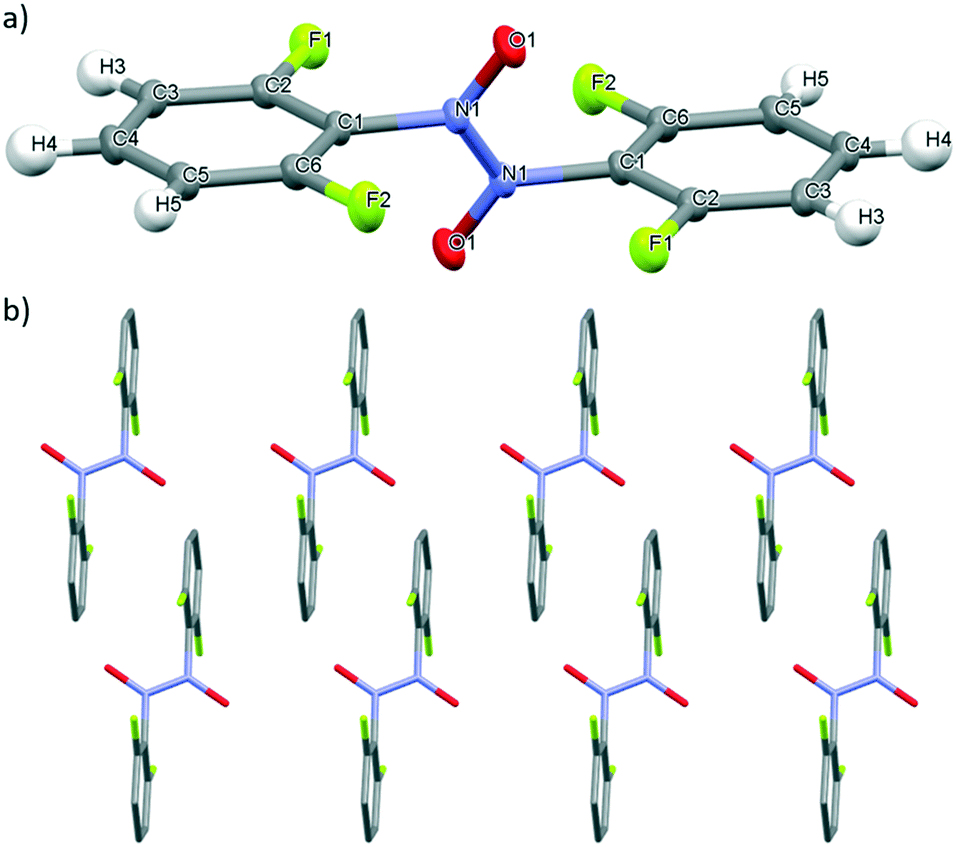 | ||
| Fig. 8 a) Mercury drawing of 5 with thermal ellipsoids drawn at 50% probability level and numbering is also shown. b) X-ray structure of 5 highlighting crystal packing arrangement with the columnar stacks of molecules running along the c axis and the N–O bond of the nitroso orientated along the c* axis. | ||
In 5, the molecules align in columnar planar stacks along the c axis, with the N–O bond orientated in a regular manner along the c* axis (Fig. 8b) and this crystal packing arrangement is supported by three types of intermolecular interactions. Firstly, charge assisted, bifurcated C–H⋯O− hydrogen-bonding interactions involving the O atom of the nitroso group and two of the aromatic protons (H3 and H5) on two different adjacent molecules (Fig. S26†). Secondly, a series of C–H⋯F hydrogen-bonding interactions,59 involving C(5)–H(5)⋯F(1)–C(1), between adjacent molecules and, thirdly, intermolecular off-set face-to-face π–π stacking interaction (see Fig. S27 and S28† respectively). As in 4, the use of chloroform for the crystallisation conditions yielded the trans-azodioxy dimer of the aromatic C-nitroso compound. Unfortunately, attempts to grow single crystals of 5 suitable for X-ray diffraction analysis, from chloroform/methanol solvent mixtures were unsuccessful, thus, preventing direct comparison of the influence of these crystallisation conditions on the solid state cis-/trans-azodioxy dimer speciation behaviour of 4 and 5.
Conclusions
We have explored the rich solid state chemistry of aromatic C-nitroso compounds, accessing four different species in the solid state, through simple modifications of the electronic and steric properties of the ortho-substituent, relative to the nitroso group on the arene ring. A diverse array of non-covalent interactions including neutral and charged assisted hydrogen bonding interactions (e.g. O–H⋯O, C–H⋯O, N–H⋯O, C–H⋯F and C–H⋯π) and π–π stacking interactions have been identified to support different supramolecular aggregates in the solid state, including one-dimensional supramolecular chains and tunnels.We have confirmed that the presence of the strongly electron-donating amide groups, acetyl and carbamate, at the ortho-position of the ring leads to the adoption of the monomer in the solid state. In the acetylated compound, 2-nitrosoacetanilide, a supramolecular one-dimensional hydrogen bonding chain is formed through intermolecular N–H⋯OC interactions of the amide groups. However, in the carbamate analogue, tert-butyl-N-(2-nitrosophenyl)carbamate, it is likely that the steric demands of the bulky Boc group prevents formation of intermolecular hydrogen-bonding interactions involving the amide group and, consequently, no supramolecular chain is observed for this compound.
The oxo-oxime tautomer was accessed through the incorporation of a hydroxyl group at ortho-position of the ring and the orthogonal stacking arrangement of the molecules observed in the crystal packing arrangement was supported by a combination of intermolecular hydrogen bonding (O–H⋯O and C–H⋯O) and π–π stacking interactions.
For nitrosotoluene, the nature of the solvent employed in the crystallisation conditions strongly influenced the species (cis- or trans-azodioxy dimer) adopted in the solid state. The presence of a polar protic solvent (methanol) in the crystallisation solvent mixture led to the preferential formation of the cis-azodioxy dimer whilst the exclusive use of chloroform generated the trans-azodioxy dimer. Distinct crystal packing behaviour was observed for these two isomers, with the cis-azodioxy dimer adopting a porous packing structure with tunnels orientated along the c axis whilst the trans isomer adopted a zigzag crystal packing arrangement where no tunnels are present. The incorporation of the weakly electron-withdrawing fluorine groups on the nitroso ring generates the trans-azodioxy dimer under the tested crystallisation conditions.
We anticipate that this study, highlighting the significance of both the ortho-substituent and the nature of the solvent type employed in the crystallisation conditions, on the speciation behaviour of this important class of compounds, will facilitate their future design for use in synthetic systems, including solid state reactions with photochromic or thermochromic material applications. Moreover, these studied aromatic C-nitroso compounds could potentially be interesting building blocks for assembling new covalent organic frameworks.
Experimental section
X-ray crystallography
Single crystal X-ray diffraction data on 1–5 were collected using a Bruker X8 diffractometer with an APEX II detector and monochromated Mo Kα radiation (λ = 0.7107 Å) at 173 K. The data was processed using Bruker SAINT, the structures determined with SHELXT60 and subsequently refined with SHELXL61 within the program Olex2.62 Crystal structures were visualised using Mercury.63 In the case of cis-4, initial attempts to solve the structure in orthorhombic space groups was unsuccessful. Reindexing as a monoclinic cell (21.7113 Å, 10.4211 Å, 15.176 Å, 90°, 134.346°, 90°) with a second data collection on a new crystal led to a successful solution in the C2/c space group. Subsequently attempts to locate higher symmetry with the ADDSYM function in platon64 generated the final pseudo-orthorhombic cell in I2/a. Challenges with the decomposition of the crystal during data collection limited the level of data that could be obtained. For all other samples possible missing symmetry was investigated through the tools in platon64 and none was located. The X-ray data for 1–5 have been deposited with the Cambridge Crystallographic Data Centre. CCDC numbers: 2003802–2003806 contain the supplementary crystallographic data for this paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. J. P. acknowledges the University of Bradford Research Development Fund for support. This work was supported by a UKRI Future Leaders Fellowship [MR/S035486/1]. S. J. P. is a UKRI Future Leaders Fellow.Notes and references
- W. Adams and O. Krebs, Chem. Rev., 2003, 103, 4134–4146 Search PubMed.
- (a) N. Momiyama and H. Yamamoto, Chem. Commun., 2005, 3514–3525 Search PubMed; (b) N. Momiyama and H. Yamamoto, Org. Lett., 2002, 4, 3579–3582 CrossRef CAS PubMed; (c) S. Carosso and M. J. Miller, Org. Biomol. Chem., 2012, 12, 7445–7468 RSC.
- J. Huang, Z. Chen, J. Yuan and Y. Peng, Asian J. Org. Chem., 2016, 5, 951–960 CrossRef CAS.
- M. Cameron and B. G. Gowenlock, Chem. Soc. Rev., 2019, 396, 124–140 Search PubMed.
- J. Lee, L. Chen, A. H. West and G. B. Richter-Addo, Chem. Rev., 2002, 102, 1019–1065 CrossRef CAS PubMed.
- K. A. Emhoff, L. Balaraman, A. M. H. Salem, K. I. Mudarmah and W. C. Boyd, Coord. Chem. Rev., 2002, 102, 1019–1065 Search PubMed.
- B. W. Griffin, Can. J. Chem., 1982, 60, 1463–1473 CrossRef CAS.
- M. R. Gunther, Free Radical Biol. Med., 2004, 36, 1345–1354 CrossRef CAS PubMed.
- (a) W. G. Rice, C. A. Schaeffer, B. Harten, F. Villinger, T. L. South, M. F. Summers, L. E. Henderson, J. W. Bess Jr, L. O. Arthur, J. S. McDougal, S. L. Orloff, J. Mendeleyev and E. Kun, Nature, 1993, 361, 473–475 CrossRef CAS PubMed; (b) W. G. Rice, C. A. Schaeffer, L. Graham, M. Bu, J. S. McDougal, S. L. Orloff, F. Villinger, M. Young, S. Oroszlan, M. R. Feseni, Y. Pommier, J. Mendeleyev and E. Kun, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9721–9724 CrossRef CAS PubMed.
- D. Beaudoin and J. Wuest, Chem. Rev., 2016, 116, 258–286 CrossRef CAS PubMed.
- (a) I. Biljan and H. Vančik, Crystals, 2017, 7, 376 CrossRef; (b) I. Halasz, E. Mestrovic, H. Cicak, Z. Mihalic and H. Vancik, J. Org. Chem., 2005, 70, 8461–8467 CrossRef CAS PubMed.
- (a) D. A. Fletcher, B. G. Gowenlock and K. G. Orrell, J. Chem. Soc., Perkin Trans. 2, 1997, 2201–2205 RSC; (b) M. Azoulay and G. Wettermark, Tetrahedron, 1978, 34, 2591–2596 CrossRef CAS.
- J. Varga, J. Volarić and H. Vančik, CrystEngComm, 2015, 17, 1434–1438 RSC.
- D. Beaudoin, T. Maris and J. D. Wuest, Nat. Chem., 2013, 5, 830–834 CrossRef CAS PubMed.
- H. Vančik, V. Šimunić-Mežnarić, I. Caleta, E. Mestrovic, S. Milovac, K. Milnaric-Majerski and J. Valjkovic, J. Phys. Chem. B, 2002, 106, 1576–1580 CrossRef.
- M. Witanowski, Z. Biedrzycka, W. Sicinska and G. A. Webb, Magn. Reson. Chem., 1997, 35, 262–266 CrossRef CAS.
- Y. Yoshimura and M. Nakahara, Ber. Bunsen-Ges., 1988, 92, 46–50 CrossRef CAS.
- M. D. Lumsden, G. Wu, R. E. Wasylisben and R. D. Curtis, J. Am. Chem. Soc., 1993, 115, 2825–2832 CrossRef CAS.
- B. G. Gowenlock, M. Cameron, A. S. F. Boyd, B. M. Al-Tahou and P. McKenna, Can. J. Chem., 1994, 72, 514–518 CrossRef CAS.
- C. Darwin and D. Crowfoot Hodgkin, Nature, 1950, 166, 827–828 CrossRef CAS PubMed.
- C. P. Fenimore, J. Am. Chem. Soc., 1950, 72, 3226–3231 CrossRef CAS.
- D. A. Dickie, I. S. MacIntosh, D. A. Ino, Q. He, O. Labeodan, M. C. Jennings, G. Schatte, C. J. Walsby and J. A. C. Clyburne, Can. J. Chem., 2008, 86, 20–31 CrossRef CAS.
- B. G. Gowenlock and K. J. McCullough, J. Chem. Soc., Perkin Trans. 2, 1989, 551–553 RSC.
- (a) E. Bosch, J. Chem. Crystallogr., 2014, 44, 98–102 CrossRef CAS; (b) J. Kozhukh, J. F. Lopes, H. F. Dos Santos and S. J. Lippard, Organometallics, 2012, 31, 8063–8066 CrossRef CAS.
- D. A. Dieterich, I. C. Paul and D. Y. Curtin, J. Am. Chem. Soc., 1974, 96, 6372–6380 CrossRef CAS.
- I. Halasz, I. Bilijan, P. Novak, E. Meŝtrović, J. Plavec, G. Mali, V. Smrečki and H. Vančik, J. Mol. Struct., 2009, 918, 19–25 CrossRef CAS.
- D. A. Fletcher, B. G. Gowenlock, K. G. Orrell, V. Šik, D. E. Hibbs, M. B. Hursthouse and K. M. A. Malik, J. Chem. Soc., Perkin Trans. 2, 1996, 191–197 RSC.
- D. A. Fletcher, B. G. Gowenlock, K. G. Orrell, D. C. Apperley, M. B. Hursthouse and K. M. A. Malik, J. Chem. Res., Synop., 1999, 202–203 RSC.
- (a) I. Halasz, E. Meŝtrović, H. Čičak, Z. Mihalić and H. Vančik, J. Org. Chem., 2005, 70, 8461–8467 CrossRef CAS PubMed; (b) I. Halasz and H. Vančik, CrystEngComm, 2011, 13, 4307–4310 RSC.
- D. A. Dieterich, I. C. Paul and D. Y. Curtin, J. Chem. Soc. D, 1970, 1710–1711 RSC.
- P. A. Lightfoot, R. G. Pritchard, H. Wan and J. E. Warren, Chem. Commun., 2002, 2072–2073 RSC.
- P. P. Rodenbough, D. P. Karothu, T. Gjorgjieva, P. Commins, H. Hara and P. Naumov, Cryst. Growth Des., 2018, 18, 1293–1296 CrossRef CAS.
- C. K. Prout, A. Coda, R. A. Forder and B. Kamenar, Cryst. Struct. Commun., 1974, 3, 39–42 CAS.
- B. G. Gowenlock, M. J. Maidment, K. G. Orrell, V. Šik, G. Mele, G. Vasapollo, M. B. Hursthouse and K. M. A. Malik, J. Chem. Soc., Perkin Trans. 2, 2000, 2280–2286 RSC.
- J. Armand, Y. Armand, L. Boulares, M. Philoche-Levisalles and J. Pinson, Can. J. Chem., 1981, 59, 1711–1716 CrossRef CAS.
- H. J. Talberg, Acta Chem. Scand., Ser. A, 1976, 30, 829–834 CrossRef.
- N. N. Dhaneshwar, S. N. Naik and S. S. Tavale, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 217–218 CrossRef.
- K. Lewinski, W. Nitek and P. Milart, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 188–190 CrossRef.
- S. Wirth, A. U. Wallek, A. Zernickel, F. Feil, M. Sztiller-Sikorska, K. Lesiak-Mieczkowska, C. Brauchle, I.-P. Lorenz and M. Czyz, J. Inorg. Biochem., 2010, 104, 774–789 CrossRef CAS PubMed.
- R. G. Pritchard, G. S. Heaton and I. M. El-Nahhal, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 829–831 CrossRef.
- H. J. Talberg, Acta Chem. Scand., 1977, 31A, 743–751 CrossRef.
- Synthetic experimental and purification details for 1–5 are provided in the ESI.† .
- J. B. F. N. Engberts, T. A. J. W. Wajer, C. Kruk and T. J. de Boer, Recl. Trav. Chim. Pays-Bas, 1969, 88, 481 Search PubMed.
- A. Quesada, A. Marchal, M. Melguizo, M. Nogueras, A. Sánchez, J. N. Low, D. Cannon, D. M. M. Farrell and C. Glidewell, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 300–315 CrossRef PubMed.
- S. Horowitz and R. C. Trievel, J. Biol. Chem., 2012, 287, 41576–41582 CrossRef CAS PubMed.
- S. Wirth, C. J. Rohbogner, M. Cieslak, J. Kazmierczak-Baranska, S. Donevski, B. Nawrot and I.-P. Lorenz, J. Biol. Inorg. Chem., 2010, 15, 429–440 CrossRef CAS PubMed.
- The unit cell data matches that which has previously been reported for cis-4 see: M. Azoulay, J. Appl. Crystallogr., 1982, 15, 245–246 CrossRef CAS ; whom employed Weissenberg photographs to identify the structure and so, accordingly, only limited crystal data and no three coordinates were provided.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- Wettermark and co-workers identified the importance of crystallisation conditions on the preparation of the cis/trans ratio of 4 in the solid state, noting that two cis-isomers of 4 could be obtained using chloroform/ethanol (1/1) solvent mixtures with slow evaporation at room temperature over 4–8 h, whilst polycrystalline trans-azodioxynitrosotoluene dimer could be access either through steam-water distillations or rapid evaporation of chloroform/ethanol (1/1) solvent mixtures of 4 in under 0.5 h, see (a) M. Azoulay, B. Stymne and G. Wettermark, Tetrahedron, 1976, 32, 2961–2966 CrossRef CAS.
- D. A. Dickie, I. S. MacIntosh, D. A. Ino, Q. He, O. A. Labeodan, M. C. Jennings, G. Schatte, C. J. Walsby and J. A. C. Clyburne, Can. J. Chem., 2008, 86, 20–31 CrossRef CAS.
- Whilst the crystal structure of trans-4 has previously been reported, see ref. 24a, no crystallisation conditions were provided .
- At the narrowest point of the tunnel in cis-4 is 3.51 Å but in this current study attempts to trap gas or water molecules into the cavity of the tunnel have not been undertaken. However, in the future it could be of interest to probe the porosity of cis-4 with different guest molecules particularly given the encapsulation potential of the crystalline covalent organic networks; see ref. 14 .
- I. Rozas, I. Alkorta and J. Elguero, J. Phys. Chem. A, 1998, 9925 CrossRef CAS.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- R. Glaser, R. K. Murmann and C. L. Barnes, J. Org. Chem., 1996, 61, 1047–1058 CrossRef CAS.
- C. Fehling and G. Fredrichs, J. Am. Chem. Soc., 2011, 133, 17912–17922 CrossRef CAS PubMed.
- S. J. Pike, A. D. Bond and C. A. Hunter, CrystEngComm, 2018, 20, 2912–2915 RSC.
- Whilst reported crystal structures of cis-azodioxy dimers of aromatic C-nitroso compounds are rare they have been identified for several species including nitrosotoluene (cis-4) and methyoxynitrosobenzene (see ref. 31) both of which bear ortho-substituents that are more sterically demanding than those found in 5. Hence, from a geometrical point of view, with respect to steric demands of the fluorinated ortho-substituents in 5, the system does have the potential to form cis-azodioxy dimers in the solid state but is not observed under the crystallisation conditions employed .
- (a) A. J. Mountford, D. L. Hughes and S. J. Lancaster, Chem. Commun., 2003, 2148 RSC; (b) E. Kryachko and S. Scheiner, J. Phys. Chem. A, 2004, 13, 2527 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS; C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray data for 1–5 has been deposited with the Cambridge Crystallographic Data Centre (CCDC: 1879457 (for 1) and 2003802–2003806 (for 2–5)). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce00728e |
| This journal is © The Royal Society of Chemistry 2020 |