
Nature of fluorine interactions in ‘wheel and axle’ topology based hexa-coordinated Sn(iv)-porphyrins: an experimental and theoretical analysis†

Abstract
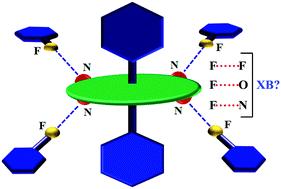
This study targets the construction of metalloporphyrin assemblies directed by fluorine-centered non-covalent interactions that can conveniently operate on a metallated tetrapyridylporphyrin system, which topologically resembles a wheel–axle duo. We chose a series of Sn(axial-L)2-(5,10,15,20-tetrapyridylporphyrin) [Sn(L)2-TPyP, where L = fluorine-substituted benzoate moiety] complexes as our building units, which have only one fluorine atom on the benzoate moiety (compounds 1 and 2) but progressively increased to 2 (compound 3) and to 5 (compound 4). This stepwise augmentation also showed a concomitant increase in fluorine-based intermolecular interactions. Four complexes with varying F : H ratios of 1 : 4 to 5 : 0 at the axle part of the molecule were structurally analyzed by single-crystal X-ray diffraction. Fluorine-centered intermolecular interactions have been investigated and found to correlate with the number of fluorine atoms present at the axial benzoato-ligand of the wheel–axle duo. The augmentation in the number of fluorine-centered interactions was theoretically supported by Hirshfeld surface analysis showing a steep increase from 11% (1/2) to 20% (3) to 39% (4) as a function of the degree of fluorine-substitution. However, electrostatic potential surface (ESP) analysis clearly disproves the formation of σ-holes at fluorine atoms and thus vitiates their role as XB donors in these complexes. The occurrence of such short-contacts as a result of the ‘Gulliver effect’ should not be ruled out in fluorine based crystal engineering attempts.
 Please wait while we load your content...
Please wait while we load your content...

