
Air exposed 1,4-bis(trimethylsilyl)-1,4-dihydropyrazine: an avant-garde carbonization precursor for multi-functionalized carbon material†‡
Shaikh Saddam
Shoukat Ali
a,
Kingshuk
Roy
ab,
Naveenkumar
Akula
a,
Satishchandra B.
Ogale
 bc and
Moumita
Majumdar
*a
bc and
Moumita
Majumdar
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research, Pune-411008, Maharashtra, India. E-mail: moumitam@iiserpune.ac.in
bResearch Institute for Sustainable Energy (RISE), TCG Centres for Research and Education in Science and Technology (TCG-CREST), Omega, BIPL Building, Blocks EP & GP, Sector V, Salt Lake, Kolkata 700091, India
cDepartment of Physics, Indian Institute of Science Education and Research, Pune-411008, Maharashtra, India
First published on 28th October 2020
Abstract
1,4-Bis(trimethysilyl)-1,4-dihydropyrazine 1 has been utilized as a small molecule precursor for carbonization to N,O-containing few-layered carbon sheets 3via the formation of a polymeric material 2 upon simple air exposure at room temperature. Without any further purification, this multi-functionalized carbon material 3 exhibited excellent anode performance in a lithium ion battery.
Bis-silylated non-aromatic organosilicon reagents have been skillfully employed as salt-free reductants for a variety of transition metal complexes1 and main group elements.2 Metal(0) nano-composites have also been obtained that show anode performance in lithium ion batteries (LIBs).3 Remarkably, organosilicon reagents enable the efficient synthesis of bis(trimethylsilyl)peroxide (BTSP) directly by the activation of O2 in air and rearomatization to the corresponding (hetero)aromatic species.4 Simple air exposure has led to a handful of exciting chemistry discoveries such as the thermal stabilization of block copolymers of polyacrylonitrile,5 thereby making the oxidized polymer an appropriate precursor for carbonization.
Carbonization of organic precursors is one of the most popular methods for carbon material syntheses.6 Carbon materials with good physicochemical properties, unique microstructures, and hetero-atom contents exhibit application potential in diverse fields.7 Traditionally, the organic precursors used for carbonization were limited to natural products8 or synthetic polymers.9 The major drawbacks of these conventional precursors are their poor control over the product composition and microstructure. In principle, more versatile small molecule precursors can potentially tailor the intrinsic properties of carbon materials.10 However, despite their common availability, low cost and desired heteroatom content, small molecules are very rarely used due to their volatility at elevated temperatures. Notably, small molecules, such as urea, cyanamide, etc., are well-known to undergo thermal decomposition in a closed vessel, yielding graphitic carbon nitride.11 The three prominent methods for preparing carbon materials are the direct carbonization of ionic liquids,12 transition metal assisted carbonization of small molecules13 and alkali metal assisted carbonization of pyrrole.14 The latter two methods require an additional step for removing the metal impurities post-carbonization. Therefore, the ab initio design of ‘smart’ organic molecules for carbonization with minimal or no purification efforts demands prompt investigations.
In this work, we have established 1,4-bis(trimethysilyl)-1,4-dihydropyrazine151 as a state-of-the-art small molecule precursor for pyrolysis to prepare a N,O-containing carbon material 3via the formation of a polymeric intermediate 2 upon simple air exposure of 1 at room temperature. This approach is based on the unique organometallic concept of O2 activation by organosilicon reagents.4 This multi-functionalized carbon material 3 shows outstanding merit as an anode material in LIBs.
1,4-Bis(trimethylsilyl)-1,4-dihydropyrazine 1 was prepared from pyrazine and Me3SiCl using Li metal according to a procedure described in the literature.15 A bright yellow solution of 1 dissolved in pentane, taken in a Petri-dish, gradually turned into an orange solid 2 within four hours at room temperature when exposed to air (Scheme 1). The as-synthesized solid 2 was characterized using a multi-nuclear NMR study (Fig. S1–S4, ESI‡), advanced polymer chromatography (APC) (Fig. S5, ESI‡), optical spectroscopy (Fig. S6 and S7, ESI‡), Fourier transform infrared (FT-IR) spectroscopy and X-ray photoelectron spectroscopy (XPS) (Fig. 1 and Fig. S8, S9, ESI‡). Solid 2 was soluble only in highly polar solvents. The relatively broad 1H spectrum and the 13C NMR spectrum of 2 in DMSO-d6 (Fig. S1 and S2, ESI‡) revealed the presence of aromatic rings, alkenyl groups, –NH and C–OH functional groups and –SiMe3 groups. The 29Si NMR spectrum of 2 taken in DMSO-d6 shows a major peak at 7.22 ppm attributed to hexamethyldisiloxane (Fig. S3, ESI‡), as a by-product from the initially formed BTSP.4 The 29Si NMR spectrum of an in situ reaction generating 2 from 1 in THF-d8 medium also detected the presence of hexamethyldisiloxane (Fig. S4, ESI‡). A high molecular weight polymeric species (Mn = 4.6 kDa, PDI = 1.18) was detected by APC of 2 (Fig. S5, ESI‡). Therefore, we confirmed the formation of polymeric intermediate 2 from the combination of NMR and APC studies. The solid-state absorbance spectrum of 2 (Fig. S6, ESI‡) displays a broad peak that was de-convoluted into three additive peaks at 255, 379 and 491 nm. The polymeric material 2 exhibits emission at 475 and 574 nm, as shown in Fig. S7 (ESI‡).
 | ||
| Scheme 1 Synthesis of N,O-containing carbon material from 1. | ||
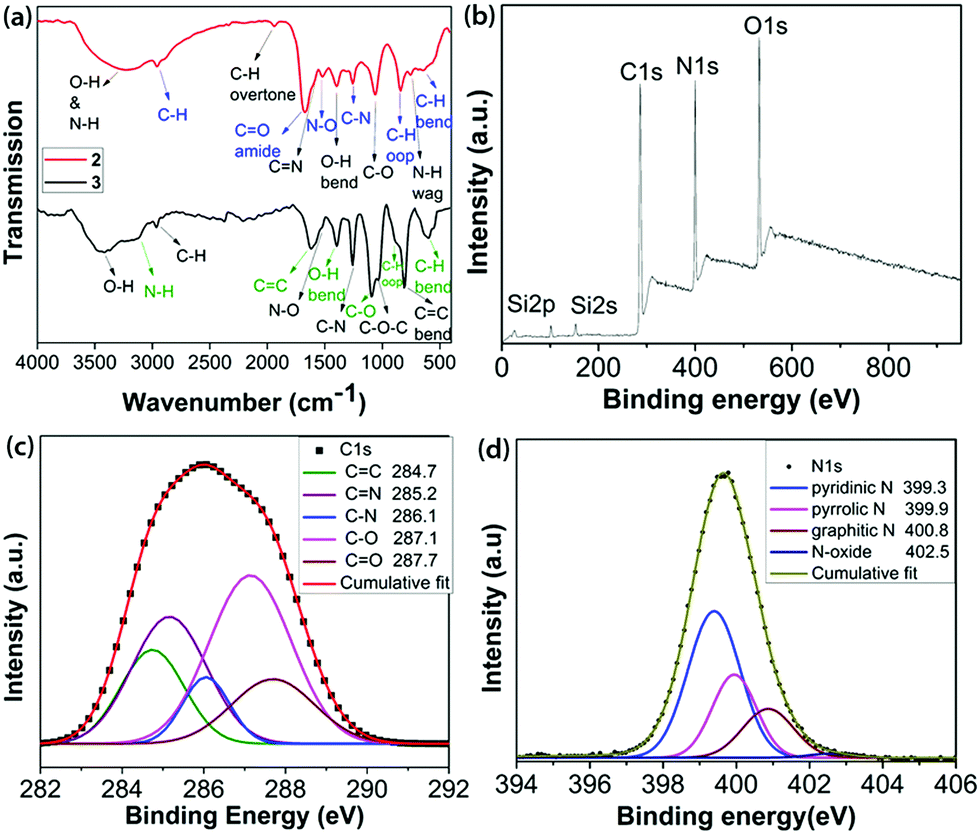 | ||
| Fig. 1 (a) FT-IR spectra of 2 and 3; (b) survey XPS; and high resolution XPS (c) C1s and (d) N1s regions of 2. | ||
FTIR analysis of 2, as shown in Fig. 1a and detailed in the ESI,‡ confirmed the availability of various types of N- and O-containing functional groups. The XPS analysis indicated the presence of 52.50% C, 22.63% N, 19.64% O and 5.23% Si in 2 by weight percentage (Fig. 1b and Table S1, ESI‡). The de-convoluted high resolution XPS spectrum of C1s (Fig. 1c) displays binding energies for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN, C–N, C–O and CO bonding configurations.16 The de-convoluted high resolution O1s spectrum (Fig. S8, ESI‡) shows a small peak at 535.3 eV assigned to N–O. Fitting the N1s spectrum (Fig. 1d) indicated the presence of pyridinic N, pyrrolic N, graphitic and N-oxide. A slight increase in the C
C, CN, C–N, C–O and CO bonding configurations.16 The de-convoluted high resolution O1s spectrum (Fig. S8, ESI‡) shows a small peak at 535.3 eV assigned to N–O. Fitting the N1s spectrum (Fig. 1d) indicated the presence of pyridinic N, pyrrolic N, graphitic and N-oxide. A slight increase in the C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N ratio to 2.32:1 in 2 compared to the ratio present in the initial pyrazine ring core is due to the small amounts of siloxanes present in 2. The C:O ratio of solid 2 is 2.67:1. The high resolution Si2p spectrum (Fig. S9, ESI‡) revealed the presence of cyclic and acyclic siloxanes.17 It is noteworthy that the wt% content of Si decreases substantially from 1 (24.08) to 2 (5.23) due to the formation of volatile siloxanes.18 The overall chemical composition was derived from a combination of XPS, field emission scanning electron microscopy (FE-SEM) and elemental analyses, as mentioned in Table S1 (ESI‡).
:N ratio to 2.32:1 in 2 compared to the ratio present in the initial pyrazine ring core is due to the small amounts of siloxanes present in 2. The C:O ratio of solid 2 is 2.67:1. The high resolution Si2p spectrum (Fig. S9, ESI‡) revealed the presence of cyclic and acyclic siloxanes.17 It is noteworthy that the wt% content of Si decreases substantially from 1 (24.08) to 2 (5.23) due to the formation of volatile siloxanes.18 The overall chemical composition was derived from a combination of XPS, field emission scanning electron microscopy (FE-SEM) and elemental analyses, as mentioned in Table S1 (ESI‡).
The reactivity of 1 in air is different from that reported for 2,3,5,6-tetramethyl-1,4-bis(trimethysilyl)-1,4-dihydropyrazine4 due to the presence of ring C–H bonds. An experiment carried out in our laboratory with the latter under similar conditions to that of 1 led to the spontaneous formation of crystalline tetramethylpyrazine, as reported in the literature,4 which was confirmed using single crystal X-ray diffraction analysis (Table S2 and Fig. S15, ESI‡). Two major types of reactions occur when 1 is exposed to air: (1) oxidative dehydrogenative C–C coupling occurs in the presence of the in situ generated oxidant BTSP and (2) oxidation of the 1,4-dihydropyrazine ring. There is a literature precedence for this kind of reactivity that usually occurs through a radical pathway.19–21 Notably, no reaction was observed when BTSP was added to the pyrazine. Based on a combined understanding of the analytical data, we have proposed a probable structure for 2 (Scheme S1, ESI‡) with three chromophores I–III. These proposed chromophores have been optimized using Gaussian 09, and their time dependent DFT calculations show absorbance wavelength values close to those experimentally obtained for 2 (Fig. S12–S14, ESI‡). Thermogravimetric analysis (Fig. S16, ESI‡) under a flow of N2 revealed that polymer 2 is thermally more stable compared to 1.
Pyrolysis of the intermediate 2 at 700 °C taken in an open crucible within a tube furnace under argon containing 10% H2 led to the formation of a black residue 3 (Scheme 1). 10% dihydrogen was introduced to remove some of the strongly bonded oxygen functionalities and consume the residual oxygen present in the furnace atmosphere, thereby preventing further reaction with the residue.22 No post-carbonization purification was done with 3. Powder X-ray diffraction (PXRD) of 3 (Fig. 2a) showed a broad peak centered at 2θ = 25°, which corresponds to the (002) reflection of graphite along with a very weak signal at ∼43°(101 plane of graphite).23 A left shift of the peak suggests a slightly larger basal plane interlayer distance compared to 0.34 nm observed in pristine graphite, arising due to the heteroatom content in 3.24 The Raman spectrum of 3 (Fig. 2b) was characterized by three bands: D at 1338 cm−1, G at 1573 cm−1 and a broad 2D band at 2636 cm−1, which are typical of chemically derived graphenic materials.16 The presence of an intense D band (ID/IG = 1.73) and the downshift of the G band with respect to pristine graphene indicated the heteroatom content and occurrence of defects in the sp2 carbon network of 3.
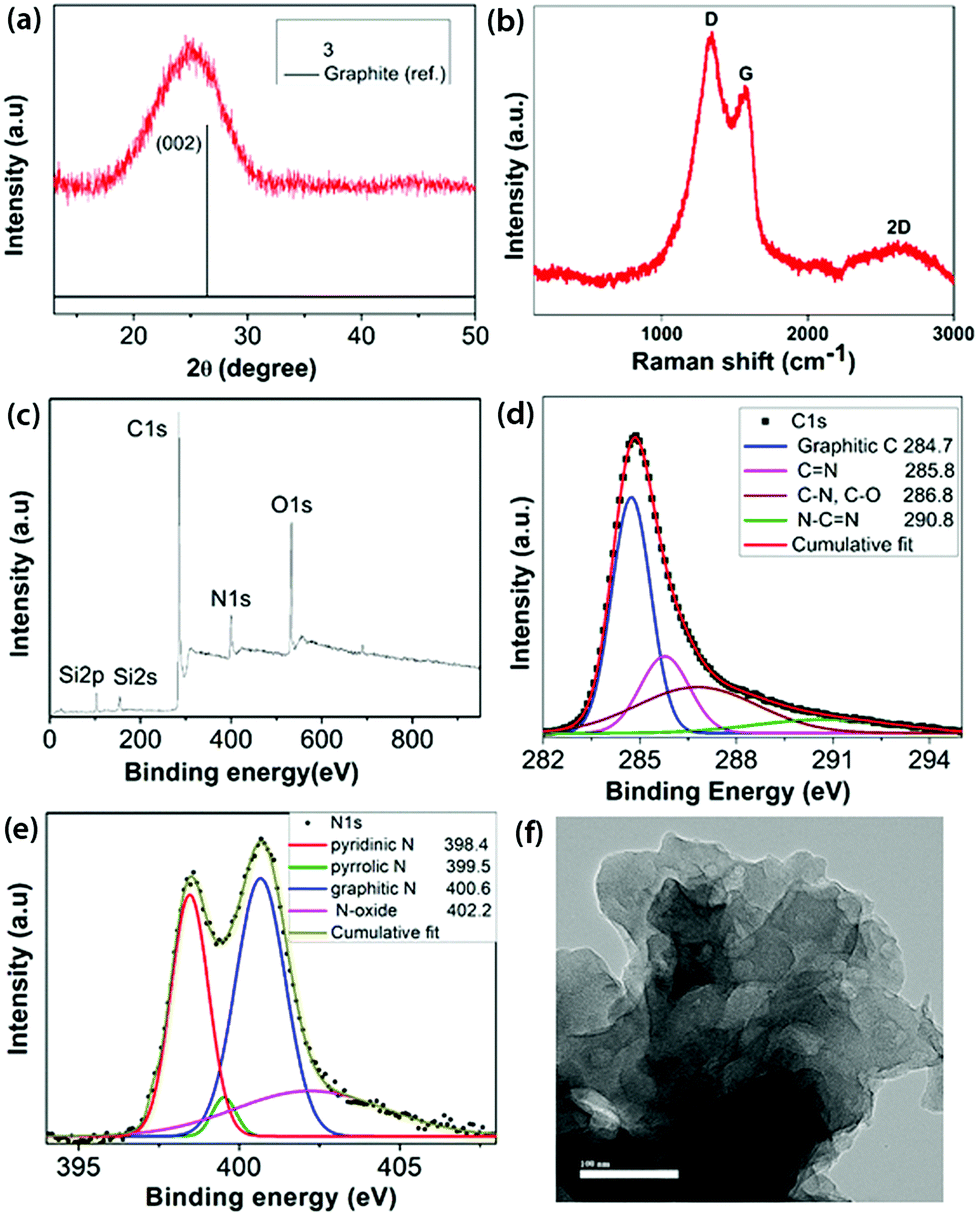 | ||
| Fig. 2 (a) PXRD pattern; (b) Raman spectrum; (c) survey XPS; high resolution XPS (d) C1s and (e) N1s regions; and (f) HRTEM image (100 nm scale bar) of 3. | ||
The XPS survey spectrum of 3 (Fig. 2c) clearly shows a significant reduction in N (10.07 wt%) content and also a reduction in O (12.26 wt%) content with a corresponding increase in its C (73.84 wt%) content. Therefore, the C:N and C:O ratios increased to 7.33 and 6.02, respectively, in 3 compared to 2. The main peak in the fitted high resolution C1s spectrum of 3 (Fig. 2d) was assigned to the graphite-like sp2 C. The other de-convoluted peaks correspond to the binding energies of CN, C–O and NC–N.16 The high resolution O1s spectrum (Fig. S10, ESI‡) was de-convoluted into three components: C–OH (532.6 eV), C–O–C (532.9 eV) and N–O (535.6 eV).25 The high resolution N1s spectrum (Fig. 2e) revealed the N environment in 3, containing 31.6% pyridinic-N, 3.2% pyrrolic-N, 45.9% graphitic-N and 19.3% N–O. The Si content in 3 is majorly present in the form of SiO2, as determined from the high resolution Si2p spectrum (Fig. S11, ESI‡).26 The chemical composition obtained from FE-SEM and CHN analysis echoes the values obtained from XPS analysis (Table S1, ESI‡). The FT-IR spectrum of 3 (Fig. 1a) clearly shows the corresponding new characteristic vibrational modes of CC stretching at 1619 cm−1 and C–O–C stretching at 1038 cm−1.
The high graphitic carbon content in 3 arises from the presence of thermally stable aromatic rings, CC bonds, and fused aromatic moieties in 2, which can undergo cross-linking through thermally assisted dehydrogenation.27 Mass analysis of the evolving gases during thermal decomposition (TG-MS) of 2 in an argon atmosphere confirmed the release of N2/CO, HCN, C2H2, H2O, H2 and volatile cyclic siloxanes (Me2SiO)3 and (Me2SiO)4 (Fig. S17, ESI‡). The oxygen functionalities (C–OH, N–O) present in 2 are more stable and hence were difficult to remove under the carbonization conditions,28 although some were lost as CO and H2O. The loss of volatile siloxane leaves behind a small amount of SiO2 in 3.18 We have proposed a structure for 3 (Scheme S2, ESI‡) taking into consideration its analytical data and the elimination of gaseous small molecules during carbonization.
FE-SEM (Fig. S19, ESI‡) revealed a disordered arrangement of the few layered carbon sheets. This was further confirmed using high resolution transmission electron microscopy (HRTEM) (Fig. 2f and Fig. S20, ESI‡), which shows the sheet-like morphology, however without any obvious ordered lattice fringe, implying poor crystallinity.29 Thus, a N- and O-functionalized carbon material 3 has been obtained by carbonization starting from a N-containing small molecule precursor 1 involving an inexpensive oxidation technique. The process of air oxidation of 1 followed by carbonization established herein is completely reproducible with the same chemical compositions and microstructure obtained each time. It is worth mentioning that 3 is not achievable starting from the commonly available pyrazine due to its reluctance to underdo ring oxidation and subliming nature. Thermogravimetric analysis of 3 in air (Fig. S18, ESI‡) suggested its thermo-oxidative stability up to 400 °C, indicating a relatively ordered graphenic network.30
N-Containing carbon materials are considered to be ideal frameworks exhibiting excellent electrochemical performance in LIBs (Table S3, ESI‡). Additionally, optimal oxygen inclusion is known to improve the electrochemical performance; however, this can be detrimental due to possible oxidation and excessive potential hysteresis (Table S3, ESI‡). Therefore, an anode was fabricated with 3 as the active material using standard fabrication procedures (see ESI‡) and the electrochemical storage capacity was evaluated on the basis of the half-cell configuration of a LIB. Since SiO2 is inactive toward Li31 and present only in very small amounts in 3, no removal efforts were made.
The galvanostatic charge–discharge voltage profile (Fig. 3a) at a current density of 100 mA g−1 gives a high initial capacity of 2072 mA h g−1, which drops to 1071 mA h g−1 in the second cycle. The large irreversible capacity, which is common for carbon materials, can be attributed to the formation of a solid–electrolyte interface (SEI) on the electrode surface.24 The presence of the plateau at around 0.6 V (Fig. 3a) in the first discharge cycle marks the formation of a stable SEI. Li ion intercalation into the graphenic layers primarily led to the capacity in the potential region below 0.5 V.24 However, the absence of distinct potential plateaus suggests electrochemically and geometrically non-equivalent Li ion sites due to the disorder present in 3.24 Reversible capacity was obtained from the second cycle onward with high coulombic efficiency. The cyclic voltammogram (Fig. S21, ESI‡) reflects this reversible electrochemical behaviour with the cathodic peak at 0.2 V due to lithiation and its delithiation counterpart at 0.4 V vs. Li/Li+. The anodic peak at around 1.2 V was attributed to the breaking of bonds of Li with heteroatoms at the defect sites during the charging process.32 The cycling performance of 3 at the current density of 100 mA g−1 (Fig. 3c) depicts a high reversible capacity of 840 mA h g−1 at the end of 150 cycles, indicating good cycling stability. The rate capability of 3 evaluated at different current densities (Fig. 3b) demonstrates the stability and reversibility of 3.
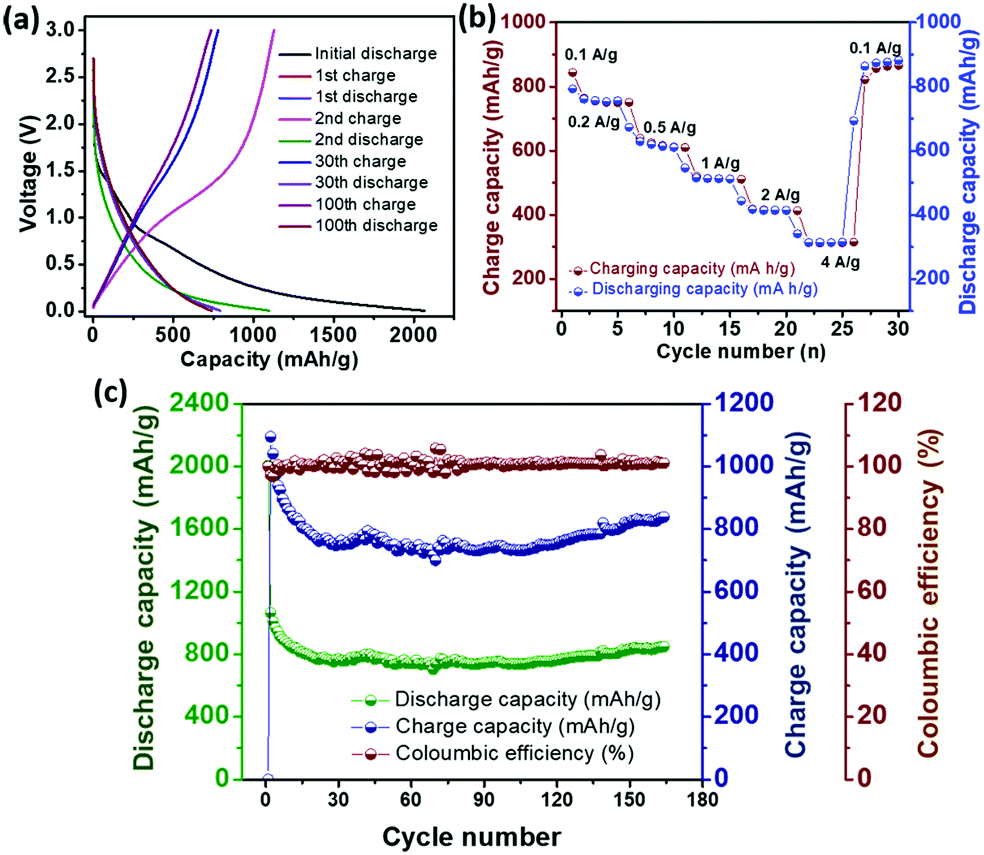 | ||
| Fig. 3 (a) Discharge–charge voltage profile, (b) rate capability and (c) cycling performance of the electrode containing 3 in the half-cell w. r. t. Li/Li+. | ||
The low charge-transfer resistance (RCT = 22 Ω) and the Warburg coefficient related to the diffusion kinetics of Li ions33 obtained from the Nyquist plot (Fig. S22 and S23, ESI‡) further explicate the excellent electrochemical activity of 3. This may arise due to the synergic effect of N and O-containing functional groups present and a favourable degree of graphitization present in 3. Heteroatoms induce defects that provide additional Li+ storage sites, stabilize solid–electrolyte interfaces, offer more channels for Li+ diffusion, and increase electrode/electrolyte wettability and inter-sheet distance,24 thus making 3 a competent and robust (Fig. S24 and S25, ESI‡) electrode material.
To summarize, we have established 1,4-bis(trimethysilyl)-1,4-dihydropyrazine 1 as a small molecule precursor for carbonization to the N,O-functionalized carbon material 3via the formation of a polymeric intermediate 2 upon air exposure. The as-obtained carbonized material 3 showed promising anode performance in a lithium ion battery. The potential of this fundamental research reported here lies in utilizing 2 to prepare functional hybrids and composites and investigating the versatility of this synthetic strategy with other analogous organosilicon reagents with a clear correlation between molecule, carbon material and performance.
M. M. thanks the Department of Science and Technology (DST), India (CRG/2018/000988); M. M. and S. O. thank DST Nano-mission Thematic Unit, India (SR/NM/TP-13/2016) for financial support. M. M. thanks Dr Anil Suryawanshi for his generous help in rate capability measurement and Prof. H. Tsurugi and Prof. K. Mashima, Osaka University, Japan, for scientific discussions. M. M. thanks all reviewers for their insightful comments and suggestions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Tsurugi and K. Mashima, Acc. Chem. Res., 2019, 52, 769 CrossRef CAS.
- M. Majumdar, R. K. Raut, P. Sahoo and V. Kumar, Chem. Commun., 2018, 54, 10839 RSC.
- N. Akula, N. Sharma, A. Lohegaonkar, S. B. Ogale and M. Majumdar, Chem. – Asian J., 2020, 15, 585 CrossRef CAS.
- K. Yamamoto, S. Tanaka, H. Hosoya, H. Tsurugi, K. Mashima and C. Copéret, Helv. Chim. Acta, 2018, 101, e1800156 CrossRef.
- M. Zhong, E. K. Kim, J. P. McGann, S.-E. Chun, J. F. Whitacre, M. Jaroniec, K. Matyjaszewski and T. Kowalewski, J. Am. Chem. Soc., 2012, 134, 14846 CrossRef CAS.
- S. Zhang, K. Dokko and M. Watanabe, Chem. Mater., 2014, 26, 2915 CrossRef CAS.
- M. R. Benzigar, S. N. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680 RSC.
- Z.-Y. Wu, H.-W. Liang, L.-F. Chen, B.-C. Hu and S.-H. Yu, Acc. Chem. Res., 2016, 49, 96 CrossRef CAS.
- E. Gottlieb, K. Matyjaszewski and T. Kowalewski, Adv. Mater., 2019, 31, 1804626 CrossRef.
- L. Borchardt, Q.-L. Zhu, M. E. Casco, R. Berger, X. Zhuang, S. Kaskel, X. Feng and Q. Xu, Mater. Today, 2017, 20, 592 CrossRef CAS.
- J. Liu, T. Zhang, Z. Wang, G. Dawson and W. Chen, J. Mater. Chem., 2011, 21, 14398 RSC.
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87 CrossRef CAS.
- Z.-Y. Wu, S.-L. Xu, Q.-Q. Yan, Z.-Q. Chen, Y.-W. Ding, C. Li, H.-W. Liang and S.-H. Yu, Sci. Adv., 2018, 4, eaat0788 CrossRef CAS.
- K. Liu, J. Zhang, R. Ding, X. Zheng, T. Yang, C. Wang and M. Chen, Carbon, 2019, 155, 326 CrossRef CAS.
- T. Saito, H. Nishiyama, H. Tanahashi, K. Kawakita, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2014, 136, 5161 CrossRef CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781 CrossRef CAS.
- D. W. Niles, H. Höchst, G. W. Zajac, T. H. Fleisch, B. C. Johnson and J. M. Meese, J. Vac. Sci. Technol., A, 1988, 6, 1584 CrossRef CAS.
- N. A. Warner, A. Evenset, G. Christensen, G. W. Gabrielsen, K. Borga and H. Leknes, Environ. Sci. Technol., 2010, 44, 7705 CrossRef CAS.
- L. Wang, W. Sha, Q. Dai, X. Feng, W. Wu, H. Peng, B. Chen and J. Cheng, Org. Lett., 2014, 16, 2088 CrossRef CAS.
- D. J. R. Brook, B. C. Noll and T. D. Koch, J. Org. Chem., 1997, 62, 6767 CrossRef CAS.
- A. Rodrigues, P. M. T. Ferreira and L. S. Monteiro, Tetrahedron, 2004, 60, 8489 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210 CrossRef CAS.
- H. Wang, Z. Xu, A. Kohandehghan, Z. Li, K. Cui, X. Tan, T. J. Stephenson, C. K. King’ondu, C. M. B. Holt, B. C. Olsen, J. K. Tak, D. Harfield, A. O. Anyia and D. Mitlin, ACS Nano, 2013, 7, 5131 CrossRef CAS.
- F. J. Sonia, M. Aslam and A. Mukhopadhyay, Carbon, 2020, 156, 130 CrossRef CAS.
- N. B. Erdal, K. H. Adolfsson, T. Pettersson and M. Hakkarainen, ACS Sustainable Chem. Eng., 2018, 6, 1246 CrossRef CAS.
- M. Ouyang, C. Yuan, R. J. Muisener, A. Boulares and J. T. Koberstein, Chem. Mater., 2000, 12, 1591 CrossRef CAS.
- B. Saha and G. C. Schatz, J. Phys. Chem. B, 2012, 116, 4684 CrossRef CAS.
- T. Konakahara and Y. Takagi, Bull. Chem. Soc. Jpn., 1977, 50, 2734 CrossRef CAS.
- X. N. Sun, D. Xu, W. Hu and X. Y. Chen, ACS Sustainable Chem. Eng., 2017, 5, 5972 CrossRef CAS.
- S.-M. Jung, E. K. Lee, M. Choi, D. Shin, I.-Y. Jeon, J.-M. Seo, H. Y. Jeong, N. Park, J. H. Oh and J.-B. Baek, Angew. Chem., Int. Ed., 2014, 53, 2398 CrossRef CAS.
- W.-S. Chang, C.-M. Park, J.-H. Kim, Y.-U. Kim, G. Jeong and H.-J. Sohn, Energy Environ. Sci., 2012, 5, 6895 RSC.
- Z. Xing, Z. Ju, Y. Zhao, J. Wan, Y. Zhu, Y. Qiang and Y. Qian, Sci. Rep., 2016, 6, 26146 CrossRef CAS.
- J. Kaspar, M. Graczyk-Zajac and R. Riedel, Electrochim. Acta, 2014, 115, 665 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Kazushi Mashima from Osaka University, Japan on his 63rd birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental, characterization and measurement details. See DOI: 10.1039/d0cc06597h |
| This journal is © The Royal Society of Chemistry 2020 |