
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA concise route to MK-4482 (EIDD-2801) from cytidine†
N.
Vasudevan
a,
Grace P.
Ahlqvist
 b,
Catherine P.
McGeough
b,
Dinesh J.
Paymode
a,
Flavio S. P.
Cardoso
a,
Tobias
Lucas
c,
Jule-Phillip
Dietz
c,
Till
Opatz
c,
Timothy F.
Jamison
b,
Frank B.
Gupton
a and
David R.
Snead
*a
b,
Catherine P.
McGeough
b,
Dinesh J.
Paymode
a,
Flavio S. P.
Cardoso
a,
Tobias
Lucas
c,
Jule-Phillip
Dietz
c,
Till
Opatz
c,
Timothy F.
Jamison
b,
Frank B.
Gupton
a and
David R.
Snead
*a
aMedicines for All Institute, 737 N. 5th St., Box 980100, Richmond, VA 23298-0100, USA. E-mail: drsnead@vcu.edu
bDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, Massachusetts 02139, USA
cDepartment of Chemistry, Johannes Gutenberg University, Duesbergweg 10-14, Mainz 55128, Germany
First published on 2nd October 2020
Abstract
A two-step route to MK-4482 (EIDD-2801, 1) was developed consisting of an esterification and hydroxamination of cytidine. The selective acylation and direct amination eliminate the need for protecting and activating groups and proceed in overall yield of 75%, a significant advancement over the reported yield of 17%. The step count is reduced from five transformations to two, and expensive uridine is replaced with the more available cytidine.
Remdesivir has presented itself as an option for COVID-19 treatment; however, improvements upon this initial solution are still desired. Fulfilling demand is complicated by issues related to raw material supply,1a price,1b and synthetic route length.2 MK-4482 (EIDD-2801)3 presents an interesting complement to remdesivir for COVID-19 treatment. It is structurally simple in comparison, and it can likely be made from abundant raw materials. These factors would be expected to alleviate supply chain difficulties and reduce costs. Encouragingly, MK-4482 shows potential to treat mice with remdesivir resistant strains of COVID-19, and the active pharmaceutical ingredient (API) is orally bioavailable. These advantages prompted Merck to license the drug candidate from Ridgeback Biotherapeutics.4
The initial disclosure of MK-4482 is the only synthesis which appears in the open literature (Fig. 1), and not unexpectedly there are significant opportunities for improvement over this early route:5
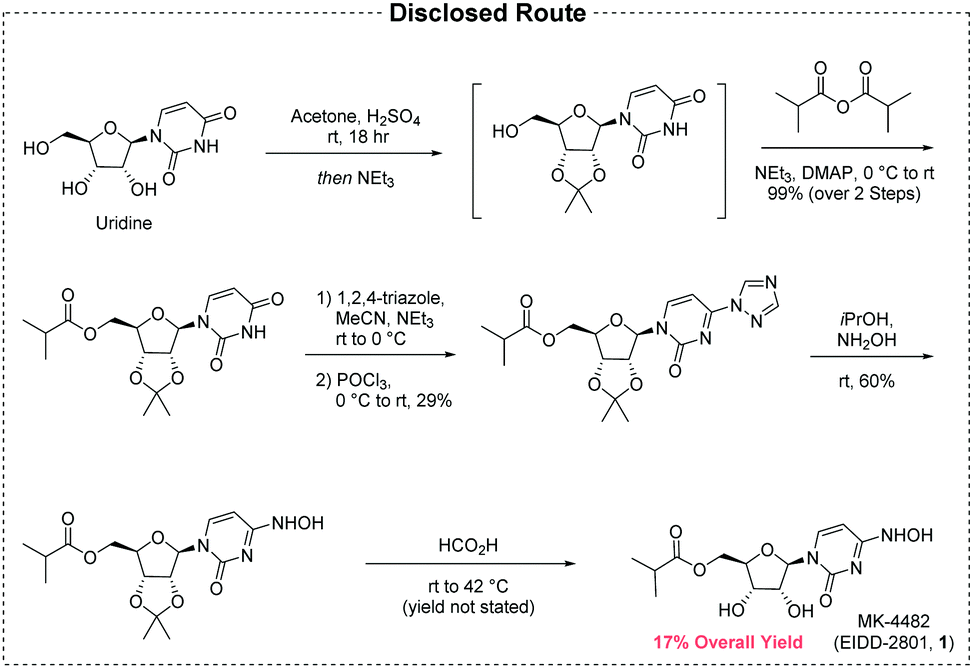 | ||
| Fig. 1 The first generation route to MK-4482 from uridine. | ||
• The API is constructed over five chemical transformations.
• The route suffers from low yield (17% maximum, yield of diol deprotection not disclosed).
• The step count is lengthened by derivatizations and protections.
• Uridine, an expensive material of limited availability, is the synthetic starting point.
Building the API from cytidine instead of uridine presents several advantages. First, raw material costs can be decreased because cytidine is ∼40% of the price of uridine.6 Secondly, there is potential to reduce the synthesis to a two-step sequence comprising esterification and transamination (Fig. 2).
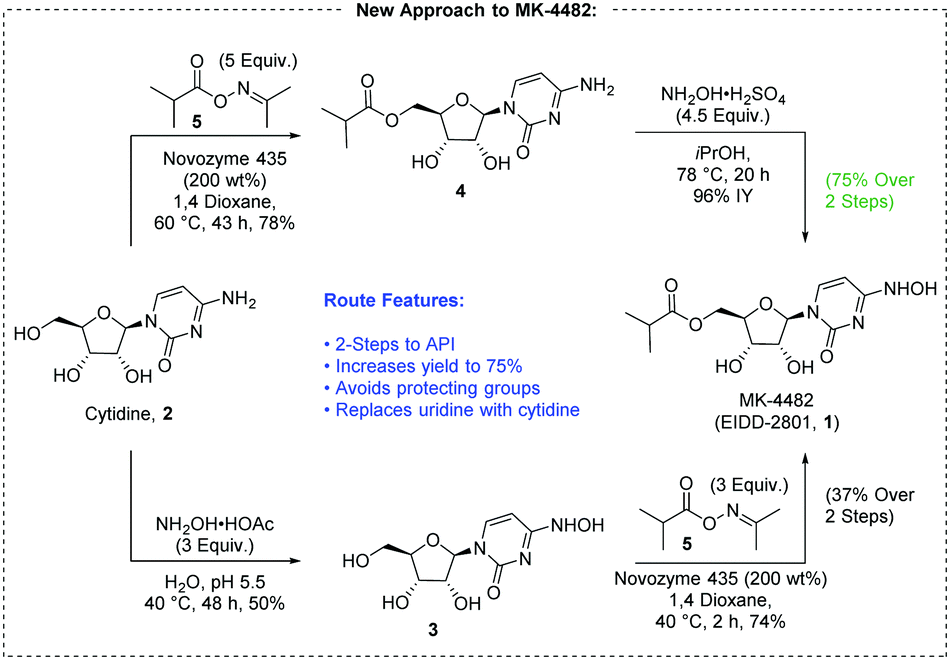 | ||
| Fig. 2 A new route to MK-4482 from cytidine. | ||
We began our exploration by examining direct transamination of cytidine with hydroxylamine.7 Older literature studies suggested that mono-hydroxamination can be achieved under the right concentration, temperature and pH, while minimizing over-reaction of the substrate.7a,b More recently this finding was repeated by Purohit with preparative HPLC separation7c while Painter claimed difficulties with the procedure leading to 20% yield.7d In our hands, with slight adjustment of the reaction conditions, N(4)-hydroxycytidine (NHC, 3) was synthesized in 70% assay yield (AY). Importantly, upon concentration, pure NHC was obtained by simple crystallization directly from the reaction mixture in 50% isolated yield.
We also explored transamination of cytidine isobutyryl ester 4, and surprisingly, with the use of NH2OH·H2SO4 in iPrOH, dihydroxamination was avoided completely. We were quite pleased to find that the ester remained intact. MK-4482 was obtained from 4 in 96% isolated yield (IY), demonstrating viability of direct hydroxamination from either cytidine reaction pathway.
Selective acylation remained as the largest technical uncertainty toward production of a shorter, protecting group free route to MK-4482. The esterification of NHC 3 would need to be selective for one of four hydroxyl groups, and the literature suggests that the N-hydroxy group is most reactive toward acylation by chemical means.7d,8 Enzyme catalyzed esterification of cytidine has achieved this goal by making use of vinyl esters and anhydride acyl donors.9 The use of oxime ester transfer agents was of particular interest due to the structural similarity with N-hydroxycytidine, 3, and excellent selectivity was observed with uridine though cytidine was unfortunately reported to give the O,N-diacylated product.10 We were curious whether this approach would work to form the desired α-branched esters.
Surprisingly immobilized CALB (Candida Antarctica Lipase B) provided the desired selectivity not only for N-hydroxycytidine but also for cytidine. Isobutyric oxime ester 5 was used as the acyl transfer agent with solid supported enzyme (200 wt%, 1.5 mol%). A sufficient excess of the oxime ester was necessary to drive the reaction to completion, and early results have been best with 1,4-dioxane. MK-4482 was isolated in 74% yield from 3, and 4 was isolated in 78% yield from cytidine. A traditional chemically catalyzed acylation was developed to provide a non-enzymatic option to reach 4.11 Though inexpensive this option might not be preferable to the enzymatic route. More reagents are added to the reaction system and a greater number of byproducts are formed which might hamper efforts to purify the intermediate at scale. Conversion was stopped at 90% to halt over-acylation of the product, resulting in an isolated yield of 76% of 4. Similarly, an alternative route to 3 was developed from uridine.11
This completes two concise routes to MK-4482, which differ in the order of synthetic transformations. When conducting esterification first, MK-4482 is obtained in 75% yield, and it is made in 37% yield when hydroxyamination is conducted first. The step count is reduced from five transformations to two, and the more expensive uridine is replaced with cytidine. The use of protecting groups and derivatization is eliminated. We plan to further report on the optimization of this preliminary result to refine catalyst loadings, solvent selection, and yield while developing process-amenable isolation sequences.
We thank the Bill and Melinda Gates Foundation for their longstanding support of our research. Grace Ahlqvist would also like to acknowledge support by the National Science Foundation Graduate Research Fellowship under Grant No. 1745302.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) An Update on COVID-19 from our Chairman & CEO, Gilead Sciences, Inc., accessed July 1, 2020, https://stories.gilead.com//articles/an-update-on-covid-19-from-our-chairman-and-ceo; (b) An Open Letter from Daniel O’Day, Chairman & CEO, Gilead Sciences, Gilead Sciences, Inc, accessed July 1, 2020, https://www.gilead.com/news-and-press/press-room/press-releases/2020/6/an-open-letter-from-daniel-oday-chairman-ceo-gilead-sciences.
- (a) L. Zhang, S. Neville, E. Carra, W. Lew, B. Ross, Q. Wang, L. Wolfe, R. Jordan, V. Soloveva, J. Knox, J. Perry, M. Perron, K. M. Stray, O. Barauskas, J. Y. Feng, Y. Xu, G. Lee, A. L. Rheingold, A. S. Ray, R. Bannister, R. Strickley, S. Swaminathan, W. A. Lee, S. Bavari, T. Cihlar, M. K. Lo, T. K. Warren and R. L. Mackman, J. Med. Chem., 2017, 60, 1648 CrossRef; (b) C. De Savi, D. L. Hughes and L. Kvaerno, Org. Process Res. Dev., 2020, 24, 940 CrossRef CAS; (c) T. Vieira, A. C. Stevens, A. Chtchemelinine, D. Gao, P. Badalov and L. Heumann, Org. Process Res. Dev., 2020 DOI:10.1021/acs.oprd.0c00172; (d) F. Xue, X. Zhou, R. Zhou, X. Zhou, D. Xiao, W. Zhong, E. Gu, W. Guo, J. Xiang, K. Wang, L. Yang and Y. Qin, Org. Process Res. Dev., 2020, 24, 1772–1777 CrossRef CAS.
- (a) T. P. Sheahan, A. C. Sims, S. Zhou, R. L. Graham, A. J. Pruijssers, M. L. Agostini, S. R. Leist, A. Schafer, K. H. Dinnon III, L. J. Stevens, J. D. Chappel, X. Lu, T. M. Hughes, A. S. George, C. S. Hill, S. A. Montgomery, A. J. Brown, G. R. Bluemling, M. G. Natchus, M. Saindane, A. A. Kolykhalov, G. Painter, J. Harcourt, A. Tamin, N. J. Thornburg, R. Swanstrom, M. R. Denison and R. S. Baric, Sci. Transl. Med., 2020, 12, 1–15 Search PubMed; (b) B. Halford, An emerging antiviral takes aim at COVID-19, Chem. Eng. News, 2020, 98, 22 CAS.
- R. Cross, Merck & Co. joins race for COVID-19 vaccines and therapies, Chem. Eng. News, 2020, 98, 12 Search PubMed.
- (a) G. R. Painter, G. R. Bluemling, M. G. Natchus and D. Guthrie, WO2019113462, 2018; (b) G. R. Painter, D. Perryman and G. R. Bluemling, WO2019173602, 2019.
- Prices obtained through analysis of Indian import/export records.
- (a) D. W. Verwoerd, H. Kohlhage and W. Zillig, Nature, 1961, 192, 1038–1040 CrossRef CAS; (b) N. K. Kochetkov and E. I. Budovskii, Organic Chemistry of Nucleic Acids, Part B, Plenum Publishing Corporation, New York, 1972, pp. 269–348 Search PubMed; (c) M. K. Purohit, E. Poduch, L. W. Wei, I. E. Crandall, T. To, K. C. Kain, E. F. Pai and L. P. Kotra, J. Med. Chem., 2012, 55, 9988 CrossRef CAS; (d) G. R. Painter, D. B. Guthrie, G. R. Bluemling and M. G. Natchus, WO2016106050A1, 2016.
- (a) M. A. Ivanov, E. V. Antonova, A. V. Maksimov, L. K. Pigusova, E. F. Belanov and L. A. Aleksandrova, Collect. Czech. Chem. Commun., 2006, 71, 1099 CrossRef CAS; (b) R. F. Schinazi, F. Amblard, B. D. Cox, L. Bassit, L. Zhou and C. Gavegnano, WO2017165489, 2016.
- (a) C. H. Wong, S. T. Chen, W. J. Hennen, J. A. Bibbs, Y. F. Wang, J. L. C. Liu, M. W. Pantoliano, M. Whitlow and P. N. Bryan, J. Am. Chem. Soc., 1990, 112, 945 CrossRef CAS; (b) X.-F. Li, M.-H. Zong and R.-D. Yang, J. Mol. Catal. B: Enzym., 2006, 38, 48–53 CrossRef CAS; (c) H. Wu, M. Zong and X. Chen, Biotechnol. Appl. Biochem., 2009, 53, 201 CAS; (d) R. Kumar, M. Kumar, J. Maity and A. K. Prasad, RSC Adv., 2016, 6, 82432 RSC.
- F. Moris and V. Gotor, J. Org. Chem., 1993, 58, 653 CrossRef CAS.
- See ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc05944g |
| This journal is © The Royal Society of Chemistry 2020 |