
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe first ring-expanded NHC–copper(I) phosphides as catalysts in the highly selective hydrophosphination of isocyanates†
Thomas M.
Horsley Downie
 a,
Jonathan W.
Hall
a,
Thomas P.
Collier Finn
a,
David J.
Liptrot
*a,
John P.
Lowe
a,
Mary F.
Mahon
*b,
Claire L.
McMullin
a and
Michael K.
Whittlesey
a
a,
Jonathan W.
Hall
a,
Thomas P.
Collier Finn
a,
David J.
Liptrot
*a,
John P.
Lowe
a,
Mary F.
Mahon
*b,
Claire L.
McMullin
a and
Michael K.
Whittlesey
a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: dl260@bath.ac.uk
bX-Ray Crystallography Suite, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: m.f.mahon@bath.ac.uk
First published on 1st October 2020
Abstract
A range of N-heterocyclic carbene-supported copper diphenylphosphides (NHC = IPr, 6-Dipp, SIMes and 6-Mes) were synthesised. These include the first reports of ring-expanded NHC–copper(I) phosphides. The compounds were characterised by NMR spectroscopy and X-ray crystallography. Reaction of (6-Dipp)CuPPh2 with isocyanates, isothiocyanates and carbon disulfide results in the insertion of the heterocumulene into the Cu–P bond. The NHC–copper phosphides were found to be the most selective catalysts yet reported for the hydrophosphination of isocyanates. They provide access to a broad range of phosphinocarboxamides in excellent conversion and good yield.
The exploitation of metal phosphides is a widespread route to organophosphorus compounds.1–4 Beyond their stoichiometric application, catalytic intermediates of the form LnMPR2 have been integral to a green revolution in organophosphorus chemistry, especially when the metals concerned are Earth-abundant first row transition metals (i.e., Fe, Co). These mid-d-block systems, however, are prone to redox-switching and single-electron transfer reactions which, coupled with the stability of phosphorus centred radicals, often result in undesirable side reactions.5–7
The propensity of copper(I) towards the well-defined isohypsic reaction steps of σ-bond metathesis, and insertion of unsaturated substrates should allow copper(I) phosphides to overcome such issues. Despite this appeal, copper phosphides often form only as complex oligomers, with a concomitant reduction in reactivity.8–17 Low-coordinate copper phosphides supported by sterically demanding ligands are far less common, as evidenced by the small number of structurally characterised (NHC)CuPR2 (NHC = N-heterocyclic carbene) complexes hitherto reported.18 Despite their relative rarity, such species hint at the potential of low-molecularity copper(I) phosphides. For example, [(NHC)CuPPh2]3 complexes (NHC = IiPr, 1,3-bis(isopropyl)-imidazol-2-ylidene; ItBu, 1,3-bis(tert-butyl)-imidazol-2-ylidene), mediate the hydrophosphination of terminal alkynes at elevated temperature.19
In contrast to the formation of alkyl and alkenyl phosphines from alkenes and alkynes, hydrophosphination of isocyanates has been far less well-studied. Often, significant competing side-reactivity6 or poor substrate scope20 hamper access to, and thus exploitation of, phosphinocarboxamide products. These products are potential synthetic intermediates and, particularly, ligands owing to their polydentate nature comprising both hard and soft donor atoms.21–23
Ring expanded N-heterocyclic carbenes (RE-NHCs) can offer increased stability and reduced molecularity to reactive metal species through their significant steric demand.24–26 RE-NHC–copper(I) complexes such as copper(I) halides and copper(I) alkoxides have been established as having enhanced catalytic activity over their five-membered ring counterparts.27–29
We thus set out to synthesise and characterise a range of low-coordinate copper phosphide species supported by both 5- and 6-membered NHCs in order to evaluate their potential as catalytic intermediates. Herein we report the rational synthesis of four (NHC)CuPPh2 compounds (NHC = IPr, 6-Dipp, SIMes and 6-Mes), including the first examples of RE-NHC copper phosphides. These compounds were fully characterised by NMR spectroscopy and X-ray crystallography and investigated for their onward reactivity towards heterocumulenes. This access to well-defined putative reaction intermediates allows us to report the most selective catalytic hydrophosphination of isocyanates found to date.
Addition of 1 equivalent of Ph2PSiMe3 to a colourless benzene solution of (IPr)CuOtBu resulted in the instantaneous appearance of a yellow colouration and the emergence of new peaks in the 1H and 31P NMR spectra. (IPr)CuPPh2, 1, was isolated from this solution (Scheme 1) in 81% yield, and characterised by the presence of a 31P{1H} NMR resonance at δ −26.1 ppm, consistent with the data previously reported by Nolan and co-workers.30 Application of these conditions to (NHC)CuOtBu (NHC = 6-Dipp, SIMes and 6-Mes) yielded (6-Dipp)CuPPh2, 2, as a yellow solid, as well as (SIMes)CuPPh2, 3, and (6-Mes)CuPPh2, 4, as orange solids (Scheme 1). Compounds 2, 3 and 4 each exhibit a single 31P{1H} NMR resonance at δ −23.8, −32.0 and −23.5 ppm respectively. The presence of peaks attributed to tBuOSiMe3 in the 1H NMR spectra of each of these reactions was interpreted as an indication that the formation of compounds 1–4 occurs via a Cu–O/P–Si σ-bond metathesis.
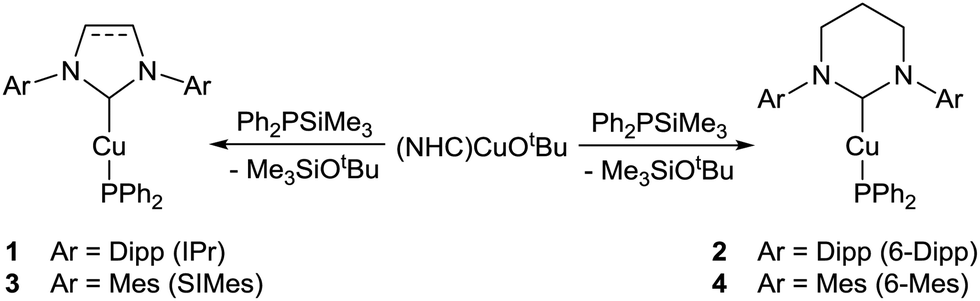 | ||
| Scheme 1 Synthesis of (NHC)CuPPh2 complexes. | ||
Crystals suitable for single crystal X-ray diffraction analysis of 1–4 were grown from benzene/hexane solutions at room temperature. Compounds 1 (Fig. S1, ESI†) and 2 (Fig. 1a), featuring bulky diisopropylphenyl groups on the flank of the NHC, exhibit monomeric structures with a near-linear geometry at the copper centre (1: C1–Cu1–P1 177.09(6)°; 2: C1–Cu1–P1 177.51(6)°). Compounds 3 (Fig. 1b) and 4 (Fig. S2, ESI†), containing less sterically-encumbering mesityl groups, adopt dimeric crystal structures in which the copper centres are three-coordinate, bonding to two bridging phosphorus atoms to give approximately rhomboid four-membered rings (3: P1–Cu1–P1i 88.712(16)°, Cu1–P1–Cu1i 91.286(16)°; 4: P1–Cu1–P2 85.61(3)°, Cu1–P2–Cu2 94.66(3)°). Previous reports of dimeric NHC–copper(I) complexes supported by less bulky NHCs feature short Cu⋯Cu distances of 2.23–2.31 Å, which were interpreted as indicating the presence of copper–copper interactions. The longer Cu⋯Cu interatomic distances of compounds 3 (3.3502(7) Å) and 4 (3.4852(5) Å) suggest there is no significant interaction between the metal centres in these two cases. Comparatively, the only other structurally characterised (NHC)CuPPh2, [(ItBu)CuPPh2]3, adopts a trimeric structure in the solid state.19
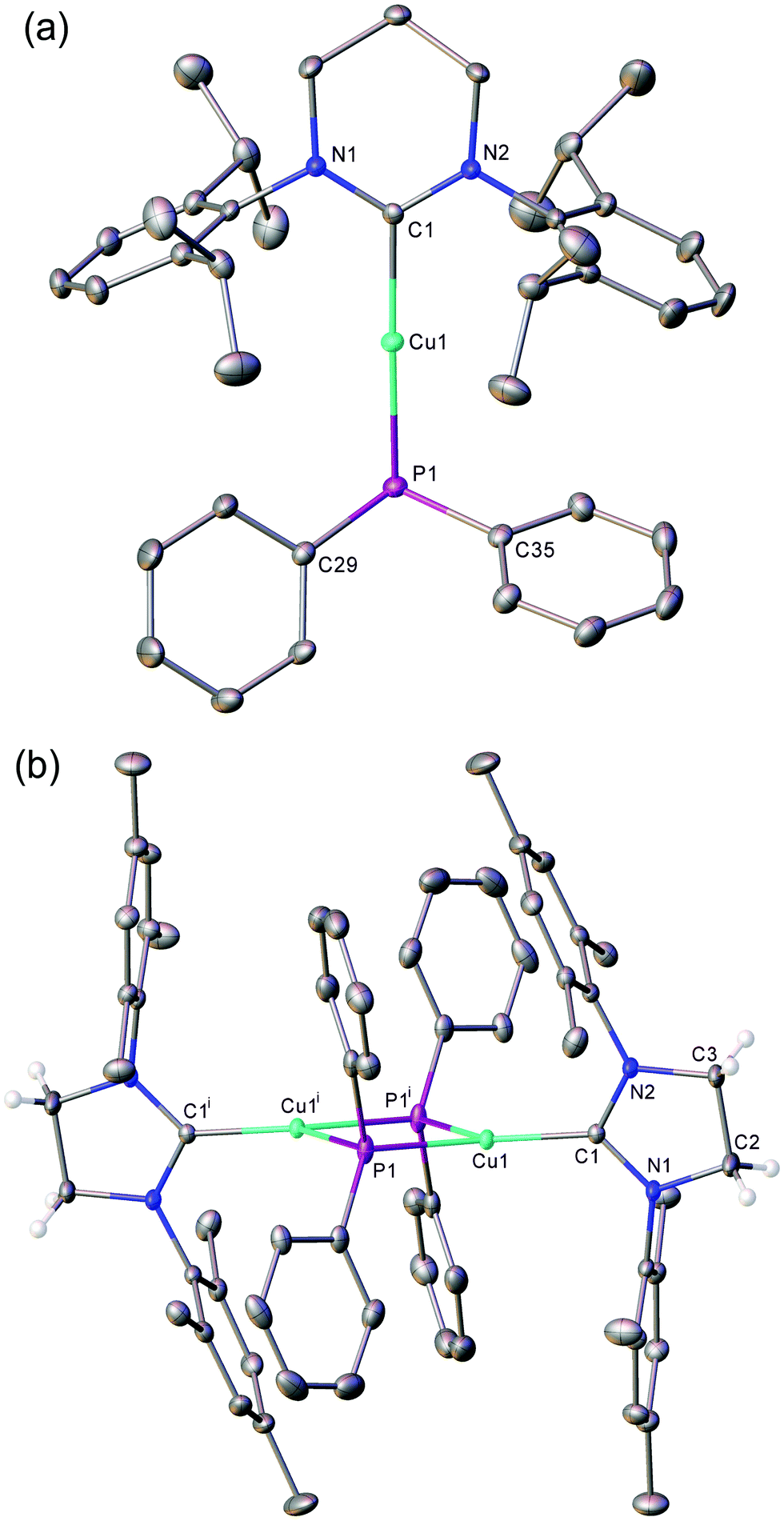 | ||
| Fig. 1 ORTEP representations (30% probability ellipsoids) of (a) compound 2 and (b) compound 3. Hydrogen atoms except those on the NHC backbone of 3 are omitted for clarity. Selected bond lengths (Å) and angles (°): (2) Cu1–P1 2.2113(5), Cu1–C1 1.9272(18), P1–C29 1.835(2), P1–C35 1.836(2), C1–Cu1–P1 177.51(6), Cu1–P1–C29 107.31(6), Cu1–P1–C35 102.92(6), C29–P1–C35 105.12(9); (3) Cu1–P1 2.3298(5), Cu–P1i 2.3558(5), Cu1–C1 1.9234(15), P1–C22 1.824(2), P1–C28 1.8208(18), C1–Cu1–P1 137.59(5), C1–Cu1–P1i 133.14(4) P1–Cu1–P1i 88.712(16) Cu1–P1–Cu1i 91.286(16), Cu1–P1–C22 110.55(5), Cu1–P1–C28 121.30(6). | ||
The Cu–CNHC bond distances of compounds 1–4 range between 1.90–1.97 Å. The monomeric complexes, 1 (1.9037(17) Å) and 2 (1.9272(18) Å), possess shorter bonds than in 3 (1.9234(15) Å) and 4 (1.969(3), 1.965(3) Å), an observation attributed to the increased steric demand provided by two bridging phosphides. These compare with the even longer Cu–CNHC bonds of 1.990–1.997 Å observed in trimeric [(ItBu)CuPPh2]3.19 Complexes 2 and 4, featuring ring-expanded NHCs, have noticeably longer Cu–CNHC bonds than 1 and 3 respectively, likely due to the enhanced steric demand of the flanking aryl groups resulting from the increased N–C–N bond angle of the NHC. These trends are conserved in the Cu–P bond distances which are longer for the RE-NHC containing compounds, 2 and 4, compared to their respective 5-membered counterparts, 1 and 3, (1: 2.2079(5) Å, versus2: 2.2113(5) Å; 3 2.3298(5) Å, versus4 2.3803(9), 2.3908(8) Å) and wherein the dimeric species 3 and 4 show greater Cu–P distances than the monomeric compounds, 1 and 2.
In order to investigate the persistence of the observed solid-state structures in solution, the compounds were interrogated by diffusion ordered 1H NMR spectroscopy (DOSY). Compounds 1 and 2 were found to have hydrodynamic radii that corresponded to their crystallographically derived monomeric structures (1: rDOSY = 5.40 Å, rcomp = 5.31 Å; 2: rDOSY = 5.27 Å, rcomp = 5.52 Å). The hydrodynamic radius observed by DOSY for compound 3 showed a close correlation with that calculated for its dimeric structure seen in the solid state (3: rDOSY = 6.03 Å, rcomp = 6.50 Å). This interpretation is corroborated by the low-temperature (235 K) 13C{1H} NMR spectrum of 3 which shows a broad triplet for the carbenic carbon from a 2J13C–31P coupling to the two bridging but equivalent phosphorus atoms. Compound 4, however, was found to have a hydrodynamic radius that was more consistent with it existing in solution as a monomer, not the dimer observed in the solid state (4: rDOSY = 5.29 Å, rcomp = 5.21 Å for monomer, rcomp = 6.55 Å for dimer).
Addition of a slight excess of isopropyl isocyanate to a yellow C6D6 solution of 2 resulted in a loss of colour. Analysis by 31P{1H} NMR spectroscopy revealed the attenuation of the resonance assigned to 2, and the appearance of a single major resonance at δ 2.2 ppm. Comparison to the 31P{1H} NMR signal of δ −4.0 ppm observed for the free phosphaurea Ph2PC(O)NH(iPr) was interpreted as implying that insertion of the isocyanate into the Cu–P bond had occurred (Scheme 2).6 The corresponding 1H NMR spectrum exhibited a new set of peaks consistent with a complex incorporating the 6-Dipp ligand and the isopropyl isocyanate.
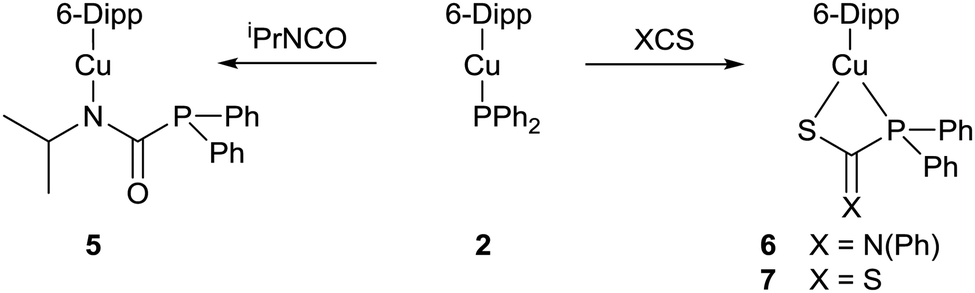 | ||
| Scheme 2 Reactivty of (6-Dipp)CuPPh2, 2, with iPrNCO, PhNCS and CS2. | ||
The formation of (6-Dipp)Cu[N(iPr)C(O)PPh2], 5, was confirmed by X-ray diffraction analysis, performed on colourless single crystals which grew from the reaction solution when left to stand at room temperature (Fig. 2). In contrast to previously reported phosphaureates which comprise a κ2-(N,O) coordination mode and delocalised π-bonding,21–235 features κ1-coordination of the nitrogen to the copper atom. This results in a relatively short Cu–N bond length (1.9053(11) Å), with C–N (1.3314(19) Å) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1.2383(17) Å) bond lengths consistent with localised single and double bonds respectively. Evidence for the persistence of this bonding mode in solution can be inferred from the 31P{1H} NMR signal at 2.2 ppm. This value is significantly downfield of known Zr(IV) and Y(III) compounds which feature a delocalised κ2-(N,O) bonding mode in the solid state and solution 31P{1H} NMR spectra containing resonances at δ −12.1 and −20.1 ppm respectively.21,22
O (1.2383(17) Å) bond lengths consistent with localised single and double bonds respectively. Evidence for the persistence of this bonding mode in solution can be inferred from the 31P{1H} NMR signal at 2.2 ppm. This value is significantly downfield of known Zr(IV) and Y(III) compounds which feature a delocalised κ2-(N,O) bonding mode in the solid state and solution 31P{1H} NMR spectra containing resonances at δ −12.1 and −20.1 ppm respectively.21,22
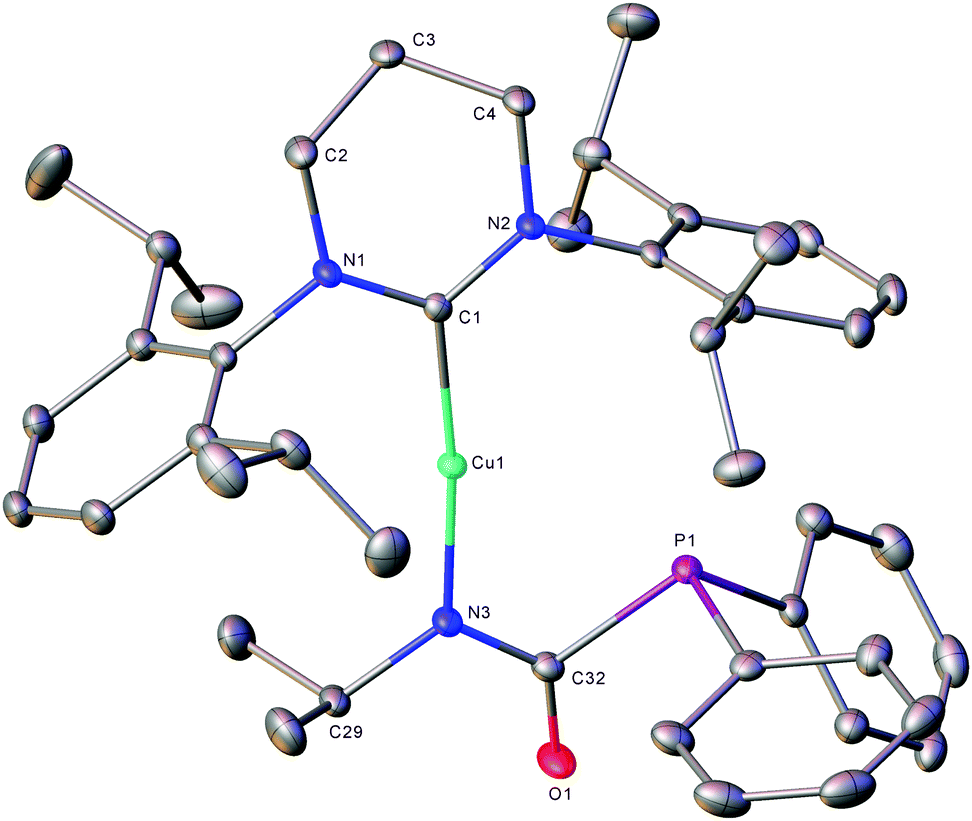 | ||
| Fig. 2 ORTEP representations (30% probability ellipsoids) of compound 5. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cu1–N3 1.9053(11), Cu1–C1 1.9283(13), N3–C29 1.4871(18), N3–C32 1.3314(19), O1–C32 1.2383(17), P1–C32 1.8826(14), C1–Cu1–N3 173.90(5), Cu1–N3–C29 124.19(9), Cu1–N3–C32 120.92(9), N3–C32–O1 128.34(13), N3–C32–P1 109.84(9), O1–C32–P1 121.76(11). | ||
The facile insertion of an isocyanate into the Cu–P bond of 2 prompted a study of other electrophiles. Reactions with excesses of phenyl isothiocyanate and carbon disulfide resulted in insertion of the respective heterocumulene into the Cu–P bond to give new compounds, 6 and 7 (Scheme 2). X-Ray diffraction analysis performed on colourless crystals of 6 (Fig. S3, ESI†) and red crystals of 7 (Fig. S4, ESI†) reveal that the copper centre is bonded to the sulfur atom in both cases. In contrast to the phosphaureate 5, the phosphorus atoms in compounds 6 and 7 also coordinate to the metal centre resulting in CuSCP four-membered rings in both cases. The 31P{1H} NMR spectra of 6 and 7 each reveal a single major resonance, at δ 28.8 and 45.2 ppm respectively.
Addition of 1 equivalent of diphenylphosphine to a colourless solution of compound 5 resulted immediately in yellow colouration, and inspection of the 31P{1H} NMR spectrum suggested that Ph2PC(O)NH(iPr) had been liberated with the regeneration of complex 2. These stoichiometric reactions could be assembled into a catalytic manifold (Fig. 3) to hydrophosphinate a range of isocyanates with diphenylphosphine in C6D6 in the presence of 1 mol% of 1–4 (Table 1).
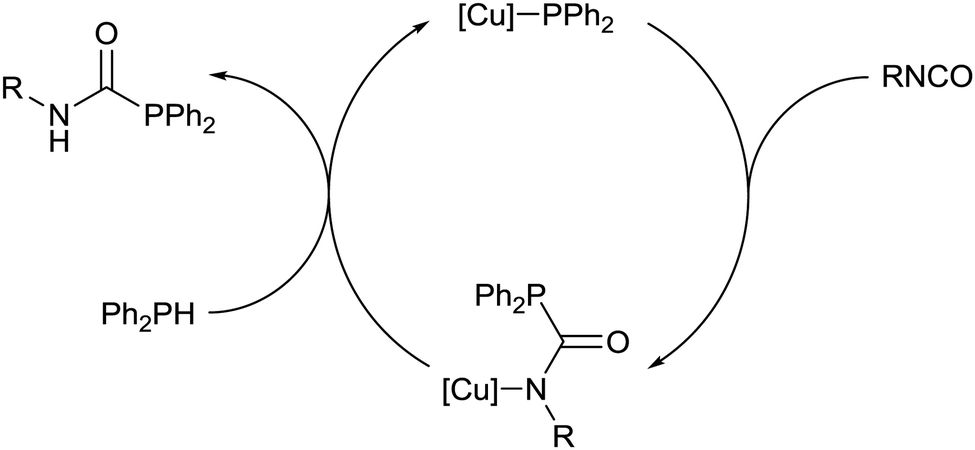 | ||
| Fig. 3 Proposed mechanism for the copper-catalysed hydrophosphination of isocyanates. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Catalyst | Time (h) | Conversiona (%) |
I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :II :II |
| a Conversions based on relative 1H and 31P NMR spectroscopic integrations of starting material and product. b Isolated yields of I from a preparative scale reaction in toluene, see ESI. | |||||
| 1 | iPr | 1 | 0.5 | 90 | 100:0 |
| 2 | 2 | 5 | 96 | 100:0 |
|
| 3 | 3 | 0.5 | >99 (70)b | 100:0 |
|
| 4 | 4 | 0.5 | >99 | 100:0 |
|
| 5 | t Bu | 1 | 2 | 94 | 100:0 |
| 6 | 2 | 4 | 81 | 100:0 |
|
| 7 | 3 | 2 | 91 (67)b | 100:0 |
|
| 8 | 4 | 2 | 96 | 100:0 |
|
| 9 | Ph | 1 | 0.5 | >99 | 100:0 |
| 10 | 2 | 0.5 | 48 | 92:8 |
|
| 11 | 3 | 0.5 | >99 (77)b | 95:5 |
|
| 12 | 4 | 0.5 | >99 | 100:0 |
|
| 13 | Cy | 3 | 0.5 | >99 (79)b | 100:0 |
| 14 | 4 | 0.5 | >99 | 100:0 |
|
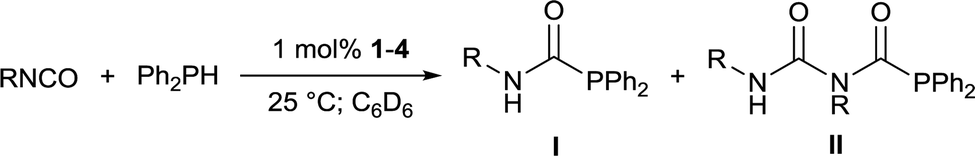
Whilst catalyst-free hydrophosphination of isocyanates can be conducted under strictly neat conditions, the reactivity was limited to a range of small aromatic isocyanates;20 cyclohexyl isocyanate exhibited no reactivity under such conditions. In contrast, 1 mol% of complexes 3 and 4 brought about full conversion of both cyclohexyl and isopropyl isocyanate within 30 minutes, and even the very sterically-congested tert-butyl isocyanate reached excellent conversions (>90%) within 2 hours. 2 was found to catalyse the reaction more slowly than 1, 3, and 4, attributable to the greater steric demands of the 6-Dipp ligand.
In contrast to the iron-catalysed hydrophosphination of isocyanates,6 only very limited amounts of double insertion of the NC bond were observed for phenyl isocyanate when catalysed by 2 or 3, and none was observed when 1 or 4 were the precatalyst. In all other cases, the reaction was specific for single insertion, and in no case was the formation of Ph2PPPh2 observed.
In conclusion, the synthesis of a series of NHC–copper(I) phosphides was accomplished by reaction of alkoxide precursors with Ph2PSiMe3. Complexes 1–4, including the first reports of RE-NHC copper(I) phosphides (compounds 2 and 4), were found to be either monomeric or dimeric in the solid- and solution-state depending on the steric bulk of the NHC ligand. Reaction of 2 with isocyanates, isothiocyanates and carbon disulfide resulted in the insertion of the heterocumulene into the Cu–P bond to give compounds 5–7. Compounds 1–4 were found to be highly active and selective precatalysts for the hydrophosphination of isocyanates. We continue to investigate the nature and scope of this catalytic reactivity, including its application in the hydrophosphination of other heterocumulenes.
This research made use of the Balena High Performance Computing (HPC) Service at the University of Bath. DJL thanks the Royal Society for the support of a University Research Fellowship. We acknowledge financial support from the EPSRC Centre for Doctoral Training in Catalysis (EP/L016443/1) for JWH.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Greenberg and D. W. Stephan, Chem. Soc. Rev., 2008, 37, 1482–1489 RSC.
- L. Rosenberg, ACS Catal., 2013, 3, 2845–2855 CrossRef CAS.
- V. Koshti, S. Gaikwad and S. H. Chikkali, Coord. Chem. Rev., 2014, 265, 52–73 CrossRef CAS.
- D. S. Glueck, J. Org. Chem., 2020 DOI:10.1021/acs.joc.0c00667.
- A. K. King, A. Buchard, M. F. Mahon and R. L. Webster, Chem. – Eur. J., 2015, 21, 15960–15963 CrossRef CAS.
- H. R. Sharpe, A. M. Geer, W. Lewis, A. J. Blake and D. L. Kays, Angew. Chem., Int. Ed., 2017, 56, 4845–4848 CrossRef CAS.
- J. Rajpurohit, P. Kumar, P. Shukla, M. Shanmugam and M. Shanmugam, Organometallics, 2018, 37, 2297–2304 CrossRef CAS.
- T. Greiser and E. Weiss, Chem. Ber., 1978, 111, 516–522 CrossRef CAS.
- G. van Koten, J. G. Noltes and A. L. Spek, J. Organomet. Chem., 1978, 159, 441–463 CrossRef CAS.
- T. A. Annan, R. Kumar and D. G. Tuck, J. Chem. Soc., Chem. Commun., 1988, 446–448 RSC.
- A. H. Cowley, D. M. Giolando, R. A. Jones, C. M. Nunn and J. M. Power, J. Chem. Soc., Chem. Commun., 1988, 208–209 RSC.
- D. J. Brauer, G. Hessler, P. C. Knueppel and O. Stelzer, Inorg. Chem., 1990, 29, 2370–2375 CrossRef CAS.
- T. A. Annan, R. Kumar and D. G. Tuck, J. Chem. Soc., Dalton Trans., 1991, 11–18 RSC.
- A. Eichhöfer, D. Fenske and W. Holstein, Angew. Chem., Int. Ed. Engl., 1993, 32, 242–245 CrossRef.
- C. Meyer, H. Grützmacher and H. Pritzkow, Angew. Chem., Int. Ed. Engl., 1997, 36, 2471–2473 CrossRef CAS.
- S. Scholz, M. Bolte, M. Wagner and H.-W. Lerner, Z. Anorg. Allg. Chem., 2007, 633, 1199–1204 CrossRef CAS.
- P. J. Harford, J. Haywood, M. R. Smith, B. N. Bhawal, P. R. Raithby, M. Uchiyama and A. E. H. Wheatley, Dalton Trans., 2012, 41, 6148–6154 RSC.
- B. Khalili Najafabadi and J. F. Corrigan, Dalton Trans., 2015, 44, 14235–14241 RSC.
- J. Yuan, L. Zhu, J. Zhang, J. Li and C. Cui, Organometallics, 2017, 36, 455–459 CrossRef CAS.
- M. Itazaki, T. Matsutani, T. Nochida, T. Moriuchi and H. Nakazawa, Chem. Commun., 2020, 56, 443–445 RSC.
- A. J. Roering, S. E. Leshinski, S. M. Chan, T. Shalumova, S. N. MacMillan, J. M. Tanski and R. Waterman, Organometallics, 2010, 29, 2557–2565 CrossRef CAS.
- W. Yi, J. Zhang, L. Hong, Z. Chen and X. Zhou, Organometallics, 2011, 30, 5809–5814 CrossRef CAS.
- R. J. Schwamm, J. R. Fulton, M. P. Coles and C. M. Fitchett, Dalton Trans., 2017, 46, 2068–2071 RSC.
- J. Li, W.-X. Shen and X.-R. Li, Curr. Org. Chem., 2012, 16, 2879–2891 CrossRef CAS.
- M. J. Page, W. Y. Lu, R. C. Poulten, E. Carter, A. G. Algarra, B. M. Kariuki, S. A. Macgregor, M. F. Mahon, K. J. Cavell, D. M. Murphy and M. K. Whittlesey, Chem. – Eur. J., 2013, 19, 2158–2167 CrossRef CAS.
- R. C. Poulten, M. J. Page, A. G. Algarra, J. J. Le Roy, I. López, E. Carter, A. Llobet, S. A. Macgregor, M. F. Mahon, D. M. Murphy, M. Murugesu and M. K. Whittlesey, J. Am. Chem. Soc., 2013, 135, 13640–13643 CrossRef CAS.
- J. K. Park, H. H. Lackey, B. A. Ondrusek and D. T. McQuade, J. Am. Chem. Soc., 2011, 133, 2410–2413 CrossRef CAS.
- J. W. Hall, D. M. L. Unson, P. Brunel, L. R. Collins, M. K. Cybulski, M. F. Mahon and M. K. Whittlesey, Organometallics, 2018, 37, 3102–3110 CrossRef CAS.
- F. Sebest, J. J. Dunsford, M. Adams, J. Pivot, P. D. Newman and S. Díez-González, ChemCatChem, 2018, 10, 2041–2045 CrossRef CAS.
- G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 3966–3972 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, X-ray analysis of compounds 1–7. DOSY experiments, details of the computational analysis and atomic coordinates of the DFT computed structures. CCDC 2014370–2014376. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc05694d |
| This journal is © The Royal Society of Chemistry 2020 |