
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefluorosilylation of trifluoromethane: upgrading an environmentally damaging fluorocarbon†
Daniel J.
Sheldon
 ,
Greg
Coates
and
Mark R.
Crimmin
*
,
Greg
Coates
and
Mark R.
Crimmin
*
Molecular Sciences Research Hub, Department of Chemistry, Imperial College London, London W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
First published on 17th September 2020
Abstract
The rapid, room-temperature defluorosilylation of trifluoromethane, a highly potent greenhouse gas, has been achieved using a simple silyl lithium reagent. An extensive computational mechanistic analysis provides a viable reaction pathway and demonstrates the unexpected electrophilic nature of LiCF3. The reaction generates a bench stable fluorinated building block that shows promise as an easy-to-use difluoromethylating agent. The difluoromethyl group is an increasingly important bioisostere in active pharmaceutical ingredients, and therefore our methodology creates value from waste. The potential scalability of the process has been demonstrated by achieving the reaction on a gram-scale.
Despite being widely employed as refrigerants, hydrofluorocarbons (HFCs) are potent greenhouse gases and important contributors to global warming.1,2 The threat posed by HFCs was highlighted by a recent amendment to the Montreal Protocol seeking to reduce HFCs by >80% by 2050.3 Trifluoromethane (HCF3, HFC-23) has a global warming potential 11
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 times greater than CO2, and an atmospheric lifetime of 264 years.4 It is produced on a vast scale (ca. 20 kilotons per year) as a by-product from a range of industrial processes, such as the manufacture of PTFE (Teflon) and refrigerant gases (e.g. ClCF2H).2,5 Despite its widespread production, there is currently little application for trifluoromethane. Consequently, it is either stored or destroyed at high cost to prevent its release into the environment.2,4
700 times greater than CO2, and an atmospheric lifetime of 264 years.4 It is produced on a vast scale (ca. 20 kilotons per year) as a by-product from a range of industrial processes, such as the manufacture of PTFE (Teflon) and refrigerant gases (e.g. ClCF2H).2,5 Despite its widespread production, there is currently little application for trifluoromethane. Consequently, it is either stored or destroyed at high cost to prevent its release into the environment.2,4
In the pharmaceutical industry, fluorine substitution is commonly used to improve drug efficiency and quality by enhancing the metabolic stability and overall bioavailability of a drug.6,7 There is a particular growing interest in the use of the difluoromethyl (CF2H) group in drug design, where it is considered a lipophilic bioisostere of the hydroxyl, thiol and amine groups.8,9 The CF2H moiety is already present in various commercialised pharmaceuticals such as Eflornithine and Pantoprazole (Fig. 1b).10,11 The growth in interest for CF2H installation has created a growing demand for an easy-to-use, mild difluoromethylating agent.12,13
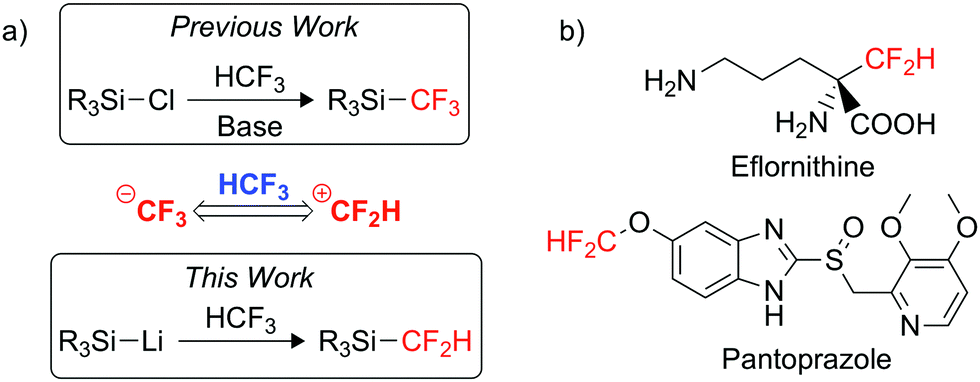 | ||
| Fig. 1 (a) Umpolung reactivity mode of HCF3. (b) Pharmaceuticals containing the CF2H moiety. | ||
We postulated the potential environmental and economic benefit in the use of trifluoromethane as a feedstock gas for the synthesis of a valuable difluoromethyl building block, by developing a process to transform the C–F bond into a reactive C–Si bond.
Much progress has been made in the field of upgrading fluorocarbons into reactive building blocks, particularly with the use of nucleophilic main group reagents.1,14,15 Fluoroalkanes remain the least reactive substrates due to high sp3 C–F bond dissociation energies and a lack of charge stabilisation in the bond-breaking transition state.1,16 Despite this, our group has in recent years demonstrated the C–F activation of simple fluoroalkanes using aluminium and magnesium nucleophiles.17,18 Furthermore, the groups of Shibata and Martin have both reported C–F activation of a range of fluoroalkanes using group 1 metal silyl nucleophiles.19,20 We also recently reported the defluorosilylation of industrially relevant hydrofluoroolefins (HFOs) with simple silyl lithium reagents,21 and in this work we sought to extend the methodology to HFCs.
Trifluoromethane itself has very limited synthetic use, stemming from its low boiling point (−83 °C) and its relatively acidic C–H bond (pKA ∼ 25 in H2O).2 The CF3− anion generated from deprotonation can decompose into difluorocarbene (:CF2) and a fluoride anion (F−),2,5,22,23 although under appropriate conditions it has been utilised in trifluoromethylation reactions (Fig. 1a).2,24 However, there are only a handful of examples where trifluoromethane is employed as a {CF2H}+ synthon through C–F functionalisation.25–30 Mikami and co-workers used a highly nucleophilic boryl lithium reagent to demonstrate the defluoroborylation of HCF3 to form an organoboron building block.28 While a mechanistic study was not carried out for this system, a related computational study by Mikami on the α-difluoromethylation of lithium enolates was utilised to propose a pathway for the defluoroborylation.26 The authors suggest initial deprotonation of HCF3 occurs to form LiCF3, before C–F cleavage then proceeds via an SN2-type attack by the nucleophilic boryl lithium at LiCF3, in a bimetallic transition state.26 While an important discovery, any application of the defluoroborylation methodology is limited by issues regarding scalability. The boryl lithium reagent is extremely difficult to synthesise and is highly susceptible to degradation, in fact it could only be synthesised in situ and required a temperature of −78 °C. The organoboron building block was reported as bench stable but its utility is unknown.28
In this paper, we report the rapid, room-temperature defluorosilylation of trifluoromethane using a simple silyl lithium reagent to form a promising difluoromethyl organosilicon building block. This methodology offers the potential to recycle a highly abundant, low-value fluorocarbon, minimising waste and environmental damage, to create a pharmaceutically relevant building block of high-value.1
Trifluoromethane (1 bar, 22 °C) was added to a C6D6 solution of the silyl lithium reagent PhMe2SiLi·PMDETA (1·PMDETA) (PMDETA = pentamethyldiethylenetriamine), and the building block PhMe2SiCF2H (2) was formed in a 90% spectroscopic yield (Fig. 2). PhMe2SiH was also formed as a by-product in a 10% yield. The optimum concentration of 1·PMDETA was found to be 0.02 M, which results in approximately 7.5 equivalents of HCF3 being added to the headspace of the reaction vessel. The yield of 2 was found to decrease with an increasing concentration of 1·PMDETA (and a consequently decreasing equivalents of HCF3). It was also found that the PMDETA ligand was crucial to the reaction, with alternative THF (1·THF) and TMEDA (1·TMEDA) (TMEDA = tetramethylethylenediamine) adducts resulting in no formation of the desired product 2. The structures of the silyl lithium nucleophiles 1·PMDETA and 1·TMEDA have previously been reported.21,31 A solvent screen showed that polar solvents such as THF were detrimental to the yield of 2, whilst low reaction temperatures (−78 °C) altered the reaction pathway to form undesired products (see ESI,† for full details of reaction optimisation).
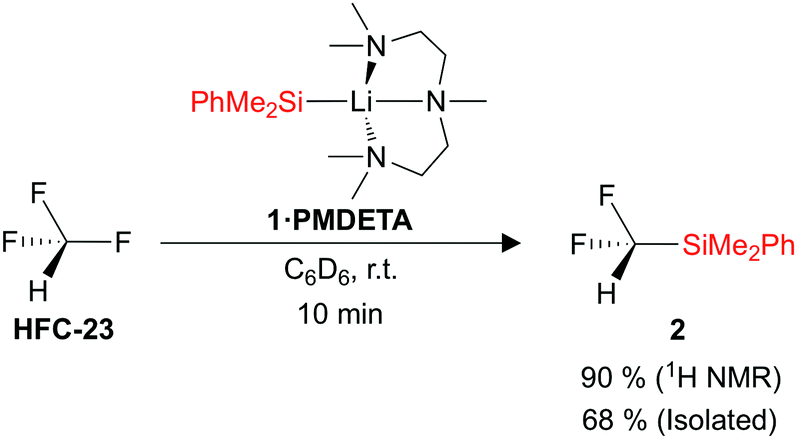 | ||
| Fig. 2 Reaction scheme for the defluorosilylation of trifluoromethane. | ||
After achieving the defluorosilylation of HCF3 in a >90% yield on an NMR scale, we sought to demonstrate the potential scalability of this methodology, and were able to achieve the transformation on a gram-scale of 1·PMDETA. The product PhMe2SiCF2H (2) was successfully isolated after work-up in a 68% yield.
In order to probe the mechanism, we set out to complete a kinetic analysis of the reaction by NMR spectroscopy. Unfortunately, we were unable to achieve this at room temperature as the reaction goes to completion within 5 minutes (as shown by 1H NMR spectroscopy). Efforts to obtain an analysis of the reaction at low temperature (−78 °C) were thwarted by a change in reaction selectivity, where the desired product 2 was produced in only an 8% yield, with PhMe2SiH and H2CF2 instead formed as the major products (see ESI,† for full details).
An extensive DFT study was carried out to explore the mechanism (Fig. 3). Our calculations support a mechanism similar to that proposed by Mikami and co-workers for the defluoroborylation of trifluoromethane.26,28 The first step is deprotonation of HCF3 by 1·PMDETA, proceeding viaTS1 (ΔG1‡ = 20.5 kcal mol−1) to form PMDETA·LiCF3 and the experimentally observed by-product PhMe2SiH. The rate-determining C–F activation step then occurs by an SN2-like attack by a further equivalent of 1·PMDETA at the PMDETA·LiCF3 carbenoid, proceeding viaTS2 (ΔG2‡ = 23.4 kcal mol−1). In this transition state, one lithium cation stabilises the fluoride leaving group, and the other stabilises the carbenoid carbon, acting as an anchor for C–Si bond formation. It has been suggested that strong Li⋯F interactions are crucial for stabilising similar transition states.25,26TS2 is a concerted, albeit highly asynchronous transition state involving early C–F cleavage with concomitant LiF formation, and late C–Si bond formation. Finally, PhMe2SiCF2Li·PMDETA undergoes protonation by a further equivalent of HCF3 to give the desired product 2 and PMDETA·LiCF3, viaTS3 (ΔG3‡ = 18.7 kcal mol−1). The reaction is therefore proposed to be catalytic in LiCF3 (Fig. 4). All three steps are exergonic processes.
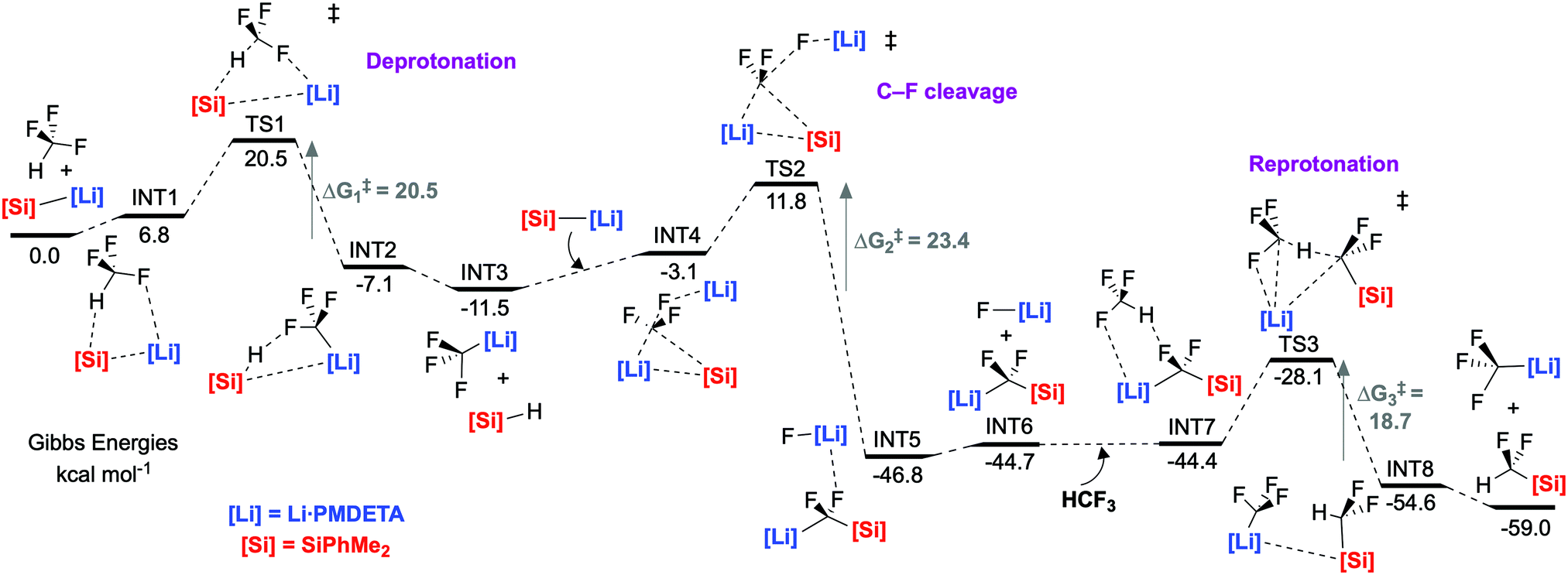 | ||
| Fig. 3 Calculated potential energy surface for trifluoromethane defluorosilylation. The B3PW91 functional was used with a hybrid basis set, 6-31g**(C, H)/6-311+g*(N, Si, Li, F). Solvation (PCM, benzene) and dispersion (GD3) were incorporated into the optimisations. | ||
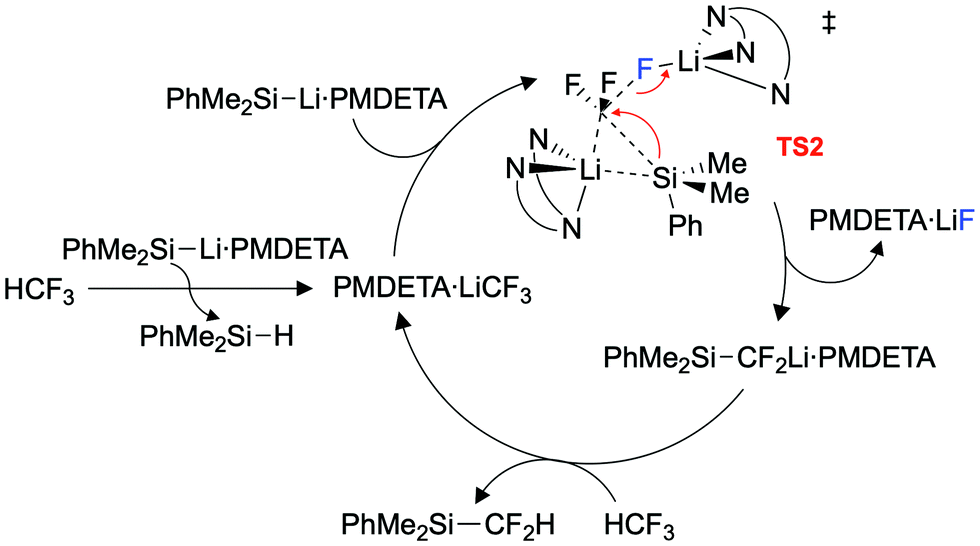 | ||
| Fig. 4 Proposed reaction cycle for trifluoromethane defluorosilyation. | ||
NBO analysis was carried out to elucidate the nature of the transition states (see ESI,† for full details). The NPA charge on the carbenoid carbon of PMDETA·LiCF3 in INT3 (and subsequently INT4 and TS2) was found to be positive, despite this species being viewed as carbanion (Fig. 5). This is due to the strong electron withdrawing effect of the three fluorine atoms, and has been noted in previous calculations on LiCF3.32 The positive NPA charge explains the electrophilic nature of PMDETA·LiCF3 and hence why it is attacked by the silicon nucleophile. Notably, the positive NPA charge on the carbenoid carbon increases from INT4 (+0.55) to TS2 (+0.64), suggesting an accumulation of positive charge approaching the transition state. This is consistent with the asynchronous nature of TS2 where C–F cleavage occurs prior to C–Si formation. Second-order perturbation analysis of TS2 suggests there is a small donation of electron density from a Si lone pair to a p orbital of the carbenoid carbon (≈6 kcal mol−1). We therefore suggest that TS2 possesses some SN1-like character, however, is overall considered a highly asynchronous SN2-like step as it is concerted in C–F cleavage and C–Si formation. The geometry of TS2 is somewhat similar to the transition-state proposed by Mikami for the attack of a THF-stabilised lithium enolate on LiCF3.26
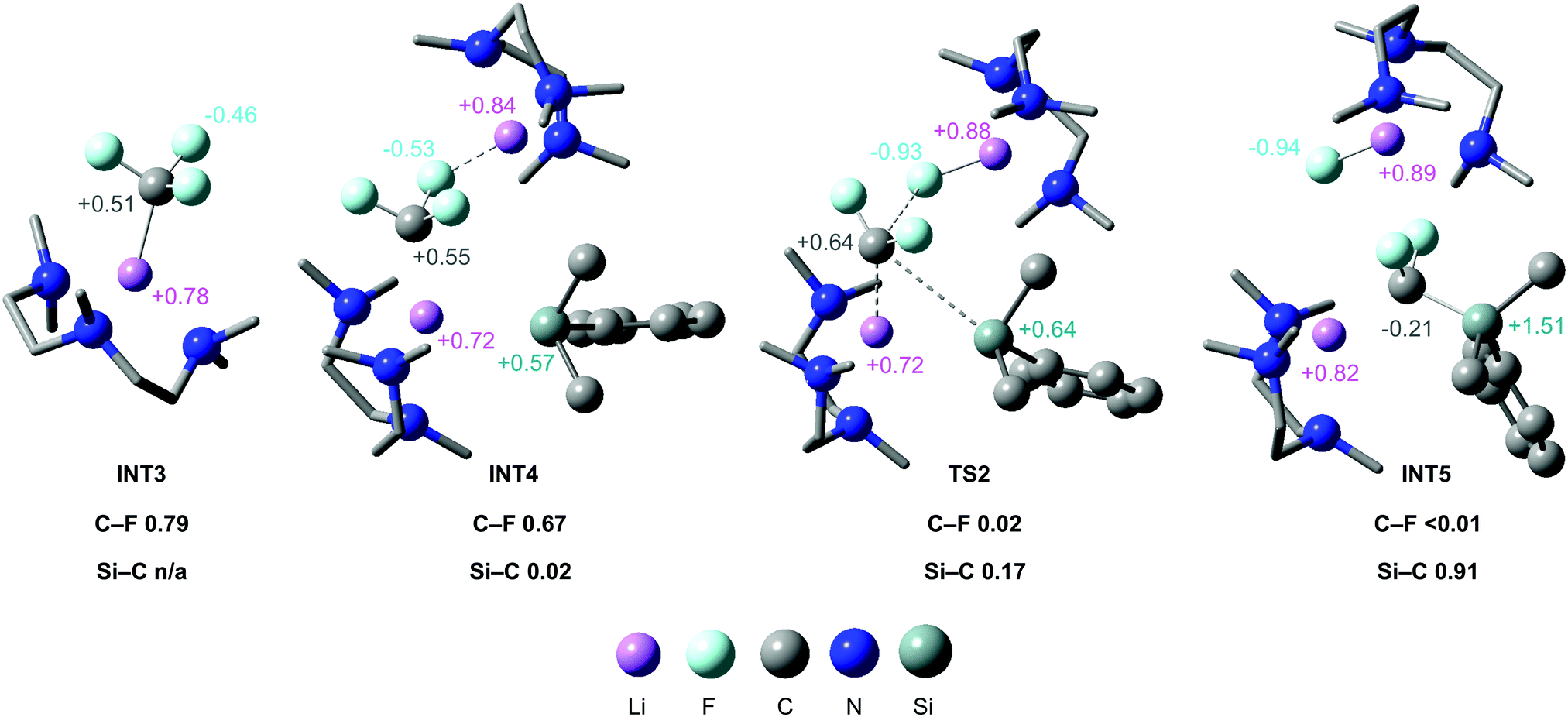 | ||
| Fig. 5 Calculated structures for the stationary points of the C–F activation step, annotated with relevant NPA charges and Wiberg Bond Indices for C–F cleavage and C–Si formation. | ||
Alternative mechanisms were explored by DFT calculations and ruled out on the basis of identifying transition states that were prohibitively high in energy. A classical, direct SN2 attack by 1·PMDETA at HCF3 was calculated to proceed viaTS4 (see ESI†) (ΔG4‡ = 53.8 kcal mol−1). A ‘frontside SN2’ approach was also considered, as this mechanism has been proposed to operate with highly fluorophilic nucleophiles,18,33 and the high-energy TS5 (see ESI†) (ΔG5‡ = 44.7 kcal mol−1) was found. We were unable to find a transition state for difluorocarbene formation from PMDETA·LiCF3.
The experimental observation of PhMe2SiH as a reaction by-product is consistent with deprotonation of HCF3 as the first step of the reaction to form LiCF3. It has been reported that LiCF3 can decompose to form LiF and :CF2.2,29,30 There was no evidence for the presence of difluorocarbene (:CF2) from several carbene trapping experiments that were carried out (see ESI,† for full experimental details). While these results cannot rule out a carbene mechanism entirely, they strongly suggest, in combination with results from DFT, that a carbene pathway is not occurring.
The difluoromethyl building block 2 has already been applied as an easy-to-use reagent for the installation of the CF2H moiety in carbonyl substrates.34,35 Its use is somewhat scarce, however, and this could be due to the difficulty or cost of its synthesis (it requires the now-banned substance HCF2Cl).36,37 Our methodology provides a simple, gram-scale synthesis of this promising difluoromethylating agent, which we believe could lead to an increase in the use of the difluoromethyl group in new pharmaceutical and agrochemical products.
In conclusion, we have developed a simple process to achieve the rapid, room- temperature defluorosilylation of an environmentally damaging fluorocarbon, trifluoromethane. The reaction generates a bench stable fluorinated building block without the need for cryogens, in a reaction that can be performed on a gram-scale. The fluorinated building block is an established difluoromethylating agent, hence the approach allows the generation of value from waste. Through an extensive computational study, we have proposed a viable mechanism for sp3 C–F bond activation, rationalising the unexpected electrophilic nature of LiCF3. The benefits of using trifluoromethane as a feedstock gas would be greatly amplified if scaled up to a continuous flow process,38 and is the subject of future work in our laboratories.
We are grateful to Dr David Mills and co-workers. The ERC is thanked for generous funding (StG: 677367).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Coates, F. Rekhroukh and M. R. Crimmin, Synlett, 2019, 2233–2246 Search PubMed.
- G. K. S. Prakash, P. V. Jog, P. T. D. Batamack and G. A. Olah, Science, 2012, 338, 1324–1327 Search PubMed.
- EU ratifies Kigali Amendment to the Montreal Protocol | Climate Actionhttps://ec.europa.eu/clima/news/eu-ratifies-kigali-amendment-montreal-protocol_en.
- W. Han, Y. Li, H. Tang and H. Liu, J. Fluorine Chem., 2012, 140, 7–16 Search PubMed.
- S. Kyasa, Synlett, 2015, 1911–1912 Search PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 Search PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 Search PubMed.
- Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 Search PubMed.
- G. K. S. Prakash, S. Krishnamoorthy, S. K. Ganesh, A. Kulkarni, R. Haiges and G. A. Olah, Org. Lett., 2014, 16, 54–57 Search PubMed.
- R. A. Casero and P. M. Woster, J. Med. Chem., 2009, 52, 4551–4573 Search PubMed.
- S. M. Cheer, A. Prakash, D. Faulds and H. M. Lamb, Drugs, 2003, 63, 101–133 Search PubMed.
- D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676–14701 Search PubMed.
- S. Monfette, Y.-Q. Fang, M. M. Bio, A. R. Brown, I. T. Crouch, J.-N. Desrosiers, S. Duan, J. M. Hawkins, C. M. Hayward, N. Peperni and J. P. Rainville, Org. Process Res. Dev., 2020, 24, 1077–1083 Search PubMed.
- T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 Search PubMed.
- W. Chen, C. Bakewell and M. Crimmin, Synthesis, 2016, 810–821 Search PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 Search PubMed.
- M. R. Crimmin, M. J. Butler and A. J. P. White, Chem. Commun., 2015, 51, 15994–15996 Search PubMed.
- G. Coates, B. J. Ward, C. Bakewell, A. J. P. White and M. R. Crimmin, Chem. – Eur. J., 2018, 24, 16282–16286 Search PubMed.
- B. Cui, S. Jia, E. Tokunaga and N. Shibata, Nat. Commun., 2018, 9, 4393 Search PubMed.
- X.-W. Liu, C. Zarate and R. Martin, Angew. Chem., Int. Ed., 2019, 58, 2064–2068 Search PubMed.
- G. Coates, H. Y. Tan, C. Kalff, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2019, 58, 12514–12518 Search PubMed.
- C. P. Johnston, T. H. West, R. E. Dooley, M. Reid, A. B. Jones, E. J. King, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2018, 140, 11112–11124 Search PubMed.
- A. García-Domínguez, T. H. West, J. J. Primozic, K. M. Grant, C. P. Johnston, G. G. Cumming, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2020, 142, 14649–14663 Search PubMed.
- P. Novák, A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2012, 134, 16167–16170 Search PubMed.
- T. Iida, R. Hashimoto, K. Aikawa, S. Ito and K. Mikami, Angew. Chem., Int. Ed., 2012, 51, 9535–9538 Search PubMed.
- K. Honda, T. V. Harris, M. Hatanaka, K. Morokuma and K. Mikami, Chem. – Eur. J., 2016, 22, 8796–8800 Search PubMed.
- K. Aikawa, K. Maruyama, K. Honda and K. Mikami, Org. Lett., 2015, 17, 4882–4885 Search PubMed.
- S. Ito, N. Kato and K. Mikami, Chem. Commun., 2017, 53, 5546–5548 Search PubMed.
- S. Okusu, E. Tokunaga and N. Shibata, Org. Lett., 2015, 17, 3802–3805 Search PubMed.
- C. S. Thomoson and W. R. Dolbier, J. Org. Chem., 2013, 78, 8904–8908 Search PubMed.
- C. Strohmann and C. Däschlein, Chem. Commun., 2008, 2791–2793 Search PubMed.
- J. Kvíčala, J. Štambaský, S. Böhm and O. Paleta, J. Fluorine Chem., 2002, 113, 147–154 Search PubMed.
- C. Strohmann, M. Bindl, V. C. Fraaß and J. Hörnig, Angew. Chem., Int. Ed., 2004, 43, 1011–1014 Search PubMed.
- T. Hagiwara and T. Fuchikami, Synlett, 1995, 717–718 Search PubMed.
- Y. Zhao, W. Huang, J. Zheng and J. Hu, Org. Lett., 2011, 13, 5342–5345 Search PubMed.
- T. Fuchikami and I. Ojima, J. Organomet. Chem., 1981, 212, 145–153 Search PubMed.
- G. J. M. Velders, A. R. Ravishankara, M. K. Miller, M. J. Molina, J. Alcamo, J. S. Daniel, D. W. Fahey, S. A. Montzka and S. Reimann, Science, 2012, 335, 922–923 Search PubMed.
- M. Köckinger, C. A. Hone, B. Gutmann, P. Hanselmann, M. Bersier, A. Torvisco and C. O. Kappe, Org. Process Res. Dev., 2018, 22, 1553–1563 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details. See DOI: 10.1039/d0cc04592f |
| This journal is © The Royal Society of Chemistry 2020 |