
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDibenzocycloheptatriene as end-group of Thiele and tetrabenzo-Chichibabin hydrocarbons†
Vicente G.
Jiménez‡
 a,
Paula
Mayorga-Burrezo‡
b,
Victor
Blanco
a,
Vega
Lloveras
b,
Carlos J.
Gómez-García
c,
Tomáš
Šolomek
d,
Juan M.
Cuerva
a,
Jaume
Veciana
*b and
Araceli G.
Campaña
*a
a,
Paula
Mayorga-Burrezo‡
b,
Victor
Blanco
a,
Vega
Lloveras
b,
Carlos J.
Gómez-García
c,
Tomáš
Šolomek
d,
Juan M.
Cuerva
a,
Jaume
Veciana
*b and
Araceli G.
Campaña
*a
aDepartment of Organic Chemistry, Unidad de Excelencia de Química aplicada a Biomedicina y Medio Ambiente, University of Granada (UGR), C. U. Fuentenueva, Granada 18071, Spain. E-mail: araceligc@ugr.es
bDepartment of Molecular Nanoscience and Organic Materials, Institut de Ciència de Materials de Barcelona (ICMAB)/CIBER-BBN, Campus Universitari de Bellaterra, Cerdanyola, Barcelona, E-08193, Spain. E-mail: vecianaj@icmab.es
cDepartment of Inorganic Chemistry and Instituto de Ciencia Molecular, Universidad de Valencia, Paterna 46980, Spain
dDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, Basel 4056, Switzerland
First published on 17th September 2020
Abstract
Thiele (Th) and tetrabenzo-Chichibabin (TBC) derivatives with terminal dibenzocycloheptatriene (DBHept) units were prepared. A clear correlation between their electronic and molecular structures was stablished. Insights into their closed- or open-shell ground states were gained, where particular contribution of the heptagonal carbocycles as end-groups was proved. Remarkably, a thermally accessible triplet diradical configuration was confirmed for the DBHept-TBC compound.
Electronic structure of polycyclic aromatic hydrocarbons1 (PAHs) with small HOMO–LUMO gap appears as halfway between closed- (CS) and open-shell (OS) with two possible spin configurations: a singlet (SOS) and a triplet (TOS). As a result, a number of intriguing properties may arise in these so-called diradicals,2 such as two-photon absorption enhancement,3 singlet fission,2,4 interesting chiroptical,5 or amphoteric redox6 behaviour. In the case when the frontier MOs are disjoint (i.e. have no atoms in common), a small singlet–triplet energy gap and thermally accessible magnetism may arise. Several π-extended diradical PAHs based on ortho- or para-quinodimethanes (o-/p-QDM),7 such as indenofluorenes, zethrenes, teranthrenes and others, have been reported.8 Among them, Thiele9 and Chichibabin10 PAHs, the first reported diradicals, are of particular interest (Chart 1A). Organic chemists have devoted great effort to analyse their peculiar CS/OS resonance while battling their high reactivity.11 The gain in steric hindrance between the (diphenyl)-methyl end-groups and the central aromatic rings extended to anthracenes12,13 (Chart 1B) twists the structure and diminishes the exchange between the two spins. This enhances the SOS character and lowers the singlet–triplet gap.
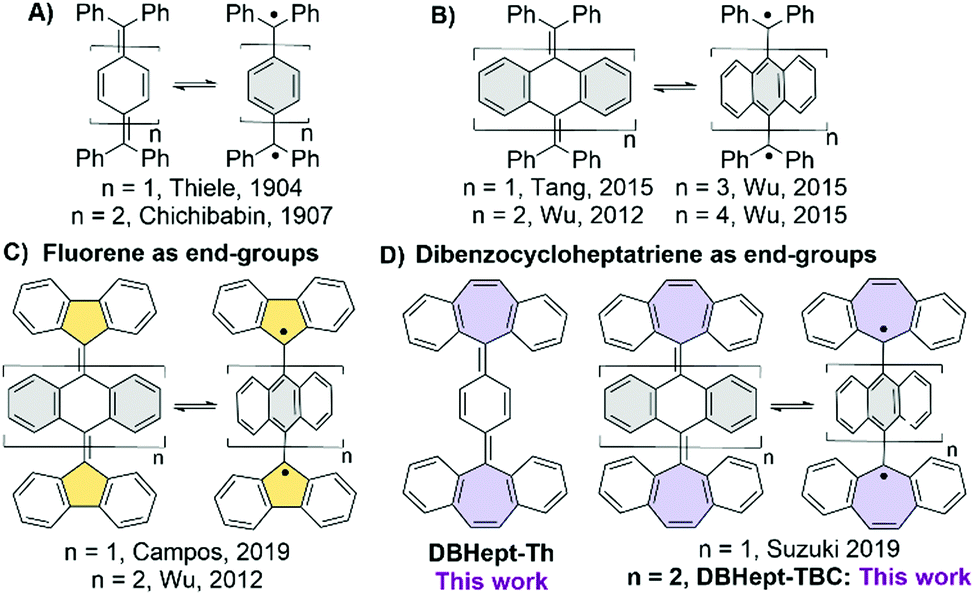 | ||
| Chart 1 CS (left) and OS (right) forms of previously reported: (A) Thiele (Th) and Chichibabin hydrocarbons, (B) tetrabenzo-Chichibabin (TBC) hydrocarbon, (C) TBC-derivatives with terminal fluorene groups. (D) Previous and new Th and TBC-derivatives incorporating dibenzocycloheptatriene (DBHept) as end-groups. | ||
By increasing the length of the spacers (n = 2 → 4, Chart 1B), Wu et al. observed a slow conversion of the OS orthogonal structure to the quinoidal CS singlet state with contorted butterfly-like geometry (n = 1).13 In the extreme case (n = 3, 4), the derivatives behaved like uncoupled radicals in the solid-state EPR experiments. In recent years, the effect of the end-groups has been explored,14 including carbenes14c–h or non-hexagonal carbocycles. Using fluorenyl end-group, Wu et al. stabilized13a the orthogonal geometry (n = 2, Chart 1C) with the OS electronic configuration and Campos et al. showed a thermal interconversion of stable CS/OS forms (n = 1, Chart 1C).15 Only the CS form with no hints of a radical behaviour was recently reported by Suzuki et al. when a heptagonal (i.e. dibenzocycloheptatriene, DBHept) end-group (n = 1, Chart 1D) was employed.16 The same observation was done by some of us with cumulenes as the conjugated bridges between two DBHept groups.17 Only few other examples of radicals stabilized using heptagonal rings can be found,18 mostly as a combination of five- and seven-membered rings.18c,d,f To further investigate the effect of heptagonal end-groups on quinoidal PAHs, herein, we report the synthesis and properties of new DBHept derivatives (Chart 1D), Thiele DBHept-Th and tetrabenzo-Chichibabin DBHept-TBC.
Firstly, DBHept-Th was prepared following the synthetic strategy described in Scheme 1A. Double lithium-halogen exchange in 1,4-dibromobenzene, and subsequent nucleophilic addition to dibenzosuberenone afforded diol 1. DBHept-Th was then achieved by reduction of 1 with SnCl2 in CH2Cl2. Noteworthy, DBHept-Th resulted to be bench stable at air and light. The CS nature of DBHept-Th was fully characterized by means of NMR and IR spectroscopies as well as high-resolution mass spectrometry (HRMS, ESI-TOF) (see ESI†). Single crystals of DBHept-Th for X-ray analysis were also grown by slow evaporation of hexane/CH2Cl2 solutions, showing the saddle curvature of the end-groups caused by the heptagonal carbocycles and the typical C–C bond distances alternation of the p-QDM core (ESI,† Fig. S8). UV-Vis spectrum of DBHept-Th in CH2Cl2 showed the lowest energetic absorption band between 340 and 430 nm (λmax = 386 nm), in good agreement with a CS ground state (ESI,† Fig. S9). Remarkably, this CS form remained stable after heating up to 373 K in C2D2Cl4, as observed by VT-NMR (ESI,† Fig. S4). As expected, no ESR response was obtained for DBHept-Th in o-DCB even after heating up to 360 K (ESI,† Fig. S11). In contrast to DBHept-Th, previous reports on the isolation of Th analogues with terminal fluorenyl units showed to be very difficult.19 Consequently, these results show the positive role of heptagonal carbocycles as end-groups for the stabilization of p-QDM units in Thiele PAHs and DBHept-Th represents the first example of a Thiele PAH analogue incorporating heptagonal carbocycles as end-groups.
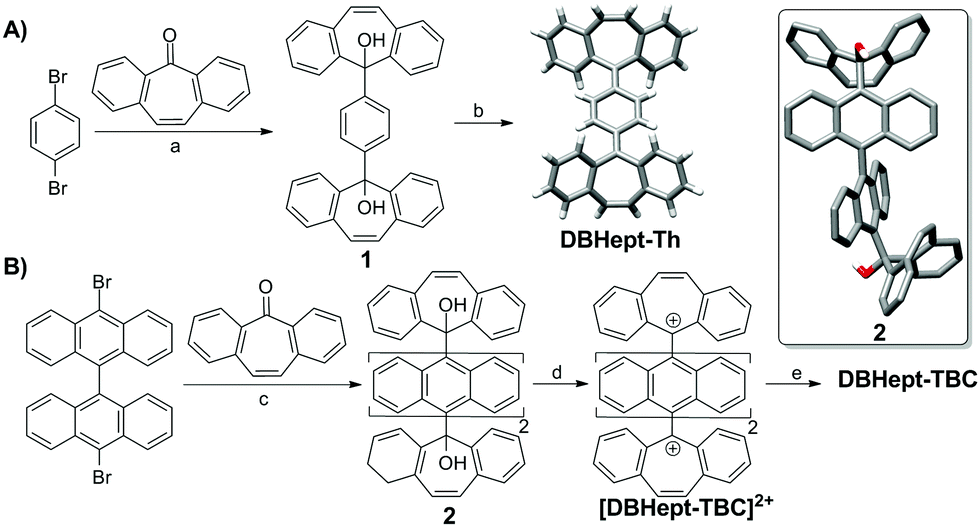 | ||
| Scheme 1 (A) Synthesis of DBHept-Th. (a) nBuLi, THF, −78 °C, 1 h; then dibenzosuberenone, THF −78 °C to r.t., 16 h, 25%. (b) SnCl2, CH2Cl2, r.t., 24 h, 62%. (B) Synthesis of DBHept-TBC. (c) nBuLi, THF, −78 °C, 1 h; then dibenzosuberenone, THF −78 °C to r.t., 16 h, 86%. (d) TFA-d, CD2Cl2, 1 min, r.t. (e) Ferrocene (Fc), CD2Cl2, 1 min, r.t. Single crystal X-ray structures of DBHept-Th and 2 (inset) are shown. | ||
Having studied the behaviour of DBHept-Th, we wondered what the influence of heptagonal rings as end-groups in a Chichibabin-hydrocarbon would be. However, due to its known high reactivity, we chose its higher kinetically stable analogue, tetrabenzo-Chichibabin hydrocarbon (TBC) with 9,9′-bianthracene central core, presented by Wu and co-workers.13a The synthesis of DBHept-TBC (Scheme 1B) started from 10,10′-dibromo-9,9′-bianthracene, by a double lithium-halogen exchange and nucleophilic addition to dibenzosuberenone giving diol 2. The resolved X-ray crystal structure of 2 showed the 9,9′-bianthracene core in an orthogonal geometry and the saddle shape of the dibenzosuberenol moiety (ESI,† Fig. S8). DBHept-TBC was generated by a stepwise protocol and monitored by 1H-NMR.
First, titration of 2 with trifluoroacetic acid-d (TFA-d) in CD2Cl2 afforded a red solution of [DBHept-TBC]2+ with two CF3COO− as counterions. 1H-NMR confirmed the generation of the dication with a pronounced downfield shift of the proton resonances in the dibenzotropylium moieties, as expected from the deshielded environment (Fig. 1A, bottom to middle). HRMS also confirmed the formation of the [DBHept-TBC]2+ (ESI,† Fig. S50). According to the DFT calculations at B3LYP/6-31G(d,p) in CH2Cl2 level, [DBHept-TBC]2+ (ESI,† Table S3) planar dibenzotropylium and anthracene units are orthogonal in good agreement with similar reported dicationic species.20
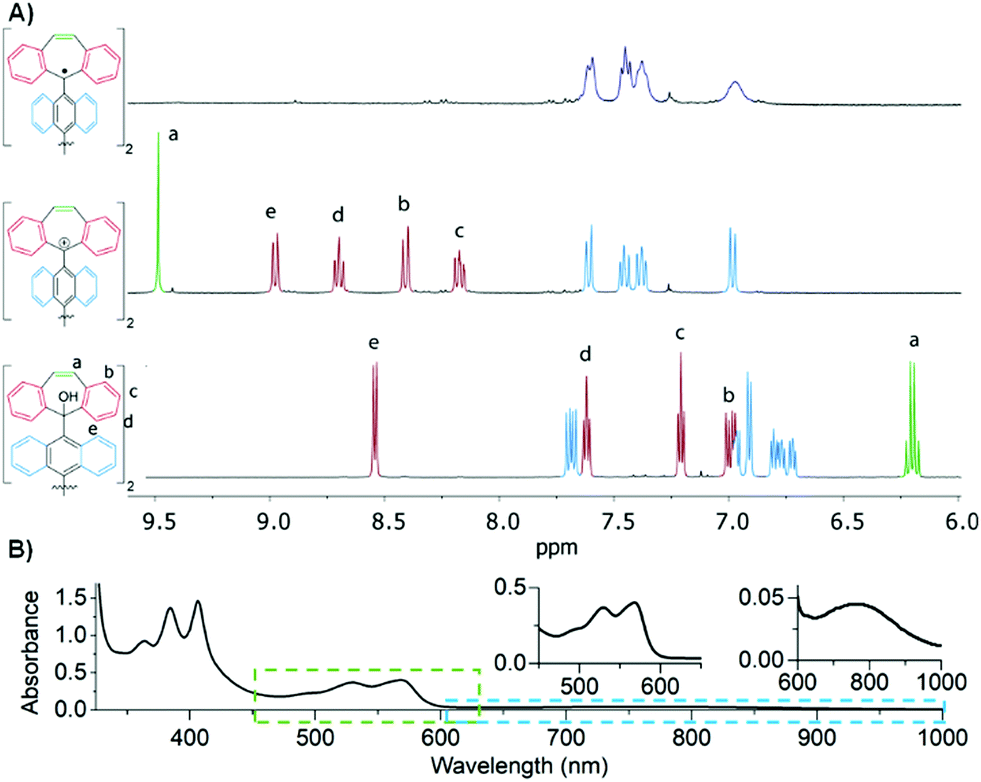 | ||
| Fig. 1 (A) Partial 1H NMR (400 MHz, CD2Cl2, 298 K, 3.2 mM) of 2 (bottom), upon addition of TFA-d to generate [DBHept-TBC]2+ (middle) and subsequent addition of Fc to DBHept-TBC (top). (B) UV-Vis-NIR absorption spectrum of DBHept-TBC (CD2Cl2, 0.1 Mm, r.t.). | ||
Electrochemical properties (ESI,† Section S8) of [DBHept-TBC]2+ were then investigated by cyclic and square wave voltammetry, where two reversible reductions (Ered1/2 = −0.25 V and −1.02 V vs. Fc/Fc+) and two quasi-reversible oxidations processes (Eox1/2 = 1.18 V and 1.44 V vs. Fc/Fc+)+, due to the oxidation of the two anthracene moieties, were observed. Consequently, DBHept-TBC could easily be obtained from [DBHept-TBC]2+ by addition of an adequate reducing agent, such as excess of ferrocene (Fc). Thus, chemical reduction of [DBHept-TBC]2+ in CD2Cl2, by adding an excess of Fc, was followed by 1H-NMR (Fig. 1A, top) showing the disappearance of the protons at the terminal DBHept units (central, red signals) and suggesting the presence of paramagnetic species. The signals associated with the protons in the 9,9′-bianthracene core of [DBHept-TBC]2+ (central, blue signals) were remarkably broad (top). In this regard, similar partial broadening has been reported for twisted OS diradicals too.21 The formation of DBHept-TBC was further confirmed by HRMS (ESI,† Fig. S52).
VT-1H-NMR experiments (ESI,† Section S3) were run for the generated DBHept-TBC. High-T experiments in C2D2Cl4 showed a marked line broadening upon heating up to 373 K while low-T experiments in CD2Cl2 (down to 237 K) afforded again the signals of [DBHept-TBC]2+. Since returning to r.t. favoured again the disappearance/broadening of the signals, a subtle equilibrium between DBHept-TBC/[DBHept-TBC]2+ in the presence of Fc/Fc+ at low temperatures was considered. Accordingly, the chemical reversibility of DBHept-TBC/[DBHept-TBC]2+ was also proved by addition of tris(4-bromophenyl)ammoniumyl hexachloroantimonate as oxidant to a CD2Cl2DBHept-TBC solution, as the [DBHept-TBC]2+ NMR signals were again restored (ESI,† Fig. S3).
Electron spin resonance (ESR) measurements (ESI,† Section S6) were conducted in o-dichlorobenzene, aiming at assessing the electronic structure of DBHept-TBC. A spectrum with five groups of partially overlapped lines (g = 2.00263) was recorded at 400 K (Fig. 2a). Remarkably, this signal was only accessible at high temperature. On the contrary, an ESR-silent response was obtained at r.t., and importantly, VT-ESR experiments showed the reversibility of the process. No half-field signal, typical of triplets, was observed since frozen solutions are needed to this purpose. The associated isotropic hyperfine coupling constants with five different pairs of H nuclei were determined by simulation from the optimized structure of the triplet diradical state (DFT//UCAM-B3LYP/6-31G(d,p) level of theory, see aH values in Fig. 2c). The obtained data are in good agreement with previous results reported by Kubo et al., for anthracene-attached tricyclic monoradicals having one seven-membered ring.18e
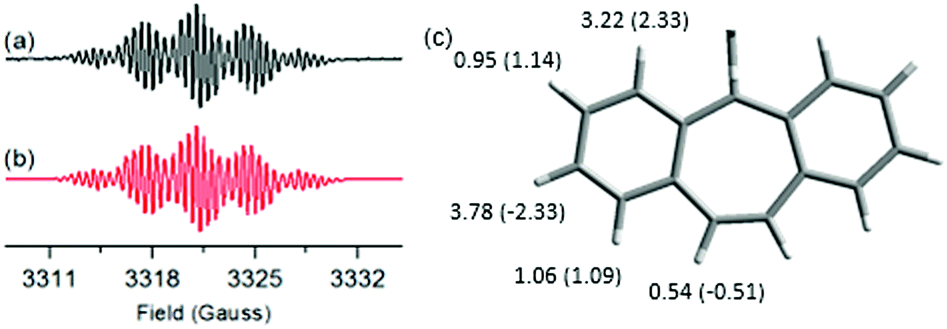 | ||
| Fig. 2 (a) Experimental (black, 10−4 M in o-DCB, 400 K, g = 2.00263) and (b) simulated (red) ESR spectra of DBHept-TBC. (c) Experimentally determined aH values by simulation (in G) for DBHept-TBC. In parentheses, the calculated values at the DFT//UCAM-B3LYP/6-31G(d,p) level of theory, over a fraction of the optimized structure for the triplet diradical state. | ||
Despite all our efforts, we could not grow suitable single crystals of DBHept-TBC for X-ray diffraction. Instead, we isolated a deep red amorphous solid by precipitation of DBHept-TBC solution with Et2O. The isolated solid displayed the same behavior as the samples generated in situ in solution. Thus, by re-dissolving this solid in CD2Cl2 or o-DCB we obtained the same NMR, UV-Vis or ESR spectra, respectively. The UV-Vis spectrum showed the typical vibronic progression of anthracene (350–400 nm, Fig. 1B) and a long absorption tail in the near-IR region typical for similar OS diradicals (Chart 1C, n = 2).13a Note that the same UV-Vis spectrum was obtained by in situ reduction of either [DBHept-TBC]2+ with Fc or diol 2 with SnCl2, (ESI,† Fig. S10).
Magnetic susceptibility data (ESI,† Section S9) of this solid also provided insight into the electronic structure of DBHept-TBC. The temperature dependence of χmT (2 to 350 K) shows that the magnetic moment steadily increases with temperature without achieving a constant value at high temperatures and attaining a value of 0.4 cm3 K mol−1 at 350 K, which is lower than the expected one of 0.75 cm3 K mol−1 for two independent S = 1/2 spins. This indicates that there is an antiferromagnetic interaction in DBHept-TBC and it has a singlet (S = 0) ground state with the first triplet (S = 1) excited state accessible thermally at high temperature. The obtained data can be fitted with the Bleaney–Bowers model and the singlet–triplet gap estimated as 0.74 kcal mol−1.
Theoretical calculations show that the contorted quinoidal closed-shell singlet, DBHept-TBC-CS, is lower in energy (Table S2, ESI†) than the triplet state in the same contorted butterfly-like or the orthogonal geometries by >45 or 10 kcal mol−1, respectively, being therefore, thermally inaccessible from DBHept-TBC-CS. Indeed, reduction of orthogonal [DBHept-TBC]2+ must lead to diradical in orthogonal geometry that is kinetically prevented by the steric bulk of the end-groups to convert to the more stable DBHept-TBC-CS for which we got no spectroscopic evidence. This agrees well with the rotation barrier (22.7 kcal mol−1) observed13 for such transformation with smaller (diphenyl)-methyl end-groups or the calculations (20 kcal mol−1) for model DBHept-Anthracene radical (ESI,† Section S7.4).18e Model studies suggest this stabilization is mostly kinetic unlike in case of fluorenyl end-groups13 where the orthogonal diradical is the thermodynamic product (ESI,† Section S7.5). This shows that the shapes of the end-groups govern the properties of diradical Chichibabin PAHs. ESR experiments, however, suggest that the DBHept-TBC ground state is singlet. Broken-symmetry DFT shows that the triplet and SOS are nearly isoergic, as expected for a diradical with fully disjoint degenerate frontier MOs (FMOs, see ESI†). In this case, two mechanisms may lead to preferential stabilization of the SOS state in the orthogonal DBHept-TBC: (i) vibronic coupling or (ii) dynamic spin polarization (DSP). Vibronic coupling was suggested to affect singlet states and their reactivity in diradicals.22 CAS(2,2) calculations do not show any admixture of excited states that might arise by lowering the D2d symmetry of the triplet DBHept-TBC and stabilize the OS singlet. Note, however, that the dynamic electron correlation missing in CASSCF calculations and solvation could provide a different result. Therefore, DSP, the reason why diradicals with disjoint degenerate FMOs possess the singlet ground state,23 stabilizes the OS singlet, while it is absent in triplet DBHept-TBC. DBHept-TBC in orthogonal geometry is thus predicted to have a singlet OS ground state as observed in the ESR and the magnetic susceptibility experiments or by broadening of the NMR resonances. Although we can not discard formation of diamagnetic oligomers at r.t.,24 clearly, paramagnetic triplet DBHept-TBC is reached upon heating. Therefore, DBHept-TBC incorporating heptagonal carbocycles is the first TBC diradical with major kinetic stabilization embedded by these end-groups.
In summary, we revealed the great impact that heptagonal carbocycles as end-groups have on the ground state in Thiele and Chichibabin diradical PAHs. The saddle-shaped structure imposed by distorted DBHept moieties led to a CS quinoidal geometry in DBHept-Th, while the size of planar DBHept enhanced the steric clash with central anthracenes in DBHept-TBC and kinetically stabilized the orthogonal diradical structure allowing to thermally reach the triplet diradical configuration.
The authors are grateful for the financial support from: MICIU/FEDER/AEI, Spain (PG2018-101181-B-I00, PGC2018-095808-B-I00, MAT2016-80826-R, FIP-2018-HECTIC-PTM, Prometeo2019/076 and the “Severo Ochoa” Programme for Centres of Excellence in R&D; SEV-2015-0496), the European Research Council (ERC) (677023), DGR (Catalunya) (2017-SGR-918), and SNSF (Switzerland, TŠ, PZ00P2_174175). We thank the CSIRC-Alhambra and SciCore (Basel, Switzerland) for supercomputing facilities and the Servei de RMN, UAB, for instrument time.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 Search PubMed; (b) A. Narita, X. Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 Search PubMed.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 Search PubMed.
- K. Kamada, K. Ohta, T. Kubo, A. Shimizu, Y. Morita, K. Nakasuji, R. Kishi, S. Ohta, S. I. Furukawa, H. Takahashi and M. Nakano, Angew. Chem., Int. Ed., 2007, 46, 3544–3546 Search PubMed.
- K. M. Felter and F. C. Grozema, J. Phys. Chem. Lett., 2019, 10, 7208–7214 Search PubMed.
- (a) P. Mayorga-Burrezo, V. G. Jiménez, D. Blasi, I. Ratera, A. G. Campaña and J. Veciana, Angew. Chem., Int. Ed., 2019, 58, 16282–16288 Search PubMed; (b) Q. Jin, S. Chen, Y. Sang, H. Guo, S. Dong, J. Han, W. Chen, X. Yang, F. Li and P. Duan, Chem. Commun., 2019, 55, 6583–6586 Search PubMed; (c) P. Mayorga-Burrezo, V. G. Jiménez, D. Blasi, T. Parella, I. Ratera, A. G. Campaña and J. Veciana, Chem. – Eur. J., 2020, 26, 3776–3781 Search PubMed.
- M. Chikamatsu, T. Mikami, J. Chisaka, Y. Yoshida, R. Azumi, K. Yase, A. Shimizu, T. Kubo, Y. Morita and K. Nakasuji, Appl. Phys. Lett., 2007, 91, 043506 Search PubMed.
- (a) A. Shimizu and Y. Tobe, Angew. Chem., Int. Ed., 2011, 50, 6906–6910 Search PubMed; (b) Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582–2591 Search PubMed.
- (a) C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977–987 Search PubMed; (b) Y. Li, W. K. Heng, B. S. Lee, N. Aratani, J. L. Zafra, N. Bao, R. Lee, Y. M. Sung, Z. Sun, K. W. Huang, R. D. Webster, J. T. López Navarrete, D. Kim, A. Osuka, J. Casado, J. Ding and J. Wu, J. Am. Chem. Soc., 2012, 134, 14913–14922 Search PubMed; (c) A. Konishi, Y. Hirao, M. Nakano, A. Shimizu, E. Botek, B. Champagne, D. Shiomi, K. Sato, T. Takui, K. Matsumoto, H. Kurata and T. Kubo, J. Am. Chem. Soc., 2010, 132, 11021–11023 Search PubMed; (d) P. Ravat, T. Šolomek, M. Rickhaus, D. Häussinger, M. Neuburger, M. Baumgarten and M. Juríček, Angew. Chem., Int. Ed., 2016, 55, 1183–1186 Search PubMed.
- J. Thiele and H. Balhorn, Chem. Ber., 1904, 37, 1463–1470 Search PubMed.
- E. Tschitschibabin, Chem. Ber., 1907, 40, 1810–1819 Search PubMed.
- T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186–2199 Search PubMed.
- Z. He, L. Shan, J. Mei, H. Wang, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, X. Gu, Q. Miao and B. Z. Tang, Chem. Sci., 2015, 6, 3538–3543 Search PubMed.
- (a) Z. Zeng, Y. M. Sung, N. Bao, D. Tan, R. Lee, J. L. Zafra, B. S. Lee, M. Ishida, J. Ding, J. T. López Navarrete, Y. Li, W. Zeng, D. Kim, K.-W. Huang, R. D. Webster, J. Casado and J. Wu, J. Am. Chem. Soc., 2012, 134, 14513–14525 Search PubMed; (b) Z. Lim, B. Zheng, K. W. Huang, Y. Liu and J. Wu, Chem. – Eur. J., 2015, 21, 18724–18729 Search PubMed.
- (a) T. Nishiuchi, R. Ito, E. Stratmann and T. Kubo, J. Org. Chem., 2020, 85, 179–186 Search PubMed; (b) Y. Ishigaki, T. Hashimoto, K. Sugawara, S. Suzuki and T. Suzuki, Angew. Chem., Int. Ed., 2020, 59, 6581–6584 Search PubMed . Some examples on singlet carbenes as end-groups, see: ; (c) D. Rottschäfer, B. Neumann, H. Stammler, D. M. Andrada and R. S. Ghadwal, Chem. Sci., 2018, 9, 4970–4976 Search PubMed; (d) D. Rottschäfer, N. K. T. Ho, B. Neumann, H. Stammler, M. Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 5838–5842 Search PubMed; (e) D. Rottschäfer, J. Busch, B. Neumann, H. Stammler, M. Gastel, R. Kishi, M. Nakano and R. S. Ghadwal, Chem. – Eur. J., 2018, 24, 16537–16542 Search PubMed; (f) J. Messelberger, A. Grünwald, P. Pinter, M. M. Hansmann and D. Munz, Chem. Sci., 2018, 9, 6107–6117 Search PubMed; (g) G. Kundu, S. De, S. Tothadi, A. Das, D. Koley and S. S. Sen, Chem. – Eur. J., 2019, 25, 16533–16537 Search PubMed; (h) A. Maiti, J. Stubbe, N. I. Neuman, P. Kalita, P. Duari, C. Schulzke, V. Chandrasekhar, B. Sarkar and A. Jana, Angew. Chem., Int. Ed., 2020, 59, 6729–6734 Search PubMed.
- X. Yin, J. Z. Low, K. J. Fallon, D. W. Paley and L. M. Campos, Chem. Sci., 2019, 10, 10733–10739 Search PubMed.
- Y. Ishigaki, Y. Hayashi and T. Suzuki, J. Am. Chem. Soc., 2019, 141, 18293–18300 Search PubMed.
- V. G. Jiménez, R. Tapia, M. A. Medel, I. F. A. Mariz, T. Ribeiro, V. Blanco, J. M. Cuerva, E. Maçôas and A. G. Campaña, Chem. Commun., 2018, 54, 3359–3362 Search PubMed.
- (a) M. P. Cava and R. H. Schlessinger, J. Am. Chem. Soc., 1963, 85, 835–836 Search PubMed; (b) J. Kolc and J. Michl, J. Am. Chem. Soc., 1973, 95, 7391–7401 Search PubMed; (c) J. Liu, S. Mishra, C. A. Pignedoli, D. Passerone, J. I. Urgel, A. Fabrizio, T. G. Lohr, J. Ma, H. Komber, M. Baumgarten, C. Corminboeuf, R. Berger, P. Ruffieux, K. Müllen, R. Fasel and X. Feng, J. Am. Chem. Soc., 2019, 141, 12011–12020 Search PubMed; (d) A. Konishi, K. Horii, D. Shiomi, K. Sato, T. Takui and M. Yasuda, J. Am. Chem. Soc., 2019, 141, 10165–10170 Search PubMed; (e) T. Nishiuchi, R. Ito, A. Takada, Y. Yasuda, T. Nagata, E. Stratmann and T. Kubo, Chem. – Asian J., 2019, 14, 1830–1836 Search PubMed; (f) X. Fu, H. Han, D. Zhang, H. Yu, Q. Ho and D. Zhao, Chem. Sci., 2020, 11, 5565–5571 Search PubMed.
- (a) G. Wittig, E. Dreher, W. G. Wittig, E. Dreher, W. Reuther, H. Weidinger and R. Steinmetz, Justus Liebigs Ann. Chem., 1969, 726, 188–200 Search PubMed; (b) J. Ipaktschi, R. Hosseinzadeh and P. Schlaf, Angew. Chem., Int. Ed., 1999, 38, 1658–1660 Search PubMed; (c) D. Beaudoin, O. Levasseur-Grenon, T. Maris and J. D. Wuest, Angew. Chem., Int. Ed., 2016, 55, 894–898 Search PubMed.
- Y. Sakano, R. Katoono, K. Fujiwara and T. Suzuki, Chem. Lett., 2014, 43, 1143–1145 Search PubMed.
- (a) T. Kubo, A. Shimizu, M. Sakamoto, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Morita and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 6564–6568 Search PubMed; (b) C. Wentrup, M. J. Regimbald-Krnel, D. Müller and P. Comba, Angew. Chem., Int. Ed., 2016, 55, 14600–14605 Search PubMed.
- T. Šolomek, P. Ravat, Z. Mou, M. Kertesz and M. Juríček, J. Org. Chem., 2018, 83, 4769–4774 Search PubMed.
- (a) P. G. Wenthold, D. A. Hrovat, W. T. Borden and W. C. Lineberger, Science, 1996, 272, 1456–1459 Search PubMed; (b) W. C. Lineberger and W. T. Borden, Phys. Chem. Chem. Phys., 2011, 13, 11792–11813 Search PubMed.
- Q. Xiang, J. Guo, J. Xu, S. Ding, Z. Li, G. Li, H. Phan, Y. Gu, Y. Dang, Z. Xu, Z. Gong, W. Hu, Z. Zeng, J. Wu and Z. Sun, J. Am. Chem. Soc., 2020, 142, 11022–11031 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, further details and additional figures. CCDC 2012097 and 2012098. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04489j |
| ‡ Both authors contributed as first authors. |
| This journal is © The Royal Society of Chemistry 2020 |