
Synthesis and characterization of low-nuclearity lantern-type porous coordination cages†
Garrett A.
Taggart
a,
Gregory R.
Lorzing
ab,
Michael R.
Dworzak
a,
Glenn P. A.
Yap
 a and
Eric D.
Bloch
*ab
a and
Eric D.
Bloch
*ab
aDepartment of Chemistry & Biochemistry, University of Delaware, Newark, Delaware 19716, USA. E-mail: edb@udel.edu
bCenter for Neutron Science, Department of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716, USA
First published on 19th June 2020
Abstract
Permanent porosity in lantern-type M4L4 paddlewheel-based cages is rare and has only been reported for naphthalene, naphthyridine, and diethynylbenzene-based linkers. This work presents the design, synthesis, and characterization of small lanterns that exhibit CO2 accessible BET surface areas in excess of 200 m2 g−1. The crystal packing and porosity of these cages can be tuned by either ligand functionalization or the choice of M2+ source used in their synthesis. Given their low nuclearity, these cages with internal M–M distances of less than 5 Å represent the lower size limit for permanently microporous coordination cages.
Porous coordination cages have received considerable recent attention,1 particularly as the number of surface areas reported for them continues to increase.2 The majority of these are higher nuclearity carboxylate-based systems that adopt either tetrahedral,3 octahedral,4 or cuboctahedral5 structures containing bi-,6 tri-,7 or tetrametallic8 building units.9 Porous paddlewheel-based cages, for example, most commonly adopt M12L12 or M24L24 structures.10 In contrast, supramolecular cages based on nitrogen-containing heterocycles, which have been used in catalysis,11 separations,12 molecular trapping,13 and drug delivery,14 have been isolated for a broad range of M
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L ratios and overall cage nuclearity from M2L2 through M48L96 and beyond.15–17 This level of tunability allows for molecular-level control of host–guest interactions.18 In this regard, there is great need for the development of permanently porous, low-nuclearity cages as their potentially small pore sizes can be tuned for selective binding or activation of small gaseous molecules.19 However, reported porosity in these systems has been rare.20–22 For these, porous lantern-type cages have been exclusively reported for carboxylate-functionalized paddlewheel-based building units incorporating either naphthalene, naphthyridine, or diethynylbenzene-type ligands (Fig. 1). Porous M4L4 lanterns have been isolated for Cu2+, Mo2+, and Rh2+ paddlewheels and ligands with varying levels of functionalization on either their interior or exterior surfaces.23–34
:L ratios and overall cage nuclearity from M2L2 through M48L96 and beyond.15–17 This level of tunability allows for molecular-level control of host–guest interactions.18 In this regard, there is great need for the development of permanently porous, low-nuclearity cages as their potentially small pore sizes can be tuned for selective binding or activation of small gaseous molecules.19 However, reported porosity in these systems has been rare.20–22 For these, porous lantern-type cages have been exclusively reported for carboxylate-functionalized paddlewheel-based building units incorporating either naphthalene, naphthyridine, or diethynylbenzene-type ligands (Fig. 1). Porous M4L4 lanterns have been isolated for Cu2+, Mo2+, and Rh2+ paddlewheels and ligands with varying levels of functionalization on either their interior or exterior surfaces.23–34
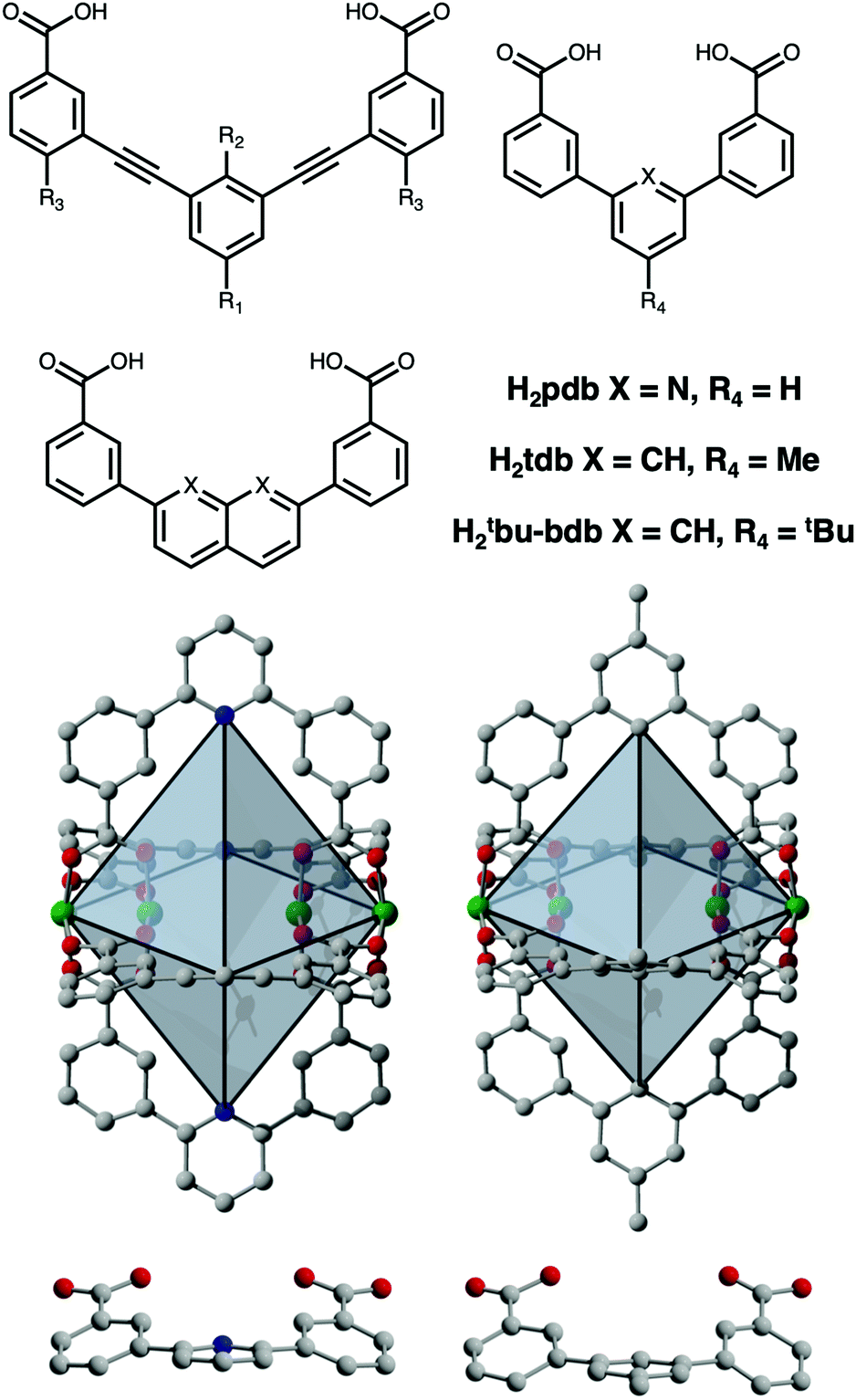 | ||
| Fig. 1 (Top Left) Prominent ligands for porous lantern-type paddlewheel cages that have been combined with Cu, Mo, or Rh to afford M4L4 structures. This study focuses on Cu4(pdb)4, Cu4(tBu-bdb)4, and M4(tdb)4 where M = Cr, Cu, Mo. (Bottom) Structures of Cu4(pdb)4 (left) and Cu4(tdb)4 (right) where green, red, gray, and blue spheres represent copper, oxygen, carbon, and nitrogen atoms, respectively. Although the pyridine-based cage has no C–H on the central ring, the ligand is significantly distorted away from planarity in the crystal structure. | ||
Lah and coworkers described the design and synthesis of a set of Cu4L4 lanterns based on functionalized 3,3′-[1,3-benzenediyldi-(ethynyl)]dibenzoic acid ligands and showed that groups on the exterior surface of the cage can be used to tune crystal packing and thus gas uptake.21 This idea has since been elaborated on by numerous researchers in subsequent studies where functionalization of 3,3′-[1,3-benzenediyldi-(ethynyl)]dibenzoic acid was leveraged to tune both inter- and intra-cage space.22,23 This has resulted in Cu2+ and Rh2+ cages with a variety of tunable catalytic and gas adsorption properties.24,25,28–32 In terms of the latter, these have included gate-opening effects, tunable adsorption/desorption hysteresis, and most recently, switchable on/off porosity. Zhou and coworkers have shown that the metal–metal spacing on the interior cavity of the cage can be tuned via the utilization of naphthalene-, naphthyridine-, or pyridine-based linkers where the benzoic acid groups are coordinated directly to the central rings (Fig. 1).25 As opposed to the 9–10 Å between metal cations on the interior of the alkyne-based cages, they isolated lanterns with an M–M distance of ∼7.5 Å in their fused-ring system and just 4.8 Å in the pyridine-based cage Mo4(pdb)4 (H2pdb = pyridinedibenzoic acid). They further showed by comparison with a MOF based on lantern-type pores, that the M–M distance in the former is optimal for CO2 binding.
These larger lantern-type cages based on extended ligands have BET surface areas that span 0 to 485 m2 g−1, and can likely be further increased by utilization of even longer bridging ligands and continued development of the ligand functionalization strategies that were previously reported. However, in terms of the smaller benzene- or pyridine-based cages, surface areas have not been reported. Herein, we discuss the design and synthesis of five novel porous M4L4 lantern-type coordination cages based on Cr2+, Cu2+, and Mo2+ (Fig. 1). Notably, these cages lack the alkyne functional groups typically used in the assembly of these systems and represent the lower limit in paddlewheel cage size. The diameter of the pore in these cages is limited by the inter-paddlewheel distance of ∼5 Å and to the best of our knowledge, are the smallest coordination cages to exhibit permanent porosity.
The syntheses of M4L4 lantern-type paddlewheel-based cages proceed similarly to those reported for octahedral or cuboctahedral cages where the solvothermal reaction of metal salts with an organic ligand in amide solvents or amide/alcohol mixtures typically affords cage in high yield. Here, a similar protocol was used for the synthesis of Cu4(pdb)4 (H2pdb = pyridinedibenzoic acid; Fig. 1) For this reaction, 168 mg (1 equiv.) of copper(II) nitrate hemipentahydrate and 207 mg (1 equiv.) of H2pdb were added to a 20 mL scintillation vial and dissolved in 17 mL N,N-dimethylformamide (DMF). Three mL of methanol (MeOH) was then layered on top of the solution and the mixture heated to 80 °C in a dry bath overnight. Over the course of the reaction, diffraction-quality single crystals of Cu4(pdb)4 formed in high yield. Structural analysis of this cages reveals it adopts the expected lantern geometry that is nearly identical to the previously reported molybdenum(II) cage, Mo4(pdb)4. Both cages crystallize in the same I4/m space group (Table S1, ESI†) with similar cell parameters where a = b = 12.324(1) and c = 24.440(4) Å for Mo4(pdb)4 and a = b = 12.121(4) and c = 23.730(1) Å for Cu4(pdb)4.23 The decrease in unit cell axes for the latter is largely a result of shorter M–L bond distances (1.96 vs. 2.10 Å) associated with the smaller ionic radius first-row metal. The most dramatic differences in the cages are also a result of the nature of the metal cation, with the d4-based molybdenum cage containing multiply-bonded bimetallic clusters with Mo–Mo distances of 2.10 Å while the corresponding Cu–Cu distance (2.587 Å) is significantly longer. As a result of this, the internal metal–metal distance in Cu4(pdb)4 is significantly shorter than in Mo4(pdb)4 (4.49 Å vs.4.84 Å). The three-dimensional packing of Cu4(pdb)4 reveals minimal pore space in the structure. In the a–b plane, each cage interacts with four adjacent cages to form layers that pack in an offset manner along c. The closest intermolecular aromatic ring centroid distances, face-to-face 4.58 Å and face-to-edge 5.89 Å, suggest that pi–pi interactions are not as significant as they are in alkyne extended structures. Instead, the cages pack in a dense conformation where all four bridging ligands occupy the window of an adjacent cage.
Given the moderate porosities previously reported for the extended lantern structures, we targeted solvent exchange and activation procedures to optimize the surface area of Cu4(pdb)4. As a result of the close-packed nature of the molecules in the solid state, the cage is completely insoluble and is thus more amenable to solvent exchange protocols commonly used for metal–organic frameworks. Ultimately, a sample was thoroughly washed with fresh DMF over the course of three days and subsequently solvent exchanged with methanol, over the course of which the sample displayed no decrease in crystallinity. A degas survey, wherein a surface area is measured after successive heating steps, indicated the optimal activation temperature for this material was 175 °C. Although Cu4(pdb)4 had very little N2 accessible surface area, it had a CO2 accessible BET (Langmuir) surface area of 195 (325) m2 g−1. The CO2 saturation capacity at 195 K and 1.0 bar of 3.55 mmol g−1 corresponds to the adsorption of 5 CO2 molecules/cage.
In order to tune the surface area or solubility of these types of cages, modifications can either be made at the metal cation site or with ligand functionalization. In terms of the latter, the functionalized dibromopyridines that are necessary for this route are significantly more expensive than the analogous benzene-based starting materials. The cages of this type that have been synthesized thus far have largely avoided directly-coupled benzene rings, presumably to avoid the C–H interactions present in these systems. However, close inspection of the crystal structures of Mo4(pdb)4 and Cu4(pdb)4 indicate that these interactions are avoided given the significant distortion away from planarity in the ligand. Rather than C–H/C–H interactions driving cage distortion this suggests that the cage structure inherently distorts the ligand. With this in mind, we prepared two functionalized, benzene-based ligands H2tdb and H2tBu-bdb (Fig. 1). However, these levels of functionalization did not have the desired effect as the former produced cage, Cu4(tdb)4, that crystallized with nearly identical crystal packing and the latter gave what was presumably cage although we were unable to obtain diffraction quality single crystals. The Cu4(tdb)4 unit cell has a slightly extended a and b axis of 12.80 Å compared to the Cu4(pdb)4 cage due to the methyl substituent forcing the cages further apart. It is important to note that in the structure of Cu4(tdb)4 the C–H groups on the center of the ligand are sufficiently separated as a result of the aforementioned cage-induced ligand distortion (Fig. 1).
To tune cage packing, we instead turned to the synthesis of other metal analogues of M4(tdb)4. Similar to the synthesis of the copper cage, both Cr4(tdb)4 and Mo4(tdb)4 were prepared via solvothermal syntheses, but with care taken to limit their exposure to O2 given the well-known redox active nature of Mo2 and Cr2 paddlewheel units. Crystal structures of M4(tdb)4 reveal all three cages adopt the expected geometry and the chromium and copper structures pack the same as M4(pdb)4. Cu4(tdb)4 and Cr4(tdb)4 have nearly identical unit cells, however the interior paddlewheel–paddlewheel distance of Cu4(tdb)4 is 5.04 Å and the Cr4(tdb)4 lantern is 5.26 Å, again a result of the shorter M–M distance from the quadruple bond in the latter. The extended structure of Mo4(tdb)4 is significantly different than the other cages reported here and the previously reported Mo4(pdb)4 phase. This lantern crystallized with two cages in the asymmetric unit, where they sit orthogonal to each other (Fig. 2). An interstitial pore exists between the ligands of the cages with a diameter of 8.76 Å. These toluene-based cages were similarly prepared for gas adsorption measurements by implementing amide and methanol washes. Degas surveys for M4(tdb)4 gave optimal activation temperatures of 25, 25, and 100 °C for the Cr, Mo, and Cu analogues, respectively. These resulted in CO2 accessible BET (Langmuir) surface areas of 145 (401), 180 (516), and 218 (527) m2 g−1 (Fig. 3). It is notable that CO2 adsorption isotherms were used to calculate these surface areas, which is more typically done with N2 adsorption. However, given the small pore sizes of these cages, they were nonporous to N2.
 | ||
| Fig. 2 Solid-state packing of Mo4(tdb)4 where cages in a given layer alternate by 90 degrees. | ||
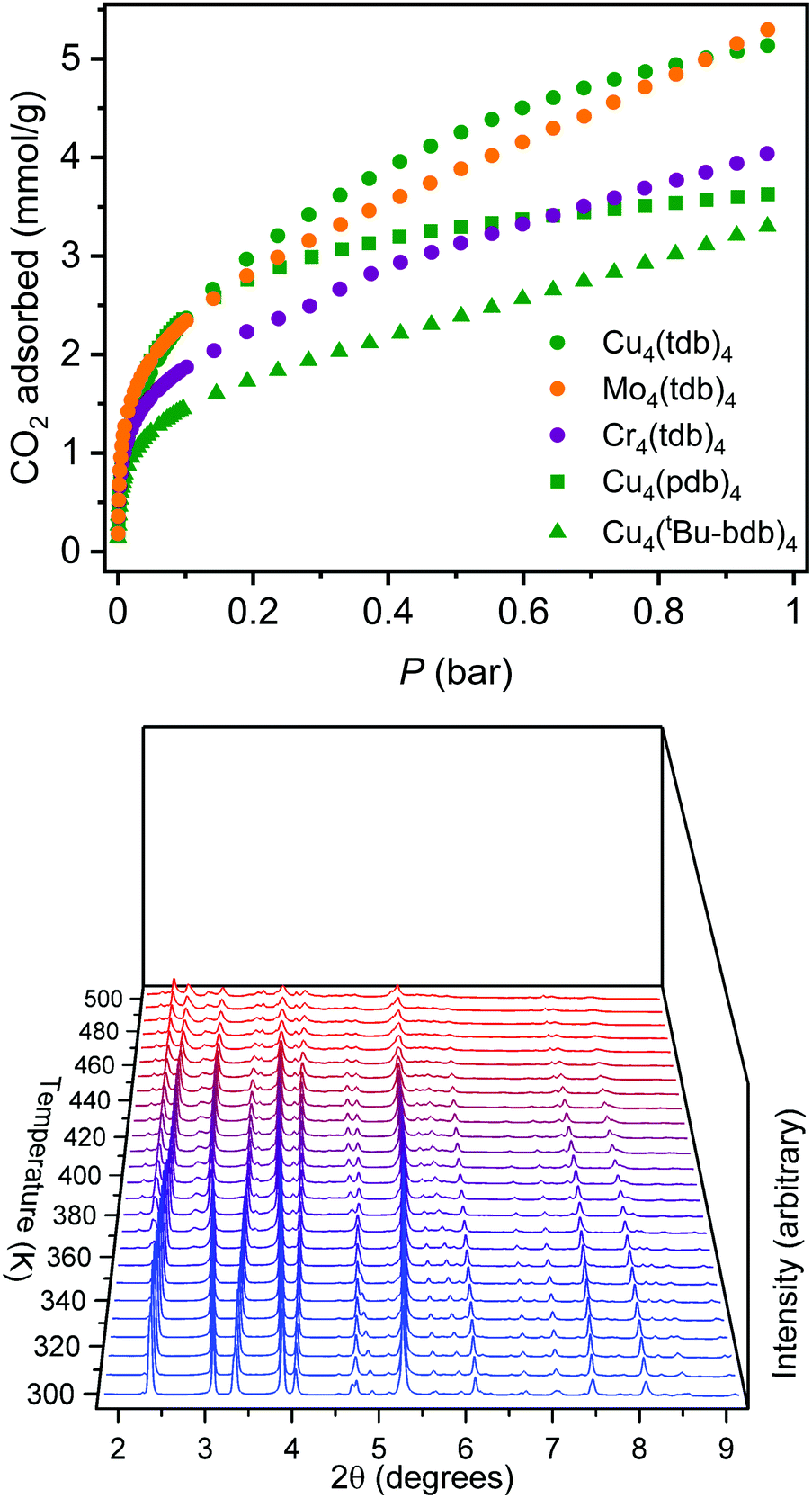 | ||
| Fig. 3 (Top) CO2 adsorption in M4(L)4 cages at 195 K. (Bottom) PXRD patterns as a function of temperature for Cu4(tdb)4 spanning the temperature range of 300–550 K. During this activation, the cage remains moderately crystalline while undergoing significant structural rearrangement. | ||
As described above, naphthalene bridged lanterns have a M–M distance calculated to be optimal for fitting a single CO2 molecule in the lantern pore. The crystal structure of these smaller lanterns with M–M distances less than 5.2 Å do not have the interior pore size capable of adsorbing CO2 in the same manner, although they feature bridging solvent molecules trapped between the metal cation sites. Due to the tight cage packing, accessible pores or channels for gas adsorption are also not expressly visible in the solvated crystal structure. Despite this, these smaller lanterns retain surface areas greater than lanterns extended by fused rings. Solvent exchanging and activating the lanterns in vacuo likely alter cage packing, potentially allowing gas accessibility. The Cu4(pdb)4 and Cu4(tdb)4 lanterns are thermally stable with optimal activation temperatures of 175 °C and 100 °C respectively, and remain crystalline upon activation. Methanol exchanged Cu4(tdb)4 was characterized with in situ PXRD experiments while under vacuum to monitor crystal structure changes (Fig. 3). As the temperature increases from room temperature to 200 °C, there are clear peak shifts and intensity changes taking place, indicating a transition to a new space group. After heating to 225 °C in vacuo, the material is assumed to be fully activated. Due to preferred orientations in the measured sample, a satisfactory space group could not be obtained, however new unit cell dimensions could be acquired via Pawley refinement. Fitting the diffraction pattern collected at 225 °C yielded a distinctly different unit cell with a = 21.91 Å, b = 16.83 Å, and c = 22.06 Å (from a = b = 12.859 Å, and c = 23.956 Å). Removal of solvent, cage rearrangement, and defects in the structure disrupt the periodicity of the sample, resulting in much larger unit cell dimensions. It is likely that as these structure changes take place, accessible pore spaces become available which explains the surprisingly high surface areas for coordination cages of this size.
The foregoing results demonstrate the importance of both ligand functionalization and metal cation selection in the synthesis of small, porous lantern-type coordination cages. These reported structures represent the smallest versions of these cages that can be synthesized and yet they display BET surface areas as high as 200 m2 g−1. It is expected that further functionalization on the backbone of this ligand scaffold can be leveraged to tune the extra-pore space in the structure and tune solubility and stability of porous lanterns. Work in our lab along these lines will focus on the utilization of ester- and amide-functionalization routes.
Crystal structures were submitted to the Cambridge Structural Database under the CCDC numbers 1998863–1998866.† This material is based upon work supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy under the Hydrogen and Fuel Cell Technologies and Vehicle Technologies Offices under Award Number DE-EE0008813. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Conflicts of interest
There are no conflicts to declare.References
- N. Ahmad, A. H. Chughtai, H. A. Younus and F. Verpoort, Coord. Chem. Rev., 2014, 280, 1–27 CrossRef CAS.
- X. X. Li, D. Zhao and S. T. Zheng, Coord. Chem. Rev., 2019, 397, 220–240 CrossRef CAS.
- A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Côté, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110–7118 CrossRef CAS PubMed.
- C. A. Rowland, G. R. Lorzing, E. J. Gosselin, B. A. Trump, G. P. A. Yap, C. M. Brown and E. D. Bloch, J. Am. Chem. Soc., 2018, 140, 11153–11157 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, J. B. Wachter, H. K. Chae, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4368–4369 CrossRef CAS PubMed.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- G. Liu, Y. Di Yuan, J. Wang, Y. Cheng, S. B. Peh, Y. Wang, Y. Qian, J. Dong, D. Yuan and D. Zhao, J. Am. Chem. Soc., 2018, 140, 6231–6234 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O’Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- D. J. Tranchemontagne, Z. Ni, M. O’Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS PubMed.
- G. R. Lorzing, B. A. Trump, C. M. Brown and E. D. Bloch, Chem. Mater., 2017, 29, 8583–8587 CrossRef CAS.
- M. Pan, K. Wu, J. H. Zhang and C. Y. Su, Coord. Chem. Rev., 2019, 378, 333–349 CrossRef CAS.
- B. Li, T. He, Y. Fan, X. Yuan, H. Qiu and S. Yin, Chem. Commun., 2019, 55, 8036–8059 RSC.
- S. Horiuchi, T. Murase and M. Fujita, J. Am. Chem. Soc., 2011, 133, 12445–12447 CrossRef CAS PubMed.
- B. Woods, M. N. Wenzel, T. Williams, S. R. Thomas, R. L. Jenkins and A. Casini, Front. Chem., 2019, 7, 1–6 CrossRef PubMed.
- L. M. Mesquita, J. Anhäuser, D. Bellaire, S. Becker, A. Lützen and S. Kubik, Org. Lett., 2019, 21, 6442–6446 CrossRef CAS PubMed.
- D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Nature, 2016, 540, 563–566 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161–171 RSC.
- D. Preston, K. F. White, J. E. M. Lewis, R. A. S. Vasdev, B. F. Abrahams and J. D. Crowley, Chem. – Eur. J., 2017, 23, 10559–10567 CrossRef CAS PubMed.
- F. A. Cotton, L. M. Daniels, C. Lin and C. A. Murillo, Chem. Commun., 1999, 841–842 RSC.
- G. R. Lorzing, E. J. Gosselin, B. S. Lindner, R. Bhattacharjee, G. P. A. Yap, S. Caratzoulas and E. D. Bloch, Chem. Commun., 2019, 55, 9527–9530 RSC.
- P. W. V. Butler, P. E. Kruger and J. S. Ward, Chem. Commun., 2019, 55, 10304–10307 RSC.
- M. J. Prakash, M. Oh, X. Liu, K. N. Han, G. H. Seong and M. S. Lah, Chem. Commun., 2010, 46, 2049–2051 RSC.
- J.-R. Li and H.-C. Zhou, Nat. Chem., 2010, 2, 893–898 CrossRef CAS PubMed.
- J.-R. Li, A. A. Yakovenko, W. Lu, D. J. Timmons, W. Zhuang, D. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2010, 132, 17599–17610 CrossRef CAS PubMed.
- W. Lu, D. Yuan, A. Yakovenko and H.-C. Zhou, Chem. Commun., 2011, 47, 4968–4970 RSC.
- J.-R. Li, J. Yu, W. Lu, L.-B. Sun, J. Sculley, P. B. Balbuena and H.-C. Zhou, Nat. Commun., 2013, 4, 1538 CrossRef PubMed.
- V. Brega, M. Zeller, Y. He, H. P. Lu and J. K. Klosterman, Chem. Commun., 2015, 51, 5077–5080 RSC.
- L. Chen, J. Kang, H. Cui, Y. Wang, L. Liu, L. Zhang and C.-Y. Su, Dalton Trans., 2015, 44, 12180 RSC.
- L. Chen, T. Yang, H. Cui, T. Cai, L. Zhang and C.-Y. Su, J. Mater. Chem. A, 2015, 3, 20201 RSC.
- S. A. Boer and D. R. Turner, Chem. Commun., 2015, 51, 17375–17378 RSC.
- G. A. Craig, P. Larpent, S. Kusaka, R. Matsuda, S. Kitagawa and S. Furukawa, Chem. Sci., 2018, 9, 6463–6469 RSC.
- G. A. Craig, P. Larpent, H. Urabe, A. Legrand, M. Bonneau, S. Kusaka and S. Furukawa, Chem. Commun., 2020, 56, 3689–3692 RSC.
- W. M. Bloch, R. Babarao and M. L. Schneider, Chem. Sci., 2020, 11, 3664–3671 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1998863–1998866. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03266b |
| This journal is © The Royal Society of Chemistry 2020 |