
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfoxide synthesis from sulfinate esters under Pummerer-like conditions†
Akihiro
Kobayashi
,
Tsubasa
Matsuzawa
,
Takamitsu
Hosoya
 and
Suguru
Yoshida
*
and
Suguru
Yoshida
*
Laboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan. E-mail: s-yoshida.cb@tmd.ac.jp
First published on 6th April 2020
Abstract
A facile synthetic method for the preparation of allyl sulfoxides by S-allylation of sulfinate esters proceeds through sulfonium intermediates without [3,3]-sigmatropic rearrangement and further Pummerer-type reactions of the resulting allyl sulfoxides. On the basis of the plausible reaction mechanism involving sulfonium salt intermediates, S-alkynylation and S-arylation were also accomplished.
Organosulfur compounds have gained attention in a broad range of research fields such as pharmaceutical sciences, agrochemistry, and materials science.1,2 The recent remarkable successes of synthetic chemistry using sulfoxides have enhanced the accessibility of highly functionalized compounds by virtue of the significant transformability of sulfoxides.3–8 For example, the preparations of diverse compounds 2–5 were achieved by a variety of transformations of allyl aryl sulfoxides 1 through C–S bond cleavage (Fig. 1A).6,7,8h,j In particular, multisubstituted aromatic sulfides 4 and 5 were synthesized from sulfoxides 1 by a reaction with aryne intermediate I in the presence of electrophiles and the [3,3]-sigmatropic rearrangement of allyl sulfonium intermediate II, respectively.6,7 Similar interrupted Pummerer reactions of sulfoxides 6 and 8 with trifluoromethanesulfonic anhydride (Tf2O) in the presence of allyltrimethylsilane were also accomplished through the [3,3]-sigmatropic rearrangement, showing the notable reactivity of allyl sulfonium intermediates III and IV (Fig. 1B and C).4b,d,e,n Herein, we describe an efficient synthesis of various allyl sulfoxides by allylation of sulfinate esters9 using allyltrimethylsilane under the Pummerer-like conditions4 through sulfonium intermediate V having a methoxy group, enabling to avoid the [3,3]-sigmatropic rearrangement and further Pummerer-type reactions of the resulting allyl sulfoxides (Fig. 1D).
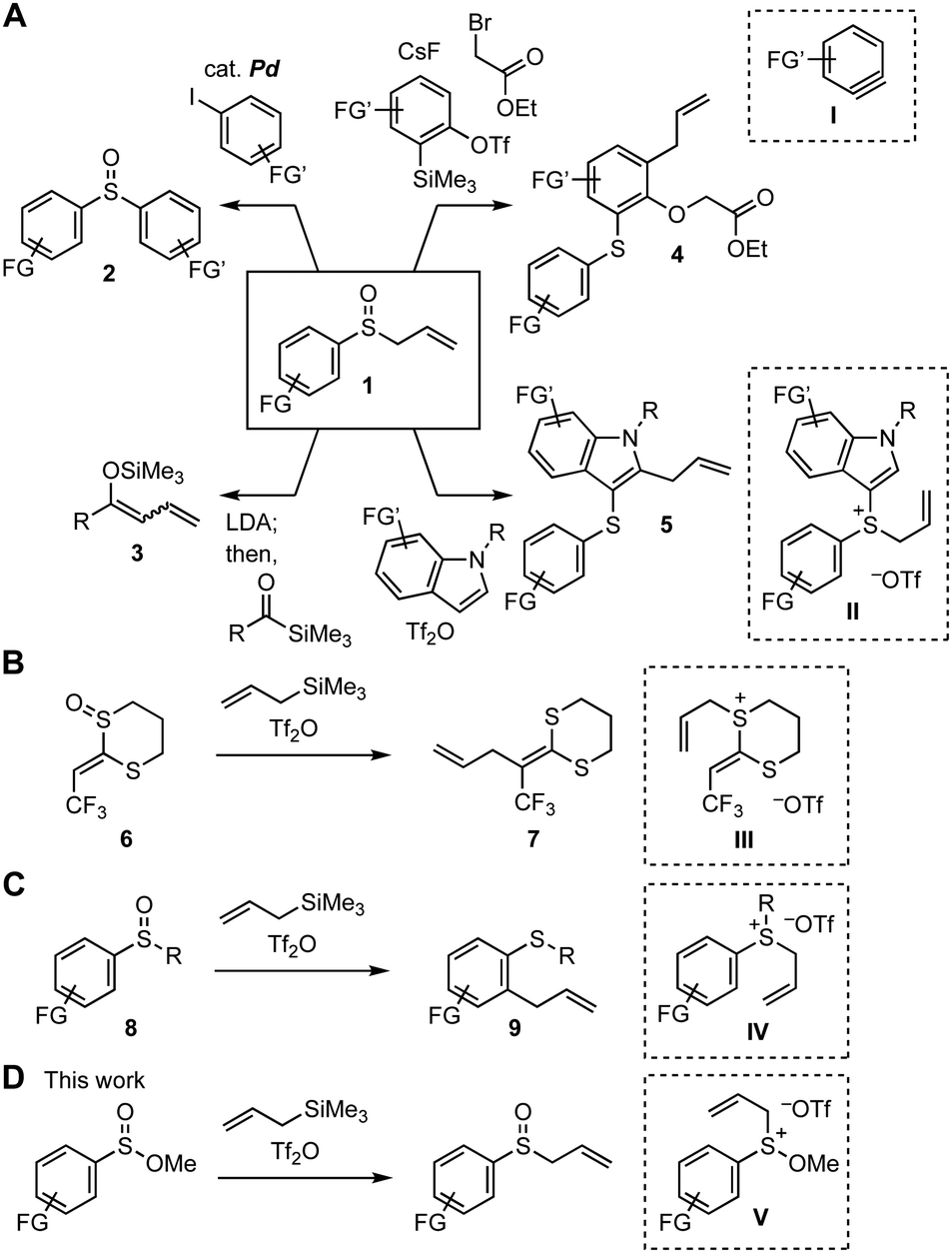 | ||
| Fig. 1 Transformations through the Pummerer-type activation of sulfoxides and sulfinate esters. (A) Versatile transformations using allyl sulfoxides 1. (B) Interrupted Pummerer reaction of ketenedithioacetal monoxide 6. (C) Interrupted Pummerer reaction of aromatic sulfoxide 8. (D) This work. | ||
Sulfinate esters hitherto have served in the sulfoxide synthesis with Grignard reagents.9a Recently, Lewis acid-mediated Friedel–Crafts-type sulfinylation of electron-rich arenes using sulfinate esters was developed.9b Taking the sulfinate ester chemistry into account, we envisioned that the Pummerer-type activation of sulfinate esters 10 in the presence of allylsilanes 11 and stability of methoxy sulfonium intermediates10 would allow for the facile synthesis of allyl sulfoxides 12, considering that the hydrolysis of methoxy sulfonium intermediates V′ can afford sulfoxides (Fig. 2A). As a result of screening the reaction conditions, we found that treatment of methyl benzenesulfinate (10a) with Tf2O in the presence of allyltrimethylsilane (11a) followed by addition of aqueous sodium bicarbonate provided allyl phenyl sulfoxide (12a) in high yield (Fig. 2A and 2B). Examinations using a variety of acid anhydrides or Lewis acids showed the remarkable reactivity of Tf2O in the S-allylation of sulfinate ester 10a.11,12 A wide range of allyl sulfoxides 12b–12j were prepared by the S-allylation under the Pummerer-like conditions, where C-allylation products through the [3,3]-sigmatropic rearrangement were not obtained. Indeed, not only electron-rich aromatic sulfinate esters bearing methyl and methoxy groups but also electron-deficient substrates with chloro and nitro groups were efficiently allylated to furnish sulfoxides 12b–12e. Sulfoxides 12f and 12g were obtained uneventfully by the reactions of bulky 2-bromo- and 2,6-dimethyl-substituted benzenesulfinate esters. Furthermore, S-allylations of 2-naphthyl-, benzyl-, and n-pentyl-substituted sulfinate esters also took place smoothly to provide sulfoxides 12h–12j.
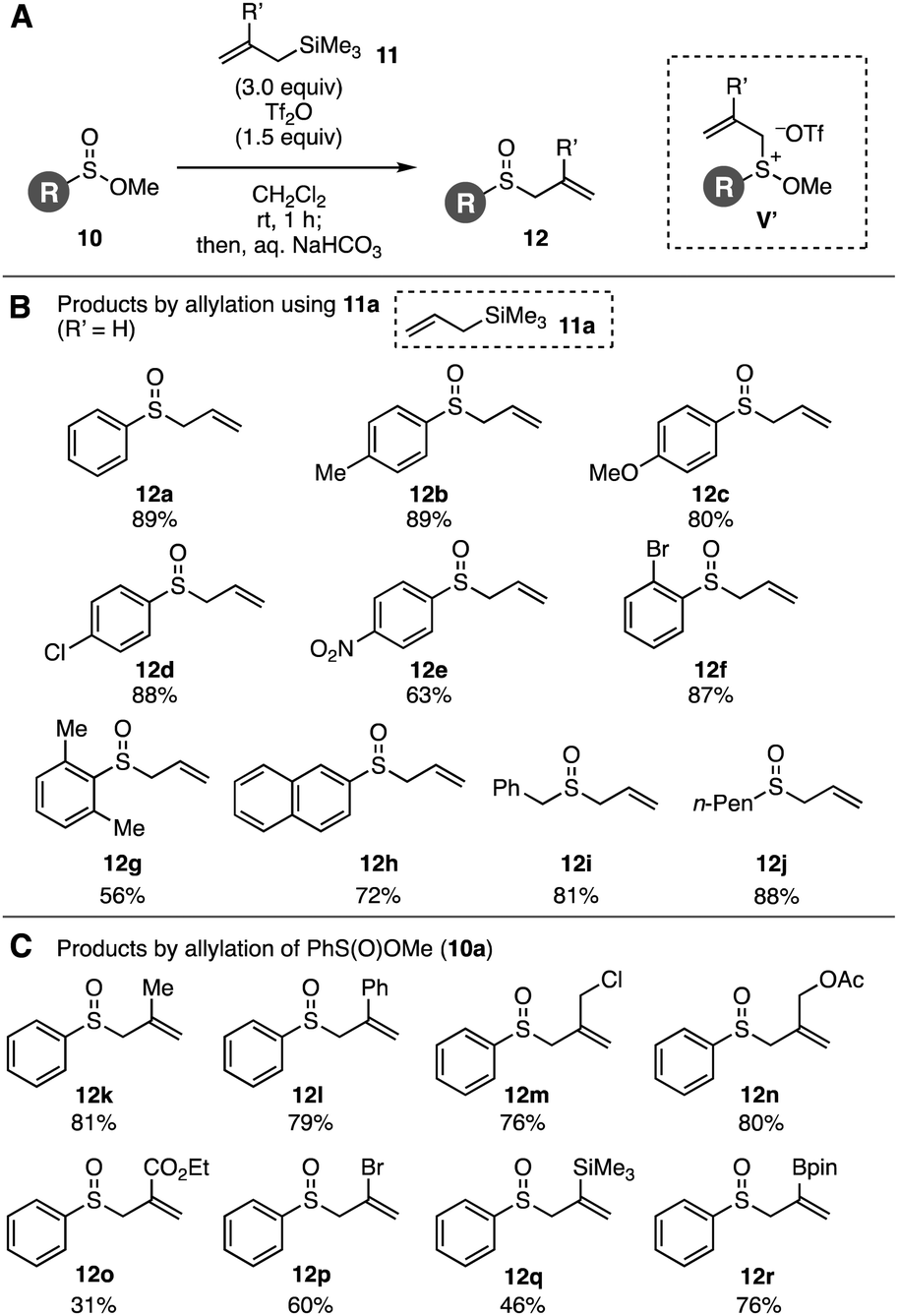 | ||
| Fig. 2 Allyl sulfoxide synthesis from sulfinate esters 10 and allylsilanes 11. (A) General scheme. (B) Results using various sulfinate esters 10 with 11a. (C) Results using allylsilanes 11 with 10a. | ||
Various functionalized allylsilanes 11 participated in the S-allylation of sulfinate ester 10a (Fig. 2A and C).13 Sulfoxides 12k and 12l were efficiently synthesized by 2-methyl- and 2-phenylallylation, respectively. It is worth noting that the C–S bond formation enabled to prepare allyl chloride 12m, allyl acetate 12n, ester 12o, and bromoalkene 12p leaving highly electrophilic functional groups untouched, while it is not easy to synthesize sulfoxides having electrophilic moieties by the conventional allyl sulfoxide synthesis via allylation of thiols and subsequent oxidation. Moreover, transformable sulfoxides 12q and 12r possessing a silyl and boryl groups were obtained in moderate to good yields without damaging these reactive functional groups.
To gain insight into the reaction mechanism of the S-allylation of sulfinate esters under the Pummerer-like conditions, we then examined control experiments (Fig. 3). Firstly, the reaction using [18O]H2O in the hydrolysis using aqueous sodium bicarbonate was conducted to clarify the origin of the oxygen atom of sulfoxide 12a (Fig. 3A). The result showed that 18O-incorporated 12a′ was obtained selectively, indicating that the sulfoxide oxygen was derived from water in the hydrolysis. We then attempted to isolate sulfonium intermediate 13 (Fig. 3B). As a result, after sulfinate ester 10a was treated with Tf2O in the presence of allylsilane 11a, an addition of solid sodium bicarbonate, filtration of the resulting mixture, removal of the solvent of the filtrate, and washing with diethyl ether afforded sulfonium salt 13 quantitatively. Hydrolysis of sulfonium salt 13 with aqueous sodium bicarbonate underwent uneventfully to give sulfoxide 12a. In addition, reduction of sulfonium salt 13 with sodium borohydride successfully provided allyl phenyl sulfide (14) in good yield.10c On the basis of these results, we proposed a reaction mechanism of the S-allylation (Fig. 3C). The Pummerer-type activation of sulfinate ester by virtue of the remarkable reactivity of Tf2O,14 and following S-allylation of the resulting sulfonium intermediate VI would furnish sulfonium intermediate 13 along with trimethylsilyl triflate. Then, hydrolysis of sulfonium salt 13 with aqueous sodium bicarbonate involving the nucleophilic attack of external water to the sulfur atom leads to sulfoxide 12a. Although the role of methoxy group is still unclear, the stability of sulfonium salt 13 would achieve the sulfoxide synthesis without [3,3]-sigmatropic rearrangement or the Pummerer-type reactions of allyl sulfoxide 12a and further Pummerer-type reactions of the resulting allyl sulfoxides.15
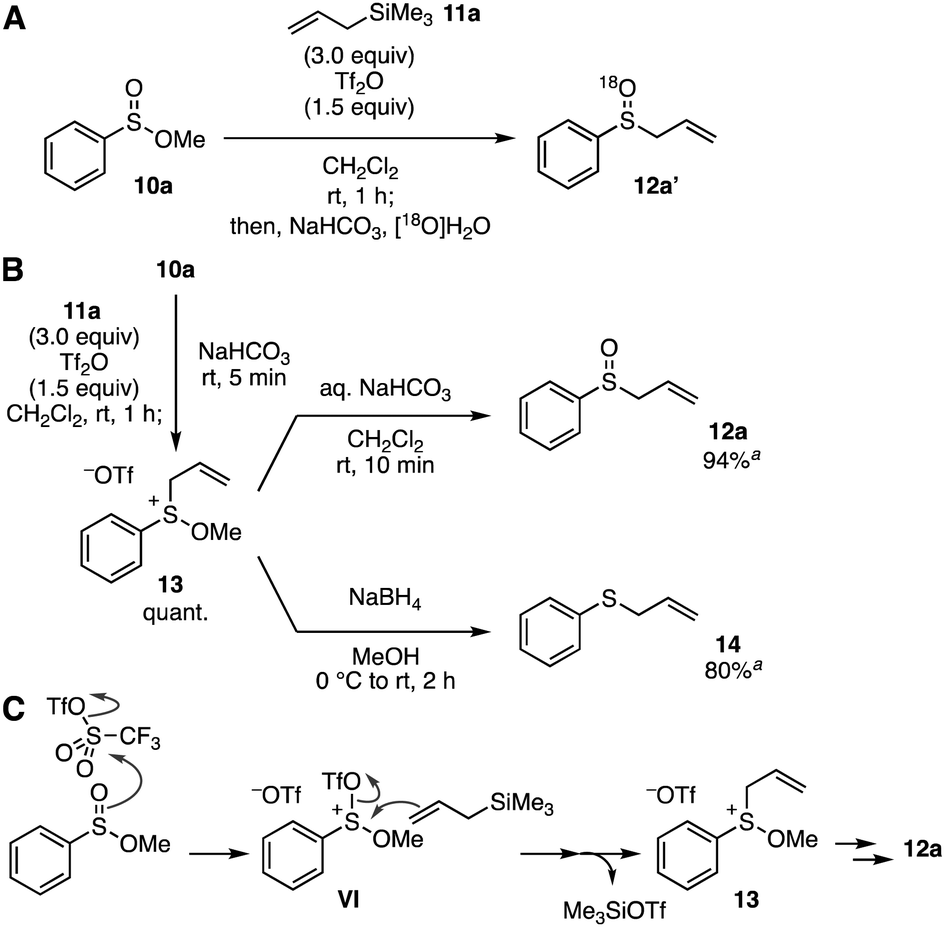 | ||
Fig. 3 Control experiments. (A) Reaction using [18O]H2O. (B) Isolation of sulfonium salt 13, hydrolysis of 13, and reduction of 13. (C) Plausible reaction mechanism. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1H NMR yield. 1H NMR yield. | ||
Our attention then directed toward novel transformations through the cationic intermediates generated by the Pummerer-type activation of sulfinate esters with Tf2O (Fig. 4). In this context, we have developed a facile synthetic method of alkynyl sulfoxides 16 using alkynyl silanes 15 (Fig. 4A). Indeed, treatment of sulfinate ester 10a dissolved in nitromethane with Tf2O in the presence of ethynylsilanes 15 at −20 °C furnished alkynyl sulfoxides 16 in moderate to high yields. This novel transformation enabled the preparation of alkynyl sulfoxides 16a–16c having a methyl, phenyl, and 4-anisyl group. Since alkynyl sulfoxides serve in a variety of reactions including carbometallation, [2+2] cycloaddition, and cyclopropanation, the alkynyl sulfoxide synthesis developed in this study would allow for the preparation of a range of organosulfur compounds.4k,16 In addition, Friedel–Crafts-type arylation of sulfinate ester 10a also took place smoothly to afford a regioisomeric mixture of diaryl sulfoxides 18 and 18′ in good yield (Fig. 4B).9b
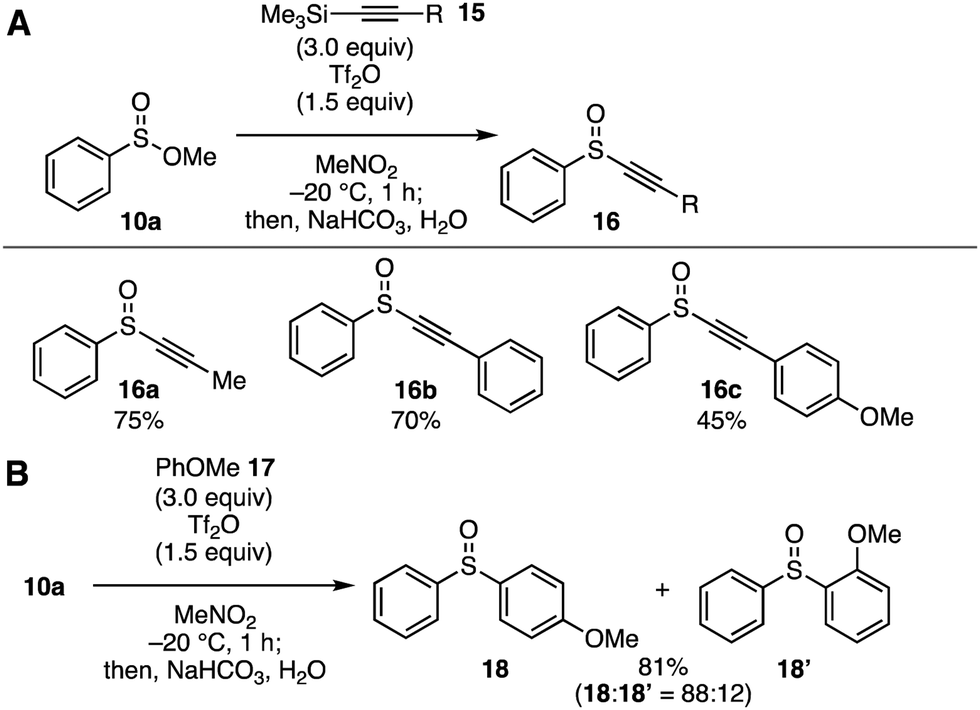 | ||
| Fig. 4 Alkynylation and arylation of sulfinate ester 10a. (A) Alkynylation with alkynylsilanes 15. (B) Arylation with anisole (17). | ||
Wide transformability of allyl aryl sulfoxides synthesized from sulfinate esters was showcased by the syntheses of multisubstituted aromatic compounds (Fig. 5). Modifying the conditions for the trifunctionalization of aryne intermediates reported by Li and coworkers6 (Fig. 1A, 1 to 4), we found that 2,3,6-trisubstituted phenol 20a was obtained in moderate yield with avoiding further arylation between phenol 20a and 3-methoxybenzyne when the aryne trifunctionalization was performed in hot 1,4-dioxane5d in the absence of electrophiles such as ethyl bromoacetate (Fig. 5A). Iodine-mediated cyclization of the resulting phenol 20a and subsequent elimination with a base successfully furnished benzofuran 21.17 Methallylation of sulfinate ester 10d followed by the aryne trifunctionalization led to the synthesis of highly functionalized phenol 20b (Fig. 5B). Furthermore, tetrasubstituted indole 23 was prepared through 2-bromoallylation of sulfinate ester 10c and following 2,3-difunctionalization of indole 22 according to the reports by Procter and coworkers7 (Fig. 5C). Functionalized allyl aryl sulfoxide 12t and indole 22 bearing o-silylaryl triflate moiety18 for the aryne generation participated in the 2,3-disubstituted indole synthesis leaving the reactive functional groups intact. Thus, a wide variety of indoles would be synthesized by S-allylation of sulfinate esters, 2,3-difunctionalization of indoles, and further transformations through indolyne intermediates with a number of arynophiles.18,19
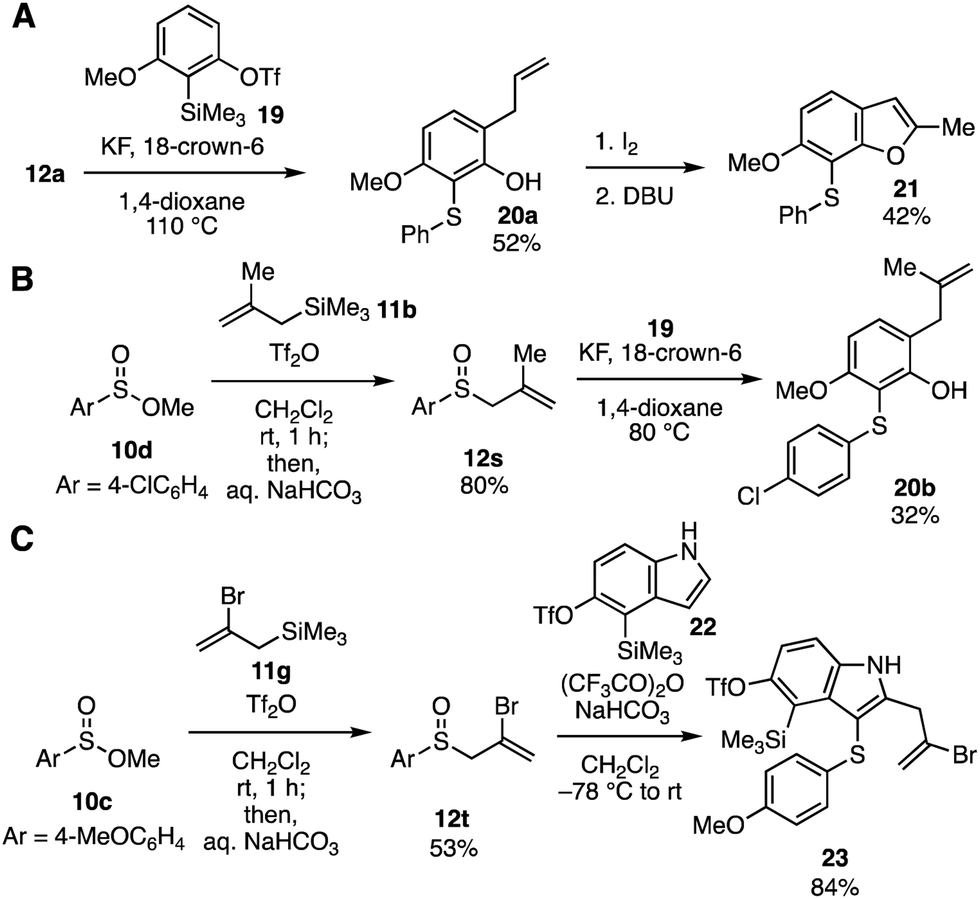 | ||
| Fig. 5 Transformations of allyl sulfoxides. (A) Benzofuran synthesis. (B) Trisubstituted phenol synthesis. (C) Multisubstituted indole synthesis. | ||
In summary, we have developed a facile synthetic method of allyl sulfoxides by S-allylation of sulfinate esters through sulfonium intermediates without [3,3]-sigmatropic rearrangement and further Pummerer-type reactions of the resulting allyl sulfoxides. On the basis of the plausible reaction mechanism, S-alkynylation and S-arylation were also accomplished. Further studies to expand the scope of these transformations using sulfinate esters under the Pummerer-like conditions, chiral sulfoxide synthesis, and the applications to the synthesis of bioactive compounds are now in progress.
The authors thank Dr Yuki Sakata at Tokyo Medical and Dental University for HRMS analyses. This work was supported by JSPS KAKENHI Grant Numbers JP19K05451 (C; S.Y.), JP18H02104 (B; T.H.), JP18H04386 (Middle Molecular Strategy; T.H.), and 19J14128 (JSPS Research Fellow; T.M.); the Naito Foundation (S.Y.); the Japan Agency for Medical Research and Development (AMED) under Grant Number JP19am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS); and the Cooperative Research Project of Research Center for Biomedical Engineering.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews of bioactive sulfur-containing compounds, see: (a) K. Pluta, B. Morak-Młodawska and M. Jeleń, Eur. J. Med. Chem., 2011, 46, 3179 CrossRef CAS PubMed; (b) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed.
- For selected reviews of sulfur-containing compounds in materials science, see: (a) A. S. Rahate, K. R. Nemade and S. A. Waghuley, Rev. Chem. Eng., 2013, 29, 471 CAS; (b) S. Dadashi-Silab, C. Aydogan and Y. Yagci, Polym. Chem., 2015, 6, 6595 RSC.
- (a) S. K. Bur and A. Padwa, Chem. Rev., 2004, 104, 2401 CrossRef CAS PubMed; (b) K. S. Feldman, Tetrahedron, 2006, 62, 5003 CrossRef CAS; (c) S. Akai and Y. Kita,, Top. Curr. Chem., 2007, 274, 35 CrossRef CAS; (d) L. H. S. Smith, S. C. Coote, H. F. Sneddon and D. J. Procter, Angew. Chem., Int. Ed., 2010, 49, 5832 CrossRef CAS PubMed; (e) X. Huang, S. Klimczyk and N. Maulide, Synthesis, 2012, 175 Search PubMed; (f) A. Shafir, Tetrahedron Lett., 2016, 57, 2673 CrossRef CAS; (g) A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842 CrossRef CAS PubMed; (h) H. Yorimitsu, Chem. Rec., 2017, 17, 1156 CrossRef CAS PubMed; (i) T. Yanagi, K. Nogi and H. Yorimitsu, Tetrahedron Lett., 2018, 59, 2951 CrossRef CAS; (j) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701 CrossRef CAS PubMed; (k) L. Zhang, M. Hu and B. Peng, Synlett, 2019, 2203 CAS.
- For selected examples, (a) S. Akai, N. Kawashita, H. Satoh, Y. Wada, K. Kakiguchi, I. Kuriwaki and Y. Kita, Org. Lett., 2004, 6, 3793 CrossRef CAS PubMed; (b) S. Yoshida, H. Yorimitsu and K. Oshima, Org. Lett., 2009, 11, 2185 CrossRef CAS PubMed; (c) X. Huang and N. Maulide, J. Am. Chem. Soc., 2011, 133, 8510 CrossRef CAS PubMed; (d) A. J. Eberhart, J. Cicoira, E. Imbriglio and D. J. Procter, Org. Lett., 2011, 13, 5882 CrossRef CAS PubMed; (e) A. J. Eberhart, C. Cicoira and D. J. Procter, Org. Lett., 2013, 15, 3994 CrossRef CAS PubMed; (f) M. Tayu, K. Higuchi, T. Ishizaki and T. Kawasaki, Org. Lett., 2014, 16, 361 CrossRef PubMed; (g) G. Hu, J. Xu and P. Li, Org. Lett., 2014, 16, 6036 CrossRef CAS PubMed; (h) L. Hu, Q. Gui, X. Chen, Z. Tan and G. Zhu, J. Org. Chem., 2016, 81, 4861 CrossRef CAS PubMed; (i) D. Chen, Q. Feng, Y. Yang, X.-M. Cai, F. Wang and S. Huang, Chem. Sci., 2017, 8, 1601 RSC; (j) L. Shang, Y. Chang, F. Luo, J.-N. He, X. Huang, L. Zhang, L. Kong, K. Li and B. Peng, J. Am. Chem. Soc., 2017, 139, 4211 CrossRef CAS PubMed; (k) D. Kaldre, I. Klose and N. Maulide, Science, 2018, 361, 664 CrossRef CAS PubMed; (l) L. Zhang, J.-N. He, Y. Liang, M. Hu, L. Shang, X. Huang, L. Kong, Z.-X. Wang and B. Peng, Angew. Chem., Int. Ed., 2019, 58, 5316 CrossRef CAS PubMed; (m) K. Okamoto, M. Hori, T. Yanagi, K. Murakami, K. Nogi and H. Yorimitsu, Angew. Chem., Int. Ed., 2019, 58, 7813 CrossRef PubMed; (n) J. Yan, A. P. Pulis, G. J. P. Perry and D. J. Procter, Angew. Chem., Int. Ed., 2019, 58, 15675 CrossRef CAS PubMed; (o) X. Meng, D. Chen, X. Cao, J. Luo, F. Wang and S. Huang, Chem. Commun., 2019, 55, 12495 RSC; (p) J. Li, Y. Chen, R. Zhong, Y. Zhang, J. Yang, H. Ding and Z. Wang, Org. Lett., 2020, 22, 1164 CrossRef CAS PubMed; (q) Z. He, G. J. Perry and D. J. Procter, Chem. Sci., 2020, 11, 2001 RSC; (r) X. Huang, Y. Zhang, W. Liang, Q. Zhang, Y. Zhan, L. Kong and B. Peng, Chem. Sci., 2020, 11, 3048 RSC.
- For our previous reports on sulfoxides chemistry, see: (a) S. Yoshida, K. Uchida and T. Hosoya, Chem. Lett., 2014, 43, 116 CrossRef CAS; (b) S. Yoshida, K. Uchida and T. Hosoya, Chem. Lett., 2015, 44, 691 CrossRef CAS; (c) S. Yoshida, F. Karaki, K. Uchida and T. Hosoya, Chem. Commun., 2015, 51, 8745 RSC; (d) T. Matsuzawa, K. Uchida, S. Yoshida and T. Hosoya, Org. Lett., 2017, 19, 5521 CrossRef CAS PubMed; (e) Y. Nakamura, Y. Miyata, K. Uchida, S. Yoshida and T. Hosoya, Org. Lett., 2019, 21, 5252 CrossRef CAS PubMed.
- Y. Li, D. Qiu, R. Gu, J. Wang, J. Shi and Y. Li, J. Am. Chem. Soc., 2016, 138, 10814 CrossRef CAS PubMed.
- M. Šiaučiulis, S. Sapmaz, A. P. Pulis and D. J. Procter, Chem. Sci., 2018, 9, 754 RSC.
- (a) R. Hunter and C. D. Simon, Tetrahedron Lett., 1986, 27, 1385 CrossRef CAS; (b) Y. Kita, O. Tamura, F. Itoh, H. Yasuda, T. Miki and Y. Tamura, Chem. Pharm. Bull., 1987, 35, 562 CrossRef CAS; (c) D. H. Hua, M. J. Coulter and I. Badejo, Tetrahedron Lett., 1987, 28, 5465 CrossRef CAS; (d) M. R. Binns, R. K. Haynes, A. G. Katsifis, P. A. Schober and S. C. Vonwiller, J. Am. Chem. Soc., 1988, 110, 5411 CrossRef CAS; (e) R. Hunter and C. D. Simon, Tetrahedron Lett., 1988, 29, 2257 CrossRef CAS; (f) E. Alonso, D. Guijarro and M. Yus, Tetrahedron, 1995, 51, 2699 CrossRef CAS; (g) W. A. Loughlin and M. A. McCleary, Synthesis, 2005, 761 CrossRef CAS; (h) E. Bernoud, G. Le Duc, X. Bantreil, G. Prestat, D. Madec and G. Poli, Org. Lett., 2010, 12, 320 CrossRef CAS PubMed; (i) S. Fustero, S. Catalán, M. Sánchez-Roselló, A. Simón-Fuentes and C. del Pozo, Org. Lett., 2010, 12, 3484 CrossRef CAS PubMed; (j) M. Honda, T. Nakajima, M. Okada, K. Yamaguchi, M. Suda, K.-K. Kunimoto and M. Segi, Tetrahedron Lett., 2011, 52, 3740 CrossRef CAS; (k) Z. Huang and J. Xu, RSC Adv., 2013, 3, 15114 RSC; (l) D. Qiu, J. Shi, Q. Guo, Q. Xu, B. Li and Y. Li, J. Am. Chem. Soc., 2018, 140, 13214 CrossRef CAS PubMed.
- For the sulfoxide synthesis with Grignard reagents, see: (a) H. Gilman, J. Robinson and N. J. Beaber, J. Am. Chem. Soc., 1926, 48, 2715 CrossRef CAS ; For recent transformations of sulfinate esters, see: ; (b) F. Yuste, A. H. Linares, V. M. Mastranzo, B. Ortiz, R. Sánchez-Obregón, A. Fraile, J. Luis and G. Ruano, J. Org. Chem., 2011, 76, 4635 CrossRef CAS PubMed; (c) J. A. Lujan-Montelongo, A. O. Estevez and F. F. Fleming, Eur. J. Org. Chem., 2015, 1602 CrossRef CAS PubMed; (d) N.-L. T. Nguyen, H.-T. Vo, F. Duus and T. X. T. Luu, Molecules, 2017, 22, 1458 CrossRef PubMed; (e) A. Mohd, T. Anitha, K. R. Reddy, J. Wencel-Delord and F. Colobert, Eur. J. Org. Chem., 2019, 7836 CrossRef CAS; (f) G.-J. Li, Y.-L. Pan, Y.-L. Liu, H.-F. Xu and J.-Z. Chen, Tetrahedron Lett., 2019, 60, 151260 CrossRef CAS; (g) L. Chen, J. Zhang, Y. Wei, Z. Yang, P. Liu, J. Zhang and B. Dai, Tetrahedron, 2019, 75, 130664 CrossRef.
- (a) C. R. Johnson and W. G. Phillips, Tetrahedron Lett., 1965, 6, 2101 CrossRef; (b) C. R. Johnson and W. G. Phillips, J. Org. Chem., 1967, 32, 1926 CrossRef CAS; (c) C. R. Johnson and W. G. Phillips, J. Org. Chem., 1967, 32, 3233 CrossRef CAS.
- See the ESI† for the details.
- The reaction at 80 °C in a sealed tube also provided sulfoxide 12a, where [3,3]-sigmatropic rearrangement did not proceed.
- The reaction using a γ-phenyl-substituted allylsilane afforded a complex mixture of products.
- For recent examples, see:
(a) Y. Unoh, K. Hirano and M. Miura, J. Am. Chem. Soc., 2017, 139, 6106 CrossRef CAS PubMed;
(b) H. Huang, J. Ash and J. Y. Kang, Org. Lett., 2018, 20, 4938 CrossRef CAS PubMed;
(c) C. R. Gonc
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) lves, M. Lemmerer, C. J. Teskey, P. Adler, D. Kaiser, B. Maryasin, L. Gonzaĺez and N. Maulide, J. Am. Chem. Soc., 2019, 141, 18437 CrossRef PubMed;
(d) J. Wang, Y.-J. Deng, X.-X. Yan, Y.-J. Liu, C.-P. Ge, Y. Yan, S. Chao and P.-X. Zhou, Org. Chem. Front., 2020, 7, 715 RSC.
lves, M. Lemmerer, C. J. Teskey, P. Adler, D. Kaiser, B. Maryasin, L. Gonzaĺez and N. Maulide, J. Am. Chem. Soc., 2019, 141, 18437 CrossRef PubMed;
(d) J. Wang, Y.-J. Deng, X.-X. Yan, Y.-J. Liu, C.-P. Ge, Y. Yan, S. Chao and P.-X. Zhou, Org. Chem. Front., 2020, 7, 715 RSC. - Although detailed studies involving the theoretical calculation should be performed, the resonance effect of the methoxy group might prevent the [3,3]-sigmatropic rearrangement leading to the sulfenate.
- (a) J. P. Marino, M. B. Rubio, G. Cao and A. de Dios,, J. Am. Chem. Soc., 2002, 124, 13398 CrossRef CAS PubMed; (b) G. Sklute, D. Amsallem, A. Shabli, J. P. Varghese and I. Marek, J. Am. Chem. Soc., 2003, 125, 11776 CrossRef CAS PubMed; (c) N. Maezaki, S. Yagi, R. Yoshigami, J. Maeda, T. Suzuki, S. Ohsawa, K. Tsukamoto and T. Tanaka, J. Org. Chem., 2003, 68, 5550 CrossRef CAS PubMed; (d) Q. Xu and X. Huang, Tetrahedron Lett., 2004, 45, 5657 CrossRef CAS; (e) G. Sklute and I. Marek, J. Am. Chem. Soc., 2006, 128, 4642 CrossRef CAS PubMed; (f) F. Sandrinelli, C. Boudou, C. Caupène, M.-T. Averbuch-Pouchot, S. Perrio and P. Metzner, Synlett, 2006, 3289 CAS; (g) G. Zhang and L. Zhang, J. Am. Chem. Soc., 2008, 130, 12598 CrossRef CAS PubMed; (h) J. Wei and Z. Sun, Org. Lett., 2015, 17, 5396 CrossRef CAS PubMed; (i) M. J. Barrett, G. F. Khan, P. W. Davies and R. S. Grainger, Chem. Commun., 2017, 53, 5733 RSC; (j) B. Alcaide, P. Almendros and C. Lázaro-Milla, Adv. Synth. Catal., 2017, 359, 2630 CrossRef CAS.
- A. K. Yadav, B. K. Singh, N. Singh and R. P. Tripathi, Tetrahedron Lett., 2007, 48, 6628 CrossRef CAS.
- G.-Y. J. Im, S. M. Bronner, A. E. Goetz, R. S. Paton, P. H.-Y. Cheong, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2010, 132, 17933 CrossRef CAS PubMed ; and references therein.
- For selected reviews of arynes, see: (a) S. Yoshida and T. Hosoya, Chem. Lett., 2015, 44, 1450 CrossRef CAS; (b) J. Shi, Y. Li and Y. Li, Chem. Soc. Rev., 2017, 46, 1707 RSC; (c) F. I. M. Idiris and C. R. Jones, Org. Biomol. Chem., 2017, 15, 9044 RSC; (d) H. Takikawa, A. Nishii, T. Sakai and K. Suzuki, Chem. Soc. Rev., 2018, 47, 8030 RSC; (e) T. Roy and A. T. Biju, Chem. Commun., 2018, 54, 2580 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization for new compounds including NMR spectra. See DOI: 10.1039/d0cc02253e |
| This journal is © The Royal Society of Chemistry 2020 |