
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicroscope laser assisted photooxidative activation of bioorthogonal ClickOx probes†
Attila
Kormos‡
 ,
Dóra
Kern‡
,
Alexandra
Egyed
,
Bianka
Söveges
,
Krisztina
Németh
and
Péter
Kele
*
,
Dóra
Kern‡
,
Alexandra
Egyed
,
Bianka
Söveges
,
Krisztina
Németh
and
Péter
Kele
*
Chemical Biology Research Group, Institute of Organic Chemistry, Research Centre for Natural Sciences, Magyar tudósok krt. 2, Budapest H-1117, Hungary. E-mail: kele.peter@ttk.hu
First published on 6th April 2020
Abstract
A photoactivatable fluorogenic tetrazine-rhodaphenothiazine probe was synthesized and studied in light-assisted, bioorthogonal labeling schemes. Experimental results revealed that the bioorthogonally conjugated probe efficiently sensitizes 1O2 generation upon illumination with green or orange light and undergoes self-oxidation leading to an intensely fluorescent sulfoxide product. An added value of the present probe is that it is also suitable for STED super-resolution microscopy using a 660 nm depletion laser.
Due to recent developments, fluorescence microscopy has become the primary technique for the visualization of cellular structures and events because of its excellent sensitivity and spatiotemporal resolution. Thus, it has grown into an inevitable tool in studies aiming at understanding biological processes.1 Its importance has become even more profound since the advent of super-resolution techniques. The emerging subdiffraction methods enable visualization of biological structures in details never seen before.2 Parallel to the evolution of optical microscopy techniques, chemical biology methods have also gone through remarkable developments, which led to advanced labeling techniques.3 Fusion of engineered fluorescent proteins (FPs) to the protein of interest (POI) is routinely accomplished and enables highly selective labeling; however, the photophysical characteristics of FPs often limit resolution. Though the use of small, chemically tailored probes with improved photophysical features is also possible through fusion tags (e.g. halo-, snap- or clip-tags), similar to FPs, the comparable size of these tags often hampers the native function of the POI. Furthermore, they also impair precise localization of the protein of interest due to linkage error. Moreover, the use of FPs and other fusion tags is limited (mostly) to the terminal labeling of proteins only. Site-specific incorporation of non-canonical amino acids bearing a bioorthogonal function, via genetic code expansion, enables much greater flexibility in terms of labeling sites and highly selective installation of small, minimally perturbing synthetic probes carrying a complementary bioorthogonal motif.4 A further advantage of bioorthogonal labeling schemes is that they are not limited to biomolecules directly encoded in the genome and they allow probing of nucleic acids, lipids, sugars, etc. as well. One of the most popular bioorthogonal reactions in labeling schemes is the inverse electron demand Diels–Alder reaction of tetrazine dienes and strained dienophiles (e.g. cyclooctynes and trans-cyclooctenes).5 An extra feature of the tetrazine moiety, besides being an excellent bioorthogonal platform, is its capability of quenching fluorescence of fluorophores. This feature can be harvested in the development of turn-on (fluorogenic) probes as a chemical transformation of the tetrazine motif results in reinstated emission intensity (reactivity-based fluorogenicity).6 When it comes to the design of such bioorthogonally applicable small synthetic probes, features such as suitable excitation and emission maxima, brightness, quantum yields, photostability, membrane permeability, etc. are all on the wish list.7a Besides these, improved probes should also address problems often associated with fluorescence imaging using small, synthetic probes. Such problems are auto- and background fluorescence.7 While the former one is relatively easily addressed by selecting frames excitable towards the red end of the spectrum, or having large Stokes shifts, reduction of background fluorescence either requires extensive washing cycles or, more elegantly, the use of fluorogenic probes, whose fluorescence intensity increases remarkably upon selective ligation to the biomolecule of interest. We and others recently reported on improved probes, where the two-in-one feature of the tetrazine motif, i.e. being bioorthogonal and a quencher of fluorescence at the same time, was exploited in the development of bioorthogonally applicable fluorogenic probes.6b,8 It was observed, however, that the fluorescence increase upon bioorthogonal ligation, i.e. fluorogenicity, dramatically decreases towards the biologically preferred red range of the spectrum. In order to access better fluorogenic probes in this spectral range, we have introduced the concept of multiple fluorogenicity, where we either increased the number of quencher units (e.g. two tetrazines)9 or combined different quenching mechanisms10 (e.g. tetrazine and polarity driven valence tautomerism). Though these multiply fluorogenic probes have improved fluorogenicities in the red regime, their applicability is limited either to special sequences or microenvironments. Thus, new approaches that could facilitate the design of generally applicable, visible-light excitable probes with improved fluorogenicity are still needed. In the quest of improved fluorogenic probes, we searched for alternative mechanisms that can strengthen the moderate reactivity based fluorogenicity of tetrazinylated probes in the green-red range. During tetrazine-based ligation schemes, the highly electron deficient tetrazine function is transformed into a dihydropyridazine or pyridazine, depending on the dienophile. Such fine changes in electronic properties can promote profound differences in reactivities, e.g. in oxidizability, between reacted and non-reacted forms. More particularly, we intended to use light to induce such oxidation. We further envisioned that transformation of an appending tetrazine moiety to a pyridazine, upon bioorthogonal ligation, leads to a less electron deficient congener that is more susceptible to light-induced oxidation. This holds promise for the primary switchability of the specifically reacted, i.e. biomolecule-conjugated, forms, enabling facile distinction from non-specifically bound, unreacted species, which overall translates to a new fluorogenic mechanism. Prompted by this, we sought frames whose structure is likely to be oxidized upon encountering light resulting in altered photophysical properties. Phenothiazines, e.g. chlorpromazine or methylene blue, are known for their singlet oxygen generating properties upon irradiation with light.11 Oliveros et al. reported on the photooxidation of 10-methyl phenothiazine. Their study revealed that the process involved self-sensitized 1O2 leading to 10-methyl phenothiazine sulfoxide, possessing distinct spectral maxima.12a Other instances reported on sensitizer (e.g. Rose-Bengal)-mediated photoxidation of N-ethylphenothiazine.12b To test our hypothesis, we designed rhodamine fused phenothiazines 1a and b (Fig. 1, and ESI†).13 We have also prepared sulfoxides 10a and 10b for control experiments. With the probes in hand, we next examined their spectral properties together with their in situ formed click-reaction products with a strained cyclooctyne ((1R,8S,9s)-bicyclo[6.1.0]non-4-yn-9-ylmethanol, BCN). Not much difference in intensities could be seen between the click-conjugates and their parent tetrazines, indicating that tetrazine-dependent, reactivity-based fluorogenicity is very poor in this spectral range (Table 1). However, a considerable blue-shift of spectral maxima and a two-orders of magnitude increase in emission intensities were seen in the case of the oxidized forms compared to their respective reduced congeners, irrespective of being clicked or not. Next, we elaborated whether the oxidized species, 10, and their BCN conjugates can be accessed upon irradiation of probes 1 with light. To this end, we have illuminated oxygenated solutions (methanol was used as solvent to obtain a reasonable concentration range necessary for the photophysical studies) of 1a and b and their respective in situ formed BCN conjugates with orange (around 600 nm) and green (around 540 nm) LEDs (for the LED emission spectra, see Fig. S1, ESI†). To our delight, in both cases, the formation of the respective oxidized products was evident, as indicated by LC-MS and the emission spectra (Fig. S2, ESI†). In the presence of a known 1O2 scavenger, sodium azide, however, no photooxidation was observed (Fig. S3, ESI†).14 At the same time, no oxidation occurred when deoxygenated samples were illuminated or when oxygenated samples were kept in the dark. These results confirm that light-triggered production of 1O2 sensitized by probes 1a and 1b or their BCN conjugates is responsible for the oxidation process (Fig. S4, ESI†). The formation of 1O2 was further confirmed using the RNO method (Fig. S5, ESI†).15 We compared the photooxidative characteristics of probes 1a and 1b with those of their BCN-conjugates. The gratifying results showed a marked difference between the photoconversion rate of the clicked conjugates and the parent tetrazines. For example, when probe 1a was irradiated with green light for 2 h, only a 20% photoproduct could be observed. However, when click conjugate 1a.BCN was irradiated with the same light source for 2 h, 90% of the starting material was converted to the respective oxidized product, as indicated by LC-MS (Fig. S6, ESI†). This confirms our hypothesis regarding electronic changes upon click reaction. Upon prolonged irradiation of 1, the appearance of so called dealkylated photobluing products16 together with unidentifiable byproducts was observed (Fig. S7 and Table S1, ESI†).
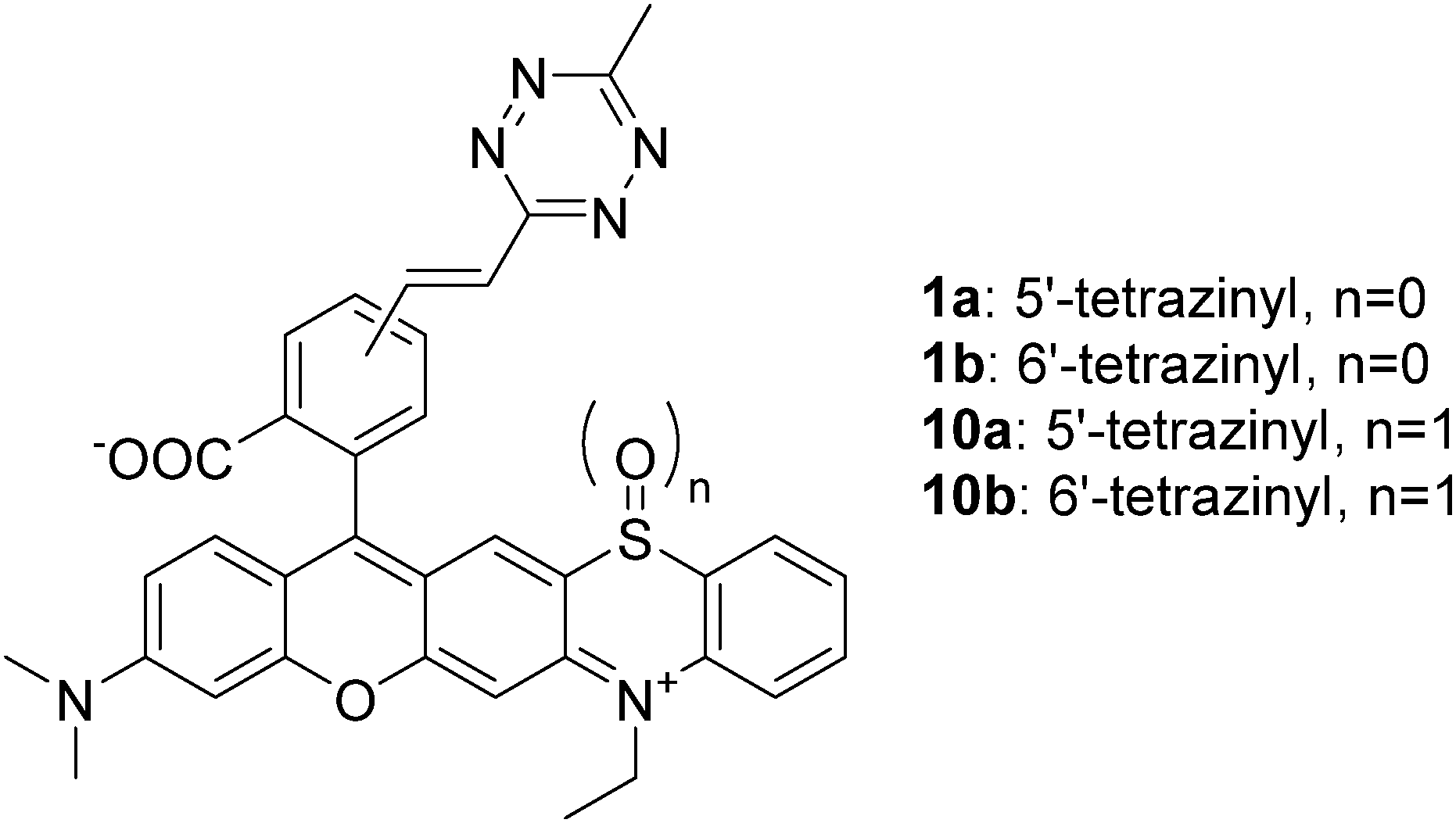 | ||
| Fig. 1 Structures of probes 1 and 10. | ||
| λ exc (nm) | λ em (nm) | ε (M−1 cm−1) | Φ | Φ BCN /Φtet | Φ 10.BCN /Φ1 | |
|---|---|---|---|---|---|---|
| a Complete consumption of tetrazines and formation of BCN conjugates were monitored by LC-MS. | ||||||
| 1a | 608 | 743 | 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
0.0037 | 1.19 | 154 |
| 1a.BCN | 608 | 743 | 32100 |
0.0044 | ||
| 10a | 554 | 588 | 41200 |
0.31 | 1.84 | |
| 10a.BCN | 553 | 586 | 42900 |
0.57 | ||
| 1b | 610 | 743 | 39300 |
0.0036 | 1.17 | 158 |
| 1b.BCN | 607 | 743 | 37100 |
0.0042 | ||
| 10b | 554 | 584 | 35600 |
0.21 | 2.7 | |
| 10b.BCN | 551 | 583 | 35800 |
0.57 | ||
The finding that BCN conjugated products were more prone to photooxidation, leading to a product emitting around 600 nm upon LED illumination, can be translated to selective photooxidation of specifically click-conjugated probes, 1a and b, in cellular labeling schemes. This implies a special fluorogenic mechanism as unreacted or non-specifically bound probes remain mainly in their weakly emissive reduced form, emitting above 650 nm, thus minimally contributing to background fluorescence upon microscope imaging in the 565–635 nm range, even under no-wash conditions. Upon examination of the excitation and emission bands of 1 and 10.BCN (Fig. 2 and Fig. S8, ESI†), we envisioned that the commercially available green laser (552 nm) used in confocal microscopes is suitable to promote photooxidation and subsequent excitation of the photoproducts for imaging. Excitation in the orange range is also suitable to carry out the oxidation step, however the product requires green excitation. Thus, excitation with a green laser seems an ideal two-in-one combination.
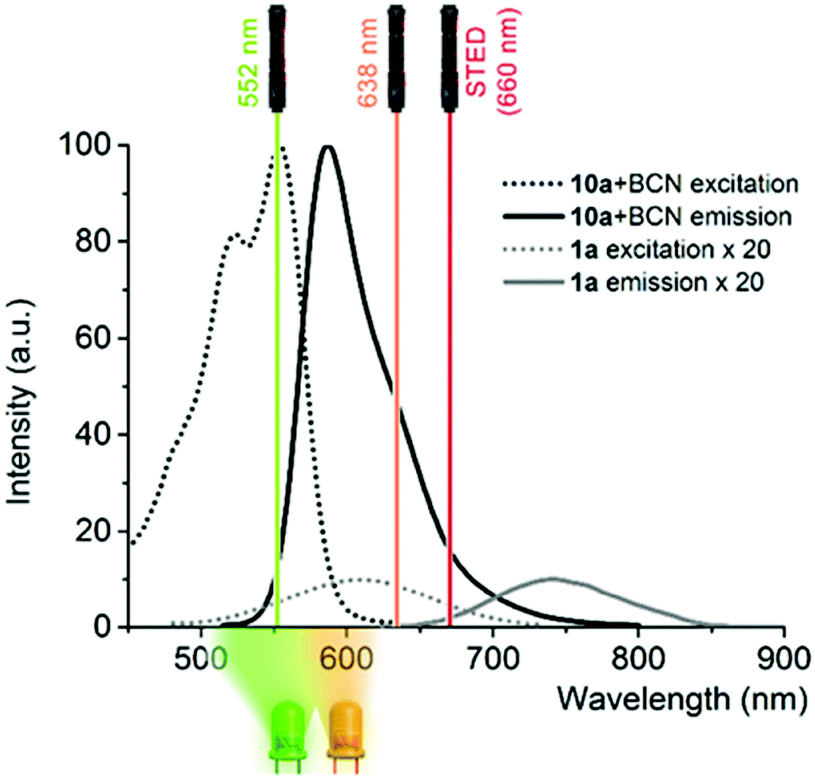 | ||
| Fig. 2 Excitation and emission spectra of 1a and 10a-BCN in PBS-SDS with the LED emission ranges used for the photooxidation experiments together with commercial microscope laser lines (note that the spectra of 1a are magnified 20× for better comparison). | ||
These considerations prompted us to carry out a proof-of-principle study with probe 1. Towards this end, bundles of actin filaments of fixed COS7 cells were tagged with a cyclooctynylated (BCN) affinity tag, BCN-phalloidin. Excess of the affinity tag was removed, then the samples were stained with probe 1a and washed. Cells were then imaged under a confocal microscope.
Excitation at 638 nm with a 652–760 nm detection window (i.e. the spectral range suitable to excite and detect probe 1a) resulted in poor image quality with no structural details. Similar image quality was obtained when the same detection window was set at 552 nm excitation. However, when the samples were excited at 552 nm and detected between 565 and 653 nm (i.e. the spectral range suitable to image probe 10), nice images of the actin filament network appeared immediately. These results confirm that the green laser used for excitation is suitable both for immediate photooxidation and subsequent excitation of the specifically conjugated, oxidized probes. Next, we wished to explore whether the marked difference in photooxidizability between the reacted and unreacted forms seen in solution allows selective activation of the specifically conjugated probes. Therefore, we repeated the above experiment; however, at this time, excess of probe 1a was not washed out (Fig. 3). The gratifying results showed similar images to the washed samples. This indicates that primarily, the clicked probes are oxidized by the laser. We also carried out the labeling experiments with the oxidized form, 10a. As expected, low quality images of the actin network were acquired due to the considerable background fluorescence of the non-specifically bound, unreacted probes (Fig. S11, ESI†). These observations further confirm that the specifically bound, clicked products are selectively photooxidized to result in intensely fluorescent products by the green laser, while unreacted tetrazine probes mostly remain in their poorly fluorescent reduced form contributing minimally to background fluorescence. The emission spectrum of 10a.BCN (Fig. 2) suggests that such clicked, oxidized probes (ClickOx) could even be suitable for super-resolution, STED microscopy imaging using a commercial 660 nm depletion laser. Indeed, when we tested the cells stained with probe 1a at their actin filament network, subdiffraction resolution was achieved with STED microscopy using a 552 nm laser for excitation and a CW 660 nm laser for depletion (Fig. 4).
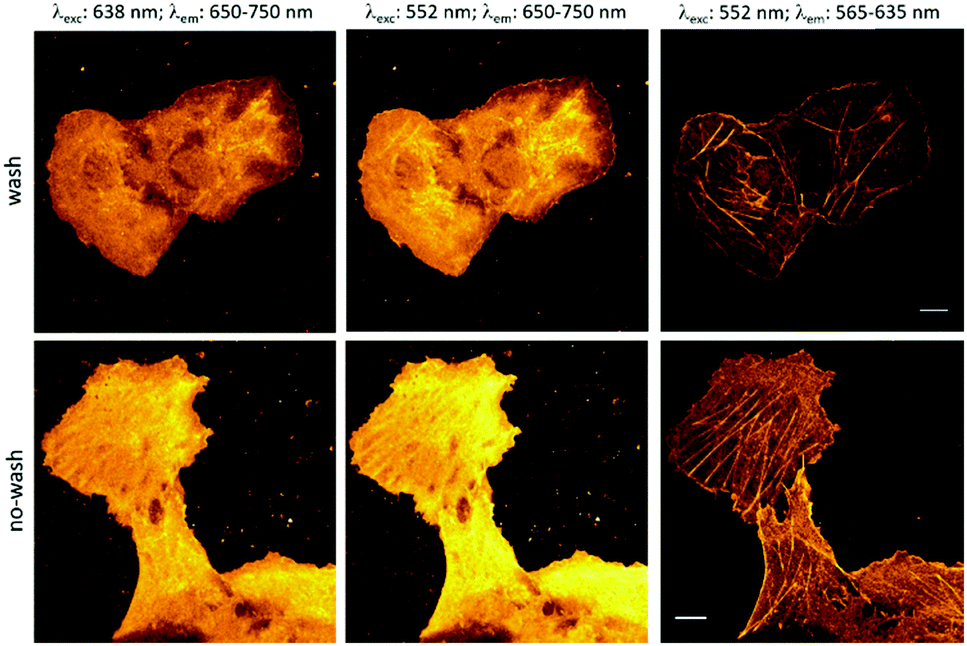 | ||
| Fig. 3 Actin filament staining. COS7 cells were tagged with cyclooctynylated phalloidin, then labeled with 1a (scale bar: 10 μm). | ||
 | ||
| Fig. 4 Confocal and STED images of BCN-phalloidin tagged actin filaments stained with 1a (λexc = 552 nm, and λSTED = 660 nm (CW); FWHM: 374 ± 127 vs. 139 ± 9 nm for confocal and STED images, respectively). | ||
In conclusion, we have designed and synthesized bioorthogonally applicable, tetrazine functionalized rhodaphenothiazines and elaborated their light-assisted photooxidation-based fluorogenic characteristics. Experimental results suggest that the probes efficiently sensitize 1O2 generation upon illumination with green or orange light. Furthermore, the produced singlet oxygen promotes self-oxidation of the probes leading to intensely fluorescent sulfoxide products. Further studies revealed a marked difference between the photooxidizability of the free tetrazine and the bioconjugated pyridazine forms, which implies a two-orders of magnitude fluorescence increase due to this new fluorogenic mechanism. This unique fluorogenic behavior allows selective photoactivation of specifically conjugated probes, as demonstrated in the labeling schemes of actin filaments with low background fluorescence even under no-wash conditions. A remarkable feature of the developed “ClickOx” probes is that the same commercial green excitation laser (552 nm) is suitable for carrying out photooxidation of the probes and subsequent excitation of the product. An added value of the present probe is that it is also suitable for STED super-resolution microscopy using a 660 nm depletion laser. Since it is the lack of availability of good probes that is considered among the limitations of further improvements of resolution, we believe that discovery of new mechanisms and design elements such as the presented photo-activation mechanism is an important step towards improved probes. Currently, we are exploring the potential of the new ClickOx probe in targeted photodynamic therapy and in light-assisted indication of oxidative stress-triggered inflammation in cells and tissues, the results of which are to be published in due course.
The present work was supported by the “Lendület” Program of the Hungarian Academy of Sciences (LP2013-55/2013) and the National Research, Development and Innovation Office (NKFIH-K-123917). DK is grateful for the support of the ÚNKP-19-2 New National Excellence Program of the Ministry for Innovation and Technology.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) E. A. Specht, E. Braselmann and A. E. Palmer, Annu. Rev. Physiol., 2017, 79, 93–117 CrossRef CAS PubMed; (b) P. O. Bayguinov, D. M. Oakley, C. C. Shih, D. J. Geanon, M. S. Joens and J. A. J. Fitzpatrick, Curr. Protoc. Cytom., 2018, 85, e39 CrossRef PubMed.
- (a) J. Vangindertael, R. Camacho, W. Sempels, H. Mizuno, P. Dedecker and K. P. F. Janssen, Methods Appl. Fluoresc., 2018, 6, 022003 CrossRef CAS PubMed; (b) D. Baddeley and J. Bewersdorf, Annu. Rev. Biochem., 2018, 87, 965–989 CrossRef CAS; (c) G. Huszka and M. A. M. Gijs, Microsyst. Nanoeng., 2019, 2, 7–28 Search PubMed; (d) S. J. Sahl, S. W. Hell and S. Jakobs, Nat. Rev. Mol. Cell Biol., 2017, 18, 685–701 CrossRef CAS PubMed.
- (a) C. Li, A. G. Tebo and A. Gautier, Int. J. Mol. Sci., 2017, 18, 1473 CrossRef PubMed; (b) G. Zhang, S. Zheng, H. Liu and P. R. Chen, Chem. Soc. Rev., 2015, 44, 3405–3417 RSC.
- (a) K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed; (b) I. Nikić, T. Plass, O. Schraidt, J. Szymański, J. A. Briggs, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2014, 53, 2245–2249 CrossRef PubMed; (c) J. W. Chin, Annu. Rev. Biochem., 2014, 83, 379–408 CrossRef CAS PubMed.
- (a) E. Kozma, O. Demeter and P. Kele, ChemBioChem, 2017, 18, 486–501 CrossRef CAS PubMed; (b) A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- (a) E. Kozma and P. Kele, Org. Biomol. Chem., 2019, 17, 215–233 RSC; (b) A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, Chem. Sci., 2017, 8, 1506–1510 RSC; (c) J. C. Carlson, L. G. Meimetis, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2013, 52, 6917–6920 CrossRef CAS PubMed.
- (a) L. Wang, M. S. Frei, A. Salim and K. J. Johnsson, J. Am. Chem. Soc., 2019, 141, 2770–2781 CrossRef CAS PubMed; (b) G. B. Cserép, A. Herner and P. Kele, Methods Appl. Fluoresc., 2015, 3, 042001 CrossRef PubMed.
- (a) L. G. Meimetis, J. C. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef CAS PubMed; (b) Y. Lee, W. Cho, J. Sung, E. Kim and S. B. Park, J. Am. Chem. Soc., 2018, 140, 974–983 CrossRef CAS PubMed; (c) S. J. Siegl, J. Galeta, R. Dzijak, A. Vázquez, M. Del Río-Villanueva, M. Dračniský and M. Vrabel, ChemBioChem, 2019, 20, 886–890 CrossRef CAS PubMed; (d) G. Knorr, E. Kozma, A. Herner, E. A. Lemke and P. Kele, Chem. – Eur. J., 2016, 22, 8972–8979 CrossRef CAS PubMed; (e) G. Knorr, E. Kozma, J. M. Schaart, K. Németh, G. Török and P. Kele, Bioconjugate Chem., 2018, 29, 1312–1318 CrossRef CAS PubMed.
- (a) O. Demeter, A. Kormos, C. Koehler, G. Mező, K. Németh, E. Kozma, L. Takacs, E. A. Lemke and P. Kele, Bioconjugate Chem., 2017, 28, 1552–1559 CrossRef CAS PubMed; (b) A. Kormos, C. Koehler, E. A. Fodor, Z. R. Rutkai, M. E. Martin, G. Mező, E. A. Lemke and P. Kele, Chem. – Eur. J., 2018, 24, 8841–8847 CrossRef CAS PubMed.
- E. Kozma, G. Estrada Girona, G. Paci, E. A. Lemke and P. Kele, Chem. Commun., 2017, 53, 6696–6699 RSC.
- (a) T. Iwaoka and M. Kondo, Bull. Chem. Soc. Jpn., 1974, 47, 980–986 CrossRef CAS; (b) R. W. Redmond and J. N. Gamlin, Photochem. Photobiol., 1999, 70, 391–475 CrossRef CAS PubMed.
- (a) T. Manju, N. Manoj, A. M. Braun and E. Oliveros, Photochem. Photobiol. Sci., 2012, 11, 1744–1755 RSC; (b) E. Baciocchi, T. Del Giacco, O. Lanzalunga, A. Lapi and D. Raponi, J. Org. Chem., 2007, 72, 5912–5915 CrossRef CAS PubMed.
- C. Liu, X. Jiao, Q. Wang, K. Huang, S. He, L. Zhao and X. Zeng, Chem. Commun., 2017, 53, 10727–10730 RSC.
- A. Gollmer, J. Arnbjerg, F. H. Blaikie, B. W. Pedersen, T. Breitenbach, K. Daasbjerg, M. Glasius and P. R. Ogilby, Photochem. Photobiol., 2011, 87, 671–679 CrossRef CAS PubMed.
- I. Kraljić and S. El Mohsni, Photochem. Photobiol., 1978, 28, 577–581 CrossRef.
- A. N. Butkevich, M. L. Bossi, G. Lukinavičius and S. W. Hell, J. Am. Chem. Soc., 2019, 141, 981–989 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of synthetic procedures and characterization data, spectroscopy experiments and protocols of actin-labeling and microscopy imaging experiments. See DOI: 10.1039/d0cc01512a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |