
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA method for designing a novel class of gold-containing molecules†
Christian A.
Celaya
 ab,
Mesías
Orozco-Ic
a,
Maria
Dimitrova
a,
Lukas N.
Wirz
a and
Dage
Sundholm
*a
ab,
Mesías
Orozco-Ic
a,
Maria
Dimitrova
a,
Lukas N.
Wirz
a and
Dage
Sundholm
*a
aDepartment of Chemistry, University of Helsinki, P.O. Box 55 (A.I. Virtanens plats 1), FIN-00014, Finland. E-mail: Dage.Sundholm@helsinki.fi
bDepartamento de Materiales de Baja Dimensionalidad, Instituto de Investigacíones en Materiales, UNAM, Apartado Postal 70-360, Ciudad de México 04510, Mexico
First published on 7th April 2020
Abstract
We propose a novel class of gold-containing molecules, which have been designed using conjugated carbon structures as templates. The sp-hybridized carbons of C2 moieties are replaced with a gold atom and one of the adjacent carbons is replaced by nitrogen. Applying the procedure to hexadehydro[12]annulene yields the well-known cyclic trinuclear gold(I) carbeniate complex. Planar, tubular and cage-shaped complexes can be obtained by taking similar sp-hybridized carbon structures as the starting point.
In crystalline form, cyclic trinuclear gold(I) carbeniate complexes exist as dimers, oligomers, or infinite molecular chains.1–4 Among them, trinuclear gold(I) carbeniate complexes [Au3(RN
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR′)3]n have been extensively studied both as solids and in solution as they can be used as emitting layers for phosphorescent organic light-emitting diodes or as phosphors in inorganic light-emitting diodes.5 The gold atoms in the [Au3(RNCR′)3]3 complexes are bridged by three RNCR′ units, forming an equilateral triangle. R is an organic substituent (e.g., methyl, n-butyl, cyclopentyl, phenyl) while R′ is often a hydroxyl group or a methoxy group.1–3,6–8 There are strong intermolecular aurophilic Au–Au interactions which determine the crystalline structure of the complex.9–13 The hexagonal polymorph of [Au3(MeNCOMe)3] in solution emits bright yellow light upon irradiation with near-UV light.1,4,6,7,14 Previous studies have shown that the dimer has a large triplet quantum yield,15 whereas the monomer does not exhibit phosphorescence, which suggests that the complex forms dimers also in solution.
CR′)3]n have been extensively studied both as solids and in solution as they can be used as emitting layers for phosphorescent organic light-emitting diodes or as phosphors in inorganic light-emitting diodes.5 The gold atoms in the [Au3(RNCR′)3]3 complexes are bridged by three RNCR′ units, forming an equilateral triangle. R is an organic substituent (e.g., methyl, n-butyl, cyclopentyl, phenyl) while R′ is often a hydroxyl group or a methoxy group.1–3,6–8 There are strong intermolecular aurophilic Au–Au interactions which determine the crystalline structure of the complex.9–13 The hexagonal polymorph of [Au3(MeNCOMe)3] in solution emits bright yellow light upon irradiation with near-UV light.1,4,6,7,14 Previous studies have shown that the dimer has a large triplet quantum yield,15 whereas the monomer does not exhibit phosphorescence, which suggests that the complex forms dimers also in solution.
In the cyclic trinuclear gold(I) carbeniate complexes, the gold atoms function as a bridge between conjugated organic moieties.16–19 Hakala and Pyykkö suggested that solid gold(I) cyanide (AuCN) can consist of 1,3,5-triazine (C3N3) rings connected to each other via gold(I) atoms, yielding linear C–Au–N bridges between the rings.19 They proposed that in solid state, gold(I) cyanide consists of two-dimensional sheets with the same C–Au–N motifs as in [Au3(RNCR′)3]3. Its structure resembles that of γ-graphyne which is a two-dimensional all-carbon structure consisting of benzene rings that are connected via –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– bridges.20–22 The mapping between the two structures is done by replacing the –C–CC–C– moieties of γ-graphyne with linear C–Au–N motifs.
C– bridges.20–22 The mapping between the two structures is done by replacing the –C–CC–C– moieties of γ-graphyne with linear C–Au–N motifs.
Here we propose that conjugated carbon structures with CC bonds can be used as templates for designing gold-containing molecules. This is the case of the aforementioned AuCN structure based on γ-graphyne, however other two-dimensional structures can be constructed using α-, β-, or δ-graphyne as a starting point.
The same principle can be used to design other gold-containing molecules with C–Au–N motifs using any all-carbon structure with conjugated –C–CC–C– moieties as a template. The size of the –CC– unit is about the same as the size of a gold atom connected via single bonds to two carbon atoms. When constructing the molecule, one of them has to be replaced by a nitrogen atom since gold has an odd number of electrons, unlike the –C–CC–C– moiety. Thus, when it is replaced by a C–Au–N unit, the obtained gold-containing molecule is a stable closed-shell molecule as the template molecule and the overall molecular structure is nearly the same.
The unsubstituted [Au3(HNCH)3] molecule can be obtained from hexadehydro[12]annulene by replacing the three –C–CC–C– units with three C–Au–N moieties. The planar molecules shown in Fig. 1 can be derived from γ-graphyne fragments.19 Calculations on the optimized molecular structures at the density functional theory level using the B3LYP23 functional including the D3-BJ24 dispersion correction yield large HOMO–LUMO gaps of 4.21 eV and 3.15 eV respectively.
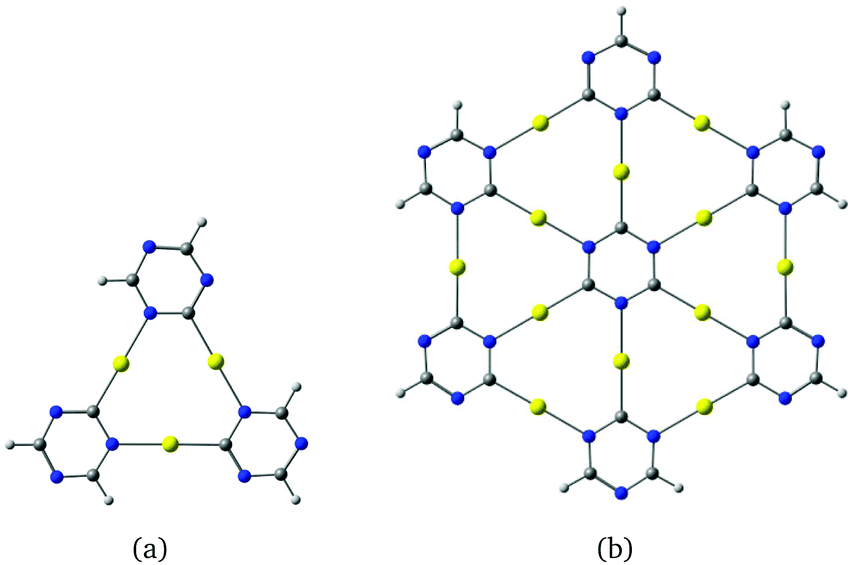 | ||
| Fig. 1 (a) The molecular structure of the cyclic trinuclear [Au3(N3(CH)2C)3] complex. An analogous molecule consisting of seven annelated [Au3(N3C3)] units is shown in (b). The dangling bonds of the outer carbons are terminated with hydrogens. The structures are obtained using γ-graphyne fragments as a template. The HOMO–LUMO gaps at the B3LYP level are 4.21 eV and 3.15 eV, respectively. | ||
The corresponding infinite two-dimensional sheet derived from γ-graphyne and its unit cell are shown in Fig. 2. It has a band gap of 1.35 eV at the PBE+vdW level. The computational details are given as ESI.†
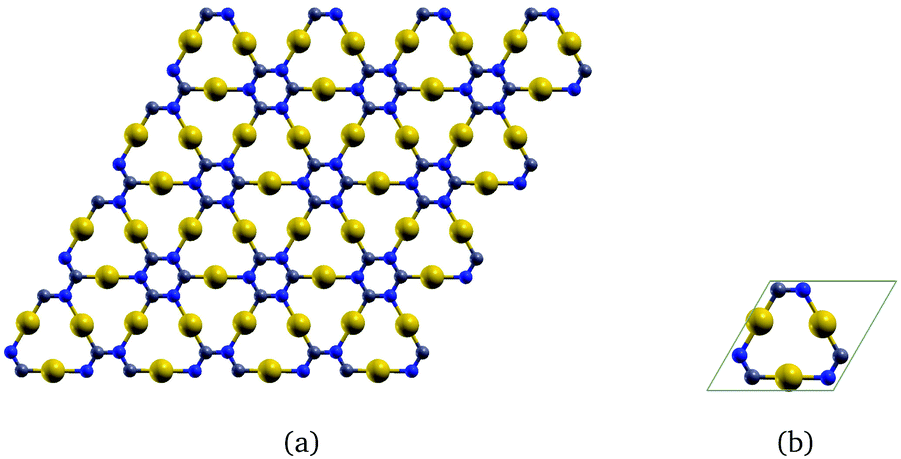 | ||
| Fig. 2 The gold-containing two-dimensional structure analogous to γ-graphyne is shown in (a) and its unit cell in (b). | ||
Molecules based on α-graphyne form six-membered rings with three carbon and three nitrogen atoms at the vertices and six gold atoms along the edges as shown in Fig. 3(a). The aurophilic interactions between the gold atoms cause slight bending in the C–Au–N units, which distorts the hexagonal structure. The planar coronene-like structure in Fig. 3(a) has a small HOMO–LUMO gap of 0.62 eV at the B3LYP level, suggesting that structures based on α-graphyne are not as stable as those based on γ-graphyne.
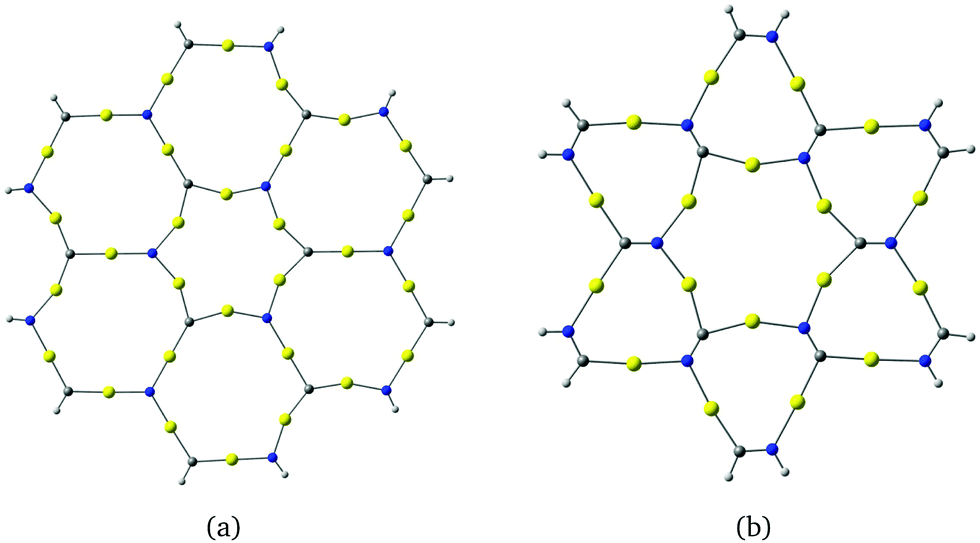 | ||
| Fig. 3 Molecular structures constructed from (a) α-graphyne and (b) β-graphyne. The HOMO–LUMO gaps calculated at the B3LYP level are (a) 0.62 eV and (b) 1.71 eV. | ||
The molecule in Fig. 3(b) is obtained by using a β-graphyne fragment as a template. Each [Au3(NC)3] moiety shares the NC groups with three annelated [Au3(NC)3] units. Its HOMO–LUMO gap of 1.71 eV at the B3LYP level is larger than for the coronene-like gold complex but smaller than that for the gold complex derived from γ-graphyne.
The two-dimensional structures based on α-graphyne are metallic. The two-dimensional structure in Fig. 4 constructed from β-graphyne has an optical gap of 1.23 eV at the PBE+vdW level.
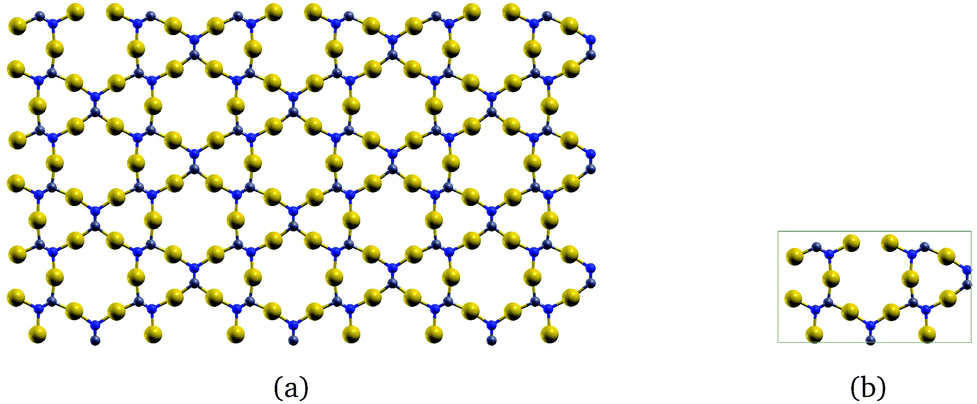 | ||
| Fig. 4 The gold-containing two-dimensional structure analogous to (a) β-graphyne and (b) its unit cell. | ||
The gold complexes shown in Fig. 5 are derived from δ-graphyne fragments. The molecular structures belonging to the C2v point group have large optical gaps of 4.23 eV and 4.85 eV, respectively. The C–Au–N moieties are linear because the shortest Au–Au distance is 3.40 Å, implying that the aurophilic interaction is rather weak. Optimization of the structures in Cs symmetry yielded bent structures as shown in the ESI.† The energy difference between the planar and bent structures is 0.5 kJ mol−1 and −3 kJ mol−1 for Fig. 5(a and b), respectively. Thus, the bent structure of Fig. 5(b) is slightly lower in energy. The small energy differences show that Fig. 5(a and b) are very flexible.
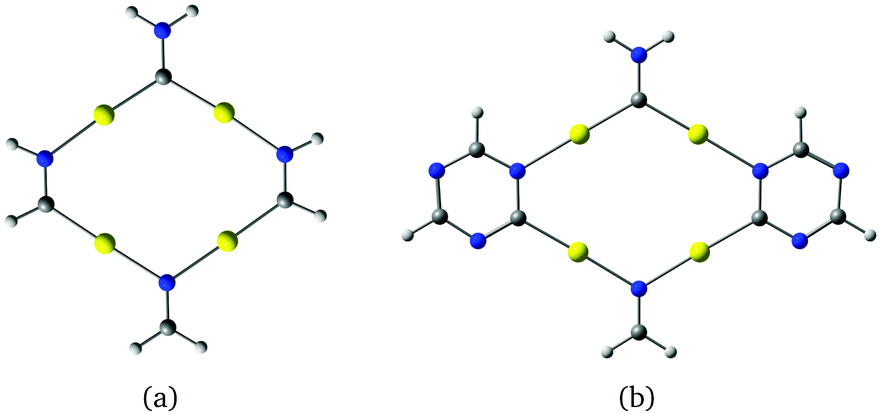 | ||
| Fig. 5 Molecular structures belonging to the C2v point group constructed from δ-graphyne. The HOMO–LUMO gaps calculated at the B3LYP level are (a) 4.23 eV and (b) 4.85 eV. | ||
The gold complex with fused triazine rings shown in Fig. 6 can be obtained using polycyclic antiaromatic hydrocarbons as the starting point.25 The structure resembles a six-pointed star with cyclic trinuclear gold(I) carbeniate moieties at the vertices connected via 1,3,5-triazine rings. In the optimized structure, the C–Au–N units are linear. The molecule has a very large HOMO–LUMO gap of 3.95 eV at the B3LYP level. The two-dimensional gold structure shown in Fig. 6(a) has an optical gap of 1.08 eV. It is obtained by using a unit cell based on the star-shaped molecule in Fig. 6(b) and periodic boundary conditions.
 | ||
| Fig. 6 The molecular structure constructed from a polycyclic antiaromatic hydrocarbon reminiscent of a β-graphyne fragment is shown in (a). The HOMO–LUMO gap at the B3LYP level is 3.95 eV. An example of a two-dimensional structure that is constructed based on the star-shaped molecule is shown in (b). | ||
Stacked cyclic trinuclear gold(I) carbeniate complexes are strongly phosphorescent in crystalline form and in solution. The phosphorescence originates from the presence of dimers which are bound through aurophilic interaction. In the crystalline form, the distances between the individual molecules alternate in a pairwise manner due to aurophilic interactions. In the solid state, the individual molecules are often shifted with respect to each other to enable stronger aurophilic interactions. The Au–Au distances in the excite-state dimers shrink significantly which leads to strong phosphorescence because the rate constants of intersystem crossing (ISC) and internal conversion (IC) from the triplet state of the dimers to the singlet ground state are very small.15 We investigated the double-decker dimers shown in Fig. 7 which exhibit prominent Au–Au interactions in addition to the van der Waals interactions between the triazine rings. The Au–Au distance between the monomers are 3.23 Å and 3.20 Å. The dimer structures of C3h and D3h symmetry of Fig. 7(a and b) are higher in energy and have smaller HOMO–LUMO gaps of 3.34 eV and 2.25 eV. The excitation energies of 3.24 eV and 3.18 eV calculated at the M06-2X level for Fig. 7(a and b) show that the lowest excited state in the near-UV region can be reached by the dipole-allowed HOMO–LUMO transition. The excitation accumulates electron density between the two molecules suggesting that the dimers have interesting optical properties as also found for stacked trinuclear gold(I) carbeniate complexes.1,6,7,14,15
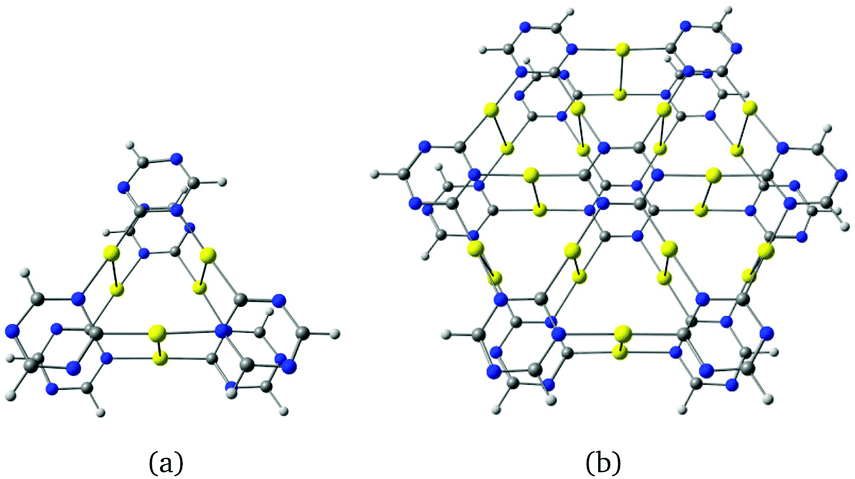 | ||
| Fig. 7 Double-decker dimer structures of gold-containing γ-graphyne fragments belonging to the D3 and D3d point groups. The HOMO–LUMO gaps at the B3LYP level are 3.55 eV and 2.68 eV for (a) and (b), respectively. | ||
The described procedure for designing gold-containing structures can also be applied to other graphyne-based carbon materials such as nanotubes and cages.20–22 The structures of the gold–nanotube fragment shown in Fig. 8 and the corresponding gold-containing nanotube in Fig. 9 were obtained using graphyne nanotubes as templates.26,27 The gold-containing cage structures in Fig. 10 were constructed using all-carbon cages with sp- and sp2-hybridized bonds (gaudienes) as a template.26–29
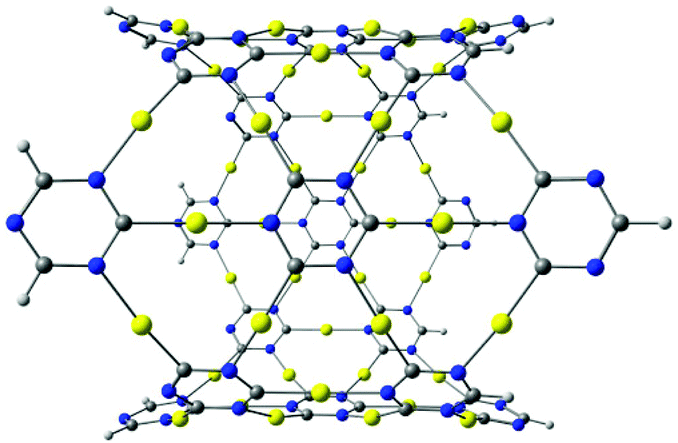 | ||
| Fig. 8 Gold–nanotube complex constructed by using a fragment of a carbon nanotube of γ-graphyne. The molecular structure belongs to the C4v point group and has a large HOMO–LUMO gap of 2.90 eV at the B3LYP level. | ||
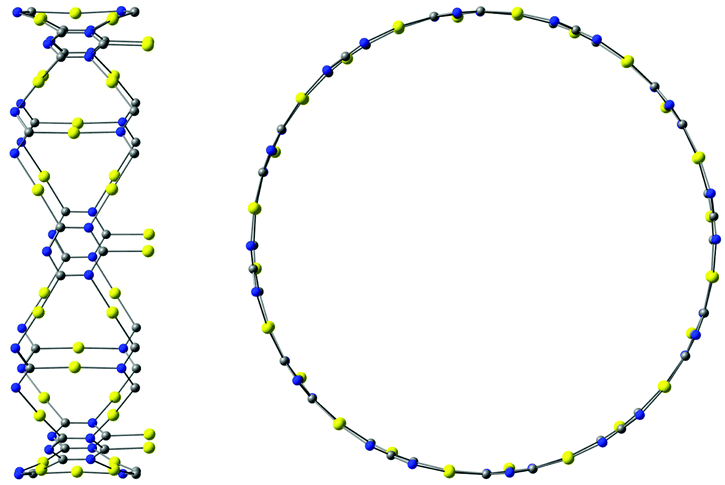 | ||
| Fig. 9 The unit cell of the gold–nanotube complex. The band gap is 1.83 eV at the PBE+vdW level. | ||
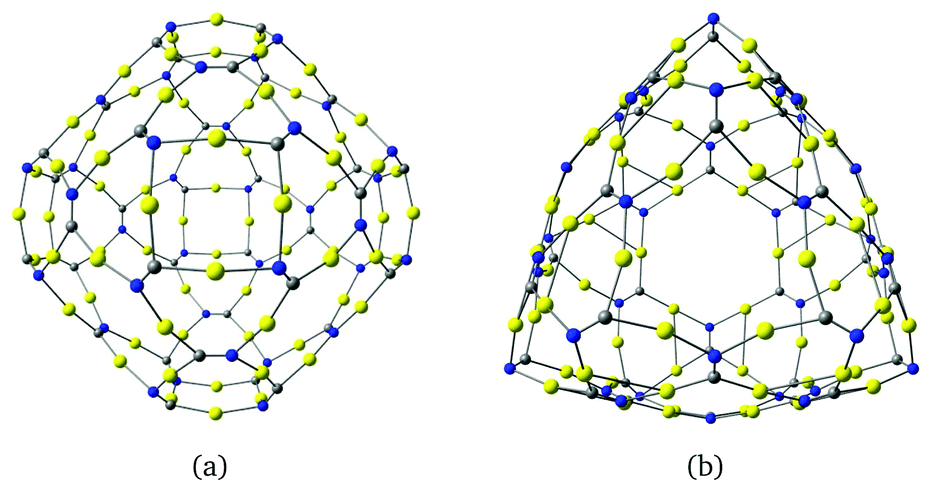 | ||
| Fig. 10 The molecular structures of (a) a gold–gaudiene cage (C36N36Au72) and (b) a gold–cage complex obtained from a β-graphyne cage. The structures belong to the Td point group. The HOMO–LUMO gaps of the molecules are 0.31 and 0.93 eV respectively at the B3LYP level. | ||
A small gold-containing cage (C12N12Au12) can be constructed from the recently proposed C48 quasi-fullerene.30 The molecular structure of the gold–cage complex shown in Fig. 11 belongs to the Td point group. It has a large HOMO–LUMO gap of 2.93 eV at the B3LYP level. The molecular structure of the cage consists of four 1,3,5-triazine rings pairwise connected with Au2 bridges.
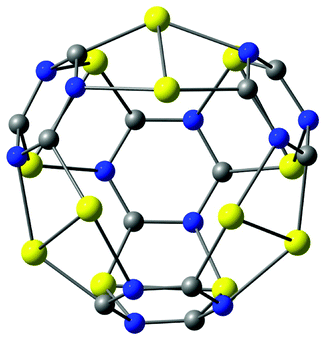 | ||
| Fig. 11 The molecular structure of a gold–cage complex obtained by using a recently suggested all-carbon C48 cage as template.30 Its HOMO–LUMO gap is 2.93 eV. | ||
In most of the studied structures, the C–Au–N motifs are linear or nearly linear with Au–C and Au–N the distances in the range of 1.99–2.06 Å and 2.09–2.14 Å, respectively. The Au–Au distances depend on the employed template. They are between 3.38 Å and 3.42 Å in the structures derived from γ-graphene, while slightly shorter Au–Au distances of 2.72 Å and 2.89 Å from are obtained for molecules that are constructed from α-graphyne and β-graphyne, because the C–Au–N motifs at the edges are slightly nonlinear. The infinite sheets exhibit Au–Au distances of the same length as obtained for the corresponding basic molecular structures. Some of the C–Au–N motifs of the cages are non-linear with Au–Au distances that are shorter than 3.0 Å. However, the Au–Au distances of the cages are in general in the range of 3.25–3.35 Å.
In this work, we have shown that the molecular structure of [Au3(HNCH)3] can be derived from hexadehydroannulene via substitution of the –C–CC–C– moieties with C–Au–N motifs. More general gold-containing molecules with C–Au–N motifs can be obtained using various graphyne-based structures as template. The method is suitable for designing large gold complexes, including nanotubes and cages. The gold atoms in the obtained complexes lie close to each other, allowing for aurophilic interactions. The dimers of the novel gold-containing molecules might have interesting phosphorescence properties as those observed for cyclic trinuclear gold(I) complexes in crystalline form and in solution.
This work has been supported by the Academy of Finland through project 314821. CSC – the Finnish IT Center for Science and the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533) are acknowledged for computer time. We thank Magnus Ehrnrooth foundation and the Swedish Cultural Foundation in Finland for financial support. C. A.-C. and M. O.-I. also thank to Conacyt for their PhD fellowships. LNW thanks Alexander von Humboldt Foundation for a Feodor Lynen fellowship.
Conflicts of interest
There are no conflicts to declare.References
- R. N. McDougald, Jr., B. Chilukuri, H. Jia, M. R. Perez, H. Rabaâ, X. Wang, V. N. Nesterov, T. R. Cundari, B. E. Gnade and M. A. Omary, Inorg. Chem., 2014, 53, 7485–7499 CrossRef PubMed.
- R. L. White-Morris, M. M. Olmstead, F. Jiang, D. S. Tinti and A. L. Balch, J. Am. Chem. Soc., 2002, 124, 2327–2336 CrossRef CAS PubMed.
- C. Yang, M. Messerschmidt, P. Coppens and M. A. Omary, Inorg. Chem., 2006, 45, 6592–6594 CrossRef CAS PubMed.
- H. Rabaâ, M. A. Omary, S. Taubert and D. Sundholm, Inorg. Chem., 2018, 57, 718–730 CrossRef PubMed.
- Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908–912 CrossRef CAS PubMed.
- J. C. Vickery, M. M. Olmstead, E. Y. Fung and A. L. Balch, Angew. Chem., Int. Ed. Engl., 1997, 36, 1179–1181 CrossRef CAS.
- R. L. White-Morris, M. M. Olmstead, S. Attar and A. L. Balch, Inorg. Chem., 2005, 44, 5021–5029 CrossRef CAS PubMed.
- K. Winkler, M. Wysocka-Zołopa, K. Rećko, L. Dobrzyński, J. C. Vickery and A. L. Balch, Inorg. Chem., 2009, 48, 1551–1558 CrossRef CAS PubMed.
- H. Schmidbaur, Gold Bull., 1990, 23, 11–21 CrossRef CAS.
- P. Pyykkö, Chem. Rev., 1997, 97, 597–636 CrossRef PubMed.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- H. Schmidbaur and H. G. Raubenheimer, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.201916255.
- E. Y. Fung, M. M. Olmstead, J. C. Vickery and A. L. Balch, Coord. Chem. Rev., 1998, 171, 151–159 CrossRef CAS.
- J. Greiner, R. R. Valiev and D. Sundholm, Phys. Chem. Chem. Phys., 2020 Search PubMed , submitted.
- P. Pyykkö and P. Zaleski-Ejgierd, Phys. Chem. Chem. Phys., 2008, 10, 114–120 RSC.
- P. Zaleski-Ejgierd, M. Hakala and P. Pyykkö, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 094104 CrossRef.
- P. Pyykkö, M. O. Hakala and P. Zaleski-Ejgierd, Phys. Chem. Chem. Phys., 2007, 9, 3025–3030 RSC.
- M. O. Hakala and P. Pyykkö, Chem. Commun., 2006, 2890–2892 RSC.
- R. H. Baughman, H. Eckhardt and M. Kertesz, J. Chem. Phys., 1987, 87, 6687–6699 CrossRef CAS.
- Q. Li, Y. Li, Y. Chen, L. Wu, C. Yang and X. Cui, Carbon, 2018, 136, 248–254 CrossRef CAS.
- J. Kang, Z. Wei and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 2692–2706 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Jusélius and D. Sundholm, Phys. Chem. Chem. Phys., 2008, 10, 6630–6634 RSC.
- V. R. Coluci, S. F. Braga, S. B. Legoas, D. S. Galvão and R. H. Baughman, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 035430 CrossRef.
- B. Kang and J. Y. Lee, Carbon, 2015, 84, 246–253 CrossRef CAS.
- D. Sundholm, Phys. Chem. Chem. Phys., 2013, 15, 9025–9028 RSC.
- D. Sundholm, L. N. Wirz and P. Schwerdtfeger, Nanoscale, 2015, 7, 15886–15894 RSC.
- C. A. Celaya, J. Muñiz and L. E. Sansores, Comput. Theor. Chem., 2017, 1117, 20–29 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The computational methods and the Cartesian coordinates of the optimized molecular structures. See DOI: 10.1039/d0cc01227k |
| This journal is © The Royal Society of Chemistry 2020 |