
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of benzyl sulfides via substitution reaction at the sulfur of phosphinic acid thioesters†
Yoshitake
Nishiyama
 ,
Takamitsu
Hosoya
and
Suguru
Yoshida
*
,
Takamitsu
Hosoya
and
Suguru
Yoshida
*
Laboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan. E-mail: s-yoshida.cb@tmd.ac.jp
First published on 16th April 2020
Abstract
An ambident electrophilicity of phosphinic acid thioesters is disclosed. Unexpected carbon–sulfur bond formation took place in the reaction between phosphinic acid thioesters and benzyl Grignard reagents. The developed method for benzyl sulfides has a wide substrate scope and was applicable for the synthesis of a drug analog.
Sulfides play significant roles in various fields such as medicinal chemistry, agrochemistry, and materials science, and thus many synthetic methods for sulfides have been developed.1,2 Especially, a group of benzyl sulfides have attracted synthetic chemists because of their utility. Benzyl sulfides are used not only as protected thiols3 or precursors of sulfenyl chlorides,4 but also as important structures of bioactive compounds such as sulconazole5 or dosulepin.6 However, the synthesis of benzyl sulfides depends almost only on the nucleophilic attack of thiols to alkyl halides, often suffering from the limited scope as well as the unpleasant odor of thiols, especially of benzyl mercaptans. Herein, we disclose a new synthetic method for benzyl sulfides via the reaction between benzyl Grignard reagents and phosphinic acid thioesters, which have a phosphorus–sulfur bond.
Previously, we found that Grignard reagents attacked the phosphorus atoms7,8 of phosphonic acid dithioesters to give phosphinic acid thioesters, which also reacted with Grignard reagents at higher temperature by further substitution reactions on the phosphorus atoms to provide a wide range of phosphine oxides.9 In the course of further investigation for the reactivity of phosphinic acid thioesters, we unexpectedly found that these compounds behaved as two-faced “ambident” electrophiles.10,11 Thus, while S-(4-tolyl) diphenylphosphinothioate (1a) reacted with phenylmagnesium bromide at the phosphorus atom to smoothly give triphenylphosphine oxide (2a), treatment of 1a with a benzyl Grignard reagent furnished the target phosphine oxide 2b only in low yield, along with benzyl sulfide 3a as the major product (Fig. 1A). This result suggested a possibility that benzyl sulfide 3a was obtained directly by the reaction of the benzyl Grignard reagent at the sulfur atom of phosphinic acid thioester 1a,12 contrary to our previous report that C–P bond formation took place using alkyl and aryl Grignard reagents.
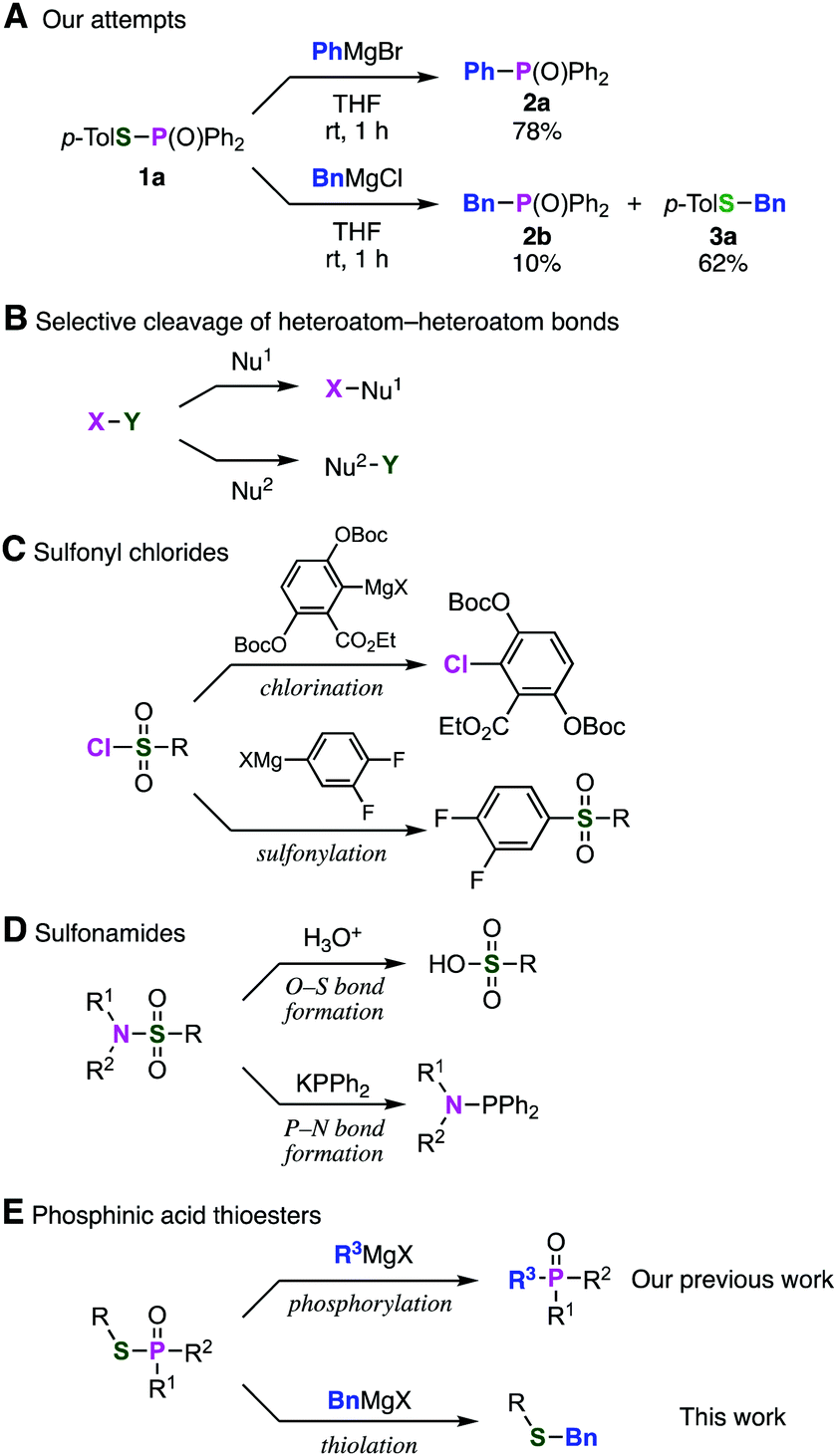 | ||
| Fig. 1 (A) Abnormal reactivity of a benzyl Grignard reagent toward a phosphinic acid thioester. (B) Nucleophilic substitution reaction of heteroatom–heteroatom compounds. (C) Sulfonyl chlorides. (D) Sulfonamides. (E) Phosphinic acid thioesters. | ||
To date, several reports switching the bond-forming site have been reported in the substitution reaction using electrophiles bearing a heteroatom–heteroatom bond depending on the nucleophiles examined (Fig. 1B). For instance, reactions of sulfonyl chlorides with aryl Grignard reagents result in C–Cl bond formation to afford aryl chlorides10a or C–S bond formation to afford sulfones10b according to the type of Grignard reagents (Fig. 1C). Two types of substitution reactions of sulfonamides have been also developed; P–N bond formation occurred by the reaction with phosphide anion,10c while various nucleophiles such as water attacked selectively on the sulfur atom (Fig. 1D). However, limited numbers of the ambident electrophiles were reported so far and controlling the ambident electrophilicity is not easy.11 These limited but significant site-selective reactions motivated us to examine the C–S bond-forming reaction of phosphinic acid thioesters with benzyl Grignard reagents in terms of the site-selectivity for efficient synthesis of benzyl sulfides (Fig. 1E).
To improve the site-selectivity and to clarify the generality of the reaction, a variety of phosphinic acid thioesters 1, which have different substituents on the phosphorus atoms, were subjected to the reaction with a benzyl Grignard reagent (Table 1). Changing the substituents on the phosphorus atom from a phenyl group to alkyl groups such as butyl or cyclohexyl group decreased the selectivities, affording the desired sulfide 3a in low yields (entries 2 and 3). On the other hand, S-benzylation of S-(4-tolyl) di(2-tolyl)phosphinothioate (1e) proceeded smoothly to afford benzyl sulfide 3a in the highest yield among tested, accompanying with a small amount of phosphine oxide 2f (entry 4). Thus, we successfully controlled the ambident electrophilicity of phosphinic acid thioesters by changing the substrates, enabling efficient S-benzylation in terms of the yield and selectivity. With a bulkier mesityl group, the selectivity rather degraded (entry 5). In addition, when using thiophosphoric acid ester 1f, benzyl sulfide 3a was obtained with perfect selectivity (entry 6).
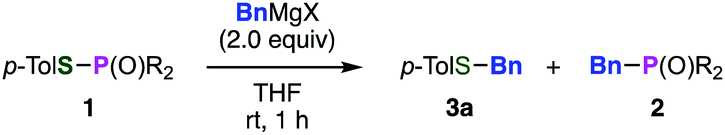
Benzylation of phosphinic acid thioester 1e using other benzyl Grignard reagents allowed for efficient preparation of various benzyl sulfides (Fig. 2). Indeed, 4-chlorobenzylation and 1-naphthylmethylation of 1e proceeded smoothly to give 3b and 3c, respectively. A benzyl Grignard reagent bearing a methyl group at the benzylic position was also applicable to the C–S bond formation, affording benzyl sulfide 3d in high yield. Furthermore, bulky (2-phenylpropan-2-yl)magnesium chloride successfully reacted with 1e to furnish sulfide 3e, which is not easy to synthesize by the conventional method using thiols and alkyl halides. In addition, a 1-phenylvinyl Grignard reagent did not participate in the C–S bond forming reaction.
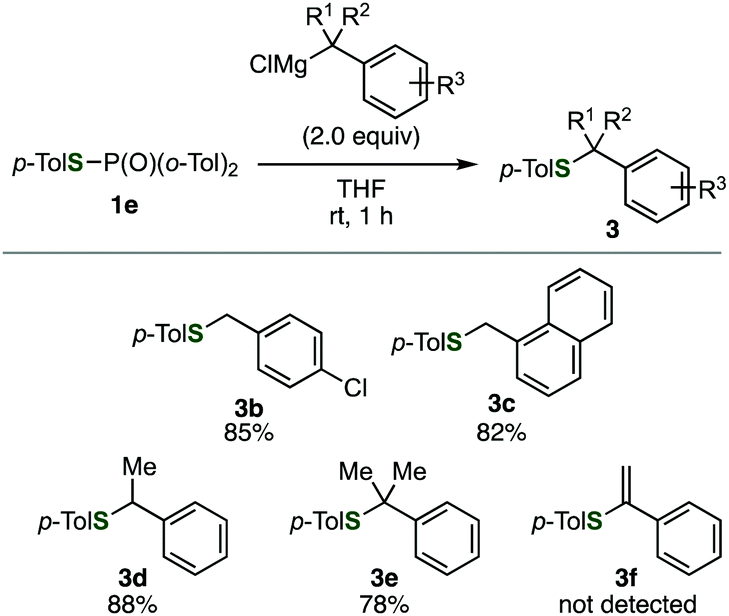 | ||
| Fig. 2 Reactions of a phosphinic acid thioester with various benzyl Grignard reagents. | ||
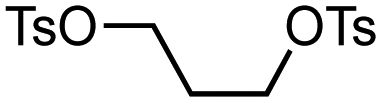
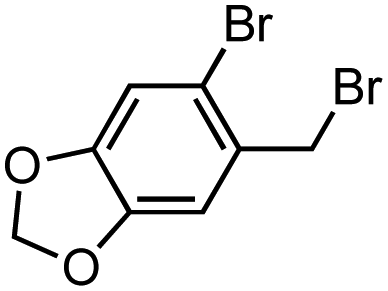
The use of readily available thiophosphinic acid 4 enabled easy access to various alkyl di(2-tolyl)phosphinothioates 1, which were also benzylated by treating with a benzyl Grignard reagent (Table 2). Alkylation of 4 with a variety of alkyl halides afforded phosphinothioates 1g–k without damaging functional groups such as amino, bromo, and tosyloxy groups (entries 1–4). When the synthesized phosphinic acid thioesters 1g–j were treated with benzylmagnesium chloride, S-benzylation proceeded smoothly to give benzyl sulfides 3g–j. Notably, a bromo group and a tosyloxy group were tolerated under the benzylation conditions. On the other hand, S-benzylation of bulky α-methylbenzyl ester prepared from 1k did not occur (entry 5).
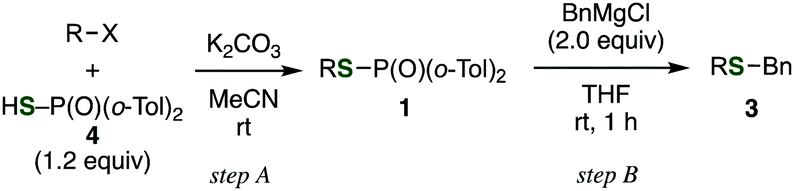
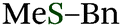

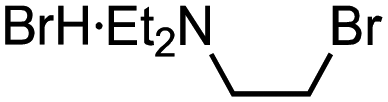

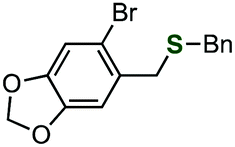
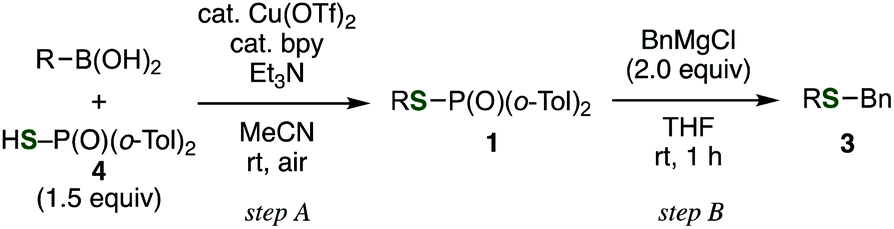
Not only alkyl phosphinothioates, an array of aryl phosphinothioates were also prepared via the Chan–Lam–Evans-type deborylthiolation (Table 3).13 Treatment of the corresponding boronic acid with thiophosphinic acid 4 in the presence of triethylamine and a catalytic amount of copper(II) triflate and bipyridyl afforded a variety of phosphinic acid thioesters, such as substituted phenyl thioesters 1l–p (entries 1–5), 2-naphthyl thioester 1q (entry 6), 3-thienyl thioester 1r (entry 7), and styryl thioester 1s (entry 8). Benzylation of these phosphinic acid thioesters proceeded smoothly to give corresponding benzyl sulfides 3l–s (entries 1–8). It is noteworthy that benzyl 3-thienyl sulfide (3r) and benzyl styryl sulfide (3s) were generally difficult to synthesize because of the lack of the availability of the corresponding thiols. Thus, these results clearly demonstrated the utility of this method via the Chan–Lam–Evans-type deborylthiolation.

We next turned our attention to the mechanism of this unusual S-benzylation (Fig. 3). Plausible reaction mechanism is shown in Fig. 3A including direct substitution reaction of phosphinic acid thioesters 1 with a benzyl Grignard reagent on the sulfur atom (path a). Other reaction mechanisms involving the ligand coupling of pentavalent organophosphorus intermediate I (path b) or the single-electron transfer (SET) (path c) could not be excluded at this stage. Considering that C–P bond formation took place using various alkyl or aryl Grignard reagents probably through pentavalent phosphorus intermediates, the S-benzylation via P-benzylation and subsequent ligand coupling14 of the resulting pentavalent phosphorus intermediate I to give the benzyl sulfide is also possible (path b). Another possibility is shown as path c through the SET mechanism, followed by S-benzylation between thiyl radical III and benzyl radical to give the benzyl sulfide.
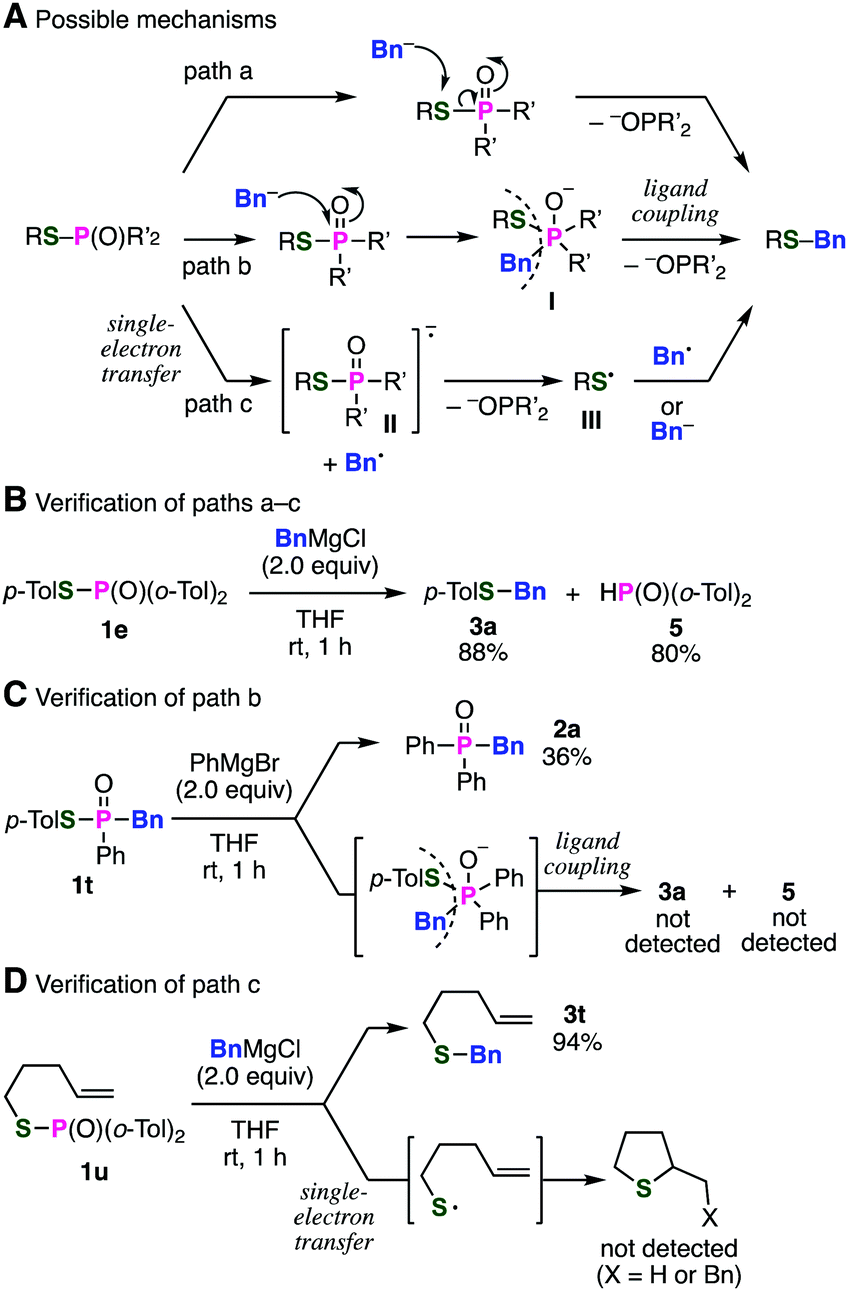 | ||
| Fig. 3 Reactions of a phosphinic acid thioester with various benzyl Grignard reagents. | ||
To gain insight into the mechanism, we at first examined the products of the reaction between di(2-tolyl)phosphinic acid thioester 1e and a benzyl Grignard reagent (Fig. 3B). The result showed that di(2-tolyl)phosphine oxide (5) was obtained after the protonation of the phosphinate anion in paths a–c as a leaving group,15 along with benzyl sulfide 3a. Then, we next treated phosphinic acid thioester 1t, equipped with a benzyl group in advance, with phenylmagnesium bromide to verify the possibility of path b involving the pentavalent phosphorus intermediate having benzyl and sulfanyl groups (Fig. 3C). As a result, phosphine oxide 2a was obtained similar to our previous report9 without generation of benzyl sulfide 3a and diarylphosphine oxide 5, suggesting that path b involving reductive elimination of the phosphorus is not plausible. In order to verify the possibility of the SET mechanism, S-(4-penten-1-yl) di(2-tolyl)phosphinic acid thioester (1u) was subjected to the S-benzylation reaction (Fig. 3D). While benzyl 4-penten-1-yl sulfide (3t) was obtained in high yield, any radical-cyclization products16 were not detected in the reaction.17 From these results, benzyl sulfides would be generated via direct attack of benzyl Grignard reagents on the sulfur atoms of phosphinic acid thioesters.
By using the developed benzyl sulfide synthesis, we synthesized an analog of tiopinac, which is a highly potent anti-inflammatory, analgesic, and anti-pyretic agent.18 Treatment of phosphinic acid thioester 1e with 2-bromo-5-methoxybenzylmagnesium bromide gave benzyl sulfide 3u without damaging the bromo group (Scheme 1). Palladium-catalyzed carbonylation of phenyl bromide in methanol afforded methyl ester 6. Hydrolysis of the ester moiety, and following intramolecular Friedel–Crafts cyclization19 of the resulting carboxylic acid provided tiopinac analog 7 in shorter steps.
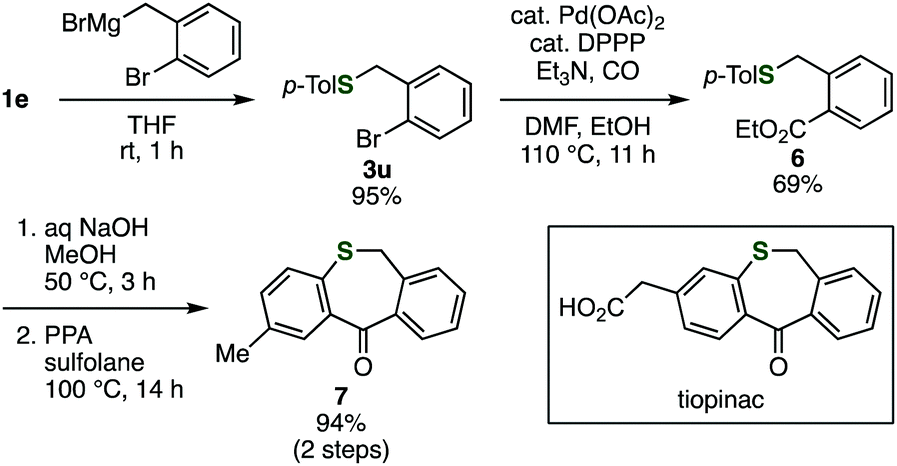 | ||
| Scheme 1 Synthesis of tiopinac analog. DPPP = 1,3-bis(diphenylphosphino)propane. PPA = polyphosphoric acid. | ||
In summary, we found an ambident electrophilicity of phosphinic acid thioesters. While the reactions using various alkyl and aryl Grignard reagents results in C–P bond formations, S-benzylation took place by treatment of phosphinic acid thioesters with benzyl Grignard reagents, probably via the direct nucleophilic S-benzylation reaction at the sulfur atom. The developed S-benzylation reaction allowed for synthesizing a variety of benzyl sulfides involving an intermediate of a drug analog from phosphonic acid thioesters and Grignard reagents. Further studies of ambident electrophiles based on the heteroatom chemistry are now underway.
This work was supported by JSPS KAKENHI Grant Numbers JP19K05451 (C; S. Y.), JP19K15711 (Young scientists; Y. N.), JP18H02104 (B; T. H.), and JP18H04386 (Middle Molecular Strategy; T. H.); the Naito Foundation (S. Y.); the Japan Agency for Medical Research and Development (AMED) under Grant Number JP19am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS); and the Cooperative Research Project of Research Center for Biomedical Engineering.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) T. Yamamoto and K. Takimiya, J. Am. Chem. Soc., 2007, 129, 2224 CrossRef CAS PubMed; (b) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed; (c) N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246 RSC.
- (a) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed; (b) A. Ghaderi, Tetrahedron, 2016, 72, 4758 CrossRef CAS; (c) Z. Qiao and X. Jiang, Org. Biomol. Chem., 2017, 15, 1942 RSC; (d) T. Matsuzawa, S. Yoshida and T. Hosoya, Tetrahedron Lett., 2018, 59, 4197 CrossRef CAS.
- J. L. Wood and V. du Vigneaud, J. Biol. Chem., 1939, 131, 267 CAS.
- N. Kharasch and R. B. Langford, Org. Synth., 1964, 44, 47 CrossRef CAS.
- F. C. Odds, C. E. Webster and A. B. Abbott, J. Antimicrob. Chemother., 1984, 14, 105 CrossRef CAS PubMed.
- E. Richelson and M. Pfenning, Eur. J. Pharmacol., 1984, 104, 277 CrossRef CAS PubMed.
- For substitution reactions of compounds bearing phosphorus–heteroatom bonds, see: (a) C. Lopin, G. Gouhier, A. Gautier and S. R. Piettre, J. Org. Chem., 2003, 68, 9916 CrossRef CAS PubMed; (b) A. Sato, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2005, 44, 1694 CrossRef CAS PubMed; (c) T. Wada, A. Kondoh, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 1155 CrossRef CAS PubMed; (d) M.-C. Lamas and A. Studer, Org. Lett., 2011, 13, 2236 CrossRef CAS PubMed; (e) Y. Sato, S.-i. Kawaguchi, A. Nomoto and A. Ogawa, Angew. Chem., Int. Ed., 2016, 55, 9700 CrossRef CAS PubMed; (f) T. Murai, Y. Maekawa, Y. Hirai, K. Kuwabara and M. Minoura, RSC Adv., 2016, 6, 15180 RSC; (g) Y. Maekawa, T. Maruyama and T. Murai, Org. Lett., 2016, 18, 5264 CrossRef CAS PubMed; (h) Y. Maekawa, K. Kuwabara, A. Sugiyama, K. Iwata, T. Maruyama and T. Murai, Chem. Lett., 2017, 46, 1068 CrossRef; (i) Y. Okugawa, Y. Hayashi, S. Kawauchi, K. Hirano and M. Miura, Org. Lett., 2018, 20, 3670 CrossRef CAS PubMed; (j) R. J. Reddy, A. Shankar and A. H. Kumari, Asian J. Org. Chem., 2019, 8, 2269 CrossRef CAS.
- D. J. Jones, E. M. O’Leary and T. P. O’Sullivan, Tetrahedron Lett., 2018, 59, 4279 CrossRef CAS.
- Y. Nishiyama, Y. Hazama, S. Yoshida and T. Hosoya, Org. Lett., 2017, 19, 3899 CrossRef CAS PubMed.
- (a) N. Hendricks, P. Himmelberg and E. Fossum, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2007, 48, 1004 CAS; (b) W. Lin, O. Baron and P. Knochel, Org. Lett., 2006, 8, 5673 CrossRef CAS PubMed; (c) S. Yoshida, K. Igawa and K. Tomooka, J. Am. Chem. Soc., 2012, 134, 19358 CrossRef CAS PubMed.
- For examples of studies on ambident electrophiles, see: (a) C. C. P. Wagener, A. M. Modro and T. A. Modro, J. Phys. Org. Chem., 1991, 4, 516 CrossRef CAS; (b) P. Tundo, L. Rossi and A. Loris, J. Org. Chem., 2005, 70, 2219 CrossRef CAS PubMed.
- For recent reviews of masked thiols as electrophilic sulfur surrogates, see: (a) H. Liu and X. Jiang, Chem. – Asian J., 2013, 8, 2546 CrossRef CAS PubMed; (b) Z. Qiao and X. Jiang, Org. Biomol. Chem., 2017, 15, 1942 RSC; (c) P. Mampuys, C. R. McElroy, J. H. Clark, R. V. A. Orru and B. U. W. Maes, Adv. Synth. Catal., 2020, 362, 3 CrossRef CAS.
- To our knowledge, this is the first example of Chan–Lam–Evans-type coupling between a boronic acid and a thiophosphinic acid. For a related reaction, Tang and coworkers reported one-pot synthesis of S-aryl phosphorothioates probably including a Chan–Lam–Evans-type process: J. Xu, L. Zhang, X. Li, Y. Gao, G. Tang and Y. Zhao, Org. Lett., 2016, 18, 1266 CrossRef CAS PubMed.
- Y. Uchida, K. Onoue, N. Tada, F. Nagao and S. Oae, Tetrahedron Lett., 1989, 30, 567 CrossRef CAS.
- (a) C. Tejo, J. H. Pang, D. Y. Ong, M. Oi, M. Uchiyama, R. Takita and S. Chiba, Chem. Commun., 2018, 54, 1782 RSC; (b) T. Yano, T. Kawasaki, T. Yuhki, N. Ishida and M. Murakami, Org. Lett., 2018, 20, 1224 CrossRef CAS PubMed.
- M. P. Winters, C. A. Teleha, F.-A. Kang, D. McComsey, J. C. O’Neill, C. Hou, T. Kirchner, P. Wang, D. Johnson and Z. Sui, Bioorg. Med. Chem. Lett., 2014, 24, 2137 CrossRef CAS PubMed.
- The possibility of the reaction pathway involving the benzyl radical generated only from benzylmagnesium chloride by the dissociation was not excluded.
- J. Ackrell, Y. Antonio, F. Franco, R. Landeros, A. Leon, J. M. Muchowski, M. L. Maddox, P. H. Nelson, W. H. Rooks, A. P. Roszkowski and J. M. Muchowski, J. Med. Chem., 1978, 21, 1035 CrossRef CAS PubMed.
- S. A. Laufer, G. M. Ahrens, S. C. Karcher, J. S. Hering and R. Niess, J. Med. Chem., 2006, 49, 7912 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization for new compounds including NMR spectra. See DOI: 10.1039/d0cc02039g |
| This journal is © The Royal Society of Chemistry 2020 |